
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDistinct thermoresponsive behaviour of oligo- and poly-ethylene glycol protected gold nanoparticles in concentrated salt solutions†
Miriam
Chávez
 ,
Ángela
Fernández-Merino
,
Guadalupe
Sánchez-Obrero
,
Rafael
Madueño
,
José Manuel
Sevilla
,
Manuel
Blázquez
and
Teresa
Pineda
*
,
Ángela
Fernández-Merino
,
Guadalupe
Sánchez-Obrero
,
Rafael
Madueño
,
José Manuel
Sevilla
,
Manuel
Blázquez
and
Teresa
Pineda
*
Department of Physical Chemistry and Applied Thermodynamics, Institute of Fine Chemistry and Nanochemistry, University of Cordoba, Campus Rabanales, Ed. Marie Curie 2a Planta, E-14014 Córdoba, Spain. E-mail: tpineda@uco.es
First published on 2nd July 2021
Abstract
In this work, the methoxy terminated oligo- and polyethylene glycol of different chain lengths (EGn, n = 7, 18, 45 and 136) is grafted on AuNP surfaces under conditions where they attain maximum grafting densities. These EGn–AuNPs gain stability relative to the pristine c-AuNPs in aqueous solutions and in a wide temperature interval and they form stable suspensions in solutions of high NaCl concentrations. To show the thermoresponsive properties of these EGn–AuNPs, temperature titration experiments are carried out in the presence of increasing amounts of salts. The concentrations of NaCl are chosen by checking the stability of EGn–AuNPs at room temperature and choosing the highest concentrations that allow them to form stable suspensions. The analysis of the temperature titration experiments monitored by UV-visible spectroscopy and dynamic light scattering allows us to establish the existence of transitions from individual to assembled nanoparticles, the reversibility of the temperature transitions and hysteretic behaviour in these systems. While EG7–AuNPs only show reversible temperature transitions in the presence of 5 mM NaCl, EG18–AuNPs do up to 1 M NaCl, becoming only partially reversible in 2 M NaCl. The titrations of EG45–AuNPs in 3 and 5 M NaCl show irreversible temperature transitions. Finally, EG136–AuNPs present a complex and interesting behaviour with two temperature transitions, the first one showing hysteresis and the second being reversible.
Introduction
Surface-grafted polymers or polymer brushes are currently used in macroscopic and nanoscopic surfaces such as gold nanoparticles (AuNPs) as they confer many interesting properties that are necessary in their applications for in vivo systems. One of these properties is biocompatibility and a very popular strategy followed to endow nanomaterials with it is by surrounding their surface with polyethylene glycol of different chain lengths (EGn), a process known as PEGylation.1,2 It is considered that the hydrophilic and uncharged nature of EGn molecules helps to avoid to a great extent the unspecific adsorption of proteins and the recognition by the immune system.3 These properties of the EGn protected AuNPs (EGn–AuNPs) are believed to be determined by the length and grafting density of the chains in the surface, and recently, it has been reported that the specific chemical properties of the ligands are the major driving force determining the composition of the protein corona that ultimately will be responsible for the prolongation of the blood circulation time and the immune response of the nanoparticles.4,5 One of the most important parameters determining the interaction of nanoparticles with proteins is the grafting density of EGn used to protect the surface and therefore its related chain conformation.6 To determine the grafting density, many approximations have been reported that include nuclear magnetic resonance spectroscopy,5,7,8 thermogravimetric analysis,6 isothermal titration calorimetry,9 analytical ultracentrifugation and total organic carbon analysis,10 among others. The chain conformation of the surface-grafted polymer dictates the interfacial properties, and as described in the Alexander-de-Gennes theory,11,12 good solvents make interactions with the solvent preferred over these with other polymer chains. Thus, under low grafting densities, there are no lateral constraints, and the polymer adopts a swollen conformation, behaving like a free polymer in solution, and the Flory radius describes the size of the random coil that is formed by such a polymer in the solution.13 This structure is called the mushroom conformation. When the grafting density is high, the polymer chains are forced into a stretched or brush conformation.14The grafting densities of EGn in AuNPs are influenced by the EGn molecular weight and the size of the nanoparticle, and are mainly responsible for the stabilization of EGn–AuNPs in the solutions of different pH, salt concentrations and temperatures.6,8 In this sense, the thermoresponsive properties of these polymers can be studied by analysing their assembly behaviour. The EGn–water system shows a phase diagram of the close-loop type with a lower critical solution temperature (LCST) at around 100 °C that is mainly due to the formation of hydrogen bonds.15 This limits the use of EGn polymers for the temperature triggered assembly of polymer grafted nanoparticles. However, the presence of salts can lower LCST16 and allows studying their thermoresponsive properties in lower temperature ranges. In a study of AuNPs coated with short EGn ligands terminated with alkyl heads, Ijiro et al. found that when the alkyl head was absent, the nanoparticles did not assemble in the range from 20 to 70 °C but, with ethyl, iso-propyl or propyl-headed EGn–AuNPs, the assembly took place at lower temperatures, indicating that the part of the ligand displayed on the outermost surface is important in the thermoresponsive behavior.17 Recently, they have demonstrated that this thermoresponsive assembly/disassembly process in water takes place through the hydration/dehydration of the EGn portion in a manner dependent on the hydrophobicity at their terminus and the surface curvature of the different size AuNPs employed. Also, by using molecular dynamics simulations they showed that the surface curvature tuned the distribution of the hydrophobic terminus in the normal direction along the surface, leading to variations in molecular configurations, and that the smaller the curvature the higher the local density, promoting the bending of the ligand molecule configuration and stabilizing the hydrophobic terminus in the relatively dense EGn moiety.18 These results are in agreement with the observed behaviour for gold nanorods that shows a two-step assembly/disassembly with temperature, induced by stepwise dehydration, that depends on the nanorod diameter but is independent of its length.19
EG18-and EG45–AuNPs form clusters at high ionic strength (K2SO4) and elevated temperature as a result of collapse of the surface-grafted EGn molecules.20,21 Another way to obtain thermoresponsive polymers at lower temperature is to form a statistical polymer constituted by EG and propylene oxide (PO) groups that are modified by a dihydrolipoic acid terminal to attach to an AuNP surface. The transition temperature of these polymers can be tuned by adjusting the ratio between EG and PO, the size of the nanoparticle and the salt concentration.22 Moreover, the steric hindrance generated by the reorganization of flexible hydrophilic polymer brushes during interparticle association is critical for the morphological selectivity in the assembly.23
Taking advantage of the hydrophobic/hydrophilic tuneable characteristics of the PEG polymers depending on the experimental conditions, Vaknin et al. have dedicated great efforts to elucidating the influence of different salt concentrations24–28 and temperature of the suspensions29–31 on the interfacial assembly of EGn–AuNPs. The addition of ions to the solution causes a poor solvent character for the polymers that could provoke the self-assembly of the EGn–AuNPs and their phase separation. The specific effect of ions, in general, follows the Hofmeister series. In fact, the presence of low level K2CO3 salt concentration makes the EG18- and EG136–AuNPs migrate to the vapor/solution interface without any specific organization but, when the salt concentration increases, short range hexagonal order develops, finally acquiring a highly ordered hexagonal crystallinity. The hydrodynamic radius of these EGn–AuNPs is found to be independent of salt concentration probably because the polymer corona can be protected by an effective semipermeable membrane that maintains a constant salt concentration up to the θ-point.24 When the concentration increases above 1 M, 3D macroscopic precipitates are induced that exhibit short range order and are consistent with a fcc symmetry with the nearest-neighbour distance in the assemblies dominated by the polymer length.25 Although monovalent salts such as NaCl and KCl do not cause phase separation of PEG at room temperature, they lead to 2D self-assembly of EGn–AuNPs at the interface, consistent with the depletion of ions in the polymer brush at concentrations lower than that necessary for bulk phase separation, generating an osmotic pressure gradient that drives the self-assembly. However, they do not induce 3D assembly into supercrystals.26 Different strategies that include salt concentration, interpolymer complexation, pH and temperature changes demonstrate that the self-assembly of structures other than spheres is also possible as it has been observed for gold nanorods29 and gold nanotriangles30 grafted with an EGn polymer corona.
In this work, we present a study of the stability of EGn–AuNPs formed with methoxy-terminated oligo- or poly-ethylene glycol of different chain lengths (Scheme 1). The EGn–AuNPs have been prepared under conditions that guarantee the highest grafting density for the different chain length employed. Characterization by UV-visible spectroscopy, dynamic light scattering (DLS) and zeta-potential measurements in an aqueous medium is first made to define the nanoparticle properties. The influence of the ionic strength and temperature on the stability of the suspensions, pointing to the reversibility of the observed changes, is reported. The monitoring of the aggregation phenomena is made by following the changes in the LSPR bands as well as the hydrodynamic sizes of the EGn–AuNPs by DLS.
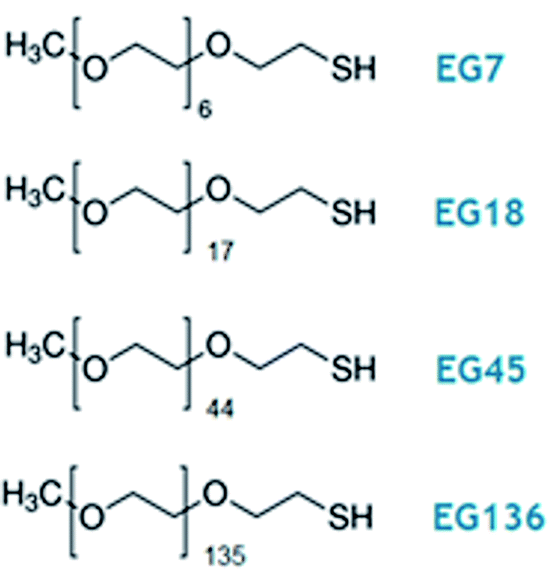 | ||
| Scheme 1 Oligo- and poly(ethylene glycol) methyl ether thiol, EGn, where n is the number of monomer units per chain, used in this work. | ||
Experimental section
Chemicals
Hydrogen tetrachloroaurate(III) trihydrate (HAuCl4), sodium citrate, O-(2-mercaptoethyl)-O′-methyl-hexa(ethylene glycol) (EG7) and poly(ethylene glycol) methyl ether thiol (MW 800, EG18; MW 2000, EG45; MW 6000, EG136) were purchased from Sigma-Aldrich (purity ≥99%). The rest of the reagents were of Merck analytical grade. All solutions were prepared with deionized ultrapure water produced by a Millipore system (Mili-Q® Direct-8).Synthesis of c-AuNPs and surface modification
The synthesis of citrate-capped gold nanoparticles (c-AuNPs) has been carried out by following the classic Turkevich method.32 In brief, 50 mL of 1 mM HAuCl4 was brought to a boil under stirring. By the addition of 5 mL 38.8 mM sodium citrate, the colour of the mixture changes from pale yellow to burgundy. Boiling was continued for 10 min and then the solution was cooled to room temperature under stirring. Finally, c-AuNPs are stored in the dark at room temperature, remaining quite stable for several weeks.The pristine c-AuNPs were diluted in Milli-Q water to reach 3.5 nM concentration and the pH was set to 7.5–8.0 with NaOH. Then a 2-fold excess of EGn–SH ligand relative to that necessary to cover the AuNP surface was added under vigorous stirring. This concentration was determined by considering the total gold atoms in the nanoparticle surfaces and using a footprint of 20 Å2 per molecule (this footprint is lower than that observed on flat surfaces33,34 and ensures that the concentration ratio is sufficient). The mixture was left overnight under shaking to allow for a maximum molecular self-assembly. To remove unreacted thiolate molecules, the mixture was submitted to three washing steps consisting of centrifugation (at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min), removal of supernatant and addition of water. The EGn–AuNPs obtained under these conditions are kept in the dark at room temperature and remain stable for weeks.
000 rpm for 15 min), removal of supernatant and addition of water. The EGn–AuNPs obtained under these conditions are kept in the dark at room temperature and remain stable for weeks.
Characterization of AuNPs
The c- and EGn–AuNPs were imaged by transmission electron microscopy (TEM), JEOL JEM 1400 instrument (from the Servicio Central de Apoyo a la Investigación (SCAI), Universidad de Córdoba) operating at 80–120 kV and the images were analysed using Image Pro Plus software. Samples were prepared onto formvar-coated Cu grids (400 mesh, Electron Microscopy Sciences). Each TEM grid was immersed in ca. 1 nM EGn-AuNP solution for an hour, then dunked in water to wash away unattached particles and finally air dried at room temperature.The grafting density of EGn–AuNPs was determined by thermogravimetric analysis (TGA), using a Mettler Toledo thermogravimetric analyser. The EGn-AuNP samples (10 mg) were prepared by first drying the washed solutions at 60 °C for 4 hours before TGA measurements. The temperature was varied between 20 °C and 800 °C at 10 °C min−1 under a nitrogen atmosphere flowing at 40 mL min−1. The amount of EGn was calculated using the ratio of weight loss occurring between 300 °C and 450 °C, considered to correspond to the degradation of EGn, to the weight remaining at 450 °C that corresponds to AuNPs.6,35 The AuNP volume, mass, and surface area are calculated assuming an ideal spherical shape with a diameter of 25 nm. The weight loss assigned to the polymer corona and the weight remaining at 450 °C are averaged by the EGn and the AuNP molar mass, respectively, to determine the amount of EGn grafted per nanoparticle. This ratio is divided by the AuNP surface area to obtain the grafting density, σ, and its reverse, the footprint of the EGn molecules.
The size, size distribution and zeta-potential of the particles were determined by dynamic light scattering (DLS) (Malvern Zetasizer Nano, ZSP) with a 633 nm He–Ne laser. The measured data are the average of at least 20 runs. The average hydrodynamic diameter (DH) and mean zeta potential of each sample were computed using the software provided by the manufacturer.
The extinction spectra were recorded using a Jasco V-670 UV-vis-NIR spectrophotometer. The studies of the temperature effects were performed by fitting a Peltier temperature controller, and a rate of 0.5 °C min−1 programme was selected.
Results and discussion
Ligand exchange of c-AuNPs by EGn polymer chains
The ligand exchange of c-AuNPs by EGn–SH molecules of different chain lengths (n = 7, 18, 45 and 136) readily takes place on nanoscopic gold surfaces. In this work, we have used c-AuNPs of 25 nm diameter synthetized by the classical Turkevich method (Fig. S1†), and the exchange reactions are carried out under the experimental conditions described in the Experimental section, that allow reaching a maximum surface coverage. UV-visible spectroscopy shows a small shift of the localized surface plasmon resonance (LSPR) band of ca. 2–3 nm relative to the c-AuNPs, regardless of the polymer chain length (Fig. S2†). This shift in the maximum wavelength is produced by the change of the refractive index of the layer immediately surrounding the AuNP surface.36 The presence of the polymer corona around the AuNP is perceived through the increase in dispersibility of the EGn–AuNPs in comparison to the c–AuNPs that become somewhat aggregated upon deposition on the TEM grid (Fig. S1†).TGA of the EGn-AuNP samples has been carried out to get information on the extent of grafting density of the different EGn molecules (see Experimental section). Table 1 gathers the molecular parameters obtained by TGA. The grafting density (σ) decreases with the increase in chain length, and consequently the footprint increases. A value of 20.7 Å2 is found for EG7, that is somewhat lower than the footprint of this molecule adsorbed on macroscopic gold34 and determined by reductive desorption of the EG7-self assembled monolayer on a gold electrode. Using the theoretical surface area of the AuNP of 25 nm diameter, the number of EGn molecules per nanoparticle is calculated.
| % weight loss | σ/nm−2 | Footprint/Å2 | EGn/AuNP | |
|---|---|---|---|---|
| EG7 | 2.9 | 4.82 | 20.7 | 9470 |
| EG18 | 5.5 | 3.90 | 25.6 | 7660 |
| EG45 | 9.9 | 3.28 | 30.5 | 6430 |
| EG136 | 9.5 | 1.04 | 96.1 | 2040 |
To evaluate the EGn shell formed around the AuNP and the existence of any aggregation phenomena, we use the DLS technique. After modification, larger DH values relative to the c-AuNPs were obtained, which can be attributed to the presence of the surface grafted EGn layer. Table 2 contains information on the EGn molecular weights, experimental hydrodynamic diameters (DH) and calculated thickness (L) for the different EGn–AuNPs. The L values increase with the chain length in a nonlinear fashion. The theoretical length values for these molecules by assuming a brush regime with the chains adopting either a helicoidal or an all-trans conformation that supposes an incremental of 2.78 Å or 3.56 Å per monomer, respectively,37 are also gathered to compare with the experimental values. Finally, the zeta-potentials measured for the different EGn–AuNPs are also included. Compared to the c-AuNPs that show a negative value of −44 mV due to the presence of citrate anions protecting the surface, the zeta-potential values are increasing from the shorter to the higher chain, as expected for the neutral characteristics of the EGn molecules. The AuNP surface modification with mercapto-derivatives is believed to take place by the exchange of the citrate anions by the thiol group and, therefore, most of the citrate anions are displaced from the surfaces and thus, an increase of the zeta-potential values38–41 that are consistent with these reported for EGn-coated AuNPs is found.6,42,43
| MW/Da | D H/nm | L/nm | Hel/nm | Trans/nm | Zeta-potential/mV | |
|---|---|---|---|---|---|---|
| EG7 | 356 | 28.4 | 1.7 | 1.9 | 2.5 | −28.5 ± 1.3 |
| EG18 | 800 | 28.4 | 1.7 | 5.0 | 6.4 | −10.8 ± 2.4 |
| EG45 | 2000 | 37.2 | 6.1 | 12.5 | 16.0 | −10.2 ± 2.3 |
| EG136 | 6000 | 59.7 | 17.3 | 37.8 | 48.4 | −6.9 ± 1.0 |
The two regimes described by polymers attached to a surface are the mushroom regime that occurs at very low grafting density and the brush regime that develops when the grafting densities are high.11,12 In the mushroom regime, the chains behave as isolated chains in solution, and can be thought as hemispheres with a size given by the Flory radius:
| RF = a × N3/5 | (1) |
When the polymer enters the brush regime, the thickness of the brush can be given by
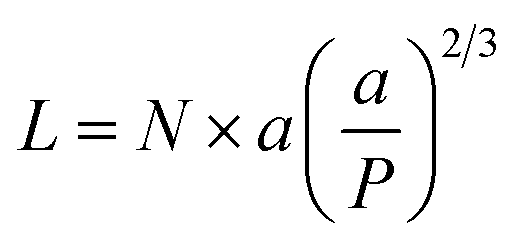 | (2) |
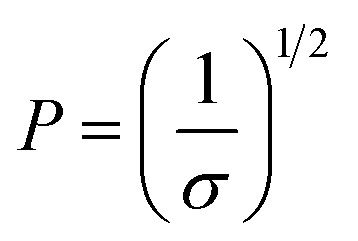 | (3) |
The conformation should be in a mushroom regime when P is greater than RF, as the chains have space to fold back over the AuNP surface, resulting in a thin layer (small L). In contrast, when P is lower than RF, the EGn should exist in a brush regime with the chains stretching out from the AuNP surface resulting a thick layer (large L).44 In the case that L > 2RF, the dense brush regime is obtained.45 Using the grafting densities σ determined by TGA (Table 1), the values of P and L are determined by eqn (2) and (3) (Table 3). As it can be observed, the values of P obtained by eqn (3) are lower than RF, indicating that the attached molecules are in the brush conformation.
On the basis of the behaviour of EGn in the presence of salts,46,47 and considering that EGn–AuNPs consist of flexible chains covalently grafted at the AuNP core of diameter D, Vaknin et al.24,25 have modelled the effective hydrodynamic diameter DH for spherical nanoparticles by eqn (4),
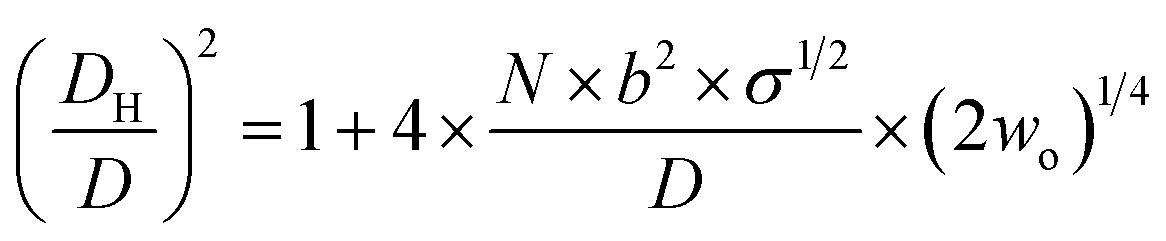 | (4) |
The hydrodynamic volumes per molecule determined by dividing the EGn corona volume, as obtained through the experimental DH values, by the number of molecules grafted on the surface, as determined from TGA, are higher than expected if we assume that the EGn molecules are in a brush conformation occupying a cylinder whose base is the footprint of the molecule, and this excess is much larger for EG136. The excess of volume must be occupied by either free or bound water molecules whose presence would be more important with increasing the distance from the AuNP surface. Recent theoretical calculations have demonstrated that the criterion for planar surfaces is insufficient to assess the achievement of the brush regime and that the curvature needs to be considered.48,49 They demonstrate that the polymer density decreases with the radial distance, and they remain well hydrated with a decrease in the fraction of free water only close to the surface. The requirement of overlapping at the height of the radius of gyration of the polymer, Rgo, leads to a modified estimate for the required grafting density to achieve the brush regime, σ* ≈ (1/Rgo + 1/R), for spherical nanoparticles of radius R, taking into account the surface curvature as well as the polymer coil dimension.48,49
Stability of the EGn–AuNPs in aqueous solutions
The obtained EGn–AuNPs have been examined in aqueous solution of different pH by UV-visible spectroscopy. The LSPR band does not shift when the pH is changed from alkaline to neutral and weakly acid media. Fig. 1 shows the variations of LSPR wavelength and extinction peak intensity for the EGn–AuNPs as a function of pH. Only in the case of EG7–AuNPs, an increase of the LSPR wavelength is observed at pH < 5 that is parallel to a decrease in extinction because of the aggregation of the nanoparticles in this medium. However, in comparison with the behaviour of the citrate-AuNPs,38,41 a higher stability is conferred by the presence of the EG7 protecting layer. Moreover, the aggregation phenomenon is reverted when the pH of the solution is changed to neutral or alkaline values.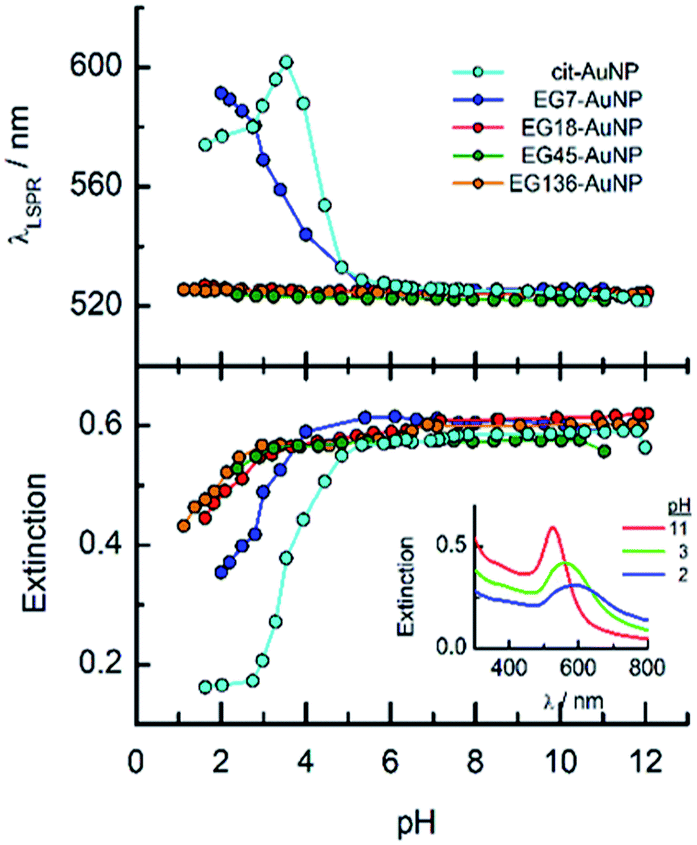 | ||
| Fig. 1 Effects of the solution pH on the LSPR wavelength and extinction of the EGn–AuNPs in aqueous solution. The pH was adjusted by adding aliquots of either NaOH or HCl diluted solutions. The experiments were run at room temperature (22 °C). Inset: spectra of EG7–AuNPs at different solution pH. | ||
The stability of the EGn–AuNPs as indicated by the position of the LSPR wavelength is preserved under these acidic media for the longer EGn chains, although a small decrease in extinction is also observed at pH < 3.
When these EGn-AuNP aqueous neutral solutions are exposed to temperature changes from 5 to 90 °C, no changes in the LSPR bands are observed for EG18-, EG45- and EG136–AuNPs. However, the EG7–AuNPs, although remain stable up to 85 °C, show an abrupt displacement from 522 nm to 536 nm with the concomitant emergence of a band near 740 nm when the temperature reaches 90 °C. Upon decreasing the temperature from this value, with the same rate as the ascending ramp, the spectrum returns to the initial shape, showing that the induced temperature aggregation of the EG7–AuNPs is completely reversible (Fig. 2).
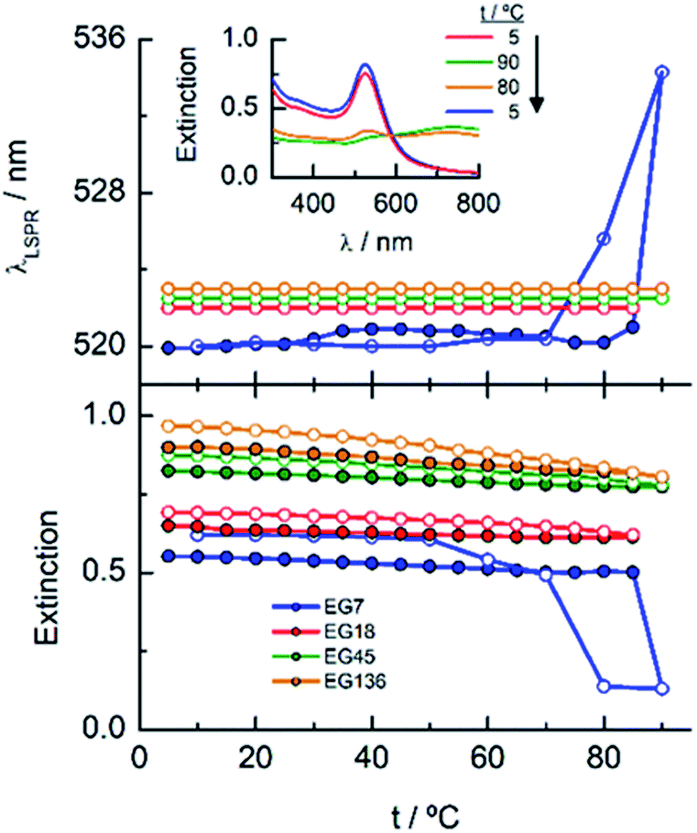 | ||
| Fig. 2 Temperature titration curves for EGn–AuNPs in neutral aqueous solutions (no additional salt is added other than that necessary to get a neutral medium). Changes in the LSPR extinction and wavelength. The direct (solid circle) and reverse (empty circle) temperature scans are plotted. Inset: spectra of EG7–AuNPs at different temperatures (the arrow indicates the direction of the temperature change). | ||
One way to check the efficiency of the protection of these AuNPs against aggregation is to study the effects of the presence of salts on the LSPR band parameters (Fig. 3). It is well known that the EGn–AuNPs remain stable in solution in the presence of 1 M NaCl when the grafting density of the chains is appropriate.50 As can be observed, either the wavelength or the extinction peak of the EG136-AuNP and EG45-AuNP spectra accomplished with these conditions does not change up to 1 M NaCl salt concentration. The behaviour observed for EG7–AuNPs in the presence of increasing NaCl concentration deserves some comments. Whereas the LSPR band obtained in water shows the typical shape for a well dispersed solution, when the concentration of salt is greater than 0.1 M, an increase of the scattering at longer wavelength is detected (Fig. 3, inset). At higher concentrations, a small band at around 800 nm appears that remains in equilibrium with that for the individual particles. These features are maintained in the presence of 1 M NaCl. Interestingly, the band at 522 nm is maintained, indicating that the individual EG7–AuNPs coexist with some big aggregates.
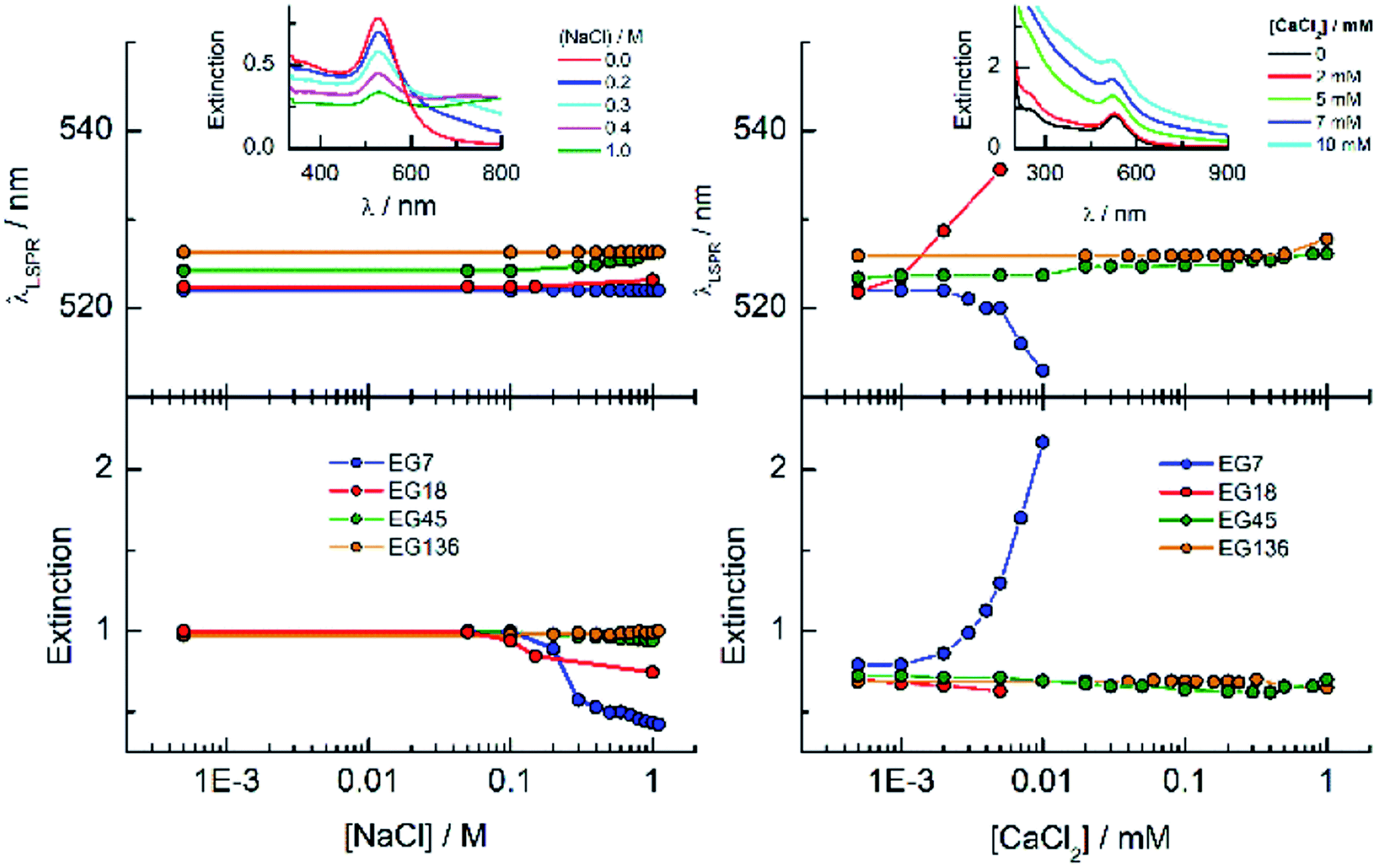 | ||
| Fig. 3 Effects of the salt addition on the stability of EGn–AuNPs as seen by UV-visible spectroscopy. The insets show the spectra of EG7–AuNPs in the presence of different concentrations of salts. The experiments were run at room temperature (22 °C). | ||
DLS measurements of EG7-AuNP dispersions in water and in the presence of 0.1 M NaCl confirm this idea. The DH in water is 28.4 nm while, in the presence of salt, a first signal at 29 nm accompanied by a second one of much higher value (∼720 nm) that should correspond to these aggregates of bigger size (Fig. S3†) is observed in the size distribution plot. However, these aggregates should exist in a low concentration as the number distribution plot of these signals shows only the peak of lower diameter, indicating that these are the most abundant in the dispersion.
EG18–AuNPs show a different and more common behaviour. The LSPR band centred at 522 nm for the dispersion in water shifts to a longer wavelength at NaCl concentrations higher than 0.2 M. This wavelength shift is accompanied by a new band at around 600 nm that overlaps with the original one. The DH value obtained in water is 28.4 nm and does not change in the presence of 0.15 M NaCl and even higher concentrations (1 M).
The influence is more pronounced for divalent salts such as CaCl2. EG18–AuNPs exhibit a displacement of the LSPR band to higher values at very low CaCl2 concentration, and this change parallels a decrease in the extinction peak following the normal behaviour of loss of stability and irreversible precipitation in the presence of salts. The EG7–AuNPs behave somewhat different. An important increase in the extinction peak concomitant with a small displacement of the band to lower wavelength is observed at concentrations higher than 2 mM. The observed wavelength displacement is however apparent and seems to be due to the influence of the scattering that is being produced in the presence of CaCl2 (Fig. 3, inset). This effect is ascribed to the decrease in transmittance due to Rayleigh scattering of the polymers amplified by the LSPR band of the AuNP when the presence of the salt makes the water a poor solvent after breaking of the polymer–water H-bonds and probably exposing a more hydrophobic surface.51
The EG7–AuNPs have the highest grafting density of the nanoparticles studied in this work, and a corona thickness that is around 90% of that theoretically calculated for an extended conformation. Moreover, the lower zeta-potential measured for these nanoparticles would indicate that most of the citrate anions remain in the surface, probably competing for the sites of water molecules (Tables 1 and 2). Although the grafting densities decrease as the EGn chains increase in length, the brush conformation seems to be adopted upon attaching to the nanoparticle surface. However, the thicknesses are lower than half of the theoretical value for the extended conformation. As the curvature of AuNPs does not change through the experiment, this difference must be ascribed to the decrease of polymer density with the radial distance and the higher hydration of the chains that can be responsible for the higher zeta-potential measured for these EGn–AuNPs.
Effect of temperature on the stability of EGn–AuNPs in the presence of high NaCl concentrations
The results described above invite us to carry out a deeper study of these systems in the presence of different salt concentrations as a function of temperature. The high LCST of the system EGn–water15 is explained by the existence of H-bonds between the polymer and water, and the loss of these interactions either with increasing temperature or in the presence of salts or both contributes to the entropic loss that makes the water a poor solvent and, consequently, to the conformational transition of the nanoparticle attached polymers that provokes the lowering of the LCST of the system. As shown in Fig. 2, the EG18-, EG45- and EG136–AuNPs present high stability in aqueous solution upon changing temperature from 5 to 90 °C. EG7–AuNPs, however, show some changes that are visible in the spectra at temperatures higher than 85 °C and in the presence of salt (Fig. 2 and 3, inset).Fig. 4 shows the analysis of the UV-visible spectra for EG7–AuNPs in the presence of 5, 50 and 100 mM NaCl as a function of temperature. At the very low concentration of 5 mM, the feature observed at 85 °C in pure water is now seen at lower temperature. The displacement of the LSPR band is concomitant with a decrease in extinction that becomes restored upon reverting the temperature. The transition temperature (TA), observed at 72 °C, is displaced by 5 °C in the decreasing temperature ramp. Suspensions of EG7–AuNPs in the presence of 50 or 100 mM NaCl follow a similar behaviour, exhibiting the expected trend of decreasing TA at 62 and 54 °C, respectively. Although the maximum LSPR band reaches its original value when returning to ambient temperature, the extinction peak does not recuperate these values. The reverse curves show transitions that are 10–12 °C lower that the direct ones, and more importantly, they do not reach the initial values (intensity loss of 20 and 40%, respectively). The shapes of the spectra representing the unstable dispersions are similar, indicating that the same phenomena are being produced under these conditions. These spectral shapes suggest the presence of a mixture of aggregates and individual particles at higher temperatures and in the transition region. Theoretical treatments52 of these nanoparticle associations point to the formation of long chains of AuNPs that start to interact as a consequence of the loss of H-bonds between the EG7 chains and water, in the presence of salt and upon increasing temperature. Following Pileni's reported equation,53 the peak with the maximum at 850 nm (Fig. 4, spectra obtained in the presence of 0.05 and 0.1 M NaCl at temperatures higher than TA) would correspond to a linear arrangement of EG7–AuNPs with an aspect ratio of 5.
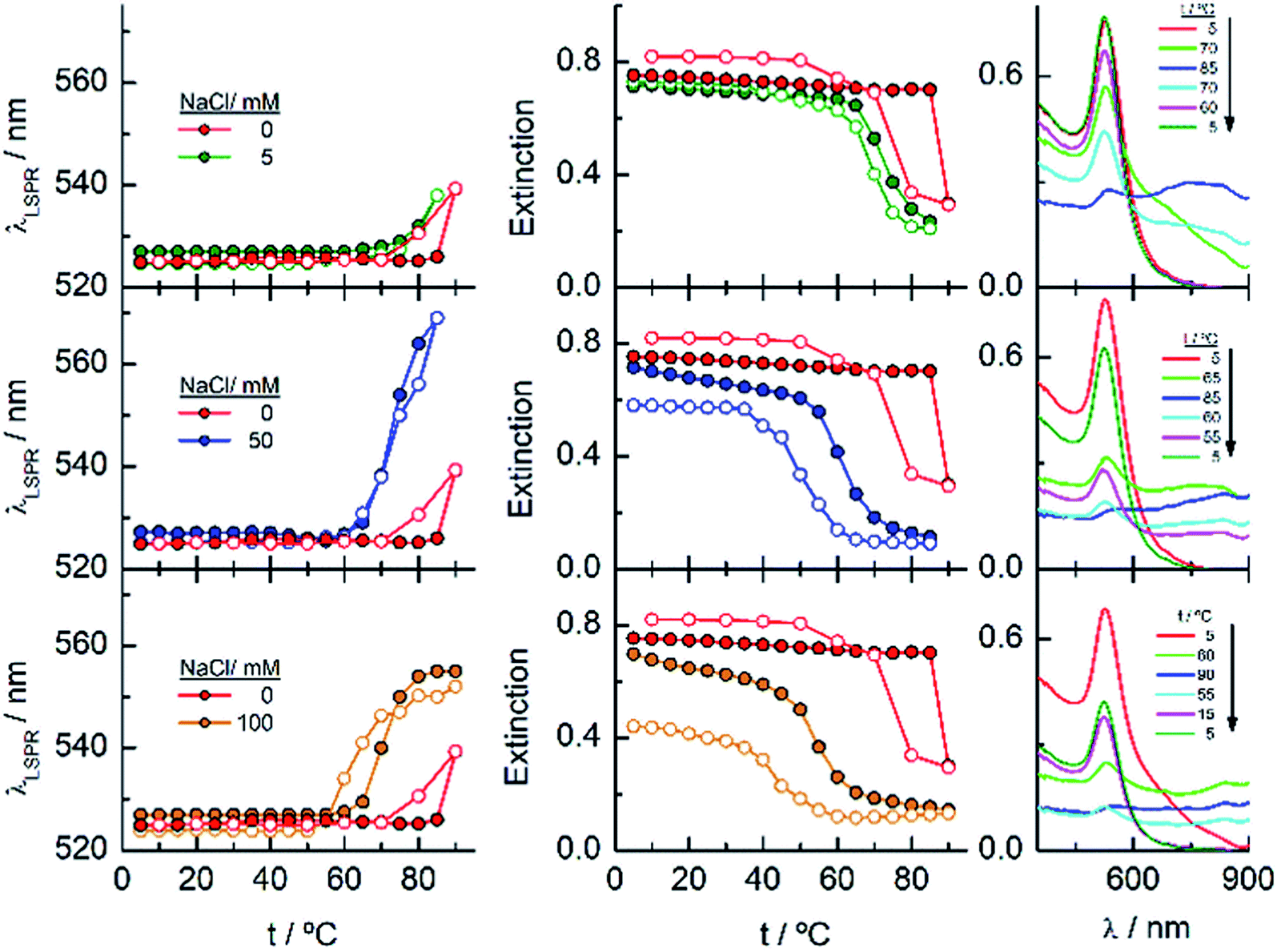 | ||
| Fig. 4 Changes in the LSPR wavelength and extinction of EG7–AuNPs as a function of temperature in the presence of different NaCl concentrations. Full circles represent the ascending temperature and empty circles, the descending temperature data. The spectra plotted in the graphs on the right correspond to these that are significative of the changes produced with temperature. | ||
As commented in relation to Fig. 2 for EG7–AuNPs, the DLS data obtained agree with this idea. In fact, whereas the DH values in water do not change with temperature up to 80 °C (the higher temperature that can be measured in our system), those obtained in the presence of 100 mM NaCl show two peaks, with the first at ∼29 nm being more important in the intensity plot than that at ∼720 nm. While the first peak does not appreciably change, the second one decreases up to values of ∼300 nm at higher temperature (Fig. S3†). These features would indicate that the firstly formed big aggregates are somewhat broken into smaller ones. Although the trend in the reverse temperature ramp is to reach the same conditions of the initial state, the decrease in size is only apparent if the 40% reduction in extinction in the UV-visible spectra is interpreted as precipitation of the big aggregates that is accelerated at high temperature and therefore, they cannot be monitored under these conditions. When the temperature goes down, the bigger aggregates have longer lifetimes and can be accounted for in the DLS measurement.
EG18–AuNPs have also been studied as a function of temperature in the presence of 0.15, 1 and 2 M NaCl (Fig. 5). While the system is stable in all the temperature ranges studied, when only 0.15 M NaCl is present, a strong displacement of the LSPR band to higher values accompanied by an extinction decrease is observed at 75 °C when the salt concentration is 1 M. The reverse scan shows a retention of these aggregates up to lower temperatures, but they terminated dissolving, and the initial state is completely recovered. When the salt concentration is increased to 2 M, a transition at 60 °C is observed that, although partially tends to go to the initial state, concludes with an extinction loss. The reverse transition determined either in the wavelength or in the extinction curve takes place at around 10 °C lower temperature than the direct one.
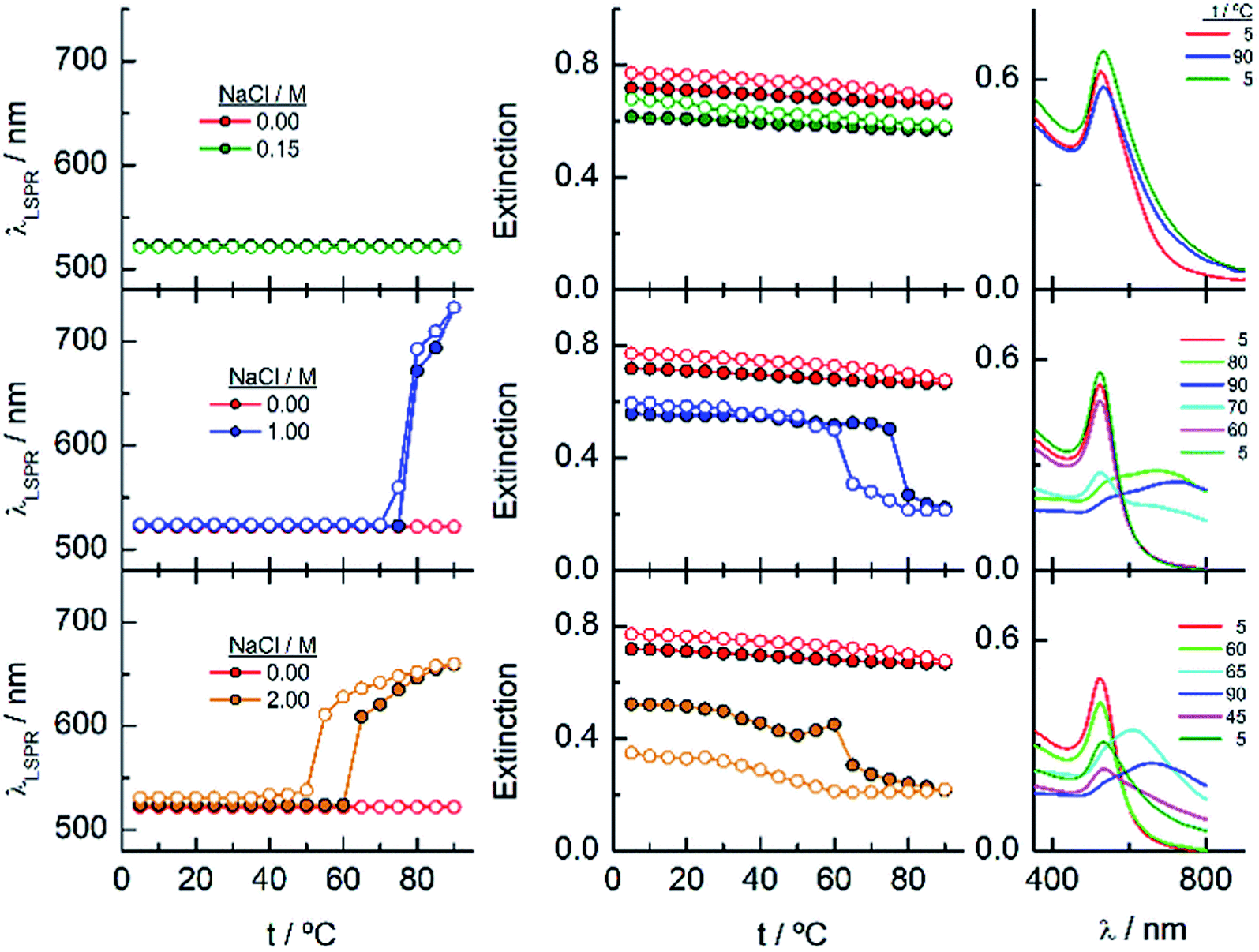 | ||
| Fig. 5 Changes in the LSPR wavelength and extinction of EG18–AuNPs as a function of temperature in the presence of different NaCl concentrations. Full circles represent the ascending temperature and empty circles, the descending temperature data. The spectra plotted in the graphs on the right correspond to these that are significative of the changes produced with temperature. | ||
To gain more insight into the different processes occurring in these temperature titrations experiments, we have studied these systems by DLS. EG18–AuNPs in the presence of 0.15 and 1 M NaCl show DH values of 29 nm, similar to these found in water, and they do not change with temperature in the measured interval (20–80 °C) (Fig. 6). However, in solutions of 1 M NaCl at 80 °C, the intensity plot shows two peaks that should be related to some assembly process under these conditions, in agreement with absorption data. The results obtained with solutions of 2 M NaCl are more complex. A mixture of peaks (with maxima at 36 and 180 nm) is obtained at the lowest temperature and they evolve with some irregularities up to 65 °C where they coalesce into a peak at higher size values (∼400–500 nm). The reverse scan shows a parallel behaviour: one peak up to 60 °C that splits into two peaks that remain in their values up to room temperature. These transitions are almost coincident with those observed by UV-visible spectroscopy. We note, however, that the plot of number distribution vs. DH shows the peak of lower DH up to the transition and after that, the one of higher DH, and a parallel behaviour in the reverse titration, thus indicating the predominance of the individual and aggregated EG18–AuNPs at lower and higher temperature, respectively. However, concomitant with these changes a precipitation of the larger aggregates should take place judging by the loss of extinction at the end of the titration.
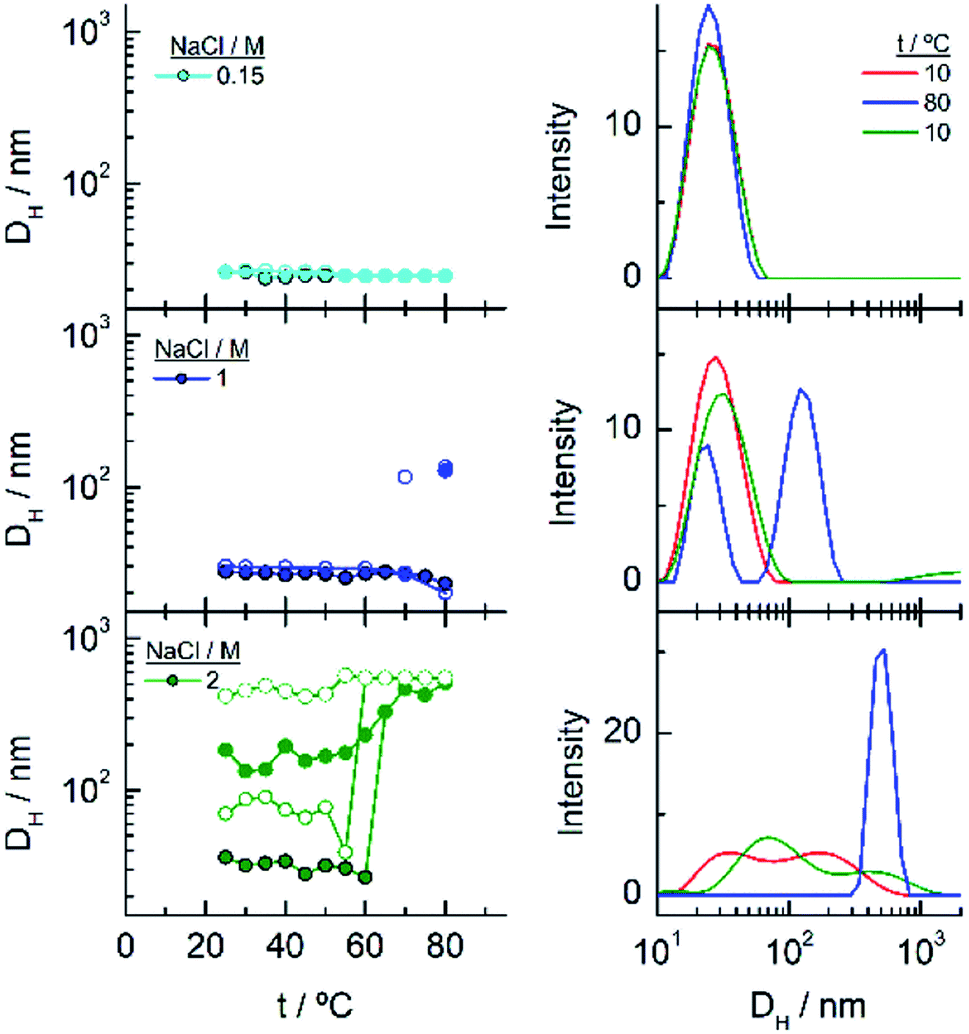 | ||
| Fig. 6 Changes in the hydrodynamic diameters of EG18–AuNPs as a function of temperature in the presence of different NaCl concentrations. Full circles represent the ascending temperature and empty circles, the descending temperature data. The intensity distribution of DH plotted in the graphs on the right correspond to these that are significative of the changes produced with temperature. | ||
When the AuNPs are protected by longer EGn chains, the dispersions present much higher stability in the presence of salt, allowing increasing the NaCl concentration up to 5 M.
Fig. 7 shows the analysis of the results obtained for EG45–AuNPs in the presence of 1, 3 and 5 M NaCl. No changes in the shape of the spectra obtained with the lowest concentration are observed in the temperature scan from 5 to 90 °C and vice versa. However, when the same experiments are run in the presence of 3 and 5 M salt concentration, transitions at 70 and 47 °C (measured in the wavelength trace), respectively, are obtained. Although in the direct scan, the curves seem similar to these observed for EG18–AuNPs, the phenomenon produced should be different. On one hand, the aggregates being formed from 45 to 90 °C do not disaggregate when the temperature is reversed (the wavelength and extinction remain almost constant in the reverse scan). On the other hand, the shape of the spectra is somewhat different to that for EG7- and EG18–AuNPs aggregates and is unchanged upon lowering the temperature to the initial value. These shapes have been related to the formation of tight clusters54 that should be more difficult to dissolve.
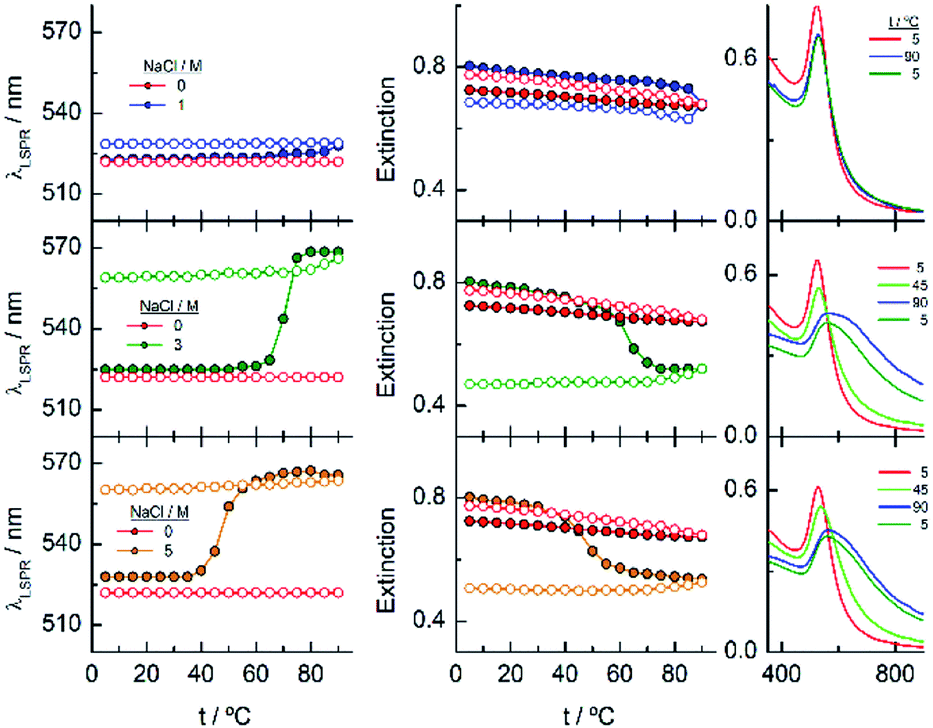 | ||
| Fig. 7 Changes in the LSPR wavelength and extinction of EG45–AuNPs as a function of temperature in the presence of different NaCl concentrations. Full circles represent the ascending temperature and empty circles, the descending temperature data. The spectra plotted in the graphs on the right correspond to these that are significative of the changes produced with temperature. | ||
In the DLS titrations of EG45–AuNPs (Fig. 8) we do not observe any transition in 1 M NaCl and a similar behaviour to that observed in 3 and 5 M salt concentration by UV-visible spectroscopy, with transitions at 57 and 42 °C, respectively. However, the observed comportment is somewhat more complex in the case of 2 M NaCl. At room temperature, the intensity distribution plot shows a broad envelope that seems to be composed of two contributions, and this feature continues up to 35 °C where they are resolved into two peaks at 45 and 195 nm. With increasing the temperature, they continue up to 60 °C where they coalesce into a signal of a size higher than 450 nm. In the reverse scan this unique peak is monitored at high temperature, and down to 45 °C, a mixture of two or three signals are observed up to room temperature. If we bear in mind the loss of extinction observed in the UV-visible titration experiment under the same experimental conditions, it can be concluded that a massive precipitation of the aggregates should occur, and the measured sizes correspond to the aggregates that remain in the suspension before they precipitate. The titration of EG45–AuNPs in the presence of 5 M NaCl is less complex as only one species is observed through the temperature scan. Moreover, the behaviour is the same as that shown by UV-visible spectroscopy. The DLS equivalent number distribution plots agree with the predominant existence of individual particles in 1 M NaCl solutions, the exchange between individual and aggregated nanoparticles upon the transition in 3 M, and in the ascending ramp in 5 M NaCl, whereas only the peaks corresponding to big aggregates are seen in the descending ramp of the latter.
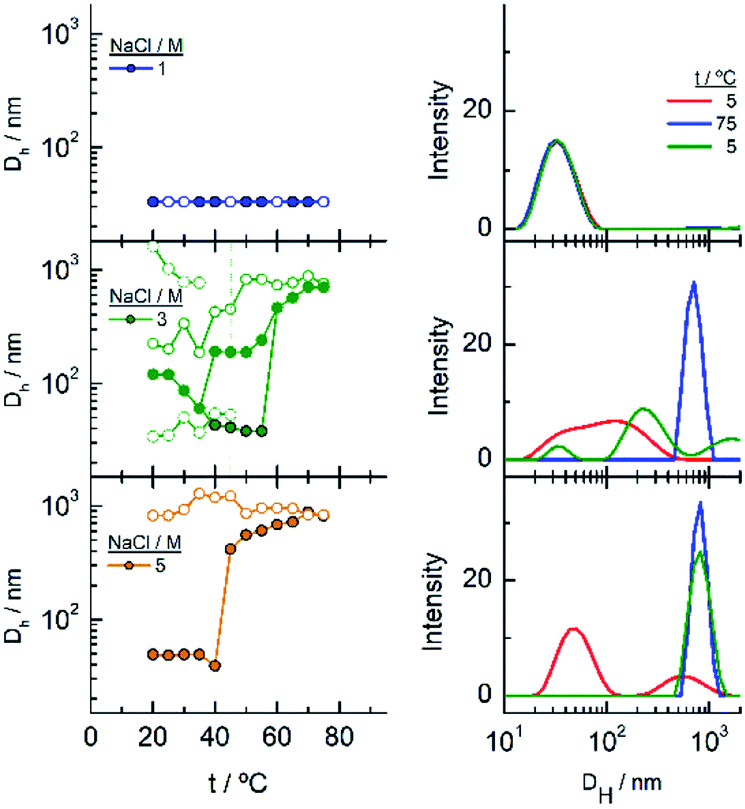 | ||
| Fig. 8 Changes in the hydrodynamic diameters of EG45–AuNPs as a function of temperature in the presence of different NaCl concentrations. The intensity distribution of DH plotted in the graphs on the right corresponds to these that are significative of the changes produced with temperature. | ||
Finally, the behaviour observed for EG136–AuNPs substantially differs from those described above (Fig. 9). Some aggregation is observed at 85 °C in 1 M NaCl solutions as detected by a displacement of the LSPR band to longer wavelength that is accompanied by some scattering at around 900 nm. The individual nanoparticles are soon obtained upon lowering the temperature to 75 °C. The presence of 3 or 5 M NaCl brings about some specific changes as a function of temperature with transitions at 60 and 45 °C in the ascending ramp, respectively, that makes the system evolve up to a state that is maintained in an interval of around 15 °C. The displacement of wavelength produced upon this transition (∼40 nm) would indicate the formation of aggregates of small size and with certain spherical symmetry.54 With increasing further the temperature, a decrease in wavelength (of around 20 nm, with transition points of 83 and 67 °C, respectively) brings the system to a new state that remains constant up to the highest temperature studied (90 °C). Interestingly, the evolution of the assemblies in the reversed ramp follows the same trend up to the second transition, where the spectra shift to the longest wavelength observed (∼590 nm) for these EG136–AuNPs, up to the first transition, to achieve the initial state at room temperature. An interesting point to note is the clear observation of a hysteresis phenomenon under these conditions, related with the first transition. Then, while the second transition is completely reversible, the first one requires lowering the temperature 10–15 °C down relative to that in the direct ramp. Again, the shape of the spectra can give some idea about the kind of aggregation that takes place in the presence of high salt concentrations. In fact, the spectrum observed in the interval of 50–65 °C would represent the formation of small clusters that, with increasing temperature, show a rise in extinction that is mainly due to Rayleigh scattering. This phenomenon is related to the behaviour of the polymer chains more than the evolution of the cluster size.51 In fact, the extinction continues increasing up to the highest temperature and follows exactly the same trend in the reverse ramp. This high temperature together with the presence of salt would make the chains undergo an additional dehydration and collapse, achieving the EG136-AuNP cloud point under these conditions (Scheme 2).
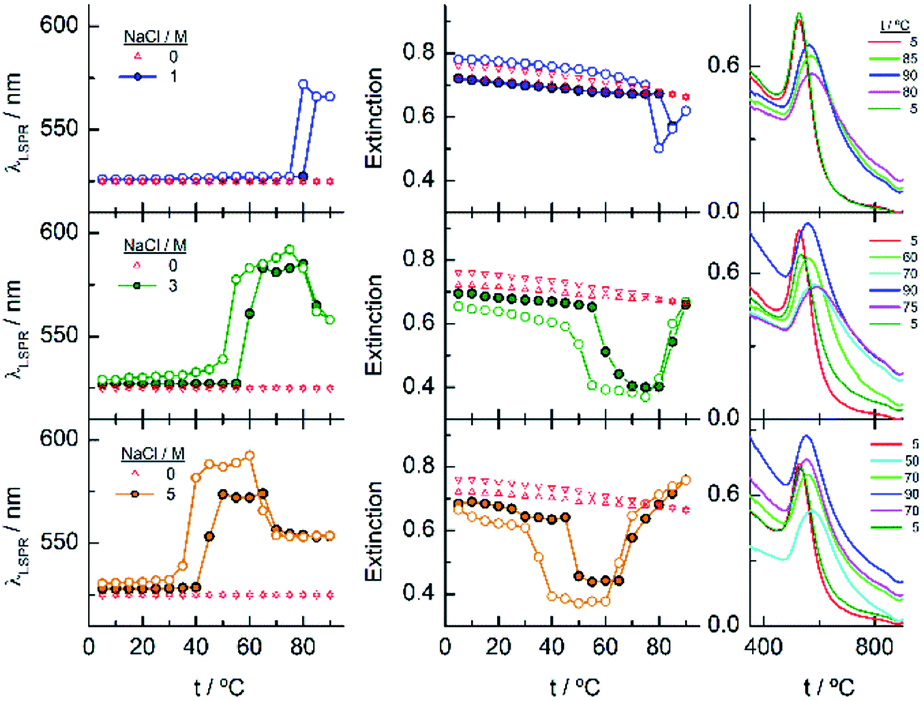 | ||
| Fig. 9 Changes in the LSPR wavelength and extinction of EG136–AuNPs as a function of temperature in the presence of different NaCl concentrations. Full circles represent the ascending temperature and empty circles, the descending temperature data. The spectra plotted in the graphs on the right correspond to these that are significative of the changes produced with temperature. | ||
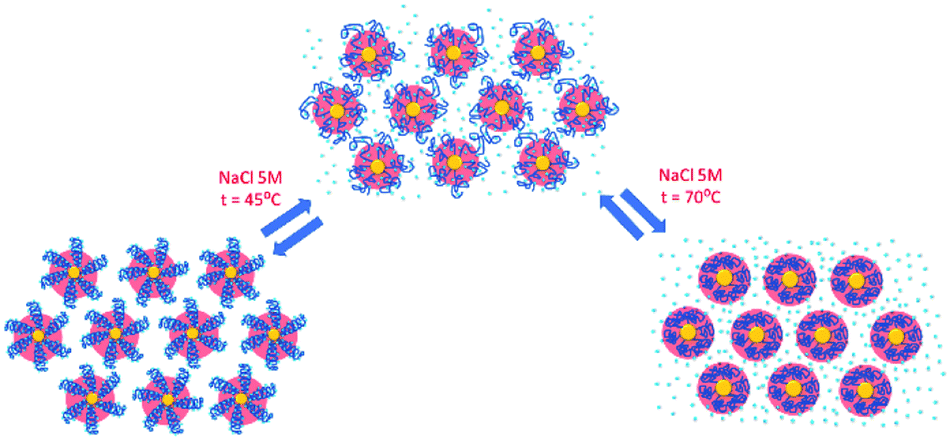 | ||
| Scheme 2 Temperature transitions of EG136–AuNPs. | ||
The phenomenon of bistability and its related hysteresis have been reported for nanoparticles functionalized with specific ionizable ligands and take place through a delicate balance between van der Waals and electrostatic interparticle interactions. The overall interaction potential features an energy barrier at finite separation that generates hysteresis when the magnitude to overcome the aggregate to the dispersed state differs from that to overcome the reverse step.55 Temperature induced hysteresis has been also observed for AuNPs during reversible clustering, pointing out that the rate of heating and cooling altered its magnitude, applying this fact as a method to distinguish the kinetics and thermodynamic regimes.56 Within the systems studied in this work, we observe this phenomenon in the case of EG136–AuNPs in the presence of 1, 3 and 5 M NaCl, although is more evident at the highest concentration. In the other EGn–AuNPs analysed there are some signs of the existence of hysteresis, but the simultaneous precipitation avoids its observation.
The observed trend with temperature in the DH values of the EG136–AuNPs in the presence of salts partially agrees with the results of UV-visible spectroscopy (Fig. 10). The only peak measured in 1 M NaCl in all the temperature scans corresponds to the findings of unchanged wavelength and extinction up to high temperature values. The suspension in 3 M NaCl shows only one peak in all the temperature ranges studied but it changes in size describing a transition at 57 °C in the direct scan to higher hydrodynamic diameters (close to 1000 nm), indicating the formation of big aggregates. These clusters of nanoparticles, however, must not strongly couple as the wavelength shift is very small. Moreover, the presence of strong Rayleigh scattering after the second transition points in this direction. In contrast, in the presence of 5 M NaCl, two peaks are observed from the lower temperature, with the largest one (at sizes higher than 600 nm) in a small ratio (not seen in the number distribution plot). After the transition, the peaks collapse, and only the larger size aggregates remain. The reverse scan shows only one peak that starts to decrease at a temperature coinciding with that observed for the first transition in UV-visible spectroscopy (∼36 °C). The feature observed at temperatures higher than 70–80 °C in absorption spectroscopy cannot be seen in DLS as the maximum temperature reached by our system is 75–80 °C. However, the high reversibility obtained in this portion of the curve would agree with the Rayleigh scattering being responsible for this effect. We have taken photographs of the EG136-AuNP suspensions at 20, 60 and 75 °C to corroborate the kind of phenomenon taking place as the temperature increases. The inset in Fig. 10 shows the changes in colour of the suspension that goes from the red wine normally observed for the individually dispersed nanoparticles (left, 20 °C) to violet when there is some aggregation (middle, 60 °C) and finally to the cloudy aspect (right, 75 °C), confirming our suggestions.
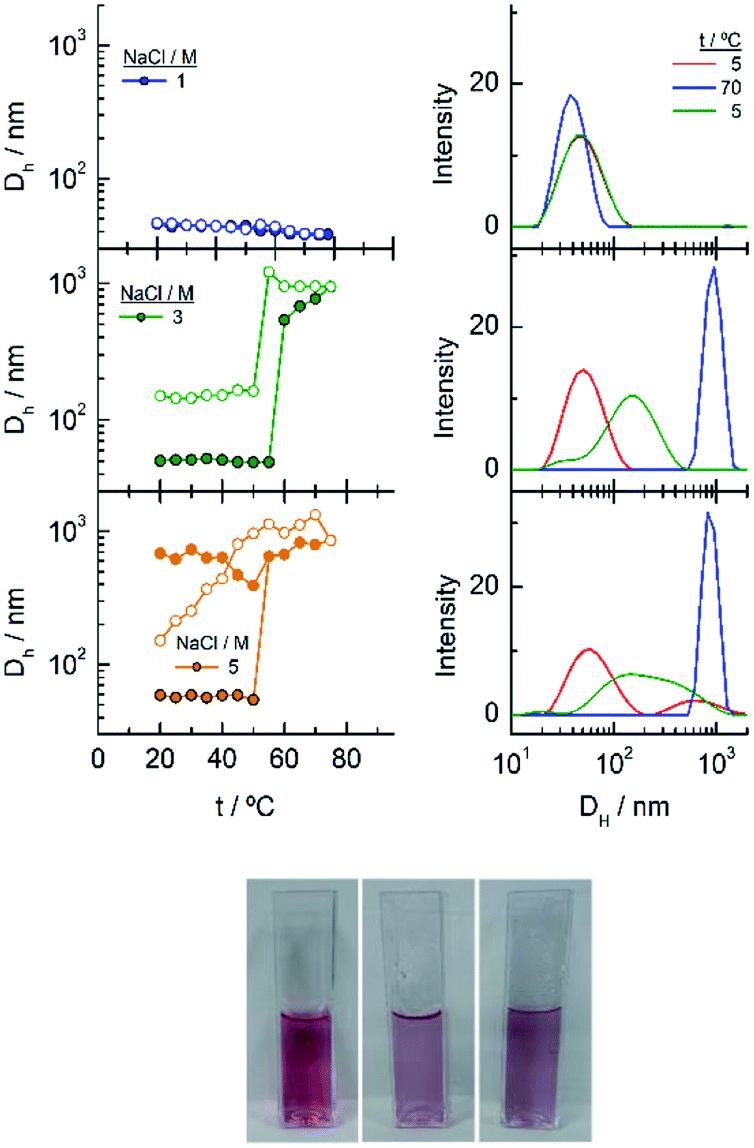 | ||
| Fig. 10 (Left) Changes in the hydrodynamic diameters of EG136–AuNPs as a function of temperature in the presence of different NaCl concentrations. The spectra plotted in the graphs on the right correspond to these that are significative of the changes produced with temperature. (Right) Photographs of the EG136-AuNP suspension in 5 M NaCl taken at 20, 60 and 75 °C (left to right). | ||
Conclusions
EGn–AuNPs prepared by ligand exchange from c-AuNPs present increased stability in aqueous suspensions in wide pH and temperature ranges. As has previously been reported, the stability of EGn–AuNPs strongly depends on the grafting density,6,8 and we showed in this work that for a specific nanoparticle core size, the chain length also has some influence. In fact, even though longer chains are grafted to the surface with larger footprints than the shorter ones, they confer higher stabilization, as it is typical for colloidal steric stabilization by neutral polymer chains. Once the optimal conditions for EGn grafting are achieved, the EGn-AuNP suspensions resist the presence of certain salt concentrations. While all the prepared nanoparticles practically withstand up to 1 M NaCl, the EG7-and EG18–AuNPs lose dispersity in the presence of relatively low concentrations of CaCl2, an effect that should be ascribed to the divalent cation.The presence of NaCl in the EGn-AuNP suspensions also influences the thermo-sensitivity differently. EG7–AuNPs show transitions from the individual to aggregated nanoparticles strongly dependent on the NaCl concentration, presenting reversibility only in very low concentration (5 mM NaCl). With increasing chain length, the suspensions resist the presence of higher salt concentrations. The EG18–AuNPs show a reversible temperature transition in 1 M NaCl but become only partially reversible in the presence of 2 M NaCl. The formation of big aggregates (as seen by DLS) and their precipitation in the later conditions should be responsible for the loss of material upon restoring the initial conditions. Although the EG45–AuNPs did not assemble in the presence of 3 and 5 M NaCl at room temperature, they suffer temperature transitions to the formation of aggregates that do not dissolve upon lowering the temperature. The bigger aggregates suddenly precipitate and cannot be monitored neither by UV-visible spectroscopy nor DLS, and the smaller ones remain suspended upon reaching the initial conditions.
The larger chain EG136 confers the most interesting behaviour to the AuNP suspensions. First, the EG136–AuNPs show a reversible transition at around 85 °C in 1 M NaCl. With increasing NaCl concentration to 3 and 5 M, the systems show two transitions in the temperature range studied. The first transition at lower temperature would lead to the formation of small aggregates that can remain suspended in a temperature interval of 15 °C and then evolve to an assembled state where the metal cores are not coupled as the LSPR wavelength decreases from the value adopted after the first transition. Under these conditions, a Rayleigh scattering is observed that is ascribed to the effects of the salt dehydrating the polymer chains and making them collapse. While this second transition is completely reversible, the first one shows a hysteretic behaviour that supposes an incremental energy barrier to overcome for the aggregates to reach the disassembled AuNPs relative to that for the assembly. These findings encourage us to go deeply into the control of the hysteretic behaviour of the EG136–AuNPs to find applications in the various fields that depend on this phenomenon.56
Author contributions
Conceptualization, M. C., A. F-M., and T. P.; methodology, M. C., A. F-M., T. P., M. B., and J. M. S.; formal analysis, M. C., A. F-M., T. P., R. M., and G. S-O.; investigation, M. C., A. F-M., T. P., M. B., J. M. S., R. M., and G. S-O.; writing-original draft preparation, M. C., A. F-M., and T. P.; funding acquisition, T. P. and M. B. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministerio de Ciencia e Innovación (Project RED2018-102412-T Network of Excellence Electrochemical Sensors and Biosensors), Junta de Andalucía and Universidad de Cordoba (UCO-FEDER-2018: ref. 1265074-2B and Plan Propio, Submod. 1.2. P.P. 2019) for financial support of this work. M. C. acknowledges Ministerio de Universidades for FPU 17/03873 grant and A. F.-M. acknowledges Universidad de Cordoba for Contrato Predoctoral grant.References
- D. Matsukuma and H. Otsuka, in Colloid and Interface Science in Pharmaceutical Research and Development, ed. H. Ohshima and K. Makino, Elsevier, Amsterdam, 2014, pp. 261–283 Search PubMed.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., Int. Ed., 2011, 50, 1980–1994 CrossRef CAS PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- R. Cai, J. Ren, Y. Ji, Y. Wang, Y. Liu, Z. Chen, Z. Farhadi Sabet, X. Wu, I. Lynch and C. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 1997–2008 CrossRef CAS PubMed.
- D. R. Hristov, H. Lopez, Y. Ortin, K. O'Sullivan, K. A. Dawson and D. F. Brougham, Nanoscale, 2021, 13, 5344–5355 RSC.
- K. Rahme, L. Chen, R. G. Hobbs, M. A. Morris, C. O'Driscoll and J. D. Holmes, RSC Adv., 2013, 3, 6085–6094 RSC.
- J. Lu, Y. Xue, R. Shi, J. Kang, C.-Y. Zhao, N.-N. Zhang, C.-Y. Wang, Z.-Y. Lu and K. Liu, Chem. Sci., 2019, 10, 2067–2074 RSC.
- M. Retout, E. Brunetti, H. Valkenier and G. Bruylants, J. Colloid Interface Sci., 2019, 557, 807–815 CrossRef CAS PubMed.
- W. Wang, Q.-Q. Wei, J. Wang, B.-C. Wang, S.-h. Zhang and Z. Yuan, J. Colloid Interface Sci., 2013, 404, 223–229 CrossRef CAS PubMed.
- D. N. Benoit, H. G. Zhu, M. H. Lilierose, R. A. Verm, N. Ali, A. N. Morrison, J. D. Fortner, C. Ayendano and V. L. Colvin, Anal. Chem., 2012, 84, 9238–9245 CrossRef CAS PubMed.
- P. G. de Gennes, Macromolecules, 1980, 13, 1069–1075 CrossRef CAS.
- S. Alexander, J. Phys., 1977, 38, 983–987 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1949, 17, 303–310 CrossRef CAS.
- P. G. de Gennes, Adv. Colloid Interface Sci., 1987, 27, 189–209 CrossRef CAS.
- S. Saeki, N. Kuwahara, M. Nakata and M. Kaneko, Polymer, 1976, 17, 685–689 CrossRef CAS.
- P. U. Kenkare and C. K. Hall, AIChE J., 1996, 42, 3508–3522 CrossRef CAS.
- R. Iida, H. Mitomo, Y. Matsuo, K. Niikura and K. Ijiro, J. Phys. Chem. C, 2016, 120, 15846–15854 CrossRef CAS.
- K. Xiong, H. Mitomo, X. Su, Y. Shi, Y. Yonamine, S.-i. Sato and K. Ijiro, Nanoscale Adv., 2021, 3, 3762–3769 RSC.
- R. Iida, H. Mitomo, K. Niikura, Y. Matsuo and K. Ijiro, Small, 2018, 14, 1704230 CrossRef PubMed.
- D. Zambo, G. Z. Radnoczi and A. Deak, Langmuir, 2015, 31, 2662–2668 CrossRef CAS PubMed.
- D. Zámbó, S. Pothorszky, D. F. Brougham and A. Deák, RSC Adv., 2016, 6, 27151–27157 RSC.
- C. Durand-Gasselin, M. Capelot, N. Sanson and N. Lequeux, Langmuir, 2010, 26, 12321–12329 CrossRef CAS PubMed.
- L. Cheng, J. Song, J. Yin and H. Duan, J. Phys. Chem. Lett., 2011, 2, 2258–2262 CrossRef CAS.
- H. H. Zhang, W. J. Wang, S. Mallapragada, A. Travesset and D. Vaknin, Nanoscale, 2017, 9, 164–171 RSC.
- H. Zhang, W. Wang, M. Akinc, S. Mallapragada, A. Travesset and D. Vaknin, Nanoscale, 2017, 9, 8710–8715 RSC.
- H. Zhang, W. Wang, S. Mallapragada, A. Travesset and D. Vaknin, J. Phys. Chem. C, 2017, 121, 15424–15429 CrossRef CAS.
- S. Nayak, M. Fieg, W. J. Wang, W. Bu, S. Mallapragada and D. Vaknin, Langmuir, 2019, 35, 2251–2260 CrossRef CAS PubMed.
- W. Wang, H. J. Kim, W. Bu, S. Mallapragada and D. Vaknin, Langmuir, 2020, 36, 311–317 CrossRef CAS PubMed.
- H. J. Kim, W. J. Wang, A. Travesset, S. K. Mallapragada and D. Vaknin, ACS Nano, 2020, 14, 6007–6012 CrossRef CAS PubMed.
- H. J. Kim, M. M. Hossen, A. C. Hillier, D. Vaknin, S. K. Mallapragada and W. J. Wang, ACS Appl. Nano Mater., 2020, 3, 8216–8223 CrossRef CAS.
- H. J. Kim, W. J. Wang, S. K. Mallapragada and D. Vaknin, J. Phys. Chem. Lett., 2021, 12, 1461–1467 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 55–75 RSC.
- G. Sanchez-Obrero, M. Chavez, R. Madueno, M. Blazquez, T. Pineda, J. M. Lopez-Romero, F. Sarabia, J. Hierrezuelo and R. Contreras-Caceres, J. Electroanal. Chem., 2018, 823, 663–671 CrossRef CAS.
- M. Chávez, G. Sánchez-Obrero, R. Madueño, J. M. Sevilla, M. Blázquez and T. Pineda, J. Electroanal. Chem., 2021, 880, 114892 CrossRef.
- C. A. Simpson, A. C. Agrawal, A. Balinski, K. M. Harkness and D. E. Cliffel, ACS Nano, 2011, 5, 3577–3584 CrossRef CAS PubMed.
- A. C. Templeton, J. J. Pietron, R. W. Murray and P. Mulvaney, J. Phys. Chem. B, 2000, 104, 564–570 CrossRef CAS.
- P. Harder, M. Grunze, R. Dahint, G. M. Whitesides and P. E. Laibinis, J. Phys. Chem. B, 1998, 102, 426–436 CrossRef CAS.
- B. Cárdenas, G. Sanchez-Obrero, R. Madueño, J. M. Sevilla, M. Blázquez and T. Pineda, J. Phys. Chem. C, 2014, 118, 22274–22283 CrossRef.
- F. Canaveras, R. Madueño, J. M. Sevilla, M. Blazquez and T. Pineda, J. Phys. Chem. C, 2012, 116, 10430–10437 CrossRef CAS.
- E. Reyes, R. Madueño, M. Blazquez and T. Pineda, J. Phys. Chem. C, 2010, 114, 15955–15962 CrossRef CAS.
- A. J. Viudez, R. Madueno, T. Pineda and M. Blazquez, J. Phys. Chem. B, 2006, 110, 17840–17847 CrossRef CAS PubMed.
- M. Schollbach, F. Zhang, F. Roosen-Runge, M. W. A. Skoda, R. M. J. Jacobs and F. Schreiber, J. Colloid Interface Sci., 2014, 426, 31–38 CrossRef CAS PubMed.
- G. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS PubMed.
- J. L. Perry, K. G. Reuter, M. P. Kai, K. P. Herlihy, S. W. Jones, J. C. Luft, M. Napier, J. E. Bear and J. M. DeSimone, Nano Lett., 2012, 12, 5304–5310 CrossRef CAS PubMed.
- V. B. Damodaran, C. J. Fee, T. Ruckh and K. C. Popat, Langmuir, 2010, 26, 7299–7306 CrossRef CAS PubMed.
- H. D. Willauer, J. G. Huddleston and R. D. Rogers, Ind. Eng. Chem. Res., 2002, 41, 1892–1904 CrossRef CAS.
- J. G. Huddleston, H. D. Willauer and R. D. Rogers, J. Chem. Eng. Data, 2003, 48, 1230–1236 CrossRef CAS.
- U. Dahal, Z. Wang and E. E. Dormidontova, Macromolecules, 2018, 51, 5950–5961 CrossRef CAS.
- U. Dahal and E. E. Dormidontova, Macromolecules, 2020, 53, 8160–8170 CrossRef CAS.
- Y. Liu, M. K. Shipton, J. Ryan, E. D. Kaufman, S. Franzen and D. L. Feldheim, Anal. Chem., 2007, 79, 2221–2229 CrossRef CAS PubMed.
- M.-Q. Zhu, L.-Q. Wang, G. J. Exarhos and A. D. Q. Li, J. Am. Chem. Soc., 2004, 126, 2656–2657 CrossRef CAS PubMed.
- R. W. Taylor, R. Esteban, S. Mahajan, R. Coulston, O. A. Scherman, J. Aizpurua and J. J. Baumberg, J. Phys. Chem. C, 2012, 116, 25044–25051 CrossRef CAS.
- A. Brioude, X. C. Jiang and M. P. Pileni, J. Phys. Chem. B, 2005, 109, 13138–13142 CrossRef CAS PubMed.
- R. W. Taylor, T.-C. Lee, O. A. Scherman, R. Esteban, J. Aizpurua, F. M. Huang, J. J. Baumberg and S. Mahajan, ACS Nano, 2011, 5, 3878–3887 CrossRef CAS PubMed.
- D. Wang, B. Kowalczyk, I. Lagzi and B. A. Grzybowski, J. Phys. Chem. Lett., 2010, 1, 1459–1462 CrossRef CAS.
- J. Kruse, S. Merkens, A. Chuvilin and M. Grzelczak, ACS Appl. Nano Mater., 2020, 3, 9520–9527 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00392e |
| This journal is © The Royal Society of Chemistry 2021 |