
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
LED-driven controlled deposition of Ni onto TiO2 for visible-light expanded conversion of carbon dioxide into C1–C2 alkanes†
Arturo
Sanz-Marco
ab,
José L.
Hueso
 abc,
Víctor
Sebastian
abc,
David
Nielsen
d,
Susanne
Mossin
d,
Juan P.
Holgado
e,
Carlos J.
Bueno-Alejo
ab,
Francisco
Balas
*abc and
Jesus
Santamaria
*abc
abc,
Víctor
Sebastian
abc,
David
Nielsen
d,
Susanne
Mossin
d,
Juan P.
Holgado
e,
Carlos J.
Bueno-Alejo
ab,
Francisco
Balas
*abc and
Jesus
Santamaria
*abc
aDepartment of Chemical and Environmental Engineering, University of Zaragoza, c/Mariano Esquillor, s/n; Campus Rio Ebro, Edificio I+D, Zaragoza, 50018, Spain. E-mail: fbalas@unizar.es
bInstitute of Nanoscience and Materials of Aragon (INMA), University of Zaragoza, Consejo Superior de Investigaciones Científicas (CSIC), c/Mariano Esquillor, s/n, 50018 Zaragoza, Spain
cNetworking Research Center in Biomaterials, Bioengineering and Nanomedicine (CIBER-BBN), C/Monforte de Lemos, 3-5, 28029 Madrid, Spain
dCentre for Catalysis and Sustainable Chemistry, Department of Chemistry, Technical University of Denmark, Kemitorvet 207, 2800 Kgs. Lyngby, Denmark
eInstituto de Ciencia de Materiales de Sevilla (ICMS, CSIC-University of Seville), Avda. Americo Vespucio, s/n, Seville, 41092, Spain
First published on 20th April 2021
Abstract
Photocatalytic gas-phase hydrogenation of CO2 into alkanes was achieved over TiO2-supported Ni nanoparticles under LED irradiation at 365 nm, 460 nm and white light. The photocatalysts were prepared using photo-assisted deposition of Ni salts under LED irradiation at 365 nm onto TiO2 P25 nanoparticles in methanol as a hole scavenger. This procedure yielded 2 nm Ni particles decorating the surface of TiO2 with a nickel mass content of about 2%. Before the photocatalytic runs, Ni/TiO2 was submitted to thermal reduction at 400 °C in a 10% H2 atmosphere which induced O-defective TiO2−x substrates. The formation of oxygen vacancies, Ti3+ centers and metallic Ni sites upon photocatalytic CO2 hydrogenation was confirmed by operando EPR analysis. In situ XPS under reaction conditions suggested a strong metal–support interaction and the co-existence of zero and divalent Ni states. These photoactive species enhanced the photo-assisted reduction of CO2 below 300 °C to yield CO, CH4 and C2H6 as final products.
Introduction
The overall depletion of fossil fuels and the overwhelming evidence of the impact of greenhouse gas emissions on the global environment are among the major challenges of the humankind today1,2 and demand a swift transition to renewable and sustainable processes. Among the strategies advocated to overcome these issues are gas-phase capture and storage, and revalorization by reduction to value-added products. In particular, the catalytic hydrogenation of CO2 for synthetic fuel production is an appealing mid-term solution to reduce the concentration of greenhouse gases, mitigate the dependence on fossil fuels and promote an overall neutral CO2 emission cycle.3–7Photocatalysis involves a greener vision of chemical processes, including the smart and selective transformation of chemicals into fuels and/or chemical intermediates of interest.8–10 The use of photocatalysts leads to an effective depletion of organic pollutants, either in the form of wastewater or as organic volatile compounds.11,12 In fact, photocatalysis takes advantage of sunlight as an abundant and inexpensive energy source, providing a sustainable approach to solving the most pressing environmental issues. Alternatively, energy-efficient artificial light systems, such as light emitting diodes (hereafter LEDs) have revealed themselves as mighty allies for inducing photocatalytic processes with enhanced effectiveness (see for instance ref. 13). Current efforts are being devoted to developing novel photocatalysts that can exploit the full-solar spectrum.14–18
Most of the current photoactive materials are based on semiconducting oxides and heterojunctions,19,20 where light irradiation promotes the formation of an electron–hole pair, or an exciton (e−–h+), in the electronic band structure of the solid, followed by the redox reaction on the surface of the catalyst. Among them, titanium dioxide, TiO2, is the most studied photocatalytic semiconductor, due to its high photoactivity, low cost, natural abundance and non-toxicity.21 However, its wide band gap (Eg = 3.2 eV for the anatase phase) limits its photo-activity to the UV region.22,23 Some of the strategies to enhance the catalytic activity of TiO2 involve the addition of nanostructured co-catalysts for improving the charge separation and the catalytic kinetics6,24 or the controlled generation of structural defects to enhance the solar absorption and the gas species adsorption on the surface.25 Since the seminal work on TiO2 as a heterogeneous photocatalyst,26 there has been an outstanding development of semiconducting TiO2-based nanostructured materials, particularly for environmental remediation applications under both liquid27,28 and gas phase conditions.29–31
A wide variety of noble metal co-catalysts have been tested for the gas phase hydrogenation of CO2 into alkanes mediated by the Sabatier's reaction in the gas phase under diverse conditions.32–35 In addition, first-row elements have been deposited on the surface of nanosized TiO2 under analogous conditions to develop efficient photocatalysts for numerous environmental applications.36 Particularly, nickel on TiO2 exhibited a high activity for the methanation reaction with dependence on the morphology, crystalline state and metal–support interactions.37 The photo-assisted catalytic activity of Ni-containing TiO2 catalysts has been tested, which showed that the work function of Ni lies in the appropriate range (5.3 eV) to enhance the charge separation of the TiO2 electron bands.38 Furthermore, Meng et al. showed that even oxidized Ni species, such as Ni(OH)2, on TiO2 nanofibers were effective for the reduction of CO2 to methane and carbon monoxide.39 Similarly, Ni nanoclusters on reduced TiO2 supports have been reported to be effective for the photocatalytic conversion of CO2 to alcohols via an aldehyde mechanism.40 To deposit metal crystallites on TiO2 a variety of methods have been used, including hydrothermal, co-precipitation and thermal evaporation procedures, yielding metal nanoparticles with sizes down to 10 nm on the surface of TiO2.41,42
Recently, the photocatalytic nature of some semiconducting metal oxides has been leveraged to reduce metal ions to nanosized metal particles upon light irradiation.43,44 This so-called photodeposition procedure is a liquid-phase reaction that uses light in the UV range to induce an exciton pair in the surface of TiO2,45 which is first trapped by surface oxygen atoms. The photogenerated holes at the Ti–OH sites act as active species for the oxidation of solvent molecules, e.g. methanol, through hydrogen evolution, and the electrons cause the reduction of metal cations present in the liquid, which precipitate as metallic particles on the surface of TiO2.46,47 The hole scavenging process allows a longer life of the e−–h+ pair and therefore a more effective metal ion reduction and deposition is achieved. Usually, the photo-assisted deposition procedure leads to metal particles, with small sizes (e.g. 5 nm), on the semiconducting oxide.42
In the present work, Ni-based nanosized particles have been deposited on the surface of TiO2 nanoparticles by a photodeposition method in methanol as a hole scavenger. These materials have been successfully tested in the gas-phase photocatalytic hydrogenation of CO2 in a fixed-bed reactor under LED irradiation at different wavelengths, including the full visible spectrum. The generation of structural defects (i.e. oxygen vacancies and Ti3+ defective centers) within the TiO2 framework was investigated with operando EPR. In situ XPS studies under reaction conditions further elucidated the coexistence of metallic and divalent Ni domains partially buried or decorating the TiO2 support, thereby suggesting a strong metal–support interaction (SMSI). We found that the ex situ thermal pre-activation of the Ni–TiO2 strongly promoted the light-driven/light-assisted/LED-driven CO2 hydrogenation towards carbon monoxide, methane and ethane.
Experimental section
Synthesis of Ni–TiO2 by LED-assisted photodeposition
A photodeposition method was used to attach Ni nanoparticles on the TiO2 surface. Briefly, 25 mg of nanosized TiO2 (Evonik P25, Dusseldorf, Germany) and 55 mg of nickel chloride hydrate (NiCl2·6H2O 98%, Sigma-Aldrich) were added to 17 ml of DI water in a 20 ml Pyrex tube, which was then closed with a rubber septum. After 20 min of degasification under nitrogen gas flow, 1 ml of methanol (CH3OH, Sigma-Aldrich) was added to the mixture. All reagents and precursors were used without purification. The suspension was subsequently irradiated with UV light using two LED lamps (OSRAM LED Engin, Wilmington MA) at 365 nm for 270 s and under continuous magnetic stirring to induce the photocatalytic reduction of Ni salts. The so-obtained suspension was further centrifuged at 7500 rpm for 5 minutes, washed several times with a mixture of double-deionized water and ethanol, and dried at 100 °C for 10 h. The final solid samples were stored in the dark for further use and characterization. Finally, some of the samples were submitted to a gas-phase ex situ reduction procedure before characterization and photocatalytic activity tests. Typically, 150 mg of the catalyst were loaded into a ceramic crucible and placed in a tube furnace (Carbolite, Hope Valley, UK) under a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 H2:N2 atmosphere at 50 sccm and heated up to 400 °C at a 10 °C min−1 heating rate. The temperature was held for 2 h before cooling to room temperature under the same atmosphere. After thermal reduction, the TiO2-based nanoparticles developed a greyish color. These nanomaterials were prepared at the Synthesis of Nanoparticles Unit 9 of the ICTS “NANBIOSIS” at the Institute of Nanoscience of Aragon and the University of Zaragoza.
:90 H2:N2 atmosphere at 50 sccm and heated up to 400 °C at a 10 °C min−1 heating rate. The temperature was held for 2 h before cooling to room temperature under the same atmosphere. After thermal reduction, the TiO2-based nanoparticles developed a greyish color. These nanomaterials were prepared at the Synthesis of Nanoparticles Unit 9 of the ICTS “NANBIOSIS” at the Institute of Nanoscience of Aragon and the University of Zaragoza.
Characterization
The crystalline structures of the TiO2 support and the photo-deposited Ni nanoparticles were identified by the X-ray diffraction (XRD) technique using a Bruker D8 Advance Diffractometer (Bruker Corporation, Billerica MA) equipped with a (002) Ge monochromator using the CuKα1 line at 1.5405 Å. The UV-vis spectra were recorded in a JASCO V-670 UV-vis/NIR spectrophotometer (JASCO, Tokyo, Japan) using the solid-state diffuse reflectance technique in a 60 mm UV-vis/NIR integrating sphere from 200 nm to 900 nm, with a scanning step of 10 nm s−1. Temperature-programmed reduction (TPR) tests were performed in a Quantachrome ChemBET Pulsar TPR/TPD analyzer (Quantachrome Instruments, Boynton Beach, FL) equipped with a Thermal Conductivity Detector (TCD). Usually, 50 mg powdered samples were loaded in one of the branches of a U-tube quartz reactor suspended between quartz wool pieces. Before every TPR test, samples were heated up to 100 °C under Ar gas flow to remove adsorbed species on the material surface. After cooling to room temperature, samples were heated from room temperature to 900 °C at 10 °C min−1 heating rate under 10:90 H2/Ar gas flow at 20 sccm. The BET surface area and porosity of catalysts were determined by means of N2 adsorption at 77 K using a Micromeritics TriStar 3000 analyzer (Micromeritics, Norcross, GA).
The TiO2 P25 and Ni/TiO2 were investigated by in situ and operando Electron Paramagnetic Resonance spectroscopy (EPR) on a Bruker EMX EPR instrument fitted with a ST4102 cavity and a Bruker variable temperature unit and quartz insert. The materials were pressed into pellets, then crushed and fractioned (150–300 μm). ∼25 mg of sample were immobilized with quartz wool in a 4 mm inner diameter quartz tube and placed in the cavity. The EPR spectra (9.45 GHz, 20 mW, 5 Gauss modulation amplitude at 100 kHz) were continuously obtained while introducing a total gas flow of 50 Nml min−1 during the sample analysis. To study the influence of light irradiation, UV light pulses in 100 s intervals (Dymax Blue Wave 75) were directed via a glass fiber to shine on the sample tube through a grid on the side of the EPR cavity. The same experimental protocol (see the ESI†) was performed on P25 and on Ni/TiO2 while monitoring with EPR to compare the responses of the photocatalysts under UV light exposure and different reaction atmospheres (see Table S1 in the ESI†). Samples were first activated in 30% H2 at 250 °C. Then the influence of UV light and of the H2/CO2 gas (separately and together) was investigated at room temperature. Then the sample was reactivated and the procedure was repeated at 250 °C. In general, the spectra exhibited lower resolution at 250 °C, as the noise level was higher at these temperatures and the changes were more difficult to discern. Overall, the same evolution was observed, but it was less clear and thus only room temperature spectra are shown in the text. The spectrum assigned to Ni species (type 1, see the main text) changed with temperature.
The Ni metal loading was evaluated using a microwave plasma-atomic emission spectrometer (4100 MP-AES, Agilent) and X-ray photoelectron spectroscopy (XPS) spectra were obtained before and after activation using a Kratos AXIS Ultra DLD surface analysis spectrometer (Kratos Analytical Ltd., Durham, UK). In situ XPS studies were performed using a customized system incorporating a hemispherical analyzer (SPECS Phoibos 100), a non-monochromatized X-ray source (Al Kα; 1486.6 eV, Mg Kα, 1253.6 eV) and a high temperature–high pressure cell (SPECS-HPC-20) that allows sample heating and gas flow. The analyzer was operated at a fixed transmission and 50 eV pass energy with an energy step of 0.1 eV. Binding energies were calibrated using C 1s or Ti 2p (284.6 eV or 458.8 eV) as an internal reference. The high temperature–high pressure cell design allowed sample heating up to 800 °C, under flow or static conditions, at pressures up to 20 bar or dynamic flows in the range 20–500 Nml min−1. This arrangement enabled a transfer of post-reaction samples from the reaction chamber to the spectrometer under UHV conditions, avoiding exposure to the laboratory atmosphere. Prior to each analysis, the samples were evacuated to 10−9 mbar at room temperature. In a typical experiment, the sample was initially placed in the sample holder (in the form of a pelletized disc) and transferred to the spectrometer chamber where XPS spectra were acquired. The sample was then transferred under vacuum to the high-pressure cell where it was exposed to the reactive gases and heated to the appropriate temperature. The flow and concentration of gases were similar to those used for catalytic testing (vide infra). After the treatment the sample was cooled to room temperature under the reaction atmosphere, evacuated down to 10−7 mbar in less than two minutes and then transferred back to the spectrometer chamber for analysis, avoiding ambient exposure.
Aberration corrected scanning transmission electron microscopy (Cs-corrected STEM) images were acquired using a high angle annular dark field detector in an FEI XFEG TITAN electron microscope operated at 300 kV equipped with a CETCOR Cs-probe corrector from CEOS Company allowing the formation of an electron probe of 0.08 nm. The geometric aberrations of the probe-forming system were controlled to allow a beam convergence of 24.7 mrad half-angle to be selected. Elemental analysis was carried out with an EDX detector for EDS experiments in scanning mode. EDX mapping was performed with an Oxford Instruments Detector and analysed with AZtec software provided by the detector manufacturer.
Photo-assisted carbon dioxide hydrogenation tests
The CO2 hydrogenation was conducted in a fixed-bed reactor as shown in Scheme 1. Typically, 120 mg of powdered catalyst was packed in a 30 × 10 × 2 mm prismatic quartz reactor that was positioned between two LED sources of different wavelengths, namely 365 nm, 460 nm and white light. Before irradiation, a gas mixture of CO2 and H2 with a 1:4 molar ratio was fed into the reactor for 30 min to ensure air removal from inside the reactor. After that, the reactor was closed and filled with the gas mixture until an absolute pressure of 1.7 bar was reached. A recirculating OEM pump (Verderflex M1500, Groeningen, NL) caused a 7.5 ml min−1 gas flow through the reactor circuit that included a 500 ml chamber to buffer pressure oscillations and provide enough total volume for gas sampling in prolonged experiments. The reaction was started by turning on the LEDs, which also caused a sharp increase in the solid temperature to reach a stable value around 250 °C. A sample of the gases was periodically analyzed using an on-line 490 micro-GC analyzer (Agilent Technologies, Santa Clara, CA, equipped with three columns: a 10 m molecular sieve column with a 5 m PPQ precolumn, a 10 m PBQ column and a 10 m CP-wax). DC current was set at 0.9 A and 12 V during LED irradiation for all tested materials.
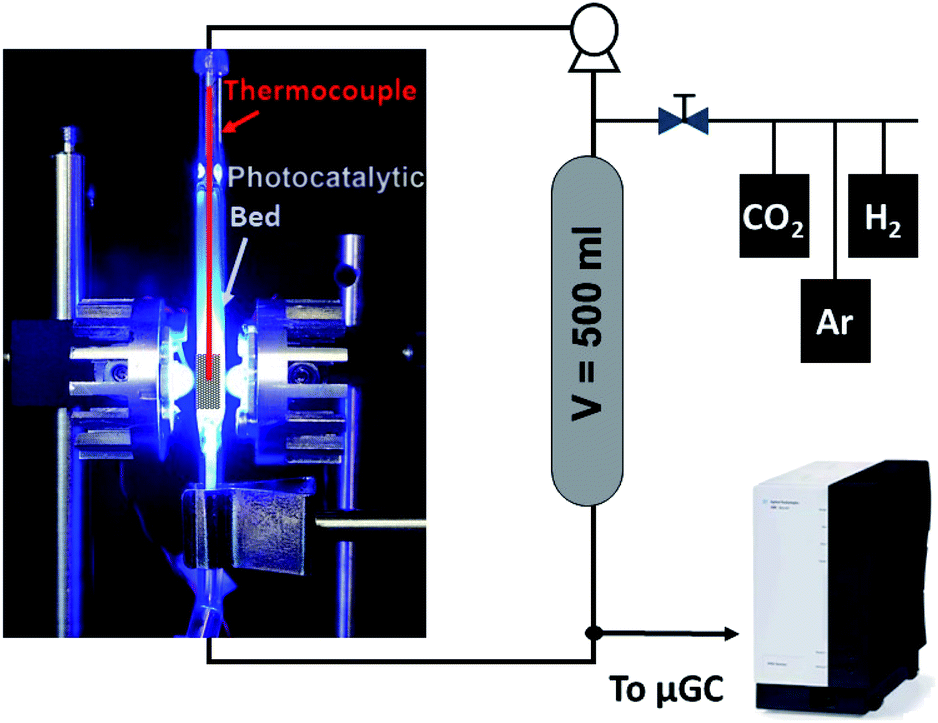 | ||
| Scheme 1 Simplified flowchart of the batch reactor system used for the photocatalytic tests. Typically, the batch reactor was filled with a 25:75 CO2/H2 mixture until a P = 1.7 bar was obtained. A 15 Nml min−1 gas flow was recirculated through the reactor. The fixed bed was operated under 9 W LED light (4.5 W per LED). A sample of the gas mixture was analyzed by gas chromatography hourly. | ||
Results and discussion
Photodeposition of Ni nanoparticles on TiO2 P25
The photo-assisted deposition method involves the photonic excitation of a semiconductor immersed in a solution that contains the ionic precursor of the material to be deposited and at least one liquid component that can act as a hole scavenger. When the incident photon energy is larger than the Eg, the valence band electrons can be excited to the conduction band.The pumped electrons are therefore used for the reduction of the metallic cations in the electrolyte, Ni2+ in this case, to deposit nanosized metallic particles on the surface of the semiconducting oxide. To avoid fast electron–hole pair recombination or the oxidation of the photodeposited metallic particles, methanol, an easy to oxidize substrate, is used, which acts as a sacrificial agent and hole scavenger when added to the electrolyte (Fig. 1a). The photo-deposition process provided white solids with a Ni mass concentration of 1.10 ± 0.11% calculated by MP-AES spectrometry (see the Experimental section). The subsequent thermal activation at 400 °C in a 10%-H2 atmosphere produced light grey-colored solids with a Ni mass concentration of 1.18 ± 0.05%, i.e., thermal activation in the hydrogen atmosphere did not alter the Ni loading of the prepared catalysts. The Ni/TiO2 catalyst particles exhibited similar size before and after thermal activation, between 20 and 25 nm. High-resolution HAADF-STEM images of individual Ni/TiO2 nanostructures showed 2 nm Ni/NiO crystallites homogeneously dispersed on the TiO2 nanoparticles (Fig. 1b). EDX analysis of individual nanoparticles confirmed the presence of Ni domains (see Fig. 1c). XRD analysis (Fig. 1d) showed the blend of anatase and rutile diffraction peaks expected for TiO2 P25 nanoparticles, without any diffraction pattern attributable to Ni or nickel oxides. This might be attributed to the concentration of deposited Ni (<2 wt%), particularly to its extraordinary dispersion and small particle size. These 2 nm sized Ni particles could be assigned to either Ni or NiO nanoparticles. Two different chemical processes could explain the presence of NiO after photodeposition: oxidation of the deposited Ni0 nanoparticles can take place upon environmental exposure, or the direct photodeposition of NiO particles may take place in a methanol-deficient medium.48,49
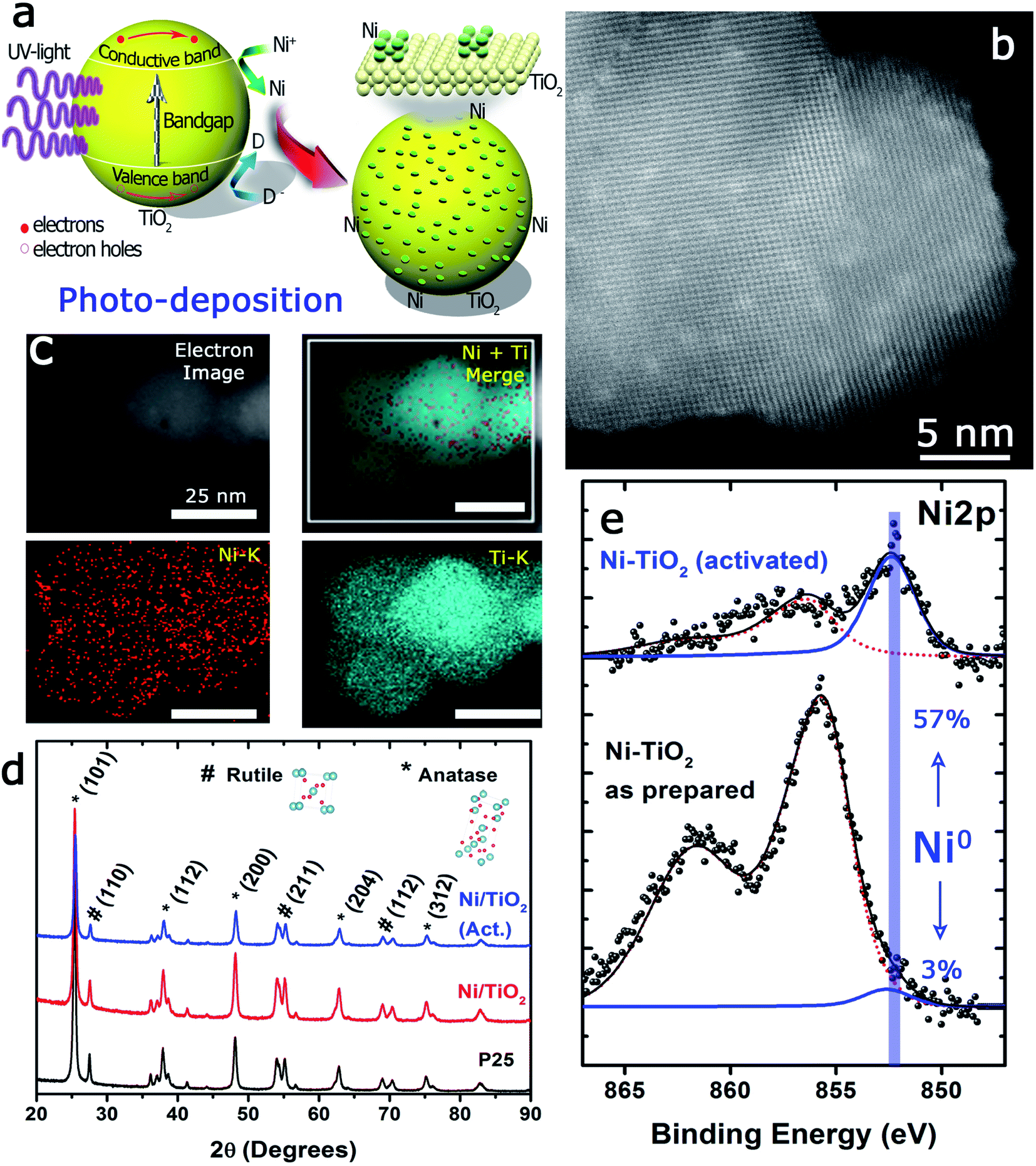 | ||
| Fig. 1 Characterization of the Ni–TiO2 photocatalyst: (a) scheme of the photo-assisted deposition process; (b) HAADF-STEM representative image of the Ni–TiO2 photocatalyst. Dots displaying lighter contrast correspond to the small Ni domains homogeneously dispersed on the TiO2 support; (c) EDX images corresponding to the selected areas shown in the inset STEM image accounting for the presence of Ti and Ni domains altogether in the catalyst nanoparticles; (d) XRD patterns of TiO2 nanoparticles after photodeposition of Ni from NiCl2 as the precursor and CH3OH as the hole-scavenging solvent. Crystalline Ni species remained undetected and only the reflections of anatase and rutile could be found. The activation procedure at 400 °C in a 10% H2 atmosphere reduced the relative height of the (100) reflection of anatase together with a slight increase in the peak width; (e) XPS spectra corresponding to the Ni 2p3/2 region before and after activation in a reducing environment. A strong increase of reduced Ni species is observed after activation. | ||
The UV-vis spectra showed very significant changes with respect to the absorption bands of TiO2 P25 (Fig. 2a), which displayed a strong absorption below 400 nm corresponding to the ground excitonic state of TiO2. The absorption bands of Ni/TiO2 were expanded towards the visible–near infrared region resulting from the formation of either metallic Ni or NiO nanoparticles.42,50,51 After thermal activation, the absorption in the visible region is greatly enhanced, which is due to the reducing conditions that led to the formation of oxygen vacancies and the incorporation of H-containing species or even Ti3+ cations on the surface of TiO2.52,53 Such structural modifications induced the formation of intermediate energy levels in the band gap, leading to broader light absorption.25 Furthermore, the optical band gap estimated using the Tauc plots (Fig. 2b) returned a value of Eg = 3.2 eV for TiO2, in good agreement with values reported elsewhere.23,25 For Ni/TiO2, a red-shift in the optical band gap could be observed which can be ascribed to an interfacial charge transfer process, consisting in the migration of electrons from the semiconducting substrate to the metallic nanoparticles directly from the valence band after excitation.54,55 This feature was slightly more noticeable after the thermal activation in a reducing atmosphere, which could be due to the formation of O2− vacancies or Ti3+ species on the TiO2 surface under those conditions.56
 | ||
| Fig. 2 UV-vis absorbance spectra (a) and Tauc plots (b) of TiO2 before and after Ni photodeposition in MeOH/H2O (Ni/TiO2) and thermal activation at 400 °C in a 5%-H2 atmosphere (Ni/TiO2-A). | ||
The TPR tests (Fig. 3) showed that Ni2+, presumably as NiO, and Ni0 were both likely present on Ni/TiO2 and Ni/TiO2-A. This was concluded after the detection of reduction peaks at TII ∼ 330 °C and TIII ∼ 370 °C which were attributed to the reduction of NiO species with low and high interaction with the support, respectively.57 The signal centered at TIV ∼ 550 °C was assigned to the reduction of smaller NiO nanoparticles dispersed in the TiO2 substrate with a very strong interaction.50,58 A noticeable decrease in the signal intensities was observed for the Ni/TiO2-A, which was attributed to the partial reduction of the Ni-based particles and TiO2 substrate after the thermal activation in a 10%-H2 atmosphere. However, the appearance of a signal at TI ∼ 230 °C after the activation suggested the presence of NiO particles with a weak or null interaction with the support,58 which is in agreement with the decrease in Ni loading observed by XPS (Table 1 and Fig. S1†). This decrease in Ni loading is also observed after a photocatalytic evaluation, due to the stronger reduction atmosphere of photocatalytic tests (80%-H2), which could lead to a better integration of Ni clusters within the TiO2 surface.
 | ||
| Fig. 3 TPR plots of TiO2 before and after Ni photodeposition in MeOH/H2O, as prepared (Ni/TiO2), and after thermal activation at 400 °C in a 10%-H2 atmosphere (Ni/TiO2-A). | ||
| BE (eV), surface concentration (at%) | ||||
|---|---|---|---|---|
| O 1s | Ti 2p | Ni 2p | C 1s | |
| Ni/TiO2 | 530.0 | 458.8 | 856.0 | 285.0 |
| 48.99% | 21.69% | 3.06% | 26.26% | |
| Ni/TiO2 (post-reaction) | 530.3 | 458.3 | 856.3 | 285.3 |
| 51.69% | 20.33% | 1.03% | 26.94% | |
| Ni/TiO2-A | 529.8 | 458.6 | 855.6 | 285.0 |
| 48.50% | 23.92% | 0.66% | 26.92% | |
| Ni/TiO2-A (post-reaction) | 529.4 | 458.4 | 855.4 | 284.4 |
| 51.45% | 21.63% | 0.14% | 26.78% | |
In situ and operando spectroscopic studies under CO2 hydrogenation conditions
With the aim of discerning the precise chemical state of the Ni and Ti on the surface of the catalyst, operando EPR spectroscopy has been used. Since both Ni and Ti paramagnetic centers, as well as potential ion defects, are present on the surface of the catalysts, EPR spectroscopy is undoubtedly a technique of choice.59 For instance, Morra et al. have used EPR to discern the active catalytic sites in Ti-containing materials, where titanium ions could be formed in low oxidation states under diverse reaction conditions.60In the present study, three different types of EPR signals were observed when performing the in situ and operando protocol. Fig. 4 shows a selection of EPR spectra and the most relevant changes observed under reaction conditions. Type 1 is observed for Ni/TiO2, but not for P25 TiO2. It is a huge broad peak centered at g ∼ 2.1 (Fig. 4a), constantly present for Ni/TiO2 but changes during activation, and is unaltered upon exposure to UV light. In order to follow the subtle change during activation, the spectra of the fresh sample were subtracted from the spectrum of the activated sample (Fig. 4a).
 | ||
| Fig. 4
In situ and operando spectroscopic studies under a CO2 hydrogenation reaction environment: (a) selected X-band EPR spectra collected at room temperature of TiO2 and Ni/TiO2 before and after thermal activation at 250 °C in 30% H2 and of Ni/TiO2-A before and after exposure to UV light and H2/CO2 at room temperature; full spectrum of Ni/TiO2 (black) and change in the EPR spectrum (×25) upon activation (red). (b) Zoom-in of the EPR spectra of TiO2 before (blue) and after activation (light blue), Ni/TiO2 after activation (dark blue) and the change after exposure to UV-light (brown) and after exposure to H2 + CO2 (light red); (c) XPS spectra corresponding to the Ni 2p3/2 and the Ti 2p regions after submitting the catalyst sample to successive thermal treatments for either the reduction/activation (10 vol% H2–Ar; flow rate: 20 Nml min−1; 400–450 °C) or carbon dioxide hydrogenation reaction (CO2:H2 – 1:4; flow rate: 5 Nml min−1; 60 mg of pelletized 1.2 wt% Ni/P25 photocatalyst; 225 °C). | ||
The spectral change corresponded to a broad EPR spectrum with a g-value of ∼2.34 and a tiny sharp peak at 2.0035. The g-value of the broad signal in the difference spectrum was significantly shifted compared to the g-value of the overall spectrum (∼2.1). Type 1 was assigned to paramagnetic Ni centers or defects in the Ni nanoparticles, which suggests the contribution from several types of Ni species, which could be either Ni2+ or Ni+. The EPR spectrum is largely featureless for a precise assignment, but it is noted that the g-value of 2.34 for the evolved species is similar to the value observed for defect sites in pure NiO nanoparticles.61 The type 2 signal was a small sharp isotropic signal centered at 2.0035, which was absent for the fresh materials but showed up after a few minutes and in comparable intensities for both TiO2 and Ni/TiO2 after activation (heating to 250 °C in 30% H2). The signal of Ni/TiO2-A increased during exposure to UV light (still under H2), but returned back to the initial state after the light was turned off (Fig. 4b). No clear development in the signal was observed for TiO2 during interaction with UV light (data not shown). The signal is not influenced by either of the materials being exposed to the H2/CO2 gas mixture. The type 2 signal was assigned to sites in the bulk of the nanoparticles: either an F-center, an electron caught in an oxygen vacancy, or a V-center, an oxygen ion caught at a cation vacancy.62,63
Finally, type 3 was a small anisotropic signal centered at 2.011 which is visible only for Ni/TiO2-A. It overlapped with the type 1 signal and was barely influenced by exposure to UV light; however, it disappeared completely after exposure to the H2/CO2 mixture (notice the negative change in the spectrum in Fig. 4b). It was tentatively assigned to oxygen-centered surface hole trapping sites in the anatase polymorph of TiO2 P25.64 The anisotropy of the signal is due to the direction dependence of surface sites. This type of site is observed in the literature on pure TiO2 at low temperatures (<100 K) where the signal to noise ratio of EPR is much better. In our catalyst such evidence suggests that the introduction of very small Ni particles by photodeposition has promoted the number of these sites to the extent that they are now observable by EPR at room temperature and above. The fact that they are sensitive to the H2/CO2 mixture confirms the assignment as surface sites and is an indication that they are involved directly in the reaction of CO2 on the surface of the material.
The state of Ni species on the TiO2 substrates was further explored using XPS analysis (Fig. 4c and Table 1). It is interesting to note that the XPS signals corresponding to the Ni 2p levels were rather complex, being deconvoluted up to three Gaussian/Lorentzian peaks (see the ESI, Table S2 and Fig. S1†). This situation is typically found in the presence of multiplet contributions and satellite structures, whose formation could be found in recent papers.50,65 For Ni2+ the satellite at about +6.0 eV might be assigned to a final state effect associated with a (core) 3d8L configuration (where L stands for ligands). However, the intensity and position of this satellite depended on many factors such as bonding with ligands, symmetry, particle size and crystallinity.66,67
Furthermore, a noticeable decrease of the atomic concentration of Ni was observed after thermal activation under a H2 atmosphere at 400 °C (Table 1). This might be attributed to the process of Ni nanoparticle growth by sintering or, alternatively, to an incomplete immersion of the Ni particles into the support due to the “decoration” effect of the TiO2 over the Ni particles. This has been previously reported for a variety of metal catalysts on different supports,68 within the context of the strong metal–support interaction (SMSI) effect.69 Both events would be enhanced in the case of nanosized particles70,71 and under treatment with hydrogen. In addition, the absence of complete reduction of Ni particles (only 57% of Ni0 after the reduction treatment) would also support this hypothesis.
In situ XPS observations were carried out to evaluate the evolution of the oxidation states of Ni and Ti under activation or CO2 hydrogenation conditions at 225 °C giving interesting and complementary insights into the operando EPR results. Although the change of the oxidation state of Ti was unclear (Fig. 4c), after the initial CO2/H2 treatment, an incomplete recovery of Ni signal intensity and a notable oxidation of the particles were noticed (Ni2+ concentration about 84%). These facts would be in agreement with the expected behavior in a SMSI scenario, with the CO2 serving as a mild re-oxidation agent. After the second reduction treatment at 450 °C, the situation was similar to that described before, and a decrease in intensity and a partial reduction of the Ni particles were noticed (Fig. 4c and Table S3†).
Nevertheless, after the second treatment with the CO2/H2 mixture, neither the Ni signal intensity nor the Ni2+/Ni0 ratios were remarkably altered. This could reflect a more permanent coverage of the Ni particles caused by the reduction at a higher temperature under hydrogen and would reinforce the close interaction between Ni and TiO2 as suggested by EPR. Actually, the HAADF-STEM images of the catalysts after reaction revealed that while a fraction of Ni domains have coarsened, the majority of the catalyst maintained similar particle size distributions (see Fig. S2†).
Light-assisted gas-phase photocatalytic CO2 hydrogenation
The photocatalytic conversion of CO2 into CO, CH4 and C2H6 alkanes was observed under LED irradiation at different wavelengths (Fig. 5). The photocatalytic performance of TiO2 P25 under UV light was evaluated before Ni photodeposition (see Fig. S4a†). Also, a control experiment was carried using Ni/TiO2 under 365 nm LED irradiation in 4/1 Ar/H2 stream. No CO, CH4 or C2H6 signals were detected after reaction for 6 h (Fig. S4b†). Results showed CO productivity around 100 μmol gcat−1 h−1 during the 6 h of reaction time. The signals attributed to both CH4 and C2H6 were also detected, at concentrations below the quantification limit. In contrast, when Ni/TiO2 catalysts were illuminated at 365 nm (Fig. 5a), the main product obtained was CH4 with a maximum initial productivity of 450 μmol gcat−1 h−1 and an increased CO productivity of 250 μmol gcat−1 h−1. Also, the production of C2H6 remained almost constant during the reaction, reaching about 2 μmol gcat−1 h−1. Catalyst activation by the high temperature hydrogen treatment produced two main effects: first, the photocatalytic activity increased significantly under UV light (Fig. 5b), and second, the catalytic response is expanded to the visible region of the solar spectrum (Fig. 5c and d). This increase in the CO and C2H6 (∼4 μmol gcat−1 h−1) productivity under UV light for the Ni/TiO2-A could be attributed to the formation of both bulk and surface oxygen vacancies (VO) on the TiO2 phase upon hydrogenation and the enhancement of the SMSI effect as suggested by the in situ XPS data. These VO induced donor intermediate energy levels in the bandgap, which might act as electron traps, facilitating charge-carrier separation and charge transfer to the adsorbed species. The irradiation under UV LEDs was more effective for the generation of CO. A similar trend with lower productivity was also found for the Ni/TiO2-A under LED illumination at blue light (460 nm, Fig. 5c) and white light (Fig. 5d). Interestingly, similar catalytic activity tests under blue or white LED irradiation using the as-synthesized Ni/TiO2 catalysts before thermal activation showed CO productivity lower than 25 μmol gcat−1 h−1 up to 4 h of reaction (data not shown). | ||
| Fig. 5 Productivity vs. time plots for Ni/TiO2 catalysts (a) under LED light irradiation at 365 nm and for Ni/TiO2-A catalysts under LED irradiation at 365 nm (b), 460 nm (c) and white light (d). | ||
The temperature at the center of the fixed bed during the reaction was about 250 °C when 460 nm and white LED units were used, but increased to 270 °C and 300 °C for the Ni–TiO2 and Ni/TiO2-A under UV illumination, respectively, which could simply be a result of a higher absorbance of the catalyst at 365 nm, but may also reflect the contribution of plasmonic effects in the Ni/NiO nanoparticles located at the surface of the TiO2,72,73 as recent studies have shown that plasmonic nanoparticles deposited on TiO2 could act as nanoheaters on the surface of the semiconductor.74
The photothermal CO2 hydrogenation is expected to proceed through the exothermic Sabatier reaction (CO2 + 4H2 → CH4 + 2H2O, ΔH0 = −165 kJ mol−1).75 Alternatively, for TiO2-based materials the reverse water gas shift (RWGS) reaction has also been proposed, yielding CO and water vapor (CO2 + H2 → CO + H2O, ΔH0 = 41 kJ mol−1).74 It has been suggested that these reactions could occur through the dissociation of physisorbed CO2 into CO intermediates that either evolve to formate76 or to carbide to finally form CH4 molecules upon further hydrogenation.77 In our case the CO2 hydrogenation progressed to the formation of CH4 on active centers, namely Ni/NiO nanoclusters, followed to a certain extent by a C–C coupling up to the formation of C2H6. At this point, it should be noted that the direct formation of ethane from CO2 over Ni was reported to be very minimal.78 On the other hand, the C–C coupling in the gas phase to generate higher hydrocarbons, such as ethane, over Ni-based catalysts was recently reported to be independent of the structure and only controlled by the hydrogenation rate.79 The photo-assisted deposition of Ni was critical for the CO hydrogenation to alkanes (Fig. 6). For Ni-containing catalysts, the initial selectivity to CH4 is close to 70%, decreasing down to 60% after 6 h of catalytic test. After activation (Ni/TiO2-A), the selectivity slightly evolved towards CH4, which can be attributed to an increase in the SMSI after the reduction treatment.
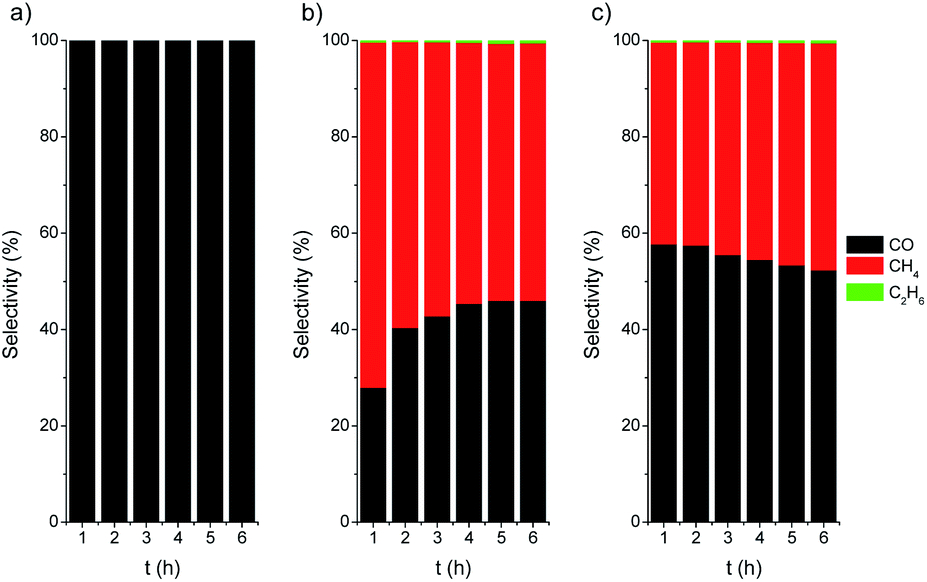 | ||
| Fig. 6 Selectivity vs. time plots for TiO2 P25 (a), Ni/TiO2 (b) and Ni/TiO2-A (c) catalysts under UV LED irradiation (365 nm). | ||
Finally, the thermally activated catalysts (Ni/TiO2-A) also exhibited a higher production of both C2 and C1 alkanes. In this case, the formed VO in the TiO2 could act as centers for the enhanced adsorption of CO2, thus improving the hydrogenation rate. The oxygen-defective TiO2 also induced the formation of Ti3+ species (see TPR data in Fig. 3), which have been reported to have a photonic response upon illumination with UV photons.25,80,81 These combined effects resulted in enhanced conversion of CO2 even under illumination in the visible range.
Conclusions
Ni/NiO nanosized particles were effectively deposited on the surface of TiO2 nanoparticles by a photodeposition procedure that used Ni2+ salts as precursors and CH3OH as a hole scavenger. The synthesis of very small Ni crystallites on TiO2 was achieved under the irradiation of LEDs at 365 nm. A thermal activation under a reducing atmosphere allowed the stabilization of Ni0 on the catalyst surface, as well as the formation of oxygen vacancies that could enhance the CO2 adsorption.Under LED illumination at different wavelengths, the Ni/TiO2 catalysts induced the photocatalytic hydrogenation of CO2 at 300 °C to produce CO, CH4 and C2H6 with a productivity of about 450 μmol gcat−1 h. The thermally reduced catalysts (Ni/TiO2-A) showed enhanced photocatalytic response in the UV region and higher CO productivities in all the studied wavelengths. As the thermal reduction induces the formation of active sites in the TiO2 support, the RWGS reaction is favored, increasing the CO selectivity. Further EPR and XPS analyses suggested that the close interaction of the metal and support plays an important role in the enhancement of photoactivity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Science and Universities through the CTQ2016-77144-R research project is gratefully acknowledged. We thank Dr Irusta for her assistance in preliminary XPS measurements. The synthesis of materials was performed by the Platform of Production of Biomaterials and Nanoparticles of the NANBIOSIS ICTS, more specifically by the Nanoparticle Synthesis Unit of the CIBER in Bioengineering, Biomaterials & Nanomedicine (CIBER-BBN). The TEM studies were conducted at the Laboratorio de Microscopias Avanzadas, Instituto de Nanociencia de Aragon, Universidad de Zaragoza, Spain. The EPR characterization was performed at the Centre for Catalysis and Sustainable Chemistry, Technical University of Denmark, supported by the Department of Chemistry for a PhD project to D. N. and by the Carlsberg Foundation for instrumentation.References
- IEA, World Energy Outlook 2018, IEA, Paris, 2018, https://doi.org/10.1787/weo-2018-en Search PubMed.
- BP p.l.c., BP Energy Outlook, 2019 Search PubMed.
- K. Li, X. An, K. H. Park, M. Khraisheh and J. Tang, Catal. Today, 2014, 224, 3–12 CrossRef CAS.
- P. R. Yaashikaa, P. Senthil Kumar, S. J. Varjani and A. Saravanan, J. CO2 Util., 2019, 33, 131–147 CrossRef CAS.
- L. Wang, M. Ghoussoub, H. Wang, Y. Shao, W. Sun, A. A. Tountas, T. E. Wood, H. Li, J. Y. Y. Loh, Y. Dong, M. Xia, Y. Li, S. Wang, J. Jia, C. Qiu, C. Qian, N. P. Kherani, L. He, X. Zhang and G. A. Ozin, Joule, 2018, 2, 1369–1381 CrossRef CAS.
- Y. Wang, J. Zhao, Y. Li and C. Wang, Appl. Catal., B, 2018, 226, 544–553 CrossRef CAS.
- F. Zhang, Y. H. Li, M. Y. Qi, Z. R. Tang and Y. J. Xu, Appl. Catal., B, 2020, 268, 118380 CrossRef CAS.
- X. Liu, X. Duan, W. Wei, S. Wang and B. J. Ni, Green Chem., 2019, 21, 4266–4289 RSC.
- Q. Mu, W. Zhu, X. Li, C. Zhang, Y. Su, Y. Lian, P. Qi, Z. Deng, D. Zhang, S. Wang, X. Zhu and Y. Peng, Appl. Catal., B, 2020, 262, 118144 CrossRef CAS.
- F. Xu, J. Zhang, B. Zhu, J. Yu and J. Xu, Appl. Catal., B, 2018, 230, 194–202 CrossRef CAS.
- O. Tursunov, L. Kustov and A. Kustov, Oil Gas Sci. Technol., 2017, 72, 30 CrossRef.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- C. J. Bueno-Alejo, J. L. Hueso, R. Mallada, I. Julian and J. Santamaria, Chem. Eng. J., 2018, 358, 1363–1370 CrossRef.
- C. Zhang, H. Q. Liang, Z. K. Xu and Z. Wang, Adv. Sci., 2019, 6, 1900883 CrossRef CAS PubMed.
- R. P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y. G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- Y. Liu, C. Miao, P. Yang, Y. He, J. Feng and D. Li, Appl. Catal., B, 2019, 244, 919–930 CrossRef CAS.
- S. Zhu, X. Chen, Z. Li, X. Ye, Y. Liu, Y. Chen, L. Yang, M. Chen, D. Zhang, G. Li and H. Li, Appl. Catal., B, 2020, 264, 118515 CrossRef.
- X. Feng, F. Pan, H. Zhao, W. Deng, P. Zhang, H. C. Zhou and Y. Li, Appl. Catal., B, 2018, 238, 274–283 CrossRef CAS.
- X. Yu, T. J. Marks and A. Facchetti, Nat. Mater., 2016, 15, 383–396 CrossRef CAS PubMed.
- M. Liras, M. Barawi and V. A. De La Peña O'Shea, Chem. Soc. Rev., 2019, 48, 5454–5487 RSC.
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217–9233 RSC.
- B. Wang, S. Shen and S. S. Mao, J. Mater., 2017, 3, 96–111 Search PubMed.
- Y. Liu, L. Tian, X. Tan, X. Li and X. Chen, Sci. Bull., 2017, 62, 431–441 CrossRef CAS.
- M. C. Ortega-Liebana, J. L. Hueso, S. Ferdousi, R. Arenal, S. Irusta, K. L. Yeung and J. Santamaria, Appl. Catal., B, 2017, 218, 68–79 CrossRef CAS.
- J. Jia, C. Qian, Y. Dong, Y. F. Li, H. Wang, M. Ghoussoub, K. T. Butler, A. Walsh and G. A. Ozin, Chem. Soc. Rev., 2017, 46, 4631–4644 RSC.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol. C Photochem. Rev., 2012, 13, 169–189 CrossRef CAS.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol. C Photochem. Rev., 2008, 9, 1–12 CrossRef CAS.
- J. C. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765–778 RSC.
- C. J. Bueno-Alejo, J. L. Hueso, R. Mallada, I. Julian and J. Santamaria, Chem. Eng. J., 2019, 358, 1363–1370 CrossRef CAS.
- W. Bi, Y. Hu, N. Jiang, L. Zhang, H. Jiang, X. Zhao, C. Wang and C. Li, Appl. Catal., B, 2020, 269, 118810 CrossRef CAS.
- Z. Deng, J. Ji, M. Xing and J. Zhang, Nanoscale Adv., 2020, 2, 4986 RSC.
- J. Xu, X. Su, H. Duan, B. Hou, Q. Lin, X. Liu, X. Pan, G. Pei, H. Geng, Y. Huang and T. Zhang, J. Catal., 2016, 333, 227–237 CrossRef CAS.
- A. Karelovic and P. Ruiz, J. Catal., 2013, 301, 141–153 CrossRef CAS.
- A. Petala and P. Panagiotopoulou, Appl. Catal., B, 2018, 224, 919–927 CrossRef CAS.
- C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, Nat. Catal., 2019, 2, 188–197 CrossRef CAS.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- R. Zhou, N. Rui, Z. Fan and C. J. Liu, Int. J. Hydrogen Energy, 2016, 41, 22017–22025 CrossRef CAS.
- B. S. Kwak, K. Vignesh, N. K. Park, H. J. Ryu, J. I. Baek and M. Kang, Fuel, 2015, 143, 570–576 CrossRef CAS.
- A. Meng, S. Wu, B. Cheng, J. Yu and J. Xu, J. Mater. Chem. A, 2018, 6, 4729–4736 RSC.
- T. Billo, F.-Y. Fu, P. Raghunath, I. Shown, W.-F. Chen, H.-T. Lien, T.-H. Shen, J.-F. Lee, T.-S. Chan, K.-Y. Huang, C.-I. Wu, M. C. Lin, J.-S. Hwang, C.-H. Lee, L.-C. Chen and K.-H. Chen, Small, 2018, 14, 1702928 CrossRef PubMed.
- W. T. Chen, A. Chan, D. Sun-Waterhouse, T. Moriga, H. Idriss and G. I. N. Waterhouse, J. Catal., 2015, 326, 43–53 CrossRef CAS.
- W. T. Chen, A. Chan, D. Sun-Waterhouse, J. Llorca, H. Idriss and G. I. N. Waterhouse, J. Catal., 2018, 367, 27–42 CrossRef CAS.
- Y. Lee, E. Kim, Y. Park, J. Kim, W. H. Ryu, J. Rho and K. Kim, J. Mater., 2018, 4, 83–94 Search PubMed.
- K. Wenderich and G. Mul, Chem. Rev., 2016, 116, 14587–14619 CrossRef CAS PubMed.
- S. Dadsetan, S. Baghshahi, F. Farshidfar and S. M. M. Hadavi, Ceram. Int., 2017, 43, 9322–9326 CrossRef CAS.
- J. L. Rodríguez, M. A. Valenzuela, F. Pola, H. Tiznado and T. Poznyak, J. Mol. Catal. A: Chem., 2012, 353–354, 29–36 CrossRef.
- A. Y. Ahmed, T. A. Kandiel, I. Ivanova and D. Bahnemann, Appl. Surf. Sci., 2014, 319, 44–49 CrossRef CAS.
- V. Iliev, D. Tomova, L. Bilyarska and G. Tyuliev, J. Mol. Catal. A: Chem., 2007, 263, 32–38 CrossRef CAS.
- A. Mills and M. A. Valenzuela, J. Photochem. Photobiol., A, 2004, 165, 25–34 CrossRef CAS.
- C. J. Bueno-Alejo, A. Arca-Ramos, J. L. Hueso and J. Santamaría, Catal. Today, 2020, 355, 678–684 CrossRef CAS.
- H. Yang, Z. Jin, K. Fan, D. D. Liu and G. Lu, Superlattices Microstruct., 2017, 111, 687–695 CrossRef CAS.
- X. Jiang, Y. Zhang, J. Jiang, Y. Rong, Y. Wang, Y. Wu and C. Pan, J. Phys. Chem. C, 2012, 116, 22619–22624 CrossRef CAS.
- Y. Guo, L. Xiao, M. Zhang, Q. Li and J. Yang, Appl. Surf. Sci., 2018, 208, 432–439 CrossRef.
- K. Fujiwara, K. Okuyama and S. E. Pratsinis, Environ. Sci.: Nano, 2017, 4, 2076–2092 RSC.
- H. Irie, S. Miura, K. Kamiya and K. Hashimoto, Chem. Phys. Lett., 2008, 457, 202–205 CrossRef CAS.
- S. Wei, R. Wu, X. Xu, J. Jian, H. Wang and Y. Sun, Chem. Eng. J., 2016, 299, 120–125 CrossRef CAS.
- X. Lin, L. Lin, K. Huang, X. Chen, W. Dai and X. Fu, Appl. Catal., B, 2015, 168–169, 416–422 CrossRef CAS.
- W. Lin, H. Cheng, L. He, Y. Yu and F. Zhao, J. Catal., 2013, 303, 110–116 CrossRef CAS.
- S. Van Doorslaer and D. M. Murphy, EPR Spectroscopy in Catalysis, in EPR Spectrosc. Appl. Chem. Biol., ed. M. Drescher and G. Jeschke, Springer, Berlin, Heidelberg, 2012, pp. 1–39 Search PubMed.
- E. Morra, E. Giamello and M. Chiesa, J. Magn. Reson., 2017, 280, 89–102 CrossRef CAS PubMed.
- M. Hashem, E. Saion, N. M. Al-Hada, H. M. Kamari, A. H. Shaari, Z. A. Talib, S. B. Paiman and M. A. Kamarudeen, Results Phys., 2016, 6, 1024–1030 CrossRef.
- A. Naldoni, M. Altomare, G. Zoppellaro, N. Liu, Š. Kment, R. Zbořil and P. Schmuki, ACS Catal., 2019, 9, 345–364 CrossRef CAS PubMed.
- T. Jia, J. Zhang, J. Wu, D. Wang, Q. Liu, Y. Qi, B. Hu, P. He, W. Pan and X. Qi, Mater. Lett., 2010, 265, 127465 CrossRef.
- D. C. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- V. Biju and M. Abdul Khadar, J. Nanopart. Res., 2002, 4, 247–253 CrossRef CAS.
- N. S. McIntyre, D. D. Johnston, L. L. Coatsworth, R. D. Davidson and J. R. Brown, Surf. Interface Anal., 1990, 15, 265–272 CrossRef CAS.
- L. E. Oi, M. Y. Choo, H. V. Lee, H. C. Ong, S. B. A. Hamid and J. C. Juan, RSC Adv., 2016, 6, 108741–108754 RSC.
- Z. Xu, Y. Li, J. Zhang, L. Chang, R. Zhou and Z. Duan, Appl. Catal., A, 2001, 210, 45–53 CrossRef CAS.
- A. Caballero, J. P. Holgado, V. M. Gonzalez-De la Cruz, S. E. Habas, T. Herranz and M. Salmeron, Chem. Commun., 2010, 46, 1097–1099 RSC.
- V. M. Gonzalez-De la Cruz, J. P. Holgado, R. Pereñíguez and A. Caballero, J. Catal., 2008, 257, 307–314 CrossRef CAS.
- R. Wang, Y. Li, R. Shi and M. Yang, J. Mol. Catal. A: Chem., 2011, 344, 122–127 CrossRef CAS.
- J. Zhao, B. Liu, L. Meng, S. He, R. Yuan, Y. Hou, Z. Ding, H. Lin, Z. Zhang, X. Wang and J. Long, Appl. Catal., B, 2019, 256, 117823 CrossRef CAS.
- L. B. Hoch, P. G. O'Brien, A. Jelle, A. Sandhel, D. D. Perovic, C. A. Mims and G. A. Ozin, ACS Nano, 2016, 10, 9017–9025 CrossRef CAS PubMed.
- K. Stangeland, D. Kalai, H. Li and Z. Yu, Energy Procedia, 2017, 2022–2027 CrossRef CAS.
- C. Heine, B. A. J. Lechner, H. Bluhm and M. Salmeron, J. Am. Chem. Soc., 2016, 138, 13246–13252 CrossRef CAS PubMed.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Nat. Catal., 2018, 1, 127–134 CrossRef CAS.
- S. Abelló, C. Berrueco and D. Montané, Fuel, 2013, 113, 598–609 CrossRef.
- C. Vogt, M. Monai, E. B. Sterk, J. Palle, A. E. M. Melcherts, B. Zijlstra, E. Groeneveld, P. H. Berben, J. M. Boereboom, E. J. M. Hensen, F. Meirer, I. A. W. Filot and B. M. Weckhuysen, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- E. Lira, S. Wendt, P. Huo, J. Ø. Hansen, R. Streber, S. Porsgaard, Y. Wei, R. Bechstein, E. Lægsgaard and F. Besenbacher, J. Am. Chem. Soc., 2011, 133, 6529–6532 CrossRef CAS PubMed.
- J. Ye, J. He, S. Wang, X. Zhou, Y. Zhang, G. Liu and Y. Yang, Sep. Purif. Technol., 2019, 220, 8–15 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of the nanocatalysts, analysis of the electron microscopy images, details of the operando EPR and additional catalytic activity tests. See DOI: 10.1039/d1na00021g |
| This journal is © The Royal Society of Chemistry 2021 |