
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-driven crystal–crystal transformation and morphology change in a 2D layered inorganic POM-based framework†
Hui-Min
Zeng
,
Chao
Wang
,
Wei-Hong
Wu
,
Wei-Tao
Mao
,
Zhan-Guo
Jiang
 * and
Cai-Hong
Zhan
*
* and
Cai-Hong
Zhan
*
College of Chemistry and Life Sciences, Institute of Physical Chemistry, Zhejiang Normal University, Key Laboratory of the Ministry of Education for Advanced Catalysis Material, No. 688, Yingbin Avenue, Jinhua, Zhejiang, China 321004. E-mail: jzg@zjnu.cn; chzhan@zjnu.cn
First published on 25th June 2021
Abstract
In this paper, a pure 2D inorganic POM-based framework underwent a single crystal to single crystal conversion when soaked in organic solvents that are miscible with water, forming a more densely packed identical framework accompanying the formation of nanowires. The change in morphology is closely related to the surface tension of water, and the lower surface tension achieved by dehydration promotes the formation of nanowires, which is revealed by SXRD, PXRD, SEM, TGA and electrochemical impedance spectroscopy (EIS).
Introduction
Layered materials, unlike porous framework materials such as metal–organic frameworks (MOFs) whose structures and corresponding properties have been greatly focused on,1–7 possess strong in-plane covalent bonds but weak out-of-plane bonds coupled by van der Waals (vdW) interactions.8 Physical or chemical methods can break these weak bonds allowing the exfoliation of 2D nanomaterials with thicknesses down to the atomically thin limit.8 The characteristic weak bonds between layers make it easy to dope foreign molecules, ions, atoms and other guests. The introduced guest can change the layer spacing, and the expansion of the layer spacing will significantly weaken the vdW interactions between the layers of two-dimensional materials, which can induce ultra-thin nanosheets in the bulk crystals.9 In 2017, Zhou et al. prepared ultra-thin 2D MOF nanosheets by intercalating 4,4′-dipyridine disulfide ligands into layered MOFs, which increased the interlayer spacing from 19.6 Å to 45.2 Å to promote stripping.10 Another effective method is to use appropriate organic solvents to assist peeling. The first successful liquid phase exfoliation of graphite was achieved in the organic solvent NMP, which reduces the surface energy of graphite to exfoliate graphene.11 However, the peeling of cluster-based materials is still rarely reported. Moreover, it is also a challenge to extract lower-dimensional materials such as 1D nanowires from three-dimensional (3D) bulk materials.Polyoxometalate (POM), a kind of anionic early transition metal oxo cluster, has been the focus of attention for researchers based on diverse structures such as cages,12,13 spheres,14 rings,15 wheel16 shapes etc.,. and intriguing properties ranging from catalysis,17,18 medicine,19 optics,20 electricity21 and magnetism.22 Herein, we report for the first time that a POM-based 2D crystalline material was transformed into low-level 1D nanowires. Firstly, a pure inorganic POM based framework with higher flexibility and weaker coordination interactions between heterometals and oxygen was synthesized. Subsequently, a single-crystal-to-single-crystal transition of the POM-based framework occurred when it was soaked in an organic solvent (methanol, ethanol, acetonitrile, acetone, and THF), obtaining an identical framework with a more densely stacking mode. Surprisingly, its morphology has changed from blocky layers to nanowires as shown in Scheme 1.
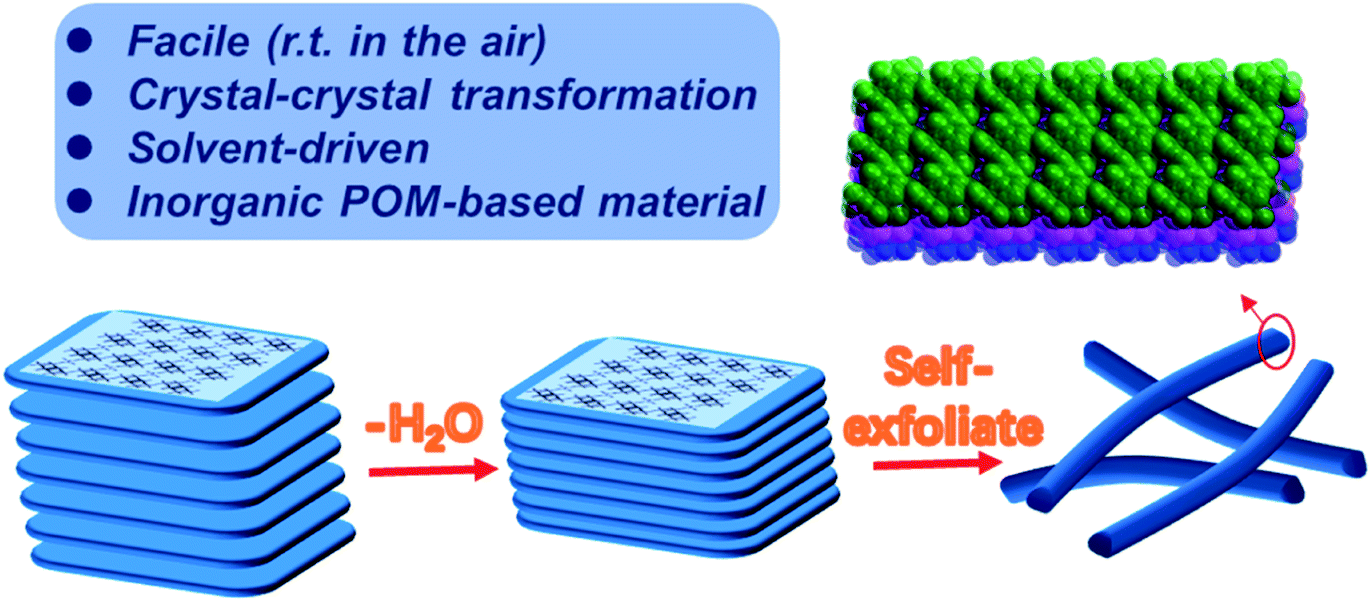 | ||
| Scheme 1 The morphology changes of the 2D layered inorganic POM-based framework. | ||
Results and discussion
Crystal structure and transformation
Compound 1 was synthesized through a one-pot method in high yield and crystallized rapidly as a kinetic product. Single-crystal X-ray diffraction analysis reveals that Compound 1 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , and its asymmetric unit consists of one {Mo4O14}, one {Mn0.5(H2O)2} and three Na+, in which two {Mo4O14} subunits stack together via edge-sharing to give rise to {Mo8O28}. The connection between Mo atoms is established via three different types of oxygen atoms: six double bridged oxygen atoms O (μ2), four three-bridging oxygen atoms O (μ3), and two four bridged oxygen atoms O (μ4) that only exists in {γ-Mo8O26}, which is the one of the eight isomers of {Mo8O26}. The difference between {γ-Mo8O28} and {γ-Mo8O26} is that the former has 16 terminal oxygen atoms and all the Mo atoms adopt a six-coordinate mode, forming eight {MoO6} octahedra, while two Mo atoms form two {MoO5} tetragonal pyramids with five O atoms in a five-coordinate mode in the latter (Fig. S1a, b†). The adjacent {γ-Mo8O28} subunits are connected into an infinite 1D chain through a single bridging oxygen atom (Fig. S2†). Each Mn atom is coordinated by six oxygen atoms forming a {MnO6} octahedron, in which four oxygen atoms are from H2O and the other two are from two octa-molybdate. Thus, octahedral Mn centers are merged into the infinite 1D chain to form the resultant 2D anionic {MnMo8O28} sheet (Fig. S3, S4†).
, and its asymmetric unit consists of one {Mo4O14}, one {Mn0.5(H2O)2} and three Na+, in which two {Mo4O14} subunits stack together via edge-sharing to give rise to {Mo8O28}. The connection between Mo atoms is established via three different types of oxygen atoms: six double bridged oxygen atoms O (μ2), four three-bridging oxygen atoms O (μ3), and two four bridged oxygen atoms O (μ4) that only exists in {γ-Mo8O26}, which is the one of the eight isomers of {Mo8O26}. The difference between {γ-Mo8O28} and {γ-Mo8O26} is that the former has 16 terminal oxygen atoms and all the Mo atoms adopt a six-coordinate mode, forming eight {MoO6} octahedra, while two Mo atoms form two {MoO5} tetragonal pyramids with five O atoms in a five-coordinate mode in the latter (Fig. S1a, b†). The adjacent {γ-Mo8O28} subunits are connected into an infinite 1D chain through a single bridging oxygen atom (Fig. S2†). Each Mn atom is coordinated by six oxygen atoms forming a {MnO6} octahedron, in which four oxygen atoms are from H2O and the other two are from two octa-molybdate. Thus, octahedral Mn centers are merged into the infinite 1D chain to form the resultant 2D anionic {MnMo8O28} sheet (Fig. S3, S4†).
Yellow rod crystals of Compound 1 were transferred to a 2 mL centrifuge tube containing acetone and soaked for 3 h. SCXRD analysis indicates that the resultant compound displays a 2D layered structure identical to that of Compound 1, and hereafter is denoted as Compound 1a, which crystallizes in the triclinic space group P (as shown in Fig. 1a, b). Compared with Compound 1, the components to construct the 2D layered structure of Compound 1a are the same, including 1D {γ-Mo8O28} chains and {MnO6} octahedra, but a slight bond contraction between each layer was observed from the crystal data (Table S2 and Fig. S5†). Fig. 1c, S6 and S7† show that the position of the {MnO6} linker has been changed as the position of the {MnO6} linker in Compound 1 or in Compound 1a can't overlap in different directions. This result shows the flexibility of the intra-layer caused by weaker coordination interactions between Mn and O.
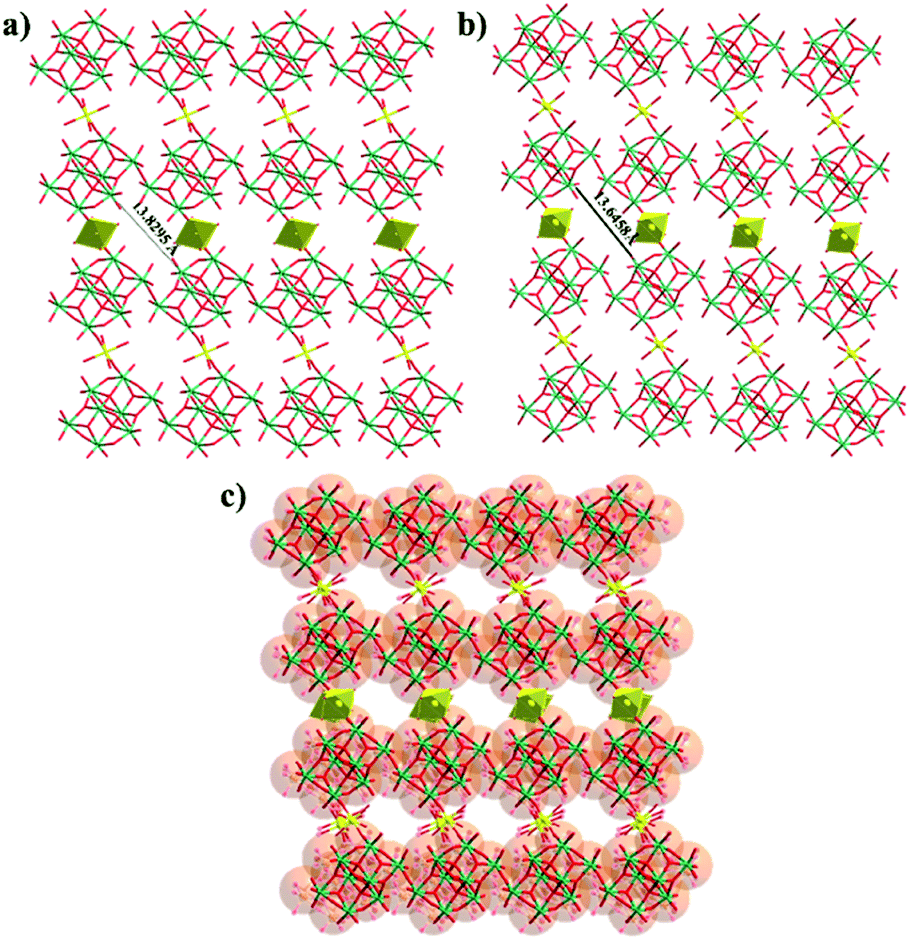 | ||
| Fig. 1 Ball and stick presentation of the 2D [MnMo8O28]n sheet of Compound 1 (a), Compound 1a (b) and the overlays of Compound 1 and Compound 1a (c) (Mo, green; Mn, yellow; O, red). | ||
Fig. S8, S9† and 1 show the 0D, 1D and 2D graphs of Compound 1 before and after conversion and the corresponding spacing change. To our surprise, the radius of the pore was shortened by 0.1837 Å from 13.8295 Å to 13.6458 Å. Moreover, a shrinkage of 2.482 Å can also be noticed between chains after transformation, as displayed in Fig. 2a, b. And the distance between adjacent layers was shortened by 2.542 Å from 9.4133 Å to 6.8713 Å (Fig. S10†).
 | ||
| Fig. 2 3D stacking pattern of the 2D [MnMo8O28]n sheets of Compound 1 (a) and Compound 1a (b). | ||
At the beginning, Compound 1 was collected by filtration and the air-dried sample was used for basic characterization. The powder X-ray diffraction (PXRD) patterns of Compound 1 can be compared with the simulated pattern obtained from the X-ray single-crystal diffraction analysis (Fig. S12†). But when Compound 1 was dried under vacuum overnight, the color slightly changed from yellow to light yellow (Fig. S13†). This aroused our interest to record their PXRD patterns because the change in color may suggest a change in structure. As we proposed, there is a big difference between their PXRD patterns and the simulated pattern (Fig. S14†). Considering that the aim of vacuum drying is to remove the solvent, we employed another method for removing the solvent. Compound 1 was soaked in acetone for 3 h, and then the intact crystal did not dissolve but shatter. This is because of water solvent reduction. And a transparent crystal was selected from the sample soaked in acetone for single-crystal X-ray diffraction analysis. The result shows that the unit cell as well as the structure changed , in which the position of the metal center and the interlayer spacing are modified. To further investigate whether only acetone can cause this change, we adopted various solvents ranging from polar solvents to non-polar solvents (methanol, ethanol, acetonitrile, DMF, THF, ether, toluene, dichloromethane, trichloromethane, and n-hexane).
When Compound 1 was soaked in methanol, ethanol and DMF, a suitable crystal for single-crystal X-ray diffraction analysis cannot be obtained, but the PXRD pattern of that soaked in methanol (Fig. S21†) and ethanol (Fig. S22†) can be compared with that soaked in acetone (Fig. 3a); while no peak can be observed from the PXRD pattern of that soaked in DMF (Fig. S20†), demonstrating that Compound 1 in DMF may be destroyed. Further experiments show that transparent crystals soaked in acetonitrile, THF, ether, toluene, dichloromethane, trichloromethane, and n-hexane can be selected for single-crystal X-ray diffraction analysis and the unit cell of the first two is the same as that of Compound 1a, while the latter five are the same as that of Compound 1.
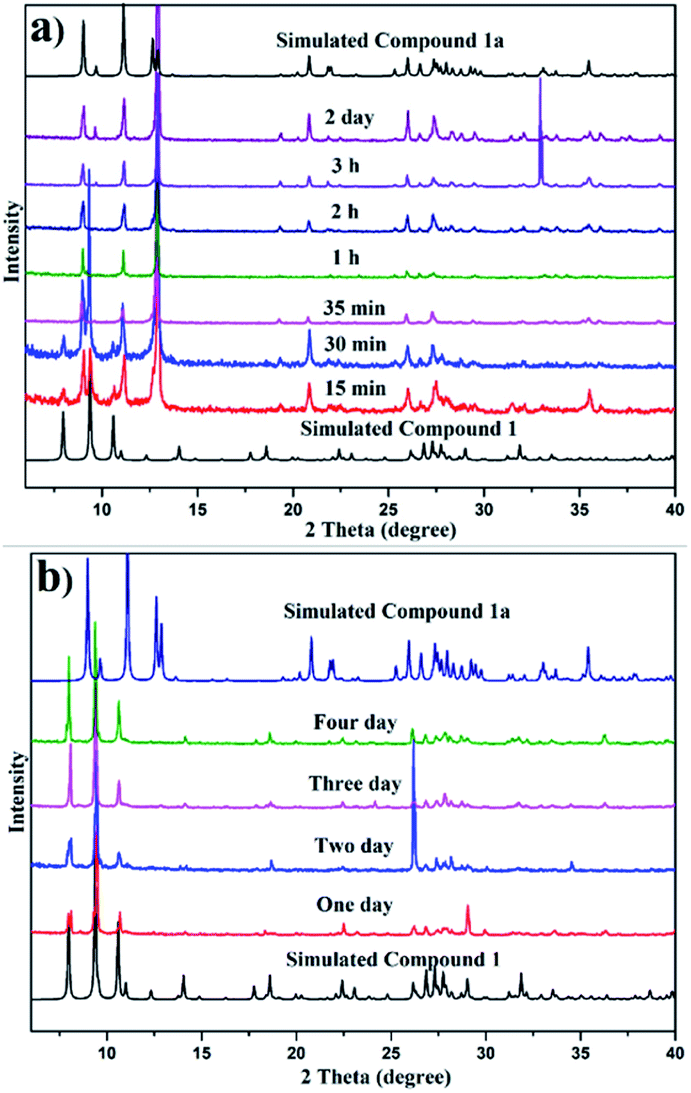 | ||
| Fig. 3 Time-dependent PXRD patterns of Compound 1 soaked in acetone (a) and soaked in trichloromethane (b). | ||
Initially, we suspected that the insufficient soaking time was the reason for the lack of transformation in the latter five solvents, and hence different soaking times for those solvents were studied. Fig. S15–S18,†3b, show that even if the soaking time is extended to 4 days, the conversion of the samples soaked in ether, toluene, dichloromethane, trichloromethane and n-hexane cannot occur. This may be attributed to the above five solvents not being immiscible with water and cannot remove water solvent, and thus are unable to drive structural transformation. Therefore, to verify this point, Compound 1 soaked in ether, toluene, dichloromethane, trichloromethane and n-hexane for 15 min, respectively, was dried under vacuum overnight and then their PXRD patterns were investigated, which can be compared with the simulated pattern of Compound 1a (Fig. S19†). Considering that the above five solvents are unable to drive the structural transformation, we attempt to restore the structure by immersing Compound 1a in the above five solvents, and the results show that Compound 1a cannot be restored in ether, toluene, dichloromethane, trichloromethane, and n-hexane solvents, suggesting that the transformation is irreversible and Compound 1a is regarded as the thermodynamically favoured product.
In order to further explore the role of solvents in conversion, different solvents were studied for various soaking times. Fig. 3a and b show the comparison of time-dependent PXRD patterns of Compound 1 soaked in acetone and trichloromethane, respectively. Fig. 3a reveals a slight contraction of the structure evidenced by the shifting of the plane from 2 θ = 7.9 to 9.0 when the soak time was extended to 35 min, while Fig. 3b shows that the structure of Compound 1 soaked in trichloromethane remains unchanged. Fig. S21–S24† display the PXRD patterns of Compound 1 in methanol, ethanol, acetonitrile and THF with different soaking times, respectively. The disappearance time of the peak at 2 θ = 7.9 for methanol, ethanol, acetonitrile and THF is 50 min, 5 min, 15 min and 4 h, respectively.
Irreversible morphology change
Considerable changes were observed in the diffraction patterns of the crystals of Compound 1 sealed in polar solvents and dried under vacuum conditions, which is probably due to the structural transformation caused by water solvent escaping from the thermodynamically unfavorable frameworks. This is verified by TGA analysis (Fig. 4a) where 15.4% weight loss for Compound 1 and 6.3% weight loss for Compound 1a are observed. The result shows that the two-dimensional layered structure will convert to another closer packing mode once the lattice solvent escapes. Considering the obvious difference of the interlayer spacing, impedance measurements of Compound 1 and Compound 1a were investigated, as shown in Fig. 4b. The resistance based on the semicircles in the Nyquist plot of Compound 1 is significantly lower than that of Compound 1a, which is consistent with the larger interlayer spacing with lower resistance. SEM images gave clear information about the morphology distinction of Compound 1 and Compound 1a (Fig. 4c–f). Compound 1 shows a large block-crystal, in which the loose layers are clearly visible. After the water escaped, slender nanowires began to appear, as shown in Fig. 4d–f. The results obtained by soaking Compound 1 in other solvents such as CH3OH, C2H5OH, CH3CN and THF are the same, as shown in Fig. S31–S34,† and the morphology of Compound 1 was not changed only by grinding, as shown in Fig. S30.† These results indicated that the change of morphology of compound 1 was driven by solvent. On one hand, the escape of water molecules reduces the interlayer spacing to obtain a close-packed structure, and on the other hand, the surface tension between layers has been reduced to make the layers spontaneously detach; also the decrease of surface tension degenerates the stability of the intra-plane interaction to break the weak Mn–O bonds of the 2D layer, thus finally obtaining nanowires.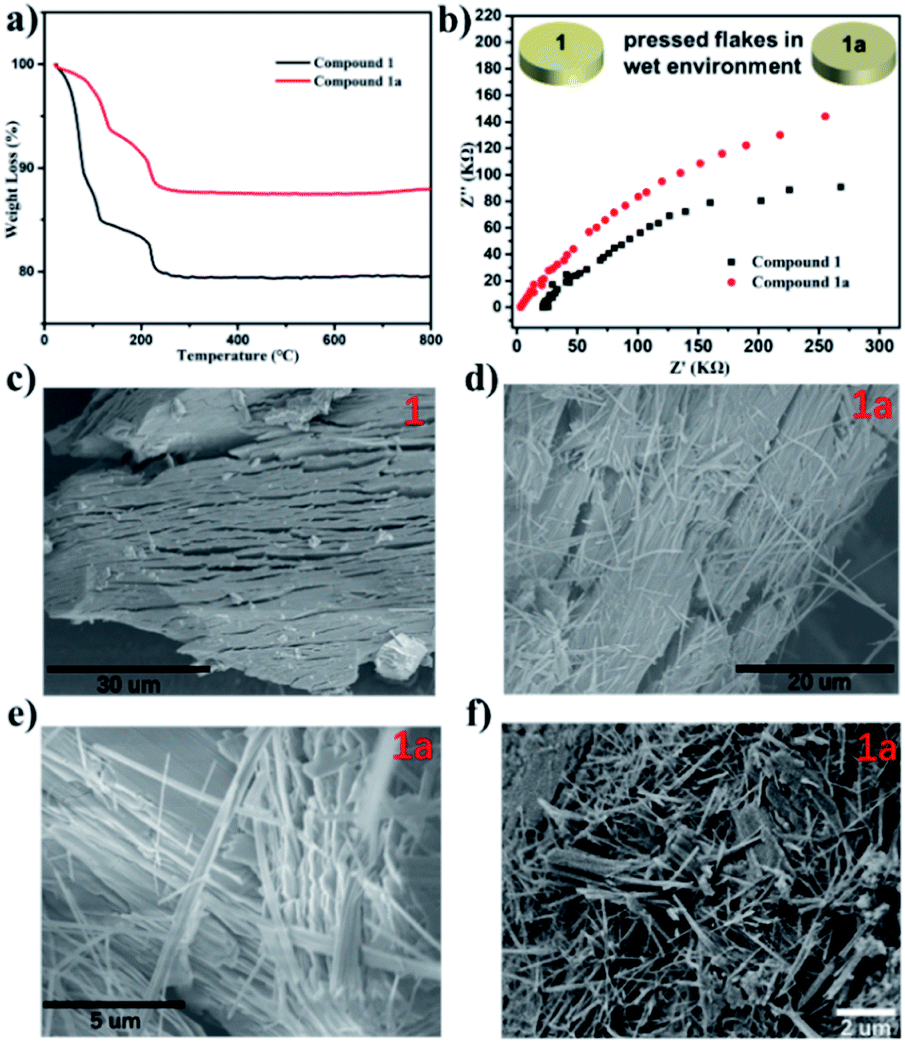 | ||
| Fig. 4 (a) The TGA curves of Compound 1 and Compound 1a. (b) Nyquist plot of Compounds 1 and Compound 1a over a frequency range from 0.1 Hz to 105 Hz. (c–f) SEM images of Compound 1 and 1a. | ||
Conclusions
In summary, a porous 2D layered structure {MnMo8O28} Compound 1 was spontaneously transformed into a thermodynamically favored closer stacking structure Compound 1a with an identical layered structure by the induction of different solvents, accompanied by a morphology change. The processes of transformation are well addressed by SCXRD, PXRD and TGA analyses. And the SEM image displays the nanowire evolutionary process of Compound 1a with smaller interlayer spacing, which may be attributed to the reduction of water with high surface tension. Moreover, the impedance measurement reveals that the conductivity of Compound 1 with larger interlayer spacing is higher than that of Compound 1a with smaller interlayer spacing. The current study provides new insights into the regulation of interlayer spacing simulated by small molecules and preparation of 1D nanowires from 2D POM materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the support of this work by the National Natural Science Foundation of China (NSFC 21801226), Zhejiang Provincial Natural Science Foundation of China (LY20B010002) and the Zhejiang Normal University Fund.Notes and references
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, EnergyChem, 2019, 1, 100006 CrossRef.
- J. He, J. Xu, J. Yin, N. Li and X. H. Bu, Sci. China Mater., 2019, 62, 1655 CrossRef CAS.
- R. F. Mendes, F. Figueira, J. P. Leite, L. Gales and F. A. Almeida Paz, Chem. Soc. Rev., 2020, 49, 9121 RSC.
- L. Zhu, D. Sheng, C. Xu, X. Dai, M. A. Silver, J. Li, P. Li, Y. Wang, Y. Wang, L. Chen, C. Xiao, J. Chen, R. Zhou, C. Zhang, O. K. Farha, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, J. Am. Chem. Soc., 2017, 139, 14873 CrossRef CAS PubMed.
- R. Zhao, Y. Wu, Z. Liang, L. Gao, W. Xia, Y. Zhao and R. Zou, Energy Environ. Sci., 2020, 13, 2386 RSC.
- Y. Wang, H. M. Zeng, W. T. Mao, X. J. Wang, Z. G. Jiang, C. H. Zhan and Y. L. Feng, Inorg. Chem. Commun., 2021, 129, 108613 CrossRef CAS.
- C. H. Zhan, D. P. Huang, Y. Wang, W. T. Mao, X. J. Wang, Z. G. Jiang and Y. L. Feng, CrystEngComm, 2021, 23, 2788 RSC.
- J. Kang, V. K. Sangwan, J. D. Wood and M. C. Hersam, Acc. Chem. Res., 2017, 50, 943 CrossRef CAS PubMed.
- X. Liu, S. Liu, L. Y. Antipina, Y. Zhu, J. Ning, J. Liu, C. Yue, A. Joshy, Y. Zhu, J. Sun, A. M. Sanchez, P. B. Sorokin, Z. Mao, Q. Xiong and J. Wei, Nano Res., 2020, 13, 1627 CrossRef CAS.
- Y. Ding, Y. P. Chen, X. Zhang, L. Chen, Z. Dong, H. L. Jiang, H. Xu and H. C. Zhou, J. Am. Chem. Soc., 2017, 139, 9136 CrossRef CAS.
- J. N. Coleman, Adv. Funct. Mater., 2009, 19, 3680 CrossRef CAS.
- J. Niu, X. Zhang, D. Yang, J. Zhao, P. Ma, U. Kortz and J. Wang, Chem.–Eur. J., 2012, 18, 6759 CrossRef CAS.
- Z. G. Jiang, W. T. Mao, D. P. Huang, Y. Wang, X. J. Wang and C. H. Zhan, Nanoscale, 2020, 12, 10166 RSC.
- A. Rezaeifard, R. Haddad, M. Jafarpour and M. Hakimi, J. Am. Chem. Soc., 2013, 135, 10036 CrossRef CAS PubMed.
- C. H. Zhan, Q. Zheng, D. L. Long, L. Vilà Nadal and L. Cronin, Angew. Chem., Int. Ed., 2019, 58, 17282 CrossRef CAS PubMed.
- A. Müller and C. Serain, Acc. Chem. Res., 2000, 33, 2 CrossRef PubMed.
- H. An, Y. Hou, S. Chang, J. Zhang and Q. Zhu, Inorg. Chem. Front., 2020, 7, 169 RSC.
- J. Lu, Y. Wang, X. Ma, Y. Niu, V. Singh, P. Ma, C. Zhang, J. Niu and J. Wang, Dalton Trans., 2018, 47, 8070 RSC.
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327 CrossRef CAS PubMed.
- Y. Hou, H. An, T. Xu, S. Zhao and J. Luo, New J. Chem., 2016, 40, 10316 RSC.
- X. Yang, P. Zhu, X. Ma, W. Li, Z. Tan and J. Sha, CrystEngComm, 2020, 22, 1340 RSC.
- C. H. Zhan, C. Busche, D. L. Long, P. I. Molina, R. S. Winter and L. Cronin, Chem. Commun., 2017, 53, 7076 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00416f |
| This journal is © The Royal Society of Chemistry 2021 |