
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled growth of ultrafine metal nanoparticles mediated by solid supports
Hongyin
Hu
a,
Shuanglong
Lu
 *a,
Ting
Li
a,
Yue
Zhang
a,
Chenxi
Guo
a,
Han
Zhu
a,
Yinghua
Jin
b,
Mingliang
Du
*a and
Wei
Zhang
*b
*a,
Ting
Li
a,
Yue
Zhang
a,
Chenxi
Guo
a,
Han
Zhu
a,
Yinghua
Jin
b,
Mingliang
Du
*a and
Wei
Zhang
*b
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, Jiangsu, China. E-mail: lushuanglong@jiangnan.edu.cn
bDepartment of Chemistry, University of Colorado, Boulder, CO 80309, USA. E-mail: wei.zhang@colorado.edu
First published on 15th February 2021
Abstract
As a unique class of nanomaterials with a high surface-area-to-volume ratio and narrow size distribution, ultrafine metal nanoparticles (UMNPs) have shown exciting properties in many applications, particularly in the field of catalysis. Growing UMNPs in situ on solid supports enables precise control of the UMNP size, and the supports can effectively prevent the aggregation of UMNPs and maintain their high catalytic activity. In this review, we summarize the recent research progress in controlled growth of UMNPs using various solid supports and their applications in catalysis.
 Hongyin Hu | Hongyin Hu will receive his bachelor's degree from the School of Chemical and Material Engineering of Jiangnan University in 2021. He has obtained the exemption to study graduate qualifications and will pursue his master's degree under the supervision of A.P. Shuanglong Lu. His current research interests focus on the synthesis of precious metal nanoparticles and their electrocatalytic application. |
 Shuanglong Lu | Shuanglong Lu received his BS and PhD degrees from Soochow University in 2013 and 2018 under the direction of Prof. Hongwei Gu. Meanwhile, he received financial support from the Chinese government to finish his joint PhD research at the University of Colorado at Boulder, USA, in Prof. Wei Zhang's group. He then joined Jiangnan University, China, as an associate professor in 2018. His current research interest mainly focuses on engineering nanostructured materials applied in both organic catalysis and electrocatalysis. |
 Mingliang Du | Mingliang Du received his PhD degree in 2007 from the South China University of Technology and he also worked there as a postdoctoral research fellow. In 2009, he joined Zhejiang Sci-Tech University in Hangzhou, as an associate professor, and was promoted to professor in 2014. In 2017, he joined the Department of Chemistry and Materials Engineering, Jiangnan University, in Wuxi. His current research focuses on synthesis of nanomaterials and carbon materials and their applications in clean energy, electrochemical biosensing, etc.; electrospinning and its applications; and controlled synthesis, characterization and application of metallic and semiconducting nanomaterials. |
 Wei Zhang | Wei Zhang received his BS in Chemistry from Peking University in 2000 and his PhD in Chemistry from the University of Illinois at Urbana-Champaign (UIUC) in 2005. After a postdoc stint at MIT, he started his independent career at the Department of Chemistry and Biochemistry at the University of Colorado Boulder in 2008 and was promoted to Associate and Full Professor in 2014 and 2018, respectively. His research is focused on utilizing dynamic covalent chemistry to develop novel organic and hybrid functional materials targeting a broad range of environmental, energy and biological applications. |
1. Introduction
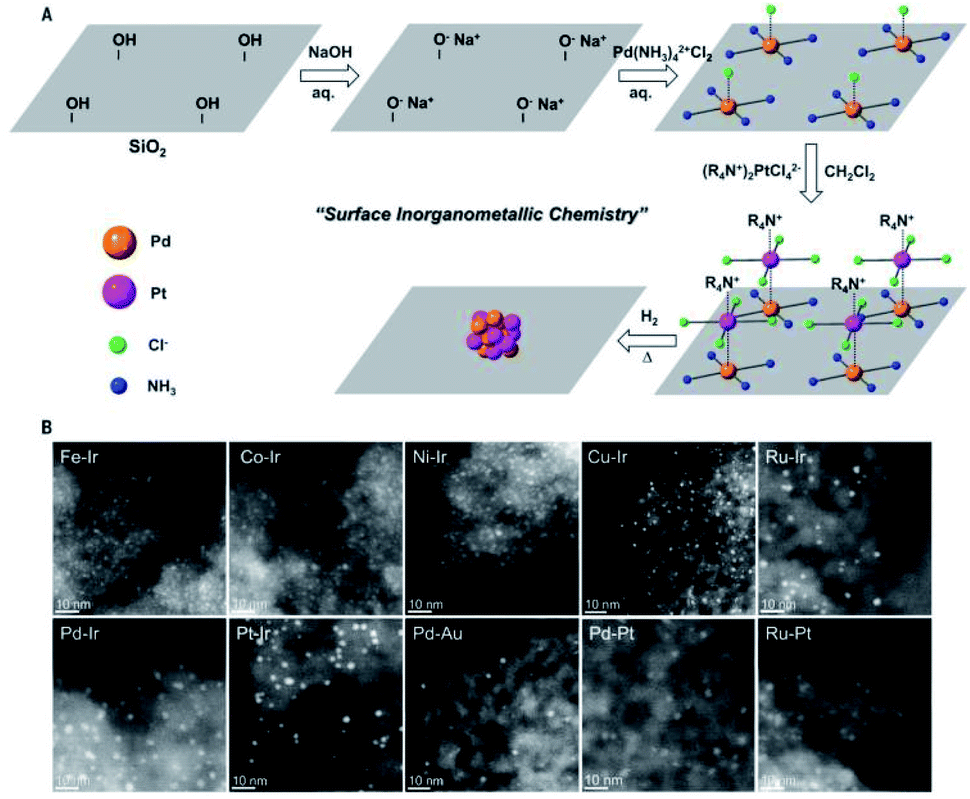
Metal nanoparticles have fascinating properties applicable in many fields, such as catalysis,1,2 sensing,3,4 energy storage,5,6 and medicine.7,8 They have made significant contributions to the development of heterogeneous catalysts, showing excellent catalytic activity and reusability. During the catalysis, the adsorption and desorption of substrates occur on the surface of metal nanoparticles. The partially filled d-orbitals can promote the adsorption of reactants and catalyze the product formation at active sites. Therefore, the number of surface metal atoms plays a critical role in the catalytic process. The smaller the diameter of metal nanoparticles, the larger the corresponding specific surface area and the larger the number of surface atoms (active sites). When the diameter of metal nanoparticles is reduced from 10 nm to 1 nm, the “effective” accessible metal atoms will increase from 20% to 90%.9 Therefore, it is necessary to control the diameter of metal nanoparticles. Ultrafine metal nanoparticles (UMNPs), which usually refer to metal nanoparticles with an average size of less than 3 nm, have shown great application potential in the field of catalysis.10 Typically, UMNPs have a high surface area and narrow size distribution, which can provide a large number of available active sites for heterogeneous catalysis. Having a large number of surface atoms, UMNPs are thermodynamically unstable.11–13 Based on the migration-coalescence and Ostwald ripening mechanism, smaller particles aggregate to form larger particles through a spontaneous thermodynamic process.14 The aggregation leads to reduced catalytic activity of UMNPs. Maintaining the dispersion and stability of UMNPs throughout the catalysis is also an important issue that must be solved to realize widespread practical applications of UMNPs in catalysis.Supports and organic capping agents are two commonly used methods for the preparation of UMNPs and preventing their aggregation.2,10,15–17 Organic capping agents, such as oleylamine and polyvinyl pyrrolidone, are often used to control the growth of metal nanoparticles.16,17 However, these capping agents usually adhere to the surface of UMNPs through strong interactions, blocking most of the catalytically active sites, thus significantly decreasing the catalytic activity of UMNPs. On the other hand, if the capping agent is removed, UMNPs tend to aggregate, which would affect their stability and also catalytic activity. Using solid supports to assist the UMNP synthesis can effectively solve the above problems. The supports can direct the growth of UMNPs and control their size through spatial confinement and electronic effects, so that different UMNPs can be prepared by varying the types and structures of supports.16,18 Moreover, by enhancing the interaction between MNPs and the support, the aggregation of MNPs can be minimized and the durability of the catalyst can be improved. Generally, metal ions or metal atoms whose d-orbitals are not filled can bind with electron-rich atoms on the support, such as S, N, O, etc., through coordination bonds.19–21 Therefore, heteroatoms or organic functional groups provided by supports are usually required for the growth and stabilization of UMNPs. In addition to the electronic effect, the confinement effect of supports on UMNPs also makes a big difference. Particularly for porous materials with aligned or interconnected nanopores, the nanopores/channels can confine the growth of metal nanoparticles.22,23 Supports such as COFs, MOFs, and CMPs can be custom-designed and synthesized to achieve molecular-level control of the size of metal nanoparticles.24–26 Simply varying the chemical nature of solid supports, a series of UMNPs with high activity, high surface area and narrow size distribution can be obtained.
In 2016, Xu et al. summarized the preparation of UMNPs immobilized on solid supports and their applications in catalysis.10 They described the roles of different supports in detail and delivered a thoughtful review on the reported UMNPs mediated by solid supports. This review focuses on the solid supports (such as COFs, metal oxides/sulfides, etc.) and the recent new research progress that has not been not covered previously. We discuss the controlled nucleation and growth of UMNPs using different solid supports, aiming to reveal the influence of different supports (scaffolds) on the growth and performance of UMNPs. These studies have demonstrated that the solid supports can not only control the size of UMNPs and prevent their aggregation, but also improve their recovery and catalytic activity through electronic and spatial confinement effects (Fig. 1).
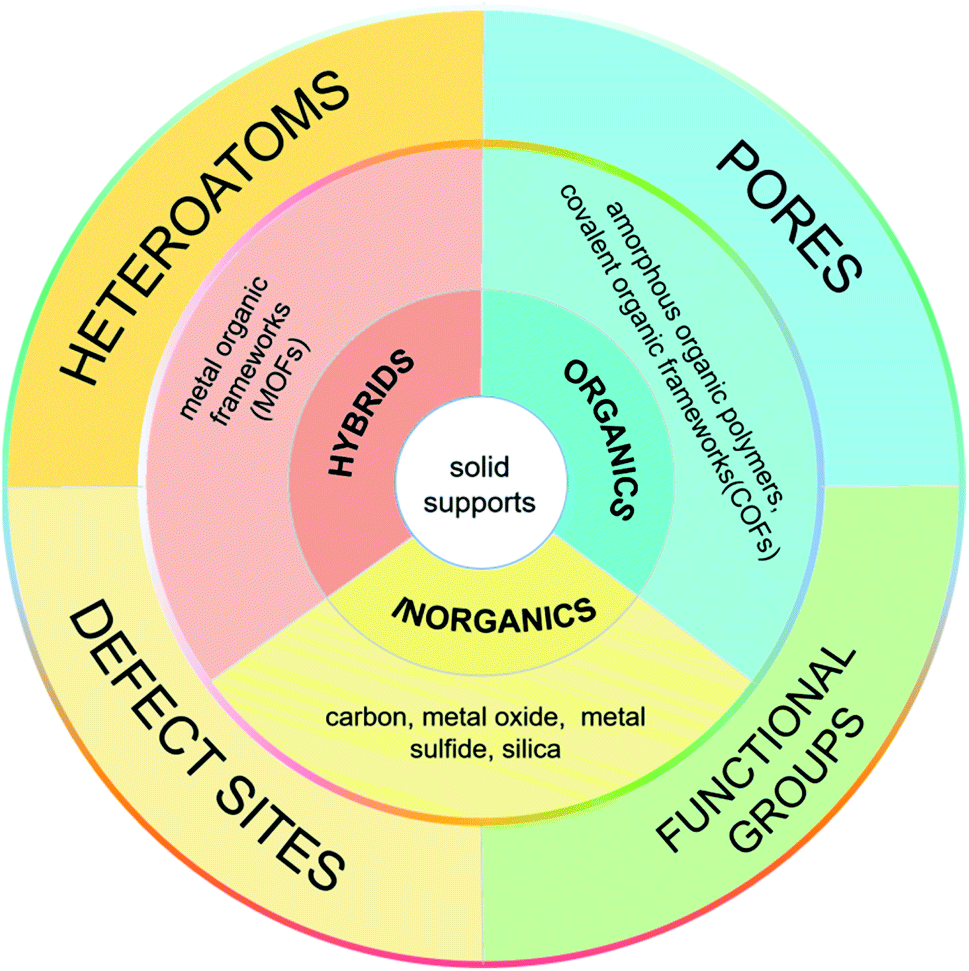 | ||
| Fig. 1 Classification of the solid supports used for controlled synthesis of UMNPs. | ||
2. The role of solid supports and design principles for UMNP preparation
UMNPs have a large number of available active sites and excellent catalytic properties, but they tend to aggregate after cycles of catalytic reaction, resulting in a significant decrease in the number of available active sites. Using a suitable solid support to anchor and isolate the UMNPs from one another is a feasible approach for maintaining the dispersion of UMNPs. The work of synthesizing metal nanoparticles using closed-shell structures as scaffolds and loading Pd nanoparticles in organic porous polymers (POP) has been systematically summarized.9,27 The structural characteristics of the solid supports play an important role in the directed synthesis (e.g., size and morphology) and stabilization of the resulting UMNPs. Spatial confinement and electronic confinement have dominated the variations of structural characteristics in solid supports.2.1. The electronic confinement effects originated from the interaction between supports and UMNPs (or the metal ion precursor)
The nucleation and growth of UMNPs on the support are highly dependent on the nucleation site. The metal ion precursors will first nucleate at the anchoring site, triggering the nanoparticle growth.28,29 If the anchoring sites in solid supports are well distributed and provide strong interactions with the “seeds”, the resulting UMNPs would remain highly dispersed during the synthesis and maintain uniformity throughout the catalysis process. There are three common ways to introduce nucleation and anchoring sites into supports: installing organic functional groups, heteroatom doping and creating defect sites.30–35 Organic functional groups can bind the metal ions in the precursor solution. It is a common method to capture metal ions and anchor the resulting UMNPs in organic or hybrid supports, such as metal organic frameworks (MOFs) and covalent organic frameworks (COFs).28,36 For example, graphene oxide is a carbon support with abundant oxygen-containing groups, which can act as nucleation and anchor sites for UMNPs.37 Heteroatom doping changes the electronic properties of the supports and provides lone-pair electrons to form coordination bonds with the surface metal atoms of UMNPs. Suitable heteroatoms for anchoring UMNPs that have been reported include N, O, S, P and B atoms.38–41 These heteroatoms may not only provide nucleation sites for UMNPs, but also improve the electron transport between UMNPs and the supports. Different heteroatoms are usually chosen for different UMNPs. Defect sites are also used as traps to capture UMNPs and prevent their aggregation. The defect sites in carbon materials, metal oxide/sulfide and other materials can be designed to control the growth of and anchor the UMNPs.42,43 Embedding UMNPs in the solid supports could also enhance their surface area (thus accessible active sites) and improve their catalytic performance.2.2 The spatial confinement effects originating from porous supports
The diameter of UMNPs loaded on the porous supports is highly dependent on the size of the pores, which enables the confined growth of UMNPs and prevents their overgrowth.25,44 By varying the diameter of the pores of the supports, the size of the UMNPs can be tuned. Such a confinement effect of the porous support can also prevent the aggregation of UMNPs.3. Synthesis of UMNPs supported by carbon materials
Carbon materials are the most economical and most widely used supports in both industrial applications and fundamental research. Typical carbon materials include graphene, diamond, porous carbon and carbon nanotubes. All of them exhibit good chemical stability, high conductivity and low density, which are all favorable features when used as supports for UMNPs. However, carbon materials often lack sites for directly anchoring MNPs. Therefore, the use of carbon materials to support UMNPs often requires surface modification or heteroatom (such as N, S, P, O, etc.) doping to introduce the functionality onto the carbon support, which provides anchoring sites for UMNPs.38–41 At the same time, the doping of heteroatoms into the carbon materials can also effectively tune the properties of UMNPs anchored on the supports, further facilitating the catalytic reaction process and improving the catalytic efficiency.3.1. Porous carbon
Porous carbon is an economical carbon material that has been mass-produced and widely used to support UMNPs. Porous carbon materials can be divided into microporous, mesoporous, and macroporous materials according to their pore size. According to International Union of Pure and Applied Chemistry (IUPAC) regulations, pores with diameters less than 2 nm are defined as micropores, those with diameters between 2 and 50 nm as mesopores, and those with diameters more than 50 nm as macropores.18 The inherent pores of porous carbon can be used as the accommodation sites of UMNPs generated in situ and prevent their agglomeration, which is beneficial to controlling the size of UMNPs. Microporous carbon usually has a large specific surface area, which can provide more anchoring sites for UMNPs. As shown in Fig. 2, Fan et al. coated microporous carbon fibers with metal precursors and polyvinylpyrrolidone (PVP).45 Metal-based nanoparticles/N-doped porous carbon hybrid films were obtained upon pyrolysis. In this process, PVP was converted into a N-doped carbon thin film, which provides binding interactions and confinement effects for UMNP formation. Nearly uniform UMNPs (2 nm) were observed. Carbon fibers were also etched by the metal at the high temperature to form micropores, increasing the specific surface area of the support with more exposed active centers. In addition, the nanoparticles are embedded in the carbon fibers. With the additional protection of microporous carbon, the UMNPs showed good stability in electrocatalysis, even at high current densities. However, the narrow channels of microporous carbon materials and the strong interactions between the solid support and the product or reactant resulted in slow mass transfer. By contrast, macroporous carbon materials have spacious pore channels, which facilitate the transportation of the product or reactant. However, the specific surface area of macroporous carbon materials is usually low with relatively limited anchoring sites. Besides, the pores of macroporous carbon are too large to provide suitable confinement for UMNPs. Therefore, porous carbon with only macropores is rarely reported as an efficient support for UMNPs. Mesoporous carbons have a relatively high specific surface area and spacious pore channels. Besides providing a large number of anchoring sites, they also possess relatively suitable pore channels for the mass transfer. Mesoporous carbons with ordered structures can be obtained by indirect template synthesis or direct synthesis. Yuan et al. have summarized the synthesis and modification methods of mesoporous carbons in detail.46 Nitrogen-doped mesoporous carbons with abundant anchoring sites for UMNPs represent one of the most promising substrates for catalysis. Wu et al. used nitrogen-doped mesoporous carbons as substrate materials and obtained Fe and FeC UMNPs through low-temperature liquid phase precipitation and heat treatment, which show excellent performance towards the electrocatalytic oxygen reduction reaction (ORR).12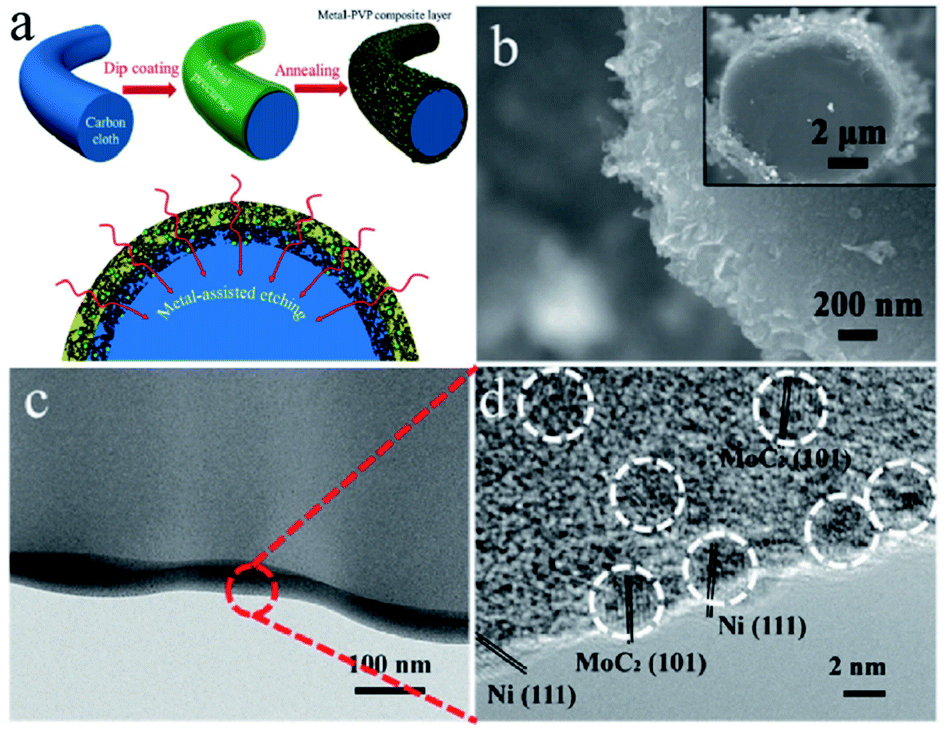 | ||
| Fig. 2 (a) Schematics of the formation of the metal-based nanoparticles/N-doped porous carbon hybrid catalysts; (b–d) characterization of NiMo UMNPs-N-doped porous carbon hybrid films: SEM (b) and TEM (c and d). Adapted with permission from ref. 45 @ copyright 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In recent years, the carbonization of metal organic frameworks (MOFs) followed by chemical etching has emerged as one of the widely used approaches for preparation of mesoporous carbons. Zhang et al. prepared a mesoporous carbon support from the commercially available Cu-MOFs (HKUST-1) through the post-synthetic method. Such a solid support was used to prepared Pt UMNPs (2–3 nm), which have shown superior performance in both catalytic methanol oxidation and nitrophenol reduction reactions.47 Beyond the two-step post-synthesis, MOFs can also serve as hosts for various precursors of UMNPs, following which one-step co-pyrolysis can be used to fabricate the UMNPs@mesoporous carbon hybrid material. Niu et al. prepared tungsten UMNPs supported on a MOF-derived mesoporous carbon through carbonizing ZIF-8 consisting of encapsulated K5BW12O40.48 The catalysts show excellent performance towards olefin epoxidation. It is worthy of noting that Zn volatilizes at high temperature, leaving the W species supported by porous carbon. Although the pyrolysis process will destroy the fine structure of MOFs, the mesoporous carbon support after pyrolysis can still protect UMNPs from aggregation (Fig. 3).
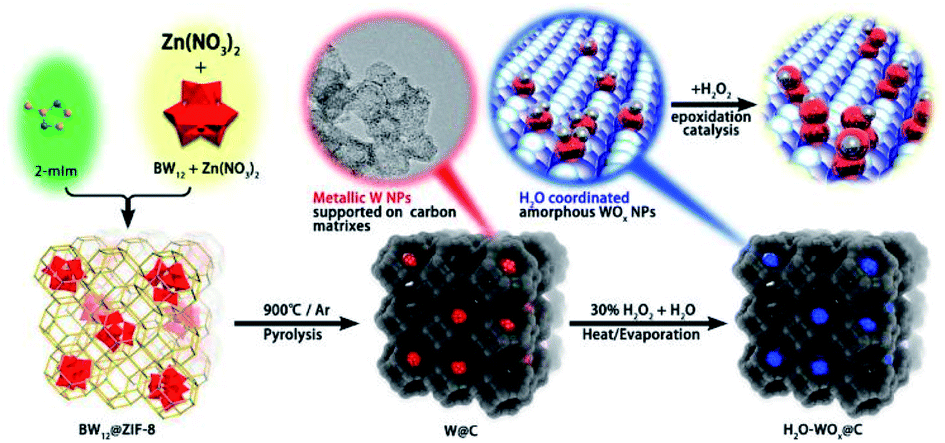 | ||
| Fig. 3 The structure of the MOFs after pyrolysis can protect UMNPs from aggregation (adapted with permission from ref. 48 @ copyright 2019 American Chemical Society). | ||
To further optimize the electron and mass transfer during the catalytic process of UMNPs, hierarchical porous carbons, which contain two or three types of micropores, mesopores and macropores, are typically used.49,50 Hierarchical porous carbons combine a high specific surface area and wide pore channels to provide abundant anchoring sites for UMNPs. At the same time, rapid mass transfer can still be achieved. Therefore, hierarchical porous carbons are very promising supports for loading UMNPs for applications in heterogeneous catalysis. Zhong et al. introduced ethylenediaminetetraacetic acid (EDTA) into MOF-808.28 The EDTA acts as ion traps to capture Pt2+. The uncaptured Pt2+ ions form PtO2 clusters. Pt UMNPs loaded in porous carbon were obtained upon pyrolysis of the MOF precursors. With the increase of the Pt2+ concentration from 10 ppm to 80 ppm, the diameter of the obtained Pt particles was increased from 2.1 nm to 4.1 nm. When the concentration was reduced to 2 ppm, the particle diameter did not decrease, but the distribution density of Pt particles was reduced. This series of Pt UMNPs showed low overpotential in catalyzing the hydrogen evolution reaction (HER).
3.2. Hollow carbon
Hollow carbon structures, such as carbon tubes and hollow carbon spheres, are commonly used as carbon materials for UMNPs. Their unique hollow structures can provide superior spatial arrangement of anchoring sites, thus maximizing the utilization of UMNPs. Carbon nanotubes (CNTs) are typical hollow carbon structures, which have a high degree of graphitization, orderly arrangement of carbon atoms, excellent electrical conductivity and superior chemical stability.51 These characteristics make them one of the most ideal support materials in various applications. The standing disadvantage of CNTs lies in their lack of anchoring sites for UMNPs. Therefore, CNTs often need to be functionalized or doped with heteroatoms to improve their interactions with UMNPs.52There are many strategies for surface functionalization of CNTs, including oxidizing defect sites of CNTs, coating CNTs with polymers, and installing organic functional groups on the surface of CNTs covalently or non-covalently.52–56 The surface functionalized CNTs have more anchoring sites for better stabilization of UMNPs. Simply linking functional groups to CNTs through chemical oxidation or covalent bonds is the most feasible method to assist the growth of UMNPs on the surface of CNTs. Liu et al. used KMnO4, concentrated sulfuric acid, and H2O2 to oxidize CNTs in multiple steps, forming many defects and oxygen-containing groups on the surface of CNTs.56 These groups increase the dispersion of CNTs in aqueous solution and assist the formation of Ag UMNPs (∼3 nm) on the surface of CNTs. However, the oxidation process causes damage to the structure of CNTs and a significant decrease in electrical conductivity, limiting their application in electrocatalysis. Chen et al. used ionic liquid polymers to uniformly introduce abundant functional groups onto the surface of CNTs through radical polymerization (Fig. 4a).52 With the functionalized CNTs, uniformly distributed Pt (1.9 ± 0.5 nm) and PtRu (1.3 ± 0.4 nm) particles were successfully prepared on CNTs. The diameter of the particles loaded on non-functionalized CNTs was significantly larger, and the size distribution was wider for both PtRu (3.5 ± 1.0 nm) and Pt (5.5 ± 1.5 nm) nanoparticles. Similarly, as shown in Fig. 4b, Ma et al. used 3,4,9,10-perylene tetracarboxylic acid-derived ionic liquids to non-covalently functionalize CNTs. Pd4Au1–P nanoparticles (2.3 nm) supported on CNTs were obtained, which showed excellent ethanol oxidation (EOR) activity.57 Since the functional groups were non-covalently connected to CNTs through supramolecular forces such as π–π stacking, the electronic structure of CNTs remained intact and the excellent electrical conductivity of CNTs was reserved.
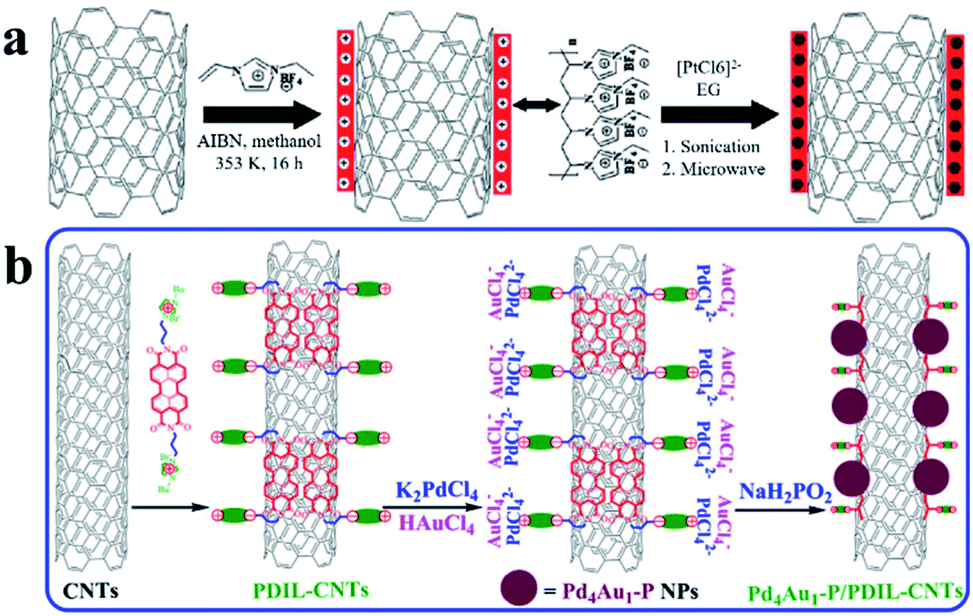 | ||
| Fig. 4 Functionalization of carbon nanotubes with an ionic liquid: (a) based on the thermal-initiation free radical polymerization of the ionic-liquid monomer 3-ethyl-1-vinylimidazolium tetrafluoroborate to form an ionic-liquid polymer on the CNT surface, which introduces a large number of surface functional groups onto the CNTs with uniform distribution to anchor and grow metal nanoparticles; (b) using ionic liquids, functional groups are non-covalently bound to CNTs with minimal change of the conductivity of CNTs (AIBN: 2,2′-azobisisobutyronitrile, EG: ethylene glycol, PDIL: 3,4,9,10-perylene tetracarboxylic acid) (adapted with permission from ref. 52 and 57 @ copyright 2017 Elsevier Inc. All rights reserved and 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
Incorporating heteroatoms into the framework of CNTs is also a common strategy for introducing UMNPs anchoring sites into CNTs, particularly the introduction of N atoms. Wang et al. reported a template method to fabricate home-made carbon tubes with uniformly doped N atoms.58 Different from conventional CNTs, these novel hollow carbon tubes are functionalized with a large number of binding sites for UMNPs. Highly dispersed Pd UMNPs (average size of 2.3 nm) were deposited on this N-doped carbon tube. Due to the synergistic effect of the support, Pd UMNPs can efficiently catalyze the reduction of 4-nitrophenol even at a low loading (Pd: 0.324 wt%).
Although carbon tubes with hollow structures have been widely used as catalyst supports, most of the UMNPs supported by carbon tubes are usually anchored on the outer surface of carbon tubes instead of the internal surface. Huang et al. encapsulated a series of noble metal UMNPs (Pd, Pt, Ru and Au) inside open-end CNTs by wet impregnation, followed by pyrolysis (Fig. 5).59 During the impregnation, metal ions enter CNTs through capillary action. When Pd UMNPs (2.5 ± 0.5 nm) were encapsulated into the CNTs, the resulting composite material showed excellent OER activity and can be used as a cathode material for Li–oxygen batteries. Other noble metal UMNPs, including Ru UMNPs (2.7 ± 0.5 nm), Pt UMNPs (1.8 ± 0.5 nm) and Au UMNPs (3.0 ± 0.5 nm) were also successfully encapsulated into CNTs.
 | ||
| Fig. 5 (a–d) HRTEM images of PdNPs@CNTs (a), RuNPs@CNTs (b), PtNPs@CNTs (c) and AuNPs@CNT (d); (e) schematic illustration of the formation of UMNPs@CNTs (adapted with permission from ref. 59 @ copyright 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
Similar to carbon tubes, hollow carbon spheres (HCNs) with a unique hierarchical structure of a spherical shell represent another type of hollow carbon material, which also has highly exposed anchoring sites for UMNPs. Nanda et al. reported PtPd–UMNPs (2 nm) dispersed in heteroatom-doped HCNs, which showed excellent catalytic performance and stability in the methanol oxidation reaction (MOR), EOR and ORR.60 The high surface area of UMNPs brings about high catalytic activity, and the surrounding carbon layer prevents the migration and aggregation of UMNPs. The carbon structure also adsorbs a small amount of –OH/–H2O species, which is conducive to the removal of CO produced during the reaction, particularly in the MOR, to reduce the risk of catalyst poisoning. Fornasiero et al. reported a new synthetic approach to prepare FeOx UMNPs on the surface of HCNs through an “inside-out” mechanism.61 They controlled the pyrolysis conditions, which can exsolve Fe species from the inside of the HCNs to the outside and form FeOx UMNPs. The different location (inside or outside) of FeOx led to the different selectivity (form H2O or H2O2) in the ORR (Fig. 6).
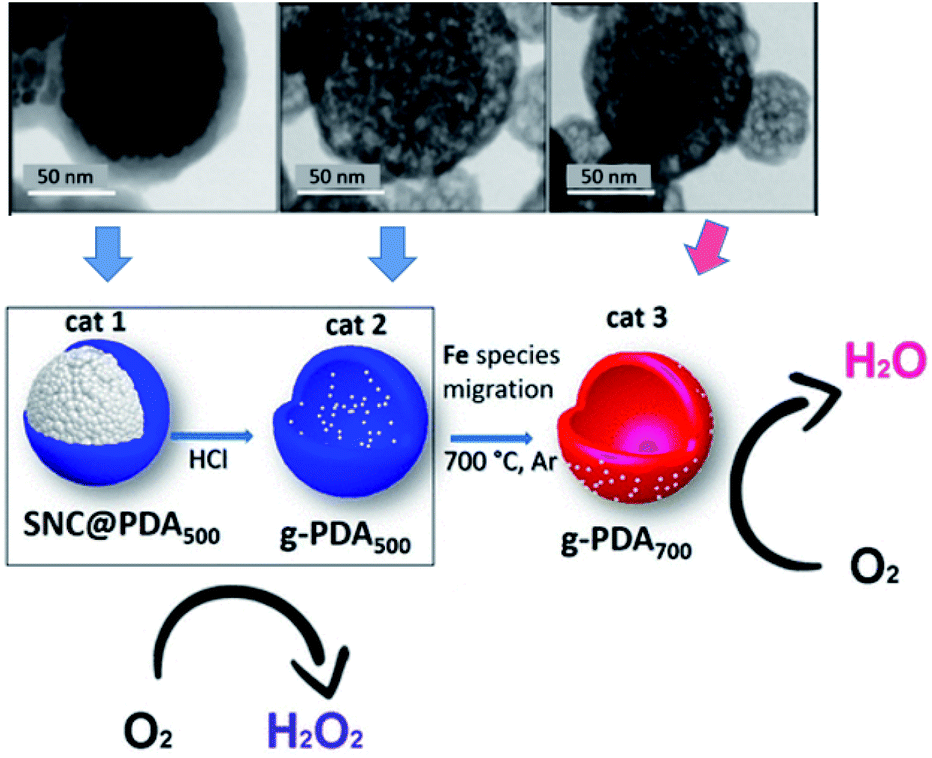 | ||
| Fig. 6 Under different pyrolysis conditions, FeOx particles were formed on different locations of CNTs as shown in the TEM images. Correspondingly, different ORR product selectivities were observed (SNC: superparamagnetic nanoparticle clusters, PDA: polydopamine) (adapted with permission from ref. 61 @ copyright 2019 American Chemical Society). | ||
3.3. Graphene and its derivatives
Graphene is a two-dimensional carbon nanomaterial with a hexagonal honeycomb lattice composed of carbon atoms and sp2 hybrid orbitals. In theory, it has a huge specific surface area up to 2600 m2 g−1. Graphene has excellent optical, electrical, and mechanical properties and has important application prospects in materials science, micro–nano processing, energy, biomedicine, and drug delivery.62,63 Graphene oxide (GO) is an important derivative obtained through oxidation of graphene. It has a large number of oxygen-containing functional groups, including hydroxyl, epoxy, carbonyl, carboxyl, etc., which are favorable anchoring sites for metal ions and UMNPs. Reduced graphene oxide (rGO) is obtained by reduction of GO. Compared with graphene, rGO not only has residual oxygen and other heteroatoms, but also has some defects, which can act as barriers for atom migration and maintain the uniform distribution of UMNPs.64Graphene only shows good dispersibility in non-polar solvents, which limits its use due to the fact that most metal precursors are dissolved in polar solvents.64 In addition, the uniform structure of graphene lacks the anchoring sites for UMNPs. In contrast to graphene, GO can be easily dispersed in water or polar organic solvents. The oxygen-containing groups of GO can facilitate exfoliation of graphene nanosheets and prevent graphene stacking. GO can be easily produced at low cost, but strong oxidants are inevitably used in the GO production process, resulting in a large number of defects in the GO network. It causes the conductivity of GO to be far inferior to that of graphene. rGO exhibits conductive properties similar to graphene and contains a small amount of unreduced oxygen-containing functional groups and defect sites.65 The most convenient way to anchor UMNPs on GO is to reduce the metal precursor and GO at the same time. Then, the metal precursor will be reduced to form UMNPs while GO transforms into rGO. Ravishankar et al. used ethylene glycol to simultaneously reduce Pt2+ and GO under microwave irradiation conditions and load Pt UMNPs (2–3 nm) on graphene.66 Liang et al. reported the formation of uniformly dispersed Cu UMNPs (2 nm) on rGO through laser ablation of a Cu target in GO solution followed by reduction treatment.67 The obtained Cu UMNPs showed excellent performance towards 4-nitrophenol reduction. However, the reduction process of metal precursors also reduces the oxygen-containing functional groups on GO. To prevent GO stacking due to the loss of oxygen-containing functional groups, Jin et al. introduced spherical carbon black as the gap between GO layers to form a stable 3D structure and maintain the high specific surface area of GO (Fig. 7).68 The carbon black and graphene doped with nitrogen were obtained upon reduction with urea. They used such a unique three-dimensional structure as the support for the growth of Pd UMNPs. Both the incorporated N sites and the initial oxygen-containing functional groups serve as the binding sites for Pd ions. Upon NaBH4 reduction, Pd UNMPs (3.2 nm) grew in situ in the retained 3-dimensional composite structure of rGO and carbon black. The hybrid catalysts showed excellent activity towards the electrocatalytic ethanol oxidation reaction.
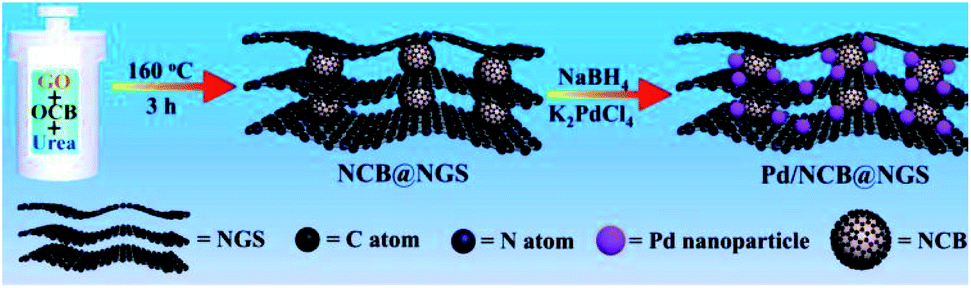 | ||
| Fig. 7 Using spherical carbon black as the gap between GO layers to maintain GO dispersion. (NCB: nitrogen-doped carbon black, NGS: nitrogen-doped graphene) (adapted with permission from ref. 68 @ copyright 2020 Elsevier B.V. All rights reserved). | ||
4. Synthesis of UMNPs supported by crystalline frameworks
Metal organic frameworks (MOFs) and covalent organic frameworks (COFs) are emerging porous materials with rigid frameworks and orderly lattice fringes. Unlike conventional crystalline materials, these framework materials are constructed from organic building blocks or through hybridization of organic building blocks and metal ions. By selecting different organic monomers, porous crystalline frameworks with different pore sizes, morphologies and functional groups can be synthesized. These materials typically have great structural tunability and high porosity, making them ideal catalyst support materials.69–71 Apart from being precursors for porous carbon which has been discussed in Section 3.1, the rigid frameworks provide stable pores without collapse for a long time, and the backbones between the cavities can prevent the agglomeration of UMNPs.4.1. MOFs
MOFs are a class of organic–inorganic hybrid materials with intramolecular pores formed by self-assembly of organic ligands and metal ions or clusters through coordination bonds. MOFs have highly tunable pore size and volume and an easily modifiable inner surface.69 In addition, their frame structure is rigid and not easy to collapse. Moreover, MOFs usually have good thermal stability. These exciting features make MOFs very promising support materials for UMNPs. Through rational design, metal ions or UMNPs can be precisely and uniformly coordinated and deposited on MOFs.72 The pore structures of MOFs separate the UMNPs one from another, which can avoid the aggregation of UMNPs during the catalytic process.73,74 The synthesis and application of MOFs is a current hot research field, and many new MOF structures are reported every year, but not all MOFs are suitable for the synthesis of UMNPs and their application in catalysis. For example, when H2O is generated or participates in the catalytic reaction pathway, the framework of MOF-177 can easily decompose and collapse.75 MOFs with suitable pore structures and high stability are required for the confined growth of UMNPs for catalysis application. Xu et al. have reviewed the controlled synthesis of UMNPs by using MOFs as supports in detail.10 Therefore, we will only briefly discuss the typical cases of the controlled synthesis of UMNPs with MOFs in recent years in this section.Li et al. demonstrated a method of using MIL-101 as a template to fix a variety of bimetal alloyed UMNPs in the pores (Fig. 8).24 The particle diameter was 1.1–2.2 nm, and the metal loading was up to 10.4 wt%. Taking Cu–Pd@MIL-101 as an example, they first introduced Cu2+ into MIL-101 by dipping and then reduced them with NaBH4 to obtain Cu particles with a diameter of 8.25 ± 2.53 nm. Subsequently, the Pd precursor was introduced, and under the action of high-intensity ultrasound, the Cu particles were effectively transformed into Pd–Cu UMNPs (diameter of about 2.16 nm) through the electric substitution reaction with Pd2+. The catalyst was used to catalyze the coupling of phenylacetylene to produce 1,4-diphenylbuta-1,3-diyne in excellent yield (up to 98%).
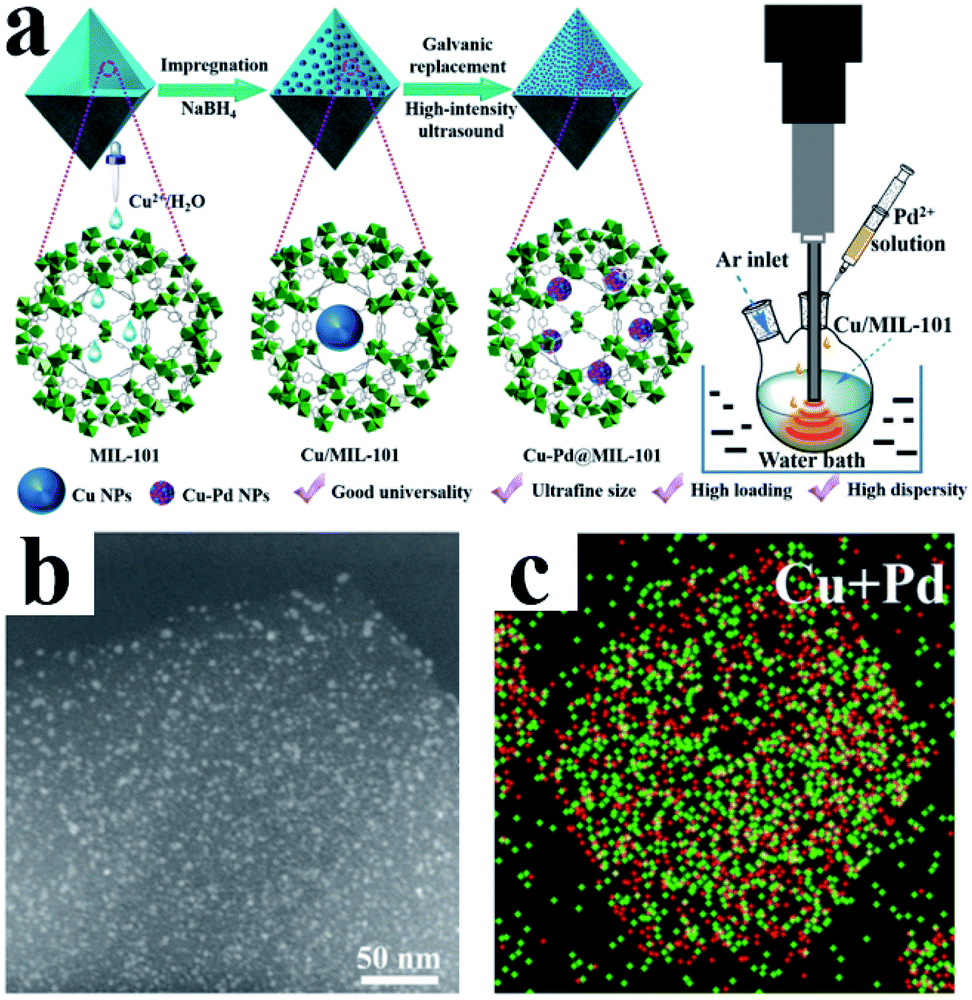 | ||
| Fig. 8 (a) Preparation of Cu–Pd@MIL-101; (b) HAADF-STEM image and (c) elemental mapping of PdCu UMNPs@MIL-101 (adapted with permission from ref. 24 @ copyright 2019 American Chemical Society). | ||
The heteroatoms incorporated into the MOF backbone or side chains can act as nucleation and anchor sites just like carbon materials. The surface micro-environment of UMNPs can be modulated by changing the functional groups and metal substitution in the MOFs to enhance their catalytic performance. Jiang et al. encapsulated Pd UMNPs (less than 1.1 nm) into the pores of UiO-66-X (X = H, OMe, NH2, 2OH, 2OH(Hf/Zr)): X represents the tunable organic groups linked to the benzene ring, Hf/Zr represents the Hf-oxo or the Zr-oxo cluster.76 Different organic linked groups in the pores of the MOFs can change the adsorption energy of Pd@UiO-66-X to a substrate and the charge transfer between Pd UMNPs and MOFs. The catalytic activity of Pd@UiO-66-2OH was around 14 times higher than that of Pd@UiO-66 in the hydrogenation of benzoic acid. In addition, changing the metal clusters (2OH, 2OH(Hf)) also led to different catalytic performances. The authors highlighted the influence of the surface microenvironment on the guest metal NPs, which can be tuned by choosing different substituents of the metal and functional groups in the host MOFs.
Embedding UMNPs into the lattice of MOFs can enhance the dispersibility of UMNPs. The electronic configuration at the interface between UMNPs and MOFs can be tuned, which may increase their catalytic performance. Zhou et al. used cobalt and benzimidazole to synthesize a MOF, (poly-[Co2(benzimidazole)4] (PCB)), with a laminar flow structure and introduced Fe3+ into the pores of the MOF by dipping (Fig. 9).77 After reduction, the generated FeCoOx UMNPs (3 nm) were embedded into the lattice of MOFs. When deposited on carbon cloth, such a composite material showed excellent activity in catalyzing the oxygen evolution reaction (OER).
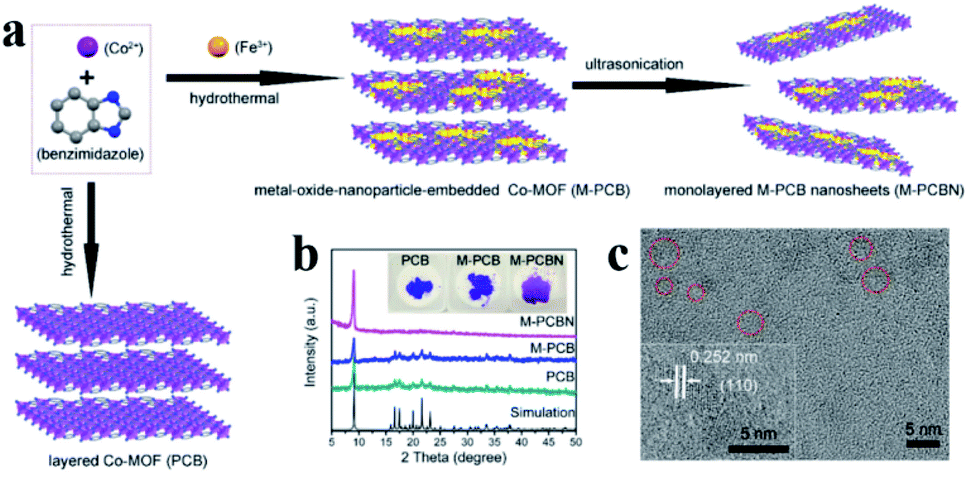 | ||
| Fig. 9 (a) Schematic of embedding FeO in the lattice of the MOF; (b) powder XRD patterns of PCB, FeCoOx UMNPs-PCB, and FeCoOx UMNPs-PCBN; and (c) HRTEM image of FeCoOx UMNPs-PCBN (adapted with permission from ref. 77 @ copyright 2020 American Chemical Society). | ||
4.2. COFs
COFs are a class of 2D or 3D crystalline organic polymers connected by covalent bonds. Similar to MOFs, COFs also have highly tunable pore size and easily modified inner surface. Therefore, the introduction of suitable functional groups into the constituent units of COFs can equip COFs with binding sites for UMNPs.78,79 Compared with MOFs, COFs have lower density without any metals in their frameworks. In addition, they generally exhibit higher stability in acid, alkali, organic and aqueous media when compared with MOFs.The introduction of anchoring sites into COFs has been mainly accomplished in two ways: (1) incorporating the sites into the backbones of COFs and (2) installing the sites as side chains on the COF backbones. The N-rich COF backbones have a large number of N atoms, which can provide abundant anchoring sites for UMNPs. Through the coordination of N atoms and the confinement of intrinsic pores, UMNPs can be well encapsulated and the aggregation of UMNPs can be prevented. A series of COFs with N-rich backbones have been successfully developed using synthetic building blocks with nitrogen-containing functional groups such as carbazole, porphyrin, amide and imine. Lu et al. used piperazine and cyanuric chloride as molecular building blocks to synthesize a COF with the N-rich backbone that can coordinate Ru ions (Fig. 10a).80 Ru ions were reduced in situ to Ru UMNPs (1.4 to 2.6 nm), which were embedded in the pores of the COF. The confinement effect from the pores and the coordination with N atoms firmly anchored the UMNPs in the COF framework. The obtained UMNPs were used to catalyze the methanolysis of ammonia borane, delivering a total turnover frequency (TOF) up to 505 min−1 at 298 K. Moreover, the catalyst showed excellent durability and maintained high dispersion during the catalytic process.
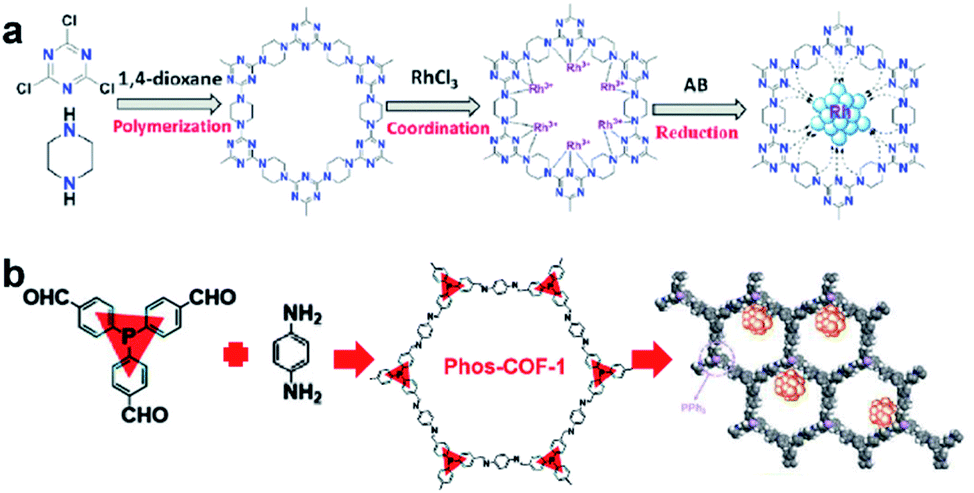 | ||
| Fig. 10 Synthesis of COF supported UMNPs: (a) N acts as the nucleation and growth sites for Ru UMNPs; (b) PPh3 provides anchoring sites for UMNPs@Phos-COF-1 (adapted with permission from ref. 33 and 80 @ copyright the Partner Organisations 2020 and 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
In addition to nitrogen-containing functional groups, other heteroatom-containing functional groups can also be used as anchor sites to coordinate UMNPs. Phosphine groups have been widely used as organic ligands for metal organic catalysts. Incorporating phosphine groups into COFs can make the phosphine groups distributed in an orderly manner and provide anchor sites for metal ions or UMNPs. Recently, a COF material (Phos-COF-1) that incorporates triphenylphosphine (PPh3) as a growth site for UMNPs was reported.33 A series of UMNPs (Pt, Pd, Au and PdAu) with narrow size distribution were successfully prepared inside Phos-COF-1 (Fig. 10b). The loading of metal precursors is also an important factor affecting the size of UMNPs. Excessive metal loading promotes the excessive random growth of UMNPs on the surface of COFs. Taking the Pd UMNPs synthesized in this work as an example, after doubling the amount of the Pd precursor, the diameter of Pd UMNPs changed from 1.62 nm to 2.74 nm on average and the size distribution range became wider. The obtained catalysts were used to catalyze a number of reactions including nitrophenol reduction, cross coupling, and tandem cross coupling–nitrophenol reduction. All of these catalysts exhibit excellent performance.
Besides directly anchoring UMNPs through the sites in the backbone of COFs, COFs can also be functionalized in their side chains to improve the binding ability towards metal ions or particles. Post synthesis and bottom-up synthesis are the two commonly used strategies to install functional groups.81–83 The post synthesis avoids functional groups participating in the polymerization of building blocks to form COF backbones. However, through post-synthesis, the crystallinity of COFs may be reduced and not all pores can be fully functionalized. The advantage of bottom-up synthesis lies in the well-maintained crystallinity of COFs and the functional groups can be evenly distributed in the framework. However, some functional groups may either be unstable under severe synthetic conditions of COF formation or interfere with the formation of crystalline COF structures.
Recently, Lu et al. reported a COF containing thiol groups (–SH) via post-synthesis strategies (Fig. 11a).84 The small pores of COFs and the strong S–Au binding energy enabled the confined growth of Au UMNPs with narrow size distribution (1.8 ± 0.2 nm). Such a composite material showed enhanced photostability and photocatalytic performance in the photodegradation of RhB. The synthesis of a COF containing thioethers (Thio-COF) using bottom-up strategies was also reported (Fig. 11b).25 Pt UMNPs (1.70 nm) and Pd UMNPs (1.78 nm) confined in Thio-COF were obtained, which showed excellent catalytic activity towards the reduction of 4-nitrophenol and the Suzuki–Miyaura coupling reaction, respectively. The thioether group with strong binding capability for metal ions or nanoparticles was pre-installed in the building block. It has been proposed the thioether groups serve as nucleation centers for the confined growth of UMNPs in COFs. When COFs without thioether in their channels were used as supports, randomly agglomerated metal particles were formed instead. In addition, the use of an amorphous COF with the same structure as the support also led to random distribution of particles. Therefore, both the crystalline structure of COFs and the suitable functional groups in the channels contribute to the high quality of the as-formed UMNPs. Similar studies using bottom-up synthesis of functionalized COFs for UNMPs have also been reported for Pd UMNPs@COF, Fe–TiO2 UMNPs@COF.19,85
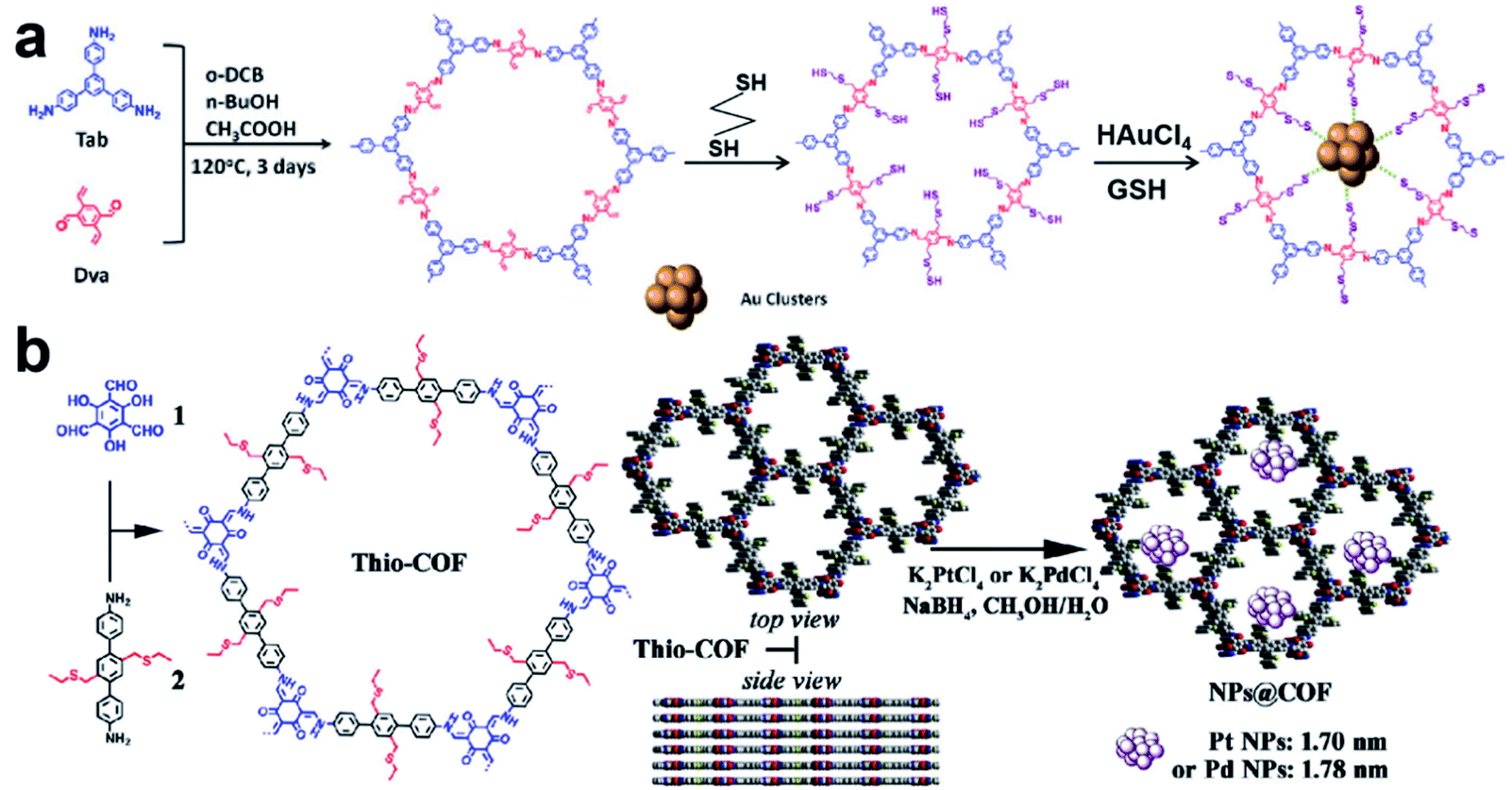 | ||
| Fig. 11 (a) Installation of thio groups through the post-synthetic approach: synthesis of Au@COF (o-DBC: 1,2-dichlorobenzene, GSH: glutathione); (b) installation of thio groups through the bottom-up synthetic approach: synthesis of Thio-COF and schematic representation of the synthesis of Thio-COF supported PtNPs@COF and PdNPs@COF (adapted with permission from ref. 25 and 84 @ copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim and 2017 American Chemical Society). | ||
5. Synthesis of UMNPs supported by amorphous organic polymers
In addition to ordered COFs, there are some other disordered polymer materials, which can also be used as supports to direct UMNP growth. Their design concept is similar to COFs, that is, modifying the polymer backbone with organic functional groups or incorporating heteroatoms.86 They can also provide a confined environment for the growth of UMNPs. Several common amorphous polymers that can be used to control the synthesis of UMNPs include conjugated micro-/meso-/macro-porous polymers (CMPs), hyper-crosslinked polymer (HCPs) and other porous organic polymers (POPs).CMPs have π-conjugated main chains and permanent porous structures, which distinguishes them from unstable porous materials, non-porous conjugated polymers, and porous carbon materials. By choosing different molecular building blocks, CMPs with different pore sizes and morphologies can be obtained.87 It is worth noting that because imine bonds can contribute to conjugation, certain imine-based COFs can also be considered as crystalline CMPs. CMPs can also be considered amorphous analogs of COFs.88 The conjugated structures of CMPs improve the electron transfer efficiency, which helps in electron transport in the catalysis process. It is even anticipated that it could be used directly in electrochemical catalysis. The work of Maji et al. gave an example of loading UMNPs onto CMPs and directly using them in the electrochemical catalysis (Fig. 12a).89 They used tris-(4-aminophenyl)amine (TPA) and perylenedianhydride (PDI) as raw materials to synthesize a CMP material with good conductivity, called TPA–PDI. Triphenylamine and its derivatives are well-recognized hole-transport agents. When coupled with electron acceptor building blocks, they can produce a CMP system with inherent conductivity. The special donor–acceptor pair and conjugation of TPA–PDI facilitate the convenient transfer of charges across the CMP structure, which makes this material promising for application in electrocatalysis. They used TPA–PDI to synthesize two UMNPs (Au and Co), called Au@TPA–PDI and Co@TPA–PDI, respectively. Au@TPA–PDI can efficiently catalyze the reduction of nitroaromatics to aminoaromatics. More interestingly, both TPA–PDI and Co@TPA–PDI exhibit considerable ORR catalytic activity without carbonization. Recently, Ag UMNPs@CMPs, Pd UMNPs@CMPs and others have also been reported.26,90
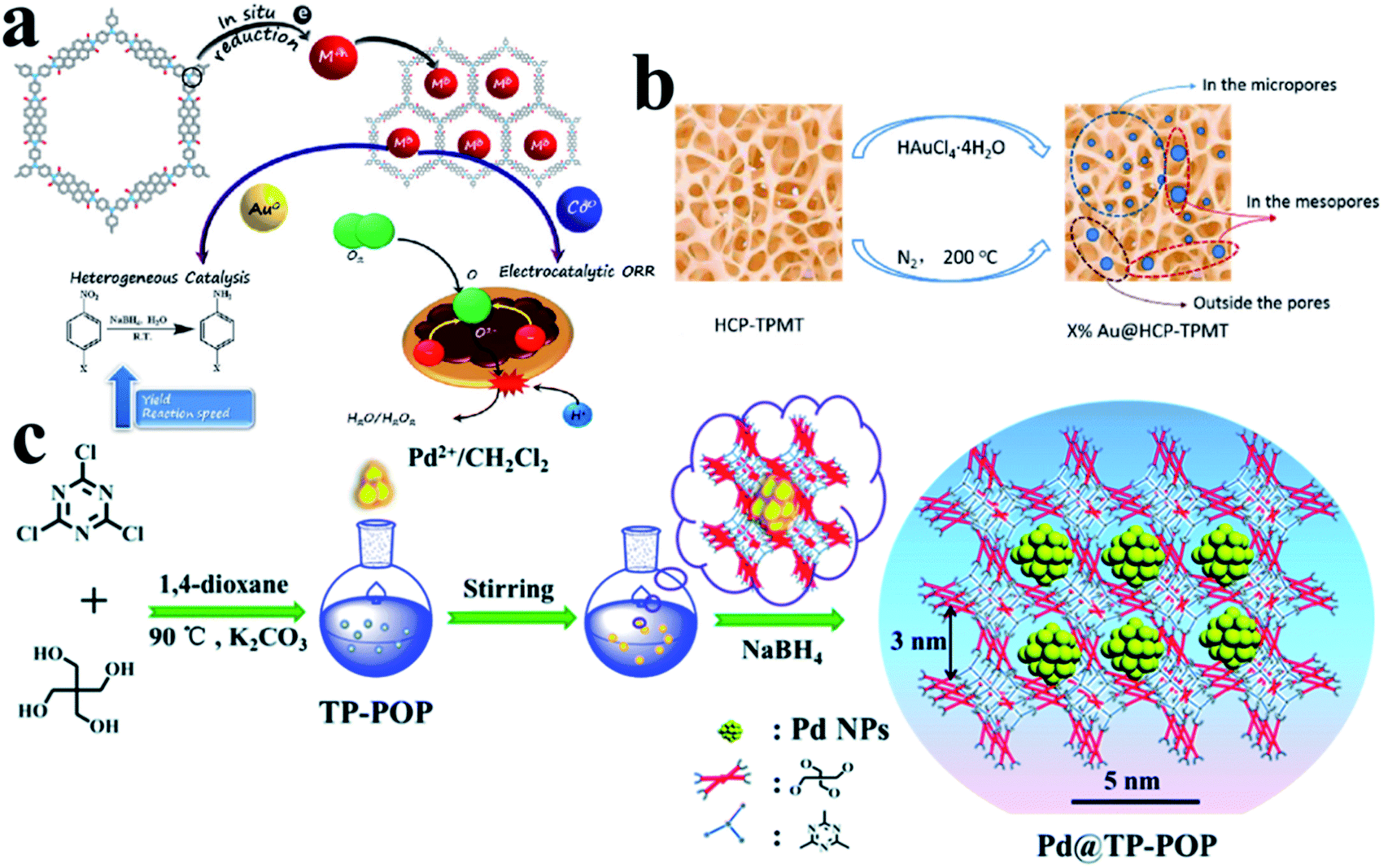 | ||
| Fig. 12 Controlling the growth of UMNPs using amorphous polymers: (a) Au@CMPs and Co@CMPs; (b) Au@HCPs; (c) Pd@TP-POP (adapted with permission from ref. 22, 32 and 89 @ copyright 2019 American Chemical Society, 2018 American Chemical Society and The Royal Society of Chemistry 2018). | ||
HCPs are a class of microporous polymers that are prepared by extensive crosslinking of linear or lightly crosslinked precursor polymers. They have high surface area, high porosity and high stability, which make them the candidate materials to support UMNPs.91 The heteroatom, organic functional groups and hyper crosslinked tubes in HCPs can prevent UMNPs aggregation.92 Dong et al. prepared thiol-containing HCPs using ferrocenecarboxaldehyde and melamine as building blocks, which were loaded with Pd UMNPs (2.89 nm).93 The resulting Pd UMNPs@HCP composites showed excellent activity and stability in the catalytic reduction of nitroarenes. Tan et al. reported a series of Au UMNPs (range of 1.7–5.1 nm) supported on a HCP (Fig. 12b).22 By simply varying the mass ratio of HAuCl4·4H2O and the HCP, the Au UMNPs with different sizes can be obtained. These composite materials exhibit high catalytic activity and recyclability toward catalytic reduction of 4-nitrophenol.
Wang et al. presented TB-POP containing triphenylamine and 2,6-bis(1,2,3-triazol-4-yl)pyridyl units.31 Adding TB-POP into CH2Cl2 solution to adsorb palladium acetate followed by reduction in a stream of H2/N2 at 200 °C provided a composite material with Pd UMNPs (1.5 ± 0.6 nm)/TB-POP. Pd UMNPs uniformly dispersed over TB-POP exhibit remarkable catalytic activity and high selectivity in the dehydrogenation of aqueous formic acid solution. Dong et al. fabricated highly dispersed Pd UMNPs (1.4–2.8 nm) loaded on a stable triazinyl-pentaerythritol porous organic polymer (TP-POP) which was synthesized through a facile polycondensation approach between the cyanuric chloride and pentaerythritol (Fig. 12c).32 The three-dimensional structure, abundant triazinyl groups and abundant pores control the growth of Pd UMNPs and prevent their agglomeration during the catalytic process. The as-prepared Pd UMNPs@TP-POP catalyst showed excellent catalytic activity and stability in the reduction of 4-nitrophenol and transfer hydrogenation of aromatic aldehydes under mild conditions.
6. Synthesis of UMNPs supported by metal oxide/sulfides
Metal oxides/sulfides (MXn, X = O or S) are also proved to be efficient support materials for UMNPs. Vacancies or defects are usually introduced into MXn to promote the interactions between the support and metal atoms or particles.94 Such strong binding interactions help UMNPs anchor in MXn tightly, stabilizing UMNPs and preventing their aggregation. In addition, oxygen vacancies in metal oxides have oxygen storage capacity, which can play an important role in promoting oxygen involved reactions, such as oxygen evolution, oxygen reduction and CO oxidation.95–97 Moreover, many MXn, such as CdS and TiO2, can not only serve as supports for UMNPs, but also have catalytic activity by themselves, synergistically acting as co-catalysts.Zhang et al. developed a way of in situ embedding Pt UMNPs (1–2 nm) into a nanorod-shaped cerium dioxide (CeO2) through a redox reaction that occurs at the solid solution interface (Fig. 13).98 CeO2 has extremely high oxygen storage performance and strong interactions with metals; therefore, is often used as a support for MNPs. Pt–CeO2 shows superior thermal stability and durability because of the strong Pt–O bonds formed between Pt UMNPs and O atoms in CeO2. The enhanced interface between Pt and CeO2 is also responsible for the excellent catalytic performance of the hybrid catalyst towards the hydrogenation of nitrophenol. Jiang et al. used Pt ion-doped Ce-MOFs to create Pt UMNPs (<2 nm) supported on CeO2 particles (s-Pt/CeO2) through ultrafast laser induced reduction.34 In this process, the MOF crystals absorb laser photons and generate high pressure and high temperature, causing the pyrolysis of organic liners and reduction of metal ions. Nanocrystalline CeO2 particles with abundant defects on their surface were in situ precipitated, which serve as anchoring sites for Pt UMNPs. Oxygen vacancies in the CeO2 support often play a role in promoting CO oxidation. As expected, the s-Pt/CeO2 can quickly convert CO to CO2 with a conversion efficiency up to 100%.
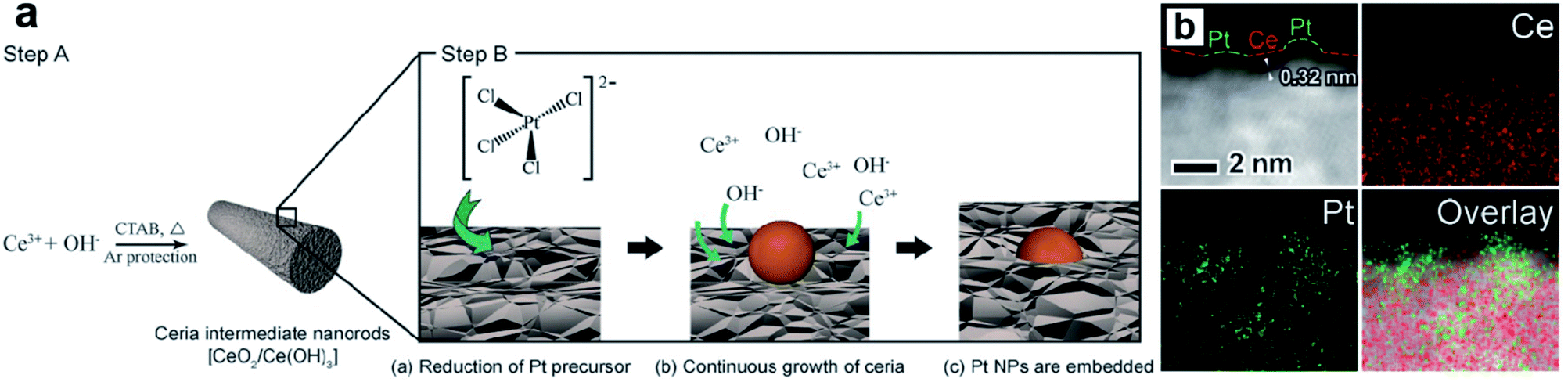 | ||
| Fig. 13 (a) Schematic illustration of the proposed mechanism of synthesizing surface-embedded Pt/CeO2 hybrid nanorods and (b) HAADF-STEMEDX mapping images of Pt/CeO2 hybrid nanorods (adapted with permission from ref. 98 @ copyright 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
The porous structures of supporting materials can increase the reaction rate by accelerating the mass transfer process of the catalytic reactions and improve the stability of UMNPs as well. Various metal oxides are featured as porous structures, which serve as ideal supports for UMNPs.99–101 Zhan et al. supported Pt UMNPs (∼3.2 nm) on mesoporous TiO2 and used them to catalyze the ORR.102 The strong metal support interaction between Pt UMNPs and porous TiO2 enhanced the stability of the catalyst, together with the spatial restriction and the anti-restriction provided by mesoporous TiO2. The catalyst exhibited a much higher stability than the commercial Pt/C after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Meijboom et al. reported Pt UMNPs (1.1–2.1 nm) on mesoporous Co3O4 for the catalytic oxidation of methylene blue.103 The similar mesoporous Co3O4 was also applied by Dai et al. to support AuPd UMNPs (2.7–4.5 nm) for catalytic methane combustion.104 In addition to improving the stability of catalysts, mesoporous Co3O4 provided a high adsorbed reactant concentration due to its porous structure and high pore volume.
000 cycles. Meijboom et al. reported Pt UMNPs (1.1–2.1 nm) on mesoporous Co3O4 for the catalytic oxidation of methylene blue.103 The similar mesoporous Co3O4 was also applied by Dai et al. to support AuPd UMNPs (2.7–4.5 nm) for catalytic methane combustion.104 In addition to improving the stability of catalysts, mesoporous Co3O4 provided a high adsorbed reactant concentration due to its porous structure and high pore volume.
Loading UMNPs on some photocatalytic catalysts, such as TiO2, is also a good approach for improving specific photocatalytic activity. In many cases, the low photocatalytic conversion efficiency is due to the rapid electron–hole recombination in the photocatalyst. Pt NPs are believed to delay electron–hole recombination by capturing electrons and promoting interface electron transfer. Biswas et al. obtained a TiO2 film through the aerosol chemical vapor deposition route and deposited Pt UMNPs (0.5–2 nm) on the TiO2 film (Fig. 14).42 The obtained composite film was used for photocatalytic reduction of CO2, which showed extremely high reduction efficiency and CH4 selectivity. The work of Lin et al. shows the superiority of CdS loaded with Pt UMNPs in the photocatalytic hydrogen evolution reaction.35 They reduced Pt4+ into Pt UMNPs (1.75 nm) through ultrasonic radiation and deposited them on CdS nanorods. When the Pt loading was 0.5 wt%, the highest H2 release efficiency (24.15 mmol h−1 g−1) was achieved. The use of Pt UMNPs as co-catalysts on the surface of CdS can effectively promote the separation of photo-generated charges on CdS, offer a low activation potential, and provide active centers for H2 generation to enhance photocatalytic H2 evolution.
 | ||
| Fig. 14 Characterization of a columnar TiO2 thin film loaded with Pt UMNPs (adapted with permission from ref. 42 @ copyright 2012 American Chemical Society). | ||
7. Synthesis of UMNPs supported by silica
Silica is one of the most abundant components in earth and possesses excellent chemical and thermal stability. It can be considered as an ideal candidate for catalyst supports because of its stability. Silica has a porous structure, especially for ordered mesoporous silica, which can disperse and isolate UMNPs to keep UMNPs stable during catalysis. There are also abundant silanol groups on the silica surface, which makes silica easy to be modified for anchoring UMNPs.105 Thus, silica has been widely used as supports for heterogeneous catalysts, especially in oxidation or reduction reactions driven by thermal energy such as semihydrogenation, CO oxidation, CH4 oxidation and so on.106–108Ding et al. synthesized a series of bimetallic UMNPs (1–3 nm) by decomposing and reducing metal salts adsorbed on silica (Fig. 15).106 These bimetallic UMNPs exhibit well-defined stoichiometry and intimacy between constituent metals and show excellent catalytic performance in semi-hydrogenation of alkynes. Fedorov et al. reported a highly efficient and selective catalyst for the semi-hydrogenation of alkynes, which is obtained by loading Cu UMNPs (2.0 ± 0.6 nm) on passivated silica.109 The competing passivation of silanol sites by Me3Si groups and low Cu precursor loading limit the density and size of Cu UMNPs on passivated silica. The size of Cu UMNPs and the IMes (1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene) anchored on the Cu surface play important roles in improving the selectivity for semihydrogenation.
 | ||
| Fig. 15 Schematic illustration and STEM of the supported bimetallic NPs (adapted with permission from ref. 106 @ copyright © 2018 American Association for the Advancement of Science). | ||
Similar to mesoporous carbon, mesoporous silica can provide a suitable confined environment for UMNPs. Yang et al. reported a novel catalyst obtained by supporting Pd UMNPs (1.1 nm) on functional mesoporous silica nanoparticles.110 Not only the walls of silica can prevent the aggregation of UMNPs, but also the unevenly distributed functional groups in porous silica can maintain the UMNPs in the same channel dispersed. Besides, the isolated silanol groups may interact with phenol through hydrogen bonding and facilitate the phenol hydrogenation process. Kao et al. synthesized Ni UMNPs (2.7 nm) by using 2D hexagonal channel mesoporous silica (SBA-15C) and 3D cage-type mesoporous silica (SBA-16C) as supports.111 Such a composite catalyst exhibits high catalytic activity and stability in the hydrogenation of nitroarenes to aminoarenes.
Even though mesoporous silica works as an efficient and stable supporting material for UMNPs, most UMNPs supported by mesoporous silica are trapped by pores near the surface rather than deeper pores inside, which may reduce the available surface area and may block the pores. As a unique type of silica, fibrous silica nanosphere (KCC-1) can provide a much higher surface area for UMNPs due to the fibrous morphology rather than pore structures.112,113 KCC-1 will not be affected by the blocking of pores and can provide faster mass transfer. Therefore, compared with mesoporous silica, KCC-1 is superior in various applications, including catalysis. Basset et al. used 3-aminopropyltriethoxysilane modified KCC-1 (KCC-1-NH2) as a support and introduced Au ions (Fig. 16).107 Then they obtained Au nanoparticles of different sizes through different reduction methods. Among them, Au UMNPs (1–2 nm) were obtained using NaBH4 as a reducing agent. It shows excellent catalytic activity for CO oxidation. The fibrous structure prevents the migration and aggregation of Au UMNPs, and also it provides a huge contact area with CO.
 | ||
| Fig. 16 (a and b) HR-TEM images, (c) HAADF-STEM images and (d) size histogram of KCC-1-NH2 supported Au UMNPs (adapted with permission from ref. 107 @ copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
8. Conclusion and perspectives
In summary, controlling the synthesis of UMNPs through the use of solid supports can control the size of the UMNPs and help the UMNPs remain well-dispersed. During the catalytic process, the supports and the UMNPs can synergistically determine the catalytic activity. Therefore, synthesizing UMNPs in situ in the presence of a support and controlling the nucleation and growth of UMNPs is a promising method for the controlled synthesis and application of UMNPs in catalysis. The structure and properties of a support play an important role in the growth and application of UMNPs. Many organic porous materials, such as COFs, MOFs, OMCs, and CMPs, can be rationally designed at the atomic level, and their pore size and shape can be precisely controlled, so as to control the size and morphology of UMNPs. The ordered porous support structure is conducive to the narrow size distribution of UMNPs. Carbon materials such as porous carbon, hollow carbon, and graphene have excellent chemical and thermal stability. At the same time, because carbon materials are cheap and easy to obtain, they are economical support materials. Carbon materials doped with heteroatoms or modified with functional groups can provide a large number of nucleation and anchoring sites for UMNPs. The introduction of UMNPs into the vacancies or defects of metal oxides/sulfides can firmly bind UMNPs to the supports through strong coupling. In addition, UMNPs can be used as co-catalysts to enhance the photocatalytic activity of metal oxides/sulfides. The controlled growth of UMNPs mediated by solid supports has been summarized in Table 1, including different types of support materials, UMNPs with different types and sizes, and variable applications with catalytic activities.| Type of supports | Support materials | Type of UMNPs | Design principles | Size | Types of catalysis | Catalytic activity | Ref. |
|---|---|---|---|---|---|---|---|
| Carbon materials | N-Doped porous carbon | Ni–Fe | Electronic effects | ∼2 nm | Electrocatalysis (OER) | η 10 = 297 mV | 45 |
| Ni–Mo | Electronic effects | ∼2 nm | Electrocatalysis (HER) | η 10 = 130 mV | |||
| Hierarchically porous carbon | Pd | Electronic effects/spatial confinement effects | 1.1 ± 0.2 nm | Dehydrogenation of formic acid | TOF achieved as 14400 h−1 at 60 °C |
114 | |
| MOF-derived mesoporous carbon | H2O−WOx | Spatial confinement effects | ∼2.24 nm | Epoxidation of cis-cyclooctene | TOF = 949 h−1 | 48 | |
| MOF-derived mesoporous carbon | Pt | Spatial confinement effects | 2–3 nm | Electrocatalysis (MOR) | 1195 mA mgPt−1 | 47 | |
| 4-Nitrophenol reduction | TOF = 0.200 s−1 g−1 L | ||||||
| MOF-derived porous carbon | Cu/Ru | Electronic effects/spatial confinement effects | 3.30 ± 0.66 nm | Ammonia borane hydrolysis | TOF achieved as 97 molH2 molcat−1 min−1 | 115 | |
| 3D N-doped porous carbon networks | Ru | Electronic effects/spatial confinement effects | ∼2.47 nm | Electrocatalysis (HER) | η 10 = 37 mV (0.5 M H2SO4), etc. | 21 | |
| N-Doped carbon nanosheets | Mo2C | Electronic effects | ∼2 nm | Electrocatalysis (ORR) | E 1/2 = 0.83 V | 116 | |
| 3-D mesoporous graphene nanosheet | PtAg | Electronic effects/spatial confinement effects | ∼2.5 nm | Electrocatalysis (ORR) | E 1/2 = 0.92 V | 117 | |
| 3-D N-doped graphene networks | Pt–Ni | Electronic effects | ∼2.24 nm | Dehydrogenation of hydrous hydrazine | TOF = 943 h−1 | 118 | |
| S-Doped graphitic carbon nitride | Ag | Electronic effects | <1 nm | Photocatalytic degradation of rhodamine B | Degradation rate is 96.5% after 50 min | 119 | |
| Nitrogen doped carbon nanosheets | Ru | Electronic effects/spatial confinement effects | ∼1.41 nm | Ammonia borane hydrolysis | TOF achieved as 440 min−1 | 120 | |
| MOF-derived carbon | Pt | Electronic effects/spatial confinement effects | 2.1–4.1 nm | Electrocatalysis (HER) | η 10 = 42.1 mV | 28 | |
| Hollow N-doped carbon tube | Ag | Electronic effects/spatial confinement effects | ∼2.19 nm | CO2 conversion | Yield achieved as 99% | 121 | |
| PDIL–carbon nanotube | Pd4Au1–P | Electronic effects | ∼2.3 nm | Electrocatalysis (EOR) | I f = 2296.3 mA mgPd−1 | 57 | |
| N, P-Codoped hollow carbon nanospheres | Ru/Ni2P | Electronic effects | ∼3 nm | Electrocatalysis (HER) | η 10 = 89 mV (0.5 M H2SO4), etc. | 122 | |
| Mesocellular graphene network | Pd | Electronic effects | ∼2.8 nm | Electrocatalysis (ORR) | E 1/2 = 0.846 V | 20 | |
| Graphene oxide | Rh | Electronic effects | 1.8 ± 0.4 nm | 4-Nitrophenol reduction | k = 7.62 × 10−3 s−1, TOF = 18 min−1 | 37 | |
| Reduced graphene oxide | Bi | Electronic effects | ∼2 nm | Electrocatalysis (CO2RR) | FE (HCOOH) = 98% | 43 | |
| N-Doped carbon microtubes | Pd | Electronic effects/spatial confinement effects | ∼2.3 nm | 4-Nitrophenol reduction | TOF achieved as 29.5 min−1 | 58 | |
| Suzuki coupling reaction | TOF achieved as 44.0 min−1 | ||||||
| Reduced graphene oxide | Pt | Electronic effects | 2–3 nm | Electrocatalysis (MOR) | 46.43 A gG-Pthybrids−1 | 66 | |
| Reduced graphene oxide | Cu | Electronic effects | 2.0 ± 0.4 nm | 4-Nitrophenol reduction | Conversion efficiency is 98.6% | 67 | |
| N-Doped carbon black embedded graphene | Pd | Electronic effects | ∼3.2 nm | Electrocatalysis (EOR) | I f = 2690.4 mA mg−1 | 68 | |
| Crystalline frameworks | MIL-101 | CuPd | Electronic effects/spatial confinement effects | ∼2.16 nm | Homocoupling reaction of phenylacetylene | Yield(1,4-diphenylbuta-1,3-diyne) = 98% | 24 |
| UiO-66 | Pt–Co | Electronic effects/spatial confinement effects | ∼2.0 nm | Hydrogenation of nitrobenzene | Conversion efficiency is more than 99% | 123 | |
| UiO-66 | Pd | Electronic effects/spatial confinement effects | 0.8–1.1 nm | Hydrogenation of benzoic acid | Yield(cyclohexanecarboxylicacid) = 100% | 76 | |
| UiO-67 | Pt | Electronic effects/spatial confinement effects | 2.5 ± 0.7 nm | CO oxidation | TOF = 0.066 s−1 | 124 | |
| UiO-66 | Ni | Electronic effects/spatial confinement effects | ∼2 nm | CO2 hydrogenation to methane | Yield(methane) = 57.6% | 13 | |
| MOF199 | Au | Electronic effects/spatial confinement effects | <2 nm | A3-Coupling reaction | Yield(propargylamine) = 93% | 125 | |
| UiO-66-NH2, etc. | Pt | Electronic effects/spatial confinement effects | ∼2 nm | CO oxidation reaction | Conversion efficiency is 100% | 126 | |
| 2D nanosheet of mixed-ligand Ni(II) MOF | Au | Electronic effects/spatial confinement effects | ∼1 nm | 4-Nitrophenol reduction | Total reacted to 4-aminophenol within 6 min, k = 0.404 min−1 | 72 | |
| Zn-Based MOFs | Cu2O | Electronic effects/spatial confinement effects | 1.61 ± 0.46 nm | Photocatalytic CO2 methanation | TOF = 50 × 10−3 s−1 | 127 | |
| Multi-layered manner inside MOFs | Pt | Electronic effects/spatial confinement effects | ∼3 nm | 4-Nitrophenol reduction | k = 0.54 min−1, conversion efficiency is near 100% | 128 | |
| MOF-74 | NiMg | Electronic effects/spatial confinement effects | 0.7 ± 0.1 nm | Carbon dioxide methanation | Yield (CH4) = 55.8% | 74 | |
| Monolayered CoN4-based MOF | CoFeOx | Electronic effects/spatial confinement effects | ∼3 nm | Electrocatalysis (OER) | η 10 = 232 mV | 77 | |
| Nitrogen-rich COFs | Ru | Electronic effects/spatial confinement effects | 1.4–2.6 nm | Methanolysis of ammonia borane | TOF achieved as 505 min−1 | 80 | |
| Triazine basic COFs | Pt | Electronic effects/spatial confinement effects | ∼2.10 nm | Electrocatalysis (ORR) | E 1/2 = 0.89 V | 129 | |
| sp3 N-rich flexible COF | CoxNiy(OH)2 | Electronic effects/spatial confinement effects | ∼2 nm | Electrocatalysis (OER) | η 10 = 258 mV | 130 | |
| Phosphine-based COFs | Pd | Electronic effects/spatial confinement effects | ∼1.62 nm | Suzuki–Miyaura coupling reaction | TOF achieved as 1648 h−1 | 33 | |
| Phenol–pyridyl COF | Cu/Cu2O | Electronic effects/spatial confinement effects | 2–3 nm | Glaser–Hay coupling | Yield achieved as 80%, TOF = 50 h−1 | 131 | |
| TpBD–Me2 COF | RuO2 | Electronic effects/spatial confinement effects | ∼1.2 nm | Formic acid dehydrogenation reaction | Yield of H2 achieved as 97% | 132 | |
| TpTa–COF | Fe doped TiO2 | Electronic effects/spatial confinement effects | 2.3 ± 0.9 nm | Photocatalytic degradation of methylene blue | Degradation efficiency over 95% | 19 | |
| Amine-functionalized COFs | Pd | Electronic effects/spatial confinement effects | 1.58 ± 0.2 nm | Benzyl alcohol oxidation | 97.0% selectivity to benzaldehyde | 85 | |
| Sulfur-containing covalent organic framework | Au | Electronic effects/spatial confinement effects | 4.2 ± 1.2 nm | 4-Nitrophenol reduction | Total reacted to 4-aminophenol within 7 min | 36 | |
| COFs with thiol chains | Au | Electronic effects/spatial confinement effects | 1.8 ± 0.2 nm | Photocatalytic degradation of RhB | Degradation achieved as 97.3% | 84 | |
| Photocatalytic degradation of bisphenol A | Degradation more than 90% | ||||||
| Thioether-containing COFs | Pt | Electronic effects/spatial confinement effects | 1.70 ± 0.2 nm | 4-Nitrophenol reduction | Total reacted to 4-aminophenol within 8 min | 25 | |
| Pd | Electronic effects/spatial confinement effects | 1.78 ± 0.2 nm | Suzuki–Miyaura coupling reaction | Yield is more than 99.0% | |||
| Amorphous organic polymers | Redox-active CMPs | Au | Electronic effects/spatial confinement effects | ∼2 nm | Reduction of nitro aryls | Yield is more than 99.0% | 89 |
| Co | Electronic effects/spatial confinement effects | ∼10 nm | Electrocatalysis (ORR) | E onset = 0.91 V (vs. RHE) | |||
| Electron-rich 3D CMP | Pd | Electronic effects/spatial confinement effects | ∼2.4 nm | Suzuki coupling reaction | Yield achieved as 96% | 90 | |
| Sonogashira cross-coupling reaction | Yield achieved as 96% | ||||||
| Stille cross-coupling reaction | Yield achieved as 97% | ||||||
| Covalent carbazole framework | Ag | Electronic effects/spatial confinement effects | ∼2.5 nm | 4-Nitrophenol reduction | Normalized rate constant achieved as 21.49 mmol−1 s−1 | 133 | |
| Aminal-based HCPs | Pd | Electronic effects/spatial confinement effects | ∼2.89 nm | Reduction of nitroarenes | Yield achieved as 99% | 93 | |
| Mesoporous HCPs | Bi | Electronic effects/spatial confinement effects | 1–3 nm | 4-Nitrophenol reduction | Reaction rate constant k = 2.2 min−1 | 134 | |
| Hypercrosslinked polystyrene | Pt | Spatial confinement effects | 2.3 ± 0.5 nm | Catalytic in wet air oxidation of phenol | Conversion efficiency is 97%, selectivity to CO2 and H2O is 94.2% | 135 | |
| HCP–PPh3 | Rh | Electronic effects/spatial confinement effects | ∼2.1 nm | Ammonia borane hydrolysis | TOF = 481 molH2 (molRh min)−1 | 136 | |
| HCPs | Au | Spatial confinement effects | 1.7–5.1 nm | Reduction of 4-nitrophenol | Conversion efficiency is near 100% | 22 | |
| PyPPh2@POP | Pd | Electronic effects/spatial confinement effects | 1.81–3.40 nm | Dehydrogenation of 3-methyl-2-cyclohexen-1-one to 3-methyl phenol | Conversion efficiency is 88.2% | 137 | |
| Phosphorus-doped POPs | Pd | Electronic effects/spatial confinement effects | ∼2.7 nm | Hydrogenation of α, β-unsaturated compound | Yield achieved as 99% | 138 | |
| Reductive cyclization of 2-nitrophenylacetonitrile to indoles | Yield achieved as 99% | ||||||
| Triazinyl-containing POP | Pt | Electronic effects/spatial confinement effects | ∼2.96 nm | Ammonia borane hydrolysis | TOF = 133.17 molH2 molPt−1 min−1 | 139 | |
| Hydrogenation of halogenated nitrobenzenes | Conversion efficiency is 100% with a selectivity of 98% | ||||||
| Gallic acid-derived POPs | Ag | Electronic effects/spatial confinement effects | ∼1 nm | Carboxylative cyclization of CO2 and propargyl alcohols | Yield achieved as 99% | 140 | |
| Triazine functionalized POP | Pd | Electronic effects/spatial confinement effects | ∼3 nm | Alkene hydrogenation | Conversion efficiency is 99% | 141 | |
| Prefunctionalized POPs | Pd | Electronic effects/spatial confinement effects | 1.6 ± 0.35 and 3.5 ± 0.35 nm | Dehalogenation reaction of chlorobenzene | Yield is near 100% | 142 | |
| Porous magnetic core–shell POP nanospheres | Pd | Electronic effects/spatial confinement effects | 1.5–2.1 nm | Hydrogenation of nitrobenzene | Yield is 100%, TOF = 106.4 h−1 | 11 | |
| Hydrogenation of alkenes and alkynes | Yield is 100%, TOF = 212.8 h−1 | ||||||
| POPs | Pd | Electronic effects/spatial confinement effects | 0.9–4 nm | Suzuki–Miyaura coupling reaction | Yield achieved as 92% | 143 | |
| Sonogashira coupling reaction | Yield achieved as 99% | ||||||
| 1,2,3-Triazolyl-containing POPs | Pd | Electronic effects/spatial confinement effects | 1.39 ± 0.31 nm | Hydrogenation of 1-hexene | Yield is 100% | 144 | |
| Boron organic polymers | Pt | Electronic effects/spatial confinement effects | ∼2.3 nm | Ammonia borane hydrolysis | TOF = 1082.5 molH2 molPt−1 min−1 | 145 | |
| Pd | Electronic effects/spatial confinement effects | ∼3.6 nm | Ammonia borane hydrolysis | TOF = 890.0 molH2 molPd−1 min−1 | |||
| Imidazolium-based organic polymers | Pd–Au | Electronic effects/spatial confinement effects | 1.50 ± 0.20 nm | Ammonia borane hydrolysis | 25.0 molH2 molcat−1 min−1 | 146 | |
| TB–POP | Pd | Electronic effects/spatial confinement effects | 1.5 ± 0.6 nm | Dehydrogenation of formic acid | TOF achieved as 1344 h−1, selectivity is 100% | 31 | |
| Triazinyl-pentaerythritol POPs | Pd | Electronic effects/spatial confinement effects | 1.4–2.8 nm | 4-Nitrophenol reduction | Yield is more than 99% | 32 | |
| Metal oxide/sulfide | CeO2 nanorods | Pt | Electronic effects | 1–2 nm | 4-Nitrophenol reduction | Similar as surface-embedded sample | 98 |
| CeO2 nanorods | Pt | Electronic effects | 1.3–2.5 nm | Oxidation of toluene | TOFPt = (7.95 ± 0.43) × 10−3 s−1 | 147 | |
| CeO2 nanorods | Pd | Electronic effects | ∼2 nm | CO oxidation | Generation rate is 1 × 1021molecules CO gPd−1 s−1 | 148 | |
| CeO2 nanocrystals | Au | Electronic effects | ∼3 nm | CO oxidation | TOF = 0.69 s−1 | 149 | |
| Mesoporous CeO2 | Au | Electronic effects/spatial confinement effects | ∼3 nm | CO oxidation | The initial conversion is of ca. 40% | 95 | |
| Ce–MOFs derived CeO2 nanoparticles | Pt | Electronic effects | <2 nm | CO oxidation | Conversion efficiency above 90% (170 °C), and 100% (650 °C) | 34 | |
| Porous CeO2 nanofibers | Pt | Electronic effects | ∼1.7 nm | Water gas shift reaction | CO conversion reached 95% under 450 °C | 150 | |
| 3D ordered macroporous TiO2 | Pd | Electronic effects/spatial confinement effects | ∼1.1 nm | Soot oxidation | Yield of CO2 = 97.6% | 151 | |
| N-Doped TiO2 | Pd | Electronic effects | ∼2.2 nm | H2O2 synthesis | H2O2 productivity = 4.1 molH2O2 gPd−1 h−1 | 152 | |
| TiO2 nanoparticles | Cu | Electronic effects | 2–4 nm | Photocatalytic hydrogen generation | Generation rate of H2 = 9.5 mmol g−1 h−1 | 153 | |
| TiO2 | Pd | Electronic effects | 1–2 nm | Photocatalytic hydrogen peroxide production | Selectivity >80% | 154 | |
| TiO2 single crystals | Pt | Electronic effects | 0.5–2 nm | Photocatalytic CO2 photoreduction | CH4 yield is 1361 μmol gcat−1 h−1 | 42 | |
| Co3O4 microflowers | Pt | Electronic effects | ∼2.3 nm | Electrocatalysis (HER) | η 10 = 34 mV | 155 | |
| CdS nanorods | Pt | Electronic effects | ∼1.75 nm | Photocatalytic hydrogen evolution | H2 evolution rate of 24.15 mmol h−1 g−1 | 35 | |
| Hollow CdS structure | Pd/PdS | Electronic effects | ∼1.5 nm | Photocatalytic hydrogen evolution | H2 evolution rate of up to 144.8 mmol h−1 g−1 | 156 | |
| MoS2 nanosheets | Pt | Electronic effects | 2–5 nm | Electrocatalysis (HER) | η 10 = 31 mV | 157 | |
| CoS2 nanosheet | Pt | Electronic effects | ∼1.7 nm | Electrocatalysis (HER) | η 10 = 24 mV | 158 | |
| Electrocatalysis (OER) | η 10 = 300 mV | ||||||
| Mesoporous TiO2 | RuO2 | Electronic effects/spatial confinement effects | ∼2 nm | CO2 methanation | 2.05 μmolCH4 gcat−1·s−1 | 159 | |
| Urchin-like mesoporous TiO2 hollow spheres | Pt | Electronic effects/spatial confinement effects | ∼3.2 nm | Electrocatalysis (ORR) | E 1/2 = 0.867 V | 102 | |
| Mesoporous Co3O4 | Pt | Electronic effects/spatial confinement effects | 1.1–2.1 nm | Oxidation of methylene blue | K 35°C = (9.23 ± 0.15) × 10−4 | 103 | |
| 3D ordered mesoporous Co3O4 | Au–Pd | Electronic effects/spatial confinement effects | 2.7–4.5 nm | Methane combustion | T 90 = 324 °C | 104 | |
| Silica | 2D silica nanosheets | Ag | Electronic effects | ∼2.77 nm | Reduction of 4-nitrophenol | TOF = 3.52 min−1 | 160 |
| Mesoporous silica nanoparticle | Pd | Electronic effects/spatial confinement effects | 0.9 ± 0.2 nm | Reduction of 4-nitrophenol | k = 0.31 min−1 | 161 | |
| Cage-type mesoporous silica | Ni | Electronic effects/spatial confinement effects | ∼2.7 nm | CO2 hydrogenation | TOFCO2 >110 s−1 | 162 | |
| Silica | Ru | Electronic effects/spatial confinement effects | ∼1.56 nm | CO2 methanation | CO2 conversion achieve as 70% | 163 | |
| Silica | Pt–Pd | Electronic effects/spatial confinement effects | 1–3 nm | Semihydrogenation of alkynes | C2H2 conversion achieve as 100% near 100 °C | 106 | |
| Passivation silica | Cu | Electronic effects/spatial confinement effects | 2.0 ± 0.6 nm | Semihydrogenation of alkynes | Selectivity to (Z)-olefins achieve as 97% | 109 | |
| Bimodal mesopore silica | Pd | Electronic effects/spatial confinement effects | ∼3 nm | Oxidation of toluene | T 90 = 228 °C | 108 | |
| –COOH functionalized SBA-16 | Ag-Doped Ni | Electronic effects/spatial confinement effects | ∼3 nm | Reduction of 4-nitrophenol | Activity parameter achieve as 3340.4 s−1 gAgNi−1 | 164 | |
| Mesoporous silica | RhOx | Electronic effects/spatial confinement effects | 1–2.5 nm | N2O decomposition | T 90 = 407 °C | 165 | |
| SBA-15 monoliths | Ni | Electronic effects/spatial confinement effects | 1–3 nm | Methane dry reforming | TOF = 1.4 s−1 | 166 | |
| Mesoporous silica | Au–Cu | Electronic effects/spatial confinement effects | ∼1.5 nm | Glycerol oxidation | Dihydroxyacetone selectivity = 90% | 167 | |
| 3D dendritic mesoporous silica nanospheres | Pd | Electronic effects/spatial confinement effects | ∼1.5 nm | Suzuki–Miyaura cross-coupling reactions | Conversion >99% | 168 | |
| Mesoporous MMT-1 silica | Pd | Electronic effects/spatial confinement effects | ∼1.1 nm | Hydrogenation of phenol to cyclohexanone | Conversion = 99%, selectivity = 98% | 110 | |
| Ordered mesoporous silicas | Ni | Electronic effects/spatial confinement effects | ∼2.7 nm | Hydrogenation of nitroarenes | Apparent reaction rate = 5.68 × 10−3 | 111 | |
| PDETA-functionalized KCC-1 | Pd | Electronic effects/spatial confinement effects | ∼2.8 nm | Dehydrogenation of formic acid | TOF = 332 h−1, selectivityH2 = 100% | 169 | |
| Aminopropyl groups functionalized KCC-1 | Pd | Electronic effects/spatial confinement effects | 1–5 nm | Suzuki coupling reaction | Yield achieved as 97% | 170 | |
| Aminopropyl groups functionalized KCC-1 | Au | Electronic effects/spatial confinement effects | 1–2 nm | CO oxidation | T 50 = 275 °C | 107 |
This review summarizes the control of the growth of UMNPs with solid supports and their applications in the field of catalysis. Through the analysis of the reported examples, we believe that the following conditions play crucial roles in controlling the growth of UMNPs, while still facing some problems and challenges:
(1) The design of supports: porous supports can provide confinement for the growth of UMNPs and prevent the accumulation of UMNPs through site isolation. At the same time, the high specific surface area provides a large number of available active sites and anchoring sites. However, in many cases, UMNPs grow outside of the pores of the porous support, which causes the pores to be blocked. Many active sites in the pores cannot participate in the catalytic process, which affects the mass transfer of the catalytic process. Through a proper design, metal precursors can be preferentially loaded inside the pores, which can effectively prevent the growth of UMNPs on the surface of supports. The hierarchical design of micropores, mesopores, and macropores can incorporate the micropores and smaller mesopores, which may help in efficient transfer mass. It is also necessary to consider the interactions between supports and UMNPs to facilitate catalytic reactions while avoiding catalyst poisoning.
(2) Synergistic catalysis provided by UMNPs and supports: some supports can adsorb reaction substrates, which increases the local concentration of reaction substrates near UMNPs and facilitates the catalytic reactions. Bai et al. prepared covalent triazine framework (CTF) nanosheets coated with Ag particles to catalyze CO2 conversion.171 CTFs have no obvious catalytic activity for CO2 conversion, but they have high CO2 capture ability. The coated Ag particles can catalyze the coupling of CO2 and phenylacetylene to produce 3-phenylpropiolic acid. The hybrid materials of Ag particles and CTFs can act synergistically to catalyze the reaction. Through the rational design of UMNPs and supports, the catalytic activity of the catalyst can be improved effectively.
(3) Shape and diameter of UMNPs: precise control of the shape and diameter of UMNPs is still challenging, which is critical to improving their performance in heterogeneous catalysis. The spatial confinement effect is more inclined to macroscopic design, which can limit the growth of nanoparticles and control the size and shape of nanoparticles to a certain extent. It is still hard to control UMNPs with a size less than 3 nm by using most of the inorganic supports, while electronic effects have a remarkable influence over the size of UMNPs. The size of UMNPs can be fine-tuned by adjusting the concentration of metal ions and the ratio of heteroatoms, functional groups or defects. The development of new supports that can combine the electronic effects and characteristic spatial shapes may be the future of realizing the precise control of UMNPs, especially for the shape control.
(4) Stability: high stability of the UMNPs and carriers, even under harsh catalytic conditions, is highly desired. Improving the stability of the carrier and UMNPs can increase the number of cycles of a catalyst, which is conducive to reducing the cost of the catalyst and enabling UMNPs for practical applications. Long-term usage or harsh conditions (such as high temperature or corrosive medium) may ruin the structure of solid supports and cause the catalyst to be deactivated. It calls for the development of more stable or even self-repairable supports.
(5) Nucleation and growth mechanism: there have been some studies on the nucleation and growth mechanism of UMNPs on the carrier interface, but the mechanism of nucleation and growth is still unclear and in-depth exploration is needed, which can provide design principles of solid supports and efficient synthetic routes to MNPs.
Although great progress has been achieved, controlling the nucleation and growth of UMNPs through the use of solid supports still faces many challenges. With continuous in-depth research, efficient preparation of UMNPs with well-controlled uniform size and high stability is anticipated, which will enable wide-spread practical applications of UMNPs in the field of catalysis and beyond.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the National Science Foundation (DMR-1420736), K. C. Wong Education Foundation, the National Natural Science Foundation of China (NSFC) (51803077, 21905115), the Natural Science Foundation of Jiangsu Province (BK20180627), the China Postdoctoral Science Foundation (2018M630517, 2019T120389, 2020M671334), the MOE & SAFEA, 111 Project (B13025), the national first-class discipline program of Light Industry Technology and Engineering (LITE2018-19), and the Fundamental Research Funds for the Central Universities (JUSRP11929) for support.Notes and references
- Q. Wang, L. Chen, Z. Liu, N. Tsumori, M. Kitta and Q. Xu, Adv. Funct. Mater., 2019, 29, 1903341 CrossRef
.
- Y. Cao, W. Geng, R. Shi, L. Shang, G. I. Waterhouse, L. Liu, L. Z. Wu, C. H. Tung, Y. Yin and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 14952–14957 CrossRef CAS PubMed
- C. Y. Hui, M. Liu, Y. Li and J. D. Brennan, Angew. Chem., Int. Ed., 2018, 57, 4549–4553 CrossRef CAS PubMed
- L. Gao, K. Cao, X. Hu, R. Xiao, B. Gan, W. Wang and Y. Lu, Appl. Mater. Today, 2020, 18, 100475 CrossRef
- P. Liu, K. Zhu, Y. Gao, H. Luo and L. Lu, Adv. Energy Mater., 2017, 7, 1700547 CrossRef
- J. Wu, A. Mahajan, L. Riekehr, H. Zhang, B. Yang, N. Meng, Z. Zhang and H. Yan, Nano Energy, 2018, 50, 723–732 CrossRef CAS
- M. Bjornmalm, K. J. Thurecht, M. Michael, A. M. Scott and F. Caruso, ACS Nano, 2017, 11, 9594–9613 CrossRef CAS PubMed
- S. Zhang, S. Zhou, H. Liu, M. Xing, B. Ding and B. Li, Adv. Funct. Mater., 2020, 30, 2002434 CrossRef CAS PubMed
- R. Tao, X. Ma, X. Wei, Y. Jin, L. Qiu and W. Zhang, J. Mater. Chem. A, 2020, 8, 17360–17391 RSC
- Q.-L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS
- J. Yang, Y. Zhu, M. Fan, X. Sun, W. D. Wang and Z. Dong, J. Colloid Interface Sci., 2019, 554, 157–165 CrossRef CAS PubMed
- R. Zhang, K. Huang, D. Wang, N. Hussain, A. Zhang, H. Wei, G. Ou, W. Zhao, C. Zhang and H. Wu, Electrochim. Acta, 2019, 313, 255–260 CrossRef CAS
- Z.-W. Zhao, X. Zhou, Y.-N. Liu, C.-C. Shen, C.-Z. Yuan, Y.-F. Jiang, S.-J. Zhao, L.-B. Ma, T.-Y. Cheang and A.-W. Xu, Catal. Sci. Technol., 2018, 8, 3160–3165 RSC
- P. W. Voorhees, J. Stat. Phys., 1985, 38, 231–252 CrossRef
- J. Jin, C. Wang, X.-N. Ren, S.-Z. Huang, M. Wu, L.-H. Chen, T. Hasan, B.-J. Wang, Y. Li and B.-L. Su, Nano Energy, 2017, 38, 118–126 CrossRef CAS
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2021, 121, 834–881 CrossRef CAS PubMed
- R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 24, 181–190 CrossRef PubMed
- K. A. Cychosz, R. Guillet-Nicolas, J. Garcia-Martinez and M. Thommes, Chem. Soc. Rev., 2017, 46, 389–414 RSC
- Y. Zhang, Y. Hu, J. Zhao, E. Park, Y. Jin, Q. Liu and W. Zhang, J. Mater. Chem. A, 2019, 7, 16364–16371 RSC
- X. Cui, Y. Xu, L. Chen, M. Zhao, S. Yang and Y. Wang, Appl. Catal., B, 2019, 244, 957–964 CrossRef CAS
- H. Li, M. Zhang, L. Yi, Y. Liu, K. Chen, P. Shao and Z. Wen, Appl. Catal., B, 2021, 280, 119412 CrossRef CAS
- J. He, S. Razzaque, S. Jin, I. Hussain and B. Tan, ACS Appl. Nano Mater., 2018, 2, 546–553 CrossRef
- R. Fang, P. Tian, X. Yang, R. Luque and Y. Li, Chem. Sci., 2018, 9, 1854–1859 RSC
- F. Chen, K. Shen, J. Chen, X. Yang, J. Cui and Y. Li, ACS Cent. Sci., 2019, 5, 176–185 CrossRef CAS PubMed
- S. Lu, Y. Hu, S. Wan, R. McCaffrey, Y. Jin, H. Gu and W. Zhang, J. Am. Chem. Soc., 2017, 139, 17082–17088 CrossRef CAS PubMed
- H. L. Cao, H. B. Huang, Z. Chen, B. Karadeniz, J. Lu and R. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 5231–5236 CrossRef CAS PubMed
- L. Qiu, R. McCaffrey and W. Zhang, Chem.–Asian J., 2018, 13, 362–372 CrossRef CAS PubMed
- J. Li, H. Huang, Y. Li, Y. Tang, D. Mei and C. Zhong, J. Mater. Chem. A, 2019, 7, 20239–20246 RSC
- D. Mullangi, D. Chakraborty, A. Pradeep, V. Koshti, C. P. Vinod, S. Panja, S. Nair and R. Vaidhyanathan, Small, 2018, 14, e1801233 CrossRef PubMed
- G. Chen, T. Wang, P. Liu, Z. Liao, H. Zhong, G. Wang, P. Zhang, M. Yu, E. Zschech, M. Chen, J. Zhang and X. Feng, Energy Environ. Sci., 2020, 13, 2849–2855 RSC
- C. Cui, Y. Tang, M. A. Ziaee, D. Tian and R. Wang, ChemCatChem, 2018, 10, 1431–1437 CrossRef CAS
- J. Yang, M. Yuan, D. Xu, H. Zhao, Y. Zhu, M. Fan, F. Zhang and Z. Dong, J. Mater. Chem. A, 2018, 6, 18242–18251 RSC
- R. Tao, X. Shen, Y. Hu, K. Kang, Y. Zheng, S. Luo, S. Yang, W. Li, S. Lu, Y. Jin, L. Qiu and W. Zhang, Small, 2020, 16, e1906005 CrossRef PubMed
- S. Guo, Y. Zhao, H. Yuan, C. Wang, H. Jiang and G. J. Cheng, Small, 2020, 16, e2000749 CrossRef PubMed
- S. Liu, Z. Guo, X. Qian, J. Zhang, J. Liu and J. Lin, Sustainable Energy Fuels, 2019, 3, 1048–1054 RSC
- Q.-P. Zhang, Y.-l. Sun, G. Cheng, Z. Wang, H. Ma, S.-Y. Ding, B. Tan, J.-h. Bu and C. Zhang, Chem. Eng. J., 2020, 391, 123471 CrossRef CAS
- X. Liu, Q. Han, Y. Zhang, X. Wang, S. Cai, C. Wang and R. Yang, Appl. Surf. Sci., 2019, 471, 929–934 CrossRef CAS
- F. Yu, L. Xie, F. Wu, B. Yuan, C. Xie, S. Yu, X. Liu, L. Wang and D. Wang, ChemCatChem, 2019, 11, 1518–1525 CrossRef CAS
- X. Liu, F. Liu, J. Yu, G. Xiong, L. Zhao, Y. Sang, S. Zuo, J. Zhang, H. Liu and W. Zhou, Adv. Sci., 2020, 7, 2001526 CrossRef CAS PubMed
- X. Du, P. Cai, W. Luo and G. Cheng, Int. J. Hydrog. Energy, 2017, 42, 6137–6143 CrossRef CAS
- Q. Jia, S. Zhang, X. Jia, X. Dong, Z. Gao and Q. Gu, Catal. Sci. Technol., 2019, 9, 5077–5089 RSC
- W. N. Wang, W. J. An, B. Ramalingam, S. Mukherjee, D. M. Niedzwiedzki, S. Gangopadhyay and P. Biswas, J. Am. Chem. Soc., 2012, 134, 11276–11281 CrossRef CAS PubMed
- Y. X. Duan, K. H. Liu, Q. Zhang, J. M. Yan and Q. Jiang, Small Methods, 2020, 4, 1900846 CrossRef CAS
- C.-W. Kung, C. O. Audu, A. W. Peters, H. Noh, O. K. Farha and J. T. Hupp, ACS Energy Lett., 2017, 2, 2394–2401 CrossRef CAS
- Y. Zhang, X. Xia, X. Cao, B. Zhang, N. H. Tiep, H. He, S. Chen, Y. Huang and H. J. Fan, Adv. Energy Mater., 2017, 7, 1700220 CrossRef
- T. Y. Ma, L. Liu and Z. Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC
- X. Q. Wu, J. Zhao, Y. P. Wu, W. W. Dong, D. S. Li, J. R. Li and Q. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 12740–12749 CrossRef CAS PubMed
- M. Zhang, V. Singh, X. Hu, X. Ma, J. Lu, C. Zhang, J. Wang and J. Niu, ACS Catal., 2019, 9, 7641–7650 CrossRef CAS
- G. Xia, L. Zhang, F. Fang, D. Sun, Z. Guo, H. Liu and X. Yu, Adv. Funct. Mater., 2016, 26, 6188–6196 CrossRef CAS
- J.-T. Ren, L. Chen, D.-D. Yang and Z.-Y. Yuan, Appl. Catal., B, 2020, 263, 118352 CrossRef CAS
- H. Tabassum, A. Mahmood, B. Zhu, Z. Liang, R. Zhong, S. Guo and R. Zou, Energy Environ. Sci., 2019, 12, 2924–2956 RSC
- B. Wu, D. Hu, Y. Kuang, B. Liu, X. Zhang and J. Chen, Angew. Chem., Int. Ed., 2009, 48, 4751–4754 CrossRef CAS PubMed
- C.-L. Chiang, C.-C. Wang and C.-Y. Chen, Mater. Sci. Eng., B, 2018, 238–239, 42–49 CrossRef CAS
- R. Sun, F. Ren, D. Wang, Y. Yao, Z. Fei, H. Wang, Z. Liu, R. Xing and Y. Du, Colloids Surf., A, 2019, 578, 123566 CrossRef CAS
- R. Wang, P. Sun, H. Wang and X. Wang, Int. J. Hydrog. Energy, 2018, 43, 17244–17251 CrossRef CAS
- N. Wang, S. Pandit, L. Ye, M. Edwards, V. R. S. S. Mokkapati, M. Murugesan, V. Kuzmenko, C. Zhao, F. Westerlund, I. Mijakovic and J. Liu, Carbon, 2017, 111, 402–410 CrossRef CAS
- H. Yang, Z. Yu, S. Li, Q. Zhang, J. Jin and J. Ma, J. Catal., 2017, 353, 256–264 CrossRef CAS
- X. Duan, M. Xiao, S. Liang, Z. Zhang, Y. Zeng, J. Xi and S. Wang, Carbon, 2017, 119, 326–331 CrossRef CAS
- X. Huang, H. Yu, H. Tan, J. Zhu, W. Zhang, C. Wang, J. Zhang, Y. Wang, Y. Lv, Z. Zeng, D. Liu, J. Ding, Q. Zhang, M. Srinivasan, P. M. Ajayan, H. H. Hng and Q. Yan, Adv. Funct. Mater., 2014, 24, 6516–6523 CrossRef CAS
- R. Nandan and K. K. Nanda, J. Mater. Chem. A, 2017, 5, 10544–10553 RSC
- S. Kralj, F. Longobardo, D. Iglesias, M. Bevilacqua, C. Tavagnacco, A. Criado, J. J. Delgado Jaen, D. Makovec, S. Marchesan, M. Melchionna, M. Prato and P. Fornasiero, ACS Appl. Nano Mater., 2019, 2, 6092–6097 CrossRef CAS
- R. Ye and J. M. Tour, ACS Nano, 2019, 13, 10872–10878 CrossRef CAS PubMed
- H. Huang, H. Shi, P. Das, J. Qin, Y. Li, X. Wang, F. Su, P. Wen, S. Li, P. Lu, F. Liu, Y. Li, Y. Zhang, Y. Wang, Z. S. Wu and H. M. Cheng, Adv. Funct. Mater., 2020, 30, 1909035 CrossRef CAS
- R. Tarcan, O. Todor-Boer, I. Petrovai, C. Leordean, S. Astilean and I. Botiz, J. Mater. Chem. C, 2020, 8, 1198–1224 RSC
- E. Yazici, S. Yanik and M. B. Yilmaz, Carbon, 2017, 111, 822–827 CrossRef CAS
- P. Kundu, C. Nethravathi, P. A. Deshpande, M. Rajamathi, G. Madras and N. Ravishankar, Chem. Mater., 2011, 23, 2772–2780 CrossRef CAS
- X. Kang, D. Teng, S. Wu, Z. Tian, J. Liu, P. Li, Y. Ma and C. Liang, J. Colloid Interface Sci., 2020, 566, 265–270 CrossRef CAS PubMed
- S. Li, J. Shu, S. Ma, H. Yang, J. Jin, X. Zhang and R. Jin, Appl. Catal., B, 2021, 280, 119464 CrossRef CAS
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed
- S. Dalapati, C. Gu and D. Jiang, Small, 2016, 12, 6513–6527 CrossRef CAS PubMed
- R. Yan, Y. Zhao, H. Yang, X.-J. Kang, C. Wang, L.-L. Wen and Z.-D. Lu, Adv. Funct. Mater., 2018, 28, 1802021 CrossRef
- L. Wang, P. Jin, S. Duan, H. She, J. Huang and Q. Wang, Sci. Bull., 2019, 64, 926–933 CrossRef CAS
- T. Zurrer, K. Wong, J. Horlyck, E. C. Lovell, J. Wright, N. M. Bedford, Z. Han, K. Liang, J. Scott and R. Amal, Adv. Funct. Mater., 2021, 31, 2007624 CrossRef CAS
- D. Saha and S. Deng, J. Phys. Chem. Lett., 2009, 1, 73–78 CrossRef
- D. Chen, W. Yang, L. Jiao, L. Li, S. H. Yu and H. L. Jiang, Adv. Mater., 2020, 32, e2000041 CrossRef PubMed
- W. Zhang, Y. Wang, H. Zheng, R. Li, Y. Tang, B. Li, C. Zhu, L. You, M. R. Gao, Z. Liu, S. H. Yu and K. Zhou, ACS Nano, 2020, 14, 1971–1981 CrossRef CAS PubMed
- Z. Li, T. He, Y. Gong and D. Jiang, Acc. Chem. Res., 2020, 53, 1672–1685 CrossRef CAS
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC
- X. Li, C. Zhang, M. Luo, Q. Yao and Z.-H. Lu, Inorg. Chem. Front., 2020, 7, 1298–1306 RSC
- S. Qiu, Q. Fang, H. Li, X. Guan and Y. Yusran, Natl. Sci. Rev., 2020, 7, 170–190 CrossRef
- H. L. Qian, C. X. Yang and X. P. Yan, Nat. Commun., 2016, 7, 12104 CrossRef CAS PubMed
- E. Tavakoli, A. Kakekhani, S. Kaviani, P. Tan, M. M. Ghaleni, M. A. Zaeem, A. M. Rappe and S. Nejati, J. Am. Chem. Soc., 2019, 141, 19560–19564 CrossRef CAS PubMed
- Y. Deng, Z. Zhang, P. Du, X. Ning, Y. Wang, D. Zhang, J. Liu, S. Zhang and X. Lu, Angew. Chem., Int. Ed., 2020, 59, 6082–6089 CrossRef CAS PubMed
- C. Xu, J. Lin, D. Yan, Z. Guo, D. J. Austin, H. Zhan, A. Kent and Y. Yue, ACS Appl. Nano Mater., 2020, 3, 6416–6422 CrossRef CAS
- Y. Zhang and J. Y. Ying, ACS Catal., 2015, 5, 2681–2691 CrossRef CAS
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC
- J. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed
- S. Bhattacharyya, D. Samanta, S. Roy, V. P. Haveri Radhakantha and T. K. Maji, ACS Appl. Mater. Interfaces, 2019, 11, 5455–5461 CrossRef CAS PubMed
- N. Huang, Y. Xu and D. Jiang, Sci. Rep., 2014, 4, 7228 CrossRef CAS PubMed
- M. Salzano de Luna, R. Castaldo, R. Altobelli, L. Gioiella, G. Filippone, G. Gentile and V. Ambrogi, Carbohydr. Polym., 2017, 177, 347–354 CrossRef CAS PubMed
- X. Cai, J. Nie, G. Yang, F. Wang, C. Ma, C. Lu and Z. Chen, Mater. Lett., 2019, 240, 80–83 CrossRef CAS
- D. Xu, F. Wang, G. Yu, H. Zhao, J. Yang, M. Yuan, X. Zhang and Z. Dong, ChemCatChem, 2018, 10, 4569–4577 CrossRef CAS
- Y. Li, P. Wang, C. Huang, W. Yao, Q. Wu and Q. Xu, Appl. Catal., B, 2017, 205, 489–497 CrossRef CAS
- J. M. López, R. Arenal, B. Puértolas, Á. Mayoral, S. H. Taylor, B. Solsona and T. García, J. Catal., 2014, 317, 167–175 CrossRef
- D. Hassen, M. A. Shenashen, S. A. El-Safty, M. M. Selim, H. Isago, A. Elmarakbi, A. El-Safty and H. Yamaguchi, J. Power Sources, 2016, 330, 292–303 CrossRef CAS
- P. Wen, F. Su, H. Li, Y. Sun, Z. Liang, W. Liang, J. Zhang, W. Qin, S. M. Geyer, Y. Qiu and L. Jiang, Chem. Eng. J., 2020, 385, 123878 CrossRef CAS
- J. S. Du, T. Bian, J. Yu, Y. Jiang, X. Wang, Y. Yan, Y. Jiang, C. Jin, H. Zhang and D. Yang, Adv. Sci., 2017, 4, 1700056 CrossRef PubMed
- K. An, S. Alayoglu, N. Musselwhite, S. Plamthottam, G. Melaet, A. E. Lindeman and G. A. Somorjai, J. Am. Chem. Soc., 2013, 135, 16689–16696 CrossRef CAS PubMed
- L. Jin, B. Liu, M. E. Louis, G. Li and J. He, ACS Appl. Mater. Interfaces, 2020, 12, 9617–9627 CrossRef CAS PubMed
- Y. Song, Y. Li, Y. Peng, C. Zhang and F. Yin, Microporous Mesoporous Mater., 2021, 314, 110866 CrossRef CAS
- S. He, C. Wu, Z. Sun, Y. Liu, R. Hu, L. Guan and H. Zhan, Nanoscale, 2020, 12, 10656–10663 RSC
- M. S. Xaba, J.-H. Noh and R. Meijboom, Appl. Surf. Sci., 2019, 467–468, 868–880 CrossRef CAS
- Z. Wu, J. Deng, Y. Liu, S. Xie, Y. Jiang, X. Zhao, J. Yang, H. Arandiyan, G. Guo and H. Dai, J. Catal., 2015, 332, 13–24 CrossRef CAS
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS PubMed
- K. Ding, D. A. Cullen, L. Zhang, Z. Cao, A. D. Roy, I. N. Ivanov and D. Cao, Science, 2018, 362, 560–564 CrossRef CAS PubMed
- Z. S. Qureshi, P. B. Sarawade, I. Hussain, H. Zhu, H. Al-Johani, D. H. Anjum, M. N. Hedhili, N. Maity, V. D'Elia and J.-M. Basset, ChemCatChem, 2016, 8, 1671–1678 CrossRef CAS
- N. Qiao, Y. Li, N. Li, X. Zhang, J. Cheng and Z. Hao, Chin. J. Catal., 2015, 36, 1686–1693 CrossRef CAS
- N. Kaeffer, H. J. Liu, H. K. Lo, A. Fedorov and C. Coperet, Chem. Sci., 2018, 9, 5366–5371 RSC
- C.-J. Lin, S.-H. Huang, N.-C. Lai and C.-M. Yang, ACS Catal., 2015, 5, 4121–4129 CrossRef CAS
- C. S. Budi, D. Saikia, C.-S. Chen and H.-M. Kao, J. Catal., 2019, 370, 274–288 CrossRef CAS
- E. Febriyanti, V. Suendo, R. R. Mukti, A. Prasetyo, A. F. Arifin, M. A. Akbar, S. Triwahyono, I. N. Marsih and Ismunandar, Langmuir, 2016, 32, 5802–5811 CrossRef CAS PubMed
- V. Polshettiwar, D. Cha, X. Zhang and J. M. Basset, Angew. Chem., Int. Ed., 2010, 49, 9652–9656 CrossRef CAS PubMed
- Q. Wang, N. Tsumori, M. Kitta and Q. Xu, ACS Catal., 2018, 8, 12041–12045 CrossRef CAS
- P. Pachfule, X. Yang, Q.-L. Zhu, N. Tsumori, T. Uchida and Q. Xu, J. Mater. Chem. A, 2017, 5, 4835–4841 RSC
- Y. Guo, J. Tang, J. Henzie, B. Jiang, H. Qian, Z. Wang, H. Tan, Y. Bando and Y. Yamauchi, Mater. Horiz., 2017, 4, 1171–1177 RSC
- Z. Li, Y. Li, C. He and P. K. Shen, J. Mater. Chem. A, 2017, 5, 23158–23169 RSC
- A. Kumar, X. Yang and Q. Xu, J. Mater. Chem. A, 2019, 7, 112–115 RSC
- K.-L. Wang, Y. Li, T. Sun, F. Mao, J.-K. Wu and B. Xue, Appl. Surf. Sci., 2019, 476, 741–748 CrossRef CAS
- F. Zhong, Q. Wang, C. Xu, Y. Yang, Y. Wang, Y. Zhang, D. Gao, J. Bi and G. Fan, Appl. Surf. Sci., 2018, 455, 326–332 CrossRef CAS
- X. Lan, Q. Li, L. Cao, C. Du, L. Ricardez-Sandoval and G. Bai, Appl. Surf. Sci., 2020, 508, 145220 CrossRef
- J.-Q. Chi, X.-Y. Zhang, X. Ma, B. Dong, J.-Q. Zhang, B.-Y. Guo, M. Yang, L. Wang, Y.-M. Chai and C. Liu, ACS Sustainable Chem. Eng., 2019, 7, 17714–17722 CrossRef CAS
- L. Chang and Y. Li, Mol. Catal., 2017, 433, 77–83 CrossRef CAS
- R. Vakili, E. K. Gibson, S. Chansai, S. Xu, N. Al-Janabi, P. P. Wells, C. Hardacre, A. Walton and X. Fan, ChemCatChem, 2018, 10, 4238–4242 CrossRef CAS
- Y. Jiang, X. Zhang, X. Dai, W. Zhang, Q. Sheng, H. Zhuo, Y. Xiao and H. Wang, Nano Res., 2017, 10, 876–889 CrossRef CAS
- S. Yoshimaru, M. Sadakiyo, A. Staykov, K. Kato and M. Yamauchi, Chem. Commun., 2017, 53, 6720–6723 RSC
- M. Cabrero-Antonino, S. Remiro-Buenamanana, M. Souto, A. A. Garcia-Valdivia, D. Choquesillo-Lazarte, S. Navalon, A. Rodriguez-Dieguez, G. Minguez Espallargas and H. Garcia, Chem. Commun., 2019, 55, 10932–10935 RSC
- Y. Liu, S. Wang, B. Yu, Y. Zhang, X. Kong, Y. Mi, J. Zhang, Z. Guo, W. Xu and X. Chen, Inorg. Chem., 2020, 59, 13184–13189 CrossRef CAS PubMed
- L. Zhai, S. Yang, X. Yang, W. Ye, J. Wang, W. Chen, Y. Guo, L. Mi, Z. Wu, C. Soutis, Q. Xu and Z. Jiang, Chem. Mater., 2020, 32, 9747–9752 CrossRef CAS
- D. Mullangi, V. Dhavale, S. Shalini, S. Nandi, S. Collins, T. Woo, S. Kurungot and R. Vaidhyanathan, Adv. Energy Mater., 2016, 6, 1600110 CrossRef
- D. Chakraborty, S. Nandi, D. Mullangi, S. Haldar, C. P. Vinod and R. Vaidhyanathan, ACS Appl. Mater. Interfaces, 2019, 11, 15670–15679 CrossRef CAS PubMed
- L. P. L. Gonçalves, D. B. Christensen, M. Meledina, L. M. Salonen, D. Y. Petrovykh, E. Carbó-Argibay, J. P. S. Sousa, O. S. G. P. Soares, M. F. R. Pereira, S. Kegnæs and Y. V. Kolen'ko, Catal. Sci. Technol., 2020, 10, 1991–1995 RSC
- W. Gong, Q. Wu, G. Jiang and G. Li, J. Mater. Chem. A, 2019, 7, 13449–13454 RSC
- H. Zhou, Q. Chen, X. Tong and H. Liu, Colloid Interface Sci. Commun., 2020, 37, 100285 CrossRef CAS
- E. M. Sulman, V. G. Matveeva, V. Y. Doluda, A. I. Sidorov, N. V. Lakina, A. V. Bykov, M. G. Sulman, P. M. Valetsky, L. M. Kustov and O. P. Tkachenko, Appl. Catal., B, 2010, 94, 200–210 CrossRef CAS
- C. Xu, M. Hu, Q. Wang, G. Fan, Y. Wang, Y. Zhang, D. Gao and J. Bi, Dalton Trans., 2018, 47, 2561–2567 RSC
- X. Chen, W. Wang, H. Zhu, W. Yang and Y. Ding, Mol. Catal., 2018, 456, 49–56 CrossRef CAS
- Z.-C. Ding, C.-Y. Li, J.-J. Chen, J.-H. Zeng, H.-T. Tang, Y.-J. Ding and Z.-P. Zhan, Adv. Synth. Catal., 2017, 359, 2280–2287 CrossRef CAS
- H. Zhao, G. Yu, M. Yuan, J. Yang, D. Xu and Z. Dong, Nanoscale, 2018, 10, 21466–21474 RSC
- C. Xie, J. Song, H. Wu, Y. Hu, H. Liu, Y. Yang, Z. Zhang, B. Chen and B. Han, Green Chem., 2018, 20, 4655–4661 RSC
- J. Mondal, Q. T. Trinh, A. Jana, W. K. Ng, P. Borah, H. Hirao and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 15307–15319 CrossRef CAS PubMed
- H. Zhong, C. Liu, H. Zhou, Y. Wang and R. Wang, Chem.–Eur. J., 2016, 22, 12533–12541 CrossRef CAS PubMed
- X. Ren, S. Kong, Q. Shu and M. Shu, Chin. J. Chem., 2016, 34, 373–380 CrossRef CAS
- L. Li, H. Zhao and R. Wang, ACS Catal., 2015, 5, 948–955 CrossRef CAS
- X. Zhao, Y. Fu, C. Yao, S. Xu, Y. Shen, Q. Ding, W. Liu, H. Zhang and X. Zhou, ChemCatChem, 2019, 11, 2362–2369 CrossRef CAS
- Y. Gong, H. Zhong, W. Liu, B. Zhang, S. Hu and R. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 776–786 CrossRef CAS PubMed
- R. Peng, S. Li, X. Sun, Q. Ren, L. Chen, M. Fu, J. Wu and D. Ye, Appl. Catal., B, 2018, 220, 462–470 CrossRef CAS
- G. Spezzati, A. D. Benavidez, A. T. DeLaRiva, Y. Su, J. P. Hofmann, S. Asahina, E. J. Olivier, J. H. Neethling, J. T. Miller, A. K. Datye and E. J. M. Hensen, Appl. Catal., B, 2019, 243, 36–46 CrossRef CAS
- H. Ha, S. Yoon, K. An and H. Y. Kim, ACS Catal., 2018, 8, 11491–11501 CrossRef CAS
- P. Lu, B. Qiao, N. Lu, D. C. Hyun, J. Wang, M. J. Kim, J. Liu and Y. Xia, Adv. Funct. Mater., 2015, 25, 4153–4162 CrossRef CAS
- Y. Wei, Q. Wu, J. Xiong, J. Liu and Z. Zhao, Chin. J. Catal., 2018, 39, 606–612 CrossRef CAS
- C. Ao, P. Tian, L. Ouyang, G. Da, X. Xu, J. Xu and Y.-F. Han, Catal. Sci. Technol., 2016, 6, 5060–5068 RSC
- D. Ni, H. Shen, H. Li, Y. Ma and T. Zhai, Appl. Surf. Sci., 2017, 409, 241–249 CrossRef CAS
- C. Chu, D. Huang, Q. Zhu, E. Stavitski, J. A. Spies, Z. Pan, J. Mao, H. L. Xin, C. A. Schmuttenmaer, S. Hu and J.-H. Kim, ACS Catal., 2018, 9, 626–631 CrossRef
- M. Gu, Q. Jia, Y. Zhu, L. Xu and Y. Tang, Chem.–Eur. J., 2020, 26, 15103–15108 CrossRef CAS PubMed
- Q. Sun, N. Wang, J. Yu and J. C. Yu, Adv. Mater., 2018, 30, e1804368 CrossRef
- W. Ren, H. Zhang and C. Cheng, Electrochim. Acta, 2017, 241, 316–322 CrossRef CAS
- X. Han, Y. Deng, J. Liu, J. Lu, C. Zhong and W. Hu, Adv. Energy Mater., 2018, 1800935 CrossRef
- A. Kim, C. Sanchez, B. Haye, C. Boissière, C. Sassoye and D. P. Debecker, ACS Appl. Nano Mater., 2019, 2, 3220–3230 CrossRef CAS
- Z. Yan, L. Fu, X. Zuo and H. Yang, Appl. Catal., B, 2018, 226, 23–30 CrossRef CAS
- W. Zhu, A. Noureddine, J. Y. Howe, J. Guo and C. J. Brinker, Nano Lett., 2019, 19, 1512–1519 CrossRef CAS PubMed
- C.-S. Chen, C. S. Budi, H.-C. Wu, D. Saikia and H.-M. Kao, ACS Catal., 2017, 7, 8367–8381 CrossRef CAS
- X. Guo, Z. Peng, A. Traitangwong, G. Wang, H. Xu, V. Meeyoo, C. Li and S. Zhang, Green Chem., 2018, 20, 4932–4945 RSC
- C. S. Budi, J. R. Deka, D. Saikia, H. M. Kao and Y. C. Yang, J. Hazard. Mater., 2020, 384, 121270 CrossRef CAS PubMed
- M. Piumetti, M. Hussain, D. Fino and N. Russo, Appl. Catal., B, 2015, 165, 158–168 CrossRef CAS
- O. Daoura, G. Fornasieri, M. Boutros, N. El Hassan, P. Beaunier, C. Thomas, M. Selmane, A. Miche, C. Sassoye, O. Ersen, W. Baaziz, P. Massiani, A. Bleuzen and F. Launay, Appl. Catal., B, 2021, 280, 119417 CrossRef CAS
- S. Schünemann, G. Dodekatos and H. Tüysüz, Chem. Mater., 2015, 27, 7743–7750 CrossRef
- D. Shen, L. Chen, J. Yang, R. Zhang, Y. Wei, X. Li, W. Li, Z. Sun, H. Zhu, A. M. Abdullah, A. Al-Enizi, A. A. Elzatahry, F. Zhang and D. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 17450–17459 CrossRef CAS PubMed
- S. Zhang, Y. Qian and W.-S. Ahn, Chin. J. Catal., 2019, 40, 1704–1712 CrossRef
- A. Fihri, D. Cha, M. Bouhrara, N. Almana and V. Polshettiwar, ChemSusChem, 2012, 5, 85–89 CrossRef CAS PubMed
- X. Lan, C. Du, L. Cao, T. She, Y. Li and G. Bai, ACS Appl. Mater. Interfaces, 2018, 10, 38953–38962 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2021 |