
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium nanoparticles inside well-defined unimolecular amphiphilic polymeric nanoreactors: synthesis and biphasic hydrogenation catalysis†
Hui
Wang
a,
Ambra Maria
Fiore
ab,
Christophe
Fliedel
 a,
Eric
Manoury
a,
Karine
Philippot
*a,
Maria Michela
Dell'Anna
b,
Piero
Mastrorilli
b and
Rinaldo
Poli
*ac
a,
Eric
Manoury
a,
Karine
Philippot
*a,
Maria Michela
Dell'Anna
b,
Piero
Mastrorilli
b and
Rinaldo
Poli
*ac
aCNRS, LCC (Laboratoire de Chimie de Coordination), Université de Toulouse, UPS, INPT, 205 route de Narbonne, BP 44099, F-31077 Toulouse Cedex 4, France. E-mail: rinaldo.poli@lcc-toulouse.fr
bDICATECh, Politecnico di Bari, via Orabona, 4, 70125 Bari, Italy
cInstitut Universitaire de France, 1 rue Descartes, 75231 Paris Cedex 05, France
First published on 18th March 2021
Abstract
Rhodium nanoparticles (Rh NPs) embedded in different amphiphilic core-crosslinked micelle (CCM) latexes (RhNP@CCM) have been synthesized by [RhCl(COD)(TPP@CCM)] reduction with H2 (TPP@CCM = core-anchored triphenylphosphine). The reduction rate depends on temperature, on the presence of base (NEt3) and on the P/Rh ratio. For CCMs with outer shells made of neutral P(MAA-co-PEOMA) copolymer chains (RhNP@CCM-N), the core-generated Rh NPs tend to migrate toward the hydrophilic shell and to agglomerate depending on the P/Rh ratio and core TPP density, whereas the MAA protonation state has a negligible effect. Conversely, CCMs with outer shells made of polycationic P(4VPMe+I−) chains (RhNP@CCM-C) maintain core-confined and well dispersed Rh NPs. All RhNP@CCMs were used as catalytic nanoreactors under aqueous biphasic conditions for acetophenone, styrene and 1-octene hydrogenation. Styrene was efficiently hydrogenated by all systems with high selectivity for vinyl reduction. For acetophenone, competition between benzene ring and carbonyl reduction was observed as well as a limited access to the catalytic sites when using CCM-C. Neat 1-octene was also converted, but the activity increased when the substrate was diluted in 1-nonanol, which is a better core-swelling solvent. Whereas the molecular RhI center was more active than the Rh0 NPs in 1-octene hydrogenation, the opposite trend was observed for styrene hydrogenation. Although Rh NP migration and agglomeration occurred for RhNP@CCM-N, even at high P/Rh, the NPs remained core-confined for RhNP@CCM-C, but only when toluene rather than diethyl ether was used for product extraction before recycling.
Introduction
Although metal nanoparticles (NPs) have been known for a long time, their controlled generation has attracted keen interest only recently,1–4 because of growing awareness that their characteristics such as size and morphology strongly influence their physical and chemical properties. Much effort is currently devoted to the synthesis of very precisely defined metal nanospecies, up to the atomic precision level.5–7 In addition to fundamental aspects, the particular properties that the metal NPs display relative to bulk metals and molecular complexes make them very attractive for applications in diverse domains, particularly in catalysis. Nanocatalysis is now a well-recognized discipline at the frontier between homogeneous and heterogeneous catalyses.8–19 Metal NPs are highly attractive because of their high surface/volume ratio, especially for diameters of one nanometer or below (subnanoparticles), thus providing a high number of potential active sites (>90% of surface atoms). Therefore, developing synthetic tools that enable the production of ultra-small NPs is of prime importance. In terms of catalytic performance, in addition to the metal nature and particle size, other important parameters are the crystalline structure, the nature and relative amounts of the exposed faces, edges and corners and the composition and architecture (e.g. core–shell) for multimetallic NPs. However, the performance in catalysis may also be influenced or even oriented by the surrounding stabilizer or by the support.20–22The choice of the stabilizing agent is critical as it controls both the NP size and dispersion and provides long-term stability during the catalytic process.23–26 Contrarily to heterogeneous catalysis where calcination is usual to suppress organic contaminants and liberate the active sites, but like in molecular catalysis, metal–ligand interactions are of paramount importance23,24 as they may improve the activity or even promote more interesting chemoselectivities.25 The challenge is to find capping ligands that at the same time stabilize the metal NPs and allow access to the metal surface for the catalytic transformation.25,26 Ligand-stabilized metal NPs may be involved in catalysis as colloidal suspensions in water, polyols or organic solvents and several strategies have been developed to facilitate the catalyst recovery.27
Among the available strategies for catalyst recovery and recycling, aqueous biphasic catalysis is particularly attractive when the catalyst can be confined in the aqueous phase, because the organic reaction products can be easily separated by decantation. However, the reaction may be severely limited by mass transport if the catalyst is totally insoluble in the organic reactant/product phase and if the reactants are totally insoluble in water, limiting the productive process to the organic/water interface. In this respect, the use of surfactants or micellar systems able to increase the organic/water interphase has been shown beneficial.28–32 Increased rates have indeed been obtained upon anchoring catalytic NPs on the hydrophilic shell of micelles.33–47 Thermoregulated processes,48,49 where the catalyst is anchored on thermosensitive macromolecules (hydrophilic at low temperature and hydrophobic at high temperature) have also been implemented in metal NP catalysis.50–52 An alternative solution is to anchor the catalyst to the hydrophobic core of micelles. This allows the reaction to take place with high local concentrations of both catalyst and substrates and each individual micelle operates as an independent nanoreactor. This strategy has been implemented for a few molecular catalysts,53–67 but examples with metallic nanoparticles are not available to the best of our knowledge. One limitation of self-assembled micelles is the disaggregation equilibrium with the individual amphiphilic molecular components, resulting in catalyst losses. However, this phenomenon may be blocked by making the object unimolecular by crosslinking. In this respect, dendrimers such as poly(amidoamine) (PAMAM) and poly(propylene imine) (PPI) have been used as unimolecular nanoreactors to embed catalytic NPs of various metals.68–70 However, one limiting factor for the application of dendrimers is their multistep synthesis, which may be rather time consuming, particularly for the higher generations.
Some of us have recently developed unimolecular amphiphilic star polymers with a triphenylphosphine-functionalized polystyrene core and a hydrophilic shell, thus ensuring a stable aqueous dispersion (latex).71–74 We have therefore named this polymeric architecture core-crosslinked micelles (CCM). These polymers, the general structure of which is shown in Fig. 1, were obtained by a convergent polymerisation procedure, using the reversible addition–fragmentation chain-transfer (RAFT) strategy. The procedure is particularly straightforward (three-step-one-pot) for the polymers with the neutral P(MAA-co-PEOMA) outer shell (CCM-N). The first step involved polymerisation of the water-soluble monomers to yield hydrophilic individual chains corresponding to the outer blocks of the final particles. These were extended with a hydrophobic block in the second step, which involved polymerisation-induced self-assembly (PISA) and yielded micelles. The triphenylphosphine (TPP) ligand was introduced during this step via copolymerisation of 4-diphenylphosphinostyrene (DPPS) as a comonomer of styrene. Finally, the third step linked all the micellar chains together at the core level by further chain extension with a crosslinking monomer (diethylene glycol dimethacrylate – DEGDMA).71 The synthesis of the polymer with a cationic P4VPMe+I− shell (CCM-C) presented a few obstacles, which required the introduction of additional steps.74 Anyhow, for both types of polymers, the good control ensured by the RAFT polymerisation method led to spherical polymer particles with quite narrow size distributions and average diameters in the 100–150 Å range. Versions of these polymers with a different TPP content, for both CCM-N and CCM-C, could be produced by using styrene–DPPS comonomer mixtures during the hydrophobic block chain growth (5–25% molar ratio of DPPS). Variants of this polymeric scaffold (different core ligand75,76 or polymer architecture77) have also been synthesized, but the most intensively investigated polymers have so far been those with the CCM architecture and core-anchored TPP ligands and the present contribution also focuses on the use of these polymers. They will be referred as CCM-N-x and CCM-C-x, where N and C refer to the type of hydrophilic shell (neutral and cationic, respectively) and x stands for the molar fraction of ligand-functionalized styrene in the hydrophobic core.
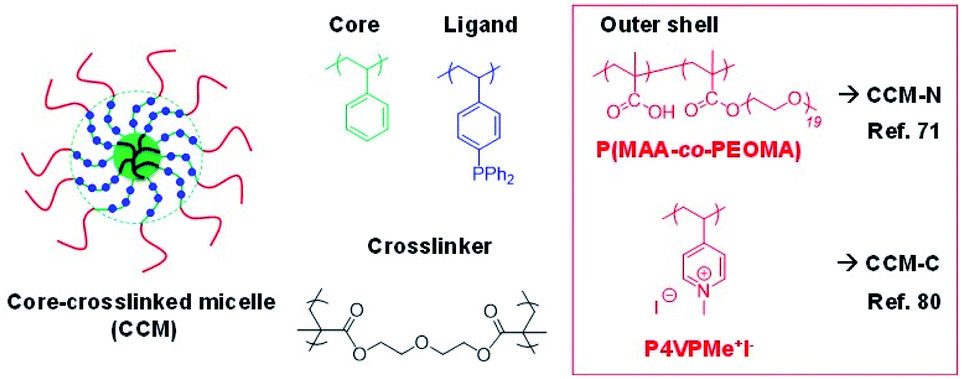 | ||
| Fig. 1 General structure of the triphenylphosphine-functionalized core-crosslinked micelles (TPP@CCM) with either a neutral or a polycationic outer shell. | ||
In our previous work, all these polymers were charged with either [Rh(acac)(CO)2] (acac = acetylacetonate) or [RhCl(COD)]2 (COD = 1,5-cyclooctadiene), which coordinated to the core-anchored triphenylphosphine ligands (referred to as TPP@CCM) by either CO replacement or Cl-bridge splitting to yield [Rh(acac)(CO)(TPP@CCM)] or [RhCl(COD)(TPP@CCM)], respectively. The resulting metal-loaded polymers were subsequently used in Rh-catalysed aqueous biphasic olefin hydroformylation71–73,75–78 or hydrogenation,79,80 showing excellent performance and recyclability with sub-ppm catalyst losses. In further exploratory investigations, the [RhCl(COD)(TPP@CCM)] precatalyst was also applied to the hydrogenation of acetophenone. However, contrarily to the observed behaviour in styrene hydrogenation, the catalytic mixture unexpectedly turned black, suggesting that the molecular Rh precatalyst was reduced to the metallic state under these conditions, with the possible formation of metal NPs. We reasoned that, in a phosphine-poor environment (P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Rh ratios of 1:1 or 2:1 were initially used), styrene might protect the RhI centre from reduction by H2 because of its π-acidity as a ligand, contrarily to acetophenone. We report herein a systematic investigation of the [RhCl(COD)(TPP@CCM)] reduction and of the effect of various parameters on the size and morphology of the produced Rh NPs. Finally, the performance of the resulting catalytic nanoreactors in acetophenone, styrene and 1-octene hydrogenation will be described, also in terms of catalyst stability and recycling, providing useful new information about the Rh NP stabilisation and mobility in the amphiphilic polymer environment.
:Rh ratios of 1:1 or 2:1 were initially used), styrene might protect the RhI centre from reduction by H2 because of its π-acidity as a ligand, contrarily to acetophenone. We report herein a systematic investigation of the [RhCl(COD)(TPP@CCM)] reduction and of the effect of various parameters on the size and morphology of the produced Rh NPs. Finally, the performance of the resulting catalytic nanoreactors in acetophenone, styrene and 1-octene hydrogenation will be described, also in terms of catalyst stability and recycling, providing useful new information about the Rh NP stabilisation and mobility in the amphiphilic polymer environment.
Results and discussion
Generation of rhodium nanoparticles in the CCM-N polymers
A first application of the [RhCl(COD)]2-loaded CCM-N latex81 to the aqueous biphasic catalysed hydrogenation of acetophenone (90 °C, 20 bar of H2) led to an unexpected colour change of the initially pale cream latex to black, suggesting metal reduction, and to a low extent of substrate reduction (see catalytic results below). On the other hand, previous work had demonstrated very efficient aqueous biphasic styrene or 1-octene hydrogenation with no colour change using the same protocol.79 Metallic rhodium, in the form of small NPs, has previously been obtained by reduction of several molecular RhI and RhIII precursors,82 including [RhCl(COD)]2.83–85 A black latex was again obtained when neat toluene, without any added acetophenone, was used to swell the polymer core, showing that acetophenone is not essential for the metal reduction. When the [RhCl(COD)(TPP@CCM)] reduction was carried out at 25 °C (either with or without acetophenone), the latex only turned light grey, suggesting that the reduction may be incomplete under these conditions. However, a black latex was again obtained at 60 °C. Reasoning that the reduction of a RhI–Cl complex by H2 also generates an equivalent amount of HCl per Rh atom, the procedure was then repeated in the presence of excess NEt3 (≥5 equiv.). Under these conditions, a black latex was obtained even at 25 °C. These initial studies were carried out with a fully metal-loaded (P/Rh = 1:1) CCM-N-0.1 latex, in which each polymer chain contains on average 30 TPP ligands in the hydrophobic PS block and 15 PEOMA/15 MAA monomers in the hydrophilic P(MAA-co-PEOMA) block. Therefore, the PEOMA/Rh ratio is ca. 0.5. It should also be pointed out that the NEt3 excess leads to the transformation of the neutral shell into an anionic one, containing triethylammonium carboxylate functions, –COO−NHEt3+. The amount of NEt3 used is sufficient for the neutralisation of all generated HCl and all the shell carboxylic functions.
The transmission electron microscopy (TEM) analyses of the recovered latexes, Fig. 2(a–c), besides confirming the formation of metal NPs, highlighted a few interesting phenomena. While the grey latex obtained in the absence of base contains isolated small size (<5 nm) NPs, see Fig. 2(a), the black latex obtained in the presence of base contains few individual small NPs together with a dominant fraction of NP agglomerates, Fig. 2(b). These agglomerates appear to accumulate mostly on the polymer particle surface (hydrophilic shell). A similar behaviour is observed for the NPs formed in the absence of base at 60 °C, Fig. 2(c), with an even more evident location of the agglomerates on the CCM outer shell. Thus, the protonation state of the shell MAA monomer does not appear to greatly affect neither the aggregation phenomenon nor the preference of the aggregates for the polymer shells. This suggests that the aggregated NPs are mostly stabilized by the shell PEO chains. Indeed, although phosphine ligands have been used as stabilizers of Rh NPs, e.g. using [Rh(acac)(COD)] and [Rh(η6-C3H5)3] as precursors,86–89 PEO has also been described as a stabilizer for the generation of Rh NPs.90–92
 | ||
| Fig. 2 TEM images of CCM-N-x polymer latexes (x = 0.1, (a)–(c); 0.05, (d) and (e)) after loading with [RhCl(COD)]2 (P/Rh = 1:1) and treatment with H2 (20 bar) under different conditions for 20 h. | ||
In separate experiments, the Rh NPs were also generated at 25 °C, both in the absence and presence of NEt3, using the fully loaded CCM-N-0.05, all other conditions being the same (H2 pressure, NEt3/Rh ratio, reaction time). The behaviour was qualitatively identical: grey and black latexes in the absence and presence of base, respectively. The TEM images of these products are shown in Fig. 2(d) and (e), respectively. The former looks rather similar to the RhNP@(CCM-N-0.1) latex obtained under the same conditions, with dispersed individual Rh NPs and no aggregates. The latter shows a few large NP aggregates located near a few polymer particles, while most other polymer particles are NP-free and there are no visible individual NPs. Fig. 2(e) also shows a few Rh NP agglomerates located outside of the CCM particles, most probably resulting from mechanical detachment from the CCM outer shells during the preparation of the TEM grid. Since the CCM-N-0.05 particles contain on average only 15 TPP functions per chain, these are located farther from each other and the PEO/TPP ratio is twice that of the CCM-N-0.1 particles. The observed trends suggest that while the Rh NPs start to form as small individual particles in the CCM core, the core TPP and the shell PEO functions subsequently compete under the influence of the PEO/TPP ratio. For comparison, we have generated Rh NPs by reduction of [RhCl(COD)]2 toluene solutions in the presence of either the PEOMA monomer or the macroRAFT chains R0-MAA15-co-PEOMA15-SC(S)SPr (macroRAFT-N), at various PEO/Rh ratios. The macroRAFT-N polymer, where R0-SC(S)SPr is the CTPPA trithiocarbonate used as RAFT agent for the polymer synthesis (see Experimental Procedures in the ESI†), is the intermediate obtained after the first step of the CCM-N synthesis as pointed out in the Introduction. The TEM images reveal particle agglomerates very much like those of Fig. 2(c) and (e), see ESI, Fig. S1.† In particular, at equivalent PEO/Rh ratios, stabilisation by the macroRAFT chains produces smaller agglomerates than the free PEOMA monomer, and the agglomerates are smaller when using a greater PEO/Rh ratio, as may be expected. The results of the experiments with the CCM in Fig. 2 also indicate mobility for the Rh NPs, with migration from the core to the shell and from one polymer particle to another. In previous work, we demonstrated that the molecular RhI complex can rapidly migrate between different particle cores via reversible interpenetration with core–core contact, in combination with phosphine exchange reactions.93 This principle can therefore be extended to the metallic NPs.
When using an incompletely Rh-loaded CCM-N latex (P/Rh = 4), no RhI reduction occurred at 25 °C, even in the presence of excess NEt3. This suggests that the excess phosphine ligand exerts a protective action against reduction to the metallic state, like the styrene and 1-octene substrates in our previous catalysed hydrogenation study,79 and confirms the principle that only a coordinatively unsaturated RhI centre, such as that obtained from [RhCl(COD)(TPP@CCM)] after removal of the COD ligand by hydrogenation in the absence of additional TPP, may be readily reduced by H2. When the same procedure was carried out at 60 °C, however, Rh NPs were once again generated. In the absence of base, NPs formed only upon warming to 90 °C. In this case, the TEM characterisation shows small NPs in all polymer particles, although they appear to be located mostly near the surface of the polymer particles rather than homogeneously dispersed in the core, see Fig. S2.† Therefore, the NPs obtained under these conditions have either reduced mobility or increased thermodynamic stability relative to the particles stabilized by the shell PEO chains. The same comparative experiments with P/Rh ratios of 1:1 and 4:1 were also carried out for the [RhCl(COD)]2-loaded CCM-N-0.2 latex (for which TPP/PEO = 4), in the presence of NEt3, yielding similar results. Large agglomerates, even larger than those obtained with the CCM-N-0.1 and 0.05 latexes, were produced when using a 1:1 P/Rh ratio at 25 °C (Fig. S3(a)†), whereas using a 4:1 ratio at 60 °C led to better dispersed and very small NPs, Fig. S3(b).† These results indicate that the Rh NP migration is strongly affected by the P/Rh ratio but not by the PEO/Rh ratio.
For comparison, Rh NPs were also generated by [RhCl(COD)]2 reduction from a homogeneous toluene solution in the presence of PPh3, using P/Rh ratio of 1 and 4, and in the presence of ≥5 equiv. of NEt3. The reduction rate followed the same trend as observed for the RhNP@CCM-N synthesis: rapid at 25 °C for a P/Rh ratio of 1:1 and no reduction at all for a 4:1 ratio, but the latter mixture yielded NPs at 60 °C. The RhNP@PPh3 obtained at 60 °C with P/Rh = 4 are significantly smaller, more narrowly dispersed, and less aggregated than those obtained at 25 °C with P/Rh = 1, see Fig. S4.† Their size is quite similar to RhNP@PPh3 previously obtained from [Rh(η3-C3H5)3].86,87 Additional control experiments carried out in the absence of H2 and in the presence of 10 equiv. of NEt3 per Rh showed no colour change over 20 h at 60 °C, whether the P/Rh ratio is 1 or 4, indicating that the amine does not act as a reducing agent for the RhI complex and that H2 is essential to accomplish the Rh NP formation. The conclusions to be drawn from these investigations are that using a low P/Rh ratio (or no phosphine at all) leads to agglomerated Rh NPs, whereas higher P/Rh ratios yield better dispersed ones. For the experiments with the Rh-loaded CCM polymers, the lower P/Rh ratio leads to extensive NP migration and accumulation as aggregates in the PEO-rich areas.
Generation of rhodium nanoparticles in the CCM-C polymers
The formation of Rh NPs has also been investigated using the cationic-shell CCM particles (CCM-C) as stabilizing matrix. The charged nature of the shell in CCM-C (see Fig. 1) stops the interpenetration of the polymer particles and the interparticle migration of the molecular RhI complexes.80 Hence, we anticipated that the Rh NP migration may also be stopped. In addition, the chemical nature of the CCM-C outer shell is not expected to strongly stabilize metal NPs, although we cannot discard a potential role of the iodide counterions.Under the same conditions (P/Rh ratios, temperature, H2 pressure, base and reaction time) the Rh NP generation in the CCM-C-0.1 latex followed the same reactivity trend as in the CCM-N latexes: reduction at 25 °C when P/Rh = 1 and only upon warming to 60 °C when P/Rh = 4. However, as anticipated, the Rh NPs remained confined in all cases within the polymer core, see Fig. 3. The amount of used base (0, 1 or 5 equiv. per Rh for P/Rh = 1) did not affect the NP morphology or their dispersion within the polymer particles, see Fig. S5.† In order to evaluate the potential of the outer shell as a stabilizer for the generation of Rh NPs, the reduction of [RhCl(COD)]2 was also carried out in the presence of the amphiphilic diblock macroRAFT agent R0-(4VPMe+I−)140-b-S50-SC(S)SPr (macroRAFT-C), using MeOH as a solvent and equivalent amounts (4VPMe+I−/Rh ratios) to those of the syntheses with the CCM-C polymer. This macroRAFT-C polymer is an intermediate of the CCM-C synthesis.74,80 The resulting Rh NPs are highly agglomerated with agglomerate sizes that are essentially independent on the macroRAFT-C/Rh ratio, in the 10–50 nm range (Fig. S6†).
 | ||
| Fig. 3 TEM images of the CCM-C-0.1 polymer latex after loading with [RhCl(COD)]2 and reduction with H2 (20 bar) in the presence of NEt3 (5 equiv. per Rh) for 20 h. (a) P/Rh = 1:1, 25 °C. (b) P/Rh = 4:1, 60 °C. | ||
Catalysed hydrogenation of acetophenone
The TEM characterisation of the RhNP@CCM shows that the P/Rh ratio and the type of CCM shell (neutral, cationic) affect the NP location (core vs. shell), whereas the presence or absence of NEt3 does not appear to induce significant changes on the synthesized NP morphology and location. The NPs are stabilized by either core-anchored TPP for the CCM-N nanoreactors at high P/Rh ratios and for the CCM-C nanoreactors under any conditions, or by the shell PEO chains for the CCM-N nanoreactors at low P/Rh ratios. Therefore, in order to properly evaluate the catalytic performance of the RhNP@CCM systems, control experiments were run with related Rh NPs, namely RhNP@PPh3, RhNP@PEOMA, RhNP@macroRAFT-N and RhNP@macroRAFT-C, generated in the presence of the corresponding stabilizers under homogeneous conditions. The catalytic experiments with the RhNP@CCM latexes were carried out under aqueous biphasic conditions using the ‘as synthesized’ latex, diluted with water, as catalyst phase and toluene as the organic carrier phase. The control runs with RhNP@PPh3, RhNP@PEOMA, RhNP@macroRAFT-N were carried out in toluene without any water phase, whereas methanol was used as a compatible solvent for the control runs with RhNP@macroRAFT-C. All results are collected in Table 1.| Entry | NP stabilizer | P/Rh | PEO/Rh | 4VPMe+I−/Rh | T/°C | Conv.c/% | TON 2c | TON 3c | TON 4c | TON MeCy c |
|---|---|---|---|---|---|---|---|---|---|---|
| a Unless otherwise stated, the Rh NPs were synthesized at 60 °C in the presence of NEt3 (10 equiv. per Rh) and the indicated support prior to catalysis. b Standard conditions: acetophenone/Rh = 200; 0.4 mL of latex, 0.5 mL of toluene; p(H2) = 20 bar; 20 h. c The figures are averages, with standard deviations in parentheses, when multiple runs were carried out. d 8.07 μmol of Rh. e 1.70 μmol of Rh. f Average and standard deviation from 3 parallel runs. g Average and standard deviation from 5 parallel runs. h Average and standard deviation from 4 parallel runs. i No NEt3 was used in the NP synthesis. j The volatility of methylcyclohexane led to escape of the product from the reaction vials and prevented a reliable measurement of its amount at the end of the reaction. k 5.09 μmol of Rh. l 1.29 μmol of Rh. | ||||||||||
| 1 | CCM-N-0.1 | 1d | 0.5 | — | 25 | 21.0 | 31.0 | 9.5 | 2.0 | 22.5 |
| 2 | 60 | 41.4 | 61.8 | 13.4 | 7.0 | 9.0 | ||||
| 3 | 90 | 100.0 | 162.1 | 16.9 | 29.1 | |||||
| 4 | 4e | 2 | — | 25 | 43.5 | 63.2 | 20.8 | 8.6 | 7.9 | |
| 5 | 60 | 100.0 | 118.6 | 33.5 | 63.8 | 44.6 | ||||
| 6 | 90 | 94.8 | 150.5 | 24.5 | 19.7 | |||||
| 7 | PPh3 | 1 | — | — | 60f | 97.2(3.7) | 146.4(7.9) | 7.6(3.1) | 44.3(13.4) | 23.9(7.7) |
| 8 | 4 | — | 60g | 78.8(6.8) | 153.8(13.2) | 0.5(0.8) | 1.3(2.1) | 2.8(0.2) | ||
| 9 | PEOMA | — | 0.5 | — | 60f | 93.6(0.5) | 75.3(47.4) | 7.9(6.5) | 105.6(54.7) | 4.3(0.9) |
| 10 | 2 | 60h | 93.6(4.7) | 152.3(11.9) | 17.5(4.5) | 19.5(2.4) | 29.5(14.0) | |||
| 11 | 8 | 60h | 96.0(0.8) | 109.9(26.7) | 21.1(4.0) | 33.0(10.8) | 3.4(0.1) | |||
| 12 | macroRAFT-N | — | 0.5 | — | 60g | 98.8(1.5) | 93.0(22.0) | 42.4(2.5) | 62.5(25.6) | 10.7(3.5) |
| 13 | 2 | 60g | 98.8(0.1) | 107.2(19.4) | 34.1(8.7) | 58.9(11.2) | 2.4(0.4) | |||
| 14 | 0.5 | 60h,i | 97.1(0.3) | 11.2(4.0) | 49.6(2.1) | 133.3(3.5) | 21.6(4.3) | |||
| 15 | 2 | 60h,i | 96.4(1.0) | 25.0(6.6) | 59.1(13.8) | 109.7(4.8) | 11.7(2.4) | |||
| 16 | CCM-C-0.1 | 1k | — | 4.7 | 25 | 12.9 | 10.2 | 4.2 | 0.0 | 1.0 |
| 17 | 60 | 9.1 | 14.0 | 4.1 | 0.0 | 60.5 | ||||
| 18 | 90 | 86.5 | 154.0 | 9.2 | 7.2 | |||||
| 19 | 4l | — | 18.7 | 25 | 5.0 | 11.5 | 0.0 | 0.0 | 10.3 | |
| 20 | 60 | 18.0 | 25.3 | 10.6 | 0.0 | 21.1 | ||||
| 21 | 90 | 23.5 | 49.8 | 0.0 | 0.0 | |||||
| 22 | macroRAFT-C | — | — | 4.7 | 60f | 20.9(6.5) | 32.0(10.0) | 6.9(1.9) | 3.2(1.5) | 2.0(0.1) |
| 23 | 18.8 | 60h | 12.5(6.9) | 17.0(7.4) | 3.8(3.5) | 2.5(1.8) | 2.0(0.2) | |||
The partial reduction products 1-phenylethanol (2, carbonyl reduction) and methyl cyclohexyl ketone (3, arene ring reduction) and the fully reduced 1-cyclohexylethanol (4) were observed in variable proportions, depending on the stabilizer and conditions (Scheme 1). A certain amount of methylcyclohexane (MeCy), produced by the ring reduction of the toluene solvent, was also observed (see Table 1). The arene ring reduction is not surprising, because arene hydrogenation is well-known to be promoted by metal NPs, particularly those of Rh.83,84,94–102 A most relevant precedent is the reported biphasic acetophenone (and other functionalized arenes) hydrogenation using buffered water, benzene or cyclohexane as organic phase, and cetyltrimethylammonium bromide or tetrabutylammonium hydrogen sulphate as phase transfer catalyst, which also produced mixtures of all the possible products 2–4.103 Although the authors of that contribution wrote “we are not certain whether the phase-transfer process described herein involves a soluble or insoluble rhodium catalyst”, their molecular precursor ([RhCl(1,5-hexadiene)]2) and catalytic conditions were quite similar to those used here. The acetophenone hydrogenation catalysed by Rh NPs has previously been reported for NPs stabilized by polyvinylpyrrolidone (RhNP@PVP)104 and by phosphine ligands, including PPh3.86 There are also a few reports on the Rh NP-catalysed transfer hydrogenation of acetophenone by isopropanol, focusing on enantioselectivity, where no ring hydrogenation was mentioned.105–107
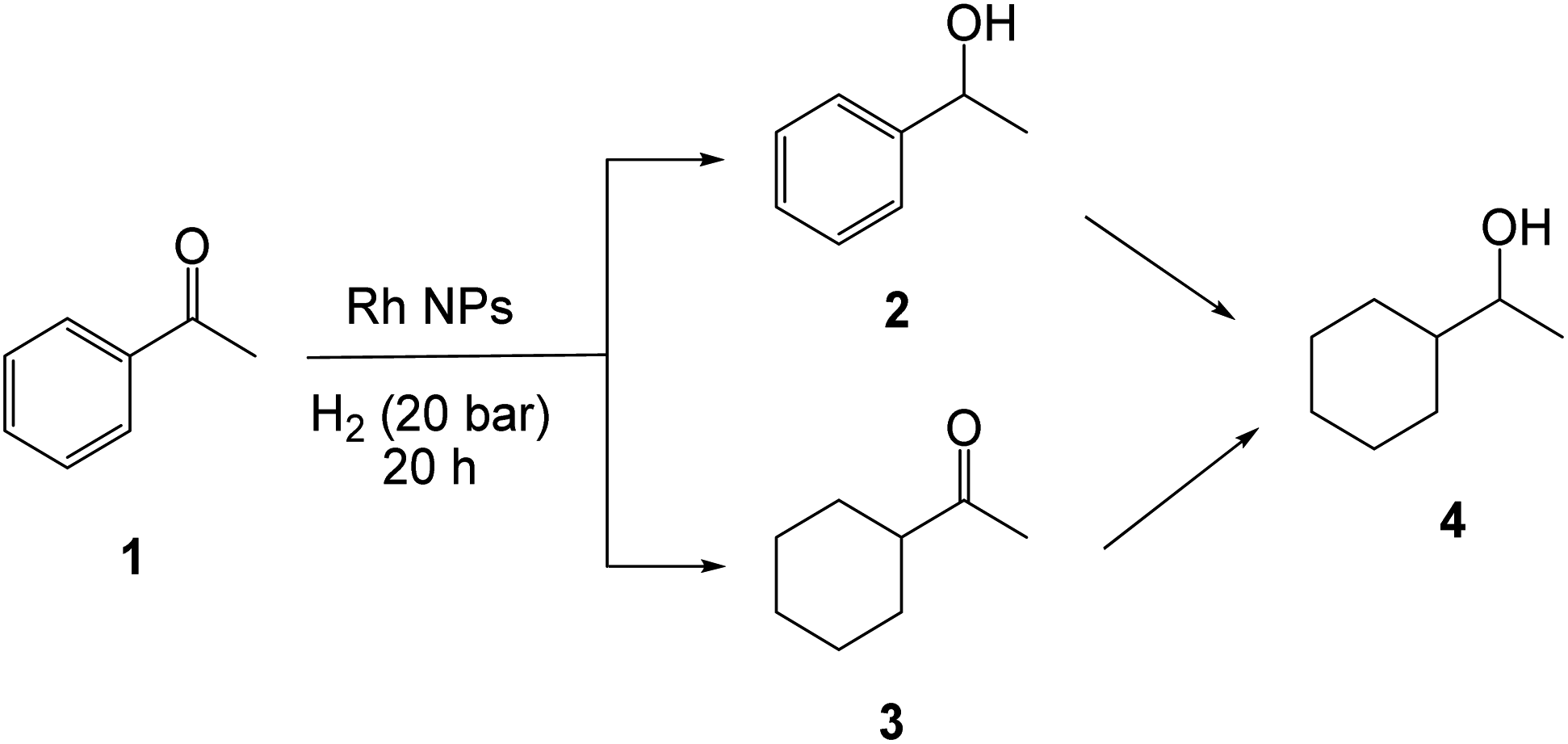 | ||
| Scheme 1 Products resulting from the hydrogenation of acetophenone catalysed by Rh NPs. | ||
For the RhNP@CCM-N-0.1 catalyst, the acetophenone reduction appears faster for P/Rh = 4 (cf. entries 4–6 with 1–3). In particular, complete substrate consumption was already achieved at 60 °C (entry 5) but only at 90 °C when P/Rh = 1. It is important to underline that, due to the uncontrolled and unreliable magnetic stirring, the conversion data (sum of all products = consumed acetophenone) for certain runs may be artificially low and should therefore be considered as a lower limit. Carbonyl reduction is faster than the arene reduction (2 > 3). This trend is opposite to that found for the above-mentioned [RhCl(1,5-hexadiene)]2-catalysed reduction under phase transfer conditions.103 In the two previous reports on the hydrogenation of acetophenone with Rh NPs in a single liquid phase, similar selectivity but greater activity were observed, e.g. up to 78 turnovers in only 5 h at 30 °C and 20 bar of H2 for the PPh3-stabilized particles86 and 71 turnovers in 2 h at 25 °C and 1 bar of H2 for the PVP-stabilized particles.104 The lower activity observed for the RhNP@CCM catalyst can be attributed, at least in part, to mass transport limitations.
All control experiments with the RhNP@PPh3, RhNP@PEOMA and RhNP@macroRAFT-N catalysts were carried out only at 60 °C. The data reported in Table 1 are averages of parallel runs with standard deviations. In all cases (entries 7–15), the observed conversions were greater (lower residual 1) than for RhNP@CCM-N-0.1 with P/Rh = 1 (entry 2) but lower than for RhNP@CCM-N-0.1 with P/Rh = 4 (entry 5). A most interesting comparison concerns the selectivity (carbonyl vs. arene ring reduction). The phosphine stabilizer (RhNP@PPh3) suppresses arene reduction, particularly for the smaller NPs obtained with P/Rh = 4 (entry 8), for which the ring reduction products 3 and 4 were obtained in very small amounts. The NPs stabilized by PEO functions (RhNP@PEOMA and RhNP@macroRAFT-N), on the other hand, gave more extensive arene reduction. The PEO/Rh ratio (from 0.5 to 8 for RhNP@PEOMA, entries 9–11; from 0.5 to 2 for RhNP@macroRAFT-N, entries 12–13) does not appear to significantly alter the activity and selectivity. The absence of NEt3 for RhNP@macroRAFT-N (hence leaving the methacrylic acid functions protonated, entries 14–15) slightly improves the arene ring reduction (3 > 2; increase of 4). These selectivities are similar to those reported in the previous studies.86,104
For the RhNP@CCM-N catalyst with P/Rh = 1, the most interesting comparison is between entries 2, 9 and 12, because the NPs in this catalyst (Fig. 2) are located near the PEO surface functions. The control catalysts (RhNP@PEOMA and RhNP@macroRAFT-N) appear more active and yield a greater fraction of arene reduction products (3 and 4). For the RhNP@CCM-N catalyst with P/Rh = 4, where the Rh NPs remain confined in the hydrophobic core, the most interesting comparison is between entries 5 and 8. While the activity compares favorably, the large selectivity difference (large amounts of arene reduction products for entry 5, traces in entry 8) suggests a different catalyst organisation, raising questions about the stability of the core confinement in RhNP@CCM-N. TEM images for this catalyst recorded after catalysis revealed large NP aggregates outside of the CCM particles, Fig. 4. Given this result, we can conclude that the CCM-N scaffold is not an appropriate support for the Rh NP confinement under these catalysis conditions.
 | ||
| Fig. 4 TEM images of the RhNP@CCM-N-0.1 (P/Rh = 4) latex before (a) and after the catalytic runs of entry 4 (b) and 6 (c) (Table 1). | ||
Moving to the cationic CCM latex (RhNP@CCM-C, entries 16–21), the substrate conversions were very poor, except at the highest temperature for P/Rh = 1 (entry 18). These results may indicate poor mass transport of the substrate towards the CCM core and suggest that the Rh NPs remain trapped within the CCM core under catalytic conditions, contrary to the RhNP@CCM-N latex examined above. A TEM analysis of the latex after catalysis (Fig. 5) confirms this conclusion. These results may seem puzzling, because both CCM-N and CCM-C previously showed excellent performance in olefin hydrogenation (notably styrene and 1-octene) by molecular RhI@CCM, which is confined within the CCM core.79,80 Indeed, the CCM-C nanoreactors performed equally well (or better) than CCM-N. Since toluene is a good solvent for polystyrene and efficiently swells the CCM core, it can vectorize substrates toward the catalyst, even those like 1-octene that do not have themselves high affinity for polystyrene. Control experiments run with RhNP@macroRAFT-C (entries 22–23) gave equally poor conversions. In this catalyst, the Rh NPs are possibly stabilized by interaction with the iodide anions associated to the polycationic P4VPMe+ chains. Therefore, the only possible way to rationalize these results is that the polycationic nature of the CCM-C and macroRAFT-C, although not affecting the mass transport of the organic solvent (toluene), have a negative effect on the mass transport of the acetophenone substrate, perhaps as a consequence of electrostatic interactions between the 4VPMe+I− functions and the substrate carbonyl group. This hypothesis is supported by the results obtained for the hydrogenation of styrene (vide infra).
 | ||
| Fig. 5 TEM image of the RhNP@CCM-C-0.1 (P/Rh = 4) latex after the catalytic run of entry 19 (Table 1). | ||
Catalysed hydrogenation of styrene
These reactions were carried out under conditions identical to those of the acetophenone hydrogenation (notably, styrene/Rh = 200), except that 1-nonanol was used as the continuous organic phase instead of toluene, for two reasons. One is to eliminate the competitive ring hydrogenation between styrene (Scheme 2) and the organic solvent. The second one is to compare the results with those of the previously published molecular hydrogenations, where 1-nonanol was also used as continuous organic phase.79,80 The Rh NP-catalysed hydrogenation of styrene has only been addressed in one recent report, with NPs stabilized by a phosphine ligand.89 Additional contributions have addressed the hydrogenation of other arenes and aromatic heterocycles with Rh NPs stabilized by PEO,90,92 cyclic iminium salts108 or phosphines.86,109 All our new results are collected in Table 2.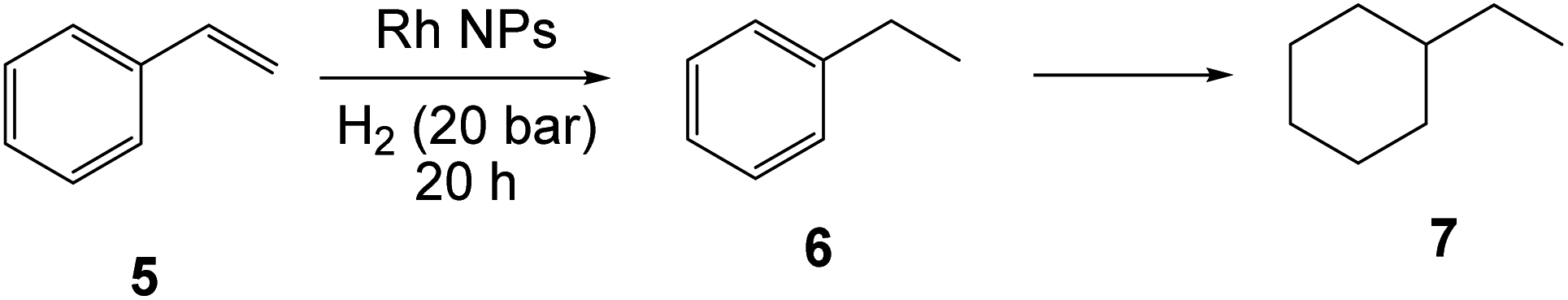 | ||
| Scheme 2 Products resulting from the hydrogenation of styrene catalysed by Rh NPs. | ||
| Entry | NP stabilizer | P/Rh | PEO/Rh | 4VPMe+I−/Rh | Styrene/Rh | T/°C | 5 /% | 6 /% | 7 /% |
|---|---|---|---|---|---|---|---|---|---|
| a Unless otherwise stated, the Rh NPs were synthesized at 60 °C in the presence of NEt3 (10 equiv. per Rh) and the indicated support prior to catalysis. b Standard conditions: 0.4 mL of latex, 0.5 mL of 1-nonanol; p(H2) = 20 bar; 20 h. c The figures are averages, with standard deviations in parentheses, when multiple runs were carried out. d 8.07 μmol of Rh. e 1.70 μmol of Rh. f Average and standard deviation from 5 parallel runs. g Average and standard deviation from 4 parallel runs. h Pure styrene was used as organic phase. i No NEt3 was used in the NP synthesis. j 5.09 μmol of Rh. k 1.29 μmol of Rh. l Average and standard deviation from 3 parallel runs. | |||||||||
| 24 | CCM-N-0.1 | 1d | 0.5 | — | 200 | 25 | 0.1 | 99.3 | 0.6 |
| 25 | 60 | 0.1 | 95.9 | 4.0 | |||||
| 26 | 4e | 2 | — | 200 | 25 | 0 | 100 | 0 | |
| 27 | 60 | 0 | 97.0 | 3.0 | |||||
| 28 | PPh3 | 1 | — | — | 200 | 60f | 0(0) | 72.7(2.4) | 27.3(2.4) |
| 29 | 4 | 60g | 0(0) | 93.1(1.4) | 6.9(1.4) | ||||
| 30 | 4 | 2000h | 60f | 0.1(0) | 98.2(0.4) | 1.7(0.4) | |||
| 31 | PEOMA | — | 0.5 | — | 200 | 60g | 0(0) | 88.0(2.3) | 12.0(2.3) |
| 32 | 2 | 60f | 0.5(0.8) | 90.6(0.9) | 8.9(0.8) | ||||
| 33 | 8 | 60g | 0(0) | 54.6(2.0) | 45.4(2.0) | ||||
| 34 | macroRAFT-N | — | 0.5 | — | 200 | 60g | 0(0) | 19.1(9.0) | 80.9(9.0) |
| 35 | 2 | 60f | 0(0) | 93.7(3.3) | 6.3(3.3) | ||||
| 36 | 0.5 | 60g,i | 5.1(3.2) | 3.6(5.2) | 91.3(4.4) | ||||
| 37 | 2 | 60g,i | 2.8(1.6) | 19.9(10.9) | 77.3(9.4) | ||||
| 38 | CCM-C-0.1 | 1j | — | 4.7 | 200 | 25 | 0 | 99.0 | 1.0 |
| 39 | 60 | 0.1 | 99.6 | 0.3 | |||||
| 40 | 2000h | 25 | 0 | 100 | 0 | ||||
| 41 | 60 | 0 | 99.8 | 0.2 | |||||
| 42 | 4k | — | 18.7 | 200 | 25 | 0 | 100 | 0 | |
| 43 | 60 | 0 | 100 | 0 | |||||
| 44 | 2000h | 25 | 0 | 100 | 0 | ||||
| 45 | 60 | 0 | 99.9 | 0.1 | |||||
| 46 | macroRAFT-C | — | — | 4.7 | 200 | 60f | 0(0) | 99.9(0.2) | 0.1(0.2) |
| 47 | 18.8 | 60g | 0(0) | 99.9(0.1) | 0.1(0.1) | ||||
| 48 | 19.2 | 2000h | 60l | 6.3(4.0) | 93.7(4.0) | 0(0) | |||
Since the RhNP@CCM-N catalytic efficiency is very high (entries 24–27), experiments were carried out only up to 60 °C. Full substrate conversion was already achieved in 20 h at 25 °C, with high selectivity for the vinyl group hydrogenation. Only traces of the final product 7 were observed at 25 °C for the NPs produced with P/Rh = 1 (entry 24) and this increased only slightly when operating at 60 °C (entry 25). The RhNP@PPh3 performed equally well (entries 28–29), with a slightly greater arene hydrogenation, particularly when P/Rh = 1. The RhNP@PEOMA also gave full conversion and similar selectivity (entries 31–33). The RhNP@macroRAFT-N also gives full conversions and an even greater proportion of 7, particularly for the sample obtained with PEO/Rh = 0.5 (entry 34). Clearly, a greater amount of stabilizer (either the phosphine or the ethylene oxide functions) negatively affects the ring hydrogenation, probably by reducing the surface accessibility. For the RhNP@macroRAFT-N catalyst, additional runs were carried out in the absence of NEt3 (both in the NP synthesis and in catalysis; same batch used for the acetophenone hydrogenation; entries 36–37). Like for the acetophenone hydrogenation, the absence of base increases the rate of ring hydrogenation. It is also interesting to compare the much lower extent of ring hydrogenation for styrene (Table 2) relative to acetophenone (Table 1). For instance, runs 1 and 24 for RhNP@CCM-N-0.1 at 25 °C and P/Rh = 1 show 27% of ring-hydrogenated products (3 + 4) for acetophenone vs. only 0.6% (7) for styrene. Likewise, these fractions are 60% and 12%, respectively, for runs 9 and 31 involving RhNP@PEOMA at 60 °C and PEO/Rh = 0.5. This difference may result from the action of the carbonyl function in acetophenone and the hydroxyl function in the phenylethanol intermediate in keeping the substrate more strongly anchored to the RhNP surface. However, because of the Rh NP migration under catalytic conditions, as already demonstrated in the previous section, interest in using this latex is limited. Indeed, a TEM analysis of the CCM-N-0.1 catalyst after styrene hydrogenation with P/Rh = 4 (run 27), shown in Fig. S7,† reveals a very similar morphology change to that observed after the acetophenone hydrogenation under the same conditions (Fig. 4(c)).
The RhNP@CCM-C catalyst also shows excellent performance (entries 38–45), like the RhNP@CCM-N catalyst, with full substrate conversion (up to 2000 equiv. vs. Rh) at both 25 and 60 °C within 20 h. This is in stark contrast with the poor performance in acetophenone hydrogenation, confirming the hypothesis of a severe mass transport limitation for the latter. High activities were also observed for the RhNP@macroRAFT-C control runs (entries 46–48). The fraction of ring-hydrogenated product is even lower than that observed for the RhNP@CCM-N-0.1 and RhNP@macroRAFT-N systems. Since neat styrene is a good solvent for polystyrene, the hydrogenation was also carried out in bulk (styrene/Rh = 2000), yielding once again full conversion in 20 h at both 25 and 60 °C for both P/Rh ratios (entries 40–41 and 44–45). High activities for the hydrogenation of neat styrene were also observed for the RhNP@PPh3 (run 30) and RhNP@macroRAFT-C (run 48) control runs.
In order to better assess the catalyst performance, a series of experiments were also carried out using shorter reaction times with the RhNP@CCM-C-0.1 (P/Rh = 4) catalyst, see Table 3. When operating in 1-nonanol (styrene/Rh = 200), quantitative conversion to ethylbenzene was still achieved down to 1 h (runs 49–53). Lowering the temperature to 25 °C and using neat styrene (conditions identical to those of entry 44 in Table 2) gave again full conversion after 2 and 1.75 h (runs 54–55). Only for shorter reaction times of 1.5, 1 and 0.5 h (runs 56–58), incomplete conversions were witnessed. From runs 55–58, average TOF values over the entire catalytic runs of 1143, 1020, 994 and 1056 h−1 can be calculated from the TON/time ratios, for an overall average of 1053 ± 46 h−1.
In order to assess the possible effect of the TPP concentration in the hydrophobic CCM core, two additional hydrogenations of neat styrene were carried out using a reaction time of 0.5 h under otherwise identical conditions, with RhNP@CCM-C-0.05 and RhNP@CCM-C-0.2 (runs 59–60). The results are quite comparable, indicating that the TPP concentration does not significantly affect the NP catalytic activity. All experiments in Table 3 show perfect selectivity in favor of the ethylbenzene product 6. In comparison with the only published example of styrene hydrogenation with Rh NPs (complete conversions in 24 h at R.T and 30 bar of H2 in isopropanol with TON up to 756),89 the activity appears much greater. However, that investigation did not report runs with shorter reaction time or greater substrate/Rh ratios. It is also of interest to compare these results with the recently reported ones carried out under the same conditions (aqueous biphasic, neat styrene, 25 °C, 20 bar of H2) with the molecular RhI system embedded in the same CCM support, where a TOF of ca. 300 h−1 was obtained.80 Thus, the catalytic activity of the Rh NPs appears superior to that of the molecular system.
| Entry | NP stabilizer | P/Rh | 4VPMe+I−/Rh | Styrene/Rh | T/°C | Time/h | 5/% | 6/% | 7/% |
|---|---|---|---|---|---|---|---|---|---|
| a The Rh NPs were synthesized at 60 °C in the presence of NEt3 (10 equiv. per Rh) and the indicated support prior to catalysis. b Standard conditions: 0.4 mL of latex; 0.5 mL of 1-nonanol (if used); p(H2) = 20 bar. c 0.96 μmol of Rh. d 2.43 μmol of Rh. e 0.45 μmol of Rh. f 1.89 μmol of Rh. g Neat styrene (no 1-nonanol). | |||||||||
| 49 | CCM-C-0.1 | 3.93c | 18.3 | 200 | 60 | 15 | 0 | 99.6 | 0.4 |
| 50 | 10 | 0 | 100 | 0 | |||||
| 51 | 5 | 0 | 100 | 0 | |||||
| 52 | 2 | 0 | 100 | 0 | |||||
| 53 | 1 | 0 | 100 | 0 | |||||
| 54 | 4.05d | 18.9 | 2000g | 25 | 2 | 0 | 100 | 0 | |
| 55 | 1.75 | 0 | 100 | 0 | |||||
| 56 | 1.5 | 23.5 | 76.5 | 0 | |||||
| 57 | 1 | 50.3 | 49.7 | 0 | |||||
| 58 | 0.5 | 73.6 | 26.4 | 0 | |||||
| 59 | CCM-C-0.05 | 4.04e | 18.8 | 2000g | 0.5 | 58.3 | 41.7 | 0 | |
| 60 | CCM-C-0.2 | 4.07f | 19.0 | 2000g | 0.5 | 56.1 | 43.9 | 0 | |
Finally, the hydrogenation of neat styrene was repeated under the same conditions of entry 58 of Table 3 (P/Rh = 4, 25 °C, 0.5 h, 20 bar of H2) with catalyst recovery and recycling. In a first series of experiments, the product recovery involved extraction of the latex phase (after decanting off the organic layer) with diethyl ether, in order to remove all product and residual substrate from the polymer hydrophobic core prior to the addition of a new substrate charge for the next catalytic run (see Experimental section). This extraction procedure is identical to that used in all individual runs of the above tables, as well as to that used for the recycling experiments with the molecular RhI catalyst embedded in the CCM.80 The results are shown in Fig. 6(a). Again, the selectivity was 100% in favor of ethylbenzene, with no trace of ethylcyclohexane. While the first run gave a higher conversion (66.4%) relative to entry 58 of Table 3, the subsequent runs indicated significant loss of activity, with a continuous drop of the conversion to less than 5% after the 5th recycle. This decrease cannot be related to metal leaching or to mechanical losses during the separation phase, because the decantation phase was rapid yielding a colourless and transparent organic phase and a sharp interface. In a separate recycle series (Fig. 6(b)), a catalyst regeneration step was operated after the 1st recycle, consisting of H2 treatment (20 bar, 80 °C, 2 h) in the absence of substrate. The activity was partially recovered in the 2nd recycle. However, it dropped again in the 3rd recycle. This suggests a NP surface deactivation process, which was only incompletely corrected by the regeneration phase. The TEM analysis of the latex recovered after the 6th recycle (first series), Fig. 6(c), reveals large Rh NP agglomerates and empty polymer particles. Therefore, the irreversible (i.e. not recovered by regeneration) activity loss can be attributed at least in part to the loss of Rh NP active surface associated to the agglomeration.
 | ||
| Fig. 6 (a) Conversion vs. recycle number for the hydrogenation of neat styrene catalysed by RhNP@CCM-C-0.1 (P/Rh = 4) and with product recovery by extraction with diethyl ether. Conditions: styrene/Rh = 2000, 25 °C, 0.5 h, 20 bar of H2 pressure. (b) Same as (a), with a catalyst regeneration step (indicated by an arrow) between recycles 1 and 2. (c) TEM image of the recovered latex after recycle 6 of the series of experiments in Fig. 6(a). | ||
The Rh NP extraction from the CCM core can be associated to the use of diethyl ether for the product separation. Indeed, the Rh NPs can be expected to interact similarly with Et2O and with the PEO functions of the CCM-N shell. In order to substantiate this hypothesis, a final series of catalytic runs with recycling was carried out using toluene instead of Et2O for product extraction, in combination with periodical NP surface regeneration. The results are shown in Fig. 7(a). A first catalyst regeneration, conducted immediately after the 1st run, led to greater activity relative to the original one (ca. 80% conversion). Without any additional regeneration, the high activity was maintained for the next two recycles and then gradually fell to ca. 20% in the 5th recycle. At this point, a second catalyst regeneration led again to a full activity recovery to ca. 80% conversion for the next two cycles. The TEM analysis of the recovered latex after the 8th recycle clearly showed that the Rh NPs remained well dispersed inside the CCM particles, see e.g.Fig. 7(b). The comparison of the activity trend for the recycles with ether and toluene washings and the TEM images of the recovered catalyst in Fig. 6 and 7 constitutes indirect proof of the Rh NP confinement in the CCM-C hydrophobic core. A precise comparison of the Rh NP size before and after catalysis is difficult, but the TEM image clearly evidences the absence of NP agglomeration. The observed behaviour confirms the surface deactivation phenomenon during the catalytic runs at 25 °C and the full reactivation by H2 treatment at 80 °C. Clearly, no surface deactivation would be expected if the catalytic hydrogenations are conducted directly at higher temperatures.
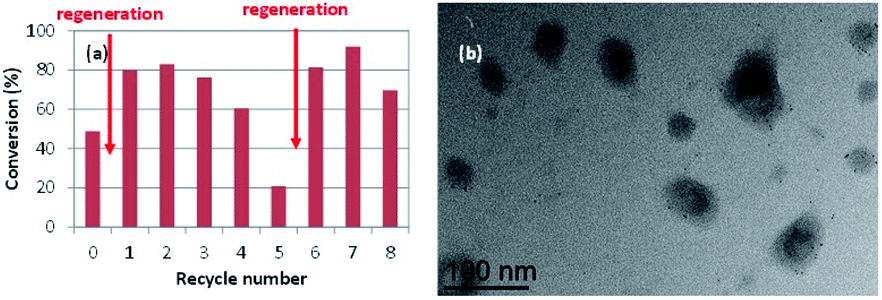 | ||
| Fig. 7 (a) Conversion vs. recycle number for the hydrogenation of neat styrene catalysed by RhNP@CCM-C-0.1 (P/Rh = 4) and with product recovery by extraction with toluene. Conditions: styrene/Rh = 2000, 25 °C, 0.5 h, 20 bar of H2 pressure. The arrows indicate additional Rh NP regeneration steps. (b) TEM image of the recovered latex after recycle 8. | ||
The marked difference in behaviour between the recycle results with diethyl ether and toluene washings provides useful information about the relative aptitude of different NP stabilizers. This difference is in line with the different NP migration behaviour observed for the RhNP@CCM-N and RhNP@CCM-C systems. Although the phosphine P lone pairs bind RhI much more tightly than the O lone pair in ethers, the same is definitely not true for the Rh0 atoms on the Rh NP surface. Thus, while diethyl ether washings did not lead to any significant RhI leaching from the supported molecular RhI@CCM (N or C) systems,79,80 the Rh NPs could be maintained in the stabilizing environment of the CCM core only in the absence of large concentrations of O-based donor stabilizers. The Rh NP extraction from the CCM-C core observed during the recycles with Et2O washings, leading to agglomeration and loss of catalytic activity, is certainly facilitated by the large diethyl ether concentration. A determination of the relative P- and O-donor affinity with respect to the Rh NP surface would require more detailed quantitative study, which is beyond the scope of the present study.
Catalysed hydrogenation of 1-octene
The main objective of these experiments was to gather additional evidence for the Rh NPs confinement in the CCM-C core. Since 1-octene is not a good solvent for polystyrene, its mass transport to the polymer core is limited unless vectorized by a good solvent such as toluene or 1-nonanol. Indeed, a previously described NMR investigation71 did not evidence any core 1-octene incorporation when added as a neat phase to a CCM-N latex, whereas this was confirmed after core swelling by toluene. All subsequent investigations of the biphasic 1-octene hydrogenation with the molecular [RhCl(COD)(TPP@CCM)] precatalyst (CCM = CCM-N or CCM-C) were carried out with 1-nonanol79,80 as vectorizing solvent.The experiments were only carried out with the CCM-C-0.1 latex, using both the in situ-activated [RhCl(COD)(TPP@CCM-C)] pre-catalyst and the RhNP-containing system, RhNP@CCM-C (see Table 4). Before catalysis implementation, the latexes were freeze-dried in order to completely remove toluene (previously used as swelling solvent to load the molecular Rh precursor) and avoid any 1-octene mass transport assistance. The experiment with the molecular catalyst (run 61) unexpectedly yielded a rather efficient hydrogenation. Since this molecular catalyst is unambiguously core-confined, this result demonstrates the occurrence of 1-octene mass transport to the polystyrene core. Consequently, the previously reported absence of 1H and 31P NMR signatures (of incorporated 1-octene and core-anchored TPP ligands, respectively) after equilibration of the CCM-N particles with neat 1-octene71 cannot be attributed to the absence of 1-octene in the core. Rather, the 1-octene amount (a poor polystyrene solvent) at equilibrium is evidently too small to confer sufficient mobility to the polymer and the correlation times remain too long for NMR observation. A new NMR investigation has confirmed the absence of 1H and 31P resonances for core-incorporated 1-octene and core-anchored TPP after equilibrating the TPP@CCM-C-0.1 latex with neat 1-octene.
| Entry | Catalyst | Substrate phase | n-Octane/% |
|---|---|---|---|
| a Conditions: 0.4 mL of latex; 0.71 μmol of Rh (P/Rh = 4); 158.9 mg of 1-octene (1-octene/Rh = 2000), p(H2) = 20 bar, 25 °C, 3 h. b The Rh NPs were synthesized at 60 °C in the presence of NEt3 (10 equiv. per Rh) prior to catalysis. c 0.4 mL of 1-nonanol. | |||
| 61 | [RhCl(COD)(TPP@CCM-C)] | Neat 1-octene | 62.6 |
| 62 | RhNP@CCM-Cb | Neat 1-octene | 31.1 |
| 63 | RhNP@CCM-Cb | 1-Octene/1-nonanolc | 100 |
As shown in entry 62, the Rh NPs also catalysed the hydrogenation of 1-octene, but the yield was ca. half that of run 61, suggesting that the Rh NPs have lower activity than the molecular catalyst for the hydrogenation of this substrate. This is opposite to the observed trend in styrene hydrogenation. This difference may be rationalized by a different relative affinity of 1-octene and styrene to bind to and be activated by a coordinatively unsaturated monometallic RhI center versus the surface of a Rh0 NP. Finally, the activity was greater (full conversion after 3 h) when 1-octene hydrogenation was carried out in the presence of 1-nonanol (run 63). This phenomenon is clearly related to an increased 1-octene mass transport, resulting from the vectorizing effect of 1-nonanol. This more than compensates the expected negative effects of substrate dilution on the kinetics and the possible 1-nonanol competition for NP surface binding. Thus, these catalysis results provide additional evidence in support of the Rh NP confinement in the CCM-C hydrophobic core environment.
Conclusions
We have extended the nanoreactor application of triphenylphosphine-functionalized core-crosslinked micelle latexes, for the first time, to metal nanoparticle catalysis. These latexes, with either neutral P(MAA-co-PEOMA)71–73,75–79,93 or polycationic P(4VPMe+I−)74,80 chains on the micelle surface (CCM-N and CCM-C, respectively) and loaded with a Rh precatalyst, [RhCl(COD)(TPP@CCM)], were previously used to support molecular catalysts. We have now shown that Rh NPs can be synthesized from these precursors by H2 reduction in the absence of olefins. For fully loaded (P/Rh = 1) latexes, the core-anchored complexes were readily reduced to Rh NPs by H2 (20 bar) at 25 °C in the presence of NEt3, whereas heating to 60 °C is needed in the absence of base. Partially loaded latexes (P/Rh = 4) yield Rh NPs only upon heating and in the presence of excess NEt3. The TEM analysis reveals migration of the produced NPs from the core to the shell for RhNP@CCM-N latexes, due to competition between the core TPP ligands and the shell PEO chains as stabilizing functions. During the catalytic applications, even the phosphine-richer (P/Rh = 4) RhNP@CCM-N latexes led to NP migration and agglomeration away from the CCM particles, invalidating the CCM-N strategy for catalyst confinement. For the cationic-shell latexes, on the other hand, the Rh NPs remained well-dispersed and core-confined for all P/Rh ratios after catalysis, but only when toluene, which displays poorer stabilizing power towards the Rh NPs than the core-anchored triphenylphosphines, was used for product recovery/catalyst recycling. These RhNP@CCM-C latexes are therefore of interest for the catalytic application of Rh NPs under aqueous biphasic conditions with catalyst recycling. The catalytic studies presented here show high activity for the reduction of styrene in bulk (TOF greater than 1000 h−1 at 25 °C and 20 bar of H2) and also of 1-octene, although the activity in the latter case is improved when the substrate is vectorized to the CCM core by 1-nonanol, which is a better polystyrene solvent. On the other hand, the polycationic nature of the CCM shell introduced mass transport limitations in the hydrogenation of acetophenone, blocking access to the catalytic NPs. Further investigations are necessary to establish the origin of this blocking effect. It should be possible to implement aqueous biphasic nanocatalysis under a wider array of experimental conditions through the development of nanoreactors with different core functions, i.e. ligands that can better stabilize the metal NPs than TPP while allowing substrate access to the NP surface.Author contributions
H. Wang: investigation, data curation; A. M. Fiore: investigation; C. Fliedel: supervision; E. Manoury: funding acquisition, validation; K. Philippot: conceptualisation, validation, writing – review & editing; M. M. Dell'Anna: supervision; P. Mastrorilli: resources, writing – review & editing; R. Poli, conceptualisation, validation, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement No. 860322 for the ITN-EJD “Coordination Chemistry Inspires Molecular Catalysis” (CCIMC), and by the Agence Nationale de la Recherche grant number ANR-11-BS07-025-01 (BIPHASNANOCAT). In addition, we wish to thank the Centre National de la Recherche Scientifique, and Politecnico di Bari (fellowship to A. M. F.) for financial support. We are also grateful to the China Scholarship Council for a PhD fellowship to H. W.Notes and references
- Nanoparticles: From Theory to Application, ed. G. Schmid, Wiley-VCH, Weinheim, 2010 Search PubMed.
- D. V. Talapin and E. V. Shevchenko, Chem. Rev., 2016, 116, 10343–10345 CrossRef CAS PubMed.
- L. H. Wu, A. Mendoza-Garcia, Q. Li and S. H. Sun, Chem. Rev., 2016, 116, 10473–10512 CrossRef CAS.
- K. D. Gilroy, A. Ruditskiy, H. C. Peng, D. Qin and Y. N. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- R. C. Jin, C. J. Zeng, M. Zhou and Y. X. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- X. Kang and M. Z. Zhu, Chem. Mater., 2019, 31, 9939–9969 CrossRef CAS.
- Y. X. Du, H. T. Sheng, D. Astruc and M. Z. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- Nanotechnology in Catalysis, ed. B. Zhou, S. Han, R. Raja and G. A. Somorjai, Springer, New York, 2003 Search PubMed.
- A. Roucoux and K. Philippot, in The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed.
- Nanocatalysis, ed. U. Heiz and U. Landman, Springer-Verlag, Berlin, Heidelberg, 2007 Search PubMed.
- Nanoparticles and Catalysis, ed. D. Astruc, Wiley Interscience, New York, 2008 Search PubMed.
- Metal Nanoclusters in Catalysis and Materials Science: The Issue of Size Control, ed. B. Corain, G. Schmid and N. Toshima, Elsevier Science, Amsterdam, 2008 Search PubMed.
- Nanoscale Materials in Chemistry: Environmental Applications, ed. L. E. Erickson, R. T. Koodali and R. M. Richards, ACS Publications, Washington D.C., 2010 Search PubMed.
- Nanomaterials in Catalysis, ed. P. Serp and K. Philippot, Wiley-VCH, Weinheim, 2013 Search PubMed.
- Nanocatalysis: Synthesis and Applications, ed. V. Polshettiwar and T. Asefa, Wiley-VCH, Weinheim, 2013 Search PubMed.
- Metal Nanoparticles for Catalysis: Advances and Applications, ed. F. Tao, Royal Society of Chemistry, Cambridge, 2014 Search PubMed.
- Nanocatalysis in Ionic Liquids, ed. M. H. G. Prechtl, Wiley-VCH, Weinheim, 2016 Search PubMed.
- Nanotechnology in Catalysis: Applications in the Chemical Industry, Energy Development, and Environment Protection, ed. B. Sels and M. Van de Voorde, Wiley-VCH, Weinheim, 2017 Search PubMed.
- D. Astruc, Chem. Rev., 2020, 120, 461–463 CrossRef CAS PubMed.
- A. R. Tao, S. Habas and P. D. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- Y. N. Xia, X. H. Xia and H. C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- G. Z. Chen, J. M. Zhang, A. Gupta, F. Rosei and D. L. Ma, New J. Chem., 2014, 38, 1827–1833 RSC.
- A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Chem. Rev., 2015, 115, 11503–11532 CrossRef CAS PubMed.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed.
- T. Y. Chen and V. O. Rodionov, ACS Catal., 2016, 6, 4025–4033 CrossRef CAS.
- Y. Yuan, N. Yan and P. J. Dyson, ACS Catal., 2012, 2, 1057–1069 CrossRef CAS.
- A. Denicourt-Nowicki and A. Roucoux, Chem. Rec., 2016, 16, 2127–2141 CrossRef CAS PubMed.
- G. Oehme, in Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook, ed. B. Cornils and W. A. Herrmann, 2nd edn, 2002, vol. 2, pp. 835–841 Search PubMed.
- V. C. Reinsborough, in Interfacial Catalysis, ed. A. G. Volkov, Marcel Dekker, New York, NY, USA, 2002, pp. 377–390 Search PubMed.
- O. Nuyken, R. Weberskirch, T. Kotre, D. Schoenfelder and A. Woerndle, in Polymeric Materials in Organic Synthesis and Catalysis, ed. M. R. Buchmeiser, Wiley-VCH, 2003, pp. 277–304 Search PubMed.
- T. Kotre, M. T. Zarka, J. O. Krause, M. R. Buchmeiser, R. Weberskirch and O. Nuyken, Macromol. Symp., 2004, 217, 203–214 CrossRef CAS.
- M. N. Khan, Micellar Catalysis, CRC Press, 2006 Search PubMed.
- G. Sharma and M. Ballauff, Macromol. Rapid Commun., 2004, 25, 547–552 CrossRef CAS.
- Y. Mei, G. Sharma, Y. Lu, M. Ballauff, M. Drechsler, T. Irrgang and R. Kempe, Langmuir, 2005, 21, 12229–12234 CrossRef CAS PubMed.
- Y. Lu, Y. Mei, M. Drechsler and M. Ballauff, Angew. Chem., Int. Ed., 2006, 45, 813–816 CrossRef CAS PubMed.
- Y. Lu, Y. Mei, R. Walker, M. Ballauff and M. Drechsler, Polymer, 2006, 47, 4985–4995 CrossRef CAS.
- Y. Lu, M. Yu, M. Drechsler and M. Ballauff, Macromol. Symp., 2007, 254, 97–102 CrossRef CAS.
- Y. Lu, Y. Mei, M. Schrinner, M. Ballauff and M. W. Moller, J. Phys. Chem. C, 2007, 111, 7676–7681 CrossRef CAS.
- M. Schrinner, F. Polzer, Y. Mei, Y. Lu, B. Haupt, M. Ballauff, A. Goldel, M. Drechsler, J. Preussner and U. Glatzel, Macromol. Chem. Phys., 2007, 208, 1542–1547 CrossRef CAS.
- S. Proch, Y. Mei, J. M. R. Villanueva, Y. Lu, A. Karpov, M. Ballauff and R. Kempe, Adv. Synth. Catal., 2008, 350, 493–500 CrossRef CAS.
- Y. B. Malysheva, A. V. Gushchin, Y. Mei, Y. Lu, M. Ballauff, S. Proch and R. Kempe, Eur. J. Inorg. Chem., 2008, 379–383, DOI:10.1002/ejic.200700825.
- M. Schrinner, S. Proch, Y. Mei, R. Kempe, N. Miyajima and M. Ballauff, Adv. Mater., 2008, 20, 1928–1933 CrossRef CAS.
- M. Schrinner, M. Ballauff, Y. Talmon, Y. Kauffmann, J. Thun, M. Moller and J. Breu, Science, 2009, 323, 617–620 CrossRef CAS PubMed.
- S. Wunder, Y. Lu, M. Albrecht and M. Ballauff, ACS Catal., 2011, 1, 908–916 CrossRef CAS.
- R. Roa, S. Angioletti-Uberti, Y. Lu, J. Dzubiella, F. Piazza and M. Ballauff, Z. Phys. Chem., 2018, 232, 773–803 CrossRef CAS.
- M. Ballauff, Prog. Polym. Sci., 2007, 32, 1135–1151 CrossRef CAS.
- Y. Lu and M. Ballauff, Prog. Polym. Sci., 2016, 59, 86–104 CrossRef CAS.
- A. Behr, in Multiphase Homogeneous Catalysis, ed. B. Cornils, W. A. Herrmann, I. T. Horvath, W. Leitner, S. Mecking, H. Olivier-Bourbigou and D. Vogt, Wiley-VCH, Weinheim, Germany, 2005, vol. 1, pp. 327–329 Search PubMed.
- A. Behr, G. Henze and R. Schomaecker, Adv. Synth. Catal., 2006, 348, 1485–1495 CrossRef CAS.
- K. X. Li, Y. H. Wang, J. Y. Jiang and Z. L. Jin, Catal. Commun., 2010, 11, 542–546 CrossRef CAS.
- W. J. Li, Y. H. Wang, M. Zeng, J. Y. Jiang and Z. L. Jin, RSC Adv., 2016, 6, 6329–6335 RSC.
- W. J. Li, Y. H. Wang, P. Chen, M. Zeng, J. Y. Jiang and Z. L. Jin, Catal. Sci. Technol., 2016, 6, 7386–7390 RSC.
- T. Terashima, M. Kamigaito, K.-Y. Baek, T. Ando and M. Sawamoto, J. Am. Chem. Soc., 2003, 125, 5288–5289 CrossRef CAS PubMed.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- T. Terashima, M. Ouchi, T. Ando, M. Kamigaito and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4966–4980 CrossRef CAS.
- T. Terashima, M. Ouchi, T. Ando and M. Sawamoto, J. Am. Chem. Soc., 2006, 128, 11014–11015 CrossRef CAS PubMed.
- A. D. Ievins, X. F. Wang, A. O. Moughton, J. Skey and R. K. O'Reilly, Macromolecules, 2008, 41, 2998–3006 CrossRef CAS.
- T. Terashima, M. Ouchi, T. Ando and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 373–379 CrossRef CAS.
- T. Terashima, A. Nomura, M. Ito, M. Ouchi and M. Sawamoto, Angew. Chem., Int. Ed., 2011, 50, 7892–7895 CrossRef CAS PubMed.
- Y. Liu, Y. Wang, Y. F. Wang, J. Lu, V. Pinon and M. Weck, J. Am. Chem. Soc., 2011, 133, 14260–14263 CrossRef CAS PubMed.
- P. Cotanda, A. Lu, J. P. Patterson, N. Petzetakis and R. K. O'Reilly, Macromolecules, 2012, 45, 2377–2384 CrossRef CAS.
- P. Cotanda, N. Petzetakis and R. K. O'Reilly, MRS Commun., 2012, 2, 119–126 CrossRef CAS.
- T. Terashima, in Encyclopedia of Polymer Science and Technology, ed. H. F. Mark, John Wiley & Sons, Inc., 4th edn, 2013, DOI:10.1002/0471440264.pst590.
- A. Lu and R. K. O'Reilly, Curr. Opin. Biotechnol., 2013, 24, 639–645 CrossRef CAS PubMed.
- J. Lu, J. Dimroth and M. Weck, J. Am. Chem. Soc., 2015, 137, 12984–12989 CrossRef CAS PubMed.
- T. Terashima and M. Sawamoto, in Effects of Nanoconfinement on Catalysis, ed. R. Poli, Springer, New York, 2017, pp. 125–146 Search PubMed.
- Effects of Nanoconfinement on Catalysis, ed. R. Poli, Springer, New York, 2017 Search PubMed.
- R. M. Crooks, M. Q. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Coord. Chem. Rev., 2013, 257, 2317–2334 CrossRef CAS.
- K. Yamamoto, T. Imaoka, M. Tanabe and T. Kambe, Chem. Rev., 2020, 120, 1397–1437 CrossRef PubMed.
- X. Zhang, A. F. Cardozo, S. Chen, W. Zhang, C. Julcour, M. Lansalot, J.-F. Blanco, F. Gayet, H. Delmas, B. Charleux, E. Manoury, F. D'Agosto and R. Poli, Chem.–Eur. J., 2014, 20, 15505–15517 CrossRef CAS PubMed.
- R. Poli, S. Chen, X. Zhang, A. Cardozo, M. Lansalot, F. D'Agosto, B. Charleux, E. Manoury, F. Gayet, C. Julcour, J.-F. Blanco, L. Barthe and H. Delmas, ACS Symp. Ser., 2015, 1188, 203–220 CrossRef CAS.
- E. Manoury, F. Gayet, F. D'Agosto, M. Lansalot, H. Delmas, C. Julcour, J.-F. Blanco, L. Barthe and R. Poli, in Effects of Nanoconfinement on Catalysis, ed. R. Poli, Springer, New York, 2017, pp. 147–172 Search PubMed.
- H. Wang, L. Vendrame, C. Fliedel, S. Chen, F. Gayet, E. Manoury, X. Zhang, M. Lansalot, F. D'Agosto and R. Poli, Macromolecules, 2020, 53, 2198–2208 CrossRef CAS.
- S. Chen, A. F. Cardozo, C. Julcour, J.-F. Blanco, L. Barthe, F. Gayet, B. Charleux, M. Lansalot, F. D'Agosto, H. Delmas, E. Manoury and R. Poli, Polymer, 2015, 72, 327–335 CrossRef CAS.
- A. Joumaa, F. Gayet, E. J. Garcia-Suarez, J. Himmelstrup, A. Riisager, R. Poli and E. Manoury, Polymers, 2020, 12, 1107 CrossRef CAS PubMed.
- E. Lobry, A. F. Cardozo, L. Barthe, J.-F. Blanco, H. Delmas, S. Chen, F. Gayet, X. Zhang, M. Lansalot, F. D'Agosto, R. Poli, E. Manoury and C. Julcour, J. Catal., 2016, 342, 164–172 CrossRef CAS.
- A. F. Cardozo, C. Julcour, L. Barthe, J.-F. Blanco, S. Chen, F. Gayet, E. Manoury, X. Zhang, M. Lansalot, B. Charleux, F. D'Agosto, R. Poli and H. Delmas, J. Catal., 2015, 324, 1–8 CrossRef CAS.
- A. Joumaa, S. Chen, S. Vincendeau, F. Gayet, R. Poli and E. Manoury, Mol. Catal., 2017, 438, 267–271 CrossRef CAS.
- H. Wang, L. Vendrame, C. Fliedel, S. Chen, F. Gayet, F. D’Agosto, M. Lansalot, E. Manoury and R. Poli, Chem.–Eur. J., 2021, 27, 5205–5214 CrossRef CAS PubMed.
- S. Chen, E. Manoury, F. Gayet and R. Poli, Polymers, 2016, 8, 26 CrossRef PubMed.
- M. Guerrero, N. T. T. Chau, S. Noel, A. Denicourt-Nowicki, F. Hapiot, A. Roucoux, E. Monflier and K. Philippot, Curr. Org. Chem., 2013, 17, 364–399 CrossRef CAS.
- Z. B. Duan, M. J. Hampden-Smith and A. P. Sylwester, Chem. Mater., 1992, 4, 1146–1148 CrossRef CAS.
- R. J. Bonilla, B. R. James and P. G. Jessop, Chem. Commun., 2000, 941–942, 10.1039/a907193h.
- E. Ramirez-Meneses, Synthèse et caractérisation de nanoparticules métalliques à base de rodhium, platine et palladium, stabilisés par des ligands, Université Paul Sabatier Toulouse, 2004, vol. 3 Search PubMed.
- J. L. Castelbou, E. Breso-Femenia, P. Blondeau, B. Chaudret, S. Castillon, C. Claver and C. Godard, ChemCatChem, 2014, 6, 3160–3168 CrossRef.
- M. Ibrahim, M. A. S. Garcia, L. L. R. Vono, M. Guerrero, P. Lecante, L. M. Rossi and K. Philippot, Dalton Trans., 2016, 45, 17782–17791 RSC.
- M. A. S. Garcia, M. Ibrahim, J. C. S. Costa, P. Corio, E. V. Gusevskaya, E. N. dos Santos, K. Philippot and L. M. Rossi, Appl. Catal., A, 2017, 548, 136–142 CrossRef CAS.
- M. Ibrahim, M. M. Wei, E. Deydier, E. Manoury, R. Poli, P. Lecante and K. Philippot, Dalton Trans., 2019, 48, 6777–6786 RSC.
- Y. D. Lu, Y. H. Wang and Z. L. Jin, Chin. Chem. Lett., 2010, 21, 1067–1070 CrossRef CAS.
- Z. Sun, Y. H. Wang, M. M. Niu, H. Q. Yi, J. Y. Jiang and Z. L. Jin, Catal. Commun., 2012, 27, 78–82 CrossRef CAS.
- M. M. Niu, Y. H. Wang, P. Chen, D. J. Du, J. Y. Jiang and Z. L. Jin, Catal. Sci. Technol., 2015, 5, 4746–4749 RSC.
- S. Chen, F. Gayet, E. Manoury, A. Joumaa, M. Lansalot, F. D'Agosto and R. Poli, Chem.–Eur. J., 2016, 22, 6302–6313 CrossRef CAS PubMed.
- K. Nasar, F. Fache, M. Lemaire, J. C. Beziat, M. Besson and P. Gallezot, J. Mol. Catal., 1994, 87, 107–115 CrossRef CAS.
- B. R. James, Y. Wang and T. Q. Hu, Chem. Ind., 1996, 68, 423–428 CAS.
- T. Q. Hu, B. R. James and C. L. Lee, J. Pulp Pap. Sci., 1997, 23, J200–J205 CAS.
- T. Q. Hu, B. R. James, S. J. Rettig and C.-L. Lee, Can. J. Chem., 1997, 75, 1234–1239 CrossRef CAS.
- K. S. Weddle, J. D. Aiken and R. G. Finke, J. Am. Chem. Soc., 1998, 120, 5653–5666 CrossRef CAS.
- B. R. James, Y. Wang, C. S. Alexander and T. Q. Hu, Chem. Ind., 1998, 75, 233–242 CAS.
- A. Roucoux, J. Schulz and H. Patin, Adv. Synth. Catal., 2003, 345, 222–229 CrossRef CAS.
- K. H. Park, K. Jang, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2007, 46, 1152–1155 CrossRef CAS PubMed.
- K. B. Zhou and Y. D. Li, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef CAS PubMed.
- K. R. Januszkiewicz and H. Alper, Organometallics, 1983, 2, 1055–1057 CrossRef CAS.
- M. Ibrahim, R. Poreddy, K. Philippot, A. Riisager and E. J. Garcia-Suarez, Dalton Trans., 2016, 45, 19368–19373 RSC.
- L. O. Nindakova, N. M. Badyrova, V. V. Smirnov and S. S. Kolesnikov, J. Mol. Catal. A: Chem., 2016, 420, 149–158 CrossRef CAS.
- L. O. Nindakova, N. M. Badyrova, V. V. Smirnov, V. O. Strakhov and S. S. Kolesnikov, Russ. J. Gen. Chem., 2016, 86, 1240–1249 CrossRef CAS.
- H. Y. Jiang, H. M. Cheng and F. X. Bian, Catal. Lett., 2019, 149, 1975–1982 CrossRef CAS.
- B. L. Tran, J. L. Fulton, J. C. Linehan, J. A. Lercher and R. M. Bullock, ACS Catal., 2018, 8, 8441–8449 CrossRef CAS.
- D. J. M. Snelders, N. Yan, W. Gan, G. Laurenczy and P. J. Dyson, ACS Catal., 2012, 2, 201–207 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and TEM images as referred to in the manuscript text. See DOI: 10.1039/d1na00028d |
| This journal is © The Royal Society of Chemistry 2021 |