DOI:
10.1039/D0NA00990C
(Paper)
Nanoscale Adv., 2021,
3, 1656-1673
Chemosensitivity enhanced by autophagy inhibition based on a polycationic nano-drug carrier†
Received
26th November 2020
, Accepted 22nd January 2021
First published on 27th January 2021
Abstract
In recent years, with the increasing understanding of the role of autophagy in tumorigenesis and development, a steady stream of studies have demonstrated that both excessive induction and inhibition of autophagy could effectively improve the therapeutic efficacy against tumors during cytotoxic or molecularly targeted drug therapy. Among them, autophagy inhibition mediated by nanomaterials has become an appealing notion in nanomedicine therapeutics, since it can be exploited as an effective adjuvant in chemotherapy or as a potential anti-tumor agent. Herein, we constructed a pH-sensitive nanoplatform loaded with epirubicin (EPI) (mPEG-b-P(DPA-b-DMAEMA)/EPI), enabling effective autophagy inhibition in the process of tumor-targeting therapy and further sensitized the tumors to EPI. It was found that polycationic nanomicelles (PEDD-Ms) displayed specific localization in lysosomes after entering tumor cells and caused the impairment of lysosomal degradation capacity through lysosomal alkalization in a dose-dependent manner. HepG2 cells treated with PEDD-Ms displayed a large-scale accumulation of autophagosomes and LC3 (an autophagosome marker protein), and the degradation of the autophagy substrate p62 was also blocked, which indicated that these functional nanomicelles could significantly inhibit autophagy. Meanwhile, the typical morphological characteristics of autophagosomes were directly visualized by TEM. In vivo results also showed that the tumor-targeted and autophagy inhibition-associated nanoplatform therapy could effectively improve the therapeutic efficiency of EPI, which may be partially attributed to the fact that autophagy inhibition could enhance the sensitivity of tumor cells to EPI. Overall, we revealed the effect of polycationic nanomicelles on autophagic processes in tumor cells and explored their possible molecular mechanism, also considering the synergistic outcome between autophagy mediated by nanomaterials and chemotherapeutic drugs to improve the therapeutic effect on tumors.
Introduction
Autophagy is a highly conservative cell biological behavior in eukaryotic cells, which plays an irreplaceable role in maintaining cellular homeostasis.1,2 According to the difference of regulation mode, the nature of the degraded substrates and the pathway of targeting lysosomes, autophagy is divided into three types: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA).3 Autophagy is generally referred to as macroautophagy, which is one of the most characteristic autophagy processes. In this dynamic process, misfolded or aggregated proteins and damaged organelles are phagocytized by autophagosomes and subsequently fuse with lysosomes to form autolysosomes. Finally, autolysosomes are disassembled, providing nutrition and energy for cells in response to many severe cellular conditions.4,5 Recent studies have shown that autophagy dysfunction is associated with tumors,6 neurodegenerative diseases,7 cardiovascular diseases8 and infections.9 Compared to other pathophysiology, the role of autophagy in cancer is more complicated because autophagy plays a “double-edged sword” role in various types of cancer,10,11 mediating carcinogenesis or anticancer effects. On the one hand, autophagy can effectively eliminate abnormal organelles and toxic unfolded proteins or promote cellular senescence to reduce self-damage, thus maintaining normal cell homeostasis and suppressing tumor growth;12 on the other hand, once malignant cancers are fully established, autophagy is additionally enhanced in response to cellular or environmental stresses such as nutrient starvation and hypoxic.13 In addition, emerging studies have revealed that cytoprotective autophagy is frequently induced in cancer cells under treatment stress, hindering the efficacy of multiple therapeutic options.14 Therefore, autophagy inhibition provides a potential way to develop new therapeutic strategies. As the key organelle at the downstream stage of autophagic flux, excessive impairment of lysosomes will directly block the autophagic flux and promote cell death. Currently, the conventional autophagy-inhibiting methods related to lysosomes (i.e., using autophagy inhibitors (chloroquine (CQ), hydroxychloroquine (HCQ) or matrine)) are widely utilized in synergistic cancer treatments, but with some side effects caused by their pharmacological activities.15–18 Notably, HCQ, as a single-agent to inhibit autophagy or combined with conventional chemotherapeutics and radiotherapy, has not only been proved to inhibit tumor growth and enhance the effectiveness of various treatment methods in human cancer cells and immunodeficient mice, but also entered the phase I/II of clinical studies to further explore the clinical efficacy of this treatment mode in multiple solid tumor patient models.19 Hence, discovering and developing molecules or nanomaterials with efficacies to suppress autophagy may provide highly promising candidates for intractable tumor destruction.
Nanomaterials have shown great potential in improving anticancer effects with unique physicochemical properties, such as good biocompatibility-controlled size and facile surface modification.20 Typically, polymeric nanoparticles with a size less than 200 nm could accumulate preferentially at tumor tissues via an enhanced permeability and retention (EPR) effect and most of them often use lysosomes as aggregation sites, which may specifically affect the autophagy-lysosome pathway. A variety of nanoparticles (NPs), such as iron oxide,21 selenium NPs22 and silica NPs,23 have been identified as a new class of autophagy inducers. Although many NPs induce survival-promoting autophagy,24 certain NPs may induce mitochondria-dependent autophagic cell death.25,26 However, increasing evidence indicates that some nanomaterials can block autophagic flux via autophagy-lysosome dysfunction. In particularly, a series of pH-sensitive polymeric NPs, as one of the most widely used drug carriers, could block the autophagic flux by accumulating in lysosomes and further impairing lysosomal function, including lysosomal alkalization and lysosomal membrane permeabilization.27 Moreover, compared with other physical properties (size and surface modification), pH sensitivity is one of the most important factors in autophagy regulation. There is an increasing number of nanotoxicological studies discussing that various types of NPs could increase the levels of ROS species and provoke oxidative stress, thus inducing mitochondrial membrane damage, which has been described to be the main cause of the cytotoxicity of various NPs by modulating autophagy.28 Furthermore, NPs that disrupt autophagy may be involved in the development of various pathologies including neurodegenerative and non-neurodegenerative diseases.29 However, autophagy dysfunction triggered by nanomaterials is not necessarily a disadvantageous scenario, which may be utilized as a sensitization tool to enhance the lethality of chemotherapeutic agents to cancer cells. More recently, Chang et al. have demonstrated that radio-labeled PEGylated liposomes with 188Re could effectively inhibit autophagy and further led to tumor destruction in two cases of recurrent ovarian cancer.30 Wang et al. reported that a kind of tumor-targeting magneto-gold @ fluorescent polymer nanoparticle, while providing T1&T2-MRI/CT/near infrared (NIR) fluorescence imaging, also contributed synergistically to the lethality effect of doxorubicin by inducing autophagy.31 Moreover, the applications of numerous smart nano-assemblies in autophagy detection and modulation increase rapidly to enhance the various modalities for disease diagnostics and therapeutics.32 Therefore, effective exploitation of nanomaterial-modulated autophagy could be an emerging field of tumor research, providing novel ideas for the treatment of refractory and drug-resistant cancer.
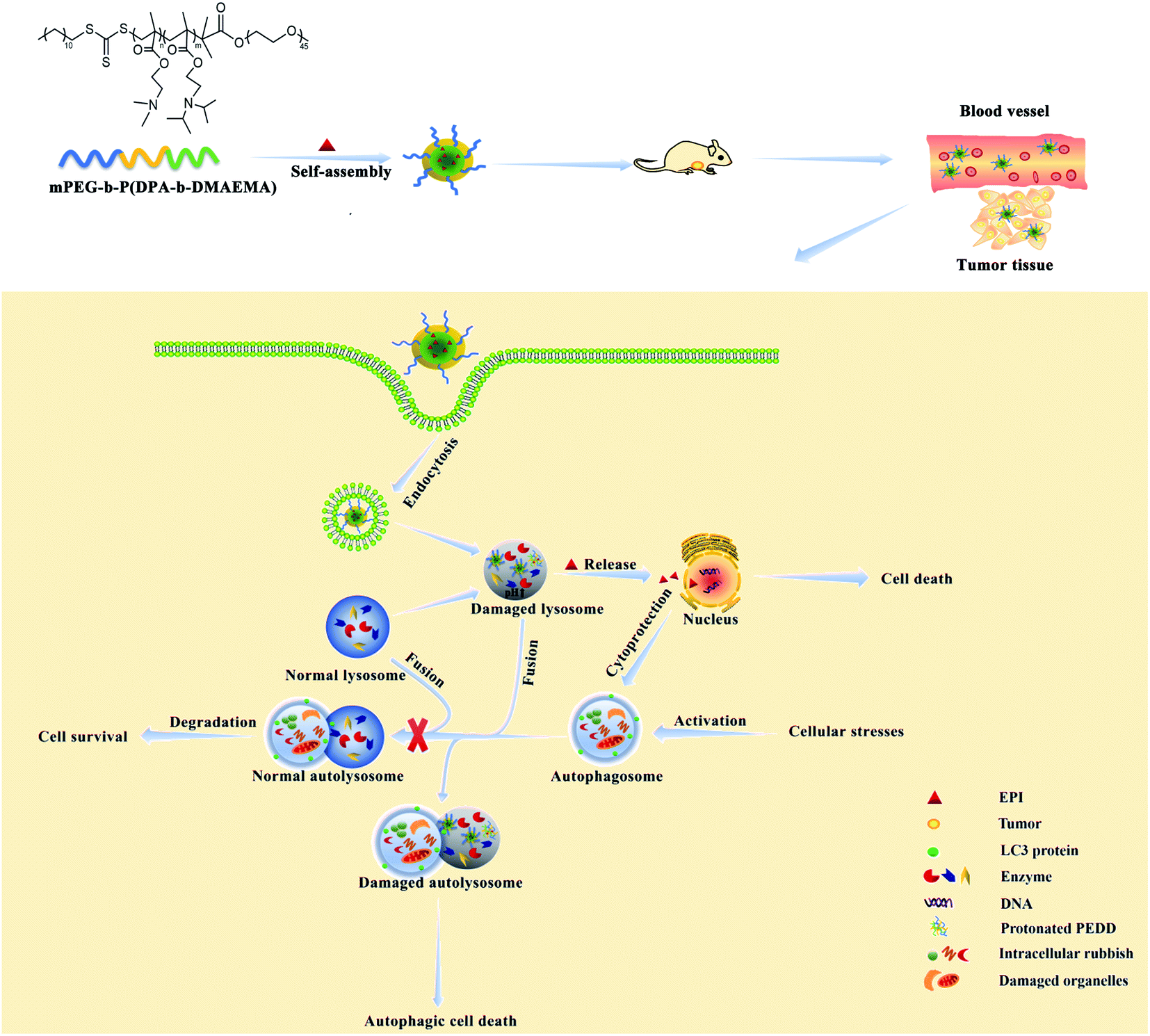
In this manuscript, we have designed a pH-sensitive polymer methoxy polyethylene glycol-b-poly(diisopropylethyl methacrylate-b-dimethylaminoethyl methacrylate) (mPEG-b-P(DPA-b-DMAEMA), PEDD polycations). PEDD polycations could be self-assembled into stable nanomicelles (PEDD-Ms) driven by the hydrophobic block of P(DPA-b-DMAEMA), and the chemotherapeutic agent epirubicin (EPI) is loaded into the nanomicelles to form drug-loaded nanomicelles (PEDD/EPI-Ms). The self-assembled nanomicelles with polyethylene glycol (PEG) as hydrophilic shells could selectively permeate and accumulate in tumors through the EPR effect and mainly accumulate in the lysosomes of tumor cells. In a lysosomal acidic environment (pH 4.5–5.5), the rapid protonation of the tertiary amines of P(DPA-b-DMAEMA) triggers the irreversible alkalization of lysosomes and eventually results in causing the impairment of lysosomal degradation capacity, thus blocking the autophagic flux. Meanwhile, the nanomicelles could effectively release EPI into the cell nucleus after dissociation, which could promote cell death more effectively while inhibiting the autophagy of tumor cells. Western blotting, transmission electron microscopy (TEM), acridine orange (AO) staining and GFP-LC3 dot formation were performed to identify autophagy behaviors in vitro. We clearly demonstrated that the treatment of PEDD-Ms could significantly inhibit autophagy in a dose-dependent manner, which was caused by the alkalization and poor degradation capacity of lysosomes. Interestingly, the inhibition of the autophagic flux by PEDD-Ms sensitizes HepG2 cells to EPI. Furthermore, this nano-drug delivery system was applied to deliver EPI in vivo for synergistic therapy, which significantly augmented drug accumulation in tumor tissues and showed marked tumor inhibition while reducing off-target systemic toxicity. The results of this manuscript may be a reliable reference for the rational design of nanomaterials for regulating autophagy and provide a promising strategy for improving the therapeutic effect of traditional cancer treatment methods (Scheme 1).
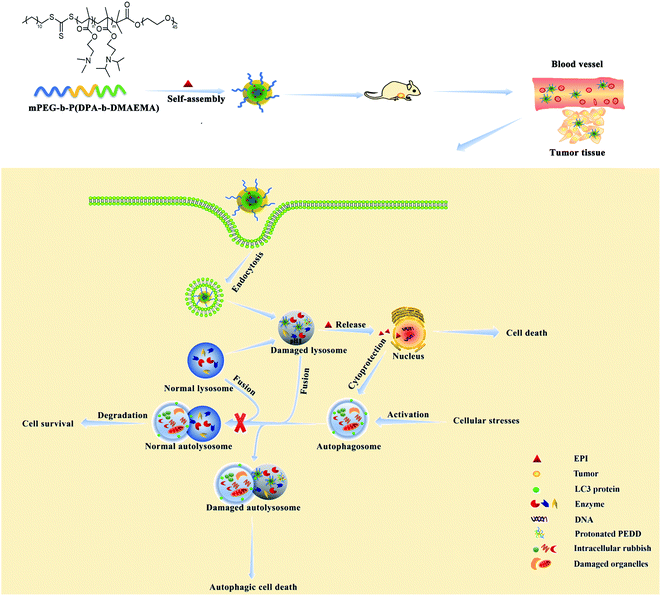 |
| | Scheme 1 Schematic illustration of PEDD/EPI-Ms further enhancing the anti-tumor effect of EPI by inhibiting autophagy. | |
Experimental section
Materials
The chain transfer agent S-1-dodecyl-S-(a,a′-dimethyl-a′′-acetic acid)-trithiocarbonate conjugated mPEG (mPEG-RAFT) was generously provided by Professor Dong Anjie from Tianjin University.33 Azobisisobutyronitrile (AIBN) and 2-diisopropylaminoethyl methacrylate (DPA) were purchased from Sigma-Aldrich (San Francisco, CA, USA). 2-(Dimethylamino)ethyl methacrylate (DMAEMA) was purchased from Aladdin Industrial Corporation (Shanghai, China). Tetrahydrofuran (THF) and N,N-dimethylformamide (DMF) were purchased from Yongda Chemical Reagent Crop (Tianjin, China). LC3 and p62 antibodies were purchased from Abcam. CQ phosphate was obtained from Sigma-Aldrich Chemical Co. (St. Louis, USA). Epirubicin hydrochloride (EPI·HCl) was supplied by Southern Shandong Pharmaceutical Group, Ltd. (Shandong, China). GFP-LC3-adenovirus were purchased from Baolaibo Technology Co. Ltd. (Beijing, China). LysoSensor™ Green DND-189 was obtained from Yeasen BioTechnologies Co. Ltd. (Shanghai, China). Acridine orange (AO) was purchased from Solarbio Corp (Beijing, China). Trypsin–EDTA, phosphate-buffered saline (PBS), Hank's balanced salt solution (HBSS) and RPMI 1640 medium were obtained from Hyclone. Fetal bovine serum (FBS) was purchased from Kangyuan Corporation (Tianjin, China). Male nude mice (4–5 weeks, 16–18 g) were purchased from Vital River Laboratory Animal Technology Co., Ltd. (Zhejiang, China).
Synthesis and characterization of amphiphilic triblock polymer mPEG-b-P(DPA-b-DMAEMA)
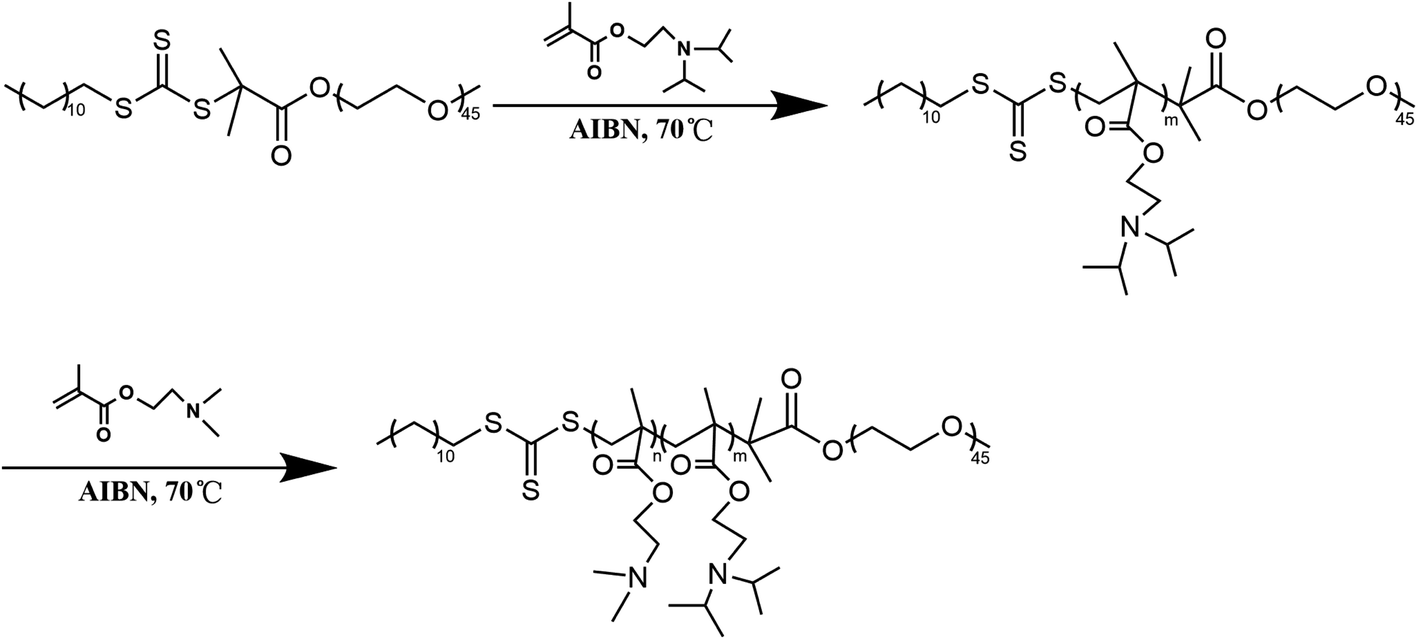
mPEG-b-P(DPA-b-DMAEMA) was synthesized by the reversible addition–fragmentation chain transfer polymerization reaction (RAFT) with mPEG-RAFT as the chain transfer agent, and DPA and DMAEMA as monomers, following a procedure reported earlier with some modification.34,35 Briefly, mPEG-RAFT and DPA (molar ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) were dissolved in 2 mL DMF in a dry reaction tube containing a magnetic stirring rotor. After the reaction content was fully dissolved, an appropriate amount of AIBN was added as an initiator. In the whole reaction process, the reaction mixture was stirred in an oil bath at 70 °C for 24 h under the protection of nitrogen. Then, DMAEMA (mPEG-RAFT/DMAEMA molar ratio = 1:50) and AIBN were added in the presence of DMF into the reaction tube and deoxygenated by purging N2 gas. The reaction tube was kept in an oil bath environment for another 24 h, and then the product was taken out to dialyze for 48 h. Finally, mPEG-b-P(DPA-b-DMAEMA) was freeze-dried for further use. The detailed synthetic route is illustrated in Scheme 2.
:100) were dissolved in 2 mL DMF in a dry reaction tube containing a magnetic stirring rotor. After the reaction content was fully dissolved, an appropriate amount of AIBN was added as an initiator. In the whole reaction process, the reaction mixture was stirred in an oil bath at 70 °C for 24 h under the protection of nitrogen. Then, DMAEMA (mPEG-RAFT/DMAEMA molar ratio = 1:50) and AIBN were added in the presence of DMF into the reaction tube and deoxygenated by purging N2 gas. The reaction tube was kept in an oil bath environment for another 24 h, and then the product was taken out to dialyze for 48 h. Finally, mPEG-b-P(DPA-b-DMAEMA) was freeze-dried for further use. The detailed synthetic route is illustrated in Scheme 2.
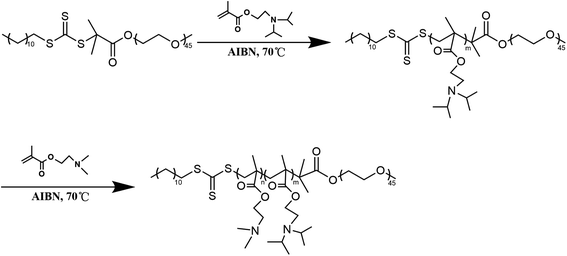 |
| | Scheme 2 The synthetic route of mPEG-b-P(DPA-b-DMAEMA). | |
The structure of mPEG-b-P(DPA-b-DMAEMA) was confirmed via1H NMR (JNM-ECP600, JEOL, Tokyo, Japan). Moreover, using THF as an eluent, the molecular weight and polydispersity (PDI) of the polymers were determined by gel permeation chromatography (GPC) at a flow rate of 1 mL min−1.36 Prior to injected into the GPC systems, all sample solutions were filtered through a 0.45 micron organic phase filter to determine their molecular weight relative to the polystyrene standard.
Preparation of the blank nanomicelles (PEDD-Ms) and EPI-loaded nanomicelles (PEDD/EPI-Ms)
PEDD-Ms were prepared by the dialysis method.37 10 mg of mPEG-b-P(DPA-b-DMAEMA) was completely dissolved in 1 mL THF and added dropwise to 10 mL bicarbonate buffer (pH 11) with continuous stirring. Finally, the micellar solution was stirred overnight and dialyzed for 24 h for further use.
The above steps were repeated to prepare EPI-loaded nanomicelles. In short, the polymers and EPI (10:1, mass ratio) were mixed in 1 mL THF and sonicated for 60 s. Under continuous stirring (1000 rpm), the above mixture was added dropwise to bicarbonate buffer (pH 11). Finally, the drug loaded polymeric nanomicelles were stirred overnight and dialyzed for 24 h to remove free EPI.
Characterization of the blank nanomicelles (PEDD-Ms) and EPI-loaded nanomicelles (PEDD/EPI-Ms)
Particle size, zeta potentials and morphological features of PEDD-Ms and PEDD/EPI-Ms.
After diluting the micellar solution with distilled water, the mean particle diameter (Z-average) and zeta potentials of the nanomicelles were determined via a Malvern Zetasizer Nano ZS90 (British Malvern Instrument Co. Ltd). The morphological features of PEDD-Ms and PEDD/EPI-Ms were observed by using a transmission electron microscope (TEM, JSM-6490LA, Japanese company JEOL). Briefly, a copper grid was immersed in a pre-diluted micellar solution for 3–5 min and then stained with 1% phosphotungstic acid after air-drying. The TEM images of the samples were taken after being dried again with an incandescent lamp.
pH-sensitivity detection of PEDD-Ms.
For the pH-dependent degradation and size changes of PEDD-Ms,38 firstly, 1 mg mL−1 of micellar solution was sufficiently dispersed in 0.1 M buffer saline solutions with a pH of 5.5 and 7.4 respectively (1:4, volume ratio). Then, the centrifuge tubes containing the mixed liquid were placed in an incubator shaker at 37 °C and shook at 200 rpm. At indicated time points, DLS was applied to record changes in the size of nanomicelles. Meanwhile, the morphology of nanomicelles in buffer saline solutions with different pH was observed by TEM, and the specific operation was the same as above.
Hemolytic toxicity study of PEDD-Ms.
To assess the potential hemolytic toxicity of PEDD-Ms, a red blood cell suspension was diluted to 4% with phosphate buffer solution (pH 7.4). 0.1, 0.2, 0.3, 0.5 and 1 mg mL−1 of PEDD-Ms were dispersed in the 4% RBC suspension (1:4, volume ratio). In addition, Triton-X-100 and physiological saline were used as the positive control and negative control, respectively. All samples were incubated in a constant temperature water bath at 37 °C for 2 h and then centrifuged at 4000 rpm for 10 min. The supernatant was collected, and its absorbance was measured at 545 nm using a microplate reader. The percentage of hemolysis was calculated by taking the absorbance of the Triton-X-100 sample as the hemolysis rate of 100%. The hemolysis rate was calculated according to the following equations:
| Hemolysis rate (%) = (APEDD-Ms − Asaline)/(ATriton-X-100 − Asaline) × 100% |
Determination of drug loading.
The drug loading capacity (DLC) and drug loading efficiency (DLE) of EPI-loaded nanomicelles were calculated by measuring the fluorescence intensity of EPI at 480 nm using a fluorescence spectrophotometer. Briefly, an appropriate amount of micellar solution was centrifuged at 4 °C with a high speed and the supernatant containing PEDD/EPI-Ms was collected. Then, EPI loaded in the PEDD/EPI-Ms nanosuspension was extracted by using methanol. The fluorescence intensity of the dissociated EPI in the above-mentioned supernatant was measured at 480 nm wavelength, and then converted to the concentration of loaded EPI by using the prepared standard calibration curve. DLC and DLE were calculated using the following equations:
| DLC (%) = Wloaded EPI/(Wloaded EPI + WPEDD) × 100% |
| DLE (%) = Wloaded EPI/Winitially added EPI × 100% |
Cell culture and cellular uptake study
HepG2 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), streptomycin (100 μg mL−1), and penicillin (100 U mL−1) at 37 °C in a sterile incubator with 5% CO2. The cells were cultured to 80% confluence and then subjected to subsequent experiments.
The cellular uptake behavior of nanomicelles on HepG2 cells was evaluated using confocal microscopy and flow cytometry.39 The HepG2 cells were digested and re-inoculated in confocal Petri dishes. After adhering for 12 h, the cells were treated with fresh RPMI 1640 medium containing free EPI and EPI loaded PEDD-Ms for 1, 2 and 4 h, respectively. Thereafter, the cells were washed with sterile PBS and then stained with pre-prepared Hoechst 33342 at 37 °C for 15 min. After washing again, the images of the cells were obtained by using a confocal laser scanning microscope (CLSM, Nikon, Japan). For quantitative analysis, the cells treated with free EPI and EPI loaded PEDD-Ms were collected and finally analyzed by using a flow cytometer (BD, ACCURI C6).
Intracellular distribution study
For the study of the intracellular release behavior of loaded PEDD-Ms in HepG2 cells, PEDD-Ms was labeled with DID (denoted as PEDD/DID-Ms) to avoid the influence of EPI on the emission wavelength of Lysotracker Green. HepG2 cells were seeded in Petri dishes at a density of 1 × 105 cells per well overnight and treated with DID-labeled PEDD-Ms for 1, 2, 4 and 8 h, respectively. After incubation, the cells were washed with cold PBS and then stained with Lysotracker Green for 15 min. Finally, the intracellular distribution of PEDD/DID-Ms was observed by using a confocal microscope after washing again.
Cell viability assay
The cell viability was analyzed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.40–42 Briefly, the well-growing cells collected by trypsinization were seeded into a 96-well plate at a density of 8 × 103 cells per well and incubated overnight in an incubator at 37 °C to reach ∼80% confluence. After exposing HepG2 cells to samples with various concentrations for 24 h, 10 μL of MTT was added to each well at a concentration of 5 mg mL−1, and the cells were further incubated for 4 h. Then, the formed purple formazan crystals were completely dissolved in DMSO (150 μL), and the absorbance of each well was measured at 492 nm using a microplate reader (Infinite M200 Pro, Swiss TECAN Company).
Effects of nanomicelles on autophagy
Acridine orange (AO) staining.
AO staining assay was performed to observe autolysosomes via a CLSM. In brief, HepG2 cells in the logarithmic growth phase were seeded onto confocal dishes at a density of 1 × 105 cells per well for 12 h and then treated with pre-warmed media containing different samples for another 24 h. Ultimately, the treated cells were washed three times with PBS before being stained with AO dye solution (2 μg mL−1, 1 mL) for 15 min. After washing with PBS, the cells were observed using a CLSM.
Transmission electron microscopy (TEM) assay.
Transmission electron microscopy (TEM) is a gold standard method to observe autolysosomes intuitively.43 In detail, after incubation with 100 μg mL−1 PEDD-Ms for 24 h, the cells were washed with PBS and then harvested using a cell scraper. After centrifugation, the collected cells were fixed with 4% glutaraldehyde overnight, and then fixed with 1% osmium tetroxide for another 1 h. Then, the cells were dehydrated with a graded series of ethanol and subsequently embedded in epoxy resin. The sections were stained with lead citrate for 10 min followed by uranyl acetate for an additional 30 min. After being washed with double distilled water, the sections could be observed under a TEM.
GFP-LC3 transfection assay.
HepG2 cells were seeded on 35 mm glass-bottom dishes overnight in an incubator with suitable conditions and then transfected with adenovirus expressing GFP-LC3 fusion protein for 24 h according to the manufacturer's instructions. The cells were then treated with various samples for the indicated time and subsequently washed with PBS. After that, the nucleus was stained with Hoechst 33342 at 37 °C for 15 min. After washing the cells again, their fluorescence images were obtained using a CLSM.
Western bolting analysis.
The treated cells were harvested and lysed on ice with cell lysis buffer containing protease inhibitors for 30 min. After centrifugation, the supernatant of cell lysates was collected and boiled at 95 °C for 10 min. The total protein content was quantified by using a bicinchoninic acid (BCA) Protein Assay Kit. An equal amount of protein was separated with 15% SDS-PAGE and then transferred onto polyvinylidene fluoride (PVDF) membranes (0.22 μm). Immediately, the PVDF membranes were blocked in TBST buffer containing 5% nonfat milk for 1.5 h with constant shaking at room temperature. After incubation with the primary antibodies (LC3, p62 and GAPDH) overnight at 4 °C, the membranes were washed three times with TBST buffer and then incubated with the secondary antibody at room temperature for 2 h, followed by being washed three times again. Finally, after incubation with an enhanced chemiluminescence (ECL) reagent, the protein bands were imaged with ChemiDoc™ XRS+ with image Lab™ Software (Bio-Rad Laboratories, Hercules, CA, USA).
Effects of nanomicelles on lysosomal stability
Detection of lysosomal volume.
It is generally accepted that lysosomes are enlarged due to lysosomal degradation defects. Lysotracker Red (LTR), which selectively aggregates in cellular acidic compartments (mainly lysosomes), is often used to visually observe the changes in lysosomal volume. After receiving different treatments for 24 h, the cells were incubated with pre-warmed PBS containing LTR for 15 min. Finally, the stained cells were washed with PBS before being observed using a confocal microscope.
Detection of acidity in the lysosomal environment.
To detect whether lysosomes in HepG2 cells exposed to PEDD-Ms were alkalized, Lysotracker Green DND-189, a specific marker of lysosomes, was used for lysosomal pH determination. HepG2 cells were treated with various samples for 24 h after being seeded overnight in confocal Petri dishes at a density of 1 × 105 cells per well. The cells were then stained with pre-warmed PBS containing 1 μM Lysotracker Green DND-189 for 30 min and observed with a CLSM, or the treated cells were resuspended and then analyzed by flow cytometry (FACS).
Acid phosphatase activity assay.
Acid phosphatase activity was quantified as described in previous studies. After being seeded in 6-well plates overnight, HepG2 cells were treated with different samples for 24 h, followed by being washed with ice-cold PBS. Then, the cells were harvested and lysed in ice-cold RIPA lysis buffer without a protease inhibitor for 30 min. After centrifugation (10000 rpm, 10 min), the supernatant of the cell lysate was collected for acid phosphatase activity assay. The total protein content in the lysate was determined by using a BCA protein assay kit. Finally, the acid phosphatase activity of other groups was compared and quantified with the untreated group.
In vivo synergistic therapy study
Male nude mice were fed in a pathogen-free, 50–70% relative humidity and temperature-controlled animal center of Qingdao University. The experiments in vivo were performed in accordance with protocols approved by the Animal Management Rules of the Ministry of Health of the People's Republic of China (document no. 55, 2001) and the examination and approval of the Medical Ethics Committee of Qingdao University Affiliated Hospital (ethical approval number: QYFYQYLL 2020-037).
For the liver cancer model establishment, HepG2 cells (1 × 107 cells) resuspended in 200 μL of sterile PBS were subcutaneously inoculated into the left anterior armpit of nude mice.
In vivo imaging.
A near-infrared (NIR) dye DID was loaded into PEDD-Ms to study the biodistribution of PEDD/DID-Ms in mice.44 The tumor-bearing mice were pre-treated with free DID and PEDD/DID-Ms via tail vein injection and then imaged using a small animal imaging system at 0, 2, 4, 6, 8, 12 and 24 h post injection. After 24 h, the mice were immediately sacrificed by cervical dislocation. Then, the tumors and major organs were collected for in vitro imaging.
In vivo antitumor assay.
On the 7th day after the implantation of HepG2 cells, the tumor-bearing mice were divided into four groups (n = 6) according to the randomized block method and received different formulations every 4 days through tail vein injection: saline, PEDD-Ms, free EPI and PEDD/EPI-Ms (the EPI concentration was 4 mg kg−1 in both groups). The whole treatment lasted 21 days. The tumor volume and body weight were recorded every other day during treatment, and the tumor volume was calculated by using the following formula: volume = (tumor length) × (tumor width)2/2. The results were expressed as the relative tumor volume V/V0, where V and V0 represent the initial tumor volume before treatment and the tumor volume measured every other day after treatment, respectively.
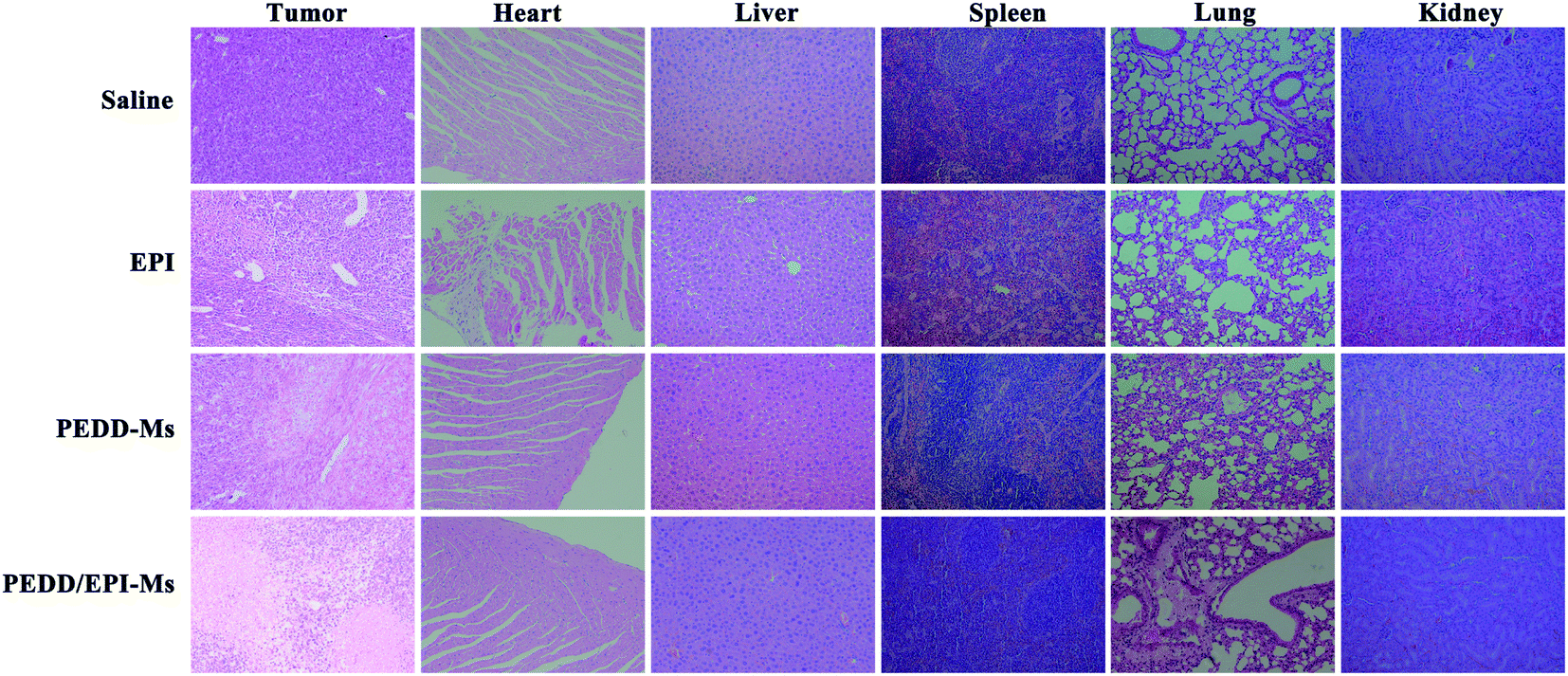
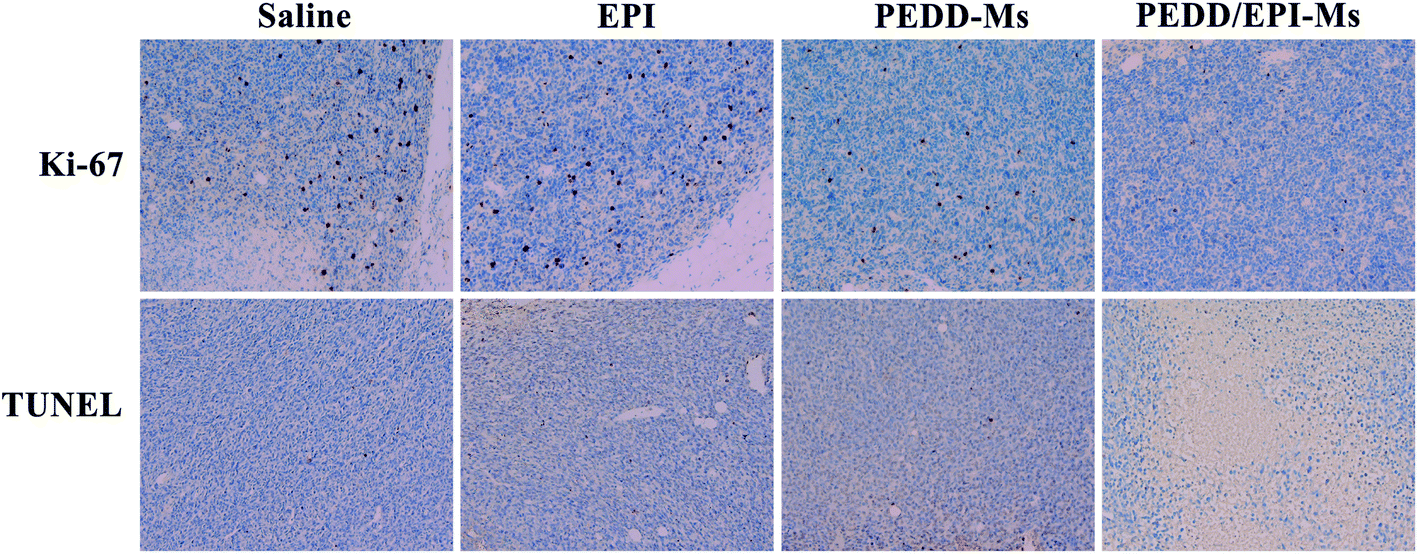
At the end of the whole treatment cycle, mice in each group were sacrificed, and all xenograft tumors were isolated and photographed. Then, the tumor tissues and other organs (heart, liver, spleen, lungs and kidneys) in each group were stained with hematoxylin and eosin (H&E) to detect the pathological changes. In addition, the therapeutic effect of each group was evaluated by immunohistochemical staining (terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) and Ki-67).
Immunofluorescence staining assay.
In addition, the expression levels of LC3 and p62 in tumor tissues were monitored by immunofluorescence staining to evaluate autophagy. As for the immunofluorescence staining assay, paraffin-embedded tissue sections were deparaffinized in xylene for 30 min and then rehydrated with graded ethanol. After repairing the antigen in 1 × citrate antigen repair solution for 30 min at 100 °C, the sections were washed with PBS (pH 7.4) for 15 min and then were blocked with double distilled water containing 5% BSA for 30 min. Subsequently, the sections were incubated with primary antibodies specific for LC3 or p62 at 4 °C overnight, followed by Cy5-labeled LC3 and p62 secondary antibodies at room temperature for 1 h. All images were obtained by using a confocal microscope.
Statistical analysis
In all experiments, all results were expressed as mean ± standard deviation (SD). Significant differences among groups were determined by one-way analysis of variance (ANOVA) and two-tailed Student's t test. A p value less than 0.05 was statistically significant.
Results and discussion
Synthesis and characterization of amphiphilic triblock polymer mPEG-b-P(DPA-b-DMAEMA)
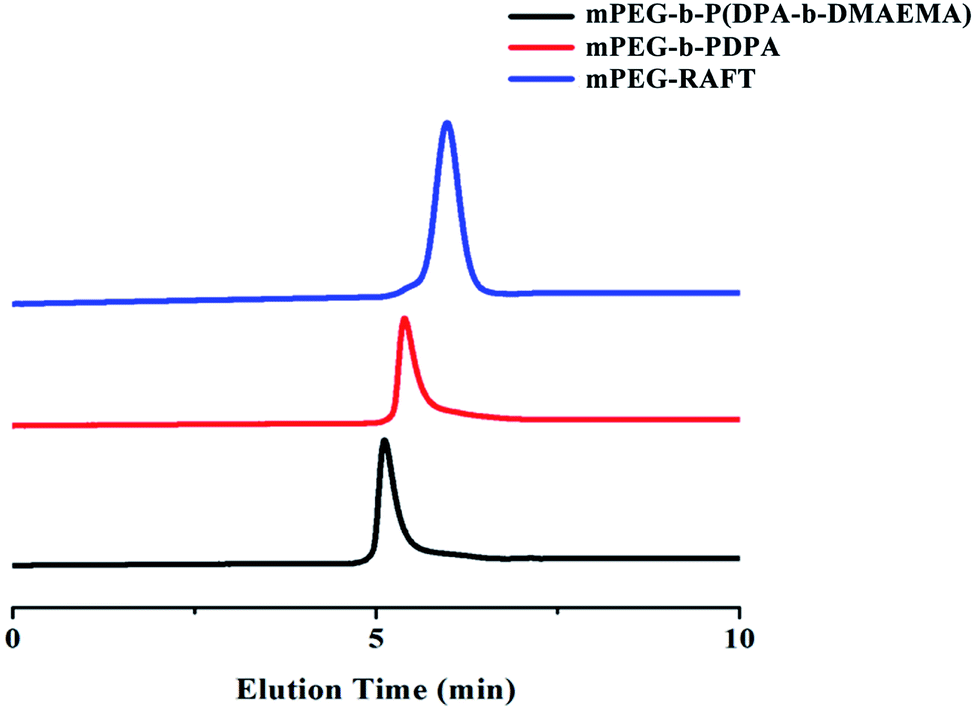
The amphiphilic triblock polymer mPEG-b-P(DPA-b-DMAEMA) was synthesized via the RAFT reaction between mPEG-RAFT, DPA and DMAEMA. The successful synthesis of the polymer was confirmed by 1H NMR and gel permeation chromatography (GPC). The 1H NMR results are shown as ESI Fig. S1.† The sharp peaks at 1.14 ppm (CH3 of NCH(CH3)2, f), 2.30 ppm (CH3 of N(CH3)2, e), and 3.05 ppm (CH of NCH(CH3)2, g) were attributed to the methyl and methylene protons of DPA and DMAEMA, which were marked in the figure. In the GPC spectrum (Fig. 1), the polymer at each stage of the reaction has a unimodal molecular weight distribution, and the elution time showed a continuous shortening trend with the extension of blocks. The molecular weights of the polymers obtained by integration are displayed in Table 1. According to the weight average molecular weight, the structure of the final product was determined to be mPEG-b-P(DPA118-b-DMAEMA36). All the above results confirmed that the triblock polymer mPEG-b-P(DPA-b-DMAEMA) was successfully synthesized with good purity.
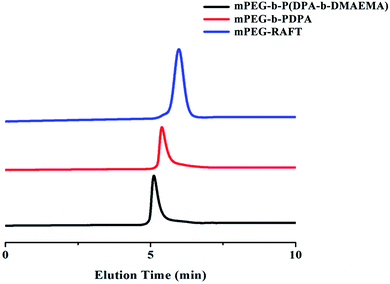 |
| | Fig. 1 The GPC chromatogram of mPEG-b-P(DPA-b-DMAEMA), mPEG-b-PDPA and mPEG-RAFT. | |
Table 1 The molecular weight of mPEG-b-P(DPA-b-DMAEMA), mPEG-b-PDPA and mPEG-RAFT
|
|
M
n
|
M
w
|
PDI |
| mPEG-RAFT |
1791 |
2149 |
1.200 |
| mPEG-b-PDPA |
23638 |
27407 |
1.159 |
| mPEG-b-P(DPA-b-DMAEMA) |
30537 |
33055 |
1.082 |
Preparation and characterization of the blank nanomicelles (PEDD-Ms) and EPI-loaded nanomicelles (PEDD/EPI-Ms)
Particle size, zeta potentials and the morphological features of PEDD-Ms and PEDD/EPI-Ms.
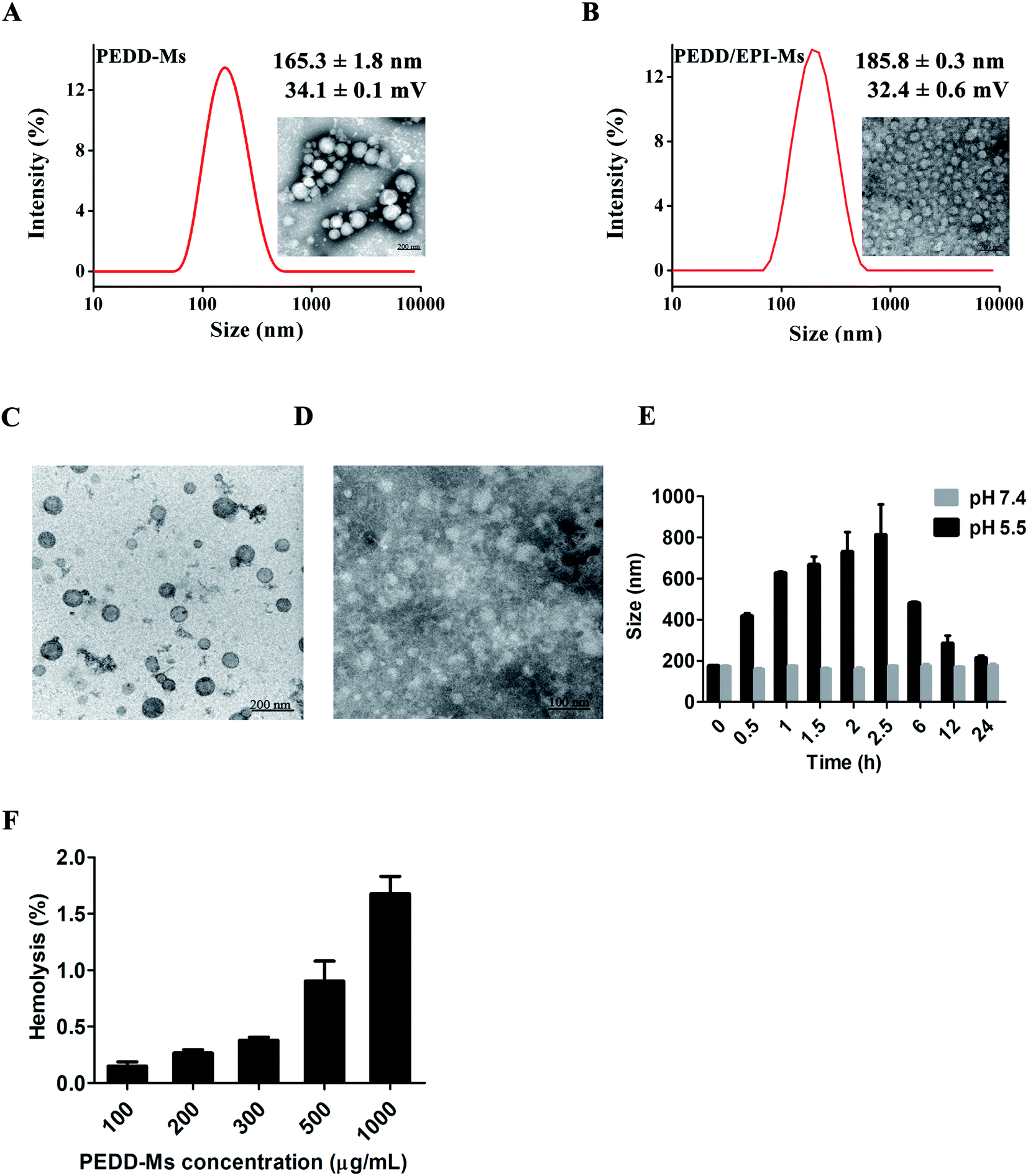
We prepared blank nanomicelles using the dialysis method, and encapsulated EPI into the hydrophobic core to form EPI-loaded nanomicelles. The morphology of PEDD-Ms and PEDD/EPI-Ms was observed by TEM analysis. As shown in Fig. 2A and B, both PEDD-Ms and PEDD/EPI-Ms clearly showed a uniform spherical morphology with an obvious core–shell structure and smooth surface. The size and zeta-potential were determined by using a Malvern Zetasizer Nano ZS90, which showed that the hydrated particle size of PEDD-Ms was 165.3 ± 1.8 nm, and the zeta potential was 34.1 ± 0.1 mV, as well as the size and zeta-potential of PEDD/EPI-Ms were 185.8 ± 0.3 nm and 32.4 ± 0.6 mV, respectively. Nano-sized particles are suitable for deep penetration and accumulation in tumor tissues, and a positive charge on the surface of particles is conducive to cellular uptake via electrostatic adsorption.45
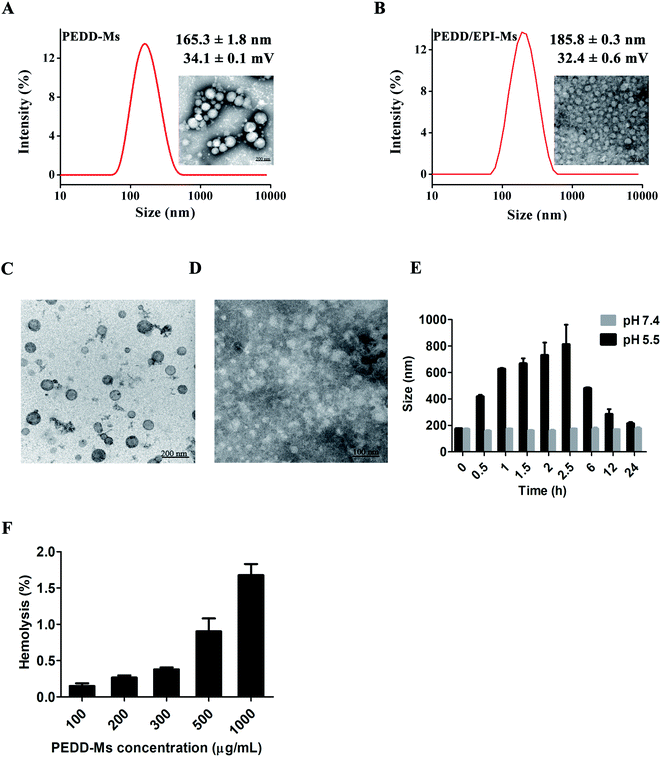 |
| | Fig. 2 Characterization of PEDD-Ms and PEDD/EPI-Ms. Particle size distribution and TEM images of PEDD-Ms (A) and PEDD/EPI-Ms (B). Typical TEM images of PEDD-Ms in different buffer solutions with pH 7.4 (C) and 5.5 (D). (E) Particle size of PEDD-Ms at different time points in buffer solutions with different pH. (F) Hemolytic toxicity study of the polymer mPEG-b-P(DPA-b-DMAEMA). | |
pH-sensitivity detection of PEDD-Ms.
To study the acid sensitivity of PEDD-Ms, the acid-triggered size change of PEDD-Ms has been detected by DLS and TEM in buffer saline solutions with a pH of 5.5 and 7.4, respectively (Fig. 2C–E). DLS analysis revealed that the hydration particle size still showed a stable distribution in a neutral environment. However, the particle size firstly increased and then decreased significantly in the buffer saline solution of pH 5.5, which may be attributed to the continuous protonation of tertiary amine groups in the core, resulting in the expansion and final breakage of the nanomicelles. Moreover, the TEM images showed that the nanomicelles still maintained a stable spherical structure immersed in neutral PBS for 24 h, while the spherical structure was destroyed and the morphology became blurred in the buffer saline solution of 5.5.
In conclusion, the above results indicated that PEDD-Ms had obvious acid sensitivity, which ensured that the carriers disintegrate rapidly and release anti-tumor drugs in the acidic microenvironment of tumors.46
Hemolytic toxicity study of PEDD-Ms.
We further investigated the hemolytic toxicity of PEDD-Ms, in which physiological saline was a negative control and Triton-X-100 was a positive control. The calculated hemolysis rate of PEDD-Ms with various concentrations is shown in Table 2. Moreover, the PEDD-Ms-induced hemolysis rate was dose-dependent (Fig. 2F). Notably, at a relatively high concentration of 1 mg mL−1, the nanomicelles exhibited the strongest hemolysis rate of 1.67%, which showed that the polymer mPEG-b-P(DPA-b-DMAEMA) had good biological safety as a drug delivery vector.
Table 2 Calculation results of the hemolysis rate of PEDD-Ms with various concentrations
| PEDD-Ms (μg mL−1) |
100 |
200 |
300 |
500 |
1000 |
| Hemolysis rate (%) |
0.15 |
0.27 |
0.38 |
0.91 |
1.68 |
Determination of drug loading.
EPI was loaded into the hydrophobic core of nanomicelles using the typical dialysis method, and the EPI-loading efficiency and entrapment efficiency were determined by measuring the fluorescence intensity of EPI using a fluorescence spectrophotometer. As shown in Table 3, the drug loading efficiency was 50.73% with a drug loading content of 4.83% at a feeding weight ratio of 1:10 (EPI/mPEG-b-P(DPA-b-DMAEMA)), suggesting that PEDD-Ms with a hydrophobic core could be employed as an ideal vector for EPI loading.
Table 3 Encapsulation efficiency and drug loading of PEDD/EPI-Ms
|
|
DLE (%) |
DLC (%) |
| PEDD/EPI-Ms |
50.73 ± 0.012 |
4.83 ± 0.001 |
Cellular uptake study
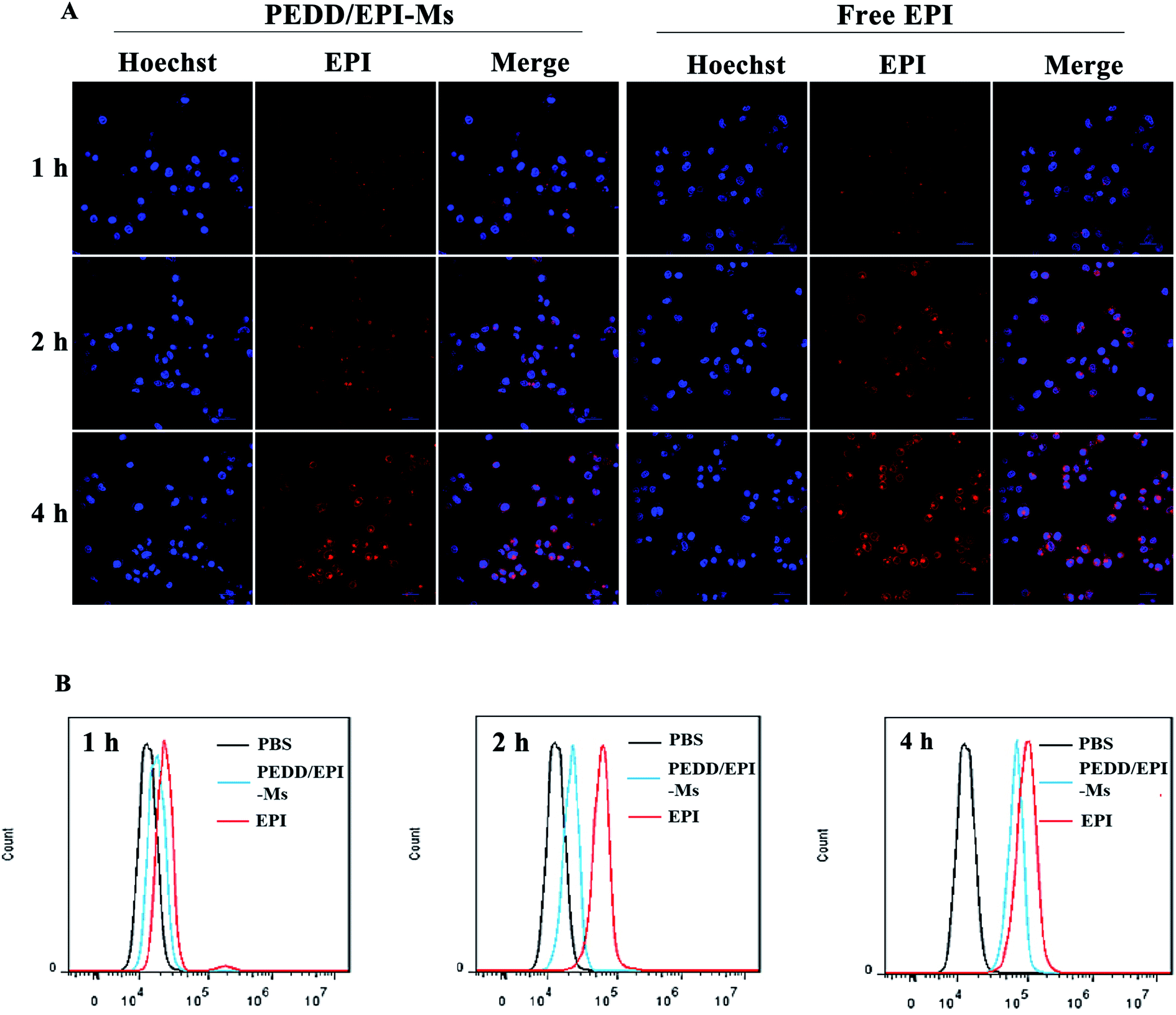
The internalization of nanomicelles by tumor cells is a prerequisite for inducing changes in cellular metabolic pathways. Herein, we evaluated the uptake of EPI-loaded PEDD-Ms and free EPI by HepG2 cells using confocal microscopy and flow cytometry (Fig. 3A and B). The CLSM images showed that the red fluorescence of EPI was observed in both free EPI and PEDD/EPI-Ms-treated cells, and the red fluorescence intensity increased gradually with the extension of incubation time, suggesting the effective internalization of PEDD/EPI-Ms and free EPI. Meanwhile, the red fluorescence could be observed in the nucleus of both cells treated with EPI-loaded PEDD-Ms and free EPI for 4 h. However, the cells treated with free EPI exhibited stronger fluorescence intensity than the cells treated with PEDD/EPI-Ms, further supporting that free EPI, as a small molecular drug, could rapidly diffuse into cells and eventually move into the cell nucleus, while EPI-loaded PEDD-Ms is internalized into cells via an endocytic pathway and could effectively release EPI into the cell nucleus after the endo/lysosomal drug release procedure.47 Additionally, the cellular uptake efficiency was quantified using flow cytometry, which was consistent with the CLSM assay.
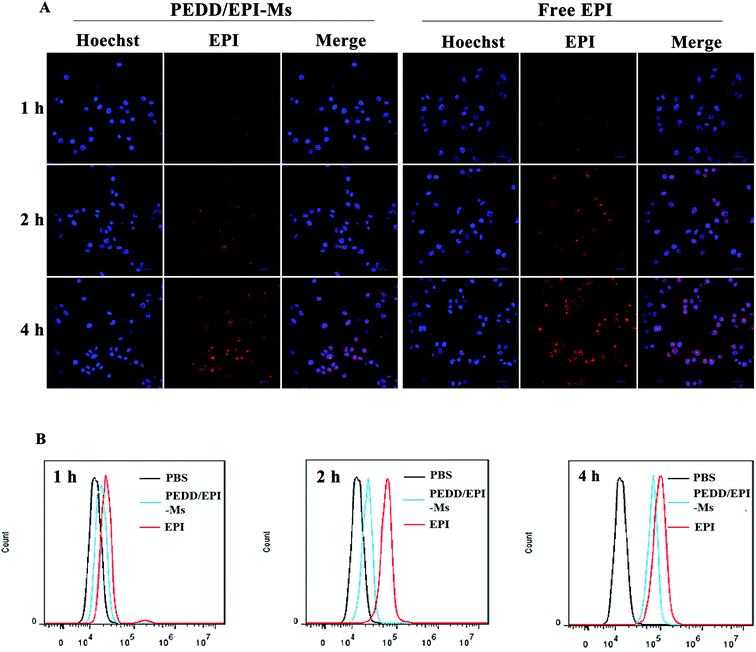 |
| | Fig. 3 Results of the cellular uptake study. (A) Confocal images of HepG2 cells after incubation with EPI-loaded PEDD-Ms and free EPI for 1, 2 and 4 h. Red: the effective internalization of EPI-loaded PEDD-Ms and free EPI; blue: cell nucleus stained with Hoechst 33342; colocalization: EPI entering the nucleus. (B) Flow cytometric analysis of HepG2 cells incubated with PEDD/EPI-Ms and free EPI for 1, 2 and 4 h, respectively. | |
Intracellular distribution study
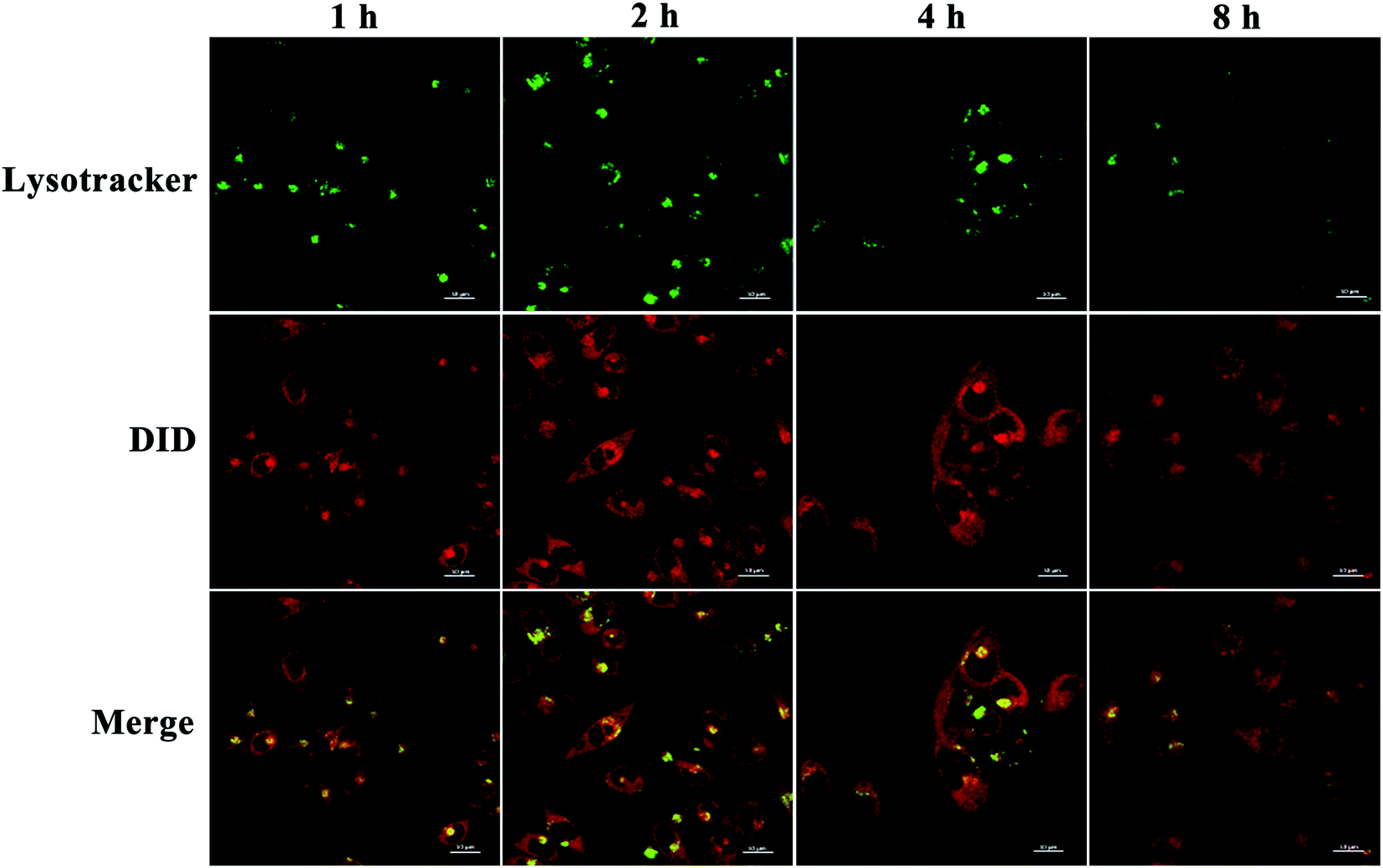
We then assessed the intracellular distribution of PEDD-Ms by CLSM.48 HepG2 cells were treated with PEDD-Ms labeled with DID for 1, 2, 4 and 8 h, respectively. Then, the treated cells were stained with LysoTracker Green DND-26. As shown in Fig. 4, the red fluorescence signals indicated that nanomicelles entered cells effectively through endocytosis. The yellow signals that red fluorescence overlapped with that of LysoTracker Green DND-26 (Green fluorescence, lysosomal labeling) indicated that nanomicelles can be trapped in lysosomes. CLSM images showed that the fluorescence intensity of yellow signals enhanced significantly upon incubation for 2 h, which indicated that nanomicelles entered cells and gradually accumulated in lysosomes. However, the gradually fading yellow signals at 2–8 h suggested that nanomicelles could effectively release loaded DID into the cytoplasm over time maybe due to the proton sponge effect.
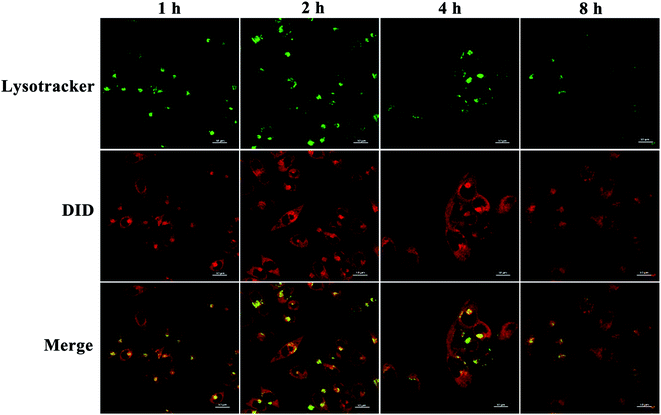 |
| | Fig. 4 Intracellular distribution study. Confocal images of HepG2 cells stained with LysoTracker Green DND-26 after incubation with DID-loaded PEDD-Ms for 1, 2 and 4 h, respectively. | |
PEDD-Ms treatment triggered autophagosome accumulation by blocking autophagic flux
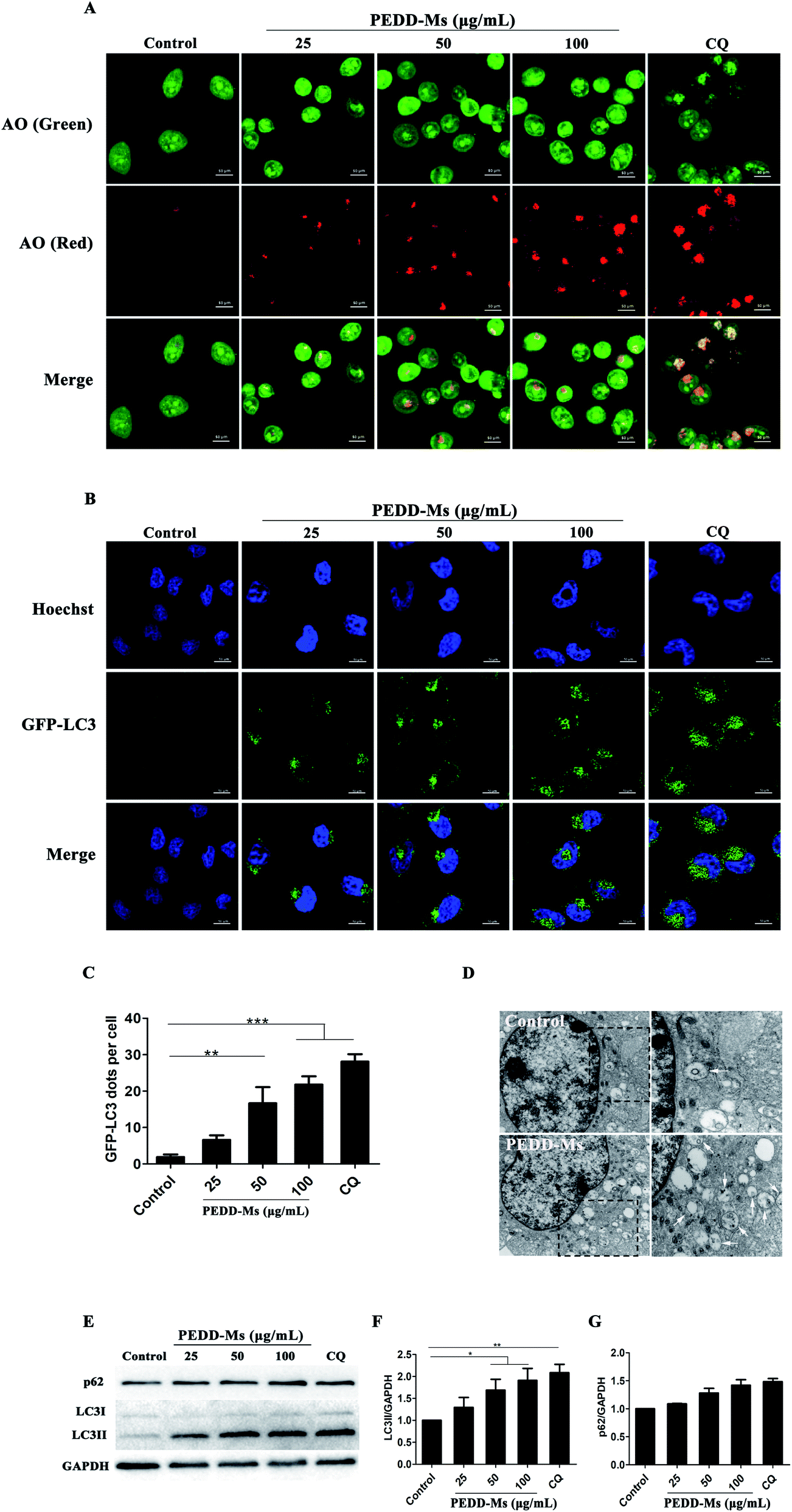
To evaluate the autophagy-modulating effects of PEDD-Ms in HepG2 cells, the fluorescent dye acridine orange (AO) was employed to detect autophagosomes. AO is a pH-sensitive dye that emits green fluorescence in DNA and cytoplasm or protonated in acid vesicular organelles such as autophagosomes to emit red fluorescence.49 CQ (50 μM), a well-known autophagy inhibitor, was used as a positive control. As shown in Fig. 5A, the bright red spots in the confocal images were significantly increased in both PEDD-Ms and CQ-treated cells compared with untreated cells, which confirmed the accumulation of autophagosomes in PEDD-Ms-treated cells and may potentially led to autophagic cell death. Interestingly, the AO-labeled vesicles in the cytoplasm were dose-dependent after exposing cells to a series of concentrations of PEDD-Ms (25, 50 and 100 μg mL−1), reflecting excellent autophagy-modulating activity. In addition, taking HEK-293T human embryonic kidney cells as a non-tumor model, we further studied whether PEDD-Ms is selective to cancer cells in the process of regulating autophagy. As shown in Fig. S2A and B† (ESI), compared with HepG2 cells treated with 100 μg mL−1 PEDD-Ms, the fluorescence intensity of red spots in HEK-293T cells was relatively weak after being exposed to 100 μg mL−1 PEDD-Ms, suggesting that autophagosomes rarely accumulated in HEK-293T cells treated with PEDD-Ms.
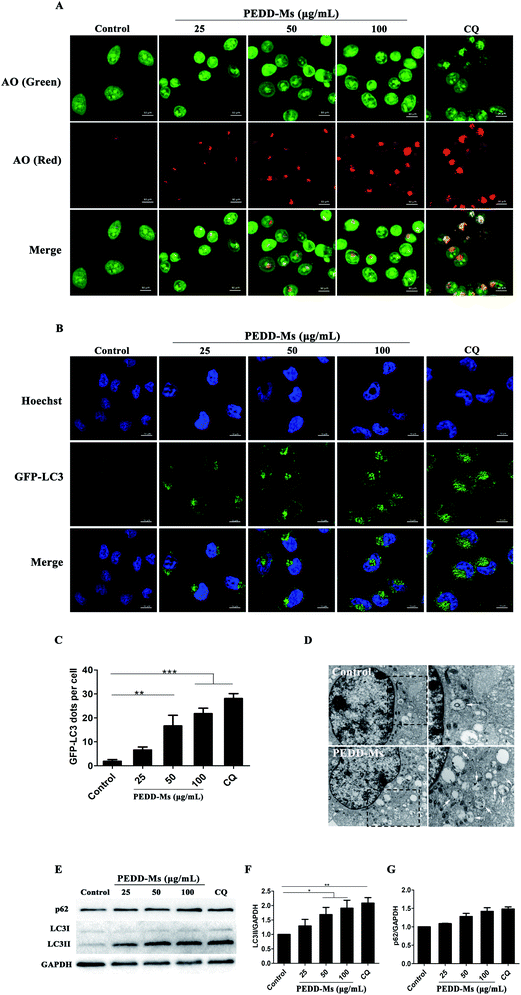 |
| | Fig. 5 PEDD-Ms could induce the accumulation of autophagosomes by blocking autophagic flux. The CLSM images of HepG2 cells treated with CQ and various concentrations of PEDD-Ms for 24 h or without treatment. (A) AO-staining confocal images of HepG2 cells treated with each sample for 24 h. After the same treatment as in (A), representative images (B) and the corresponding quantitative analysis (C) of GFP-LC3 positive puncta of non-treated and treated HepG2 cells. (D) Biological TEM images of HepG2 cells treated with 100 μg mL−1 PEDD-Ms for 24 h or without treatment. The right panel shows a high magnification image of the indicated portion. White arrows indicate autophagosomes and autolysosomes. (E) After HepG2 cells were treated, western blotting was performed with an anti-LC3 antibody and anti-p62 antibody. GAPDH served as the loading control. The semi-quantified results of LC3 (F) and p62 (G) after normalization against GAPDH (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001. | |
We further employed a specific HepG2 cell line and HEK-293T human embryonic kidney cell line that stably expressed green fluorescent protein (GFP)-tagged LC3 (GFP-LC3) to detect the ability of PEDD-Ms to manipulate autophagy (Fig. 5B and C). GFP-LC3 is distributed homogeneously in the cytoplasm of normal growing cells. During autophagy, microtubule-associated protein light chain 3 (LC3-I) can be processed to LC3-II and recruited for autophagosome formation, which appears as green fluorescence puncta on the autophagosome membrane. The CLSM images revealed that both PEDD-Ms and CQ indeed induced the accumulation of GFP-LC3-positive puncta in HepG2 cells compared with untreated cells, indicating a high degree of autophagosome aggregation. Moreover, the PEDD-Ms-induced GFP-LC3 dots were dose-dependent, and the higher concentration of PEDD-Ms induced more bright spots in HepG2 cells. In addition, the green fluorescence puncta in HEK-293T cells treated with 100 μg mL−1 PEDD-Ms were fewer than those of PEDD-Ms-treated HepG2 cells (Fig. S2C and D†). Consistent with AO staining, these results further confirmed that PEDD-Ms had a superior autophagy-regulating function in HepG2 cells that triggered a large amount of autophagosome accumulation in a dose-dependent manner.
TEM is a gold standard method to observe the autophagic ultrastructural features of autophagosomes.50 As shown in Fig. 5D (left panel), TEM images revealed an abnormal accumulation of autophagosomes and other autophagic vesicles in HepG2 cells treated with 100 μg mL−1 PEDD-Ms for 24 h compared with untreated cells. A larger image clearly showed the presence of double-membraned structures containing partially degraded cytoplasmic material in the cytoplasm (Fig. 5D, right panel).
As an autophagosome marker protein, LC3 includes two main forms: LC3-I and LC3-II. During autophagy, soluble LC3-I combines with phosphatidylethanolamine (PE) in the presence of Atg7 and Atg3 to form membrane-associated LC3-II, which is located on both the outer and inner membranes of autophagosomes. Thus, the conversion of LC3 from LC3-I to LC3-II is widely used to monitor autophagic activity.51 Western blot analysis showed that the LC3-II/GAPDH ratio was obviously increased in a dose-dependent manner in PEDD-Ms-treated HepG2 cells (Fig. 5E and F), which further confirmed the accumulation of autophagosomes after PEDD-Ms exposure. However, the accumulation of autophagosomes may be the result of inducing autophagy or inhibiting the degradation of downstream autophagosomes, so we further investigated the expression changes of autophagic protein p62 to distinguish the two possibilities. p62 (Sequestosome-1, SQSTM1) is a ubiquitin-binding protein that preferentially degrades in lysosomes as a selective substrate of autophagy.52 An enhanced p62 protein level is employed as a marker for autophagy inhibition. The results showed that the degradation of the substrate p62 was blocked in PEDD-Ms-treated HepG2 cells, similarly to CQ treatment (Fig. 5E and G), which indicated possible impairment of lysosomal degradation capacity, resulting in autophagic dysfunction. In addition, compared with 100 μg mL−1 PEDD-Ms-treated HepG2 cells, the LC3-II/GAPDH ratio in HEK-293T cells treated with 100 μg mL−1 PEDD-Ms increased by a small margin, and the degradation of p62 was also slightly blocked, reflecting relatively poor autophagy-inhibiting activity (Fig. S2E–G†).
Taken together, the above-described studies proposed that PEDD-Ms treatment could cause the accumulation of autophagosomes by inhibiting autophagic flux in HepG2 cells and may have a certain selectivity toward cancer cells, which is highly conducive to in vivo applications. As for this cell-specific autophagy inhibition, we speculated that it may be due to the higher uptake capacity of cancer cells,53 which leads to the large-scale accumulation of PEDD-Ms in cancer cells. Moreover, cancer cells have vigorous metabolism and a higher anti-apoptosis signal level,54 and any intracellular environmental stress might result in a higher autophagy level than noncancerous cells. Different doses and types of nanomaterials may lead to different autophagy effects. Similarly, pH-sensitive polymeric nanoparticles with different doses also play a paradoxical role in either inducing or inhibiting autophagic activity. A low dose of pH-sensitive nanoparticles induces autophagy in an mTOR dependent manner, but a high dose of pH-sensitive nanoparticles could block autophagic flux and eventually cause autophagic cell death.55 In our study, the autophagic flux could be blocked by 25 μg mL−1 PEDD-Ms after incubation with HepG2 cells for 24 h.
PEDD-Ms treatment could induce autophagic cell death
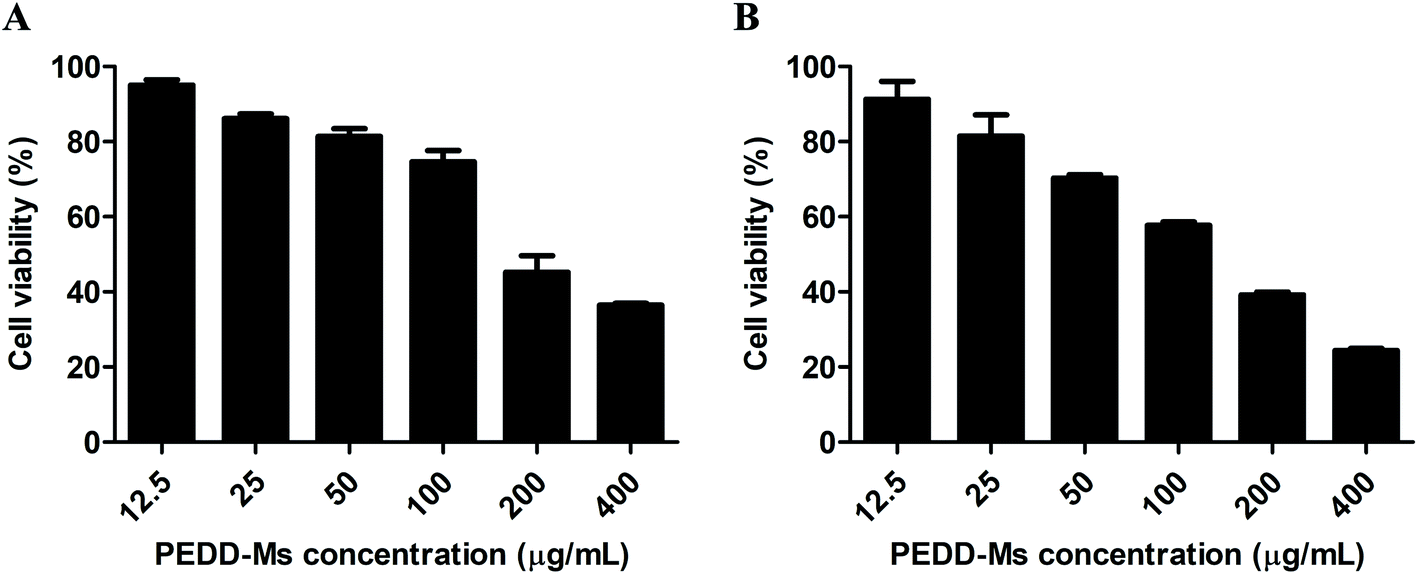
Autophagy plays an irreplaceable role in promoting cell survival and death. In most cases, autophagy usually promotes cell survival during tumorigenesis and chemotherapy, but the inhibition of the autophagy pathway increases cell death. To evaluate the potential cytotoxicity of PEDD-Ms on HepG2 cells, the cell viability was determined by MTT assay after cells were exposed to various concentrations of PEDD-Ms (12.5, 25, 50, 100, 200 and 400 μg mL−1) for 24 h. As shown in Fig. 6A, PEDD-Ms significantly induced cell death in a dose-dependent manner after incubation for 24 h. Starvation is the most widely used stimulus to induce autophagy, so we also treated cells with starvation to assess the possible role of autophagy in PEDD-Ms-induced cytotoxicity (Fig. 6B). Upon starved stimulation, the survival rate of HepG2 cells was significantly lower compared to the PEDD-Ms treatment alone, which indicated that PEDD-Ms-induced cytotoxicity indeed resulted from the autophagy inhibition.
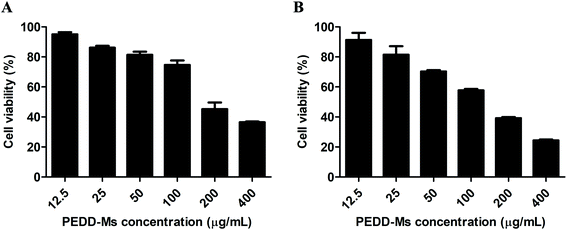 |
| | Fig. 6 PEDD-Ms induced significant autophagic cell death. (A) Cytotoxicity of HepG2 cells incubated with PEDD-Ms under normal conditions for 24 h (n = 3). (B) Upon starved stimulation, the cytotoxicity of HepG2 cells incubated with PEDD-Ms for 24 h (n = 3). | |
PEDD-Ms treatment blocked autophagic flux by impairing lysosomes
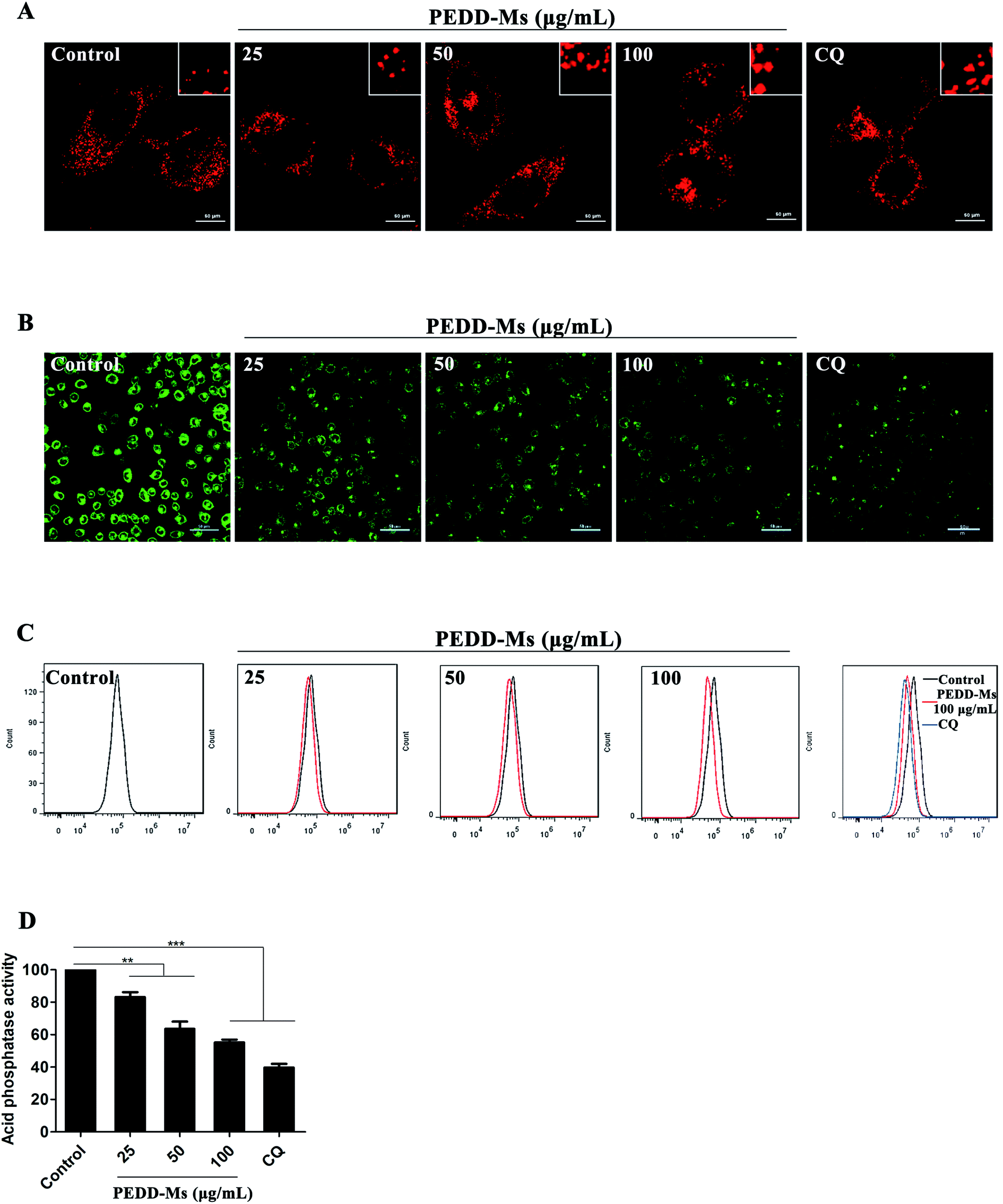
As the major catabolic and recycling centers of eukaryotic cells, lysosomes are closely related to the autophagy process in which they specifically combine with the cytoplasmic components swallowed by autophagosomes for catabolism.56 Mounting evidence strongly confirms that a variety of nanomaterials exhibit remarkable influence on lysosomes because they could accumulate highly in lysosomes through the intracellular endocytosis pathway and cause lysosomal disfunction, eventually resulting in the blockage of autophagic flux.57,58 To further confirm that PEDD-Ms could inhibit autophagic degradation by interfering with lysosomal function, we first analyzed the changes in lysosomal volume by staining HepG2 cells with LysoTracker Red (LTR, a dye specific for lysosomes). CLSM images exhibited that lysosomes in PEDD-Ms-treated cells enlarged clearly in a dose-dependent manner, similarly to CQ-treated cells (Fig. 7A). Lysosomal enlargement is a common marker for lysosomal dysfunction.59 Therefore, we further speculated that PEDD-Ms might impair lysosomal degradation capacity.
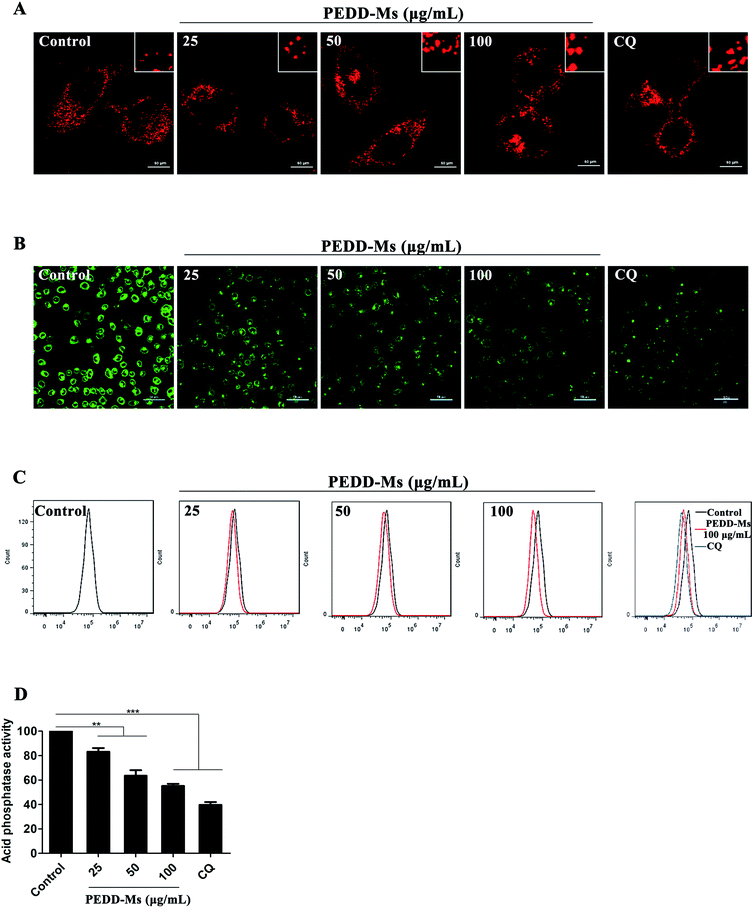 |
| | Fig. 7 Impairment of lysosomes by PEDD-Ms. (A) Confocal images of LTR staining. HepG2 cells were treated with PBS (control), CQ and various concentrations of PEDD-Ms for 24 h and then stained with LTR (red). Inset: close-up of the enlarged lysosomes. (B) Representative fluorescence images of HepG2 cells. After incubation with various samples, the cells were exposed to 1 μg mL−1 LysoSensor Green DND-189 for 30 min. (C) FACS analysis of HepG2 cells treated with 25, 50 and 100 μg mL−1 PEDD-Ms. The rightmost panel shows a comparison of the lysosomal acidity of HepG2 cells treated with PBS, CQ and 100 μg mL−1 PEDD-Ms, respectively. Black line, cells treated with PBS; red line, cells treated with 100 μg mL−1 PEDD-Ms; blue line, cells treated with CQ. (D) With CQ as a positive control, the acid phosphatase activity in HepG2 cells was determined after treatment with 25, 50 and 100 μg mL−1 PEDD-Ms, respectively (n = 3). **p < 0.01, and ***p < 0.001. | |
As it is well known that the acidity of lysosomes determines the lysosomal degradation capacity, we systematically studied the changes in the lysosomal acidic environment. LysoSensor Green DND-189, a specific dye that exhibits green fluorescence in intracellular acidic organelles (mainly lysosomes), was employed to evaluate lysosomal acidity. The fluorescence intensity positively correlates with lysosomal acidity. As shown in Fig. 7B, CQ, a typical autophagy inhibitor that blocks autophagic flux by alkalizing lysosomes, induced a significant reduction of fluorescence intensity. Similarly, the fluorescence intensity of lysosomes treated with PEDD-Ms decreased significantly in a dose-dependent manner, which revealed the dose-dependent alkalization of lysosomes in PEDD-Ms-treated cells. The results were further confirmed by flow cytometry analysis (Fig. 7C).
Lysosomes contain a variety of proteolytic enzymes with an optimal activity pH of 4.5. As a marker enzyme, the proteolytic activity of acid phosphatase can accurately reflect the changes of lysosomal acidity. We found that PEDD-Ms significantly reduced the degradation activity of acid phosphatase in a dose-dependent manner (Fig. 7D), which was consistent with the lysosomal acidity assay results described above, further confirming the conclusion that PEDD-Ms could impair lysosomal degradation capacity and block autophagic flux. Many studies have reported that various pH-sensitive nanoparticles or gold nanoparticles could also cause the accumulation of autophagosomes by alkalizing lysosomes and decreasing proteolytic capacity, thus inducing autophagic cell death, which further support our results.
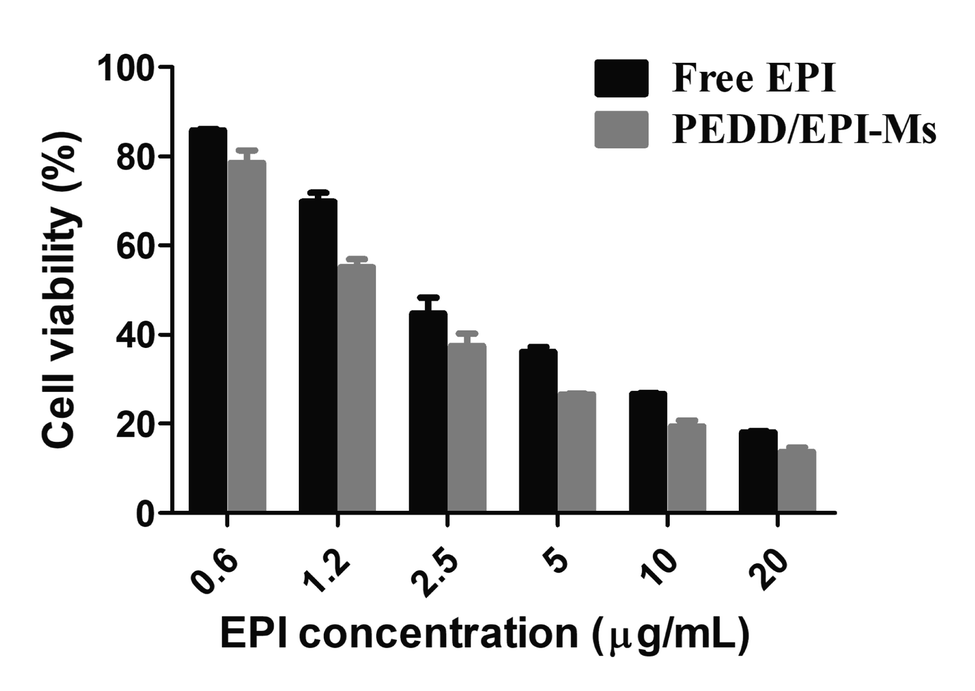
Inhibition of autophagy by PEDD-Ms enhanced the cytotoxicity of EPI
In the above studies, we strongly confirmed that PEDD-Ms could cause the impairment of lysosomal degradation capacity in a dose-dependent manner through lysosomal alkalization when exposed to HepG2 cells, thus blocking autophagic flux. In addition, PEDD-Ms can be employed as an excellent vector for EPI loading, which could be internalized by cells through endocytosis and effectively release EPI into the cell nucleus after the procedure of endo/lysosomal escape. We then evaluated whether the inhibition of autophagy induced by PEDD-Ms could potentiate the lethal effect of EPI on HepG2 cells (Fig. 8). The results showed that compared with the cells treated with free EPI, the survival rate of the cells treated with PEDD/EPI-Ms was significantly decreased, and the combination index (CI) calculated by the Chou-Talalay method was 0.83 ± 0.12, showing an obvious synergistic effect,60 which should be attributed to the fact that PEDD-Ms enhanced the sensitivity of tumor cells to EPI by inhibiting autophagy.
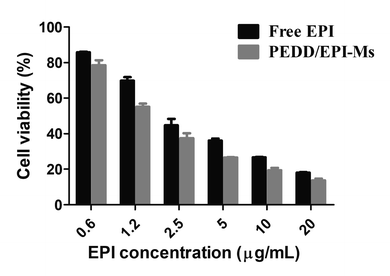 |
| | Fig. 8 Inhibition of autophagy by PEDD-Ms enhanced the cytotoxicity of EPI in HepG2 cells. Cytotoxicity of HepG2 cells incubated with PEDD/EPI-Ms or free EPI for 24 h, respectively (n = 3). | |
In conclusion, the above results supported the view that PEDD-Ms could be utilized as a sensitizer for the chemo-therapeutic agent EPI to enhance the susceptibility of HepG2 cells to death through modulation of autophagy.
In vivo imaging and antitumor efficiency
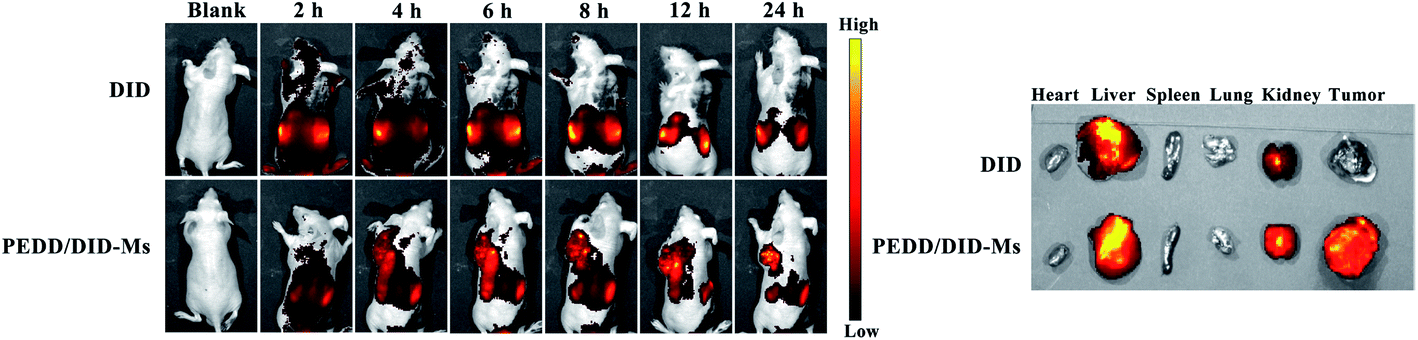
PEDD-Ms delivers EPI specifically to tumor target sites to achieve rapid blocking of autophagic flux and fixed-point release of EPI, which is the key to determine its in vivo anti-tumor effect. As shown in Fig. 9, the biological distribution of PEDD/DID-Ms nanomicelles was monitored in nude mice. Here, free DID was used as a control. The fluorescence images of the nude mice at different time points after tail vein injection of PEDD/DID-Ms and free DID were recorded by using a multispectral imaging system. As shown in mice injected with PEDD/DID-Ms, the DID signals at tumor sites could be distinctly observed over time, and the tumor still exhibited strong signals up to 24 h, whereas free DID tended to accumulate in the abdomen due to the lack of tumor-targeting capacity. The mice were executed at 24 h, and tumors or major organs were dissected for imaging. Compared with free DID, PEDD/DID-Ms not only accumulated in livers and kidneys, but also indeed significantly enhanced fluorescence signals in tumor, indicating the specific tumor-targeting capacity of PEDD/DID-Ms through the EPR effect based on the nanoscale size.
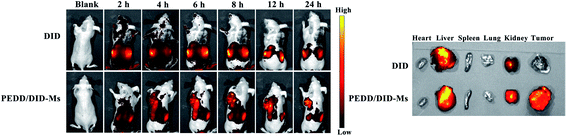 |
| | Fig. 9 PEDD-Ms could promote tumor targeting in vivo. Fluorescence imaging in vivo at 2, 4, 6, 8, 10, 12 and 24 h after tail vein injection of free DID and PEDD/DID-Ms in tumor bearing mice (left) and in vitro tissue imaging at 24 h (right). | |
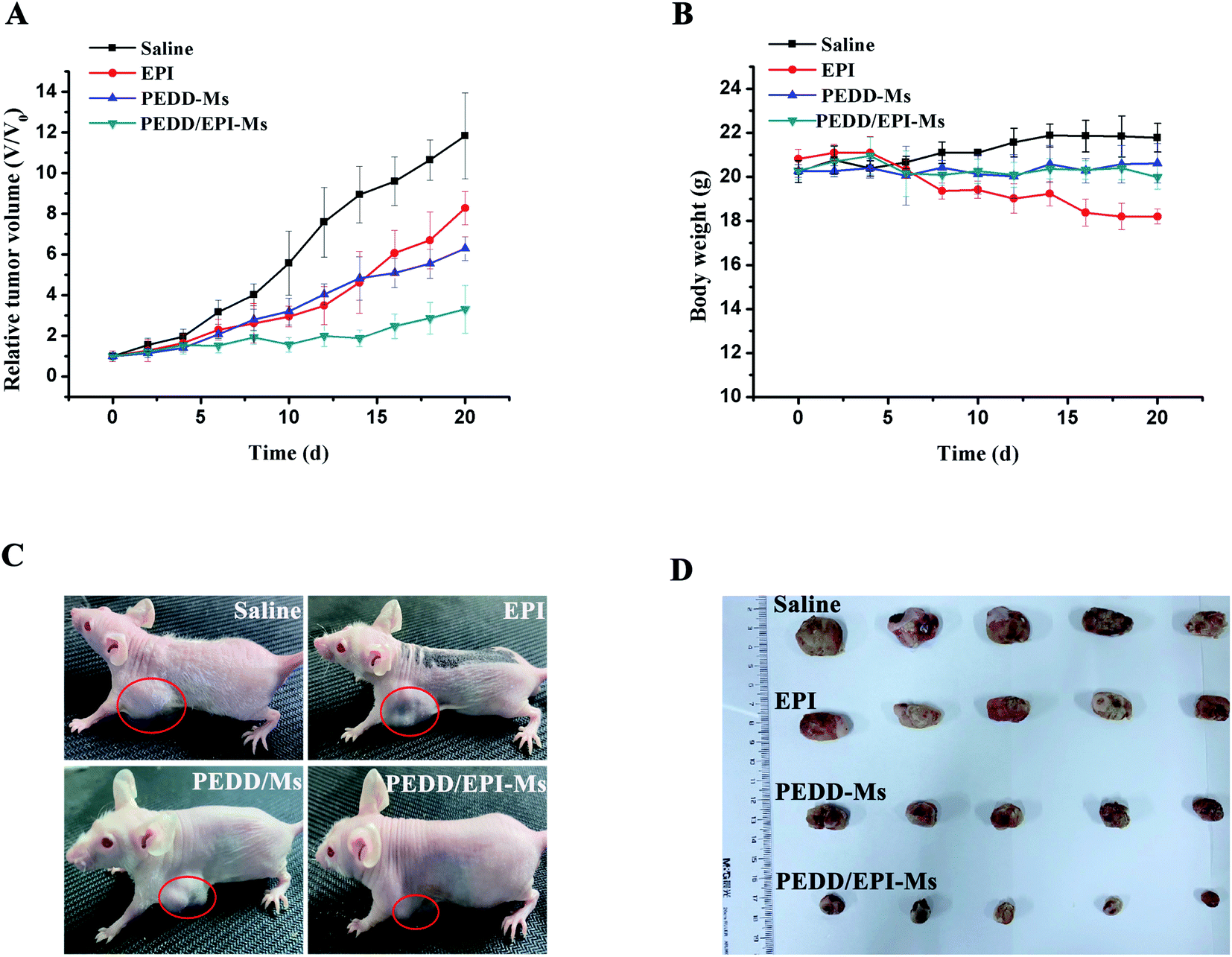
Subsequently, we evaluated the anti-tumor efficacy of PEDD/EPI-Ms in HepG2 tumor-bearing mice. The mice were randomly divided into 4 groups and then treated with various treatments. The volume of tumors is shown in Fig. 10A. Compared with the saline group, the individual EPI-treated group exhibited a slight inhibition in tumor growth, maybe due to the fact that free EPI lacked tumor-targeting ability and was metabolized quickly in vivo. Furthermore, the PEDD-Ms group also exhibited certain anti-tumor efficacy due to its specific tumor-targeting and autophagy-inhibiting ability. Interestingly, the PEDD/EPI-Ms group showed the best therapeutic efficacy during the whole treatment, which may be attributed to the fact that PEDD/EPI-Ms could specifically deliver EPI to tumor sites and further inhibit EPI-induced cellular protective autophagy. The representative photographs of the mice and excised tumors after 21 days are shown in Fig. 10C and D, and there were no significant changes in the body weight except for the free EPI group with severe toxicity (Fig. 10B), demonstrating the good biological safety of these treatments. Histological examination of main organs and tumor tissues was also evaluated by H&E staining assay (Fig. 11). No significant histological changes were observed in the main organs after receiving different treatments except for a typical myocardial injury caused by free EPI. In contrast, PEDD/EPI-Ms induced the most severe damage in tumor tissues compared to saline, indicating the efficient tumor suppression of combined therapy. TUNEL assay and Ki-67 results also validated that PEDD/EPI-Ms could effectively inhibit the proliferation of tumor cells and induce their apoptosis (Fig. 12).
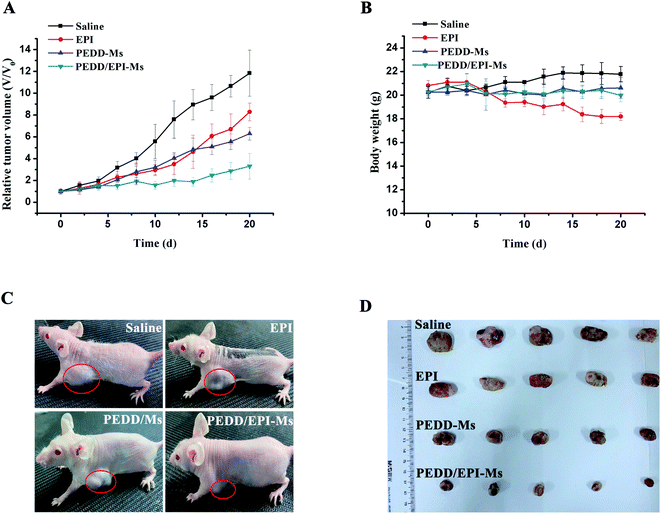 |
| | Fig. 10 PEDD/EPI-Ms improved EPI-based therapy in vivo. Relative tumor volume (A) and body weight (B) curves obtained during treatment (n = 5). Photographs of tumor-bearing mice (C) and excised tumor tissues (D) after intravenous administration of different formulations. | |
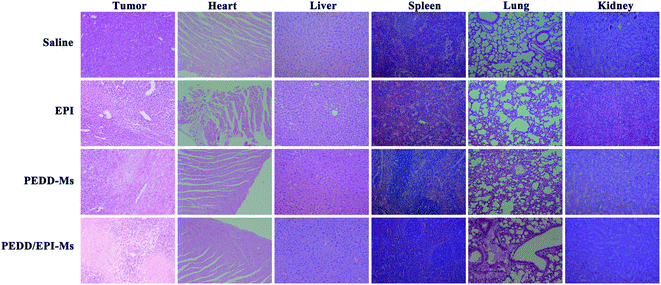 |
| | Fig. 11 Pathological features of tumor tissues and organs. The isolated tumors and organs were stained with hematoxylin and eosin (H&E). | |
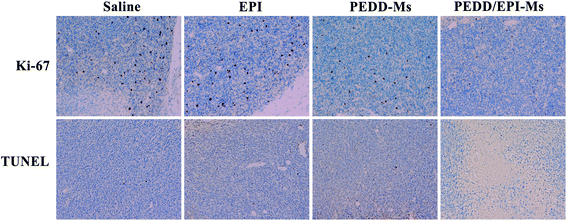 |
| | Fig. 12 Additional treatment characteristics induced by saline, EPI, PEDD-Ms and PEDD/EPI-Ms. Immunohistochemistry analysis (Ki-67 and TUNEL) of a xenograft tumor. The brown area represents proliferation and apoptosis in Ki-67 and TUNEL images, respectively. | |
Immunofluorescence staining assay
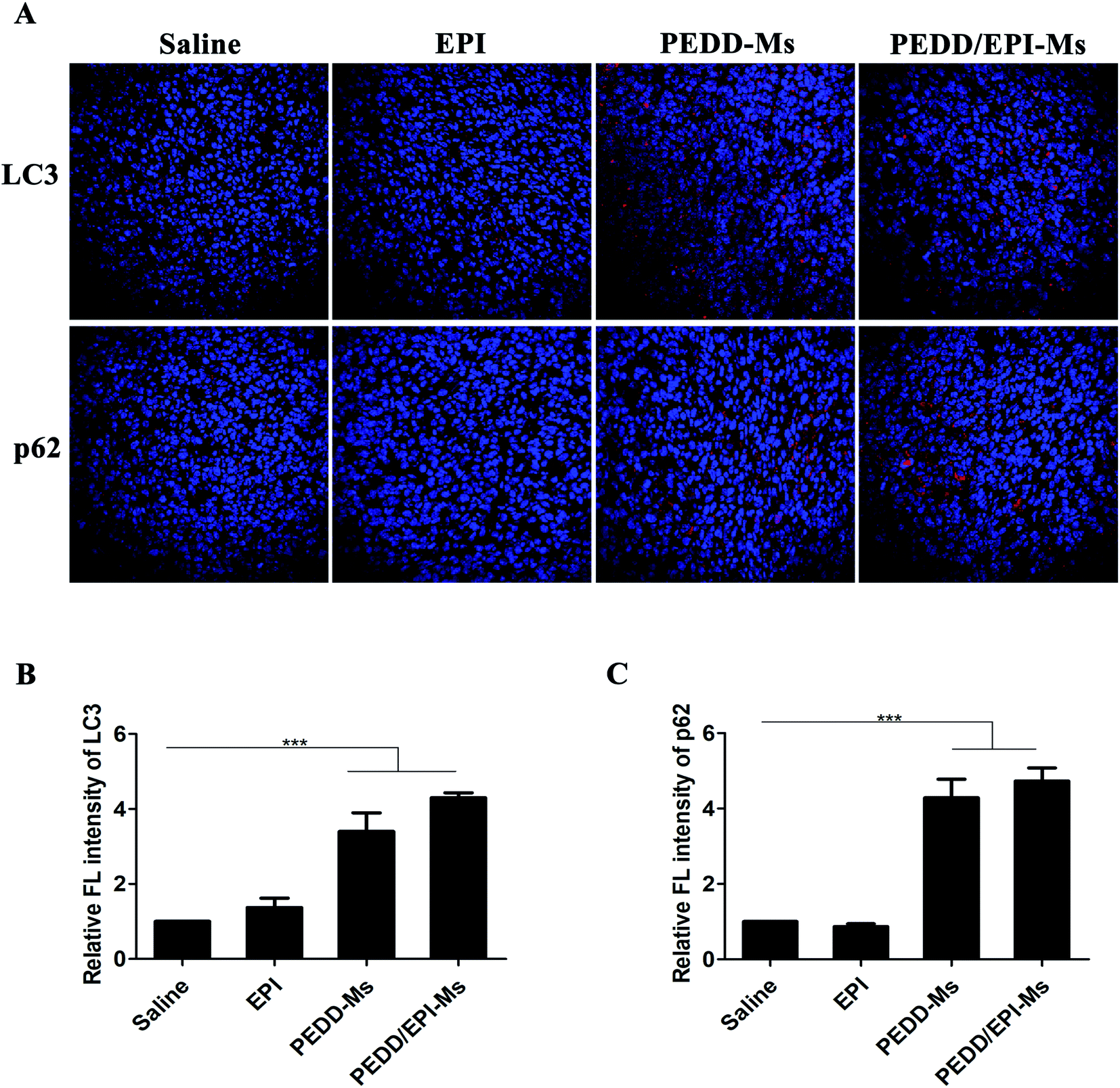
To determine the effect of PEDD/EPI-Ms on the autophagy of tumor sites in vivo, immunofluorescence analysis was performed to detect the expression levels of LC3 and p62 proteins in tumor tissues (Fig. 13). The expression of LC3 increased significantly in the PEDD-Ms and PEDD/EPI-Ms groups and slightly in the free EPI treatment group, which might be due to the autophagy induction caused by the low enrichment of free EPI in tumors. The results of immunofluorescence staining of p62 suggested that PEDD-Ms and PEDD/EPI-Ms could effectively increase the expression of P62 in tumor tissues, which combined with the expression of LC3 indicated that the autophagic flux was blocked.
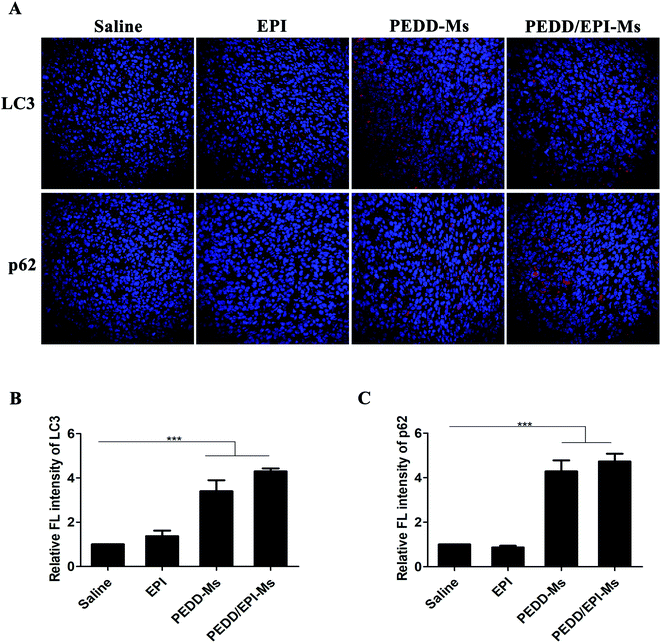 |
| | Fig. 13 Autophagic features triggered by saline, EPI, PEDD-Ms and PEDD/EPI-Ms in vivo. (A) Immunofluorescence staining of tumor tissues. Red fluorescence: LC3 and P62; blue fluorescence: nucleus. The quantitative fluorescence intensity of LC3 (B) and p62 (C) after normalization against the control group (n = 3). ***p < 0.001. | |
All in vivo results indicated that PEDD/EPI-Ms may be a nano-platform to achieve enhanced autophagy inhibition and tumor destruction. PEDD/EPI-Ms could effectively improve the anti-tumor effect, largely because PEDD/EPI-Ms could selectively deliver EPI to tumors through the EPR effect and specifically accumulate in lysosomes after endocytosis into tumor cells. The rapid protonation of PEDD polycations in the acidic environment of lysosomes leads to the alkalization of lysosomes and the blockage of autophagic flux, which further enhanced the anti-tumor effect of EPI and induced more apoptosis of tumor cells.
Conclusions
Autophagy is a lysosome-based degradation pathway to maintain cellular homeostasis. Increasing evidence suggests that autophagy is an effective target for cancer diagnosis and therapy. In this manuscript, we designed a polycationic delivery carrier to address two major issues: (i) the accuracy of targeted tumor administration and (ii) the synergistic treatment of nanomaterial-mediated autophagy inhibition and chemotherapeutic agents. It was found that the nano-carriers could significantly inhibit autophagic flux by alkalizing lysosomes in a dose-dependent manner, enhancing the lethal effect of EPI on tumors, which indicated that a pH triggered drug delivery system with effective autophagy-regulating ability in combination with chemotherapeutic agents may provide a promising strategy for improving the efficacy of tumor treatment. In conclusion, our results systematically revealed the potential of nanomaterials in the regulation of autophagy and enhancement of antitumor efficacy, which enhanced our understanding of the autophagy mediated by polycationic nanomaterials and may provide an important complement to existing tumor therapies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [81673360], Major Science and Technology Innovation Projects of Shandong Province [2018CXGC1408].
Notes and references
- L. Galluzzi and D. R. Green, Cell, 2019, 177, 1682–1699 CrossRef CAS.
- N. Mizushima and M. Komatsu, Cell, 2011, 147, 728–741 CrossRef CAS.
- N. Mizushima and L. O. Murphy, Trends Biochem. Sci., 2020, 45, 1080–1093 CrossRef CAS.
- A. Fleming and D. C. Rubinsztein, Trends Neurosci., 2020, 43, 767–779 CrossRef CAS.
- J. Liu, Y. Kang, S. Yin, A. Chen, J. Wu, H. Liang and L. Shao, Small, 2019, 15, e1901073 CrossRef.
- A. V. Onorati, M. Dyczynski, R. Ojha and R. K. Amaravadi, Cancer, 2018, 124, 3307–3318 CrossRef.
- M. Damme, T. Suntio, P. Saftig and E. L. Eskelinen, Acta Neuropathol., 2015, 129, 337–362 CrossRef CAS.
- W. E. Hughes, A. M. Beyer and D. D. Gutterman, Basic Res. Cardiol., 2020, 115, 41 CrossRef.
- Y. Xia, N. Liu, X. Xie, G. Bi, H. Ba, L. Li, J. Zhang, X. Deng, Y. Yao, Z. Tang, B. Yin, J. Wang, K. Jiang, Z. Li, Y. Choi, F. Gong, X. Cheng, J. J. O'Shea, J. J. Chae, A. Laurence and X. P. Yang, Autophagy, 2019, 15, 960–975 CrossRef CAS.
- H. T. Chen, H. Liu, M. J. Mao, Y. Tan, X. Q. Mo, X. J. Meng, M. T. Cao, C. Y. Zhong, Y. Liu, H. Shan and G. M. Jiang, Mol. Cancer, 2019, 18, 101 CrossRef.
- Z. Yang and D. J. Klionsky, Nat. Cell Biol., 2010, 12, 814–822 CrossRef CAS.
- F. Nazio, M. Bordi, V. Cianfanelli, F. Locatelli and F. Cecconi, Cell Death Differ., 2019, 26, 690–702 CrossRef CAS.
- L. Poillet-Perez, X. Xie, L. Zhan, Y. Yang, D. W. Sharp, Z. S. Hu, X. Su, A. Maganti, C. Jiang, W. Lu, H. Zheng, M. W. Bosenberg, J. M. Mehnert, J. Y. Guo, E. Lattime, J. D. Rabinowitz and E. White, Nature, 2018, 563, 569–573 CrossRef CAS.
- J. M. M. Levy, C. G. Towers and A. Thorburn, Nat. Rev. Cancer, 2017, 17, 528–542 CrossRef CAS.
- X. Chen, Q. Yu, Y. Liu, Q. Sheng, K. Shi, Y. Wang, M. Li, Z. Zhang and Q. He, Acta Biomater., 2019, 99, 339–349 CrossRef.
- F. Qu, P. Wang, K. Zhang, Y. Shi, Y. Li, C. Li, J. Lu, Q. Liu and X. Wang, Autophagy, 2020, 16, 1413–1435 CrossRef CAS.
- H. Zhang, Y. Ren, F. Cao, J. Chen, C. Chen, J. Chang, L. Hou and Z. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 29641–29654 CrossRef CAS.
- Z. Wang, J. Zhang, Y. Wang, R. Xing, C. Yi, H. Zhu, X. Chen, J. Guo, W. Guo, W. Li, L. Wu, Y. Lu and S. Liu, Carcinogenesis, 2013, 34, 128–138 CrossRef CAS.
- L. Galluzzi, J. M. Bravo-San Pedro, S. Demaria, S. C. Formenti and G. Kroemer, Nat. Rev. Clin. Oncol., 2017, 14, 247–258 CrossRef CAS.
- X. Zhao, F. Bian, L. Sun, L. Cai, L. Li and Y. Zhao, Small, 2020, 16, e1901943 CrossRef.
- Q. Wu, R. Jin, T. Feng, L. Liu, L. Yang, Y. Tao, J. M. Anderson, H. Ai and H. Li, Int. J. Nanomed., 2017, 12, 3993–4005 CrossRef CAS.
- G. Huang, Z. Liu, L. He, K. H. Luk, S. T. Cheung, K. H. Wong and T. Chen, Biomater. Sci., 2018, 6, 2508–2517 RSC.
- J. Wang, Y. Li, J. Duan, M. Yang, Y. Yu, L. Feng, X. Yang, X. Zhou, Z. Zhao and Z. Sun, Autophagy, 2018, 14, 1185–1200 CrossRef CAS.
- Y. Zhang, R. Sha, L. Zhang, W. Zhang, P. Jin, W. Xu, J. Ding, J. Lin, J. Qian, G. Yao, R. Zhang, F. Luo, J. Zeng, J. Cao and L. P. Wen, Nat. Commun., 2018, 9, 4236 CrossRef.
- M. A. Moosavi, M. Sharifi, S. M. Ghafary, Z. Mohammadalipour, A. Khataee, M. Rahmati, S. Hajjaran, M. J. Łos, T. Klonisch and S. Ghavami, Sci. Rep., 2016, 6, 34413 CrossRef CAS.
- M. Cordani and Á. Somoza, Cell. Mol. Life Sci., 2019, 76, 1215–1242 CrossRef CAS.
- Y. X. Lin, Y. Wang, S. L. Qiao, H. W. An, R. X. Zhang, Z. Y. Qiao, R. P. Rajapaksha, L. Wang and H. Wang, Small, 2016, 12, 2921–2931 CrossRef CAS.
- K. Peynshaert, B. B. Manshian, F. Joris, K. Braeckmans, S. C. De Smedt, J. Demeester and S. J. Soenen, Chem. Rev., 2014, 114, 7581–7609 CrossRef CAS.
- E. Panzarini and L. Dini, Mol. Pharm., 2014, 11, 2527–2538 CrossRef CAS.
- C. M. Chang, K. L. Lan, W. S. Huang, Y. J. Lee, T. W. Lee, C. H. Chang and C. M. Chuang, Int. J. Mol. Sci., 2017, 18, 903 CrossRef.
- G. Wang, K. Qian and X. Mei, Nanoscale, 2018, 10, 10467–10478 RSC.
- J. Liu, Y. Qu, T. Zheng and Y. Tian, Chem. Commun., 2019, 55, 9673–9676 RSC.
- C. Wang, L. Du, J. Zhou, L. Meng, Q. Cheng, C. Wang, X. Wang, D. Zhao, Y. Huang, S. Zheng, H. Cao, J. Zhang, L. Deng, Z. Liang and A. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 32463–32474 CrossRef CAS.
- S. Zhong, C. Chen, G. Yang, Y. Zhu, H. Cao, B. Xu, Y. Luo, Y. Gao and W. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 33697–33705 CrossRef CAS.
- E. Chen, S. Han, B. Song, L. Xu, H. Yuan, M. Liang and Y. Sun, Int. J. Nanomed., 2020, 15, 6311–6324 CrossRef CAS.
- Q. Huo, Y. Liang, W. Lu, X. Peng, C. Du, Y. Sun, S. Han, J. Cao, Y. Sun and B. He, J. Biomed. Nanotechnol., 2019, 15, 1673–1687 CrossRef CAS.
- Y. Li, Y. Wu, J. Chen, J. Wan, C. Xiao, J. Guan, X. Song, S. Li, M. Zhang, H. Cui, T. Li, X. Yang, Z. Li and X. Yang, Nano Lett., 2019, 19, 5806–5817 CrossRef CAS.
- L. Jiang, Y. Liang, Q. Huo, Y. Pu, W. Lu, S. Han, J. Cao, B. He and Y. Sun, J. Biomed. Nanotechnol., 2018, 14, 1409–1419 CrossRef CAS.
- X. Chen, Q. Yu, Y. Liu, Q. Sheng, K. Shi, Y. Wang, M. Li, Z. Zhang and Q. He, Acta Biomater., 2019, 99, 339–349 CrossRef.
- Z. Zhou, Y. Yan, L. Wang, Q. Zhang and Y. Cheng, Biomaterials, 2019, 203, 63–72 CrossRef CAS.
- Y. Sun, Y. Liang, N. Hao, X. Fu, B. He, S. Han, J. Cao, Q. Ma, W. Xu and Y. Sun, Nanoscale, 2020, 12, 5380–5396 RSC.
- L. Jiang, Y. Liang, Q. Huo, Y. Pu, W. Lu, S. Han, J. Cao, B. He and Y. Sun, J. Biomed. Nanotechnol., 2018, 14, 1409–1419 CrossRef CAS.
- Y. X. Lin, Y. Wang and H. Wang, Small, 2017, 13, 1700996 CrossRef.
- C. Du, Y. Liang, Q. Ma, Q. Sun, J. Qi, J. Cao, S. Han, M. Liang, B. Song and Y. Sun, J. Nanobiotechnol., 2019, 17, 113 CrossRef.
- W. Heni, L. Vonna and H. Haidara, Nano Lett., 2015, 15, 442–449 CrossRef CAS.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef CAS.
- C. Pichon, L. Billiet and P. Midoux, Curr. Opin. Biotechnol., 2010, 21, 640–645 CrossRef CAS.
- R. Ge, J. Cao, J. Chi, S. Han, Y. Liang, L. Xu, M. Liang and Y. Sun, Int. J. Nanomed., 2019, 14, 4931–4947 CrossRef CAS.
- E. Tasdemir, L. Galluzzi, M. C. Maiuri, A. Criollo, I. Vitale, E. Hangen, N. Modjtahedi and G. Kroemer, Methods Mol. Biol., 2008, 445, 29–76 CrossRef CAS.
- E. L. Eskelinen, F. Reggiori, M. Baba, A. L. Kovács and P. O. Seglen, Autophagy, 2011, 7, 935–956 CrossRef CAS.
- T. Kaizuka, H. Morishita, Y. Hama, S. Tsukamoto, T. Matsui, Y. Toyota, A. Kodama, T. Ishihara, T. Mizushima and N. Mizushima, Mol. Cell, 2016, 64, 835–849 CrossRef CAS.
- B. Rathore, K. Sunwoo, P. Jangili, J. Kim, J. H. Kim, M. Huang, J. Xiong, A. Sharma, Z. Yang, J. Qu and J. S. Kim, Biomaterials, 2019, 211, 25–47 CrossRef CAS.
- S. J. Soenen, D. Vercauteren, K. Braeckmans, W. Noppe, S. De Smedt and M. De Cuyper, Chembiochem, 2009, 10, 257–267 CrossRef CAS.
- M. L. Hartman and M. Czyz, Cancer Lett., 2013, 331, 24–34 CrossRef CAS.
- H. Y. Wan, J. L. Chen, X. Zhu, L. Liu, J. Wang and X. M. Zhu, Adv. Sci., 2017, 5, 1700585 CrossRef.
- S. T. Stern, P. P. Adiseshaiah and R. M. Crist, Part. Fibre Toxicol., 2012, 9, 20 CrossRef CAS.
- J. Wang, Y. Yu, K. Lu, M. Yang, Y. Li, X. Zhou and Z. Sun, Int. J. Nanomed., 2017, 12, 809–825 CrossRef CAS.
- W. Zheng, M. Wei, S. Li and W. Le, Nanomedicine, 2016, 11, 1417–1430 CrossRef CAS.
- E. J. Parkinson-Lawrence, T. Shandala, M. Prodoehl, R. Plew, G. N. Borlace and D. A. Brooks, Physiology, 2010, 25, 102–115 CrossRef CAS.
- T. C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00990c |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Yong
Sun
a and
Yong
Sun