
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoprobes to investigate nonspecific interactions in lipid bilayers: from defect-mediated adhesion to membrane disruption†
Nicolò
Razza
 a,
Alessio D.
Lavino
a,
Giulia
Fadda
bc,
Didier
Lairez
cd,
Andrea
Impagnatiello
d,
Daniele
Marchisio
a,
Marco
Sangermano
a and
Giancarlo
Rizza
*d
a,
Alessio D.
Lavino
a,
Giulia
Fadda
bc,
Didier
Lairez
cd,
Andrea
Impagnatiello
d,
Daniele
Marchisio
a,
Marco
Sangermano
a and
Giancarlo
Rizza
*d
aDepartment of Applied Science and Technology, Politecnico di Torino, Torino, Italy
bCSPAT UMR 7244, Université Sorbonne Paris Nord, 74 rue Marcel Cachin, 93017, Bobigny, France
cLaboratoire Léon Brillouin, CNRS, CEA, Université Paris-Saclay, 91191, Gif-sur-Yvette Cedex, France
dLaboratoire des Solides Irradiés (LSI), Institut Polytechnique de Paris, CEA/DRF/IRAMIS, CNRS, 91128, Palaiseau Cedex, France. E-mail: giancarlo.rizza@polytechnique.edu
First published on 8th July 2021
Abstract
When a lipid membrane approaches a material/nanomaterial, nonspecific adhesion may occur. The interactions responsible for nonspecific adhesion can either preserve the membrane integrity or lead to its disruption. Despite the importance of the phenomenon, there is still a lack of clear understanding of how and why nonspecific adhesion may originate different resulting scenarios and how these interaction scenarios can be investigated. This work aims at bridging this gap by investigating the role of the interplay between cationic electrostatic and hydrophobic interactions in modulating the membrane stability during nonspecific adhesion phenomena. Here, the stability of the membrane has been studied employing anisotropic nanoprobes in zwitterionic lipid membranes with the support of coarse-grained molecular dynamics simulations to interpret the experimental observations. Lipid membrane electrical measurements and nanoscale visualization in combination with molecular dynamics simulations revealed the phenomena driving nonspecific adhesion. Any interaction with the lipidic bilayer is defect-mediated involving cationic electrostatically driven lipid extraction and hydrophobically-driven chain protrusion, whose interplay determines the existence of a thermodynamic optimum for the membrane structural integrity. These findings unlock unexplored routes to exploit nonspecific adhesion in lipid membranes. The proposed platform can act as a straightforward probing tool to locally investigate interactions between synthetic materials and lipid membranes for the design of antibacterials, antivirals, and scaffolds for tissue engineering.
1. Introduction
The current understanding of cell-materials/nanomaterials has remarkably changed the way we precisely target cell regions1 and deliver therapeutics,2 the design criteria of biomaterials for tissue engineering and synthetic biological platforms.3 Control over cell-material interactions can allow the mitigation of adverse toxicological effects4 and the design of antiviral and antibacterial systems.5 Membranes are ubiquitous in biological systems, from cells and bacteria to sub-cellular regions and enveloped viruses, and are responsible for their protection from the surrounding environment. Material–cell interactions are mediated by adhesion phenomena which can be either mediated directly via membrane components (i.e. nonspecific interaction) or via membrane protein receptors and/or glycosaminoglycans (i.e. specific interaction). Membrane-mediated adhesion is directly driven by hydrophobic and electrostatic interactions between the phospholipids of the membrane and an external system. In contrast to protein-mediated interactions, the exploitation of membrane-mediated nonspecific adhesion has attracted relatively little interest. Indeed, for most material/nanomaterial–membrane interaction scenarios, nonspecific interactions lead to a loss of membrane integrity which may result in its disruption.6 Nevertheless, nonspecific interactions in the lipid bilayer are commonly exploited by biological systems in precise ways, such as in the case of membrane proteins tethered to the lipid bilayer via lipid anchors.7 Clearly, the poor exploitation potential of nonspecific interactions in lipid bilayers is not an intrinsic feature of the system but it is related to our inability to advantageously use them. Within this frame, our approach seeks to capture the fundamental aspects regulating the interaction outcome due to nonspecific binding with the lipid bilayer by providing in vitro and in silico evidence of the parameters affecting them. Additionally, we provide a probing method allowing the interrogation of nonspecific interaction ideally for any lipid membrane composition.To directly probe nonspecific interaction in lipid bilayers, a stable platform is required allowing the tailoring of its surface chemistry with hydrophobic and electrostatic contributions. In this sense, proteins have too complex surface chemistry and they can be easily denatured when changing their hydrophobicity and charge. Lepowsky and coworkers have suggested that nanoparticles with Janus-like surface heterogeneity (i.e. compartmentalized adhesive and nonadhesive domains) are always partially engulfed by a lipid bilayer (i.e. anchored) similarly to membrane proteins.8 Therefore, nanoparticles with anisotropic surface chemistry could potentially be used as a biomimetic probing tool to provide fundamental insights into the role of electrostatic and hydrophobic contributions in nonspecific interactions with lipid bilayers. However, experimental pieces of evidence of anisotropic nanoparticles interacting with lipid bilayers have exclusively shown disruptive interactions leading to the formation of microscopic defects on supported planar lipid bilayers, already at picomolar concentrations.9 Additionally, although studies have suggested that hydrophobicity and charge in nonspecific interactions are responsible for defect formation, the answer has been limited to the magnitude of defect formation rather than a mechanistic understanding of their specific contribution in the interaction outcome.10 Currently, the mechanism responsible for defect formation remains still unclear especially in the early stages of its onset. Additionally, currently there are no tools available to investigate locally cell membrane–material nonspecific interactions.
In the present work, which combines experiments and computational simulations, we developed a nanoprobe consisting of two compartmentalized lipid-adhesive/lipid-nonadhesive domains to investigate the role of hydrophobicity and charge in nonspecific interactions with the lipid membrane. Hereafter, adhesiveness and nonadhesiveness refer exclusively to membrane-mediated nonspecific interactions. Due to the presence of a spatially defined nonadhesive domain, endocytic pathways can be excluded, allowing the nanoprobe to localize at the surface of the lipid membrane and enabling it to investigate lipid integrity in terms of defect formation and their evolution (Fig. 1A). The nanoprobe surface consists of a pH-tunable polymer brush with an architecture designed to tailor the cationic electrostatic and hydrophobic contribution by simply playing on the pH of the medium. The choice of deploying cationic charges lies in the fact that most of the biological systems such as proteins and cells are negatively charged at physiological pH. Therefore, under these conditions, anionic charges would lead to nonspecific repulsive interactions which are out of the scope of the present work. In the first part of the manuscript, we will probe, in real time, the electrical behavior of lipid membranes to experimentally identify regimes leading to either destructive or conservative interactions. In particular, the destructive or conservative nature of such interactions will be directly visualized through electron microscopy techniques to question the structural changes in terms of membrane integrity upon nonspecific interaction, as well as the structural effect when transitioning from one regime to another. This will be done by using conventional TEM and in situ liquid-phase TEM. In the second part of the manuscript, coarse-grained molecular dynamics (CGMD) simulations and free-energy calculations will be employed to unveil the interplay between hydrophobicity and charge in the interaction. This will be done by looking at the onset of defect formation and its evolution in different scenarios. Finally, energy landscape analyses by using the Umbrella Sampling technique will allow the calculation of the system free-energy, upon nonspecific adhesion in different scenarios, providing thermodynamic insights into the interaction events. Our findings indicate that – although nonspecific adhesion requires the formation of nano-defects in the lipid bilayer structure – a system interacting nonspecifically with a lipid membrane does not necessarily compromise the membrane structural integrity. Indeed, a rational design of cationic electrostatic and hydrophobic contributions could make it possible to enable nonspecific adhesion in a conservative fashion, redefying the boundaries of exploitation potential for nonspecific interactions in lipid assemblies. The nanoprobes developed in this work can potentially be employed to explore nonspecific interactions in lipid membranes with any composition.
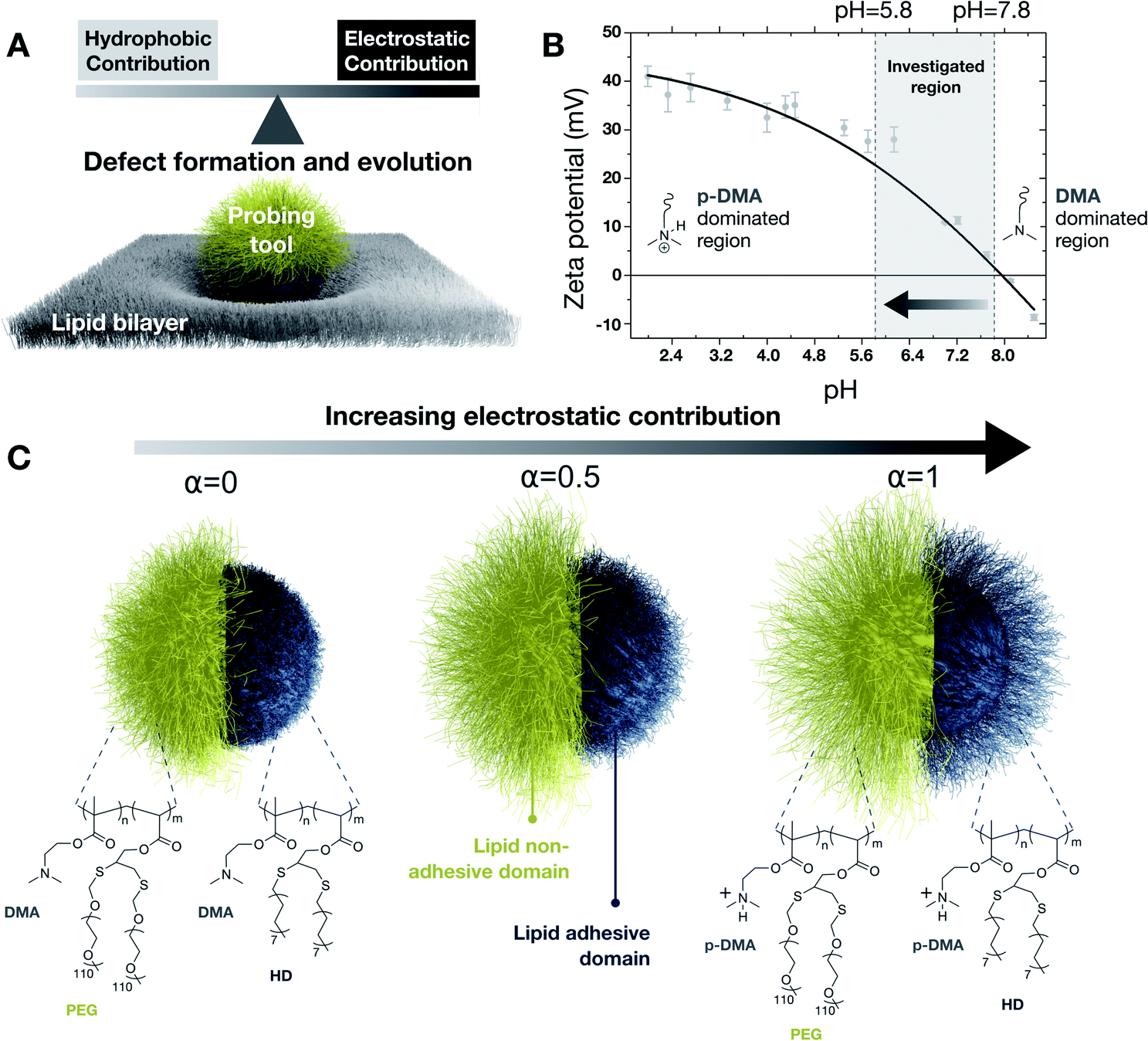 | ||
| Fig. 1 Illustration of a probing tool partially engulfed in a lipid bilayer used in this work to investigate nonspecific interaction by tuning cationic and hydrophobic contributions (A). Nanoprobe zeta potential profile illustrating the pH-tailorable surface charge through protonation and deprotonation of the dimethylamine functionalities, DMA (B). Example of representation at different protonation states of the nanoprobe complex brush architecture in compartmentalized lipid adhesive and lipid nonadhesive compartmentalized domains (C). | ||
2. Results and discussion
Probing tool
Our experimental probing tool consists of an inorganic core grafted with polybase brushes bearing a pH-tunable cationic charge. The cationic charge is chosen for the electrostatic contribution as being responsible for more severe lipid membrane damage in comparison to the anionic charge.11 The brush architecture is further functionalized via post-polymerization reactions into distinct domains, lipid adhesive (blue in particle sketches of Fig. 1) and lipid nonadhesive (yellow in particle sketches of Fig. 1). The adhesiveness is introduced by using hydrophobic alkanethiols,12 whereas the nonadhesiveness is introduced by limiting nonspecific interactions by using thiolated poly(ethylene glycol).13 More specifically, starting from silica nanoparticles (19.5 ± 2.5 nm in radius), clickable polymer brushes of poly(dimethylaminoethyl methacrylate – propargyl acrylate), P(DMAEMA-ran-PA), were uniformly grafted by light-induced surface-initiated polymerization. Subsequently, compartmentalized domains were introduced by using a supracolloidal-assisted toposelective modification method, as previously developed in our laboratories,14 in combination with thiol-yne click reactions. First, a lipid nonadhesive domain was photogenerated by anchoring thiolated poly(ethylene glycol) chains (PEG) on polymer-grafted nanoparticles immobilized on colloidal scaffolds. Subsequently, a lipid adhesive domain was photogenerated by using 1-hexadecanethiol molecules (HD) exploiting the unreacted clickable functionalities of the pristine brush domain (i.e. previously protected in the supracolloidal assembly). Detailed characterization of the as-fabricated system is reported in the ESI.† The resulting probing tool is characterized by its complex compartmentalized brush architecture for which hydrophobicity and charge can be tuned by changing the protonation state of the dimethylamine groups (with DMA indicating the unprotonated state and p-DMA the protonated one). Indeed, as a consequence of the DMA protonation, the nanoprobe exhibits a surface charge dependent on pH, with an isoelectric point of about 8, as in Fig. 1B. We selected a window of pH ranging from 5.8 to 7.8 as most relevant for biological environments because zeta potential measurements highlighted most of the charge changes within this range. As a consequence of the pH-driven tuning of the cationic charge, the brush conformation is also affected (Fig. 1C).15 The pH-induced structural changes affecting the lipid-adhesive domain were studied by using specular neutron reflectivity on planar brushes of the same composition and thickness as those on nanoprobes. From reflectivity profiles, we evaluated a change from 12.6 nm to 6.4 in thickness and from 6.2 nm to 5.4 nm in roughness, when going from a fully charged to a fully uncharged state. Such structural changes were also supported by CGMD simulations on the adhesive domain at different degrees of protonation. Changing the pH of the medium not only allows the adjustment of the cationic electrostatic contribution (i.e. availability of p-DMA) of the nanoprobe but also allows the modulation of the available hydrophobic functionalities (i.e. exposed HD) as will be highlighted by CGMD in the following sections. Detailed characterization of the anisotropic nanoprobes is reported in Section 1 of the ESI.†Interrogating membrane structural integrity upon nonspecific interactions
The engineered nanoprobes were employed to target and investigate nonspecific interactions by using a free-standing planar lipid bilayer as a cell membrane model. Phosphatidylcholine lipids were chosen as model phospholipids since they are an essential component of all cell membranes.16 In lipid bilayers, individual phospholipids are assembled so that the polar head groups are oriented outward to an aqueous solution, whereas the hydrophobic aliphatic tails are assembled facing each other inward, to the aqueous solution. Because of this assembly, lipid bilayers behave electrically as a capacitor in parallel with a resistor. When an electric potential is applied between the two membrane sides, ions accumulate leading to layers of opposite charge separated by less than 10 nm by a high dielectric constant region made of assembled aliphatic tails. Interestingly, the electrical behavior of a lipid bilayer is affected by interaction with other objects such as proteins, genetic material and nanoparticles.17 Such an electrical change due to interactions can be detected by measuring the membrane capacitance before and after the interaction. This can be done by the measurement of the electrical characteristics of two chambers separated by a micrometer-sized aperture sealed with a free-standing planer lipid membrane. Electrical experiments were conducted at various pH values (within a range of interest for biological systems, 5.8–7.8) to modulate the cationic contribution of the nanoprobe. To do so, we employed a cell apparatus to evaluate the transmembrane electrical feature of the free-standing planer lipid bilayer used as a membrane model. In a typical experiment, the transmembrane electrical properties were evaluated over time before and after the addition of nanoprobes. Nanoprobes are added in the cis compartment of the electrical apparatus (see the ESI†) so that a final concentration of 230 pM is achieved. Historically, electrical measurements on the lipid membrane are performed using “Direct Current” (DC) focusing exclusively on capacitance changes. In our approach, we used low frequency (1 Hz) “Alternative Current” (AC), for membrane electrical measurements, and we followed the evolution of complex transmembrane impedance over time, in both its real (i.e. membrane resistivity Rm) and imaginary (i.e. membrane capacitive reactance Xm) components.18 By doing so, we could probe both nanoprobe adsorption phenomena related to Xm and defect formation related to Rm. Changes in Xm and Rm are normalized by their values before nanoprobe addition, Rm 0, and Xm 0. To verify that adhesive interactions arise from the adhesive domain of the nanoprobe, we synthesized homogeneously nonadhesive nanoprobes. The overtime evaluation of the transmembrane impedance upon the addition of nonadhesive nanoprobes did not show any change in transmembrane resistivity and capacitive reactance, indicating no interaction between the nanoprobes and lipid bilayer (see ESI, Fig. S10†). The examinations of the lipid bilayer upon addition of adhesive/nonadhesive nanoprobes are summarized in Fig. 2. We can identify two different regimes when varying the cationic electrostatic contribution. For pH values above 6.6 (zeta potential <10 mV), a non-disruptive interaction pattern between the nanoprobes and lipid membrane can be identified. Upon nanoprobe addition, we can identify a decrease of the transmembrane reactance Xm (Fig. 2A), which can be traced back to the adsorption phenomena of nanoprobes on the lipid bilayer. The drop in reactance occurred within dozens of seconds from the nanoprobe addition. Additionally, this interaction resulted in a slight decrease of resistance (Fig. 2B), suggesting that the adsorption is mediated by the creation of nano-defects. Notice that, within the non-disruptive regime, we cannot identify any statistically significant difference between the different pH values in terms of transmembrane impedance variation. Overall, the impedance phase shift (Fig. 2C) indicated that the system remains electrically mainly capacitive, even after interaction phenomena have occurred, thus suggesting that such interactions do not alter irreversibly the bilayer structural integrity, representing only a tolerable perturbation for the membrane stability. On the other hand, as the cationic contribution increases, for pH values below 6.2 (zeta potential >20 mV), the interaction between the nanoprobes and the lipid bilayer led to an irreversible alteration of the membrane integrity and its final disruption. This is indicated in Fig. 2D–F by a rapid drop of transmembrane complex impedance and an impedance phase shift indicating a transition from a mainly capacitive to a mainly resistive system. These results can be associated with the disruption of the lipid bilayer, which in turn results in the creation of an ionic communication between the cis and trans compartments of the cell apparatus.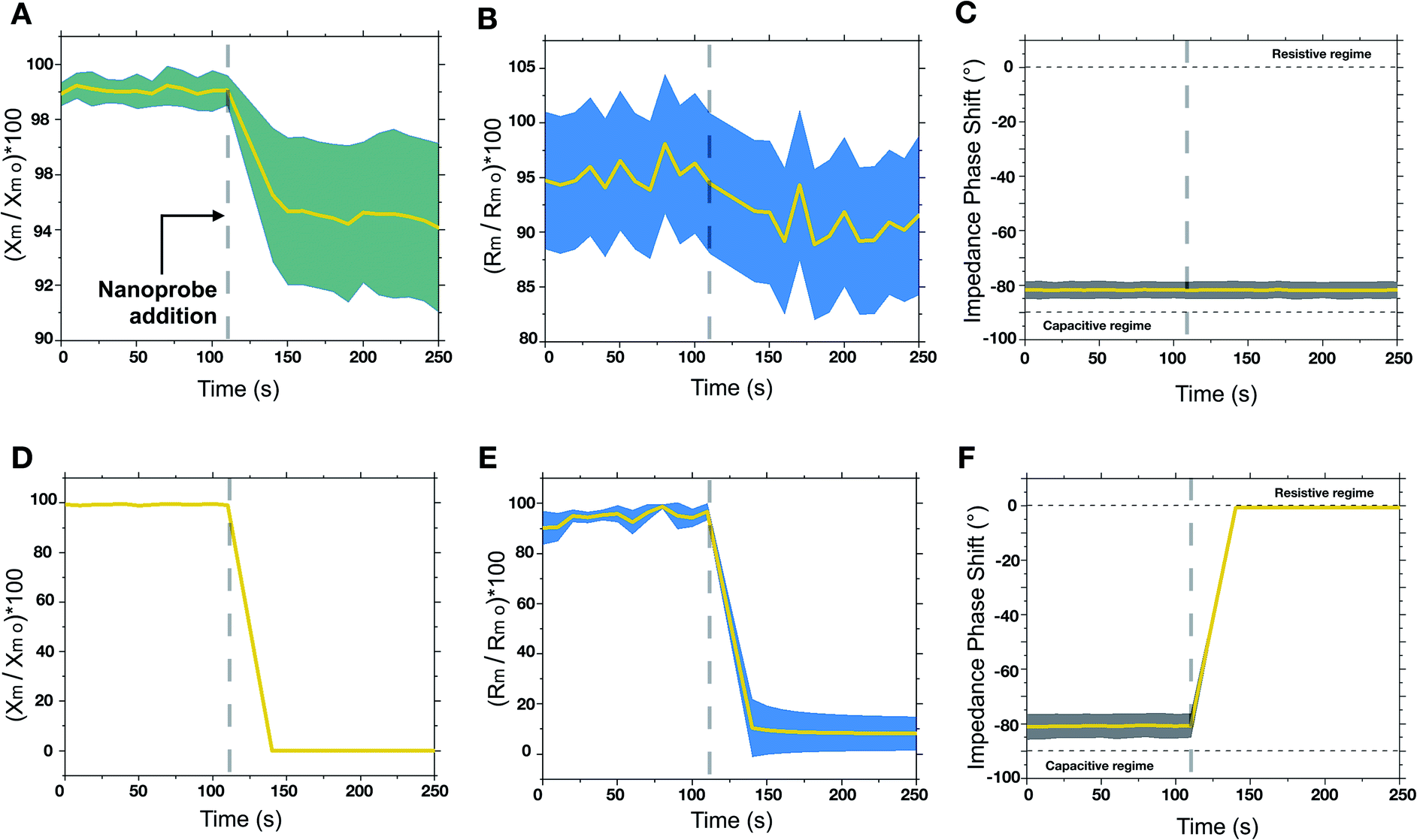 | ||
| Fig. 2 Temporal evaluation of transmembrane complex impedance via membrane electrical measurements, before and after nanoprobe addition. Averaged results for the membrane conservative regime at pH ≥6.6 in terms of normalized capacitive reactance (Xm/Xm 0) × 100 (A), resistance (Rm/Rm 0) × 100 (B) and impedance phase shift (C). Averaged results for the membrane disruptive regime at pH ≤6.2 in terms of normalized capacitive reactance (Xm/Xm 0) × 100 (D), resistance (Rm/Rm 0) × 100 (E) and impedance phase shift (F). No statistically significant differences were identified within each regime. A final average concentration of 230 pM was used in all the experiments. | ||
To achieve a deeper understanding of the interaction scenarios between nanoprobes and lipid membranes, we adopted TEM-based analyses by using liposomes (i.e. large unilamellar vesicles, LUVs) as a membrane model. Negative staining with uranyl acetate was used to visualize those interactions.19 For this purpose, liposomes were fabricated by extrusion in the same buffer composition used for electrical experiments. After letting nanoprobes and LUVs to interact for 30 minutes, negative stain TEM was used to visualize the interaction patterns. For highly charged regimes (pH ≤6.2), the interaction led to the disruption of the lipid bilayer, resulting in lipid molecule disassembly and aggregation with the nanoprobes. This leads eventually to the formation of microscopic aggregates. On the other hand, for mildly charged regimes (pH ≥6.6), the visualization of the interaction patterns revealed nanoprobe anchoring at the lipid bilayer. In Fig. 3A, an exemplificative negatively stained TEM image for the non-disruptive interaction pattern is reported in false colors. In the snapshots (i)–(iv) of Fig. 3A, it is shown that when nanoprobes interact with the lipid bilayer a partial membrane wrapping is observed. This indicates the existence of adhesion phenomena between the membrane and nanoprobe as schematically represented in Fig. 3B.20 So far, our findings suggest that a mildly charged hydrophobic domain can interact nonspecifically with lipid bilayers without compromising structural integrity. On the other hand, when the hydrophobicity is associated with a high cationic electrostatic contribution the interaction with the nanoprobe leads to the disruption of the lipid membrane. To evaluate the nanoscale dynamics of this transition, we directly visualize in real time the bilayer-nanoprobe interaction by using in situ liquid phase TEM (LP-TEM). In a typical experiment, the system in an aqueous solution is confined within a microfluidic cell composed of two silicon microfabricated chips, separated by 50 nm gold spacers. Electron transparency is provided by two 550 × 20 μm2 large and 50 nm thick silicon nitride (Si3N4) windows. To directly visualize the dynamical processes occurring in the liquid medium, the cell is sealed to separate the solution from the external environment. It should be mentioned that the real thickness of the liquid layer is generally larger than its nominal value due to the difference in pressure between the liquid and the vacuum of the microscope; this is known as the bulging effect. The two e-chips, mounted together, are placed in a dedicated TEM sample holder (Protochips 520) allowing the solution containing the nanoprobes to flux through a microfluidic cell under a constant flux of 100 μL per hour. For LP-TEM experiments, unilamellar vesicles were used as the membrane model. This choice is dictated by the thickness of the liquid in the microfabricated cell used for the in situ visualization.
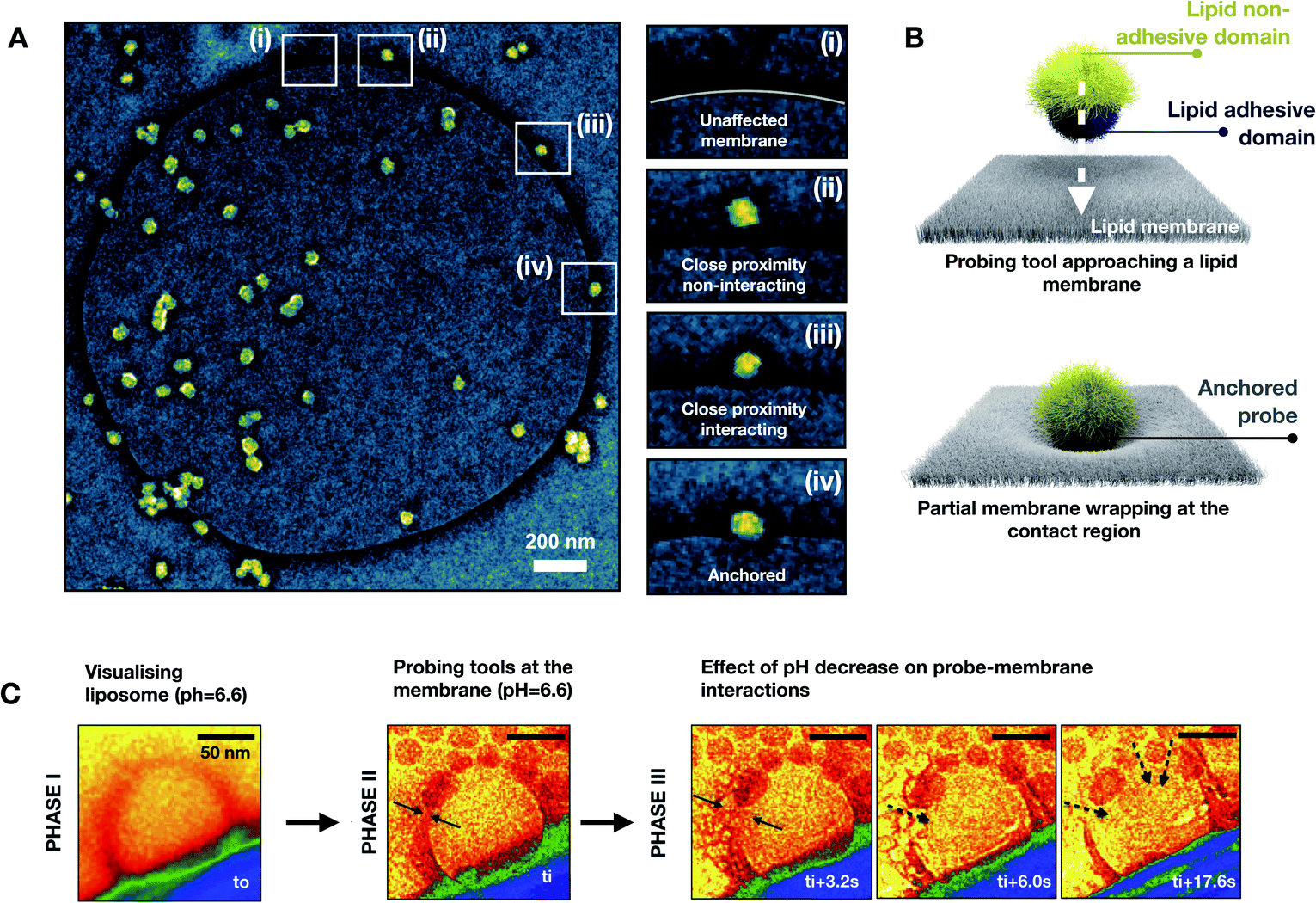 | ||
| Fig. 3 Representative image of a typical interaction scenario followed by negative staining with uranyl acetate for the membrane conservative regime (after 30 min of incubation; pH = 6.6). (A) Magnification of a single interacting nanoprobe anchored or approaching the lipid membrane (i–iv). (B) Illustration of a nanoprobe approaching a lipid bilayer and inducing membrane wrapping as a consequence of nonspecific adhesiveness. In situ LP-TEM direct visualization of the disruptive nonspecific interactions between the lipid membrane and nanoprobes, highlighting the effects due to the increase of charge contribution in the interactions (C). All TEM images are reported on a false-color scale to guide the eye. | ||
To maximize the resolution, we focused our attention on liposomes close to the window edges (phase I). Note that for beam current below about 80 electrons nm−2 sec−1, liposomes could be visualized for dozens of minutes without any beam-induced effect. Once the liposome was identified, the nanoprobes were introduced by using an external flux (100 μL per hour) of colloids. The flux was stopped when the nanoprobes reached the field of view. At this point, the interactions were visualized (phase II). A pH of 6.6 was chosen for the solution so that very little variation of pH could rapidly lead to a transition from non-disruptive to disruptive interactions. Starting from this condition, by beam-induced acidification, the pH of the liquid environment21 could be decreased and therefore the cationic contribution in the nanoprobe–membrane interaction could be increased (phase III). In Fig. 3C, representative frames of each mentioned phase are reported in a false-color representation to guide the eye. In the frame of phase I, a liposome with a diameter of about 120 nm is visible in red color in the proximity of the window edge. This moment is defined as t0 and it corresponds to a state in which the nanoprobes were still not introduced. In the frame of phase II, the nanoprobes reach the liposome and anchor to the bilayer. This moment corresponds to the time ti from which the evolution of the lipid membrane started being monitored. It is important to note that this time does not correspond exactly to the instant at which the nanoprobes reached the liposome. Indeed, there is an experimental delay of a few seconds due to the focus adjustments needed to correctly visualize the nanoprobes. From the phase III frame of Fig. 3C, the effect of the acidification of the liquid environment is directly visible. As a matter of fact, within seconds the liposome membrane integrity started being compromised in the proximity of the anchored nanoprobes. The induced defects grow, till when the lipid membrane is completely damaged. The membrane rupture is coupled with the translocation of the nanoparticles toward the interior of the vesicle structure.
The effect of the interplay between hydrophobicity and charge on the membrane fate upon interaction
Molecular dynamics simulations were used to better understand the effect of the interplay between hydrophobicity and charge on the structural integrity of the lipid bilayer upon nonspecific interactions. Coarse-grained molecular dynamics (CGMD) simulations were employed to minimize the degree of freedom by grouping a given number of atoms in a unique particle called “bead”, allowing us to simulate larger interaction volumes and for longer time-scales. Among the different methods, the MARTINI force field, developed by Marrink and colleagues,22 was chosen as being among the most reliable and optimized methods for lipid systems.23 Additionally, the MARTINI force field can be tailored specifically for polymer systems such as the adhesive brush domain of the probing tool interacting with the lipid bilayer by combining polar, nonpolar, and charged beads. All details about the employed CGMD methodology are reported in Section 3 of the ESI.†As reported in Fig. 4A, due to its small size a probing tool approaching a lipid bilayer will begin to interact utilizing a portion of chains close to the membrane. Because of the required computational load, we limited the investigation to a simulation box including only the interaction volume representing the point of the first contact between the membrane and the lipid-adhesive brush of the nanoprobe. Such an interaction volume can be limited to a portion of the lipid bilayer interacting with the polymer brush of the adhesive domain of the nanoprobe. The dimensions of the simulation box are 11 × 11 × 30 nm3 and the lipid bilayer lies on the x y plane, which means that an exposed surface area at the nanoprobe's surface of about 120 nm2 is studied by MD. These dimensions represent the smallest size of the interaction volume that allows neglecting the curvature effects. Experimentally, for the adhesive domain, a thickness of 12.6 nm and a roughness of 6.2 nm were estimated from neutron reflectivity when fully charged. To take into account the real brush dynamics, each polymer chain has been parametrized with 60 repeating units resulting in a length of about 12 nm when fully charged and stretched. Hereafter, any reference to the probe brush should be referred to the adhesive domain.
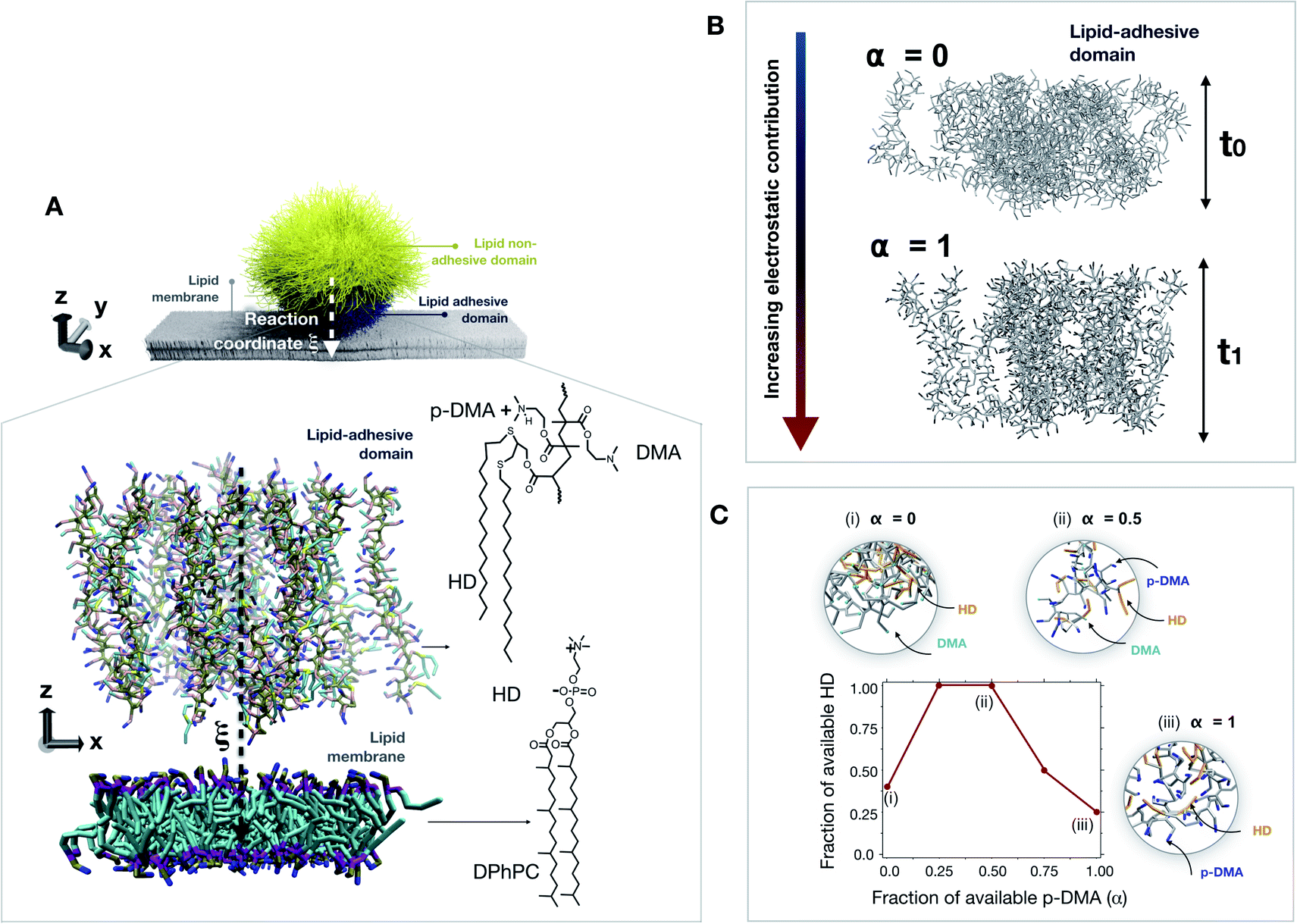 | ||
| Fig. 4 Illustration of a nanoprobe approaching a lipid bilayer including a snapshot of the relevant interaction volume for the point of first interaction used in the molecular dynamics simulation box (A). Snapshots from coarse-grained molecular dynamics simulation for the equilibrium configuration of the lipid adhesive brush domain in two extreme protonation states corresponding to a p-DMA/DMA ratio (i.e. α) equal to zero and one (B). Evaluation of the available exposed charge (p-DMAs) and hydrophobes (HDs) from the equilibrium configurations of the lipid-adhesive brush domain with increasing degree of protonation (C). | ||
To explore the probe–membrane interaction, the anchored chains are then moved along the reaction coordinate ξ (i.e., along the z coordinate) towards the lipid bilayer (Fig. 4A, close-up). In total, about 50 configurations have been evaluated with the Umbrella Sampling (US) technique, where a sampling window/configuration step Δξ = 0.2 nm was employed, representing a good trade-off value in leading to a continuous free energy profile at a reasonable computational cost, as more extensively reported in the ESI.† The total thermodynamic pathway (i.e., the overall length of the reaction coordinate ξ) investigated through US, therefore, turned out to be 10 nm, defined as the center of mass (COM) distance between the polymer brush and lipid membrane. As a common routine in US calculations, an independent set of simulations are conducted at each configuration. The system is therefore equilibrated for 1 ns and simulated for an additional 10 ns to evaluate the structural changes affecting the membrane. To adjust the contribution of the hydrophobicity and charge of the probing tool, the degree of protonation of the probe brush has been adjusted by playing on the p-DMA/DMA ratio for each configuration (i.e. α). Overall, five degrees of protonation have been evaluated ranging from zero (no charge) to one (fully charged system). It should be noted that the p-DMA/DMA ratio does not only affect the overall charge contribution taking part in the probe–membrane interaction. Indeed, since the p-DMA/DMA ratio influences the brush architecture by affecting the chain–chain interactions, this ratio affects as well the exposed hydrophobics contributing to the probe–membrane contact. For this reason, the brush conformation was deeply investigated at different degrees of protonation as reported in Fig. 4B. CGMD simulations reveal that the brush thickness decreases together with the protonation degree in the molecular scale simulations. This is consistent with our experimental findings in neutron reflectivity for which the brush goes from 12.6 nm to 6.4 nm in thickness and from 6.2 nm to 5.4 nm in roughness, when going from a fully protonated status at t0 to a fully deprotonated status at t1. Interestingly, when looking at the exposed hydrophobic (HD) and charged (p-DMA) groups available for the probe–membrane contact, we identified a non-monotonic trend (Fig. 4C). As the p-DMA/DMA ratio (α) increases, the exposed p-DMA increases whereas the exposed HD first increases and then starts decreasing for a p-DMA/DMA ratio above 0.5. Following the current understanding, the strength of hydrophobic interactions is enhanced when non-polar domains (such as HDs) are near charged functional groups (such as p-DMA).24 Under these conditions, the intrachain interactions between HD groups and the polymer backbone reduce the availability of exposed HDs for the probe–membrane contact.
To understand how the onset of nonspecific interactions takes place, we investigate the evolution of the membrane structural integrity at the point of the first contact between the probe and lipids, corresponding to a reaction coordinate ξ of the first contact. For α = 0, this configuration corresponds to a Δξ = 3 nm. After 10 ns of simulation, the system is governed by attractive chain–chain interactions within the brush since no charge is present at this point. This means that the brush structure interacts with itself rather than with the lipid membrane. As a matter of fact, α = 0 is a scenario dominated by hydrophobicity, and a probing tool under these conditions would experimentally aggregate and precipitate. Note that α = 0 represents a protonation condition outside the experimentally investigated range. When the degree of protonation is increased to α = 0.5, the brush structure is partially charged and slightly swollen. This is a scenario in which both hydrophobicity and charge have a comparable contribution to the features of the nonspecific interactions. In this scenario, each couple of hydrophobic non-polar chains (HDs) is accompanied by 4.5 charged groups (p-DMA). Under these conditions, the first contact with the lipid bilayer occurs at a reaction coordinate of Δξ = 3 nm. As reported in Fig. 5A, we observed that, at first, the charged functionalities extract lipids from the bilayer and at the same time hydrophobic functionalities protrude inside the hydrophobic compartment of the membrane. As visible from the simulation snapshot in Fig. 5B, corresponding to 10 ns of simulation, the structural integrity of the bilayer assembly is not compromised by this interaction. This is in line with the fact that the lipid extraction is, on average, compensated for by the insertion of hydrophobic functionalities (represented in orange). When the protonation is increased to α = 1, the overall character of the nonspecific interactions is unbalanced toward the cationic contribution. This is a scenario in which each couple of hydrophobic non-polar chains (HDs) is accompanied by nine charged groups (p-DMA) and on average only about 25% of the HDs are exposed and available (refer to Fig. 4C). All this leads to the lipid extraction process which is not compensated for by HD insertion (Fig. 5C). After 10 ns of the simulation, the structural integrity of the membrane is already compromised as highlighted in Fig. 5D by the membrane thinning.
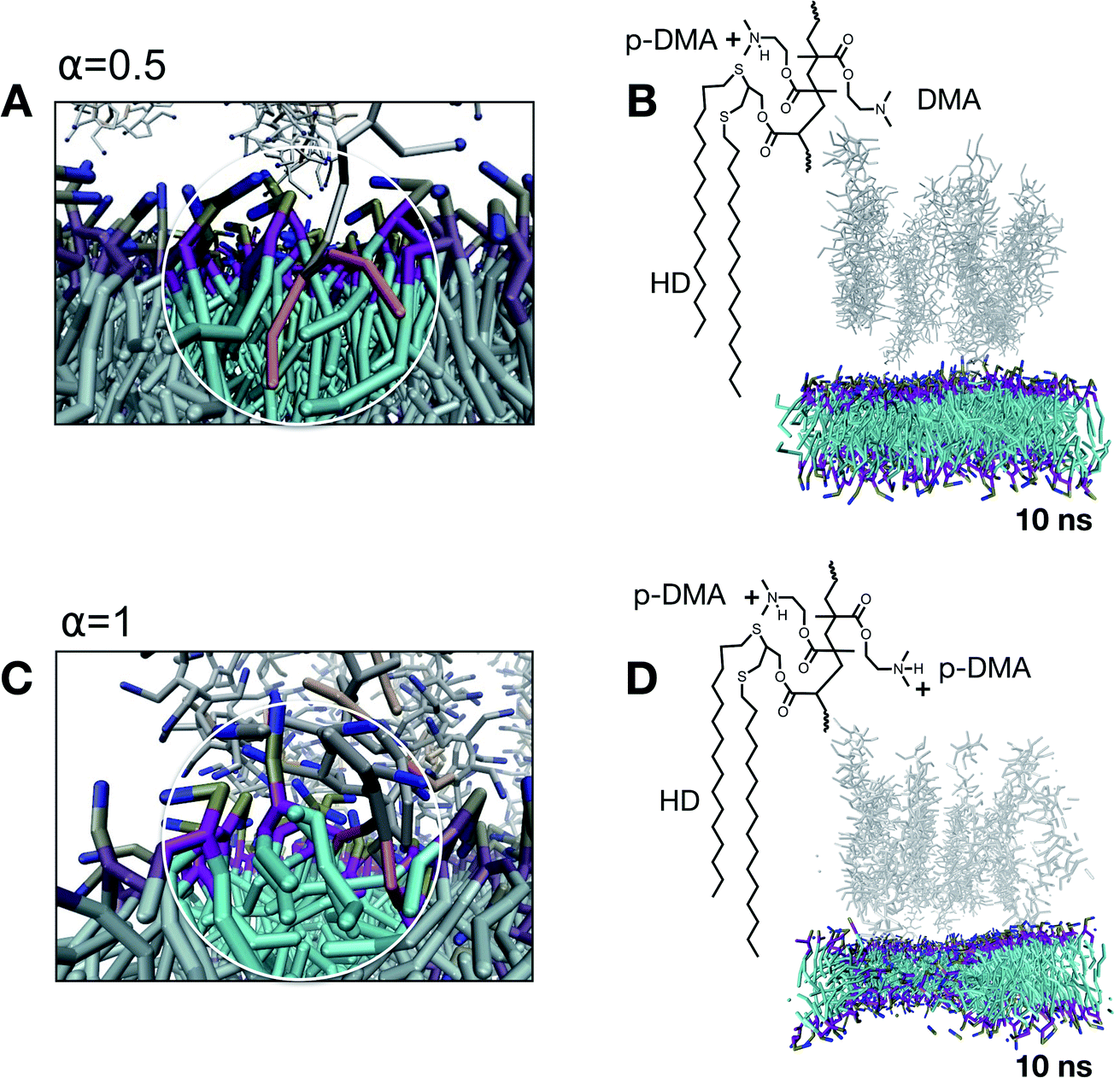 | ||
| Fig. 5 Mechanistic insights into the first contact between the probe and lipid bilayer mediated by nonspecific interactions. Close-up snapshot for α = 0.5 (A) highlighting the lipid extraction coupled with hydrophobic chain protrusion. Lipid membrane structural evolution after 10 ns of simulated evolution confirming the preservation of the structural integrity upon interaction (B). Close-up snapshot for α = 1 (C) highlighting hydrophobic chain protrusion associated with unbalanced lipid extraction. Lipid membrane structural evolution after 10 ns of simulated evolution showing major changes in the membrane integrity upon interaction (D). | ||
Insights into the structural damage at high degrees of protonation were gained by running MD simulations at α = 1 for the very first instant of the defect formation (below a simulation time of 1 ns) after an equilibration time of 1 ns. As reported in Fig. 6A and B, the initial stages of defect onsets are characterized by (i) the lipid being extracted and moving away from the membrane assembly, and this local extraction seems to lead (ii) to other lipid molecules to be attracted by the just created space. This seems to generate a hole (dotted circle in the bottom views in Fig. 6C and D) that will expand following the same mechanism over time leading to membrane thinning after 10 ns of evolution (Fig. 5D).
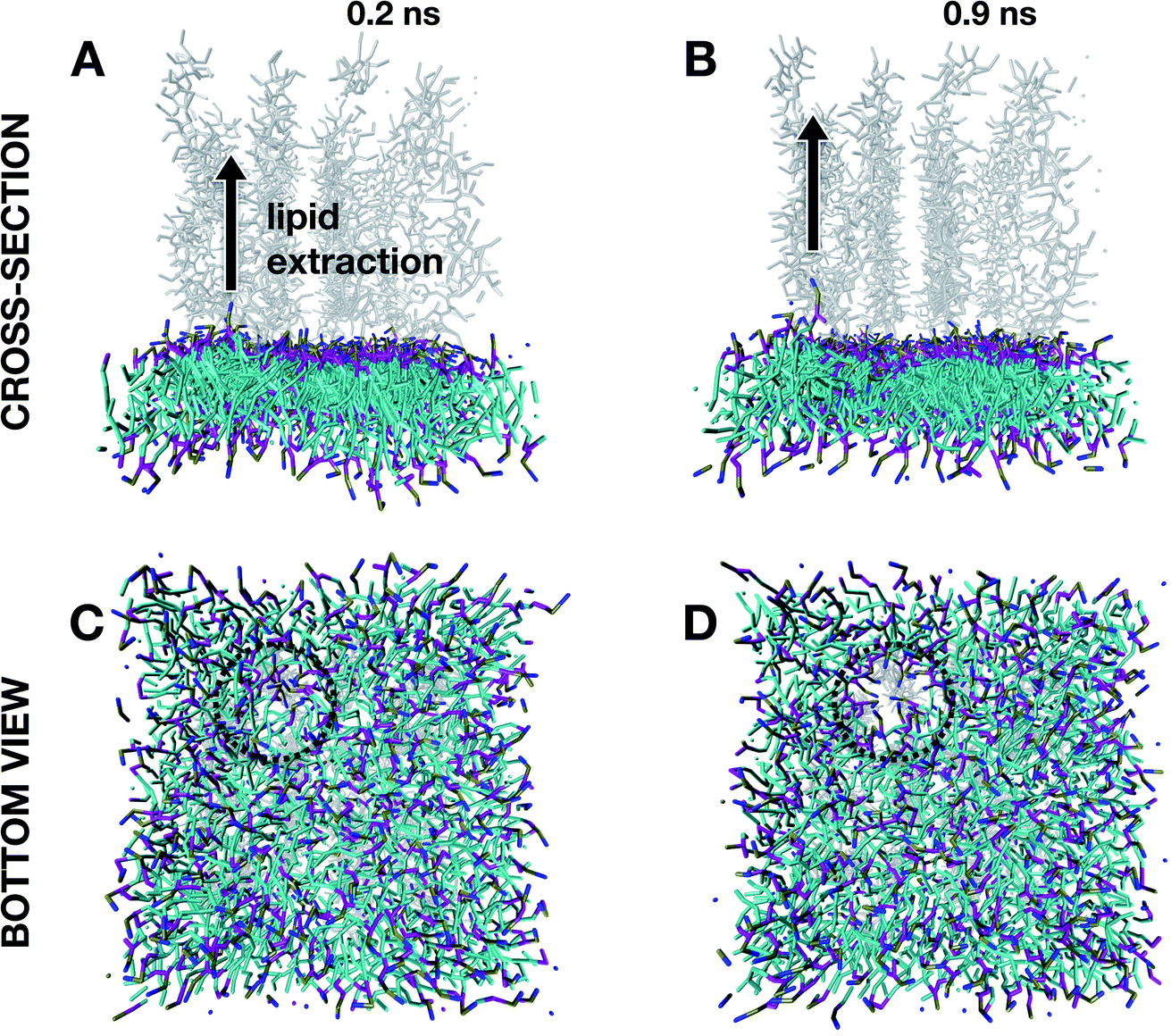 | ||
| Fig. 6 Early stages of interaction onset for a high degree of protonation (corresponding to a p-DMA/DMA ratio α equal to one) resulting in defect nucleation. Cross-sectional views (0.2 ns evolution for A and 0.9 ns evolution form B) highlighting lipid extraction and bottom views (0.2 ns evolution for C and 0.9 ns evolution form D) highlighting hole formation due to lipid rearrangement. Note that all evolution time values refer to evolution post equilibration. | ||
From the simulation of the first stage of contact between the probe and membrane, we have highlighted a different fate for the structural integrity of the membrane depending on the balance between cationic and hydrophobic contributions to the character of the nonspecific interactions. Where such an interaction will lead once the lipid-adhesive domain of the probe comes in more intimate contact with the lipid bilayer remains to be clarified. This configuration was evaluated for both α = 0.5 and α = 1 at the reaction coordinate of Δξ = 4 nm in which the brush chains are partially inserted in the lipid membrane. For α = 0.5 (Fig. 7A and C), the insertion of the chains in the bilayer is associated with the formation of defects at the membrane levels. These defects seem to be hydrophobically stabilized by the chains themselves. This finding is consistent with the slight reduction of transmembrane resistivity, observed in electrical measurements upon nanoprobe addition, which can be explained by an increased passage of ions through the chain-stabilized hole. On the other hand, for α = 1 (Fig. 7B and D), the lipid extraction is so intense that along with chain-stabilized holes (in black), we could observe non-stabilized holes (in yellow) which might be responsible for membrane rupture.
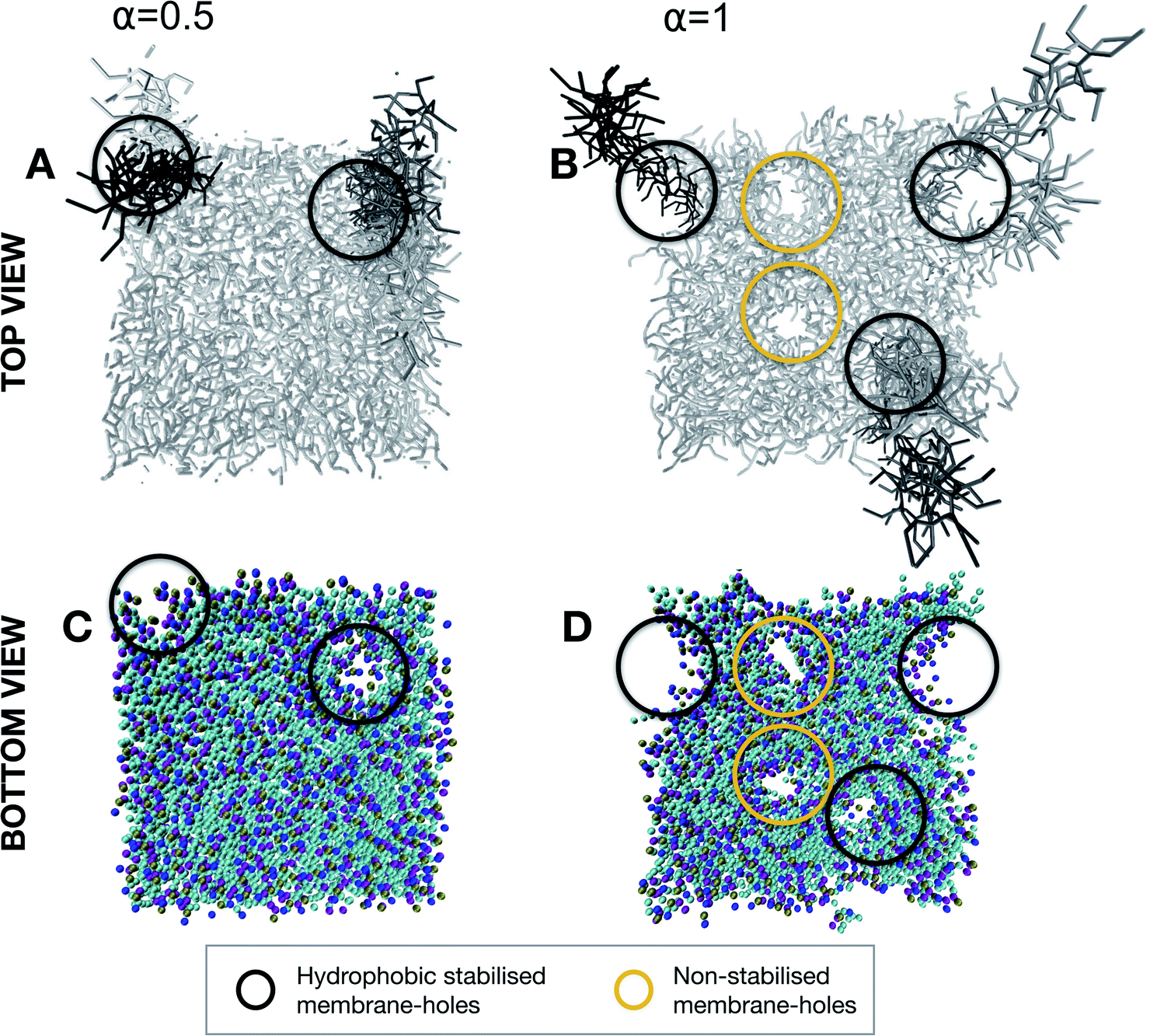 | ||
| Fig. 7 Defects after 10 ns of evolution (after equilibrium) in a configuration of intimate contact with the adhesive brush domain, corresponding to a reaction coordinate of Δξ = 4 nm. Membrane top view (A) and bottom view (C) for a mild degree of protonation (α = 0.5) showing the presence of hydrophobic stabilized membrane holes (circled in black). Membrane top view (B) and bottom view (D) for a high degree of protonation (α = 1) showing the presence of hydrophobic stabilized membrane holes (circled in black) along with non-stabilized holes (circled in yellow) responsible for membrane disruption. | ||
Thermodynamic considerations in different interaction scenarios
To gain thermodynamic insights into the interaction events, we conducted energy landscape analyses by using the Umbrella Sampling technique to calculate the system free-energy in terms of the potential of mean force (PMF).25 The value of the PMF can be extracted from a series of Umbrella Sampling simulations. To do so, a series of initial configurations are generated, each corresponding to a location wherein the polymer chains of the adhesive brush of the probing tool are harmonically restrained at decreasing brush center-of-mass (COM) distances from the lipid bilayer, via an Umbrella biasing potential. The Umbrella (harmonic) potential is applied to the COM of the brush. The heads of the brush are further restrained at a certain z0 for each configuration, using another spring (harmonic potential) to mimic the fact that they are anchored on one side of the nanoprobe core. This restraint allows the polymer chains to sample the configurational space in a defined region along the reaction coordinate ξ defined between the two groups. The extension of sampling windows/configurations must be chosen to guarantee a phase space overlap of the thermodynamic pathway or, equally, to lead to a continuous PMF curve profile. Results for the PMF at different degrees of protonation are reported in Fig. 8. The maximum of each PMF profile represents the activation energy required for the first contact in each specific scenario. Since the first contacts require the extraction of lipids, it is not consistent that such activation energy increases when increasing the degree of protonation. Indeed, as explained in the previous section, as the degree of protonation increases (i.e. the cationic electrostatic contribution increases) more and more lipids are extracted upon interaction. Interestingly, when looking at the overall change in the free energy of the system (see local minima in the PMF profiles) in the different interaction scenarios, it was possible to spot meaningful differences. For example, in a system in which interactions are dominated by cationic electrostatic contributions (α = 1), the overall change in free energy is positive. This finding corroborates the experimental observation of membrane rupture at a high degree of protonation. On the other hand, for the scenario in which there is an interplay between hydrophobic and cationic electrostatic contributions, the overall change in free energy is negative. Furthermore, among the different scenarios, in the proximity of α = 0.5 the negative change in free energy is the highest. This not only suggests that hydrophobic and cationic electrostatic contributions play a role in the interaction fate, but also means that there is a thermodynamic optimum in the interplay between the two contributions.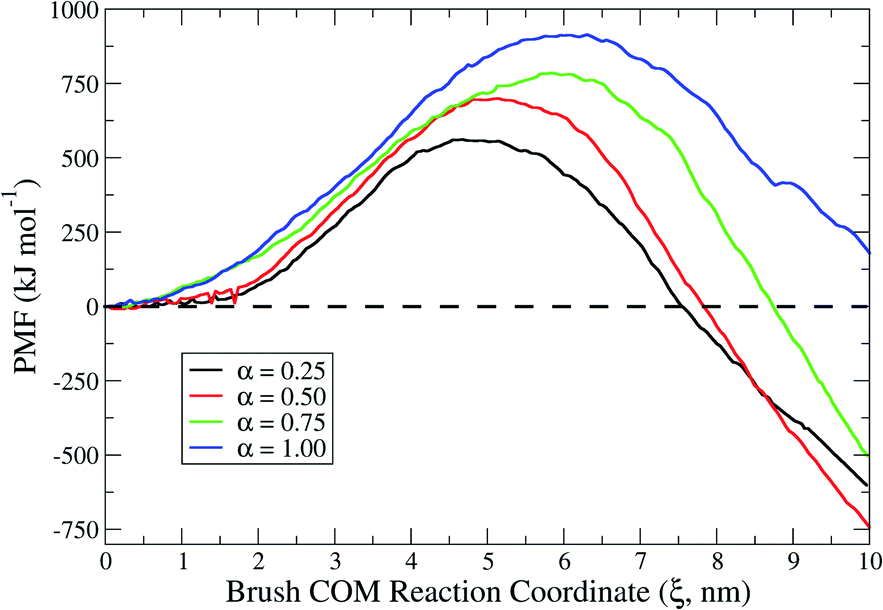 | ||
| Fig. 8 Potential of mean force as a function of the reaction coordinate for lipid-adhesive brushes with varying degrees of protonation (i.e. α ranging from 0.25 to 1) approaching and interacting with a lipid bilayer. | ||
3. Conclusion
In this study, we unveiled the role of hydrophobicity and charge contribution in nonspecific interactions of lipid membranes with external objects by providing experimental and computational findings. To do so, we developed a nanometric probing tool based on nanoparticles with anisotropic lipid adhesive/nonadhesive domains. This probing tool enabled the examination of the structural integrity of a zwitterionic lipid bilayer by modulating the balance of hydrophobicity and electrostatics. We measured the complex impedance of a free-standing lipid bilayer via electrical measurements, showing that when a mild level of charge is involved in the interactions, the structural integrity of the lipid bilayer is not compromised. On the other hand, when high cationic contributions are involved in the interaction, the structural integrity of the lipid bilayer can be compromised and lead to membrane disruption. These observations were supported by direct visualization of the interaction regimes via negative stain TEM. In situ LP-TEM also allowed shedding light on the nanoscale dynamics of the membrane disruption in real time. Furthermore, computational methods based on coarse-grained molecular dynamics (CGMD) simulations – by using the MARTINI force field – were employed to gain a mechanistic view of the nonspecific interactions in different scenarios. We found out that any interaction with the lipid bilayer is defect-mediated involving a (i) lipid extraction (electrostatic-driven) and a (ii) chain protrusion (hydrophobic-driven). Nevertheless, highly charged systems when interacting with the lipid bilayer extract many more lipids than what are replaced by chain insertion in the hydrophobic compartment of the bilayer. This unbalanced interplay between hydrophobic and cationic electrostatics is responsible for compromising the membrane structural integrity already from the initial onsets of the interaction. Simulations of more intimate interactions showed an even more pronounced effect jeopardizing the membrane integrity by the formation of non-stabilized holes on the lipid bilayer. Energy landscape evaluations have furthermore clarified the significance of the interplay between cationic and hydrophobic interactions in determining the interaction fate and in turn the structural integrity of the bilayer. We identified the existence of a thermodynamic optimum in the charge/hydrophobic balance responsible for interactions that are conservative for the structural integrity of the membrane. Based on these results, we demonstrated that nonspecific interactions do not necessarily affect the structural integrity of lipid bilayers meaning that they can be potentially exploited as tailorable adhesion mechanisms in a similar fashion to what nature does in the case of lipid-anchored proteins. Additionally, the anisotropic nanoprobes developed in this study pave the way for a straightforward method to investigate nonspecific interactions. The experimental evaluation of these interactions will be a critical asset for the future development of antiviral and antibacterial materials, and platforms for tissue engineering and cellular expansion such as scaffolds and microcarriers.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Thomas Griesser for his help in conducting and interpreting the XPS measurements. We thank Hansjörg Grützmacher for having kindly provided the trimethoxy silane-based photoinitiator used in this work.References
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC.
- A. Parodi, R. Molinaro, M. Sushnitha, M. Evangelopoulos, J. O. Martinez, N. Arrighetti, C. Corbo and E. Tasciotti, Biomaterials, 2017, 147, 155–168 CrossRef CAS PubMed.
- S. Runa, M. Hussey and C. K. Payne, J. Phys. Chem. B, 2018, 122, 1009–1016 CrossRef CAS PubMed.
- (a) B. B. Hsu, S. Y. Wong, P. T. Hammond, J. Chen and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 61–66 CrossRef CAS PubMed; (b) V. Cagno, P. Andreozzi, M. D'Alicarnasso, P. J. Silva, M. Mueller, M. Galloux, R. Le Goffic, S. T. Jones, M. Vallino and J. Hodek, Nat. Mater., 2018, 17, 195–203 CrossRef CAS PubMed.
- (a) J. Chen, J. A. Hessler, K. Putchakayala, B. K. Panama, D. P. Khan, S. Hong, D. G. Mullen, S. C. DiMaggio, A. Som and G. N. Tew, J. Phys. Chem. B, 2009, 113, 11179–11185 CrossRef CAS PubMed; (b) K. L. Chen and G. D. Bothun, Environ. Sci. Technol., 2014, 48(2), 873–880 CrossRef CAS PubMed.
- J. E. Johnson and R. B. Cornell, Mol. Membr. Biol., 1999, 16, 217–235 CrossRef CAS PubMed.
- (a) J. Agudo-Canalejo and R. Lipowsky, Soft Matter, 2017, 13, 2155–2173 RSC; (b) R. Lipowsky, Biol. Chem., 2014, 395, 253–274 CAS; (c) R. Lipowsky, in Physics of Biological Membranes, Springer, 2018, pp. 3–44 Search PubMed.
- (a) K. Lee, L. Zhang, Y. Yi, X. Wang and Y. Yu, ACS Nano, 2018, 12, 3646–3657 CrossRef CAS PubMed; (b) K. Lee and Y. Yu, Langmuir, 2018, 34, 12387–12393 CrossRef CAS PubMed.
- K. Lee and Y. Yu, Soft Matter, 2019, 15, 2373–2380 RSC.
- (a) Y. Zhang, N. V. Hudson-Smith, S. D. Frand, M. S. Cahill, L. S. Davis, Z. V. Feng, C. L. Haynes and R. J. Hamers, J. Am. Chem. Soc., 2020, 142(24), 10814–10823 CrossRef CAS PubMed; (b) H. H. Yu, K. Sakamoto, M. Akishiba, N. Tamemoto, H. Hirose, I. Nakase, M. Imanishi, F. Madani, A. Gräslund and S. Futaki, Pept. Sci., 2020, 112, e24144 CAS; (c) A. Mecke, I. J. Majoros, A. K. Patri, J. R. Baker Jr, M. M. Banaszak Holl and B. G. Orr, Langmuir, 2005, 21, 10348–10354 CrossRef CAS PubMed.
- A. L. Plant, Langmuir, 1993, 9, 2764–2767 CrossRef CAS.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. Müller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS PubMed.
- N. Razza, G. Rizza, P.-E. Coulon, L. Didier, G. C. Fadda, B. Voit, A. Synytska, H. Grützmacher and M. Sangermano, Nanoscale, 2018, 10, 14492–14498 RSC.
- D. E. Santos, D. Li, M. Ramstedt, J. E. Gautrot and T. A. Soares, Langmuir, 2019, 35, 5037–5049 CrossRef CAS PubMed.
- G. Van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS PubMed.
- (a) H. T. Tien and A. Ottova-Leitmannova, Membrane biophysics: as viewed from experimental bilayer lipid membranes, Elsevier, 2000 Search PubMed; (b) R. P. Carney, Y. Astier, T. M. Carney, K. Voïtchovsky, P. H. Jacob Silva and F. Stellacci, ACS Nano, 2013, 7, 932–942 CrossRef CAS PubMed.
- G. Fadda, D. Lairez, Z. Guennouni and A. Koutsioubas, Phys. Rev. Lett., 2013, 111, 028102 CrossRef CAS PubMed.
- L. E. Franken, E. J. Boekema and M. C. Stuart, Adv. Sci., 2017, 4, 1600476 CrossRef PubMed.
- A. H. Bahrami, M. Raatz, J. Agudo-Canalejo, R. Michel, E. M. Curtis, C. K. Hall, M. Gradzielski, R. Lipowsky and T. R. Weikl, Adv. Colloid Interface Sci., 2014, 208, 214–224 CrossRef CAS PubMed.
- N. M. Schneider, Liquid cell electron microscopy, 2016, pp. 140–163 Search PubMed.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. De Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- S. J. Marrink, A. H. De Vries and A. E. Mark, J. Phys. Chem. B, 2004, 108, 750–760 CrossRef CAS.
- C. D. Ma, C. Wang, C. Acevedo-Vélez, S. H. Gellman and N. L. Abbott, Nature, 2015, 517, 347–350 CrossRef CAS PubMed.
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: materials, synthesis protocol, characterization techniques, TEM, liquid-cell TEM, and molecular dynamics simulations. See DOI: 10.1039/d1na00360g |
| This journal is © The Royal Society of Chemistry 2021 |