
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic nanoparticle mediated-gene delivery for simpler and more effective transformation of Pichia pastoris†
Seyda
Yildiz
 a,
Kubra
Solak
b,
Melek
Acar
a,
Ahmet
Mavi
bc and
Yagmur
Unver
*a
a,
Kubra
Solak
b,
Melek
Acar
a,
Ahmet
Mavi
bc and
Yagmur
Unver
*a
aDepartment of Molecular Biology and Genetics, Atatürk University, Erzurum 25240, Turkey. E-mail: yunver@atauni.edu.tr; yagmurunver@yahoo.com
bDepartment of Nanoscience and Nanoengineering, Atatürk University, Erzurum 25240, Turkey
cDepartment of Chemistry Education, Kazım Karabekir Faculty of Education, Atatürk University, Erzurum 25240, Turkey
First published on 7th June 2021
Abstract
The introduction of exogenous DNA into a cell can be used to produce large quantities of protein. Here, we describe a novel gene delivery method for Pichia pastoris based on recombinant DNA delivery using magnetic nanoparticles (MNPs) under magnetic forces. For this purpose, a linear plasmid (pGKB-GFP) containing the Green Fluorescent Protein (GFP) gene is loaded on polyethyleneimine-coated iron oxide (Fe3O4@PEI) MNPs at doses that are non-toxic to the yeast cells. The pGKB-GFP loaded MNPs combined with enhancer PEI (Fe3O4@PEI + pGKB-GFP + PEI) are directly transferred to non-competent cells. An effective GFP expression was observed by the selection of antibiotic-resistant yeast cells and heterologous gene integration into the P. pastoris genome was provided. This method, which is very simple, effective, and advanced equipment-free compared to traditional methods, uses smaller amounts of DNA and the process can be performed in a shorter time. The suggested method might also be adapted for the transformation of other yeast species.
Introduction
Proteins are used in a number of industrial applications such as medicine, food, detergent, biofuel, textile, paper, etc.1,2 Recombinant protein production, which comprises a considerable research field both in academia and industry, represents a multibillion-dollar market. The increasing demand for recombinant proteins is leading to substantial growth in these markets, which offers a large diversity of applications from industrial enzymes to biopharmaceutical protein complexes.3,4 Generating a specific protein in an organism is typically achieved by the manipulation of gene expression in the organism. Production of desired proteins can be possible in different hosts with recombinant DNA technology which can result in much higher expression levels than those produced naturally.5 The selection of a suitable host is crucial to produce a recombinant protein. The yeast Pichia pastoris (Komagataella phaffii) is widely accepted as one of the most efficient and versatile expression platforms among the different host organisms commonly used for recombinant protein production.3 Up to now thousands of different recombinant proteins have been produced in the P. pastoris expression system, containing either 30% of the total cell protein or 80% of the total secreted protein6 [http://www.pichia.com]. There are over 300 licensed industrial processes using this yeast and more than 70 commercial products are produced in P. pastoris. This powerful eukaryotic expression system is applied in different fields including animal feed additives (e.g. phytase), industrial enzymes (e.g. nitrate reductase), and biopharmaceutical proteins (e.g. human insulin).7–9Spheroplast generation, electroporation, polyethylene glycol (PEG), alkali cation (LiCl), gene gun, etc. methods are used to introduce DNA into P. pastoris for recombinant protein production. These methods make it possible to integrate the vectors into the P. pastoris genome or introduce them as autonomously replicating elements. However, challenges such as high toxicity, low delivery efficiency, high cost of reagents or equipment and the complexity of the application became the key drawbacks in the gene delivery to yeast. The rapid development of nanobiotechnology suggests innovative solutions to overcome these problems.
The process of nucleic acid delivery to target cells by using magnetic nanocarriers through a magnet is defined as magnetofection.10 It is known as a highly efficient and ideal method that uses magnetic force to support the uptake of DNA.11,12 Furthermore, cell membrane permeability is increased efficiently by magnetic force.13 Current applications of magnetic nanoparticles (MNPs) as gene carriers have rapidly progressed in medical research and animal science, especially in the area of gene therapy for various diseases.14–16 Currently, magnetofection has been used for the introduction of DNA into bacteria,17 mammalian cells,16,18,19 and plant pollens.20 Studies showing that high doses of Fe3O4 MNPs significantly inhibit the growth of Saccharomyces cerevisiae have been found in the literature21 However, no study has yet been reported concerning the gene delivery of yeast cells like P. pastoris by MNPs.
Here, a rapid and effective method for DNA delivery into P. pastoris cells was suggested based on using polyethylenimine (PEI)-coated iron oxide (Fe3O4@PEI) MNPs. The DNA-loaded Fe3O4@PEI MNPs called magnetofectin could pass through the P. pastoris cell wall under the influence of a magnetic field. The gene reaching the yeast cell is integrated into the yeast genome and permanent expression is provided. A schematic representation of the suggested magneto-transformation method for P. pastoris is given in Fig. 1.
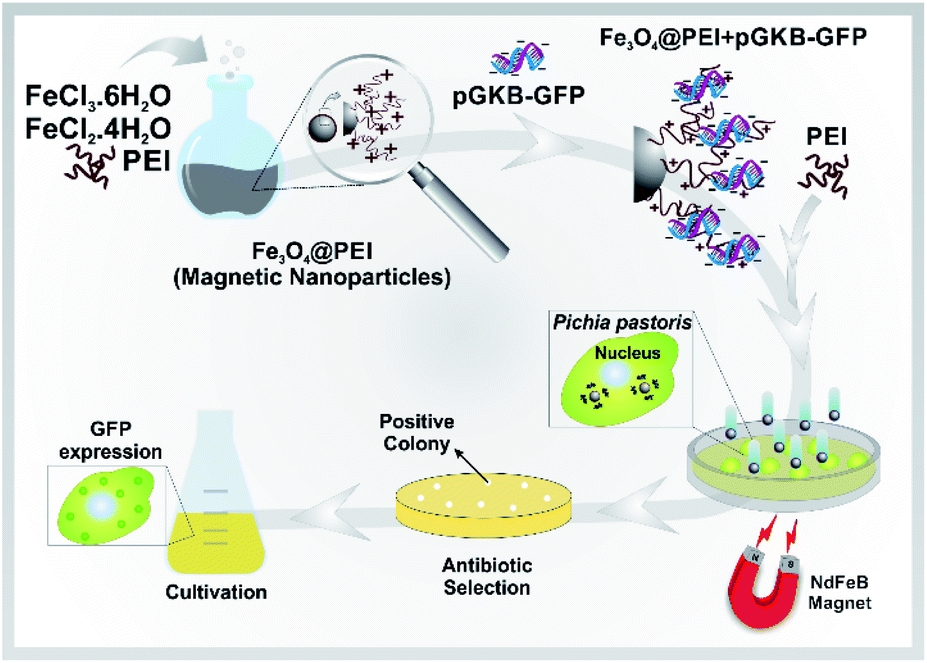 | ||
| Fig. 1 Schematic representation of the method for gene delivery to P. pastoris cells using MNPs. Fe3O4@PEI MNPs together with extra PEI have been used as a carrier platform for linear pGKB-GFP plasmid to form magnetofectin (Fe3O4@PEI + pGKB-GFP + PEI). Magnetofectin was delivered to cells with the help of a magnet. Antibiotic-resistant transformants were selected on YPD–geneticin agar. P. pastoris colonies containing the integrated green fluorescence protein (GFP) coding sequence in their genome were confirmed by a series of PCR analyses. Then, GFP expression in the cells was confirmed. | ||
Experimental section
The preparation of magnetic nanoparticles
The Fe3O4@PEI MNPs were obtained using a modified procedure from the literature by the co-precipitation method.22 Briefly, in a three-necked flask, 2.4 g FeCl3·6H2O (Sigma-Aldrich) and 1.0 g FeCl2·4H2O (Sigma-Aldrich) were dissolved in 30 mL deionized water. The solution was stirred vigorously for 30 minutes under an inert atmosphere, and heated to 80 °C. Then, 6 mL of ammonium hydroxide (25 wt%, Sigma-Aldrich) was rapidly added and the color of the mixture turned black immediately. The solution was continuously stirred for 30 minutes, following which 5% PEI aqueous solution (20 mL, ∼25 kDa, Sigma-Aldrich) was added. The mixture was stirred for 3 hours at 80 °C and then cooled naturally to room temperature. The black precipitated MNPs were collected using a magnet and washed with deionized water several times by centrifuging (at 9000 rpm for 15 minutes). The Fe3O4@PEI MNPs were stored under inert conditions in 20 mL PBS (Phosphate Buffered Saline; Fluka).Acidification was performed to increase the water dispersibility and reduce the aggregation of Fe3O4@PEI MNPs. Basically, 100 mg of Fe3O4@PEI MNPs were re-dispersed in water (15 mL). The pH value was adjusted to 2 with 0.5 M HCl (Sigma-Aldrich) and then 7 with 0.5 M NaOH (Sigma-Aldrich). The mixture was stirred for 10 minutes in each step.23 Finally, the Fe3O4@PEI MNPs (4 mg mL−1) were stored in an inert atmosphere at +4 °C.
Fluorescently tagged Fe3O4@PEI MNPs were prepared according to the manufacturer's protocol to investigate cellular uptake of the MNPs by yeast cells. Briefly, 6 mg Fe3O4@PEI MNPs in 0.5 mL of 2-propanol (Sigma-Aldrich) and 0.2 mg TRITC (5/6-tetramethylrhodamine isothiocyanate, Thermo Scientific™) in 200 μL of DMSO (Merck) were added in 5 mL of 2-propanol. The milieu was kept under nitrogen gas for 10 minutes and then shaking at room temperature in the dark overnight. TRITC-labeled Fe3O4@PEI MNPs were collected by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300g for 10 minutes and washed with 2-propanol. The TRITC-labeled MNPs were stored in an inert atmosphere and under dark conditions at −20 °C.
300g for 10 minutes and washed with 2-propanol. The TRITC-labeled MNPs were stored in an inert atmosphere and under dark conditions at −20 °C.
Characterization
The size and morphology of the Fe3O4@PEI MNPs were obtained via transmission and scanning electron microscopy (TEM, Hitachi HighTech HT7700, and SEM, Carl Zeiss), respectively. A carbon-coated copper grid was used to take TEM images, and SEM images were recorded after gold coating. The crystalline phase of the dry Fe3O4@PEI MNPs was examined by X-ray diffraction (XRD, D2 Bruker). The magnetic hysteresis loops of the sample were determined at room temperature (T = 305 K) by using a Vibrating Sample Magnetometer (VSM Quantum Design PMS9T), with an applied magnetic field from −20 kOe to +20 kOe (Oe = oersted). Fourier transform infrared spectroscopy (FTIR) was used to determine the surface functional groups by scanning dry samples in the range of 400–4000 cm−1 wavelength. Dynamic light scattering (DLS, Malvern Zetasizer Nano Zs) was performed in buffer (Na2HPO4–NaH2PO4, 10 mM) at different pH (5.00–8.00) values to determine hydrodynamic diameter (Rh), surface charge (ζ-potential) and polydispersity index (PDI) of Fe3O4@PEI MNPs.Cell strains, media, and growth conditions
Escherichia coli One Shot TOP10 and Pichia pastoris strain X-33 were purchased from Invitrogen (USA) and incubated on LB (Luria Bertani) agar (Miller) and Yeast extract Peptone Dextrose (YPD) agar to maintain the culture, respectively. YPD contains 1% yeast extract (Merck), 2% peptone (Merck), 2% glucose (Sigma-Aldrich) and 2% agar (Lab M). Bacteria cells were grown in LB broth (Miller) on a rotary shaker at 160 rpm at a temperature of 37 °C while yeast cells were grown in YPD medium (1% yeast extract, 1% peptone, and 2% glucose) on a rotary shaker at 180 rpm at a temperature of 28 °C.Cloning of GFP gene
To construct the recombinant plasmid, green fluorescent protein (GFP) gene was cloned into the pGKB plasmid which was a gift from Sheng Yang (Addgene plasmid #85077; http://n2t.net/addgene:85077;RRID:Addgene_85077).24 The GFP gene was codon-optimized for P. pastoris by replacing the low-use S, L, G and V (serine, leucine, glycine, and valine) codons in the original GFP gene (GenBank L29345.1) with optimal codons (https://eu.idtdna.com/codonopt). Before and after optimization, codon adaptation indexes (CAIs) were determined using GenScript Rare Codon Analysis Tool software (GenScript, Piscataway, NJ, USA). The codon-optimized gene was obtained synthetically. pGKB and the gene amplified using Phusion High-Fidelity DNA Polymerase (Thermo Scientific, USA) by PCR were digested with EcoRI and KpnI. Then, they were incubated for ligation at 16 °C for 16 hours. The ligated products were transferred to competent E. coli TOP10 cells. LB agar containing 50 μg mL−1 of kanamycin was used for the screening of transformants. Correct transformants were verified by colony PCR and gene sequencing analysis. The constructed expression plasmid was named pGKB-GFP.Cellular uptake of Fe3O4@PEI MNPs
P. pastoris cells were grown in YPD medium and 6 mL of the culture medium (OD600 ∼ 0.6–0.9) was centrifuged at 10000 rpm for 5 minutes. The pellet was resuspended in 600 μL YPD medium and 100 μL of cell suspension was transferred to a sterile 24-well plate. Yeast cells were treated with TRITC-labeled MNPs (250 μg/200 μL) and then the plate was held on a magnet for a predetermined time (0, 15, 30, and 60 minutes). The cells treated with Fe3O4@PEI MNPs on a magnet for 15 or 30 minutes were used as the control group. The cells were incubated at 28 °C at 180 rpm in the dark for 4 hours and 200 μL of YPD medium was added for further incubation. After 24 hours, the cells were centrifuged at 10000 rpm for 5 minutes and washed with PBS (×2). The cells were incubated with 4% PFA for 30 minutes at room temperature for fixation and washed with PBS (×2). De-permeabilization of cells was achieved with Triton X-100 treatment for 5 minutes and washing with PBS (×2). The cell suspension and DAPI (1 μg mL−1 in methanol) were mixed in equal volume (50 μL) and spread on the coverslip. After incubation at 37 °C for 15 minutes, the coverslips were washed two times with PBS. The yeast cells were visualized using a confocal laser microscope (ZEISS).
Toxicity of Fe3O4@PEI MNPs on yeast
The cell growth and proliferation of P. pastoris were evaluated via measuring the optical density (OD) by spectroscopy. The cells (∼108 cells per mL) were treated with Fe3O4@PEI MNPs (0–100 μg mL−1). Following the treatment, the cells were kept on a magnetic field for 15 minutes and incubated at 180 rpm at 28 °C. Non-treated and 15% DMSO treated cells were used as controls. After incubation for 2-, 24-, 48-, and 72 hours, 750 μL of samples were taken from all groups, and incubation was continued until the next measurement by adding an equal volume of YPD medium. OD600 of the collected samples was measured by using a spectrophotometer (BioRad). The effect of Fe3O4@PEI MNPs was calculated using the following formula:| OD600 = ODControl or Sample − ODYPD or YPD containing MNPs |
On the other hand, a spot test was performed to check the growth rate of yeast cells on Fe3O4@PEI MNP containing media. The yeast cells were exposed to Fe3O4@PEI MNPs under a magnetic field as in the proliferation test. Serial dilutions (10−3, 10−6, and 10−9) were prepared with 0.85% physiological water after 72 hours of incubation with MNPs. 3 μL of cell suspension and serial dilutions were dropped onto YPD agar. The cells were allowed to grow in YPD agar and incubated at 30 °C. Spots formed on the agar were photographed.
Magnetofectin preparation
High concentration salmon sperm DNA (Thermo Scientific, USA) was used to consume less plasmid DNA (pGKB-GFP) in the experiments of magnetofectin optimization. The DNA binding capacity of Fe3O4@PEI MNPs was investigated by altering the incubation time and concentrations of the MNP and PEI separately. In the study, an equal volume (12 μL) of each material in sterile distilled water (∼pH 6.0) was incubated at room temperature to prepare magnetofectin. First, the incubation time of DNA with MNPs and DNA-loaded MNPs with enhancer PEI was changed (0–30 minutes) and the incubation time was determined to achieve the highest DNA loading on MNPs. The ratio of enhancer PEI to DNA (N/P ratio) was also studied as it affects both DNA loading and transfection efficiency. Different N/P ratios (2, 4, 6, 8, 10, 15, 20 and 30) were calculated based on the knowledge that 43 ng PEI contains 0.25 nmol of primary amine nitrogen and 1 μg DNA contains 3 nmol of phosphate.25 Eventually, the magnetofectins prepared for use in magneto-transformation were obtained by 30 minute incubation of 1 μg DNA (12 μL) with 7.5 μg MNP (12 μL) and 30 minute incubation of enhancer PEI (12 μL) added to this mixture at a ratio of N/P 4. Measurement of the amount of unbound DNA was carried out in the supernatant after magnetofectins were collected with a magnet for 15 minutes for all experimental steps. All experiments were carried out under sterile conditions in a sterile cabinet. All results belong to the results of the experiment groups, which were repeated three times. A Nanodrop (BioTek) was used for the determination of the DNA concentration in the solution.After optimization of the magnetofectin preparation using salmon sperm DNA, magnetofectin was prepared with pGKB-GFP plasmid DNA. Magnetofectins were collected using a magnet for 15 minutes and the supernatant was used for agarose gel electrophoresis. Also, DLS analysis was used to determine the stability, dispersion, and surface charge of the pGKB-GFP loaded MNPs in water.
Transformation of cells
The prepared magnetofectins were added to the yeast cells (OD600 ∼ 0.7) and the cellular uptake of magnetofectins was accelerated through a magnet application (15 minutes). Expression of the GFP gene was examined in P. pastoris cells incubated for certain periods to clearly present transformation success.The enhancer PEI and DNA ratio (N/P) was investigated owing to its effect on transformation. Cells to which untreated, pGKB treated and treated with magnetofectins containing circular pGKB-GFP at different N/P ratios (0, 2, 4, 6, and 8) were incubated in a rotary shaker at 180 rpm at 28 °C. After incubation for 48 hours, cell lysates were prepared with acid-washed glass beads to compare the amount of GFP produced in the cells.26 A fluorescence spectrophotometer was used for the measurement of the GFP amount (excitation wavelength; Ex, 488 nm; emission filter; Em, 507 nm). Cell lysate of magneto-transformed with pGKB was used as blank for the measurement of the GFP amount in cell lysates of magneto-transformed with pGKB-GFP. Furthermore, to determine whether the magnetofectins prepared with pGKB and pGKB-GFP at N/P 4 have a toxic effect on yeast cells, transformed cells were transferred to YPD agar after 24 hours and the colonies were photographed.
Integration of pGKB-GFP into the P. pastoris genome was carried out after the recombinant plasmid linearization with PagI (BspHI) from the GAP promoter region and a combination of the linearized plasmid with MNPs as mentioned in magnetofectin preparation. Magneto-transformation was administered with pGKB without GFP for validation of the method. Transformed cells were spread separately on YPD plates containing 200 μg mL−1 geneticin (G418) for antibiotic selection. After incubation in antibiotic selection media, colony PCR was performed with selected yeast colonies. Gene and vector-specific primers (Table S1†) were used for PCR.
Integration of the gene into the yeast genome was also confirmed by performing PCR which used genomic DNAs, as templates, and gene and vector-specific primers. Genomic DNAs (gDNA) belonging to randomly selected three yeast colonies were purified using GeneJET Genomic DNA Purification Kit (Thermo Scientific, USA) and used as templates for PCR. For final confirmation of GFP production in the selected transformants, these cells were cultivated in YPD medium for 120 h. After incubation, the cells were collected and visualized in white and GFP excitation light using confocal laser scanning microscopy (ZEISS).
Statistics
All results were analysed with Origin Pro Lab 8.5 data analysis and graphing software. Statistical significance was considered when ρ < 0.05 (n > 3). ImageJ software was used for statistical analysis of confocal images.Results and discussion
The magnetic vector
Superparamagnetic iron oxide magnetic nanoparticles have been used as gene delivery agents for many years. Here the Fe3O4@PEI MNPs were synthesized by using a one-pot reaction due to the basic characteristics of the method such as simple and fast production conditions, requiring low reaction temperature, not using organic solvents in the synthesis phase, being energy efficient, etc. PEI is a synthetic organic macromolecule that has a high cationic charge density due to the presence of primary, secondary, and tertiary amino groups. A highly branched PEI neutralizes the excess anionic colloids. It has a buffering capacity at especially acidic and neutral pH which helps in DNA binding and release that makes it an ideal vector candidate for the delivery of plasmids.27 The negatively charged DNA molecules bind to positively charged MNPs by electrostatic interactions.28 Conversely, highly branched-PEI might promote the aggregation of MNPs due to the long-chain structure of PEI (Fig. 2A). Several groups have reported that acidification and then neutralization of PEI-coated MNPs cause irreversible de-aggregation of MNPs.29 It was observed that the non-acidified and acidified Fe3O4@PEI MNPs exhibited different behavior when a temporal magnetic field was applied (Fig. S1†). Faster accumulation of acidified Fe3O4@PEI MNPs under a magnetic field might assist efficient gene transfer owing to the protection of the gene and avoiding aggregation of MNPs in a complex media environment. | ||
| Fig. 2 Characterization of the polyethyleneimine (PEI)-coated Fe3O4 MNPs. (A) Depiction of non-acidified and acidified MNP dispersion. The MNPs were analyzed by (B) TEM, (C) XRD, (D) VSM, and (E) FTIR for morphological and structural investigation and determination of the crystal structure, magnetization, and functional groups, respectively. | ||
The structure of non-acidified and acidified MNPs was evaluated by TEM which demonstrated that distribution of MNPs improved after acidification (Fig. 2B and S2A†). The morphology of the MNPs was close to spherical based on TEM and SEM (Fig. S2B†) images and the MNPs have an average size of 20 nm. The absence of PEI on the surface of MNPs in TEM images is due to the transparency of PEI. Typical diffractions of the Fe3O4 crystallite were observed from the XRD patterns which are presented at [220], [311], [400], [511] and [440]22 (Fig. 2C). Acidified Fe3O4@PEI MNPs exhibited superparamagnetic behavior based on magnetic measurements by VSM (Fig. 2D). The saturation magnetization value of acidified Fe3O4@PEI MNPs was 40 emu per g. A curve that does not show any hysteresis loops characteristic for superparamagnetic MNPs was observed. The presence of PEI on the surface of MNPs was confirmed by FTIR (Fig. 2E). The stretching of the amine structure in the PEI structure appeared at 3218 cm−1. The peaks appearing between 2800 and 3000 cm−1 belong to the chain structure in the polymer and indicate the sp3 –C–H structure. The peak at 1000–1350 cm−1 belongs to –C–N– stretching being present. Finally, the Fe–O bond in the structure appeared at 543 cm−1. DLS analysis was performed to determine the zeta potential (ζ-potential), hydrodynamic size (Rh), and the polydispersity index (PDI) of Fe3O4@PEI MNPs at various pH values. DLS results (Table 1) showed that the Rh of MNPs was 195 nm which is not an excessive size for gene delivery and the ζ-potential of MNPs was 16.1 mV at pH 6.0, indicating the positive net charge of the MNPs and better dispersibility at pH 6.0. However the results of MNPs at pH 8.0 showed the Rh of 1011 nm and the ζ-potential of −12.5 mV indicating negative charge of the MNPs. It was found that the point of zero charge of the PEI-coated MNPs is at pH 7.4–8.0.
Obtaining the recombinant plasmid
Green fluorescent protein (GFP) was preferred as a target gene for gene delivery due to its easy monitoring provided by its fluorescence feature. The codon-optimized GFP sequencing was purchased according to the codon bias of P. pastoris to increase the expression level of GFP in yeast cells. The codon adaptation index (CAI) of the natural GFP gene was increased from 78% to 91% after codon-optimization. The codon-optimized GFP gene was amplified by PCR and integrated into the pGKB plasmid by a ligation reaction (Fig. S3†). Colony PCR and DNA sequence analysis were performed after selecting the bacteria transformed by the ligation product. Results of the colony PCR (Fig. S3B and C†) and gene sequence analysis (Fig. S4†) have proved that the GFP gene was successfully inserted into the correct position on pGKB. The recombinant plasmid map is presented in Fig. S5.† The plasmid isolation was performed to obtained highly concentrated pGKB-GFP for further use in the study.Cellular uptake of Fe3O4@PEI MNPs
TRITC-labeled Fe3O4@PEI MNPs were produced to observe whether yeast cells take up the MNPs. Cellular uptake of TRITC-labeled Fe3O4@PEI MNPs was observed by confocal microscopy after the MNPs were applied to the yeast with a magnet at different time intervals. Fig. 3 A indicates that TRITC-labeled Fe3O4@PEI MNPs are received by yeast cells even in the absence of a magnet under cells. The intensity of the red signal from cells treated with TRITC-labeled MNPs increases as the retention time of the cells on the magnet increases. The cells of the control and treated with non-TRITC labeled MNPs have no signal as expected. The statistical analysis of the images revealed that low fluorescence intensity was determined in cells incubated with TRITC-labelled MNPs without magnet (Fig. 3B). There was no statistically significant difference between the groups that were kept on the magnet for 30 and 60 minutes. 3D scanning was done to make sure that the MNPs were taken into the cell and not placed on the cell surface (Fig. 3C). The recorded z-stack images prove that the signals which are indicating MNPs because of TRITC and the cell nucleus because of DAPI staining, are coming from inside the cell. Eventually, it was predicted that leaving the cells treated with MNPs on the magnet for 15 minutes might be sufficient for further study.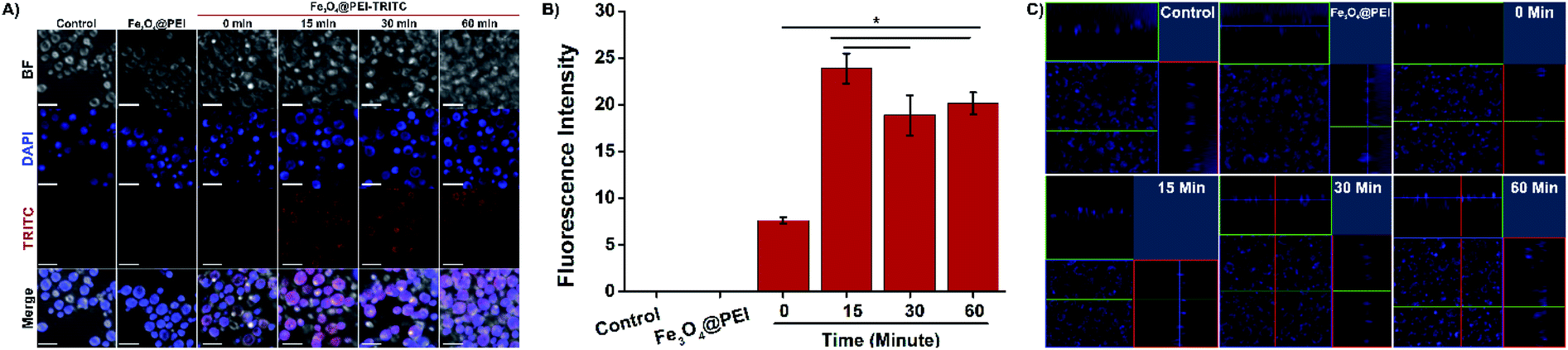 | ||
| Fig. 3 Confocal microscopy demonstration of cellular uptake of MNPs in yeast treated with TRITC-labelled MNPs. (A) 2D confocal microscope images. The scale bar is 5 μm. (B) Quantitative analysis of confocal images. (C) Z-Stack images from yeast treated with TRITC-labelled MNPs. | ||
Investigation of Fe3O4@PEI MNP toxicity on yeast cells
PEI exhibits dose-dependent cytotoxicity on both prokaryotic and eukaryotic cells at high molecular weight due to its protonation attributes. Azevedo et al. revealed that a lower concentration of PEI is able to inhibit yeast growth compared with bacteria strains.30 Therefore, the determination of non-cytotoxic concentration of Fe3O4@PEI MNPs on yeast cells was an essential matter in this study. Yeast cells were grown under normal growth conditions supplemented with Fe3O4@PEI MNPs at various concentrations (0–100 μg mL−1) and then the absorbance measurement and spot test were applied (Fig. 4). The optical density was decreased in the DMSO treated group (15%)31 while it did not significantly change in the groups of the MNPs treated even for a longer incubation time (Fig. 4A).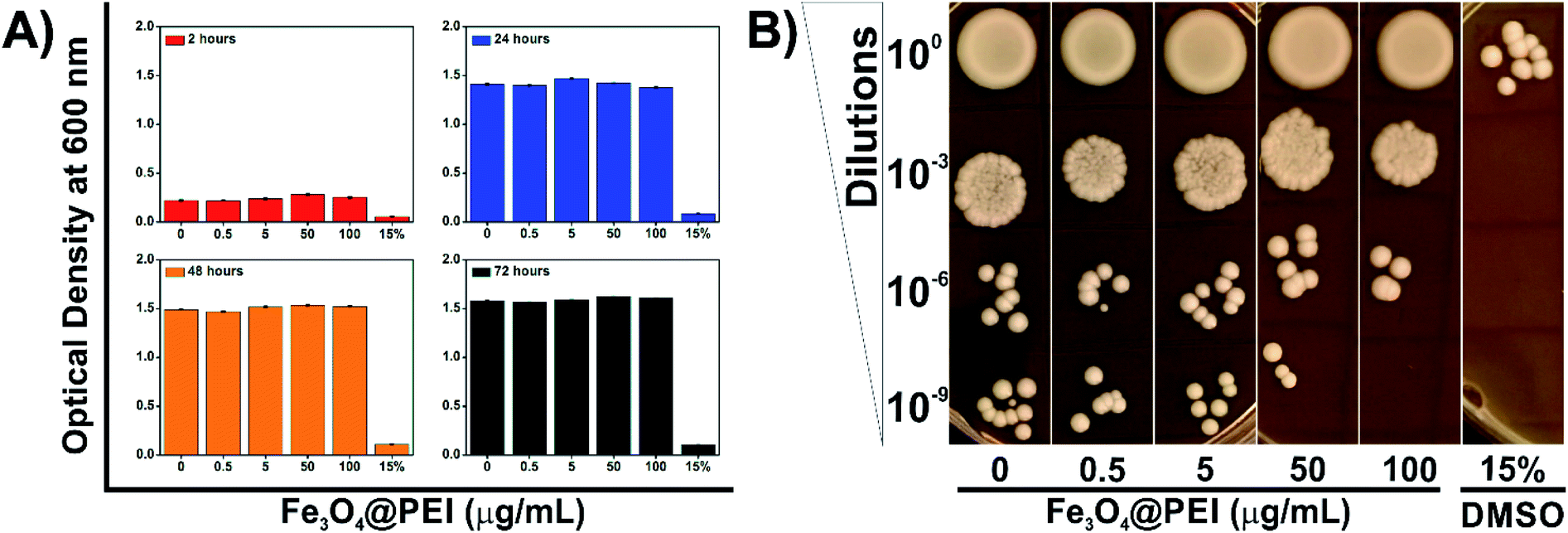 | ||
| Fig. 4 The toxicity tests for yeast cells. (A) The OD600 results recorded after 2, 24, 48 and 72 hour incubation of yeast cells in growth media containing different concentrations of Fe3O4@PEI MNPs and (B) spot test results of samples incubated with MNPs for 72 hours. | ||
The spot test was constructed to check the growth rate of yeast cells by using different media containing serial dilutions of Fe3O4@PEI MNP treated cells (Fig. 4B). The results of the spot analysis demonstrated that Fe3O4@PEI MNPs had no adverse effects at lower than 50 μg mL−1. The absence of colonies only in the highest dilution (10−9) of the groups treated with 100 μg mL−1 MNPs may indicate that high dose MNPs have a slight effect on yeast cells compared to other concentrations. In DMSO treated groups, which are known to be cytotoxic, colonies were observed only in the first series in the spot test, consistent with the OD measurement.
Preparation of magnetofectin
An enhancer material is generally used to increase the yield of DNA-binding and transfection efficiency. In this study, PEI was used both in surface modification of MNPs and as an enhancer like it is used in many studies.23,32 All conditions were carefully and step by step investigated to protect the association of Fe3O4@PEI MNPs, DNA, and enhancer PEI, and to determine suitable ratios of the components. DNA binding capabilities of Fe3O4@PEI MNPs were demonstrated by investigating varying concentrations and incubation times. Optimization studies were performed with commercially obtained salmon sperm DNA due to its low cost to create high concentration solutions. Then, pGKB-GFP loading to MNPs was demonstrated with the optimum magnetofectin conditions determined using salmon sperm DNA. The binding of plasmid DNA to MNPs was demonstrated by agarose gel electrophoresis.The results presented in Fig. 5 A demonstrate that the amount of loaded DNA on MNPs increases by time. Likewise, the longer incubation time of the DNA-loaded MNPs with the enhancer PEI causes increased DNA loading (Fig. 5A). The highest amount of DNA loading was observed at N/P 2 and 4 when the enhancer PEI effect was investigated (Fig. 5B). Here, N represents the number of nitrogen atoms in the structure of PEI and P represents the number of phosphate atoms in the structure of DNA. Fig. 5C shows that 7.5–25 μg of Fe3O4@PEI MNPs can load the most at 1 μg of salmon sperm DNA. To determine the validity of the results obtained with salmon sperm DNA for plasmid DNA, the supernatants were collected from magnetofectins containing pGKB-GFP and different concentration of MNPs using a magnet and were run in agarose gel electrophoresis. In accordance with the nanodrop results, agarose gel images show that as the MNP concentration decreases, the amount of measured pGKB-GFP in the supernatant increases (Fig. 5D). In addition, when samples containing pGKB-GFP incubated with different amounts of PEI in the MNP-free solution were loaded directly into an agarose gel, the visibility of pGKB-GFP in the gel decreased with the increase of the amount of PEI (Fig. 5E). This indicates that positively charged PEI can bind to negatively charged pGKB-GFP in a dose-dependent manner and the interaction of recombinant DNA with nucleic acid staining dye decreases with the increasing amount of PEI. Based on these results, 7.5 μg Fe3O4@PEI MNP concentration and the N/P 4 ratio were suggested to bind large amounts of DNA. Finally, magnetofectins prepared under conditions containing 7.5 μg of MNP, 1 μg of DNA (pGKB-GFP or salmon sperm DNA) and enhancer PEI at N/P 4 were magnetically collected, and supernatants were used for agarose gel electrophoresis (Fig. 5F). Since salmon sperm DNA contains different sizes of DNA pieces, the rate of binding to MNPs is higher. On the other hand, it is seen that a large part of the plasmid DNA used is also loaded especially when enhancer PEI was used. The ζ-potential and Rh results of MNPs and magnetofectin prepared with pGKB-GFP confirm the presence of DNA on MNPs (Fig. 5G and H). It was observed that when DNA was present on the surface of MNPs, the zeta potential becomes negative. Enhancer PEI and DNA existing on the MNPs caused increased hydrodynamic diameter. The negative surface charge of the magnetofectin despite the use of enhancer PEI and the absence of DNA bands when PEI is used at the rates specified in the gel results may indicate that all DNA is bound to MNPs, and that there is no extra PEI coating on the surface.
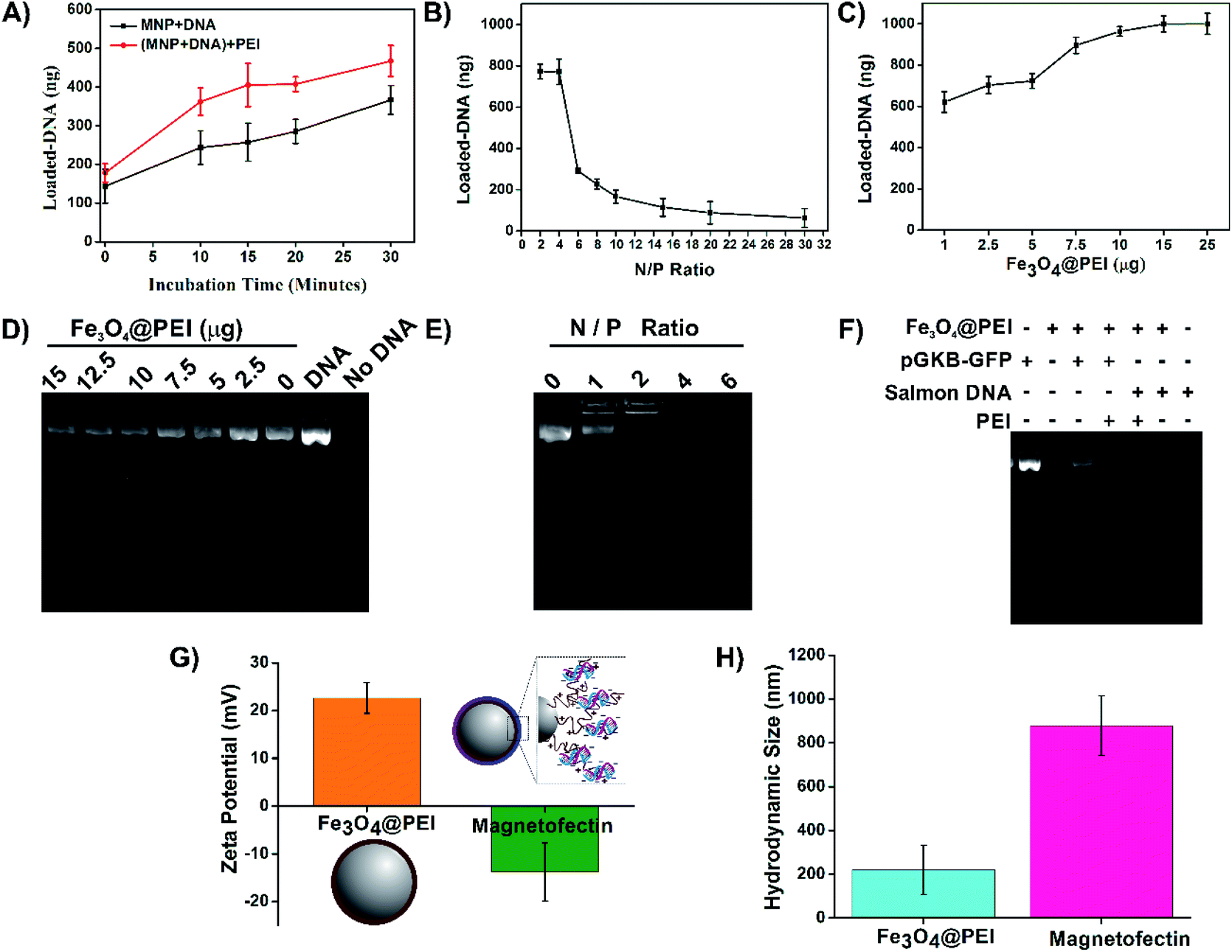 | ||
| Fig. 5 Determination of magnetofectin conditions. (A) Investigation of the incubation time of MNPs, salmon sperm DNA, and PEI. While investigating the incubation time of MNP and DNA, the incubation time was 15 minutes for PEI incubation (MNP + DNA). Then the incubation time of DNA loaded MNPs with PEI was changed (MNP + DNA + PEI). (B) Investigation of the N/P ratio. 1 μg of salmon sperm DNA and 1 μg Fe3O4@PEI MNPs were used in all experiments. (C) The effect of Fe3O4@PEI MNP concentration on salmon sperm DNA binding. Agarose gel electrophoresis image of magnetofectin containing pGKB-GFP prepared with different (D) MNP concentrations and (E) N/P ratios. (F) The image of the supernatants collected from magnetofectins containing salmon sperm DNA or pGKB-GFP. Salmon sperm DNA appears as a smear on the gel due to its content consisting of DNA fragments. (G) Surface charge and (H) hydrodynamic size of MNPs or magnetofectin which is containing pGKB-GFP, MNPs and enhancer PEI. | ||
Magneto-transformation
Methods such as spheroplast–PEG transformation, lithium, electroporation, glass bead, biolistic, etc. have been developed for transformation in yeast and their efficiency has been improved.33 For a successful yeast transformation, DNA must pass through the cell wall and reach the nucleus.34 The glass bead method has low transformation efficiency.35 Since the electroporation and biolistic methods require special equipment, methods that do not require special equipment have become more interesting.36,37 The results of the lithium method, one of them, vary according to the species and the reaction mixture is simple. However, it requires the use of large amounts of plasmid DNA.26,38 Therefore, in this study, a novel method was developed that does not require special equipment and can provide transformed cells with a low amount of plasmid DNA. Basically, magnetofectins, containing MNPs, pGKB-GFP, and enhancer PEI, were applied to the yeast cells with the help of a magnet and this process was called magneto-transformation (Fig. 6A).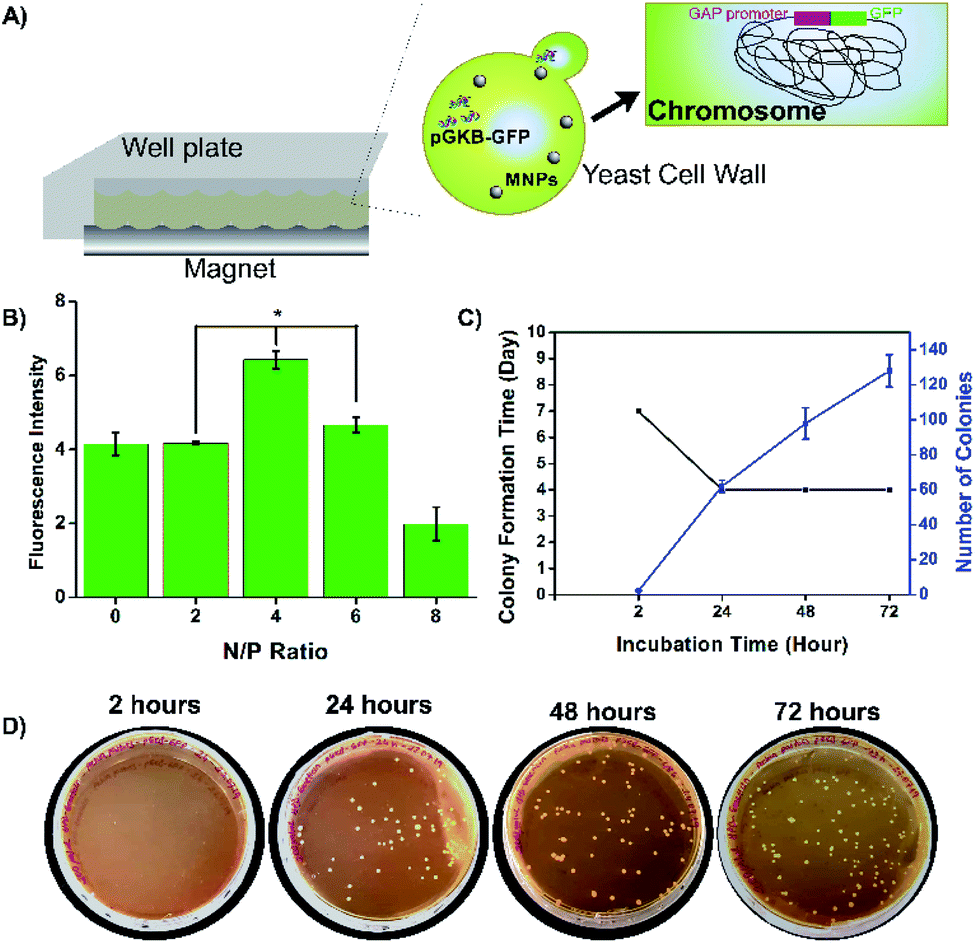 | ||
| Fig. 6 Magneto-transformation results. (A) Schema of magneto-transformation, (B) N/P ratio effect for GFP expression after magneto-transformation, (C) chart of colony formation time and numbers of colonies after magneto-transformation, (D) growing of antibiotic-resistant transformants on YPD–geneticin agar. | ||
It is known that enhancer PEI has a terrific effect on DNA binding as well as transfection efficiency. In this study, the effect of enhancer PEI on magneto-transformation was examined. Magnetofectin with the circular pGKB-GFP prepared using the conditions determined in the magnetofectin preparation step were delivered to P. pastoris cells with a magnet. The codon-optimized GFP gene was chosen as the reporter for monitoring of the heterologous gene integration and expression. The amount of produced GFP in cell lysates was determined by a fluorescence spectrophotometer. The highest fluorescent density of the recombinant protein was observed at N/P 4 (Fig. 6B) which is consistent with DNA binding results in the magnetofectin preparation (Fig. 5B). It can be said that if magnetofectin has more DNA when it was prepared at the N/P 4 ratio, the GFP expression will be high after magneto-transformation. Yeast cells to which pGKB-GFP was delivered by magneto-transformation were transferred to YPD agar and their growth was observed. No harmful effects were observed on the growth of treated yeast cells with Fe3O4@PEI + pGKB + PEI or Fe3O4@PEI + pGKB-GFP + PEI which were prepared at the N/P 4 ratio and used in prescribed amounts (Fig. S6A†).
For molecular genetic manipulation, DNA sequences of interest are introduced into and maintained in the host organism. Vectors that carry the heterologous genes can be integrated into the P. pastoris genome or can include autonomously replicating elements.39,40 Sequences shared by the yeast genome and the transforming vector provide the site for gene integration using homologous recombination.39 Hence, the controllable integration of vectors with heterologous genes can be easily performed.41 So, in the suggested method, the stable expression of the target gene, permanent gene integration and passing of target gene on to daughter cells were achieved by using the linearized pGKB-GFP to prepare magnetofectin (with enhancer PEI at the N/P 4 ratio) and the magnetofectin was delivered to P. pastoris cells by magneto-transformation with the help of a magnet for 15 minutes. After incubation of transformed cells for different times, the colony-forming time and number of the colonies were determined on YPD plates including 200 μg mL−1 geneticin. The image of the selection plates of antibiotic-resistant colonies and the times of colony formation are given in Fig. 6C and D. As the incubation time of transformed cells increased, the colony formation time became shorter and remained constant after 24 hours. The number of colonies forming also increased in proportion to the incubation time. It is obvious that enough antibiotic-resistant transformants can be obtained in the plates that spread 24 hours after the transformation. After the transformed cell growth, the second selection was performed by transferring randomly selected colonies onto YPD agar without antibiotics, and then, by transferring growing cells onto YPD–geneticin agar. We observed that all the transferred colonies were grown in plates (Fig. S6B†). Colony PCRs performed with gene (Fig. 7A) and vector-specific primers (Fig. 7B) confirmed that the randomly selected yeast clones carried GFP genes in their genomes. The sizes of the products obtained after the reaction were 731 bp and 963 bp, respectively, as expected. Furthermore, integration of the gene into the yeast genome was also confirmed by PCR reactions performed using genomic DNAs belonging to randomly selected three colonies (Fig. 7C). The PCR products were observed where expected in the gel. So, the integration of the GFP gene into the yeast genome was achieved.
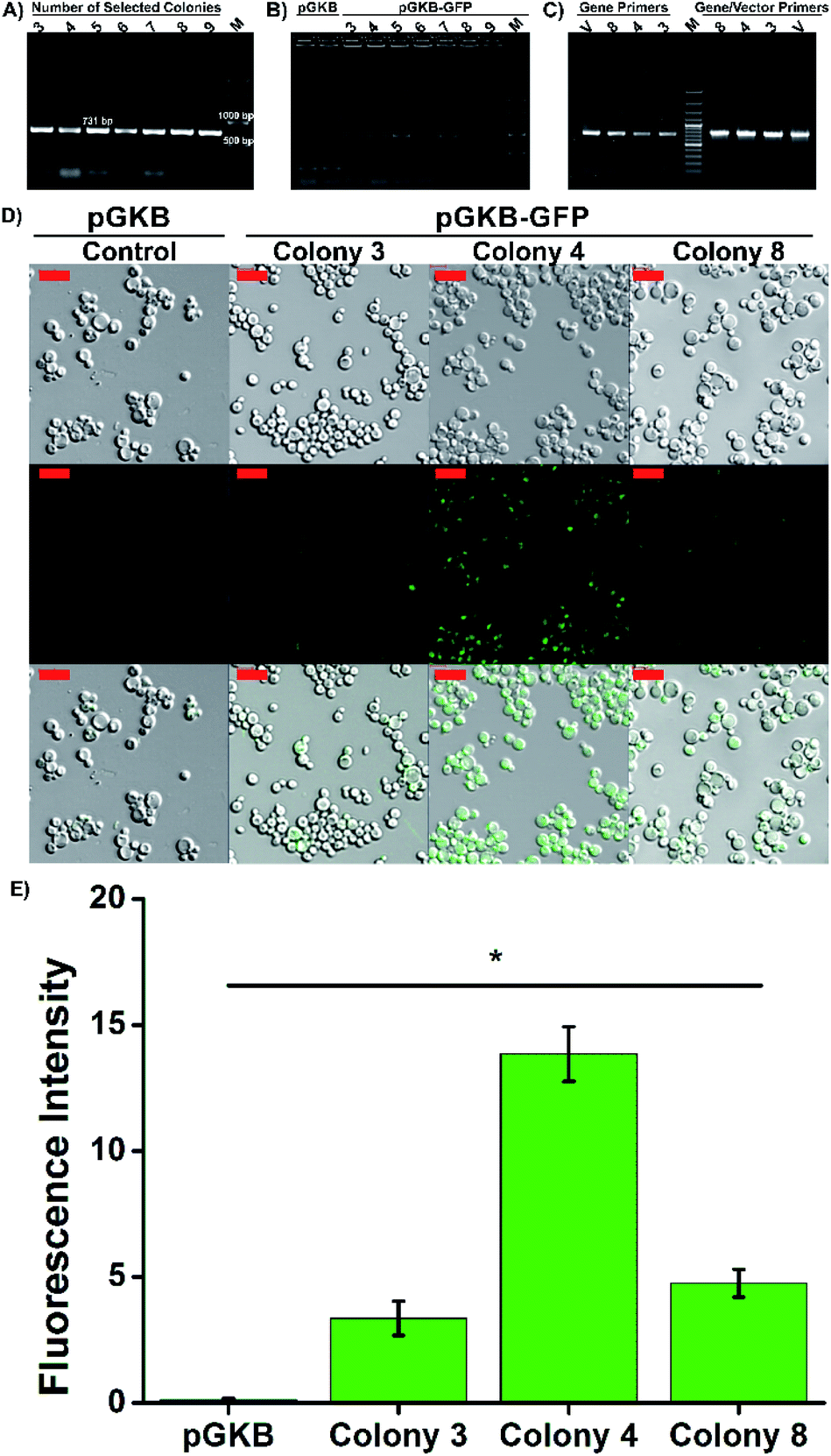 | ||
| Fig. 7 Integration of GFP coding sequence into the P. pastoris genome. Agarose gel electrophoresis image of PCR products performed with (A) the gene specific primers (GFP forward and GFP reverse; 731 bp) and (B) the vector specific primers (pGAP forward and 3′AOX1; 963 bp). Also, magneto-transformation was performed with pGKB plasmid (276 bp). (C) gDNA and gene specific or/gene and vector specific primers (pGAP forward and GFP reverse; 767 bp). M: marker and V: vector, pGKB-GFP. (D) The images of GFP produced colonies after magneto-transformation by confocal microscopy. The scale bar is 10 μm. (E) The fluorescence intensity result was calculated from confocal images. | ||
Three selected yeast colonies were grown in YPD medium and GFP gene expression was determined using confocal laser scanning microscopy. The presence of intracellular GFP was confirmed by merging the brightfield and fluorescence images. As seen in Fig. 7D, while the GFP expression was not observed in the control group cells which were transformed with pGKB by magneto-transformation, the cells forming the 4th colony, transformed with pGKB-GFP by magneto-transformation, had the best GFP production (Fig. 7E). The cells forming the 8th and 3rd colonies, respectively, had less GFP expression than the cells forming the 4th colony. This situation might be attributed to occur multi-copy integration of GFP gene into the genomes of yeast cells forming the 4th colony (Fig. 7E).42,43 Compared with the LiCl or electroporation method (Table 2), the suggested magneto-transformation system achieved an efficient transformation and resulted in sufficient numbers of transformants. Although it is known that the expression of the geneticin resistance gene requires at least two hours,44 a sufficient number of transformants with geneticin resistance were obtained after 24 hours of the transformation. Transformation efficiency was determined to be 1.3 × 103 transformants per μg DNA when the cells were spread 24 hours after the transformation. Also, the number of transformants increased as this time increased. This value was reported as nearly 102 to 103 transformants per μg DNA with LiCl or 103 to 104 transformants per μg DNA with the electroporation method.26
| Transformation method | DNA (μg) | Equipment | Transformation efficiency | Reference |
|---|---|---|---|---|
| Magneto-transformation | 1 | Magnet | High | This study |
| Spheroplast–PEG transformation | 20 | — | High | 34 |
| Lithium method | 5–10 | — | High | 26 and 38 |
| Electroporation | 10 | Gene pulser apparatus | High | 26 and 36 |
| Glass bead method | 1 | Glass bead | Very low | 35 |
| Biolistic method | 10 | Macro carrier holder | Medium | 37 |
Conclusions
In this study, we described an efficient gene delivery method for the P. pastoris expression system. We used Fe3O4@PEI MNPs as a gene carrier and achieved integration of the heterologous gene into the genome. So far, although there are plentiful methods to obtain recombinant yeast cells such as LiCl and electroporation,45–48 this is the first study to describe gene delivery to P. pastoris by Fe3O4@PEI MNPs under the influence of a magnetic field.This novel method based on Fe3O4@PEI MNPs offers some important advantages for the transformation of P. pastoris cells as follows; this gene delivery method is (i) simple, advanced equipment free and nontoxic to yeasts, (ii) Fe3O4@PEI MNPs as a gene carrier platform can simply be obtained by the co-precipitation method in a regular laboratory, (iii) both destroying the cell wall and the preparation of competent cells are performed before the transformation in other methods, but is not required in the suggested method, (iv) the suggested transformation process is carried out in a short time (magnetofectin preparation: 60 minutes, transformation: 15 minutes), (v) an extra carrier, such as salmon sperm DNA, is not required to integrate the recombinant DNA into the yeast genome, (vi) gene integration can successfully be accomplished using 5–10 times less linear recombinant plasmid DNA (1 μg) in comparison to conventional methods and (vii) we envision that, potentially, our method can be used for transformation of other yeasts, such as Saccharomyces cerevisiae, Hansenula polymorpha, Kluyveromyces lactis and Yarrowia lipolytica used in the production of recombinant proteins.
Author contributions
Y. U. is reponsible for the project and planning of the work; S. Y., K. S., and M. A. performed experimental studies, data collection and processing; A. M. edited the publication, and all authors are responsible for the writing of the publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Scientific Research Foundation of Atatürk University (FYL-2018-6854). The authors would like to acknowledge East Anatolia High Technology Application and Research Center (DAYTAM, Erzurum, Turkey) for the technical support.References
- A. L. Demain and P. Vaishnav, Biotechnol. Adv., 2009, 27(3), 297–306 CrossRef CAS PubMed.
- R. M. Bill, Front. Microbiol., 2014, 5, 1–5 Search PubMed.
- X. García-Ortega, E. Cámara, P. Ferrer, J. Albiol, J. L. Montesinos-Seguí and F. Valero, Nat. Biotechnol., 2019, 53, 24–34 Search PubMed.
- G. Walsh, Nat. Biotechnol., 2014, 32, 992–1000 CrossRef CAS PubMed.
- J. L. Adrio and A. L. Demain, Bioengineered Bugs, 2010, 1, 116–131 CrossRef PubMed.
- G. Potvin, A. Ahmad and Z. Zhang, Biochem. Eng. J., 2012, 64, 91–105 CrossRef CAS.
- M. Ahmad, M. Hirz, H. Pichler and H. Schwab, Appl. Microbiol. Biotechnol., 2014, 98, 5301–5317 CrossRef CAS PubMed.
- B. Gasser, R. Prielhofer, H. Marx, M. Maurer, J. Nocon, M. Steiger, V. Puxbaum, M. Sauer and D. Mattanovich, Future Microbiol., 2013, 8, 191–208 CrossRef CAS PubMed.
- J. L. Corchero, B. Gasser, D. Resina, W. Smith, E. Parrilli, F. Vázquez, I. Abasolo, M. Giuliani, J. Jäntti, P. Ferrer, M. Saloheimo, D. Mattanovich, S. Schwartz, M. L. Tutino and A. Villaverde, Biotechnol. Adv., 2013, 31, 140–153 CrossRef CAS PubMed.
- C. Plank, O. Zelphati and O. Mykhaylyk, Adv. Drug Delivery Rev., 2011, 63, 1300–1331 CrossRef CAS PubMed.
- F. Scherer, M. Anton, U. Schillinger, J. Henke, C. Bergemann, A. Kruger, B. Gänsbacher and C. Plank, Gene Ther., 2002, 9, 102–109 CrossRef CAS PubMed.
- J. Dobson, Gene Ther., 2006, 13, 283–287 CrossRef CAS PubMed.
- Z. Shankayi, S. M. P. Firoozabadi, M. Mansourian and A. Mahna, Electromagn. Biol. Med., 2014, 33, 154–158 CrossRef CAS PubMed.
- H. Rui, R. Xing, Z. Xu, Y. Hou, S. Goo and S. Sun, Adv. Mater., 2010, 22, 2729–2742 CrossRef PubMed.
- P. Tseng, J. W. Judy and D. Di Carlo, Nat. Methods, 2012, 9(11), 1113–1119 CrossRef CAS PubMed.
- P. T. Yin, S. Shah, N. J. Pasquale, O. B. Garbuzenko, T. Minko and K. B. Lee, Biomaterials, 2016, 81, 46–57 CrossRef CAS PubMed.
- J. Y. Chen, Y. L. Liao, T. H. Wang and W. C. Lee, Enzyme Microb. Technol., 2006, 39, 366–370 CrossRef CAS.
- S. I. Jenkins, M. R. Pickard, N. Granger and D. M. Chari, ACS Nano, 2011, 5, 6527–6538 CrossRef CAS PubMed.
- T. Buerli, C. Pellegrino, K. Baer, B. Lardi-Studler, I. Chudotvorova, J. M. Fritschy, C. Fuhrer and I. Medina, Nat. Protoc., 2007, 2, 3090–3101 CrossRef CAS PubMed.
- X. Zhao, Z. Meng, Y. Wang, W. Chen, C. Sun, B. Cui, J. Cui, M. Yu, Z. Zeng, S. Guo, D. Luo, J. Q. Cheng, R. Zhang and H. Cui, Nat. Plants, 2017, 3, 956–964 CrossRef CAS PubMed.
- Q. Peng, D. Huo, H. Li, B. Zhang, Y. Li, A. Liang, H. Wang, Q. Yu and M. Li, Chem.-Biol. Interact., 2018, 287, 20–26 CrossRef CAS PubMed.
- W. Jiang, J. Wu, R. Tian and W. Jiang, J. Porous Mater., 2017, 24, 257–265 CrossRef CAS.
- L. Zhang, Y. Li, J. C. Yu, Y. Chen and K. Ming, J. Mater. Chem. B, 2014, 2, 7936–7944 RSC.
- J. Yang, L. Nie, B. Chen, Y. Liu, Y. Kong, H. Wang and L. Diao, Yeast, 2014, 31, 115–125 CrossRef CAS PubMed.
- W. Wang, W. Li, L. Ou, E. Flick, P. Mark, C. Nesselmann, C. A. Lux, H. H. Gatzen, A. Kaminski, A. Liebold, K. Lützow, A. Lendlein, R. K. Li, G. Steinhoff and N. Ma, J. Cell. Mol. Med., 2011, 15, 1989–1998 CrossRef CAS PubMed.
- L. T. Invitrogen, Cat. no. K1740-01, Manual part no. 25-0172.
- A. Popat, S. B. Hartono, F. Stahr, J. Liu, S. Z. Qiao and G. Q. Lu, Nanoscale, 2011, 3, 2801–2818 RSC.
- M. Arsianti, M. Lim, C. P. Marquis and R. Amal, Langmuir, 2010, 26, 7314–7326 CrossRef CAS PubMed.
- X. Wang, L. Zhou, Y. Ma, X. Li and H. Gu, Nano Res., 2009, 2, 365–372 CrossRef CAS.
- M. M. Azevedo, P. Ramalho, A. P. Silva, R. Teixeira-Santos, C. Pina-Vaz and A. G. Rodrigues, J. Med. Microbiol., 2014, 63, 1167–1173 CrossRef CAS.
- I. Sadowska-Bartosz, A. Paczka, M. Moloń and G. Bartosz, FEMS Yeast Res., 2013, 13, 820–830 CrossRef CAS PubMed.
- O. Mykhaylyk, Y. S. Antequera, D. Vlaskou and C. Plank, Nat. Protoc., 2007, 2, 2391–2411 CrossRef CAS PubMed.
- S. Kawai, W. Hashimoto and K. Murata, Bioengineered Bugs, 2010, 1, 395–403 CrossRef PubMed.
- Y. Oguro, H. Yamazaki, Y. Shida, W. Ogasawara, M. Takagi and H. Takaku, Biosci., Biotechnol., Biochem., 2015, 79, 512–515 CrossRef CAS PubMed.
- J. Wang, P. Jiang, Y. Cui, X. Guan and S. Qin, Phycologia, 2010, 49, 355–360 CrossRef CAS.
- K. Moridi, M. Hemmaty, M. R. Akbari Eidgahi, M. Fathi Najafi, H. Zare, K. Ghazvini and A. Neshani, Gene Reports, 2020, 21, 100900 CrossRef.
- S. D. M. Arras and J. A. Fraser, PLoS One, 2016, 11, 1–14 Search PubMed.
- Y. Ünver, E. B. Kurbanoğlu and O. Erdoğan, Turk. J. Biol., 2015, 39, 649–655 CrossRef.
- J. M. Cregg, K. J. Barringer, A. Y. Hessler and K. R. Madden, Mol. Cell. Biol., 1985, 5, 3376–3385 CAS.
- H. R. Waterham, Y. de Vries, K. A. Russel, W. Xie, M. Veenhuis and J. M. Cregg, Mol. Cell. Biol., 1996, 16, 2527–2536 CrossRef CAS PubMed.
- J. M. Cregg, K. R. Madden, K. J. Barringer, G. P. Thill and C. A. Stillman, Mol. Cell. Biol., 1989, 9, 1316–1323 CAS.
- R. Shirvani, M. Barshan-tashnizi and M. Shahali, Biologicals, 2020, 65, 10–17 CrossRef CAS PubMed.
- X. Song, C. Shao, Y. Guo, Y. Wang and J. Cai, BMC Biotechnol., 2019, 19, 1–9 CrossRef CAS.
- O. G. Guerra, I. G. S. Rubio, C. G. da Silva Filho, R. A. Bertoni, R. C. dos Santos Govea and E. J. Vicente, J. Microbiol. Methods, 2006, 67, 437–445 CrossRef CAS.
- S. Liang, C. Zou, Y. Lin, X. Zhang and Y. Ye, Biotechnol. Lett., 2013, 35, 1865–1871 CrossRef CAS PubMed.
- Y. Unver, B. Sensoy Gun, M. Acar and S. Yildiz, Prep. Biochem. Biotechnol., 2020, 1–8 CrossRef PubMed.
- K. Hori, S. Okano and F. Sato, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- Y. Lu, C. Fang, Q. Wang, Y. Zhou, G. Zhang and Y. Ma, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00079a |
| This journal is © The Royal Society of Chemistry 2021 |