
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time surface-enhanced Raman scattering-based live cell monitoring of the changes in mitochondrial membrane potential†
Ji Hye
Lee
,
Hyeon Jeong
Shin
,
Yong Duk
Kim
and
Dong-Kwon
Lim
 *
*
KU-KIST Graduate School of Converging Science and Technology, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul, South Korea. E-mail: dklim@korea.ac.kr
First published on 7th April 2021
Abstract
Obtaining molecular information on cells in real time has been a critical challenge in studying the interaction between molecules of interest and intracellular components. Fluorescence-based methods have long served as excellent tools to study such important interactions. In this paper, we introduce a Raman scattering-based method as a promising platform to achieve the real-time monitoring of subtle molecular changes occurring within cells. We found that the Raman scattering-based method enabled monitoring changes in the mitochondrial membrane potential at the single-cell level in rheumatoid arthritis synovial fibroblasts induced by tumor necrosis factor-alpha (TNF-α) protein, various chemicals (MgCl2, FCCP, and sodium pyruvate), and a non-chemical stimulus (i.e., light). The triphenylphosphine-modified gold nanoparticles were selectively localized in the mitochondria and showed the characteristic Raman spectrum of cytochrome C and other Raman spectra of molecular components inside the cell. The surface-enhanced Raman spectrum originating from mitochondria was sensitively changed over time when mitochondrial depolarization was induced by the addition of TNF-α, or chemicals known to induce mitochondrial depolarization. The Raman-based signal changes were well matched with results of the conventional fluorescence-based analysis. However, in contrast to the conventional approach, the Raman-based method enables monitoring such changes in real time and provides detailed molecular information in terms of the interaction of molecules. Therefore, these results highlight the possibility of surface-enhanced Raman scattering-based live cell analysis for future proteomics or drug-screening applications.
Introduction
Recent developments in cell biology have strongly relied on fluorescence-based microscopy using organic fluorescent molecules (e.g., rhodamine, fluorescein, and green fluorescent protein) or inorganic nanoparticles (i.e., quantum dots) to monitor various cellular functions, morphological changes, and the expression levels of specific markers.1,2 The main challenge of fluorescence-based methods is generally considered to be the limited ability to obtain molecular information from cells and the substantial photobleaching of fluorescence signals. Spectroscopic analysis using Raman scattering has been proposed as a promising method to overcome these limitations of fluorescence, since it can provide fingerprint peaks of molecules inside a cell without interfering with water molecules.3–7 However, Raman scattering also suffers from disadvantages such as an extremely low signal intensity. To overcome this limitation, several new optical systems have been developed, including a line-by-line scanning system with a slit-scanning Raman microscope, as well as the development of non-linear phenomena such as stimulated Raman spectroscopy, including coherent anti-Stokes Raman scattering for live single-cell Raman-based analysis.8 Another approach to overcome the low signal intensity of Raman scattering is surface-enhanced Raman scattering (SERS) using plasmonic nanoparticles, which can help obtain molecular information in single living cells.9–11 Austin et al.12 demonstrated the use of plasmonically enhanced Raman scattering-based single-cell imaging spectroscopy to evaluate the activity of an anti-cancer drug using nuclear-targeting gold nanoparticles (AuNPs). The plasmonically enhanced Raman scattering in cells also showed the capability of monitoring complete cell cycle changes, showing spectrum changes at 511 cm−1, 658 cm−1, 844 cm−1, and 1322 cm−1, corresponding to the –S–S– vibration, guanine, sugar–phosphate and adenine, and guanine and RNA, respectively.13,14Furthermore, mitochondria have attracted substantial attention for Raman-based analyses owing to their key roles in metabolic regulation, cellular energy production, and programmed cell death such as apoptosis.15,16 The function of mitochondria is strongly dependent on the redox state of cytochrome C (Cyt C) in the electron transport chain (ETC), which induces changes in the inner mitochondrial membrane potential (ΔΨm).17,18 Most previous studies to obtain information on the redox state of ETC complexes were performed using fluorescence microscopy, which provides only an indirect estimation of the redox state of cytochromes and the related potential changes of ΔΨm. Direct information about the redox state of Cyt C and changes in membrane potential in living cells remains difficult to obtain because the redox state and the conformation of Cyt C are highly dynamic, thereby affecting diffusion in the intermembrane space and electron transfer process.18
Several Raman spectroscopic methods have been reported to monitor the changes in molecular dynamics in the mitochondria with or without relying on SERS.19–23 Fujita et al.23 reported the label-free Raman observation of Cyt C translocation during apoptosis with a 10 min temporal resolution. Brazhe et al. and Sarycheva et al. reported the use of silver nanostructured surfaces or silica nanoparticles to obtain detailed molecular information on mitochondria based on Raman spectra in living cells, isolated mitochondria, or isolated Cyt C.21,24 Collectively, these previous results have highlighted Raman spectroscopy as a promising tool for non-invasive investigations of complex molecular changes in mitochondria. However, to utilize the Raman-based spectroscopic method for the real-time investigation of molecular interactions between potential molecules and components in the mitochondria, a more extensive and systematic comparison between Raman spectral changes and fluorescence-based results should be established.
In our recent paper, we reported an in situ Raman-based method to monitor the changes in the Raman spectrum of disulfide bonds in the cytosol and Cyt C in the mitochondria during apoptosis that was induced by applying physical and cytotoxic chemicals to live cancer cells.25
In this study, we focused on the systematic comparison between Raman spectral changes and fluorescence-based results when inducing the ΔΨm of rheumatoid arthritis synovial fibroblasts (RASFs) with tumor necrosis factor-alpha (TNF-α), light, and nontoxic chemicals (Fig. 1). We found that measuring the Raman spectrum from cells can provide detailed and diverse molecular information during changes in the redox state of cytochromes and mitochondrial membrane potential.
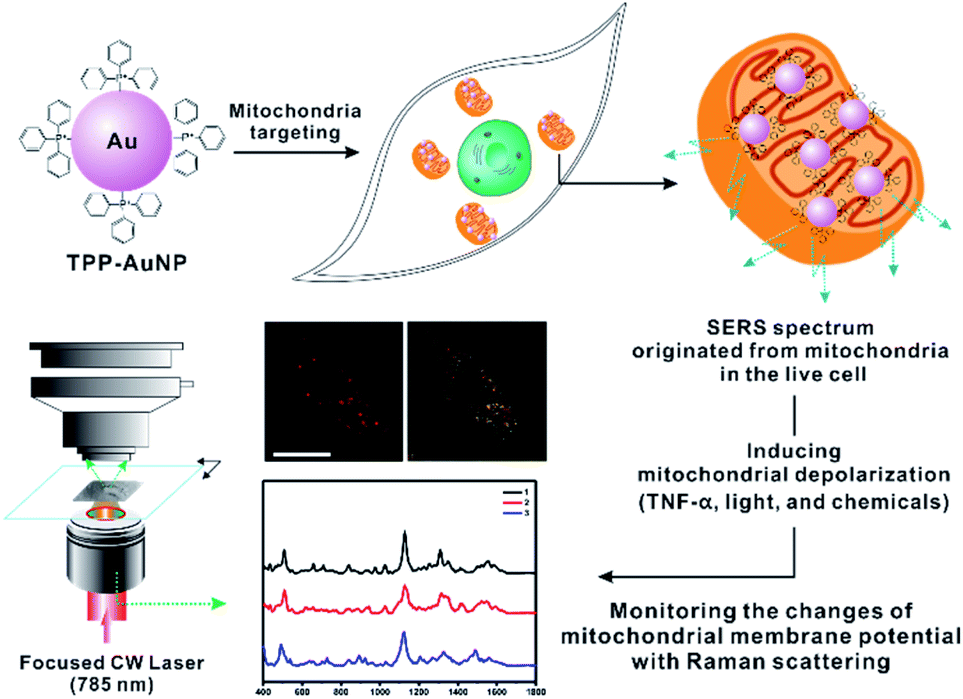 | ||
| Fig. 1 Schematic description of the experimental procedures. Triphenylphosphine-modified AuNPs (TPP-AuNPs) were used for mitochondrial targeting and Raman spectra were acquired from live cells in real time (s) with various stimuli that can cause changes in the mitochondrial membrane potential (scale bar = 20 μm). | ||
Results and discussion
Synthesis of triphenylphosphine (TPP)-AuNPs and mitochondrial targeting capability
Given the well-known mitochondria-targeting capability of TPP, TPP ligands were used for the surface modification of AuNPs following previously reported procedures.26–28 To prepare TPP-AuNPs, 1.0 μL of TPP solution in N,N-dimethylformamide (0.1 mg/10 mL) was added to 1.0 mL of tannic acid (TA)-modified gold nanoparticles (optical density = 1.0, 50 nm).25 After shaking for 2 h at room temperature, the solution was centrifuged (3400 × g, 15 min) to obtain a precipitate, and the precipitate was redispersed in distilled water (1.0 mL). The changes in the AuNP surface were confirmed by transmission electron microscopy (TEM), ultraviolet-visible (UV-VIS) spectroscopy, Raman spectroscopy, and zeta potential analysis, as shown in Fig. S1.† The prepared TPP-AuNPs were well-dispersed in distilled water, as shown by the TEM and UV-VIS spectra (Fig. S1-A and B†). Raman spectroscopy analysis further demonstrated ligand changes in TPP-AuNPs from those of TA-AuNPs (Fig. S1-C†). The TA-AuNPs had a strong negative surface charge (−50.01 mV) at neutral pH, while a relatively more positive surface charge (−30.04 mV) was observed after changing TA to TPP ligands on AuNPs (Fig. S1-D†). The hydrodynamic size of TPP-AuNPs was 50 nm (Fig. S1-E†). These results are in good agreement with our previous results and confirmed the successful formation of TPP-AuNPs.25 Moreover, the more positive charge of TPP-AuNPs can facilitate nanoparticle uptake into cells through lipid bilayers and accumulate in the mitochondria due to the large membrane potential of 150–180 mV (negative inside) across the mitochondrial membrane potential.26–28 The cell viability test showed that TPP-AuNPs had no significant cytotoxicity in RASFs (Fig. S2†).The uptake efficiency and targeting capability of the TA-AuNPs and TPP-AuNPs to mitochondria were confirmed by monitoring the particle distribution inside the cells using a blocking test with carbonylcyanide p-trifluoromethoxy-phenylhydrazone (FCCP), which is known to induce mitochondrial depolarization29 (Fig. S3†). After incubating TA-AuNPs (0.1 nM) or TPP-AuNPs (0.1 nM) with RASFs, the particle distribution was monitored using bright-field (B/F) microscopy and dark-field (D/F) microscopy (Fig. S3†). The efficient uptake of TA-AuNPs and TPP-AuNPs inside RASFs was observed after incubation for 3 h (Fig. S3-A and B†). When the RASFs were incubated with FCCP (20 μM) for 10 min before the addition of TPP-AuNPs, decreased uptake efficiency of TPP-AuNPs was observed, indicating that FCCP altered the mitochondrial membrane potential (Fig. S3-C†).29
The distribution of TA-AuNPs and TPP-AuNPs in RASFs was further investigated using B/F microscopy, D/F microscopy, single-cell Raman imaging, and Raman spectrum analysis (Fig. 2A and B for TA-AuNPs, and Fig. 2C and D for TPP-AuNPs) (the exact same experimental dataset is also presented in Fig. S4†). The cells were stained with Mito-Tracker (100 nM, 30 min) to examine the distribution of particles within the mitochondria (Fig. 2A-iv and C-iv). TA-AuNPs are expected to be localized within intracellular vesicles, likely endosomes, or the cytosol, while TPP-AuNPs would be distributed in the mitochondria.30 The B/F and D/F images shown in Fig. 2A-i and ii and C-i and ii reveal the distribution of particles inside the cells. Because of the plasmonically enhanced Raman signal, high-speed Raman imaging for single cells was possible (Fig. 2A-iii and C-iii). It took 5 min to obtain single-cell Raman images with a 30 ms exposure time per pixel (100 × 100 pixels) and a 2.98 mW cm−2 input laser power (785 nm). Interestingly, the Raman spectra obtained from cells incubated with TA-AuNPs varied from each other owing to the random distribution of TA-AuNPs in the cytosol (Fig. 2B, Raman spectra obtained from points 1, 2, and 3 in Fig. 2A-iii).30 However, the Raman spectra of all of the cells incubated with TPP-AuNPs were nearly identical (Fig. 2D, Raman spectra obtained from points 1, 2, and 3 in Fig. 2C-iii). The Raman shift peaks located at 505 cm−1, 840 cm−1, and 1000 cm−1 were assigned to the stretching vibration mode of the disulfide bond (–S–S–), symmetrical stretches of the lipid component (–O–C–C–N–) and tyrosine, and a benzene ring stretching vibration of phenylalanine, respectively.12,14 The Raman shift peaks located at 750 cm−1, 1127 cm−1, 1313 cm−1, and 1581 cm−1 were assigned to the typical vibrational modes of Cyt C upon 532 nm laser excitation.20–24,31 However, the Raman band at 750 cm−1 was not clear in the spectrum. This is because of the difference in the excitation wavelength (it should be noted that a 785 nm laser was used in the current study).21–23 The Raman shift at 1581 cm−1 also showed low signal intensity when applying 785 nm excitation (Fig. S5-A and B†). Therefore, it is believed that the Raman spectra presented in Fig. 2D are the typical Raman spectra of mitochondria of RASFs, which are similar to the Raman spectrum obtained from HSC-3 cells.25 Based on these initial Raman spectra, we investigated the time-dependent changes of the single-cell SERS spectrum after inducing changes in the mitochondrial membrane potential with TNF-α, photothermal damage, or diverse chemicals (i.e., MgCl2, FCCP, and sodium pyruvate). Before these stimulus treatments, all RASF single cells were incubated with 0.1 nM TPP-AuNPs for 3 h.
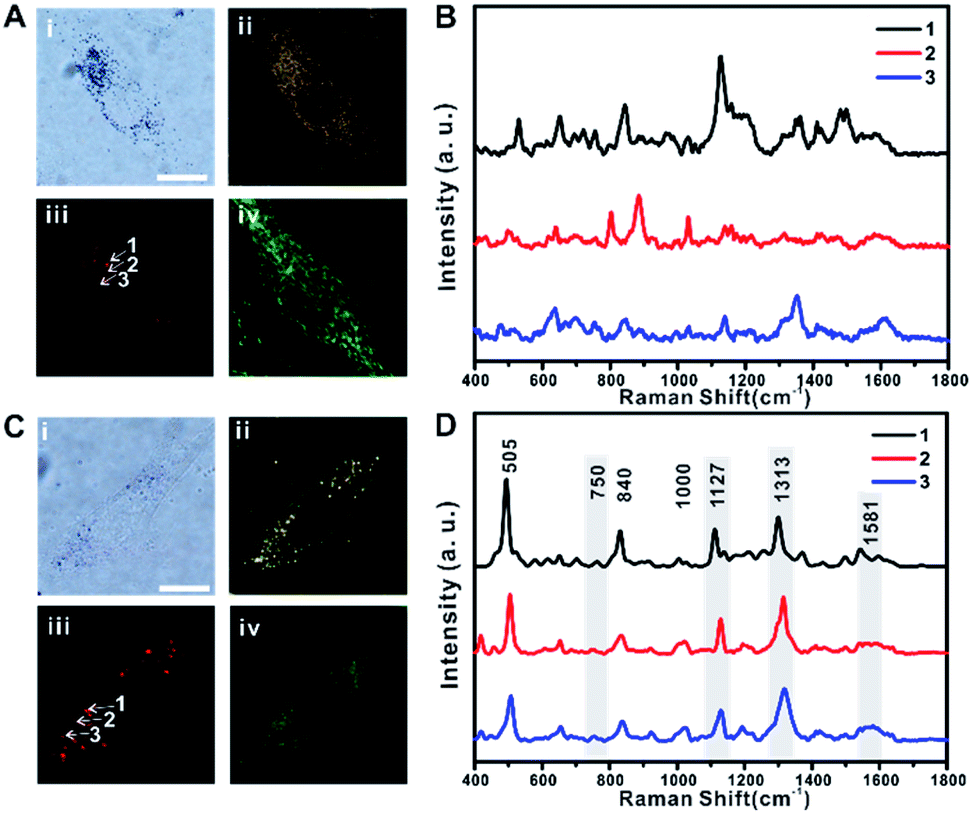 | ||
| Fig. 2 (A) Intracellular distribution of TA-AuNPs in rheumatoid arthritis synovial fibroblasts (RASFs) observed with bright field microscopy (i), dark field microscopy (ii), Raman mapping (iii), and fluorescence images (iv) stained with Mito-Tracker. (B) Raman spectra obtained from inside cells (A-iii, points 1, 2, and 3). (C) Intracellular distribution of TPP-AuNPs in rheumatoid arthritis synovial fibroblasts observed with bright field microscopy (i), dark field microscopy (ii), Raman mapping (iii) and fluorescence images (iv) stained with Mito-Tracker. (D) Raman spectra obtained from inside cells (C-iii, points 1, 2, and 3). Raman signals at 750, 1127, 1313, and 1581 cm−1 are assigned to the vibration mode of cytochrome c. Scale bars = 20 μm (objective: 100×, laser power: 3 mW, exposure time: 0.03 s (Raman mapping), 1 s (single spectrum)). | ||
TNF-α induced mitochondrial depolarization
TNF-α is known to induce the reduction of NADH-ubiquinone oxidoreductase and ATP synthase in the ETC of the mitochondrial membrane, which leads to membrane potential changes, apoptosis, and inflammation.32–34 The decreased electron movement in the ETC inhibits the production of a proton gradient across the inner mitochondrial membrane. The change in mitochondrial membrane potential associated with the release of proapoptotic factors such as Cyt C is the key mechanism for the inflammation process caused by TNF-α in RASFs.32–35 To demonstrate the feasibility of Raman scattering in monitoring the changes in mitochondrial membrane potential induced by TNF-α, we investigated the changes of Raman scattering in the mitochondria and cytosol when mitochondrial depolarization was induced by treatment of RASFs with various concentrations of TNF-α (0, 10, 20, 40, 60, and 80 ng mL−1) (Fig. 3, S6 and S7†).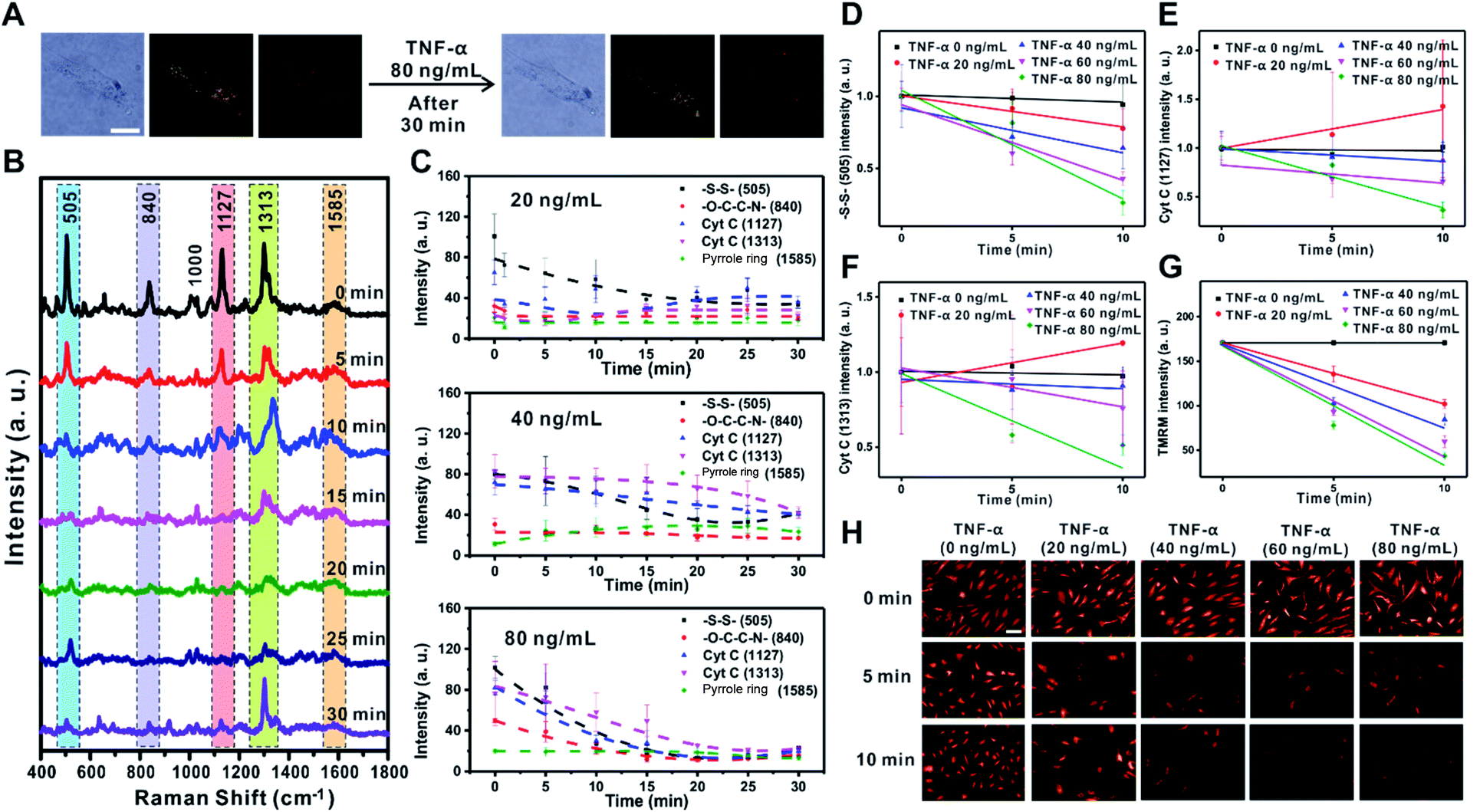 | ||
| Fig. 3 TNF-α induced mitochondrial depolarization in rheumatoid arthritis synovial fibroblasts (RASFs). (A) Bright-field, dark-field, and Raman images of RASFs before and after treatment with TNF-α (80 ng mL−1). Scale bar = 20 μm. (B) Time-dependent changes in Raman spectra during TNF-α (80 ng mL−1)-induced mitochondrial depolarization. (C) Intensity changes of specific Raman shifts at 505 cm−1 (–S–S–), 840 cm−1 (–O–C–C–N–), 1127 cm−1 (Cyt C), and 1313 cm−1 (Cyt C) after treatment of RASFs with TNF-α (20, 40, and 80 ng mL−1). The changes of Raman signal intensity at 505 cm−1 (–S–S–) (D), 1127 cm−1 (Cyt C) (E), and 1313 cm−1 (Cyt C) (F) from the initial state to 10 min after treating RASFs with TNF-α (0, 20, 40, 60, and 80 ng mL−1). (G) Fluorescence intensity changes 10 min after treating RASFs with TNF-α (0, 20, 40, 60, and 80 ng mL−1). (H) Time-dependent fluorescence images after treating RASFs with TNF-α (0, 20, 40, 60, and 80 ng mL−1). Scale bar = 100 μm. | ||
Fig. 3A shows the B/F, D/F, and Raman images of single RASFs before and after treatment with TNF-α (80 ng mL−1) for 30 min. No clear morphological changes were observed; however, the molecular signals from inside cells were significantly changed (Fig. 3B), reflecting the time-dependent change of Raman spectra measured from inside cells after treatment with TNF-α (80 ng mL−1). Interestingly, the intensity of the Raman peaks at 505 cm−1 (–S–S–), 1127 cm−1 (Cyt C), and 1313 cm−1 (Cyt C) decreased significantly, while the intensity of the Raman bands at 840 cm−1 (–O–C–C–N–) and 1585 cm−1 (pyrrole ring) showed relatively small intensity changes over time. The significant decrease in intensity of the Raman shift at 505 cm−1 (–S–S–) is related to the defense mechanism of cells to overcome the oxidative stress induced by TNF-α.35–37Fig. 3C shows the quantitative changes in Raman scattering at 505 cm−1 (–S–S–), 840 cm−1 (–O–C–C–N–), 1127 cm−1 (Cyt C), 1313 cm−1 (Cyt C), and 1585 cm−1 (pyrrole ring) depending on TNF-α concentration (20 ng mL−1, 40 ng mL−1, and 80 ng mL−1) and time. At low concentrations of TNF-α (20 ng mL−1), the Raman intensities related to Cyt C showed a slow recovery after 10 min, indicating the recovered state of membrane potential. The addition of TNF-α at concentrations higher than 20 ng mL−1 resulted in a rapid decrease in the intensity of Raman bands and very slow recovery (80 ng mL−1) (Fig. 3C). Among Raman peaks from cells, Raman scattering at 505 cm−1 (–S–S–) showed a highly quantitative response to the concentration of TNF-α (Fig. 3D). The Raman peaks corresponding to Cyt C (1127 cm−1 and 1313 cm−1) also showed an obvious decrease with a high TNF-α concentration in RASFs, which reflects the significant changes of Cyt C during mitochondrial membrane potential changes. However, greater variations in intensity were observed compared with Raman scattering at 505 cm−1 (–S–S–) (Fig. 3D–F), which is attributed to the complex dynamic process of Cyt C in the mitochondrial membrane during potential changes.18,23,38
The changes in membrane potential were also confirmed with conventional fluorescence-based methods after treating RASFs with the same concentrations of TNF-α (20, 40, 60, and 80 ng mL−1) (Fig. 3G and H). We used tetramethylrhodamine methyl ester (TMRM) to compare the results observed with Raman scattering,36 which is a well-known lipophilic dye that enters the negatively charged mitochondria where it accumulates in an inner membrane potential-dependent manner.36 After treating RASFs with TNF-α (20 ng mL−1, 40 ng mL−1, 60 ng mL−1, and 80 ng mL−1), the cells were stained with TMRM (100 nM, 30 min), and the fluorescence images were obtained every 5 min (Fig. 3G and H). With the increase in TNF-α concentrations (>20 ng mL−1), a significant decrease in fluorescence intensity was observed. A nearly perfect correlation between fluorescence intensity and TNF-α concentration was observed (Fig. 3G). These results are in good agreement with the changes monitored with Raman scattering. Although the changes in Raman signal intensities from a cell were plotted every 5 min (Fig. 3C), the exposure time for the time-dependent Raman spectrum is only 0.03 s; therefore, the temporal resolution of Raman-based analysis is high enough to be compared with that of the fluorescence-based method, which can provide more detailed information on the cell. Raman scattering can provide information on molecular changes for a sufficiently long time period without experiencing any photobleaching problem.
Photothermal damage induced changes in mitochondrial membrane potential
Next, we investigated the changes in membrane potential and cellular signals after applying a physical stimulus to the cells. We used photothermal heating as the physical stimulus in this case, as photothermal damage to cells can lead to cell death by altering the signaling pathway of mitochondria, which includes changes in mitochondrial membrane potential.39–43 We monitored the Raman spectral changes after applying the photothermal stimulus to a single RASF using a 785 nm focused laser (60× objective, NA = 0.7) for 1 s with three different laser powers (0 mW, 12 mW, and 24 mW) (Fig. 4 and S8†).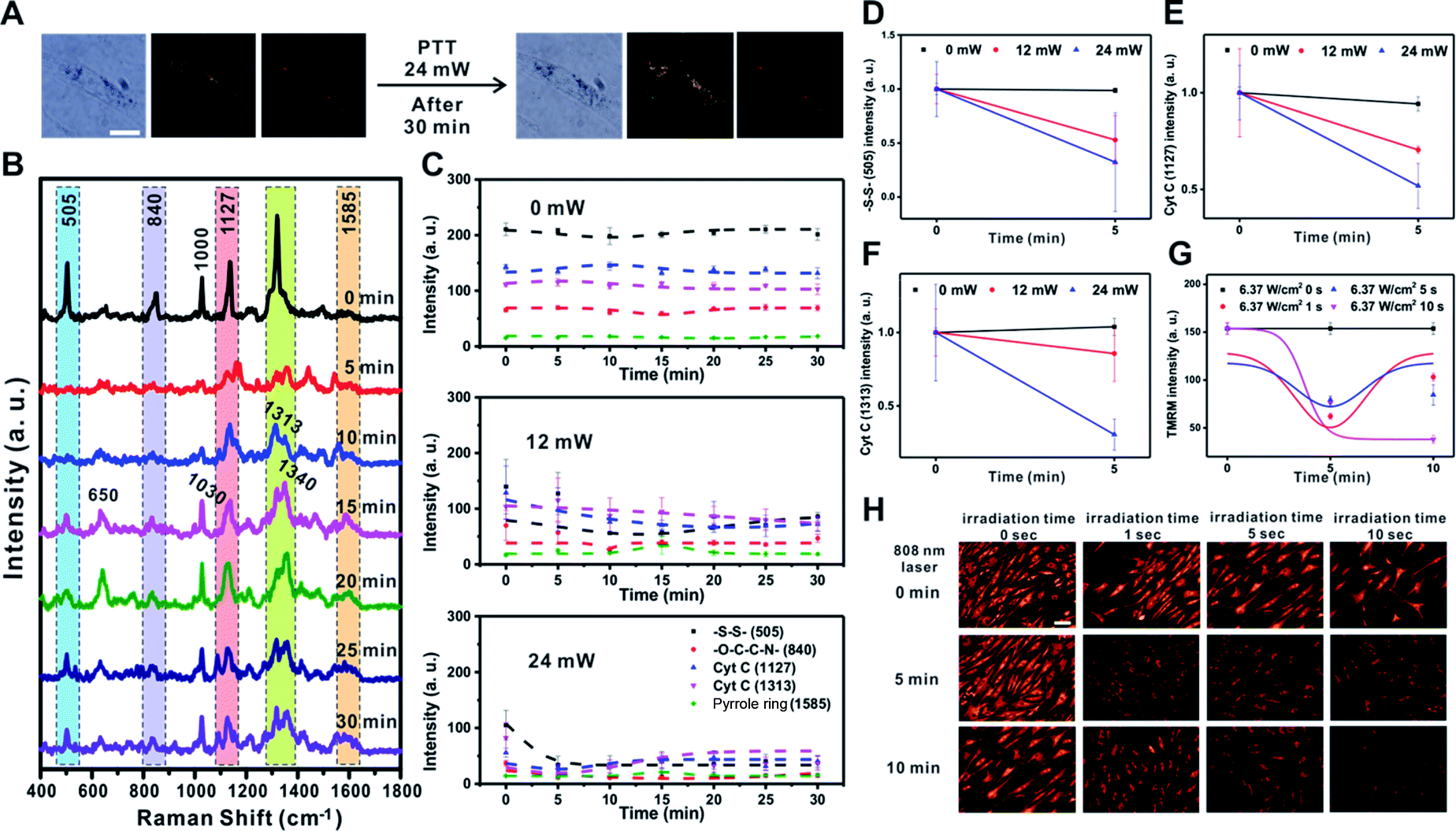 | ||
| Fig. 4 Photothermal damage induced changes of mitochondrial membrane potential in rheumatoid arthritis synovial fibroblasts (RASFs). (A) Bright-field, dark-field, and Raman images before and after inducing photothermal damage in RASFs with a focused laser (785 nm, 24 mW, 1 s exposure). Scale bar = 20 μm. (B) Time-dependent changes of Raman spectra after applying the focused laser (24 mW). (C) Intensity changes of specific Raman shifts at 505 cm−1 (–S–S–), 840 cm−1 (–O–C–C–N–), 1127 cm−1 (Cyt C), 1313 cm−1 (Cyt C), and 1585 cm−1 (pyrrole ring) after applying 0 mW, 12 mW, and 24 mW laser illumination. The SERS intensity variations of (D) –S–S– (505 cm−1), (E) Cyt C (1127 cm−1), and (F) Cyt C (1313 cm−1) until 5 min with different irradiation intensities (0 mW, 12 m, and 24 mW), respectively. (G) TMRM fluorescence intensity changes under near-infrared (808 nm) laser exposure at 5 W for 0 s, 1 s, 5 s, and 10 s. (H) TMRM fluorescence images under near-infrared (808 nm) laser exposure at 5 W for 0 s, 1 s, 5 s, and 10 s. Scale bar = 100 μm. | ||
Fig. 4A shows the B/F, D/F, and Raman images of single RASFs before and after treatment with a laser (24 mW) for 30 min. There were no clear morphological changes; however, changes in the Raman spectra could be clearly observed (Fig. 4B). The –S–S– bond observed at 505 cm−1 rapidly disappeared from 5 min after laser exposure and recovered slowly. This is because the photothermal damage induced acute oxidative stress and activated the cellular defense system by the reduction of glutathione.37,41 However, the intensity of Raman scattering assigned to –O–C–C–N– (at 840 cm−1) did not change over time. The Raman peak at 1000 cm−1, which is the ring-breathing mode of phenylalanine in the protein, disappeared and rapidly shifted to a Raman shift at 1030 cm−1, which is the C–H in-plane bending of phenylalanine. The Raman peaks corresponding to Cyt C (1127 cm−1 and 1313 cm−1) showed a slight decrease in intensity with time.19,44,45 In particular, the new Raman peaks at 650 cm−1 and 1340 cm−1 after 5 min were expected to be the thymine and adenine bases of DNA, respectively, which showed a significant increase with time. It is expected that the double-helix structure of DNA present in the mitochondria changed into an unordered single chain by the photothermal effect (Fig. 4B).44 This was the most noticeable characteristic of photothermal damage compared with the changes induced by TNF-α addition. The local heating effect can significantly induce changes in DNA in live cells. However, the Raman peak at 1585 cm−1 (pyrrole ring) did not significantly change with time.
Fig. 4C shows the time-dependent changes in the Raman bands at 505 cm−1 (–S–S–), 840 cm−1 (–O–C–C–N–), 1127 cm−1 (Cyt C), 1313 cm−1 (Cyt C), and 1585 cm−1 (pyrrole ring) depending on the input laser power. If there was no photothermal damage to the cells, the Raman spectrum from the mitochondria in RASFs did not change significantly (Fig. 4C – 0 mW and Fig. S8† – 0 mW). However, when the cell was exposed to stronger photothermal damage, more evident changes in the Raman signal intensity were observed over time (Fig. 4C 12 mW vs. 24 mW and Fig. S8† 12 mW vs. 24 mW). It is confirmed by additional plotting that the intensity of Raman shifts at 505 cm−1 (–S–S–), 1127 cm−1 (Cyt C), and 1313 cm−1 (Cyt C) decreased according to the input laser powers (Fig. 4D–F).
The changes in membrane potential due to the photothermal effect were also confirmed using the TMRM-based fluorescence method. The RASFs were illuminated with an 808 nm laser (5 W, 1 cm beam diameter) for 1, 5, and 10 s, and then the fluorescence intensity of the cells was monitored (Fig. 4G and H). Upon photothermally induced hyperthermia, RASFs were stained with TMRM (100 nM, 30 min). Fig. 4H shows the TMRM fluorescence images at 5 min intervals. We found that the fluorescence intensity of TMRM decreased until 5 min after photothermal damage induced mitochondrial depolarization. However, the mitochondrial membrane potential of RASFs recovered after resting for 10 min, except for those irradiated for 10 s (Fig. 4G and H).
MgCl2 induced changes of mitochondrial membrane potential
Cellular energy production processes are composed of many Mg2+-dependent enzymatic reactions, and mitochondria are known as major intracellular Mg2+ stores.46,47 Importantly, several studies have reported that Mg ions facilitate the Cyt C release in disrupted cells48 and mediate [MgADP]-dependent mitochondrial ATP synthase activity.49 Therefore, we also investigated the effect of MgCl2 (1, 3, and 6 mM) on the changes in mitochondrial membrane potential using Raman spectroscopy and the fluorescence-based method (Fig. 5 and S9†). Fig. 5A shows that the morphology of RASFs did not change during the treatment with MgCl2 (6 mM). Raman spectra observed every 5 min showed a tendency for the intensity of the –S–S– bond (505 cm−1) and Cyt C (1127 cm−1 and 1313 cm−1) to recover to the initial states (Fig. 5B). After treating RASFs with 1 mM MgCl2, the intensity of Raman peaks derived from the mitochondria of the cells was unchanged, demonstrating that a concentration of 1 mM Mg ions is maintained in the cytosol (Fig. 5C and S9,† 1 mM).46 After the addition of higher MgCl2 concentrations to RASFs, the decrease in Raman spectral intensity attributed to the –S–S– bond (505 cm−1) and Cyt C (1127 cm−1 and 1313 cm−1) occurred rapidly. The intensity of SERS bands corresponding to disulfide bonds and Cyt C recuperated from the instant reduction of [MgADP]-dependent activity induced by excessive mitochondrial ATP synthase Mg ions in the cytosol (Fig. 5C and S9†).49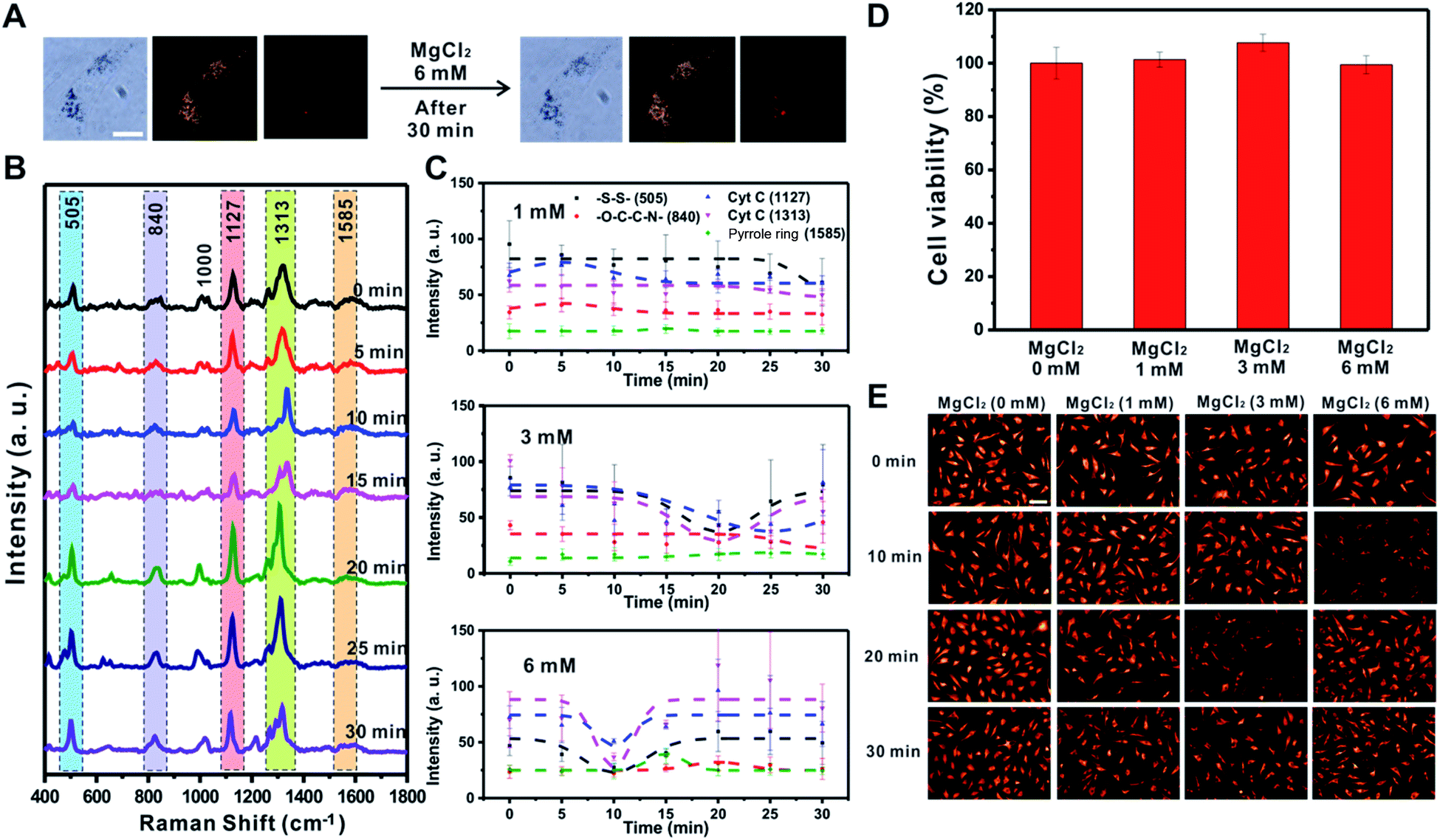 | ||
| Fig. 5 MgCl2 induced changes of mitochondrial membrane potential in rheumatoid arthritis synovial fibroblasts (RASFs). (A) Bright-field, dark-field, and Raman images before and after treatment of RASFs with 3 mM MgCl2. Scale bar = 20 μm. (B) Time-dependent changes in Raman spectra during MgCl2 (6 mM) treatment. (C) Intensity changes of specific Raman shifts at 505 cm−1 (–S–S–), 840 cm−1 (–O–C–C–N–), 1127 cm−1 (Cyt C), 1313 cm−1 (Cyt C), and 1585 cm−1 (pyrrole ring) after treatment with 1 mM, 3 mM, and 6 mM MgCl2. (D) MTS assay after MgCl2 treatment (1 mM, 3 mM, and 6 mM) for 30 min. (E) TMRM fluorescence images after RASFs were incubated with 0, 1, 3, and 6 mM MgCl2 for 0, 10, 20 and 30 min. Scale bar = 100 μm. | ||
As shown in Fig. 5D, a cell viability test was performed to confirm whether Mg ions directly cause Cyt C release and cell death. The cell survival remained nearly unchanged, even in the presence of more than 1 mM Mg ions inside cells. Fig. 5E shows the decreased fluorescence intensity at various points (3 mM treatment for 20 min and 6 mM treatment for 10 min), which are the same conditions that showed decreased Raman signals of the –S–S– bond (505 cm−1) and Cyt C (1127 cm−1 and 1313 cm−1), indicating the start of mitochondrial depolarization. Based on both Raman-based analysis and the fluorescence-based results, MgCl2 was confirmed to be a substance that causes instant changes in membrane potential in mitochondria without affecting cell viability.
FCCP induced mitochondrial depolarization
FCCP is known to disrupt ATP synthesis by interfering with the proton gradient and inducing mitochondrial depolarization.21 Therefore, we examined changes in the Raman signal over time with different concentrations of FCCP (1 μM, 10 μM, and 20 μM) after incubating RASFs with TPP-AuNPs (Fig. 6 and S10†). As mentioned earlier, the cell efficiency of TPP-AuNPs decreased after treatment with a high concentration of FCCP (20 μM, 10 min). However, we performed Raman-based experiments under the same incubation conditions (TPP-AuNPs, 0.1 nM, 3 h). Since the changes of Raman spectra from FCCP-induced mitochondrial depolarization were monitored at a specific point within a single cell, we determined that cellular uptake would not have a significant effect. Fig. 6A shows that there were no morphological changes in RASFs using B/F microscopy, D/F microscopy, and Raman imaging after treatment with the highest FCCP concentration (20 μM). Fig. 6B shows typical changes in the Raman spectra measured at 5 min intervals during FCCP-mediated mitochondrial depolarization in RASFs (20 μM). The Raman peak corresponding to the disulfide bond (505 cm−1) rapidly decreased with time due to the reduction of disulfide residues of the protein by oxidative stress.37 The intensity of the Raman peaks related to Cyt C (1127 cm−1 and 1313 cm−1) significantly decreased in proportion to the concentration of FCCP after 10 min. This is because the mitochondrial depolarization induced by FCCP leads to a reduction in the relative amount of reduced electron carriers, including Cyt C.22 Along with the changes in both the disulfide peak and Cyt C peaks, the spectra also showed significant changes in the Raman scattering at 1000 cm−1 (Phe ring breathing) and 1030 cm−1 (Phe C–H in-plane bending) after 10 min. Interestingly, the intensity of the Raman peak at 1000 cm−1 decreased, but the intensity at 1030 cm−1 slightly increased, although these peaks are both assigned to Phe.44,45 From this result, we assumed that the changes of phenylalanine states occurred via a mechanism in which the activity of the ETC was reduced due to oxidative stress induced by FCCP.44 These spectral changes were evidently related to FCCP concentration, as displayed in Fig. 6C. When RASFs were treated with a low FCCP concentration (1 μM), three Raman bands at the disulfide bond (505 cm−1) and Cyt C (1127 cm−1 and 1313 cm−1) were changed after 15 min, while their intensity slightly increased after 30 min. It is believed that the 1 μM concentration of FCCP inside cells was not too harmful to prevent recovery of the activity of the ETC in mitochondria (Fig. 6C and S10,† 1 μM). At a concentration of more than 10 μM FCCP, the Raman peaks at 505 cm−1, 1127 cm−1, and 1313 cm−1 changed very rapidly, and their decreased intensity was maintained without activating the recovery system in RASFs (Fig. 6C and S10,† 10 μM and 20 μM). In comparison, in the fluorescence-based analysis, a reduction in mitochondrial membrane potential was observed after the addition of various concentrations of FCCP (1 μM, 10 μM, and 20 μM) to RASFs (Fig. 6D); as the FCCP concentration in RASFs increased, the TMRM intensity decreased. The Raman peaks corresponding to both disulfide bonds and Cyt C showed excellent correlations with those of the fluorescence-based method.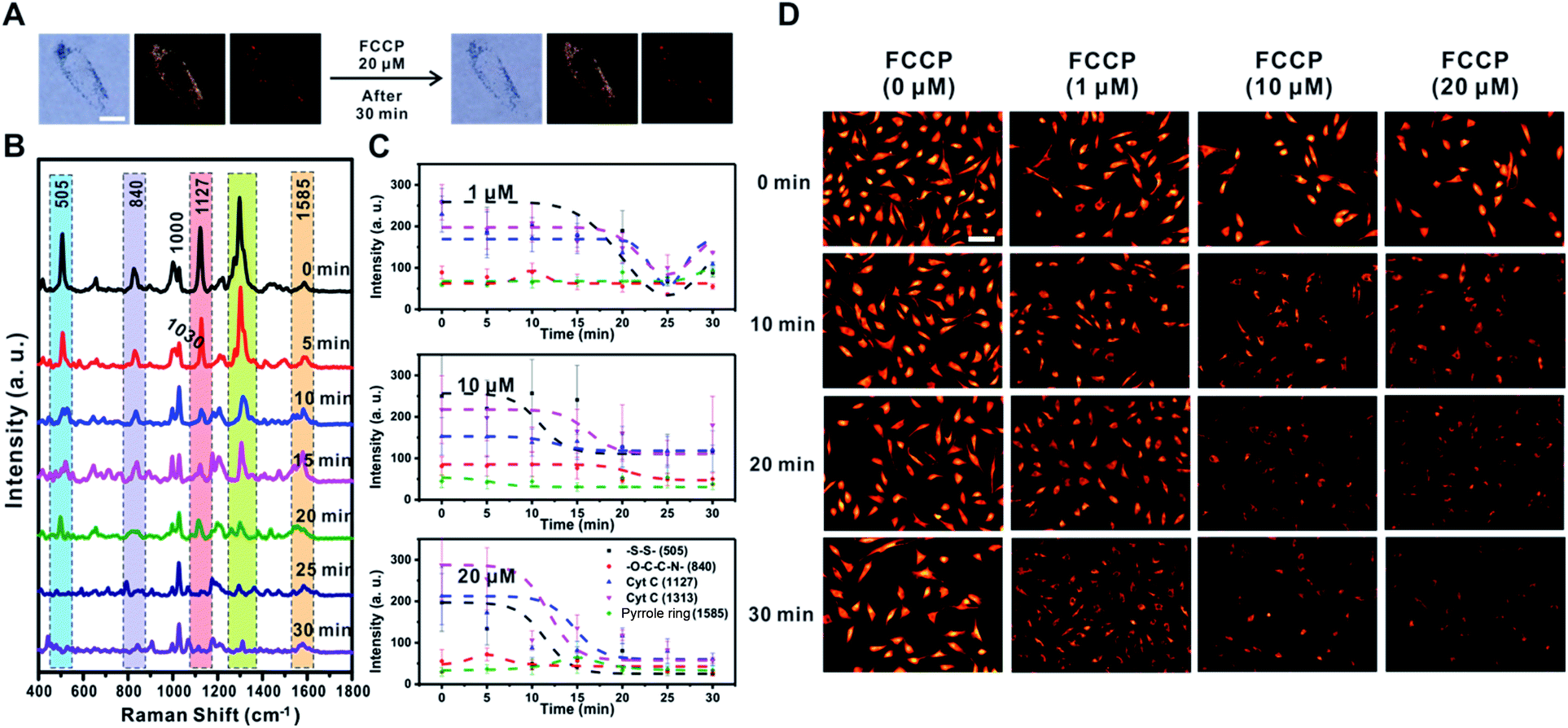 | ||
| Fig. 6 FCCP induced mitochondrial depolarization in RASFs (20 μM). (A) Bright-field, dark-field, and Raman images before and after the addition of 20 μM FCCP to RASFs. Scale bar = 20 μm. (B) Time-dependent changes in Raman spectra during FCCP-induced mitochondrial depolarization (20 μM). (C) Intensity changes of specific Raman shifts at 505 cm−1 (–S–S–), 840 cm−1 (–O–C–C–N–), 1127 cm−1 (Cyt C), 1313 cm−1 (Cyt C), and 1585 cm−1 (pyrrole ring) after treatment with FCCP (1 μM, 10 μM, and 20 μM). (D) TMRM fluorescence images after RASFs were incubated with 0, 1, 10, and 20 μM FCCP for 0, 10, 20, and 30 min. Scale bar = 100 μm. | ||
Sodium pyruvate induced changes of mitochondrial membrane potential
Pyruvate oxidized in the mitochondria results in the hyperpolarization of the inner mitochondrial membrane by helping to circulate the tricarboxylic acid cycle and activate the ETC.21 However, a previous study showed that if sodium pyruvate is present in the cytosol with a concentration greater than 3 mM inside cells, the non-oxidized pyruvates could be released to the outer mitochondrial membrane through uncoupling protein transporters expressed in human and rodent tissues, such as immune cells, causing mitochondrial depolarization.50 Therefore, the changes in mitochondrial membrane potential depending on the concentration of sodium pyruvate (0.4 mM, 1 mM, and 3 mM) present in the cytosol were investigated using the Raman-based method and fluorescence-based method (Fig. 7 and S11†).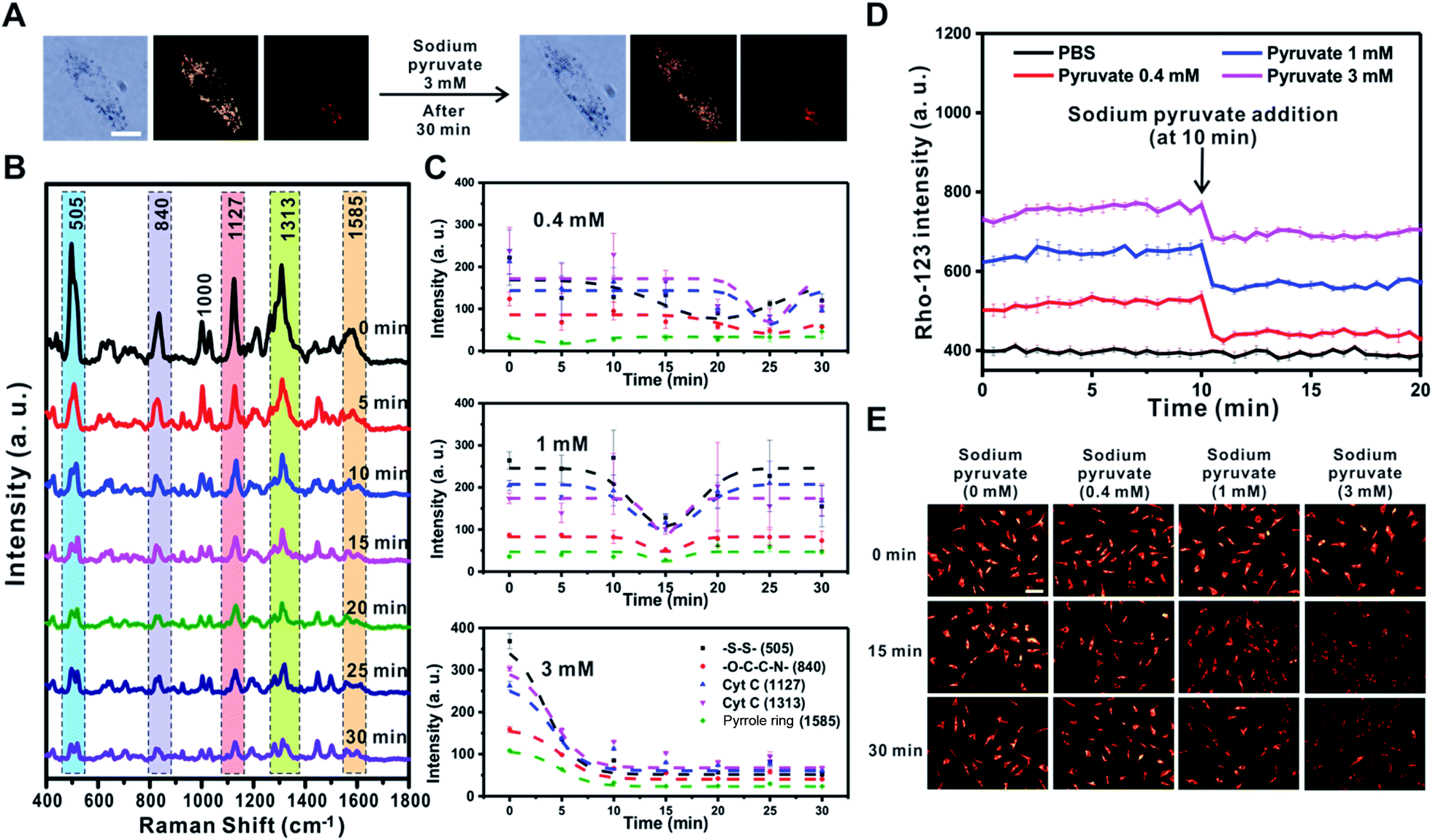 | ||
| Fig. 7 Sodium pyruvate induced changes of mitochondrial membrane potential in rheumatoid arthritis synovial fibroblasts (RASFs) (3 mM). (A) Bright-field, dark-field, and Raman images before and after the addition of 3 mM sodium pyruvate to RASFs. Scale bar = 20 μm. (B) Time-dependent changes in Raman spectra during sodium pyruvate induction (3 mM). (C) Intensity changes of specific Raman shifts at 505 cm−1 (–S–S–), 840 cm−1 (–O–C–C–N–), 1127 cm−1 (Cyt C), 1313 cm−1 (Cyt C), and 1585 cm−1 (pyrrole ring) after treatment with 0.4 mM, 1 mM, and 3 mM sodium pyruvate. (D) Real-time fluorescence intensity changes after treatment with pyruvate at 0.4 mM, 1 mM, and 3 mM. (E) TMRM fluorescence images after RASFs were incubated with 0, 0.4, 1, and 3 mM sodium pyruvate for 0, 15, and 30 min. Scale bar = 100 μm. | ||
After treatment with sodium pyruvate at the highest concentration (3 mM), the cell morphology was unchanged, and five peaks rapidly shifted within 5 min (Fig. 7A and B). As shown in Fig. 7C, the change tendency of the Raman peak was confirmed to vary depending on the concentration of sodium pyruvate. With the addition of the highest concentration of sodium pyruvate to RASFs, all five major peaks derived from the mitochondria disappeared at once and did not recover to the initial intensity of the Raman peaks. However, the intensities of the Raman peaks at –S–S– (505 cm−1) and Cyt C (1127 cm−1 and 1313 cm−1) were initially reduced and then recovered after the addition of 0.4 mM or 1 mM sodium pyruvate to RASFs (Fig. 7C and S11,† 0.4 mM and 1 mM), showing the same tendency as the effect of Mg2+ ion treatment on RASFs.
In addition, a fluorescence-based method (rhodamine-123) was used to monitor the changes in the ETC after treatment of RASFs with sodium pyruvate. For energy production in mitochondria to cause the quenching of rhodamine-123, the increase in the mitochondrial membrane potential is proportional to the degree of reduction in fluorescence intensity.36Fig. 7D shows the changes in intensity of rhodamine-123 after the addition of 0.4, 1, and 3 mM sodium pyruvate to RASFs. Before treating RASFs with sodium pyruvate, the stability of the fluorescence intensity of rhodamine-123 for 10 min was monitored. The results indicated that mitochondrial hyperpolarization occurred immediately after the addition of sodium pyruvate, and the energy generation of the mitochondria was inversely proportional to the pyruvate concentration. The TMRM intensity changes after the addition of various concentrations of sodium pyruvate (0.4 mM, 1 mM, and 3 mM) to RASFs were also identified, as displayed in Fig. 7E. Overall, the TMRM fluorescence intensity only significantly decreased after treatment of RASFs with 3 mM sodium pyruvate for 15 min and 30 min.
Conclusions
In this study, we demonstrated that SERS-based platforms can be used to sensitively monitor changes in molecular signals during changes in mitochondrial membrane potential induced by protein (TNF-α), physical damage (photothermal effect), or chemicals (MgCl2, FCCP, and sodium pyruvate) in RASFs. The use of TPP-AuNPs is essential to enhance the Raman signal in the vicinity of mitochondria. The localization of TPP-AuNPs was confirmed with microscopy observations (B/F, D/F, single-cell Raman imaging). After inducing changes in the mitochondrial membrane potential with five different stimuli, the SERS-based platform was used to obtain a high temporal resolution for the Raman spectrum of molecules inside single cells, including protein, Cyt C, and DNA. The intensity of the Raman peaks at 505 cm−1 (assigned to –S–S–) reflected the homeostasis status of the cell. These peaks showed a significant decrease in intensity when a high degree of stimulus was applied (80 ng mL−1 TNF-α, 24 mW photothermal treatment, 20 μM FCCP). The Raman peaks at 840 cm−1 (assigned to –O–C–C–N–) also showed a decrease or increase depending on the degree of stimulus applied. However, the Raman peaks at 840 cm−1 remained relatively unchanged compared to the other peaks. Dramatic changes in the Raman peaks at 1127 cm−1 and 1313 cm−1 (assigned to Cyt C) were observed in the case of treatment with TNF-α (80 ng mL−1) or FCCP (20 μM). However, photothermal damage or relatively mild polarizing agents (i.e., MgCl2 and sodium pyruvate) did not induce acute changes in the Raman peaks at 1127 cm−1 and 1313 cm−1. Interestingly, the photothermal damage induced the production of a new peak at 1340 cm−1, which reflects the change in the DNA state. The Raman peak at 1581 cm−1 can reflect the redox state of Cyt C, but no clear changes dependent on the mitochondrial potential change were observed because of the low signal intensity in case of applying 785 nm laser excitation. Reversible changes of molecular states inside the cell were observed upon treatment with mild polarizing agents (3 mM MgCl2, 1 μM FCCP, and 1 mM sodium pyruvate). Although the fluorescence-based measurement with TMRM showed a decrease or increase in the intensity in the fluorescence images, it is not possible to obtain detailed information related to the membrane potential change from the fluorescence-based method. Although the spectrum was displayed every 5 min, the exposure time for a single spectrum was 30 ms which is comparable with that of the fluorescence-based imaging method. It should also be noted that the Raman spectrum is not bleached out with continuous monitoring. However, continuous monitoring with fluorescence is not easily attainable with the fluorescence-based method. In this regard, Raman spectroscopy is a promising analytical tool for monitoring real-time changes occurring in a cell over a long period of time.10,39,51Experimental section
Chemicals and materials
TA-stabilized gold colloids (50 nm) were purchased from BBI Solutions (Madison, WI, USA). TPP was obtained from Sigma Aldrich (St. Louis, MO, USA). Dulbecco's phosphate-buffered saline (DPBS) was purchased from Mediatech, Inc. (Manassas, VA, USA). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from HyClone (Waltham, MA, USA). Antibiotic solution and 0.25% trypsin–EDTA were purchased from Gibco (Waltham, MA, USA). Mito-Tracker Green FM and Image-iT TMRM Reagent (mitochondrial membrane potential indicator) were obtained from Invitrogen, Inc. (Carlsbad, CA, USA). Rhodamine-123 and MgCl2 were acquired from Sigma Aldrich (St. Louis, MO, USA). Human TNF alpha Recombinant Protein, eBioscience was purchased from Invitrogen, Inc. (Carlsbad, CA, USA). Sodium pyruvate was obtained from Gibco (Waltham, MA, USA). FCCP was acquired from Cayman Chemical (Ann Arbor, MI, USA).Instrumentation
An H-7100 electron microscope (Hitachi, Tokyo, Japan) was used for TEM analysis. Extinction spectra were obtained using a UV-Vis spectrometer (SCINCO, South Korea). The zeta potential was evaluated with a zeta potential and particle size analyzer (ELSZ-1000, Otsuka Electronics, Tokyo, Japan). B/F, D/F, and fluorescence images were recorded using a microscope (Olympus DP80, Tokyo, Japan) equipped with a dark-field condenser [U-DCD (N.A 0.8–0.92), Tokyo, Japan]. A near-infrared laser (Sanctity Laser SSL-808-6000-10TM-MF, Shanghai, China; 808 nm) with an output power of 5 W (beam diameter 1 cm) was used to induce photothermal damage. A Nanosight LM 10 instrument (Malvern Instruments, Worcestershire, UK) equipped with a 642 nm laser was used to measure the particle size.Cell culture and viability test
RASFs were cultured in DMEM supplemented with 10% v/v FBS and 1% v/v antibiotic. Cells were maintained in a 5% CO2 atmosphere in a humidified 37 °C incubator during Raman analysis. In the cell viability test, RASFs were seeded on a 96-well plate at a density of 5k cells per well and incubated overnight. After 3 h exposure to TPP-AuNP (0.01 nM, 0.1 nM, and 1 nM) in colorless DMEM, the cells were washed with DPBS twice. The MTS assay (Promega, Madison, WI, USA), as a colorimetric method, was used to determine the number of viable cells in proliferation. 20 μL of MTS solution reagent was added to each well of the 96-well plate in 100 μL of colorless DMEM. The plate was incubated at 37 °C for 4 h in a humidified, 5% CO2 atmosphere. Quantification of live cells was performed at an absorbance of 490 nm using a Cytation 3 Cell Imaging MultiMode Reader (Bio Tek).SERS measurements
Time-dependent SERS spectra from the cells were acquired using an inverted Raman microscope (NOST, South Korea) with a 60× objective (NA 0.7) and an oil immersion lens (100×, NA 1.3) (Olympus, Tokyo, Japan). The sample was excited with a diode laser (785 nm, IPS, USA). The scattered Raman signal was detected with a confocal motorized pinhole (100 μm) directed to a spectrometer (FEX-MD, NOST, South Korea) (600 g mm−1 grating) and finally to a charge-coupled device camera spectrometer [Andor (DV401A-BVF), Belfast, Northern Ireland]. RAON Scan (NOST, South Korea) software was used to acquire the Raman images. Spectra were recorded in 0.5 μm X/Y steps for all samples. The laser beam diameter was 684.07 nm [785 nm, 60× objective (NA = 0.7)]. The laser power was set to 3 mW and an integration time of 0.03 s was chosen to ensure fast mapping and to avoid cell damage. The required time to obtain a single-cell Raman image (50 × 50 μm) was 196 s (total pixel number: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, integration time/pixel = 0.03 s, time to move to the next pixel = 0.01 s).
000, integration time/pixel = 0.03 s, time to move to the next pixel = 0.01 s).
Fluorescence imaging of mitochondrial membrane potential
For quantification of mitochondrial membrane potential in living cells, RASFs were loaded with the mitochondrial membrane potential-sensitive dye TMRM. For dye loading, RASFs were incubated with 100 nM TMRM for 30 min at 37 °C in a humidified atmosphere containing 5% CO2. The cells were gently washed once with DPBS. Fluorescent imaging was conducted with an Olympus DP80 microscope equipped with a 20× air objective. For the measurements of TMRM signals, the cells were illuminated with an excitation wavelength of 555 nm and the TMRM was detected at 572–638 nm.Real-time monitoring of the changes of mitochondrial membrane potential using rhodamine-123
The RASFs seeded in a 96-well plate were incubated in colorless DMEM containing 1 ng mL−1 rhodamine-123 for 30 min and washed once with DPBS before starting the experiment. The mitochondrial membrane potential was monitored with excitation and emission filters set at 485 nm and 520 nm, respectively, using a Cytation 3 Cell imaging multi-mode reader (Bio Tek). Sodium pyruvate was added to each well at the indicated times. The rhodamine-123 fluorescence intensity was measured in real time at intervals of 20 s.Data analysis
Five Raman spectra were obtained from a single cell by illuminating with a 785 nm laser (3 mW, 0.03 s exposure time) every 5 min, and the average Raman intensity of the selected Raman scattering peaks at 505, 840, 1127, 1313, and 1585 cm−1 was obtained. Data fitting was performed using a nonlinear curve fit and Gaussian fitting methods in the Origin program.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (2017M3D1A1039421 and 2018R1A2A3075499), Korea University, and the KU-KIST Research Fund.Notes and references
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- A. M. Smith, H. Duan, A. M. Mohs and S. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef CAS PubMed.
- Y. Kumamoto, Y. Harada, T. Takamatsu and H. Tanaka, Acta Histochem. Cytochem., 2018, 51, 101–110 CrossRef CAS PubMed.
- K. Klein, A. M. Gigler, T. Aschenbrenner, R. Monetti, W. Bunk, F. Jamitzky, G. Morfill, R. W. Stark and J. Schlegel, Biophys. J., 2012, 102, 360–368 CrossRef CAS PubMed.
- R. Smith, K. L. Wright and L. Ashton, Analyst, 2016, 141, 3590–3600 RSC.
- M. Li, S. R. Banerjee, C. Zheng, M. G. Pomper and I. Barman, Chem. Sci., 2016, 7, 6779–6785 RSC.
- W. J. Tipping, M. Lee, A. Serrels, V. G. Brunton and A. N. Hulme, Chem. Sci., 2017, 8, 5606–5615 RSC.
- C. L. Evans, E. O. Potma, M. Puoris'haag, D. Côté, C. P. Lin and X. S. Xie, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16807–16812 CrossRef CAS PubMed.
- K. A. Willets, Anal. Bioanal. Chem., 2009, 394, 85–94 CrossRef CAS PubMed.
- C. Zong, M. Xu, L.-J. Xu, T. Wei, X. Ma, X.-S. Zheng, R. Hu and B. Ren, Chem. Rev., 2018, 118, 4946–4980 CrossRef CAS PubMed.
- L. A. Lane, X. Qian and S. Nie, Chem. Rev., 2015, 115, 10489–10529 CrossRef CAS PubMed.
- L. A. Austin, B. Kang and M. A. El-Sayed, J. Am. Chem. Soc., 2013, 135, 4688–4691 CrossRef CAS PubMed.
- B. Kang, L. A. Austin and M. A. El-Sayed, Nano Lett., 2012, 12, 5369–5375 CrossRef CAS PubMed.
- M. Aioub and M. A. El-Sayed, J. Am. Chem. Soc., 2016, 138, 1258–1264 CrossRef CAS PubMed.
- N. M. Sakhrani and H. Padh, Drug Des., Dev. Ther., 2013, 7, 585–599 CAS.
- M. P. Murphy and R. C. Hartley, Nat. Rev. Drug Discovery, 2018, 17, 865 CrossRef CAS PubMed.
- L. D. Zorova, V. A. Popkov, E. Y. Plotnikov, D. N. Silachev, I. B. Pevzner, S. S. Jankauskas, V. A. Babenko, S. D. Zorov, A. V. Balakireva, M. Juhaszova, S. J. Sollott and D. B. Zorov, Anal. Biochem., 2018, 552, 50–59 CrossRef CAS PubMed.
- H. Xin, W. J. Sim, B. Namgung, Y. Choi, B. Li and L. P. Lee, Nat. Commun., 2019, 10, 3245 CrossRef PubMed.
- G. Qi, B. Wang, Y. Zhang, H. Li, C. Li, W. Xu and Y. Jin, Anal. Chem., 2019, 91, 9571–9579 CrossRef CAS PubMed.
- T. Morimoto, L.-D. Chiu, H. Kanda, H. Kawagoe, T. Ozawa, M. Nakamura, K. Nishida, K. Fujita and T. Fujikado, Analyst, 2019, 144, 2531–2540 RSC.
- N. A. Brazhe, A. B. Evlyukhin, E. A. Goodilin, A. A. Semenova, S. M. Novikov, S. I. Bozhevolnyi, B. N. Chichkov, A. S. Sarycheva, A. A. Baizhumanov, E. I. Nikelshparg, L. I. Deev, E. G. Maksimov, G. V. Maksimov and O. Sosnovtseva, Sci. Rep., 2015, 5, 13793 CrossRef PubMed.
- N. A. Brazhe, M. Treiman, B. Faricelli, J. H. Vestergaard and O. Sosnovtseva, PLoS One, 2013, 8, e70488 CrossRef CAS PubMed.
- M. Okada, N. I. Smith, A. F. Palonpon, H. Endo, S. Kawata, M. Sodeoka and K. Fujita, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 28–32 CrossRef CAS PubMed.
- A. S. Sarycheva, N. A. Brazhe, A. A. Baizhumanov, E. I. Nikelshparg, A. A. Semenova, A. V. Garshev, A. E. Baranchikov, V. K. Ivanov, G. V. Maksimov, O. Sosnovtseva and E. A. Goodilin, J. Mater. Chem. B, 2016, 4, 539–546 RSC.
- H. J. Shin, J. H. Lee, Y. D. Kim, I. Shin, T. Sim and D.-K. Lim, ACS Omega, 2019, 4, 8188–8195 CrossRef CAS PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS.
- M. P. Murphy, Biochim. Biophys. Acta, 2008, 1777, 1028–1031 CrossRef CAS PubMed.
- Z. Wang, W. Guo, X. Kuang, S. Hou and H. Liu, Asian J. Pharm. Sci., 2017, 12, 498–508 CrossRef PubMed.
- H. Yamakoshi, A. F. Palonpon, K. Dodo, J. Ando, S. Kawata, K. Fujita and M. Sodeoka, Chem. Commun., 2014, 50, 1341–1343 RSC.
- E. A. Untener, K. K. Comfort, E. I. Maurer, C. M. Grabinski, D. A. Comfort and S. M. Hussain, ACS Appl. Mater. Interfaces, 2013, 5, 8366–8373 CrossRef CAS PubMed.
- O. F. Karatas, E. Sezgin, O. Aydin and M. Culha, Colloids Surf., B, 2009, 71, 315–318 CrossRef CAS PubMed.
- L. Kastl, S. W. Sauer, T. Ruppert, T. Beissbarth, M. S. Becker, D. Suss, P. H. Krammer and K. Gulow, FEBS Lett., 2014, 588, 175–183 CrossRef CAS PubMed.
- M. J. Lopez-Armada, B. Carames, M. A. Martin, B. Cillero-Pastor, M. Lires-Dean, I. Fuentes-Boquete, J. Arenas and F. J. Blanco, Osteoarthr. Cartil., 2006, 14, 1011–1022 CrossRef CAS PubMed.
- B. S. Polla, M. R. Jacquier-Sarlin, S. Kantengwa, E. Mariethoz, T. Hennet, F. Russo-Marie and A. Cossarizza, Free Radical Res., 1996, 25, 125–131 CrossRef CAS PubMed.
- W. D. Thomas, X. D. Zhang, A. V. Franco, T. Nguyen and P. Hersey, J. Immunol., 2000, 165, 5612 CrossRef CAS PubMed.
- S. W. Perry, J. P. Norman, J. Barbieri, E. B. Brown and H. A. Gelbard, BioTechniques, 2011, 50, 98–115 CrossRef CAS.
- H. Sies, Free Radical Biol. Med., 1999, 27, 916–921 CrossRef CAS.
- T. Morimoto, L.-D. Chiu, H. Kanda, H. Kawagoe, T. Ozawa, M. Nakamura, K. Nishida, K. Fujita and T. Fujikado, Analyst, 2019, 144, 2531–2540 RSC.
- L. A. Austin, B. Kang and M. A. El-Sayed, Nano Today, 2015, 10, 542–558 CrossRef CAS.
- X. M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912–920 RSC.
- D. Jaque, L. Martínez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martín Rodríguez and J. García Solé, Nanoscale, 2014, 6, 9494–9530 RSC.
- S. Xiang, K. Zhang, G. Yang, D. Gao, C. Zeng and M. He, Nanoscale Res. Lett., 2019, 14, 211 CrossRef PubMed.
- C. Yue, Y. Yang, J. Song, G. Alfranca, C. Zhang, Q. Zhang, T. Yin, F. Pan, J. M. de la Fuente and D. Cui, Nanoscale, 2017, 9, 11103–11118 RSC.
- G. Qi, Y. Zhang, S. Xu, C. Li, D. Wang, H. Li and Y. Jin, Anal. Chem., 2018, 90, 13356–13364 CrossRef CAS PubMed.
- J. Yue, Y. Shen, L. Liang, X. Guan, X. Zhang, S. Xu, C. Liang, W. Shi and W. Xu, Analyst, 2019, 144, 5521–5527 RSC.
- I. Pilchova, K. Klacanova, Z. Tatarkova, P. Kaplan and P. Racay, Oxid. Med. Cell. Longevity, 2017, 2017, 6797460 Search PubMed.
- R. Yamanaka, S. Tabata, Y. Shindo, K. Hotta, K. Suzuki, T. Soga and K. Oka, Sci. Rep., 2016, 6, 30027 CrossRef CAS PubMed.
- R. Eskes, B. Antonsson, A. Osen-Sand, S. Montessuit, C. Richter, R. Sadoul, G. Mazzei, A. Nichols and J. C. Martinou, J. Cell Biol., 1998, 143, 217–224 CrossRef CAS PubMed.
- E. Gout, F. Rebeille, R. Douce and R. Bligny, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4560–E4567 CrossRef CAS PubMed.
- P. B. de Andrade, M. Casimir and P. Maechler, FEBS Lett., 2004, 578, 224–228 CrossRef CAS PubMed.
- Y. Wang, B. Yan and L. Chen, Chem. Rev., 2013, 113, 1391–1428 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na01076f |
| This journal is © The Royal Society of Chemistry 2021 |