
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotothermal release of an encapsulated therapeutic agent from polymer-wrapped gold nanoparticles†
Daina V.
Baker
,
Khaled M.
Arafeh
,
Amir
Asadirad
,
Brahmjot
Kaur
,
Rameez
Raza
and
Neil R.
Branda
 *
*
4D LABS, Department of Chemistry, Simon Fraser University, 8888 University Drive, Burnaby, BC V5A 1S6, Canada. E-mail: nbranda@sfu.ca
First published on 2nd July 2021
Abstract
Visible light is used to generate heat from gold nanoparticles wrapped in an amphiphilic polymer shell and trigger a reverse Diels–Alder reaction of a ‘caged’ tyrphostin therapeutic agent. The hydrophilic nature of the released agent results in it travelling from the polymer to the bulk medium, while the byproduct of the reaction is trapped in the hydrophobic layer of the nano-assembly.
Introduction
Molecular systems that use light to trigger the activation of small compounds from ‘blocked’ or ‘caged’ forms conveniently allow the user to choose when and where the release and delivery of therapeutics and other important molecular species occurs.1,2 This type of ‘on-command’ release has many appealing features including dose control, reduced administration frequency and being able to match the many pulsatile functions occurring in the body.3,4 One major issue that needs to be overcome is the fact that most of the existing ‘release-on-command’ systems rely on high-energy photons which are not compatible with sensitive environments because of the damage they can cause and their inability to penetrate deeply into materials.A variety of delivery ‘vehicles’ have been developed to sequester molecules until they can be delivered to the site of interest using less-damaging stimuli such as ultrasound and low-energy light.5 Among these examples, metal nanoparticles have been one of the preferred ‘vehicles’ of choice.6–8 The ability of metallic nanostructures to absorb light and convert it into heat is one solution to overcome the reliance on UV or high-energy visible light. Many types of metal nanoparticles, such as those made up of gold or silver, undergo this photothermal effect9 and have can be used to carry out important chemical reactions close the surface of the particles without heating the bulk environment. Illustrative examples include those that release therapeutics,10 single strands of nucleic acids11 and imaging agents12,13 decorated onto the nanoparticle's surface. In all cases, the release is achieved without damaging surrounding cells or tissue.
Gold nanoparticles are particularly appealing candidates for the photothermal release of molecules within sensitive environments, such as those found in biology, because of their biocompatibility, and ease of synthesis and surface functionalization.14–17 The examples cited in the previous paragraph all have the molecular agent of interest directly tethered onto the surface of the nanoparticles through thiol-modified linkers. While proven successful, this surface-modified approach has several disadvantages. The first is based on the need for the agent of interest to displace some of the ligands originally decorated onto the metal surface to enhance the stability, which may interfere with properties of the nanosystem (e.g. biocompatibility and aggregation-resistance). This issue is particularly relevant when the agent of interest has low water-solubility. Another issue with this system is the tendency for the undesirable cleavage of the Au–S bond anchoring the molecule to the nanoparticle11 resulting in a further decreased stability of the nanoparticle.14
A more versatile and universal approach is to encapsulate the agent of interest within an amphiphilic shell surrounding the nanoparticle. We18,19 and others20,21 have used this approach to trap hydrophobic molecules near the metal surface because the hydrophobic layer of the polymeric shell is near the nanoparticle, while the hydrophilic layer of the shell projects away from the nanoparticle imparting water solubility. In this report, we illustrate how this spontaneous encapsulation approach can be used to trap a bicyclic molecule 1, which is the Diels–Alder product of a 1,3-cyclohexadiene and (4-hydroxybenzylidene)malononitrile. The latter compound is an important target as it is known to the biochemical community as tyrphostin A8 (T8), a simple version of a class of chemotherapeutic agents.22–25 This compound is also an ideal candidate for our approach due to its short circulation time in the bloodstream so controlled release from an inactive form would be beneficial.26,27
Our concept is illustrated in Fig. 1 where three unique environments are depicted. The one on the far left represents the state of the nano-assembly prior to any irradiation. In this case, the bicyclic compound (1) is trapped within the hydrophobic layer of the encapsulating shell close the nanoparticle's surface. The middle ‘cell’ would be the result when the nano-assembly is exposed to visible light and the photothermal effect has generated enough heat to trigger the retro-Diels–Alder reaction producing tyrphostin A8 and the cyclohexadiene. The far right ‘cell’ shows the ultimate outcome. Because the tyrphostin A8 is less hydrophobic, it should be released from the nano-assembly where it can be effective as a chemotherapeutic agent. The cyclohexadiene retains its hydrophobic nature and should remain trapped in the nano-assembly. This last point is an additional advantage of our approach as it prevents any detrimental effect the byproducts of the release event may cause and differs from the traditional photo-cages.28
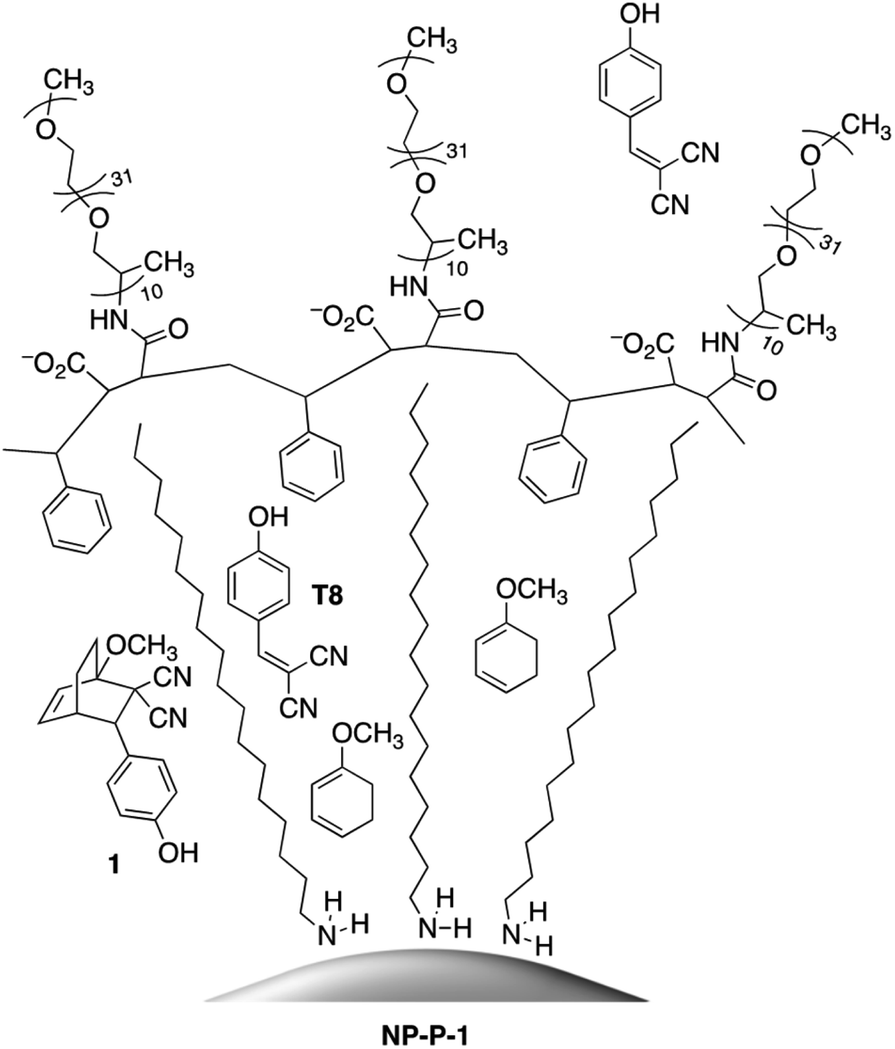 | ||
| Fig. 1 Left ‘cell’: bicyclic compound 1 encapsulated within the hydrophobic shell surrounding the octadecylamine-coated gold nanoparticles. Middle ‘cell’: the retro-Diels–Alder reaction of bicyclic compound 1 releasing tyrphostin A8 (T8) and the diene. Right ‘cell’: release of T8 into the bulk aqueous environment and retention of 1-methoxy-1,3-cyclohexadiene within the hydrophobic layer of the nano-assembly. | ||
Results and discussion
Preparation of encapsulated nanoassembly NP-P-1
The nano-assemblies are prepared as shown in Scheme 1. The bicyclic compound 1 can be prepared in one step through the Diels–Alder reaction of commercially available tyrphostin A8 ((4-hydroxybenzylidene)malononitrile) and 1-methoxy-1,3-cyclohexadiene in 1,4-dioxane. Using these conditions, the product is exclusively the exo-isomer.†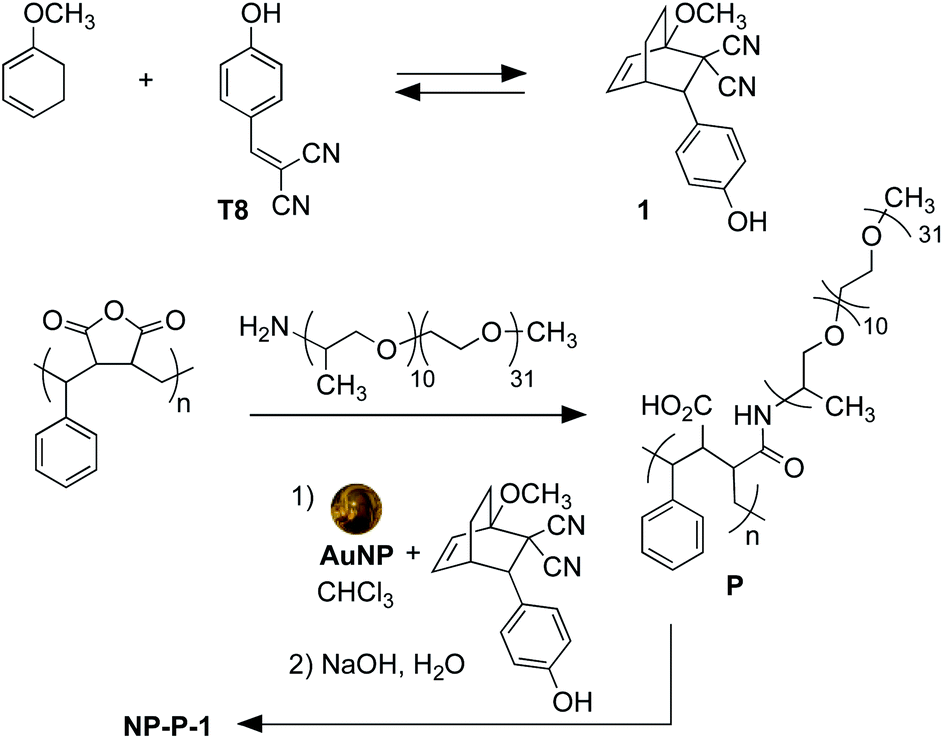 | ||
| Scheme 1 Synthesis of bicyclic compound 1 from tyrphostin A8 (T8) (top) and wrapping and encapsulation of the nano-assemble NP-P-1 (bottom). | ||
A single step produces amphiphilic polymer P by ring-opening poly(styrene-co-maleic anhydride) with O-(2-aminopropyl)-O′-(2-methoxyethyl)polypropylene glycol (JeffAmine® M-2070). These two polymeric systems ensure that the final nano-assemblies have good water dispersibility while maintaining a lipid-like core structure to trap hydrophobic molecules.
The initial water-dispersible gold nanoparticles are best prepared as an aqueous dispersion following the literature procedure,29 which uses sodium citrate as both the reducing and capping agent. In our hands, this procedure generates gold nanoparticles with an average diameter of 11.2 nm ± 1.2 nm as measured from the TEM images.† The UV-visible absorption spectrum of the citrate-coated gold nanoparticles shows its characteristic, sharp surface plasmon resonance (SPR) band centred at 519 nm (black line in Fig. 2a). The necessary hydrophobic gold nanoparticles can be readily prepared using the common ligand exchange method where the citrate ligands are replaced by octadecylamine (ODA) rendering nanoparticles that are dispersible in CHCl3.30 As anticipated, this ligand exchange results in a red small shift in the SPR band (blue line in Fig. 2a), which can be attributed to the increase in the refractive index of the medium in which they are dispersed (CHCl3nD20 = 1.4459, water nD20 = 1.3334).31
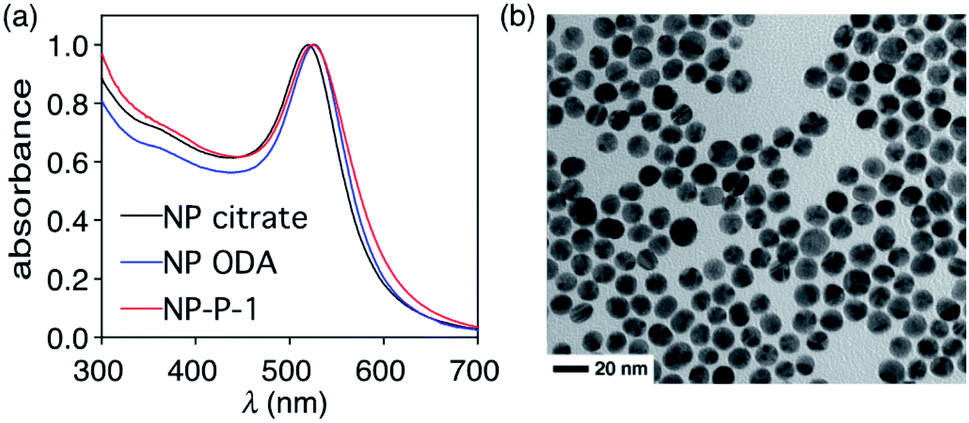 | ||
| Fig. 2 (a) Normalized UV-vis absorption spectra of dispersions of citrate-coated gold nanoparticles AuNP (NP citrate) in water (2.55 mg mL−1) (black line), ODA-coated gold nanoparticles (NP ODA) in chloroform (0.39 mg mL−1) (blue line), and NP-P-1 in water (2.66 μg μL−1) (red line). (b) TEM image of the final nano-assemblies NP-P-1. | ||
The final nano-assembly (NP-P-1) is prepared starting from a CHCl3 solution of the ODA-coated nanoparticles by sequential addition of the amphiphilic polymer (P) followed by a solution of bicyclic compound 1 in the same solvent. After removing the organic solvent, the nano-assembly can now be dispersed in alkaline water, which promotes the self-assembly process. The UV-visible absorption spectrum of the aqueous dispersion of NP-P-1 shows a bathochromic shift in the SPR band which is now centred at 524 nm (red line in Fig. 2a). The average diameter of the NP-P-1 nano-assembly is 10.7 nm ± 0.6 nm as measured using TEM (Fig. 2b) which is consistent with the diameter of the original citrate-coated nanoparticles (11.2 nm ± 1.2 nm). In contrast, the DLS measurements (Fig. S2, left†), which provide an estimated size of the entire nano-assembly including the organic layer around the inorganic core, show an increase in size of the nano-assembly NP-P-1 after encapsulation to be approximately 21.3 nm.‡ This difference in size measurements of the nano-assembly NP-P-1 from TEM and DLS can be attributed to the addition of the amphiphilic organic shell with fully extended PEG chains around the inorganic core in aqueous environments: the organic chains appear only faintly on TEM. The loading of compound 1 trapped within the nano-assembly NP-P-1 can be estimated using UV-visible absorption spectroscopy and was estimated by determining the amount of released tyrphostin A8 (in the photothermal experiment) and comparing it to an authentic sample of 1. The number of molecules of adduct 1 per nanoparticle is approximately 92.†
Thermolysis of bicyclic compound 1
The heat-induced retro-Diels–Alder reaction of the bicyclic compound can be examined by heating at 90 °C a 4.1 mM solution of 1 in water containing 40 vol% aqueous DMSO and monitoring the progress using UV-vis absorption spectroscopy (Fig. 3a and b) by taking 20 μL aliquot amounts of reaction mixture and immediately quenching them with ice-cold 1 vol% aqueous DMSO to obtain a final concentration of 82 μM.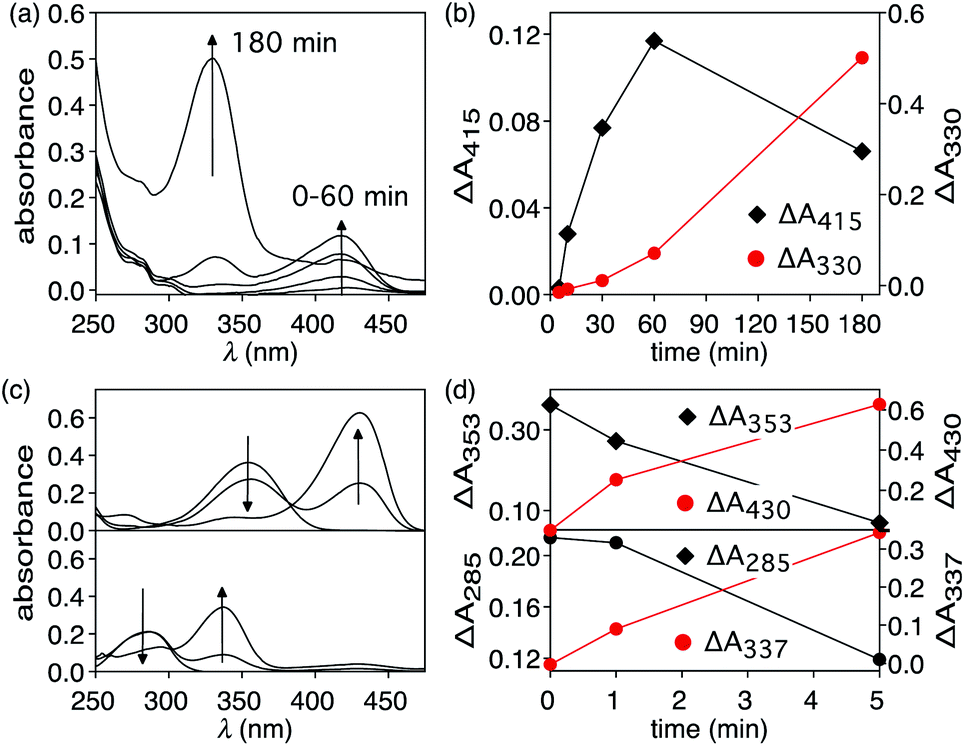 | ||
| Fig. 3 (a) Changes in the UV-vis absorption spectrum when a 4.1 mM 40% aqueous DMSO solution of 1 is heated at 90 °C for 3 hours. (b) Changes in absorption intensities at 415 nm and 330 nm corresponding to T8 and p-hydroxybenzaldehyde, respectively. (c) Changes in the UV-vis absorption spectrum when 40% aqueous DMSO solutions of T8 (top) and p-hydroxybenzaldehyde (bottom) are heated at 50 °C for 5 minutes. (d) Changes in absorption intensities at 353 nm and 430 nm for T8 (top) and 285 nm and 337 for p-hydroxybenzaldehyde (bottom). | ||
Within the first ten minutes of heating, a broad absorption band centered at 415 nm appears. After this time, an additional absorption band centered at 330 nm emerges. Both bands continue to grow, however the one at longer wavelength eventually decreases in intensity and is barely observable after 3 hours. This band corresponds to the released T8 from the bicyclic compound 1. The higher-energy band corresponds to p-hydroxybenzaldehyde showing that the reverse Knoevenagel condensation reaction occurs to produce the aldehyde and malononitrile (Scheme 2).32
 | ||
| Scheme 2 Retro-Diels–Alder reaction of bicyclic compound 1 and subsequent hydrolysis to 4-hydroxybenzaldehyde. | ||
The appearance of the bands corresponding to T8 and p-hydroxybenzaldehyde in regions of the spectrum that are shifted to longer wavelengths than expected can be explained by the solvatochromic behavior of both chromophores. When freshly prepared 10 μM 40 vol% aqueous DMSO solutions of T8 and p-hydroxybenzaldehyde are compared, the absorption bands appear, as expected, at 353 nm and 285 nm, respectively as shown in Fig. 3. Heating these solutions at 50 °C for 5 minutes results in the disappearance of both bands and the simultaneous appearance of bands that are red-shifted to 430 nm and 337 nm (Fig. 3c and d). This solvatochromic behavior has been reported already33 and can be attributed to the heat-promoted solvation of the hydroxyl group on the phenol by water through hydrogen bonds. The result is a pseudo-phenolate. The hydrolysis and production of the aldehyde must occur from the released (4-hydroxybenzylidene)malononitrile (T8) because the bicyclic compound 1 does not contain a dicyanoethene functional group.
Photothermolysis of nano-assembly NP-P-1
When an aqueous dispersion of the nano-assembly NP-P-1 (2.66 μg μL−1) is irradiated with a nanosecond pulse laser (10 ns, 532 nm) at 800 mW cm−2,§ an increase in a band centred at 412 nm in the UV-vis absorption spectrum appears after only 1 minute of exposure (Fig. 4). This band corresponds to the tyrphostin A8 released from bicyclic compound 1 through a retro-Diels–Alder reaction as illustrated in the thermolysis experiments described previously. The changes in the spectrum stop after 5 min, at which time centrifugation of the nanoassembly using a spin filter with a 10 kDa cutoff shows that the flow-through solution contains the released T8. High-resolution mass spectrometry analysis of this flow-through solution confirms the presence of tyrphostin A8 (HRMS-ESI: m/z 169.0403 (M − H) calc. C10H5N2O− 169.0407). No evidence of 1-methoxy-1,3-cyclohexadiene is observed by HRMS indicating that, as designed, this species remains trapped within the polymer shell surrounding the nanoparticle.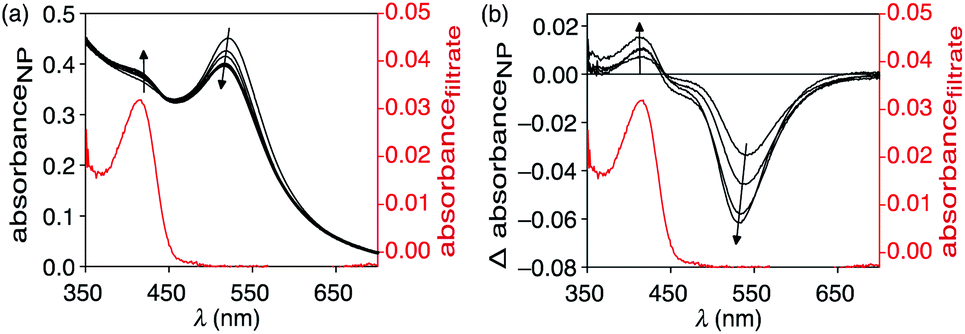 | ||
| Fig. 4 (a) Changes in the UV-vis absorption spectrum when an aqueous solution of NP-P-1 (2.66 μg μL−1) is irradiated with a nanosecond pulse laser (10 ns, 532 nm) at 800 mW cm−2. The red line corresponds to released T8 in the supernatant after centrifugation. (b) The difference spectra of the same data clearly showing the growth of an absorption band at 412 corresponding to the released T8 and a slight blue-shift of the SPR band of the nanoparticle. | ||
Another change that occurs during the irradiation is the decrease in intensity and hypsochromic shift of the band corresponding to the nanoparticle's plasmon resonance centered at 525 nm. This change also stops by the ends of the exposure period and ends up at 517 nm. This shift towards shorter wavelengths can be attributed to possible fragmentation and vaporization of gold nanoparticles, which is a direct consequence of the fact that the gold nanoparticles absorb laser energy leading to high temperatures inside them with low heat transfer to the surrounding solvent.34–36 However, encapsulating the nanoparticles within the amphiphilic polymer (P) significantly reduced the laser-induced thermal deformation of gold nanoparticles as compared with the non-capsulated ones as shown in Fig. S5.† This enhanced stability is likely due to the thermal stability of the polymer shell compared with thermally labile ligands, which detach can from the surface during to photothermal process. It seems that the polymer shell improves the heat dissipation from gold surface, keeps the particles isolated from each other and reduces thermal fusion and aggregation.
Under the conditions employed in our experiments, no observable evidence of hydrolysis of the photothermally-released (4-hydroxybenzylidene)malononitrile (T8) to p-hydroxybenzaldehyde was present. This observation implies that, despite the system generating sufficient heat to initiate the retro-Diels–Alder reaction, which in our thermal experiments is enough to hydrolyze T8, the interior of the amphiphilic shell lacks enough water for this reaction. By the time the released T8 migrates to the bulk aqueous medium, the heat is reduced below the point where it can trigger hydrolysis because the photothermal effect is highly localized.
Conclusions
In this report we illustrate a more versatile approach to trapping ‘caged’ molecules for subsequent photothermal release using visible light. The advantages of this encapsulation approach are increased stability during exposure to pulsed lasers, the avoidance of potentially unstable thiol groups to anchor the agent onto the nanoparticle surface, and the retention of the byproduct of the release within the hydrophobic shell.Our choice to demonstrate this approach using the simplest tyrphostin A8 is because it represents a broader class of electron-poor dienophiles. Other molecules with similar electronic characteristics could be used analogously. In order for the byproduct to remain in the polymer shell, a change in hydrophobicity must occur after the photothermal bond breaking, which is a consideration that must be appreciated when designing alternative systems. Additionally, it is expected that the inclusion of any polymer shell that competes for the excitation light would reduce the amount of light able to reach to the surface of the nanoparticle and reduce the photothermal effect efficiency. Further research should consider the polymer chain length, which will affect the shell thickness, and the laser power required to trigger a kinetically meaningful reaction. Finally, the exploration of the efficiency of this system compared to systems with molecules tethered to the polymer would of additional interest.
Author contributions
D. V. B. and B. K. conducted most of the experimentation, characterization, prepared the original draft and worked on the revisions. R. R. assisted with the validation of preliminary UV-visible experiments and preparation of the first draft. K. M. A. conducted the TEM and LASER experiments for the photothermolysis, designed the experiment methodology, analyse the results and assisted with revision of drafts. A. A. formulated the idea of the project and developed the methodology. N. R. B. worked on all drafts of the manuscript and supervised the overall project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada. This work made use of 4D LABS shared facilities supported by the Canada Foundation for Innovation (CFI), British Columbia Knowledge Development Fund (BCKDF), and Simon Fraser University. The authors thank Dr Saeid Kamal, manager of the LASIR facility at 4D LABS for his help with setting up the laser experiments, and Hongwen Chen, mass spectrometer technican of the SFU Chemistry department.Notes and references
- Y. Tao, H. F. Chan, B. Shi, M. Li and K. W. Leong, Adv. Funct. Mater., 2020, 30, 2005029 CrossRef CAS.
- E. Mathiowitz and M. Cohen, J. Membr. Sci., 1989, 40, 67 CrossRef CAS.
- J. R. Robinson and L. J. Gauger, J. Allergy Clin. Immunol., 1986, 78, 676 CrossRef CAS.
- S. D. Brown, P. Nativo, J. A. Smith, D. Stirling, P. R. Edwards, B. Venugopal, D. J. Flint, J. A. Plumb, D. Graham and N. J. Wheate, J. Am. Chem. Soc., 2010, 132, 4678 CrossRef CAS PubMed.
- S. Yamashita, H. Fukushima, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Langmuir, 2011, 27, 14621 CrossRef CAS PubMed.
- C. S. Brazel, Pharm. Res., 2009, 26, 644 CrossRef CAS PubMed.
- D. Pissuwan, T. Niidome and M. B. Cortie, J. Controlled Release, 2011, 149, 65 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed. Engl., 2010, 49, 3280 CrossRef CAS PubMed.
- C. M. Cobley, J. Chen, E. C. Cho, L. V. Wang and Y. Xia, Chem. Soc. Rev., 2011, 40, 44 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS PubMed.
- L. Poon, W. Zandberg, D. Hsiao, Z. Erno, D. Sen, B. D. Gates and N. R. Branda, ACS Nano, 2010, 4, 6395 CrossRef CAS PubMed.
- A. B. S. Bakhtiari, D. Hsiao, G. Jin, B. D. Gates and N. R. Branda, Angew. Chem., Int. Ed., 2009, 48, 4166 CrossRef CAS PubMed.
- J. F. Hainfeld, H. M. Smilowitz, M. J. O'Connor, F. A. Dilmanian and D. N. Slatkin, Nanomedicine, 2013, 8, 1601 CrossRef CAS PubMed.
- I. Fratoddi, I. Venditti, C. Cametti and M. V. Russo, J. Mater. Chem. B, 2014, 2, 4204 RSC.
- G. F. Paciotti, L. Myer, D. Weinreich, D. Goia, N. Pavel, R. E. McLaughlin and L. Tamarkin, Drug Delivery, 2004, 11, 169 CrossRef CAS PubMed.
- O. S. Muddineti, B. Ghosh and S. Biswas, Int. J. Pharm., 2015, 484, 252 CrossRef CAS PubMed.
- E. E. Conner, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325 CrossRef PubMed.
- T. Wu, C. J. Boyer, M. Barker, D. Wilson and N. R. Branda, Chem. Mater., 2013, 25, 2495 CrossRef CAS.
- T. Wu, J. Oake, Z. Liu, C. Bohne and N. R. Branda, ACS Omega, 2018, 3, 7673 CrossRef CAS PubMed.
- S. Kashyap, N. Singh, B. Surnar and M. Jayakannan, Biomacromolecules, 2016, 17, 384 CrossRef CAS PubMed.
- X. Wang, C. Liu, Z. Li, C.-Y. Tang, W.-C. Law, X. Gong, Z. Liu, Y. Liao, G. Zhang, S. Long and L. Chen, J. Phys. Chem. C, 2019, 123, 10658 CrossRef CAS.
- A. Gazit, N. Osherov, I. Posner, P. Yaish, E. Poradosu, A. Levitzki, A. Gazit and C. Gilon, J. Med. Chem., 1991, 34, 1896 CrossRef CAS PubMed.
- A. Gazit, P. Yaish, C. Gilon and A. Levitzki, J. Med. Chem., 1989, 32, 2344 CrossRef CAS PubMed.
- C. Wu, S. H. Hsiao, M. Murakami, M. J. Lu, Y. Q. Li, C. H. Hsieh, S. V. Ambudkar and Y. S. Wu, Cancer Lett., 2017, 409, 56 CrossRef CAS PubMed.
- S. H. Pang, Z. R. Guo and X. T. Liang, Yaoxue Xuebao, 1997, 32, 515 CAS.
- R. Robinson, J. P. Bertram, J. L. Reiter and E. B. Lavik, J. Microencapsulation, 2010, 27, 263 CrossRef CAS PubMed.
- M. Chorny, I. Fishbein, H. D. Danenberg and G. Golomb, J. Controlled Release, 2002, 83, 401 CrossRef CAS.
- G. C. R. Ellis-Davies, Nat. Methods, 2007, 4, 619 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hiller, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- L. Polavarapu and Q. H. Xu, Nanotechnology, 2009, 20(18), 185606 CrossRef PubMed.
- S. K. Ghosh, S. Nath, S. Kundu, K. Esumi and T. Pal, J. Phys. Chem. B, 2004, 108, 13963 CrossRef CAS.
- L. F. Meyer and D. K. Shah, J. Pharm. Anal., 2019, 9, 163 CrossRef PubMed.
- S. M. Panja, J. Mol. Liq., 2020, 299, 112194 CrossRef CAS.
- A. Takami, H. Kurita and S. Koda, J. Phys. Chem. B, 1999, 103, 1226 CrossRef CAS.
- S. Inasawa, M. Sugiyama and Y. Yamaguchi, J. Phys. Chem. B, 2005, 109, 9404 CrossRef CAS PubMed.
- M. Maciulevičius, A. Vinčiunas, M. Brikas, A. Butsen, N. Tarasenka, N. Tarasenko and G. Račiukaitis, Appl. Phys. A: Mater. Sci. Process., 2013, 111, 289 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00289a |
| ‡ This value was obtained directly from the DLS instrument using Malvern General Purpose & Multiple Narrow Mode NNLS (non-negative least squares) algorithm to treat the autocorrelation equation of the intensity trace describing sample intensity fluctuation recorded during the experiment. |
| § Not all of the light is absorbed by the sample. The absorbed power is 230 mW cm−2. The remainder of the light is transmitted or absorbed by the cuvette. |
| This journal is © The Royal Society of Chemistry 2021 |