
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in formic acid electro-oxidation: from the fundamental mechanism to electrocatalysts
Zhongying
Fang
 ab and
Wei
Chen
*ab
ab and
Wei
Chen
*ab
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, Jilin, China. E-mail: weichen@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei, 230029, Anhui, China
First published on 9th November 2020
Abstract
Direct formic acid fuel cells have attracted significant attention because of their low fuel crossover, high safety, and high theoretical power density among all the proton-exchange membrane fuel cells. Much effort has been devoted to the study of formic acid oxidation, including the reaction processes and electrocatalysts. However, as a model reaction, the anodic electro-oxidation process of formic acid is still not very clear, especially regarding the confirmation of the intermediates, which is not helpful for the design and synthesis of high-performance electrocatalysts for formic acid oxidation or conducive to understanding the reaction mechanisms of other small fuel molecules. Herein, we briefly review the recent advances in investigating the mechanism of formic acid electro-oxidation and the basic design concepts of formic acid oxidation electrocatalysts. Rather than an exhaustive overview of all aspects of this topic, this mini-review mainly outlines the progress of this field in recent years.
 Zhongying Fang | Zhongying Fang received her B.S. degree from the Department of Chemical and Chemical Engineering, Northeast Forestry University, China in 2017. She is currently pursuing a Ph.D. degree in electroanalytical chemistry under the supervision of Prof. Wei Chen at Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. Her current research includes the design, synthesis, and structure-dependent catalytic property studies of catalysts for electrochemical energy storage and conversion. |
 Wei Chen | Wei Chen received his Ph.D. in electrochemistry from Xiamen University in 2003. Following his graduate studies, he began working as a postdoctoral associate in the area of synthesis and the property studies of metal nanoclusters at the University of California-Santa Cruz, USA. He is currently a full professor at Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. His research interests include electroanalytical chemistry, surface electrochemistry, electrocatalysis, photoelectrocatalysis, and the controlled synthesis, characterization, and application of nanomaterials in energy storage and conversion. |
1. Introduction
Environmental pollution and the pending energy crisis are two major problems that need to be solved urgently.1 Due to the outstanding advantages of cleanliness and high energy density, proton-exchange membrane fuel cells (PEMFCs) can effectively aid alleviating the above-mentioned problems.2,3 However, although PEMFCs have been studied intensively since the 1960s, inherent limitations still exist.4 For example, the anode has the following prominent problems: unsatisfactory safety, high fuel crossover, high cost of catalysts, low durability, and a great loss of power density and cell efficiency due to the polarization and slow reaction kinetics of organic small molecular fuels in the electro-oxidation process.5,6 Recent studies have demonstrated that formic acid can be used as a suitable anodic fuel to overcome the safety problems and fuel crossover issues.7–9 The main reasons for this are the following. First, formic acid can be used as an additive in food and medicine and is not flammable, and therefore it is safe to store and transport.10,11 Second, formic acid can be ionized into its formic acid root, which is mutually repulsive with the sulfonic acid root of Nafion film, thereby partially hindering the crossover of formic acid and thus reducing the probability of producing mixed potential at the cathode. The low formic acid crossover allows using a high concentration of formic acid as fuel, which compensates for the lower energy density of formic acid compared to other liquid fuels.12–14 Moreover, the theoretical open circuit voltage (OCV) of direct formic acid fuel cells (DFAFCs) is 1.48 V, which is higher than other existing PEMFCs.15,16 Such a high voltage can bring high power density, which enhances the PEMFC's practical application in the fields of electric vehicles, portable electronic equipment, and others.17,18At present, the cost, durability and catalytic activity of the catalysts are mutually restrictive problems that commonly exist in almost all PEMFCs.19,20 Therefore, the design and synthesis of high-performance electrocatalysts are one area that represents one of the most critical research content aspects in fuel cells.21,22 The effective design and preparation of an excellent electrocatalyst for formic acid oxidation should be based on the following two aspects. Firstly, the reaction mechanism of formic acid oxidation should be explored, including the reaction path, reaction intermediates, species adsorption states, and adsorption configurations. Based on the understanding of the reaction mechanism, the compositions and surface structures of the electrocatalysts can then be rationally designed and adjusted. Although formic acid is a relatively simple type of fuel molecule among the commonly used organic fuels for fuel cells, the electro-oxidation process is still complicated. During the past decades, much work has been done to elucidate the reaction mechanism. To this end, single-crystal catalysts with different surface planes have been used as model catalysts for formic acid. Meanwhile, different in situ spectroscopic techniques combined with electrochemical methods have been widely used to detect the surface adsorbed species during formic acid oxidation, with an aim to elucidate the possible reaction mechanism.23
In recent years, with the development of in situ and operando techniques, great advances have been achieved in the above-mentioned two aspects of formic acid oxidation, i.e., the reaction mechanism and electrocatalysts. However, a detailed summary of the advance in the reaction mechanisms and electrocatalysts related to formic acid electro-oxidation (FAEO) is insufficient but imperative.24,25 Consequently, in this mini-review, the work and progress on the electrocatalytic oxidation mechanism, electrocatalyst design, and synthesis for formic acid oxidation in recent years are summarized. Furthermore, the fundamental chemistry of DFAFCs and FAEO are also discussed. Finally, a brief conclusion is provided. We hope this review is helpful for readers interested in this field to understand the current research situation of formic acid electro-oxidation.
2. Fundamental electrochemistry of DFAFCs and FAEO
PEMFC is an energy conversion device that converts chemical energy from broken bonds in small fuel molecules into electrical energy.26,27 For PEMFCs, the energy density, power density, and cycles are the important performance-evaluation parameters.3 Energy density is defined as the amount of energy stored and released by devices of the same mass/volume, so the theoretical energy density of DFAFCs can be described as: n × F × OCV × (MW)−1. Based on its reaction electron-transfer number (n = 2), the Faraday constant (F = 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), open circuit potential (OCV = 1.48 V), and molecular weight (MW = 0.04603 kg mol−1), formic acid has the theoretical energy density of 1725 W h kg−1 and 2104 W h L−1 (the liquid density is 1.22 kg L−1). On the other hand, power density is the rate at which energy is stored and released by devices of the same mass/volume, with the measuring units of W kg−1 and W L−1. Meanwhile, power is equal to the product of the current and voltage, and at a constant current, power is directly proportional to the voltage. The energy density and power density of different energy storage systems are compared in Fig. 1a.28
485 C mol−1), open circuit potential (OCV = 1.48 V), and molecular weight (MW = 0.04603 kg mol−1), formic acid has the theoretical energy density of 1725 W h kg−1 and 2104 W h L−1 (the liquid density is 1.22 kg L−1). On the other hand, power density is the rate at which energy is stored and released by devices of the same mass/volume, with the measuring units of W kg−1 and W L−1. Meanwhile, power is equal to the product of the current and voltage, and at a constant current, power is directly proportional to the voltage. The energy density and power density of different energy storage systems are compared in Fig. 1a.28
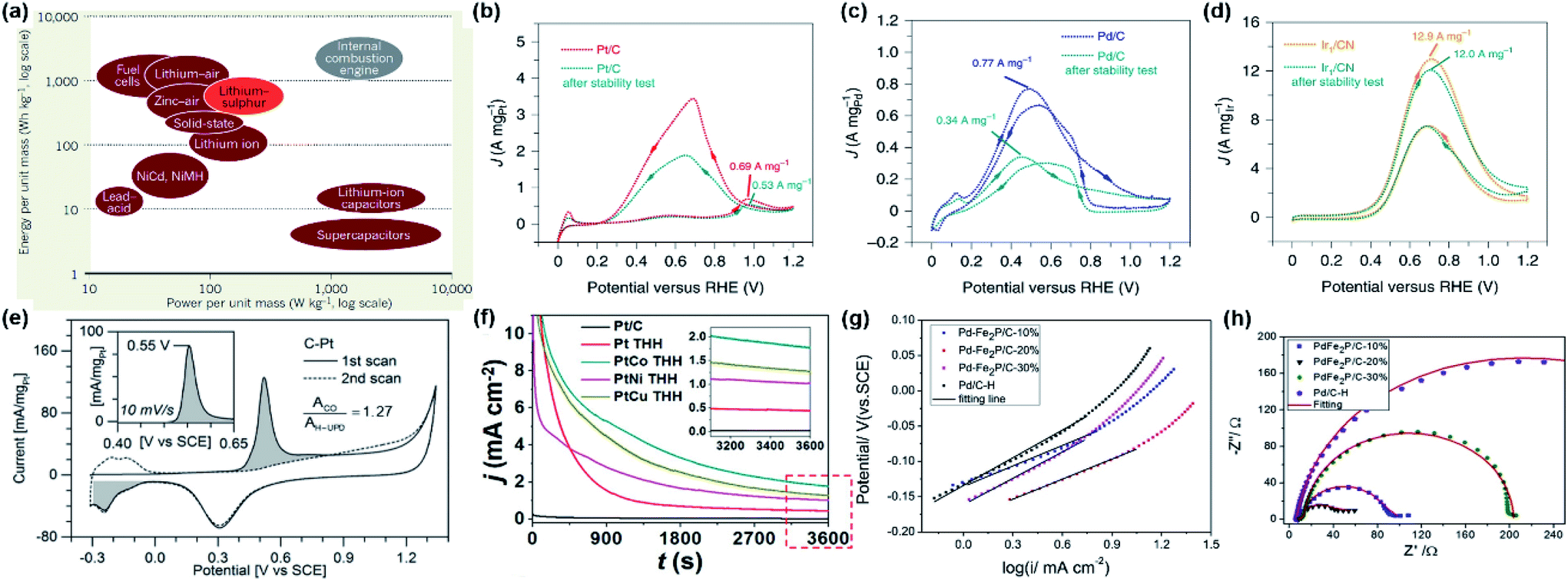 | ||
| Fig. 1 (a) Energy densities and power densities of different energy storage systems. Reproduced from ref. 28. Copyright 2013, Nature. (b–d) CVs of Pt/C, Pd/C and Ir/CN in 0.5 M H2SO4 + 0.5 M HCOOH at a scanning rate of 50 mV s−1. Reproduced from ref. 31. Copyright 2020, Nature. (e) CVs from CO-saturated C–Pt surface (solid line), and from a clean, CO-free C–Pt surface (dashed line). The charges associated with CO and hydrogen desorption at 0.55 V and −0.2 V were indicated with the filled area. Cyclic voltammograms in 0.1 M H2SO4 were recorded at 50 mV s−1, while the inset represents CO oxidation at 10 mV s−1. Reproduced from ref. 21. Copyright 2018, Wiley-VCH. (f) Chronoamperometry curves of formic acid electro-oxidation on different catalysts at 0.5 V in Ar-saturated 0.5 M HCOOH + 0.5 M H2SO4. Reproduced from ref. 5. Copyright 2020, Wiley-VCH. (g) Tafel plots of the different catalysts drawing from CV curves in 0.5 M H2SO4 + 0.5 M HCOOH with a scan rate of 5 mV s−1. (h) Nyquist plots for the different catalysts in 0.5 M H2SO4 + 0.5 M HCOOH at 0 V. Reproduced from ref. 8. Copyright 2018, Elsevier. | ||
In DFAFCs, the electro-oxidation of formic acid on the anode can be described as: HCOOH → CO2 + 2H+ + 2e− (E0 = −0.25 V vs. RHE). The oxidation process and the electrocatalytic performances of catalysts are usually investigated by cyclic voltammetry (CV), CO stripping, chronoamperometry (CA), Tafel plot, electrochemical impedance spectroscopy (EIS), and other electrochemical techniques. Among these methods, CV can provide fundamental information of FAEO. As illustrated in Fig. 1b, for formic acid oxidation on a Pt/C catalyst, the oxidation current peak I at ∼0.55 V (vs. RHE) corresponded to the direct oxidation of HCOOH to CO2. Obviously, because the surface of Pt/C has a relatively constant coverage of CO poisoning species below 0.85 V (vs. RHE), only a very low current density could be observed. Meanwhile, the peak II at ∼1.0 V (vs. RHE) was assigned to the electro-oxidation of CO intermediates accumulated in the indirect oxidation pathway. Anodic sweep is often used to evaluate the activity of the used catalyst, and the intensity ratio of peak I and peak II is used to determine the pathway of FAEO on catalysts, i.e., a direct or indirect pathway. On the other hand, based on the oxidation currents in positive-going and negative-going scans in CV measurements, the CO anti-poisoning ability of the catalyst could be qualitatively evaluated.29,30 By comparing the CVs of formic acid oxidation on Pt/C and Pd/C (Fig. 1b and c), Pt/C showed higher catalytic activity, while Pd/C showed better anti-poisoning ability. On the other hand, both Pt/C and Pd/C have poor electrochemical stability. From Fig. 1d, the Ir/CN showed relatively higher stability than Pt/C and Pd/C, although it showed a higher overpotential for HCOOH oxidation.31 From this example, a simple CV curve can provide abundant information about the HCOOH oxidation on different catalysts.
In addition to CV, CO stripping (Fig. 1e) is generally employed to assess the electrochemical active surface area (ECSA) and the catalytic active sites,21 and CA (Fig. 1f) is often used to examine the electrochemical stability.5 Meanwhile, Tafel plots (Fig. 1g) can reveal the reaction rate and mechanism during the catalytic process, where a smaller Tafel slope suggests a faster charge-transfer kinetics.13 Moreover, EIS (Fig. 1h) is always used to investigate the charge-transfer kinetics during the catalytic process, wherein a larger diameter of the semicircle corresponds to a larger charge-transfer resistance (Rct).8
From the above, although simple, CV, CO stripping, CA, Tafel plot, and EIS are the fundamental methods to study the electrochemical processes of FAEO on different electrocatalysts. Therefore, in all the reported studies on formic acid oxidation, these electrochemical techniques are used to characterize the catalytic properties of the electrodes.
3. Reaction mechanism of FAEO
Since formic acid is a kind of simple organic small molecule, it is suitable for use as a model molecule to study the reaction mechanism of the electro-oxidation of organic molecule fuels (HCOOH, CH3OH, C2H5OH, etc.) on different electrodes.32–40 Although simple, the electro-oxidation of HCOOH has a complicated reaction processes and various possible routes to generate the CO2 product, depending on the different electrode surface structures, as shown in Fig. 2a. Parsons et al. proposed the widely recognized two-pathway mechanism as early as 1973. That is, formic acid dehydrogenates in two steps and simultaneously electrons transfer to the electrode to form CO2 and H+ in the direct pathway: HCOOH → active intermediate → CO2 + 2H+ + 2e−. In the indirect pathway, formic acid first dehydrates to form CO, which is further oxidized to CO2 at a higher potential: HCOOH → COads + H2O, COads + OHads → CO2 + 2H+ + 2e−.11,41,42 The product of CO2 and the intermediate of CO and their transformation can be directly observed by using in situ attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR). As shown in Fig. 2b, for HCOOH oxidation on Pd black catalyst, in addition to the clear absorption signal of the CO2 product at 2345 cm−1, the peaks at 1869 cm−1 can be assigned to the poisonous CO intermediate.18 Recently, Wang et al. reported an in situ electrochemical shell-isolated nanoparticle-enhanced Raman spectroscopy (EC-SHINERS) technique that could monitor the electro-oxidation processes of CO to CO2.43 As shown in Fig. 2c, for CO absorbed on a Pt monolayer, the intensity of the band at 472 cm−1, corresponding to the Pt–C stretching vibration, decreased drastically as the potential increased and then disappears after 0.6 V, suggesting the desorption and oxidation of CO at 0.6 V. Meanwhile, two peaks appeared when the potential rose up to 0.9 V, one broad band at 570 cm−1, and another sharp peak at 330 cm−1. These two peaks were attributed to the Pt–O stretch with the top and bridge configurations of CO2. It should be noted that due to the complicated process, the possible oxidation mechanism of formic acid is under debate and the related research is still underway.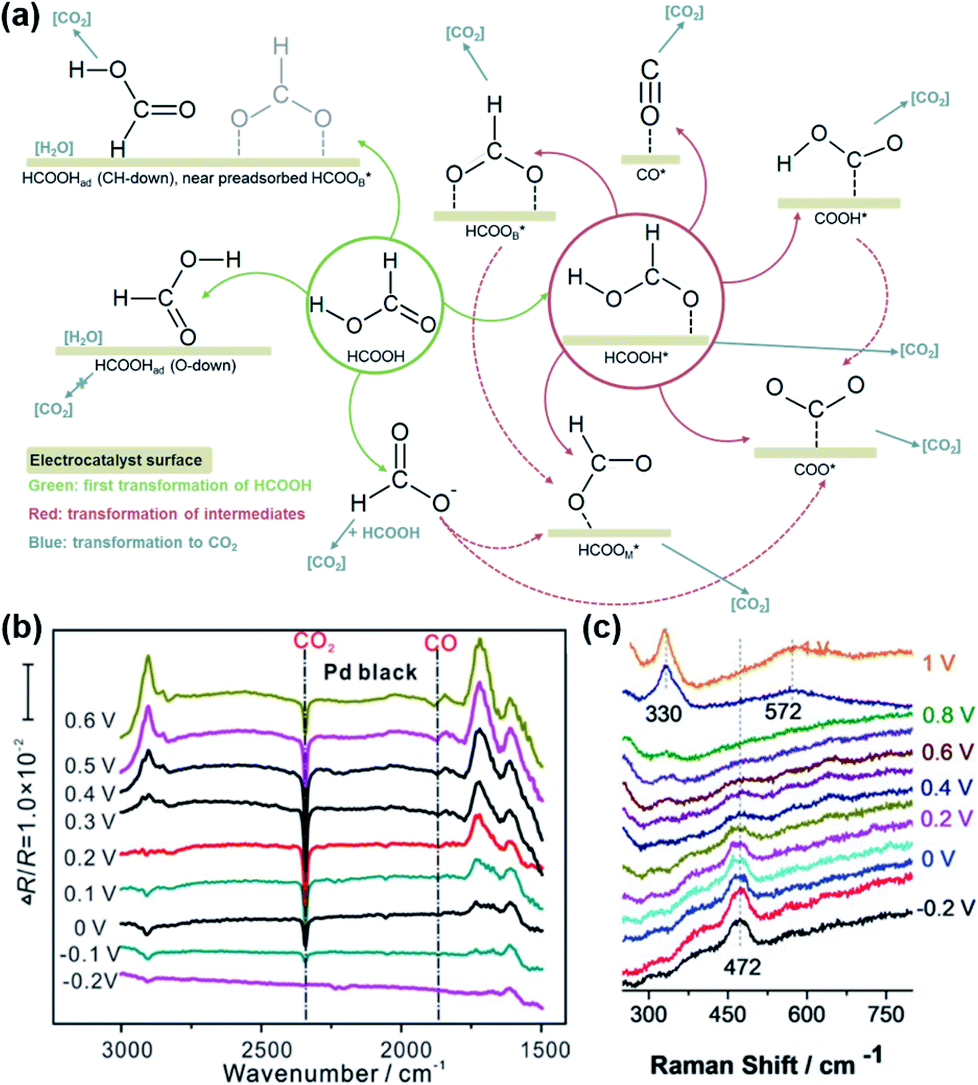 | ||
| Fig. 2 (a) The proposed mechanisms of HCOOH electro-oxidation (green: first transformation of HCOOH, red: transformation of intermediates, blue: transformation to CO2). (b) In situ ATR-FTIR spectra of electro-oxidation of formic acid on commercial Pd black in 0.25 M HCOOH + 0.5 M H2SO4 solution at different potentials from −0.20 to 0.60 V, with the reference spectra acquired at −0.25 V. 200 interferograms (resolution: 8 cm−1) were collected and combined for each spectrum. Reproduced from ref. 18. Copyright 2018, Elsevier. (c) In situ electrochemical shell-isolated nanoparticle-enhanced Raman spectroscopy (EC-SHINERS) spectra of CO electro-oxidation on Au(111)@Pt monolayer in 0.1 M HClO4 saturated CO. Reproduced from ref. 43. Copyright 2018, Wiley-VCH. | ||
3.1 Active intermediate identification
Willsau et al. first demonstrated the direct pathway of HCOOH oxidation by combining the electrochemical differential mass spectrometry technique with the isotopic method.44 Usually, COH, CHO, –HCOO, and –COOH were considered as active intermediates until Osawa's research group detected the bridge-bonded formate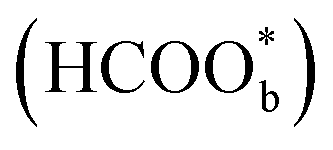 with two oxygen atoms adsorbed on the platinum electrode surface by attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS).42,45–48 At the beginning, adsorbed formate
with two oxygen atoms adsorbed on the platinum electrode surface by attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS).42,45–48 At the beginning, adsorbed formate 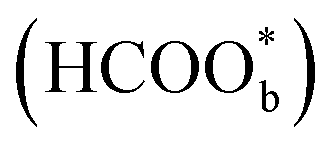 was regarded as a reactive intermediate in the direct pathway through ATR-SEIRAS.49 Then objections were raised. Chen et al. proposed that bridge-bonded formate
was regarded as a reactive intermediate in the direct pathway through ATR-SEIRAS.49 Then objections were raised. Chen et al. proposed that bridge-bonded formate 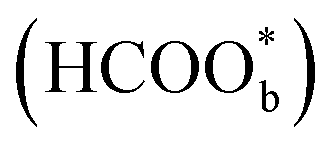 was not a reactive intermediate but a triple path for FAEO. It was also predicted that in a direct pathway, the weakly adsorbed HCOOHads precursor (HCOOH*) is directly oxidized to CO2 and the adsorbed formate
was not a reactive intermediate but a triple path for FAEO. It was also predicted that in a direct pathway, the weakly adsorbed HCOOHads precursor (HCOOH*) is directly oxidized to CO2 and the adsorbed formate 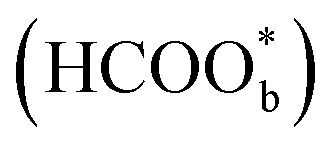 acts as a site-blocking spectator. These conclusions were based on the results of the ATR-FTIR technique with a thin-layer electrochemical flow cell.50,51 The speculation that
acts as a site-blocking spectator. These conclusions were based on the results of the ATR-FTIR technique with a thin-layer electrochemical flow cell.50,51 The speculation that 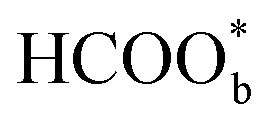 is just a spectator was supported by Neurock et al. and they showed that *COOH might be the reactive intermediate.52 In addition, Wang et al. used density functional theory (DFT) calculations with a Pt/H2O model to show that HCOOHads(O-down) is difficult to be oxidized to CO2 and that HCOOHads(CH-down) could be oxidized to CO2 in the presence of the near pre-adsorbed formate
is just a spectator was supported by Neurock et al. and they showed that *COOH might be the reactive intermediate.52 In addition, Wang et al. used density functional theory (DFT) calculations with a Pt/H2O model to show that HCOOHads(O-down) is difficult to be oxidized to CO2 and that HCOOHads(CH-down) could be oxidized to CO2 in the presence of the near pre-adsorbed formate 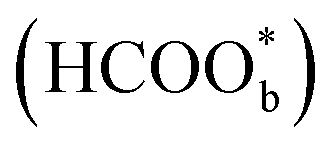 . Such a result means that formate
. Such a result means that formate 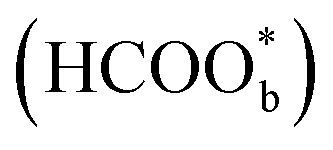 is neither the active intermediate nor the site-blocking specie, but rather a catalyst for the direct oxidation of formic acid.53 In another work, by using DFT calculations, Jacob's group studied the mechanism of HCOOH electro-oxidation on Pt (111) and found that dual pathways were involved in the process, including a formate pathway via the adsorbed HCOO* intermediate and a direct pathway from the adsorbed HCOOH* via a highly transient
is neither the active intermediate nor the site-blocking specie, but rather a catalyst for the direct oxidation of formic acid.53 In another work, by using DFT calculations, Jacob's group studied the mechanism of HCOOH electro-oxidation on Pt (111) and found that dual pathways were involved in the process, including a formate pathway via the adsorbed HCOO* intermediate and a direct pathway from the adsorbed HCOOH* via a highly transient 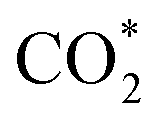 intermediate. The two pathways were strongly dependent on the applied potentials.54 Later, Osawa's group observed a volcano-shaped pH-activity diagram with the maximum current obtained near the pKa, and they revised the early view that HCOO* is the reactive intermediate in the direct pathway, and suggested that formate ions (HCOO−) were the main reactive intermediate.55 Both platinum and palladium were involved in the mechanisms mentioned above. Herron et al. pointed out that Pt and Pd are in the middle of the transition metals and balance the requirements of the two dehydrogenation steps and offer a compromise between activity and stability.56
intermediate. The two pathways were strongly dependent on the applied potentials.54 Later, Osawa's group observed a volcano-shaped pH-activity diagram with the maximum current obtained near the pKa, and they revised the early view that HCOO* is the reactive intermediate in the direct pathway, and suggested that formate ions (HCOO−) were the main reactive intermediate.55 Both platinum and palladium were involved in the mechanisms mentioned above. Herron et al. pointed out that Pt and Pd are in the middle of the transition metals and balance the requirements of the two dehydrogenation steps and offer a compromise between activity and stability.56
The above determinations of the active intermediates were from spectral results or classical theoretical calculations. More recently, the identity of the reactive intermediate has been studied by different methods. By using a combined experimental and computational approach, Vilaplana et al. proposed that the reactive intermediate is monodentate-adsorbed formate 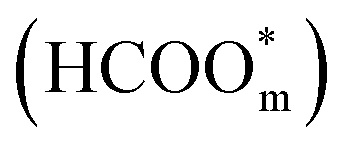 and the presence of adjacent adsorbates inhibits the transition from
and the presence of adjacent adsorbates inhibits the transition from 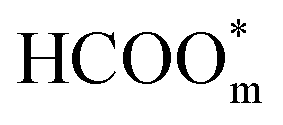 to
to 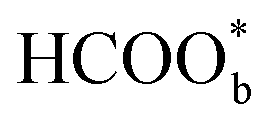 .57 Consistently, through DFT computations, Huang's group found that
.57 Consistently, through DFT computations, Huang's group found that 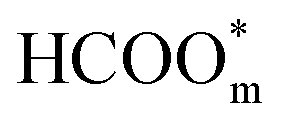 was the main active intermediate with HCOO− as the main precursor, and
was the main active intermediate with HCOO− as the main precursor, and 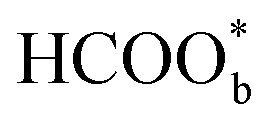 was the site-blocking species with HCOOH as the precursor.29 In recent years, based on single-crystalline electrochemistry and in situ electrochemical Fourier transform infrared spectroscopy (EC-FTIR), Chen and co-workers proposed that in the direct pathway of FAEO, formate is the site-blocking species instead of the active intermediate.58 By combining ATR-SEIRAS with ultraviolet (UV) reflectance measurements, another interesting observation made by Hartl et al. showed that the galvanostatic electro-oxidation of formic acid involves a complex autocatalytic loop.59 In the process, all of the species mentioned above play roles, and the results come from the oscillations during the galvanostatic oxidations. On the other hand, bimetal catalysts have been used recently in discussing the mechanism of FAEO. For example, through DFT calculations, Meng et al. proposed that the *COOH intermediate plays different roles for palladium atoms and palladium ensembles on the surface of copper; the presence of Pd ensembles mainly results in CO2 and isolated Pd atoms can catalyze HCOOH dissociation to form CO.60 Additionally, Yang et al. suggested that for the Mcore@Pdshell (M = Cu, Au, Co, Ni, Ag, Al) core@shell bimetal catalysts, the M@Pd (M = Au, Co, Ni, Ag), especially Ag, are highly selective for CO2 formation that goes through the COOH or HCOO intermediate, depending on the types of core metal. Meanwhile, among the studied catalysts, Al@Pd is favorable for CO formation via the *COOH intermediate instead of the HCO* intermediate.61
was the site-blocking species with HCOOH as the precursor.29 In recent years, based on single-crystalline electrochemistry and in situ electrochemical Fourier transform infrared spectroscopy (EC-FTIR), Chen and co-workers proposed that in the direct pathway of FAEO, formate is the site-blocking species instead of the active intermediate.58 By combining ATR-SEIRAS with ultraviolet (UV) reflectance measurements, another interesting observation made by Hartl et al. showed that the galvanostatic electro-oxidation of formic acid involves a complex autocatalytic loop.59 In the process, all of the species mentioned above play roles, and the results come from the oscillations during the galvanostatic oxidations. On the other hand, bimetal catalysts have been used recently in discussing the mechanism of FAEO. For example, through DFT calculations, Meng et al. proposed that the *COOH intermediate plays different roles for palladium atoms and palladium ensembles on the surface of copper; the presence of Pd ensembles mainly results in CO2 and isolated Pd atoms can catalyze HCOOH dissociation to form CO.60 Additionally, Yang et al. suggested that for the Mcore@Pdshell (M = Cu, Au, Co, Ni, Ag, Al) core@shell bimetal catalysts, the M@Pd (M = Au, Co, Ni, Ag), especially Ag, are highly selective for CO2 formation that goes through the COOH or HCOO intermediate, depending on the types of core metal. Meanwhile, among the studied catalysts, Al@Pd is favorable for CO formation via the *COOH intermediate instead of the HCO* intermediate.61
3.2 Poisoning intermediate identification
It is widely accepted that adsorbed carbon monoxide (COads) is the main poisonous species during the formic acid oxidation. Very early, infrared spectroscopy studies identified that COads is a poisoning intermediate in the indirect pathway of FAEO.62 And therefore, catalysts with high activity for CO electro-oxidation can efficiently remove the surface adsorbed CO and thus can significantly promote the FAEO.43 Just like with the identification of reactive intermediates, bridge-bonded formate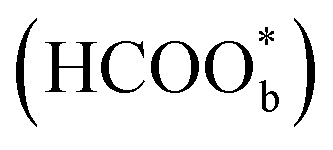 , dimeric formic acid, carboxylate (*COOH), CO2, and other species were once thought to be the precursors to CO.57,63–65 As for the formation origination of COads, Cuesta et al. proposed that the presence of adjacent platinum atoms would lead to the formation of COads. This view has been widely accepted, and many efforts have been made to construct discontinuous platinum sites to mitigate the poisoning of platinum.14,66,67 Except for FAEO, palladium is less reactive than platinum in most electrochemical reactions.68 The reasons for this are not yet known, perhaps because palladium has a more specific affinity for hydrogen, allowing the rapid dehydrogenation of formic acid molecules on the palladium surface.13,22,69,70 Meanwhile, the severe inactivation of palladium is often attributed to agglomeration, surface oxidation state change, component migration and loss, and CO-like toxic species gathering on the palladium surface. For CO-like toxic species, there is considerable evidence that CO is produced by the reduction of the product CO2 at a low oxidation potential, but there are other unknown reasons for the inactivation of palladium catalysts.71–73
, dimeric formic acid, carboxylate (*COOH), CO2, and other species were once thought to be the precursors to CO.57,63–65 As for the formation origination of COads, Cuesta et al. proposed that the presence of adjacent platinum atoms would lead to the formation of COads. This view has been widely accepted, and many efforts have been made to construct discontinuous platinum sites to mitigate the poisoning of platinum.14,66,67 Except for FAEO, palladium is less reactive than platinum in most electrochemical reactions.68 The reasons for this are not yet known, perhaps because palladium has a more specific affinity for hydrogen, allowing the rapid dehydrogenation of formic acid molecules on the palladium surface.13,22,69,70 Meanwhile, the severe inactivation of palladium is often attributed to agglomeration, surface oxidation state change, component migration and loss, and CO-like toxic species gathering on the palladium surface. For CO-like toxic species, there is considerable evidence that CO is produced by the reduction of the product CO2 at a low oxidation potential, but there are other unknown reasons for the inactivation of palladium catalysts.71–73
To reveal the mechanism of FAEO, electrochemical methods cannot provide direct information about the reaction parameters or intermediates, nor can they provide information about the electrode/solution interface at the molecular level. On the other hand, there is a gap between the density functional theory (DFT) simulated environment and the real electrode/solution interface. Therefore, it is necessary to combine some other technologies to achieve in situ characterizations, just like differential electrochemical mass spectrometry (DEMS), in situ infrared spectroscopy (in situ IR), surface-enhanced Raman spectroscopy (SERS), and so on.
For the identification of intermediates, the reported conclusions are quite different due to the differences in the experimental apparatus and conditions, and the systems selected by researchers. Based on the current research, perhaps the most well-accepted two-pathway mechanism would be a good place to start, by using the same experimental method to study various forms of monometals and alloys in various specific systems, which involves single atoms, diatoms, clusters, monolayer atoms, nanoparticles, and films composed of different index facets. After a systematic study and summary, a cross comparison should be carried out to find the commonalities and the differences. Moreover, some possible intermediates during the FAEO have a short life and the present instruments may not be sensitive enough to detect them. Therefore, developing new devices, improving existing instruments, detecting breakdown products that are specific to the short-lived intermediates, and finding suitable probe molecules are very important for clearly and correctly revealing the reaction mechanism of FAEO. One step further, there are two main solutions to the problem of electrocatalyst poisoning caused by carbon monoxide: one is the introduction of oxyphilic active sites to rapidly oxidize carbon monoxide, and the other is to eliminate certain surface sites to inhibit carbon monoxide production.
4. Electrocatalysts for FAEO
The electrocatalysts for formic acid oxidation need to meet the following requirements: safety, low cost, high electrocatalytic activity, high tolerance to poisoning species, high electrical conductivity, high physical and electrochemical stability without particle agglomeration, surface oxidation state change, component migration and loss, carrier corrosion, and so on.8,16Base on the above desired requirements, the basic design concept of electrocatalysts for formic acid oxidation is to regulate the electronic structure and surface structure, which complement each other. However, the electronic structure effect should be considered first when designing the catalysts. In other words, the activation energy of the FAEO reaction could be reduced by selecting suitable electrocatalytic materials. The effect of the surface structure on the reaction rate and mechanism should be considered after selecting a suitable electrocatalyst. Currently, the surface structure manipulation includes the chemical structure (composition and valence), geometric structure (shape and form), surface atoms arrangement, electronic structure, and others. Among these, surface ligand effects, lattice strain effects, and synergistic effects are often used to explain the enhancement of catalytic activity, and meanwhile ensemble effects and third-body effects are often used to analyze the reasons that inhibit the formation of carbon monoxide.18,20,24,25,74
4.1 Pt- and Pd-based electrocatalysts for formic acid oxidation
Both platinum- and palladium-based materials are the traditional and the most widely used catalysts for the electro-oxidation of formic acid, but their research challenges are a little different.6,9 For platinum-based materials, inhibition of the indirect pathway and carbon monoxide adsorption are the core problems.9,21,75–77 However, improving the stability is the most critical issue for palladium-based catalysts.13 Up to now, a lot of efforts have been made by researchers to resolve the above-mentioned problems.For example, controlling the shape and exposing high-index facets (Fig. 3a and b) with a low coordination number, open structure, and high surface energy can significantly enhance the catalytic activity and stability for FAEO.2,5,78 Through chemical vapor deposition (CVD) with an alloying–dealloying shape-regulating process, Huang et al. synthesized tetrahexahedron (TTH)-shaped Pt–M (M = Sb, Bi, Pb, or Te), Pd–Bi nanocrystals on carbon black, and TTH-shaped Pt/C–Bi catalyst. The synthesized catalysts with high-index {210} planes (Fig. 3a) showed higher electrocatalytic activity and stability than commercial Pt/C and Pd/C catalysts for FAEO. With the prepared catalysts, the FAEO followed a direct reaction pathway. In addition to the foreign metal modification, the enhanced catalytic performance was mainly attributed to the high-index facets of the nanocatalysts.7 It should be noted that not all metals with high-index facets will promote the catalytic performance for the electro-oxidation of formic acid. It has been found that the catalytic properties of metal nanocrystals are related to the surface structures. For Pt and Pd single-crystals, the catalytic activities of their basal planes for formic acid oxidation follow the order: Pt (110) > Pt (111) > Pt (100) and Pd (100) > Pd (111) > Pd (110).79,80 For high-index facets of Pt and Pd nanocrystals, there is still no accurate conclusion due to the large number of studies carried out in different systems.81 Tetrahexahedron Pd {hk0} facets have been studied in detail and the oxidation current density of FAEO was found to have the following order: Pd (730) < Pd (830) < Pd (310) < Pd (1030) < Pd (1130) < Pd (100).82 Furthermore, Elnabawy et al. found that the open facets of Au, Ag, Cu, Pt, and Pd required less overpotentials for the FAEO when compared to their close-packed facets, and the opposite was true for Ni, Ir, and Rh metals.65
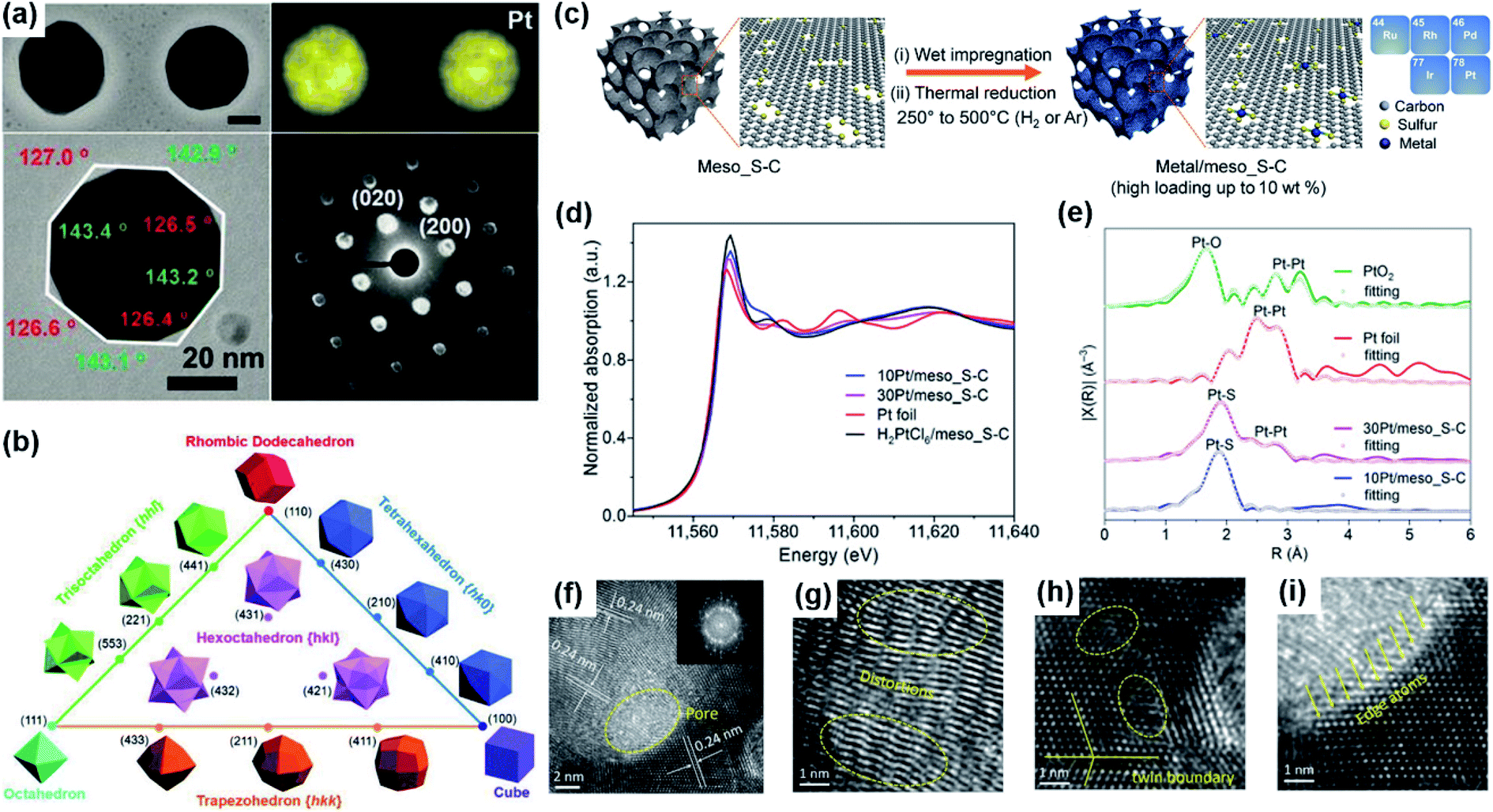 | ||
| Fig. 3 (a) STEM images, EDS elemental maps, TEM images, and corresponding diffraction patterns of the Pt–Bi (96.5% Pt, 3.5% Bi) nanocrystals. Reproduced from ref. 7. Copyright 2019, Science. (b) Triangular diagram showing the correlations between convex polyhedron with different crystallographic facets. Reproduced from ref. 78. Copyright 2019, Wiley-VCH. (c) Schematic illustration of the preparation and model structure of the atomically dispersed noble metal catalysts. Five atomically dispersed noble metal (Ru, Rh, Pd, Ir, and Pt) catalysts were prepared on the meso_S–C support with a high metal loading of up to 10 wt%. (d) Normalized XANES (X-ray absorption near-edge structure) spectra of 10Pt/meso_S–C, 30Pt/meso_S–C, H2PtCl6/meso_S–C, and Pt foil at the Pt L3-edge. (e) EXAFS (extended X-ray absorption fine structure) spectra of 10Pt/meso_S–C, 30Pt/meso_S–C, Pt foil, and PtO2. Reproduced from ref. 83. Copyright 2019, Science. (f–i) TEM images of Pd3Pt half-shells at different magnifications, inset in (f) is the corresponding FFT pattern. Reproduced from ref. 22. Copyright 2018, Wiley-VCH. | ||
The catalytic performance for the electro-oxidation of formic acid can also be improved by taking advantage of the interaction between the metal and a suitable substrate.12 For example, Wang et al. reported the atomic dispersion of Pt on mesoporous sulfur-doped carbon (meso_S–C) with Pt loading as high as up to 10% by a H2 thermal reduction process (Fig. 3c–e). The prepared hybrid showed enhanced catalytic performance for the FAEO. It was also found that the formation of Pt clusters in Pt/meso_S–C could reduce the catalytic performance. However, both of the prepared samples exhibited higher catalytic activity and stability than the commercial Pt/C. The authors ascribed the improved catalytic properties to the large surface area and mesoporous structure containing abundant and easily accessible sulfur-containing sites to accommodate a high mass percentage of Pt atoms. In fact, the isolated Pt sites and high Pt atom utilization were favorable for following a direct pathway.83 Ye et al. prepared Pd/cubes-CeO2 composite via a chemical reduction method by using ethylene glycol as a reducing agent.12 The presence of CeO2 could increase the metal dispersion and the ECSA. Meanwhile, with Pd dispersed on cubic ceria with a polar (100) surface and oxygen vacancies, the high degree of oxygen storage in CeO2 and the fast oxygen mobility at the noble metal–ceria interface could effectively resist Pd from being oxidized and could remove CO or other related poisons efficiently. In another work, Wang et al. used a microwave-assisted ethylene glycol method to synthesize a Pd–Fe2P/C catalyst in which Pd nanoparticles were deposited onto an Fe2P–carbon hybrid support.8 The fast charge-transfer kinetics and the appropriate down-shift of the d-band center of the Pd–Fe2P/C catalyst significantly enhanced the catalytic performance for FAEO. The performance improvement was mainly attributed to the contribution from Fe2P in the composite since Fe2P can make the adsorbed CO and other toxic intermediates be oxidized at extremely low potentials.
In addition, constructing special nanostructures with a high specific surface area, edge areas, and surface defects can also enhance the catalytic activity and stability for the FAEO.84,85 For instance, Ding et al. synthesized perpendicular Pd nanotube arrays (P-PdNTA) via a dual template electrodeposition method.19 Meanwhile, the massive Pd nanoparticles served as building blocks for the mesoporous walls of the nanotubes. The perpendicular and mesoporous structure provided a large electrochemical active surface area and active sites that were not easily aggregated. Additionally, there were anisotropic substructures on the catalyst surface as step edges and lattice defects (grain boundary, twin, etc.). These unique structures could accelerate the transportation, adsorption, and oxidation of formic acid. Recently, a one-pot hydrothermal synthesis of porous half-shells Pd3Pt in the presence of urea was reported by Tang and co-workers.22 Here, urea was used as a guiding surfactant to produce NH3 and CO2, which could act as a bubble template for building a hollow or porous structure. The enhanced catalytic activity and stability for FAEO was attributed to the high porosity, large electrochemical active surface area, fast mass transfer, abundant edge atoms, and a mass of crystal defects, such as distortions, a twin boundary, and atomic holes, as shown in Fig. 3f–i. Our group used a one-phase synthesis method to prepare Pt1Au24(SC12H25)18 nanoclusters, which had a similar geometry with the Au25 nanoclusters.86 It was found that although Au25 nanoclusters exhibited very poor electrocatalytic activity for FAEO, a single Pt atom doping in the center of the Au25 cluster could significantly enhance the catalytic performance. In the Pt1Au24(SC12H25)18 nanocluster, the single Pt atom could decrease the CO production while the gold atoms could protect the catalyst from CO poisoning. Therefore, the electro-oxidation of formic acid followed a direct pathway with COOH* as the preferred reactive intermediate.
Finally, alloying Pt or Pd with other transition metals could change the d-band center, thus changing the interaction between the adsorbed species and the surface of the electrocatalysts.31 In such studies, high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM), X-ray powder diffraction (XRD), and energy-dispersive X-ray spectroscopy (EDX) are commonly used methods to investigate the structure of alloys.26 For example, Duchesne et al. reported a colloidal method to prepare a series of platinum–gold (Pt–Au) nanoparticles by using ethylene glycol as a reducing agent.75 In these nanoparticles, different contents of platinum were modified on the surface of gold, and these NPs were categorized in terms of their surface structure (Fig. 4a): Au with single-atom Pt sites (Pt7Au93 and Pt4Au96), Au with single-atom and few-atom Pt sites (Pt17Au83), and Au-core/Pt-shell (Pt78Au22 and Pt53Au47). It was found that the lower the number of platinum atoms, the better the electrocatalytic performance for formic acid oxidation. The characterization results showed that the high catalytic activity and stability were derived from the optimized elemental composition, low coordination numbers of Pt atoms, and the isolated Pt atoms preventing the catalyst from self-poisoning via CO generation. The most critical role was played by single-atom Pt surface sites surrounded by Au atoms. In addition, Luo et al. synthesized intermetallic PtSnBi nanoplates (Fig. 4b–f) via a one-pot sequential complexing–reducing–ordering process.14 In this work, Bi played a key role in the formation of 2D nanoplates with an hcp crystalline structure and in suppressing the dehydration path during HCOOH oxidation. On the other hand, Sn could promote the CO oxidation on Pt sites by reacting with H2O to form Sn–OHads. Therefore, the high FAEO catalytic activity of the PtSnBi nanoplates originated from the improved catalytic selectivity with direct oxidation from HCOO* to CO2, rather than the anti-poisoning ability. Even further, Yang et al. used a seeded growth method with a self-assembly process to prepare a sea urchin-like Aucore@Pdshell electrocatalyst (Fig. 4g and h).13 The prepared material had a high ECSA with more exposed active sites and an obvious change of the d-band center of Pd. Meanwhile, the mismatched lattice constants between the Au core and Pd shell resulted in lattice strain. These factors could adjust the adsorption strength of formic acid molecules, intermediate species, and hydroxide radicals on Pd, which optimized the dehydrogenation steps and promoted the oxidation of COOHads to CO2. More Pt-based and Pd-based electrocatalysts that have been developed for FAEO in recent years are summarized in Table 1.
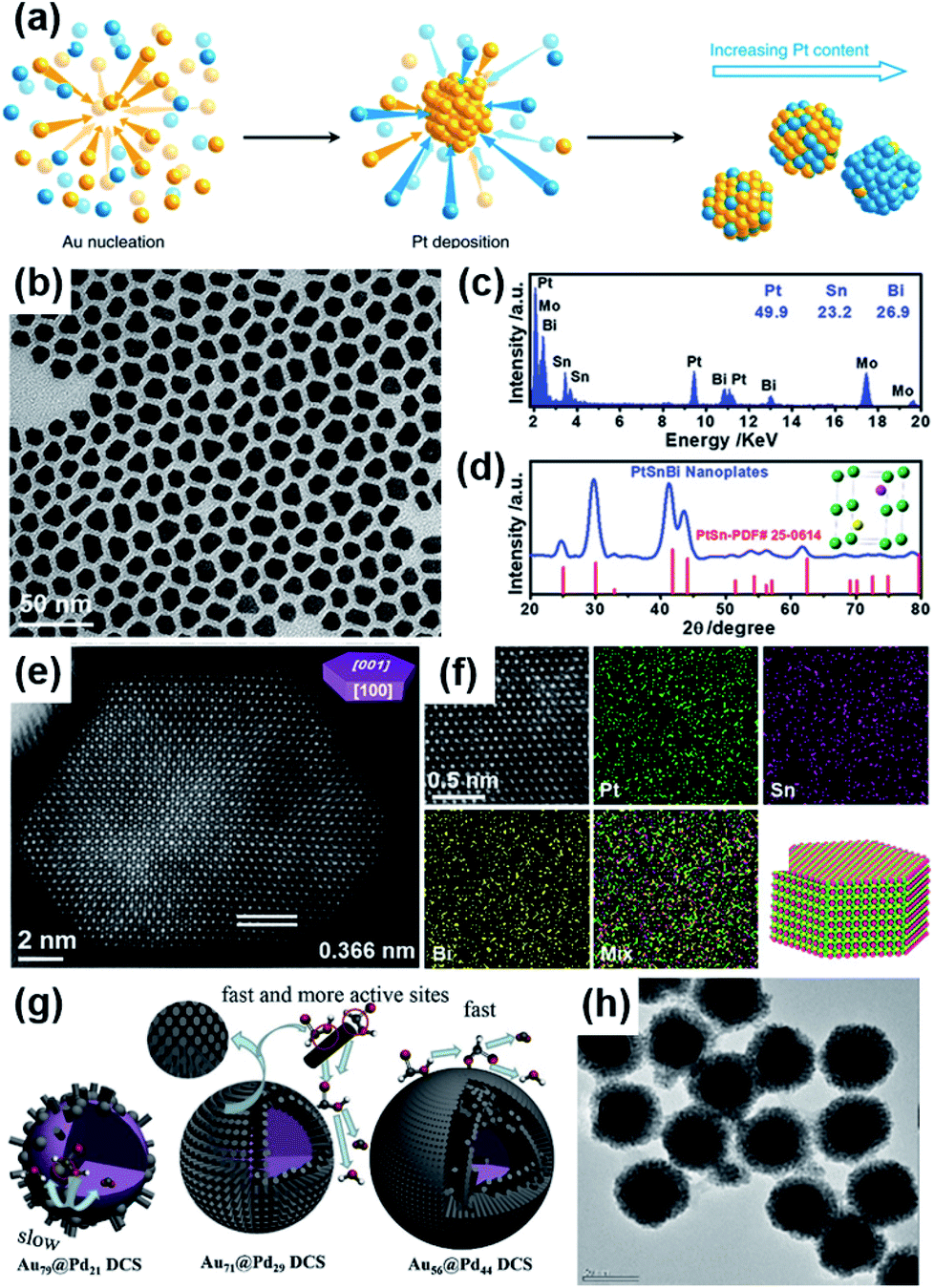 | ||
| Fig. 4 (a) Synthesis of platinum–gold (Pt–Au) electrocatalysts. Reproduced from ref. 75. Copyright 2018, Nature. (b–d) TEM image, EDX spectrum, and XRD pattern of Pt45Sn25Bi30 nanoplates. The inset in (d) shows the unit cell of intermetallic Pt50Sn25Bi25, in which green, purple, and yellow spheres represent Pt, Sn, and Bi atoms, respectively. (e) Aberration-corrected HAADF-STEM image of a typical hexagonal Pt45Sn25Bi30 nanoplate. (f) High-resolution HAADF-STEM image and the corresponding EDX maps of the representative area in Pt45Sn25Bi30 nanoplate, and the schematic illustration of the atomic arrangement in the nanoplate. Reproduced from ref. 14. Copyright 2019, Wiley-VCH. (g and h) The reaction process of Au79@Pd21DCS, Au71@Pd29DCS, and Au56@Pd44DCS, and TEM image of Au56@Pd44DCS. Reproduced from ref. 13. Copyright 2020, Elsevier. | ||
| Electrocatalyst | Preparation | Electrolyte | Peak potential (vs. RHE) | Peak current density | Current density after stability test | Ref. | |
|---|---|---|---|---|---|---|---|
| Controlling the shape and exposing high-index facets | |||||||
| Pt–M (M = Ni, Co, Cu) (NPs, (210)) | Colloidal synthesis and CVD | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.55 V | 13.25 mA cm−2 (Pt THH NPs) | 0.5 mA cm−2 (Pt THH NPs) (0.5 V, 3600 s) | 5 | |
| Pt-M (M = Sb, Bi, Pb, Te), Pd–Bi, Pt/C–Bi (NPs, (210)) | CVD with alloying–dealloying | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.55 V | 13.25 mA cm−2 (Pt–Bi) | 1.49 A mgPt−1 (Pt–Bi), 1.44 A mgPd−1 (Pd–Bi) | 0.44 mA cm−2 (Pt–Bi) (0.5 V, 7200 s) | 7 |
| PdHx (nanocrystal) | Treating commercial Pd black | 0.5 M H2SO4 + 0.25 M HCOOH | ∼0.28 V | 5.12 mA cm−2 | 1.06 A mgPd−1 | 0.4 A mgPd−1 (0.34 V, 400 s) | 18 |
|
|||||||
| Metals supported on suitable substrates | |||||||
| Pd–Fe2P/C (NPs–metal phosphide/C) | Microwave-assisted ethylene glycol method | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.45 V | 1526 mA mgPd−1 | 108.24 mA mgPd−1 (0.44 V, 3600 s) 70 mA mgPd−1 (0.44 V, 7200 s) | 8 | |
| Pt1/ATO (single atom/ATO) | H2 thermal reduction | 0.1 M HClO4 + 0.5 M HCOOH | ∼0.6 V | 9 A mgPt−1 | 11 | ||
| Pd/cubes-CeO2 (NPs/metal oxide) | Chemical reduction | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.4 V | 11.38 mA cm−2 | 1060 A gPd−1 | 12 | |
| PdSn/SnOx/TiC (triple junction structure) | NaBH4 reduction method | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.45 V | 0.23 mA cm−2 | 0.029 mA cm−2 (0.3 V, 3600 s) | 16 | |
| Pt/S–C (single atom/meso_S–C) | H2 thermal reduction | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.85 V | 3.5 A mgPt−1 | 0.1 A mgPt−1 (0.4 V, 7200 s) | 83 | |
| Pd/BG–CN (NPs/hybrid of B–N–C) | H2 thermal reduction | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.45 V | 31.7 mA cm−2 | 2215 mA mg−1 | 400 A g−1 (0.44 V, 10000 s) |
88 |
| Pd/NrGO@SiO2 (nanoclusters/N–C@SiO2) | NaBH4 reduction method | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.36 V | 2.37 A mg−1 | 0.1 A mg−1 (0.25 V, 3600 s) | 89 | |
| Pd–Mo2N/rGO (NPs–metal nitride/C) | Assembly-immobilization + NaBH4 reduction method | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.45 V | 532.7 mA mgPd−1 | 50 mA mgPd−1 (0.2 V, 3600 s) | 90 | |
|
|||||||
| Special nanostructures | |||||||
| Pd (periodically ordered mesoporous membranes) | Template electrodeposition method | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.49 V | 3.6 mA cm−2 | 3.34 A mg−1 | 6 | |
| Pd (nanotube arrays/ITO) | Dual template electrodeposition method | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.5 V | 4.41 mA cm−2 | 3.65 A mgPd−1 | 19 | |
| Pd3Pt (porous half-shells) | Hydrothermal one-pot synthesis | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.07 V | 318 mA mg−1 | 70 mA mg−1 (0.2 V, 3500 s) | 22 | |
| Pt1Au24/MCNTs (nanocluster/C) | One-phase synthesis | 0.1 M HClO4 + 0.5 M HCOOH | ∼0.6 V | 3.7 A mgPt+Au−1 (89 A mgPt−1) | 86 | ||
| Pd 2D nanoframes (low-dimensional Pd nanocrystals) | Seeded growth method and solvothermal synthesis | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.45 V | 0.95 mA μg−1 | 91 | ||
| Pd nanosheets (perforated crystalline/amorphous heterostructures) | Carbon monoxide reduction method | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.46 V | 2582.7 mA mgPd−1 | 80 mA mgPd−1 (3000 s) | 92 | |
| Sub-5 nm Pd tetrahedrons | One-pot synthesis | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.42 V | 413.89 A g−1 | 192 A g−1 (0.4 V, 3500 s) | 93 | |
|
|||||||
| Alloys | |||||||
| Pd–M (M = Ru, Pt, Cu, Au, Ag) (NPs) | Successive reduction method | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.4 V | 5.12 mA cm−2 (Pd–Cu/C) | 0.4 mA cm−2 (Pd–Cu/C) (0.26 V, 2000 s) | 3 | |
| Au@Pd (core with shell) | Seeded growth method and self-assembly process | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.47 V | 23.33 A m−2 (Au56@Pd44) | 1405 A gPd−1 (Au71@Pd29) | 170 A gPd−1 (Au71@Pd29) (0.47 V, 3600 s) | 13 |
| PtSnBi (intermetallic nanoplate) | Sequential complexing–reducing–ordering processes | 0.5 M H2SO4 + 1 M HCOOH | ∼0.65 V | 105 mA cm−2 | 4394 mA mgPt−1 (Pt45Sn25Bi30) | 22 mA cm−2 (Pt45Sn25Bi30) (0.55 V, 3600 s) | 14 |
| Pd59Cu30Co11 (nanoalloy) | Solvothermal synthesis | 0.1 M HClO4 + 0.5 M HCOOH | ∼0.95 V | 9.06 mA cm−2 | 5.32 A mgPd−1 | 0.25 A mgPd−1 (0.5 V, 2500 s) | 20 |
| CoPtAu (NPs) | One-pot synthesis method | 0.1 M HClO4 + 0.1 M HCOOH | ∼0.4 V | 3.4 A mgPt−1 | 24 | ||
| PdRuBP | One-pot synthesis method | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.64 V | 4.15 mA cm−2 | 1.71 mA μgPd−1 | 0.17 mA μgPd−1 (0.5 V, 4000 s) | 25 |
| PtnAum/C (NPs/C) | Colloidal method | 0.1 M HClO4 + 0.1 M HCOOH | ∼0.6 V | 3.9 mA cm−2 (Pt7Au93) | 2.77 A mgPt−1 (Pt7Au93) | 0.4 A mgPt−1 (0.55 V, 600 s) | 75 |
| Pd–Fe–Pt (2D nanomeshes) | Wet chemical reaction | 0.5 M H2SO4 + 0.5 M HCOOH | ∼0.7 V | 2.45 A mgPt−1 | 0.15 A mgPt−1 (0.85 V, 3600 s) | 94 | |
| PdFe/C (ordered intermetallic) | Conventional borohydride method | 0.1 M HClO4 + 0.1 M HCOOH | ∼0.62 V | 18.97 A m−2 | 352.64 A gPd−1 | 3.79 A m−2 (0.4 V, 3600 s) | 95 |
| Pt–Au/C (NPs/C) | Ultrasound-assisted method | 0.5 M H2SO4 + 1 M HCOOH | ∼0.55 V | 14.5 A mgPt−1 | 96 | ||
4.2 Non-Pt and non-Pd electrocatalysts for formic acid oxidation
Although Pt and Pd are the best electrocatalysts so far for formic acid oxidation, their high cost and limited reservation largely hinder their wide application in fuel cell catalysis. Therefore, developing Pt- and Pd-free electrocatalysts for FAEO is paramount, crucial, and has become a hot research topic in recent years.Many studies have found that downsizing metal nanoparticles can bring about excellent catalytic properties that conventional catalysts don't possess.10,87 Li et al. reported iridium single atoms supported on nitrogen-doped carbon (Ir1/CN) by pyrolyzing a metal precursors@MOF composite at high temperature.31 The electrocatalytic performance of the Ir1/CN for formic acid oxidation was higher than those of commercial Pt/C, Pd/C, Ir/C and Ir1Irn/CN catalysts with mixed single atoms and nanoparticles. The enhanced catalytic activity and stability stemmed from the more favorable COOH* pathway and the easily activated O–H bond of the generated COOH* on the Ir–N4 active centers. Meanwhile, the isolated Ir sites, high Ir atom utilization, and the interaction between Ir and the highly electronic conductive CN support were also important factors for the improved catalytic activity. However, Pt1/CN and Pd1/CN showed no electrocatalytic activity for the oxidation of formic acid, which broke with the stereotype. In another study, Xiong et al. synthesized atomically dispersed Rh on N-doped carbon (SA-Rh/CN) via a high-temperature pyrolysis of the metal precursors@Zn-MOF composites, while Zn ion sites were substituted by Rh atoms (Fig. 5a–g).84 The obtained SA-Rh/CN exhibited promising catalytic properties for formic acid electro-oxidation, but SA-Pd/CN and SA-Pt/CN synthesized by the same procedure and the as-prepared Rh nanoparticles on carbon black (Rh/C) were found to be inert toward FAEO. It was proposed that the SA-Pd/CN and SA-Pt/CN lost their catalytic abilities for FAEO due to their weak bonding ability to the intermediates. The high catalytic activity and stability of SA-Rh/CN arose from the interaction between the Rh and CN support to form RhN4, the isolated metal atoms with high metal utilization, and the porous structure with the large surface area. This study also demonstrated that CO is difficult to generate and easy to remove. Meanwhile, the SA-Rh/CN was thermodynamically and kinetically favorable for formic acid oxidation via a formate route, and the C–H cleavage of HCOO* was the rate-determining step.26
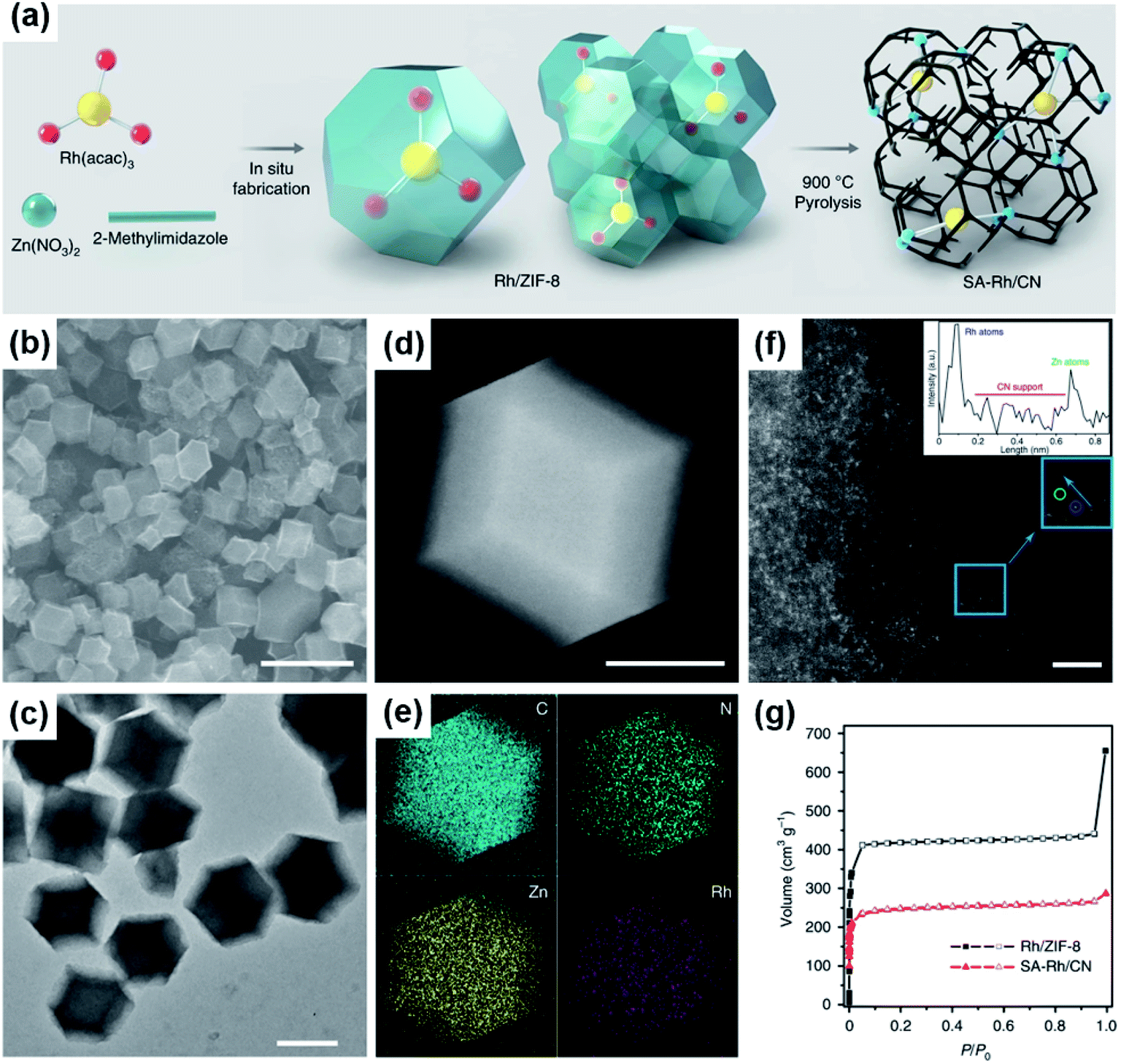 | ||
| Fig. 5 (a) Schematic illustration of the preparation strategy for SA-Rh/CN. (b) SEM and (c) TEM images of SA-Rh/CN. (d) HAADF-STEM image and (e) the corresponding EDS maps of SA-Rh/CN showing the dispersion of C (light blue), N (green), Zn (yellow), and Rh (purple), respectively. (f) AC-HAADF-STEM (aberration-corrected HAADF-STEM) image and the corresponding Z-contrast analysis. The confirmed Rh atom and Zn atom are marked by blue and green circles, respectively. Because the atomic numbers of Rh and Zn are different, the Z-contrast of the Rh atoms in AC-HAADF-STEM images is higher than that of Zn atoms. (g) N2 adsorption and desorption isotherms for SA-Rh/CN (red) and Rh/ZIF-8 (black). P/P0 is the relative pressure, where P is the pressure of the test point and P0 is the atmospheric pressure. Scale bars: 1 μm (b), 500 nm (c), 200 nm (d), 2 nm (f). Reproduced from ref. 84. Copyright 2020, Nature. | ||
Transition metals, such as platinum and palladium, contain unoccupied d orbitals and unpaired d electrons. Therefore, when the catalysts containing transition metals contact with the reactant molecules, a variety of chemical adsorption bonds with various characteristics will be formed, thus achieving the purpose of molecular activation and a reduction of the activation energy for complex reactions. However, to obtain a high catalytic performance, the structure and composition of the materials need to be optimized, such as by reducing the size of nanoparticles with high-index facets, selecting suitable supports, and adjusting the pore size, effectively improving the structural stability, efficiently reducing the dissolution of non-precious metals during the electrochemical reactions, and applying the conclusions obtained from the study of model catalysts such as metal single-crystal surfaces to the actual catalyst systems. It should be pointed out that currently there are very few high-performance non-Pt and non-Pd electrocatalysts for FAEO. Therefore, further detailed research and investigations are urgently needed.
5. Conclusions and outlook
As a typical electrocatalytic reaction in liquid fuel cells, studies on formic acid electro-oxidation have attracted great attention, including the reaction mechanism and designing high-performance electrocatalysts. However, many problems and challenges still need to be resolved before formic acid electro-oxidation systems offer sufficient performance to satisfy basic research and practical application demands. The details are as follows: (1) to reveal the reaction mechanism and identify the active intermediates are still challenging. In addition to the traditional electrochemical methods and density functional theory (DFT) calculations, developing high-resolution and ultrafast in situ or operando spectroscopic techniques is necessary to detect the intermediates and analyze the reaction mechanism of formic acid oxidation. (2) Based on the reaction mechanism, rationally designing and adjusting the structures of electrocatalysts are an ideal strategy to explore high-performance catalysts for formic acid oxidation. Both the thermodynamics and kinetics should be taken into account to improve the current density and to reduce the overpotential of formic acid electro-oxidation. In addition, investigations on effective non-Pt and non-Pd electrocatalysts are significantly important to explore cost-efficient anode catalysts for fuel cells. (3) Finally, the costs, durability, and activity should be balanced reasonably so as to realize the commercialization of direct formic acid fuel cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 21575134, 21633008, 21773224), National Key Research and Development Plan (2020YFB1506001), K. C. Wong Education Foundation.References
- M. Liu, R. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117–5160 CrossRef CAS.
- W. Chen, J. Kim, S. Sun and S. Chen, Langmuir, 2007, 23, 11303–11310 CrossRef CAS.
- S. Hu, F. Che, B. Khorasani, M. Jeon, C. W. Yoon, J.-S. McEwen, L. Scudiero and S. Ha, Appl. Catal., B, 2019, 254, 685–692 CrossRef CAS.
- Y. Lu and W. Chen, J. Phys. Chem. C, 2010, 114, 21190–21200 CrossRef CAS.
- L. Huang, C. Y. Zheng, B. Shen and C. A. Mirkin, Adv. Mater., 2020, 32, 2002849 CrossRef CAS.
- J. Ding, Z. Liu, X. Liu, B. Liu, J. Liu, Y. Deng, X. Han, W. Hu and C. Zhong, Angew. Chem., Int. Ed., 2020, 132, 5130–5139 CrossRef.
- L. Huang, M. Liu, H. Lin, Y. Xu, J. Wu, V. P. Dravid, C. Wolverton and C. A. Mirkin, Science, 2019, 365, 1159–1163 CrossRef CAS.
- F. Wang, H. Xue, Z. Tian, W. Xing and L. Feng, J. Power Sources, 2018, 375, 37–42 CrossRef CAS.
- W. Chen, J. Kim, S. Sun and S. Chen, Phys. Chem. Chem. Phys., 2006, 8, 2779–2786 RSC.
- J. T. L. Gamler, H. M. Ashberry, S. E. Skrabalak and K. M. Koczkur, Adv. Mater., 2018, 30, 1801563 CrossRef.
- J. Kim, C.-W. Roh, S. K. Sahoo, S. Yang, J. Bae, J. W. Han and H. Lee, Adv. Energy Mater., 2017, 8, 1701476 CrossRef.
- L. Ye, A. H. Mahadi, C. Saengruengrit, J. Qu, F. Xu, S. M. Fairclough, N. Young, P.-L. Ho, J. Shan, L. Nguyen, F. F. Tao, K. Tedsree and S. C. E. Tsang, ACS Catal., 2019, 9, 5171–5177 CrossRef CAS.
- L. Yang, G. Li, J. Chang, J. Ge, C. Liu, F. Vladimir, G. Wang, Z. Jin and W. Xing, Appl. Catal., B, 2020, 260, 118200 CrossRef CAS.
- S. Luo, W. Chen, Y. Cheng, X. Song, Q. Wu, L. Li, X. Wu, T. Wu, M. Li, Q. Yang, K. Deng and Z. Quan, Adv. Mater., 2019, 31, 1903683 CrossRef CAS.
- W. Chen and S. Chen, J. Mater. Chem., 2011, 21, 9169–9178 RSC.
- Q. Dong, M. Wu, D. Mei, Y. Shao, Y. Wang, J. Liu, H. Li and L. Hong, Nano Energy, 2018, 53, 940–948 CrossRef CAS.
- W. Chen, J. Kim, L.-P. Xu, S. Sun and S. Chen, J. Phys. Chem. C, 2007, 111, 13452–13459 CrossRef CAS.
- J. Zhang, M. Chen, H. Li, Y. Li, J. Ye, Z. Cao, M. Fang, Q. Kuang, J. Zheng and Z. Xie, Nano Energy, 2018, 44, 127–134 CrossRef CAS.
- J. Ding, Z. Liu, X. Liu, J. Liu, Y. Deng, X. Han, C. Zhong and W. Hu, Adv. Energy Mater., 2019, 9, 1900955 CrossRef.
- C. Li, Q. Yuan, B. Ni, T. He, S. Zhang, Y. Long, L. Gu and X. Wang, Nat. Commun., 2018, 9, 3702 CrossRef.
- N. Seselj, C. Engelbrekt, Y. Ding, H. A. Hjuler, J. Ulstrup and J. Zhang, Adv. Energy Mater., 2018, 8, 1702609 CrossRef.
- X. Yan, X. Hu, G. Fu, L. Xu, J.-M. Lee and Y. Tang, Small, 2018, 14, 1703940 CrossRef.
- N. Tian, Z.-Y. Zhou and S.-G. Sun, J. Phys. Chem. C, 2008, 112, 19801–19817 CrossRef CAS.
- J. Li, S. Z. Jilani, H. Lin, X. Liu, K. Wei, Y. Jia, P. Zhang, M. Chi, Y. J. Tong, Z. Xi and S. Sun, Angew. Chem., Int. Ed., 2019, 58, 11527–11533 CrossRef CAS.
- Y. Xu, S. Yu, T. Ren, C. Li, S. Yin, Z. Wang, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2020, 8, 2424–2429 RSC.
- D. Kim and M. Cargnello, Nat. Nanotechnol., 2020, 15, 346–347 CrossRef CAS.
- W. Chen, L.-P. Xu and S. Chen, J. Electroanal. Chem., 2009, 631, 36–42 CrossRef CAS.
- R. V. Noorden, Nature, 2013, 498, 416–417 CrossRef.
- X. Zhu and J. Huang, J. Electrochem. Soc., 2020, 167, 013515 CrossRef CAS.
- F. J. Vidal-Iglesias, R. M. Arán-Ais, J. Solla-Gullón, E. Garnier, E. Herrero, A. Aldaz and J. M. Feliu, Phys. Chem. Chem. Phys., 2012, 14, 10258–10265 RSC.
- Z. Li, Y. Chen, S. Ji, Y. Tang, W. Chen, A. Li, J. Zhao, Y. Xiong, Y. Wu, Y. Gong, T. Yao, W. Liu, L. Zheng, J. Dong, Y. Wang, Z. Zhuang, W. Xing, C.-T. He, C. Peng, W.-C. Cheong, Q. Li, M. Zhang, Z. Chen, N. Fu, X. Gao, W. Zhu, J. Wan, J. Zhang, L. Gu, S. Wei, P. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. Duan, Y. Huang, X.-M. Chen, D. Wang and Y. Li, Nat. Chem., 2020, 12, 764–772 CrossRef CAS.
- C. Zhang, R. Zhang, X. Li and W. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 29623–29632 CrossRef CAS.
- W. Wei and W. Chen, J. Power Sources, 2012, 204, 85–88 CrossRef CAS.
- Y. Lu, Y. Jiang, H. Wu and W. Chen, J. Phys. Chem. C, 2013, 117, 2926–2938 CrossRef CAS.
- Y. Lu, Y. Jiang and W. Chen, Nanoscale, 2014, 6, 3309–3315 RSC.
- P. Li, C. Du, X. Gao, Z. Zhuang, D. Xiang, C. Zhang and W. Chen, Nanoscale, 2020, 12, 13688–13696 RSC.
- M. Liu, S. He and W. Chen, Electrochim. Acta, 2016, 199, 218–226 CrossRef CAS.
- C. Du, S. He, M. Liu, X. Gao, R. Zhang and W. Chen, CrystEngComm, 2016, 18, 6055–6061 RSC.
- C. Du, X. Gao, Z. Zhuang, C. Cheng, F. Zheng, X. Li and W. Chen, Electrochim. Acta, 2017, 238, 263–268 CrossRef CAS.
- Z. Zhuang and W. Chen, J. Colloid Interface Sci., 2019, 538, 699–708 CrossRef CAS.
- A. Capon and R. Parsons, J. Electroanal. Chem., 1973, 44, 1–7 CrossRef CAS.
- W. Gao, J. E. Mueller, Q. Jiang and T. Jacob, Angew. Chem., Int. Ed., 2012, 51, 9448–9452 CrossRef CAS.
- Y.-H. Wang, M.-M. Liang, Y.-J. Zhang, S. Chen, P. Radjenovic, H. Zhang, Z.-L. Yang, X.-S. Zhou, Z.-Q. Tian and J.-F. Li, Angew. Chem., Int. Ed., 2018, 57, 11257–11261 CrossRef CAS.
- J. Willsau and J. Heitbaum, Electrochim. Acta, 1986, 31, 943–948 CrossRef CAS.
- S. Wilhelm, T. Iwasita and W. Vielstich, J. Electroanal. Chem., 1987, 238, 383–391 CrossRef CAS.
- A. Capon and R. Parsons, J. Electroanal. Chem., 1973, 45, 205–231 CrossRef CAS.
- S. G. Sun and J. Clavilier, J. Electroanal. Chem., 1988, 240, 147–159 CrossRef CAS.
- A. Miki, S. Ye and M. Osawa, Chem. Commun., 2002, 1500–1501 RSC.
- G. Samjeské and M. Osawa, Angew. Chem., Int. Ed., 2005, 44, 5694–5698 CrossRef.
- Y. X. Chen, M. Heinen, Z. Jusys and R. J. Behm, Angew. Chem., Int. Ed., 2006, 45, 981–985 CrossRef CAS.
- Y.-X. Chen, M. Heinen, Z. Jusys and R. J. Behm, Langmuir, 2006, 22, 10399–10408 CrossRef CAS.
- M. Neurock, M. Janikb and A. Wieckowskic, Faraday Discuss., 2008, 140, 363–378 RSC.
- H.-F. Wang and Z.-P. Liu, J. Phys. Chem. C, 2009, 113, 17502–17508 CrossRef CAS.
- W. Gao, J. A. Keith, J. Anton and T. Jacob, J. Am. Chem. Soc., 2010, 132, 18377–18385 CrossRef.
- J. Joo, T. Uchida, A. Cuesta, M. T. M. Koper and M. Osawa, J. Am. Chem. Soc., 2013, 135, 9991–9994 CrossRef CAS.
- J. A. Herron, J. Scaranto, P. Ferrin, S. Li and M. Mavrikakis, ACS Catal., 2014, 4, 4434–4445 CrossRef CAS.
- A. Ferre-Vilaplana, J. V. Perales-Rondón, C. Buso-Rogero, J. M. Feliu and E. Herrero, J. Mater. Chem. A, 2017, 5, 21773–21784 RSC.
- W. Chen, A. Yu, Z.-J. Sun, B.-Q. Zhu, J. Cai and Y.-X. Chen, Curr. Opin. Electrochem., 2019, 14, 113–123 CrossRef CAS.
- F. W. Hartl, H. Varela and A. Cuesta, J. Electroanal. Chem., 2019, 840, 249–254 CrossRef CAS.
- F. Meng, M. Yang, Z. Li and R. Zhang, Appl. Surf. Sci., 2020, 511, 145554 CrossRef CAS.
- M. Yang, B. Wang, Z. Li, L. Ling and R. Zhang, Appl. Surf. Sci., 2020, 506, 144938 CrossRef CAS.
- B. Benen, A. Bewick and C. Lamy, J. Electroanal. Chem., 1983, 150, 505–511 CrossRef.
- A. Cuesta, G. Cabello, M. Osawa and C. Gutiérrez, ACS Catal., 2012, 2, 728–738 CrossRef CAS.
- Y. Qi, J. Li, D. Zhang and C. Liu, Catal. Sci. Technol., 2015, 5, 3322–3332 RSC.
- A. O. Elnabawy, J. A. Herron, J. Scaranto and M. Mavrikakis, J. Electrochem. Soc., 2018, 165, J3109–J3121 CrossRef CAS.
- A. Cuesta, M. Escudero, B. Lanova and H. Baltruschat, Langmuir, 2009, 25, 6500–6507 CrossRef CAS.
- K. Jiang, H.-X. Zhang, S. Zou and W.-B. Cai, Phys. Chem. Chem. Phys., 2014, 16, 20360–20376 RSC.
- N. Uwitonze, D. Zhou, J. Lei, W. Chen, X. Q. Zuo, J. Cai and Y.-X. Chen, Electrochim. Acta, 2018, 283, 1213–1222 CrossRef CAS.
- E. Herrero and J. M. Feliu, Curr. Opin. Electrochem., 2018, 9, 145–150 CrossRef CAS.
- Y. Lu, R. Jin and W. Chen, Nanoscale, 2011, 3, 2476–2480 RSC.
- J.-Y. Wang, H.-X. Zhang, K. Jiang and W.-B. Cai, J. Am. Chem. Soc., 2011, 133, 14876–14879 CrossRef CAS.
- O. A. Petrii, Russ. J. Electrochem., 2019, 55, 1–33 CrossRef CAS.
- Y. Wang, Y. Qi, D. Zhang and C. Liu, J. Phys. Chem. C, 2014, 118, 2067–2076 CrossRef CAS.
- X. Li, C. Li, D. Xiang, C. Zhang, L. Xia, X. Liu, F. Zheng, X. Xie, Y. Zhang and W. Chen, Appl. Catal., B, 2019, 253, 263–270 CrossRef CAS.
- P. N. Duchesne, Z. Y. Li, C. P. Deming, V. Fung, X. Zhao, J. Yuan, T. Regier, A. Aldalbahi, Z. Almarhoon, S. Chen, D.-E. Jiang, N. Zheng and P. Zhang, Nat. Mater., 2018, 17, 1033–1039 CrossRef CAS.
- Y. Lu and W. Chen, ACS Catal., 2012, 2, 84–90 CrossRef CAS.
- Y. Lu and W. Chen, Chem. Commun., 2011, 47, 2541–2543 RSC.
- M. Iqbal, Y. V. Kaneti, J. Kim, B. Yuliarto, Y.-M. Kang, Y. Bando, Y. Sugahara and Y. Yamauchi, Small, 2019, 15, 1804378 CrossRef.
- S.-G. Sun and Y.-Y. Yang, J. Electroanal. Chem., 1999, 467, 121–131 CrossRef CAS.
- N. Hoshi, K. Kida, M. Nakamura, M. Nakada and K. Osada, J. Phys. Chem. B, 2006, 110, 12480–12484 CrossRef CAS.
- X. Chen, L. P. Granda-Marulanda, I. T. McCrum and M. T. M. Koper, Chem. Sci., 2020, 11, 1703–1713 RSC.
- N.-F. Yu, N. Tian, Z.-Y. Zhou, T. Sheng, W.-F. Lin, J.-Y. Ye, S. Liu, H.-B. Ma and S.-G. Sun, ACS Catal., 2019, 9, 3144–3152 CrossRef CAS.
- L. Wang, M.-X. Chen, Q.-Q. Yan, S.-L. Xu, S.-Q. Chu, P. Chen, Y. Lin and H.-W. Liang, Sci. Adv., 2019, 5, eaax6322 CrossRef CAS.
- Y. Xiong, J. Dong, Z.-Q. Huang, P. Xin, W. Chen, Y. Wang, Z. Li, Z. Jin, W. Xing, Z. Zhuang, J. Ye, X. Wei, R. Cao, L. Gu, S. Sun, L. Zhuang, X. Chen, H. Yang, C. Chen, Q. Peng, C.-R. Chang, D. Wang and Y. Li, Nat. Nanotechnol., 2020, 15, 390–397 CrossRef CAS.
- C. Zhang, F. Zheng, Z. Zhang, D. Xiang, C. Cheng, Z. Zhuang, P. Li, X. Li and W. Chen, J. Mater. Chem. A, 2019, 7, 9059–9067 RSC.
- Y. Lu, C. Zhang, X. Li, A. R. Frojd, W. Xing, A. Z. Clayborne and W. Chen, Nano Energy, 2018, 50, 316–322 CrossRef CAS.
- C. Du, Y. Gao, H. Chen, P. Li, S. Zhu, J. Wang, Q. He and W. Chen, J. Mater. Chem. A, 2020, 8, 16994–17001 RSC.
- L. Yang, X. Wang, D. Liu, G. Cui, B. Dou and J. Wang, Appl. Catal., B, 2020, 263, 118304 CrossRef CAS.
- J. Shan, T. Zeng, W. Wu, Y. Tan, N. Cheng and S. Mu, Nanoscale, 2020, 12, 12891–12897 RSC.
- H. Yan, Y. Jiao, A. Wu, C. Tian, L. Wang, X. Zhang and H. Fu, J. Mater. Chem. A, 2018, 6, 7623–7630 RSC.
- Y. Yan, X. Li, M. Tang, H. Zhong, J. Huang, T. Bian, Y. Jiang, Y. Han, H. Zhang and D. Yang, Adv. Sci., 2018, 5, 1800430 CrossRef.
- L. Y. Zhang, Y. Ouyang, S. Wang, D. Wu, M. Jiang, F. Wang, W. Yuan and C. M. Li, Small, 2019, 15, 1904245 CrossRef CAS.
- H. Zhang, X. Qiu, Y. Chen, S. Wang, S. E. Skrabalak and Y. Tang, Small, 2020, 16, 1906026 CrossRef CAS.
- X. Luo, C. Liu, X. Wang, Q. Shao, Y. Pi, T. Zhu, Y. Li and X. Huang, Nano Lett., 2020, 20, 1967–1973 CrossRef CAS.
- Y. S. Kang, D. Choi, J. Cho, H.-Y. Park, K.-S. Lee, M. Ahn, I. Jang, T. Park, H. C. Ham and S. J. Yoo, ACS Appl. Energy Mater., 2020, 3, 4226–4237 CrossRef CAS.
- H. Fan, M. Cheng, L. Wang, Y. Song, Y. Cui and R. Wang, Nano Energy, 2018, 48, 1–9 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |