
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInduction heating induced self-healing of nanocomposites based on surface-functionalized cationic iron oxide particles and polyelectrolytes†
Bastian
Oberhausen
 and
Guido
Kickelbick
*
and
Guido
Kickelbick
*
Saarland University, Inorganic Solid-State Chemistry, Campus, Building C4.1, 66123 Saarbrücken, Germany. E-mail: guido.kickelbick@uni-saarland.de
First published on 5th August 2021
Abstract
Supramolecular interactions represent versatile, reversible, and intrinsic mechanisms for bond formation after the failure of materials. Ionic interactions excel through high flexibility and binding strength. In this study, ionic interactions between polymer matrices and inorganic nanoparticles were used to induce self-healing properties. Random, anionic polyelectrolyte copolymers consisting of di(ethylene glycol) methyl ether methacrylate and sodium-4-(methacryloyloxy)butan-1-sulfonate were synthesized by atom transfer radical polymerization. Differential scanning calorimetry measurements confirmed the adjustability of the glass transition temperature via the polymer composition. Within the glass transition temperature window of the homopolymers from −23 °C to 126 °C, the range between −18 °C to 50 °C was examined, generating suitable matrices for self-healing. Superparamagnetic iron oxide nanoparticles with a size of 8 nm were synthesized by thermal decomposition of iron(III) acetylacetonate and used as the inorganic filler. Positive surface charges were introduced by functionalization with N,N,N-trimethyl-6-phosphonhexan-1-aminium bromide. Functionalization was confirmed with FTIR, TGA, and zeta potential measurements. Ionic interactions between filler and polymer promote a uniform particle dispersion within the material. Self-healing experiments were performed at 80 °C and without the addition of further healing agents. Utilizing the magnetic properties induced by the iron oxide nanoparticles, spatially resolved healing within an alternating magnetic field was achieved on a μm scale.
Introduction
Inspired by numerous biological models, the long-term reliability of self-healing materials has made their investigation a rapidly growing field of research. Self-healing mechanisms represent a potential solution to enhance the lifetimes of materials. The chemical design of intrinsic self-healing materials allows the optimization of healing rates whilst retaining the desired material properties.1 Whereas this concept is well-known for polymeric systems, pure inorganic materials lack potential healing mechanisms, or require high energy input to work due to the bonding situation in inorganic solids.2,3 In polymeric systems the reversible bond formation via Diels–Alder chemistry (DA) and supramolecular interactions, such as hydrogen bonding and electrostatic interactions, as well as metal–ligand interactions are regularly used healing mechanisms.4–7 However, many of the polymeric materials lack mechanical stability due to the necessary high backbone flexibility realized by low Tg materials. Inorganic–organic nanocomposites reveal property improvements particularly in their mechanical and thermal stability compared to conventional materials.8–12 For these materials lifetime improvements are possible by the introduction of self-healing properties. Self-healing ability can be induced by hydrogen bonds and hydrophobic interactions, allowing high healing efficiencies in short times and ambient conditions.13 Other healing mechanisms are based on reversible bond formation, for example in DA reactions or the reversible formation of disulfide-, imine- and boronate ester bonds.14–16 Especially for soft materials like gels and elastomers, the incorporation of inorganic nanofillers helps to overcome their often low mechanical and thermal stability making brought application areas accessible.17,18 In some cases the nanofillers not only act as components to improve the mechanical or thermal properties, but also deliver additional functions. As an example, CNTs have been used as nanoscale heaters due to their ability of near-IR induced heating and high thermal conductivity to promote self-healing properties.18In our group several self-healing nanocomposites were already studied, for example based on surface-functionalized silica particles embedded in acrylate and siloxane polymers.19 In these previous experiments we were able to show that the self-healing efficiency depends on the backbone mobility and flexibility and the degrees of freedom of the functional groups on the nanoparticle surface.20,21 We also systematically studied variations in the composition of such materials to gain a deeper understanding of the limiting parameters for the reaction of furan and maleimide modified SiO2 particles with their respective counterparts.22 Additionally, we investigated organophosphonate modified iron oxide particles as building blocks for self-healing nanocomposites based on DA/rDA (retro Diels–Alder) reactions, triggered by conventional heating.23 Recently we were able to increase the healing capacity of octahedral spherosilicates containing nanocomposites applying two different healing mechanisms in one material.21
Electrostatic interactions can also be applied as a healing mechanism. In polymer chemistry such systems were already extensively studied. However, there are only a few examples covering this type of interaction in self-healing nanocomposites. Zhong et al. used iron(III) as an ionic crosslinker between polymer strands consisting of vinyl hybrid silica nanoparticles (VSNPs) covalently bound to a poly(acrylic acid) network. The obtained hydrogel exhibits high tensile strength (860 kPa) and the ability to self-repair in the μm length scale.24 Odent et al.25 applied an imidazolium based cationic polymer as a matrix. Negatively charged TiO2 particles were synthesized by functionalization with 3-(trihydroxysilyl)-1-propane sulfonate at 70 °C under basic conditions (pH = 5). Compared to the pristine polymer, significant improvements in toughness and strain at break are reported for this nanocomposite, with a maximal stability at 20 wt% particle loading. With a Tg of −55 °C, this material already has a high ductility at room temperature and is therefore limited in its application to areas where low Young's moduli are required, although it has good self-healing and shape memory properties. While alkoxysilanes show some drawbacks in surface modification, phosphonic acids are a promising alternative for stable ionic organic group attachment to transition metal oxide surfaces.26
Nanocomposites based on iron oxide are attractive materials because of their thermal stability and increase in tensile strength.27 In addition to other nano building blocks, iron oxide nanoparticles offer a distinct advantage in that they can induce magnetism into the material. Important in terms of magnetic properties are composition, size, morphology, and surface functionalization.28–31 Superparamagnetism is limited to magnetite particles at the nanoscale, particularly below 25 nm.32 This introduction of superparamagnetic properties, potentially offers the opportunity for spatially resolved healing with an alternating magnetic field (AMF). The nanoparticles respond with a physical particle rotation (Brownian relaxation) and/or an internal dipole inversion (Néel relaxation), dependent on particle size, magneto crystalline anisotropy and matrix viscosity.33 Both mechanisms are associated with an increase in temperature, which triggers the self-healing process. Corten and Urban were the first to take advantage of the heating of iron oxide nanoparticles in an alternating magnetic field to trigger self-healing of a methyl methacrylate based polymer.34 Since then the approach was carried over to other polymer systems, including styrene, ethylene-vinyl acetate and propylene-ethylene-1-butene.35–37 Hohlbein et al. have also shown that the heat generated in the alternating magnetic field accelerates supramolecular healing mechanisms, in this case based on complex formation.38 The same effect could also be observed with hydrogen bridge-based healing systems made from gelatin/agarose hydrogels.39 In addition, the incorporation can induce DC electrical conductivity by forming conductive pathways throughout the polymer that enable charge transfer between two electrodes. Furthermore, the nanocomposites can be used as light-shielding coating materials due to their high specific absorption rate.40
In this study, we expand the AMF improved self-healing concept for nanocomposites to ionic interactions. In contrast to previous work the supramolecular interactions responsible for self-healing occur between the particles and the polymer matrix. We synthesized di(ethylene glycol) methyl ether methacrylate based polymers (DEGMA) with anionic functionalities via atom transfer radical polymerization (ATRP). Surface-functionalized superparamagnetic iron oxide nanoparticles with diameters smaller than 10 nm were used as inorganic nano building blocks. Surface-functionalization was achieved with cationic phosphonic acid derivatives. The resulting self-healing nanocomposites feature ionic interactions between the inorganic nanoparticles and the organic matrix providing an as homogeneous as possible distribution of the particles in the polymer. Self-healing experiments showed the ability to repair scratches in the μm scale.
Experimental section
Materials & methods
Fe(acac)3 (97%), benzyl ether (98%), 1,2-dodecandiol (90%), oleic acid (≥99%), oleyl amine (70%), CuBr2 (99%), di(ethylene glycol) methyl ether methacrylate (95%), 1,6-dibromhexane (96%), triethyl phosphite (98%), trimethylamine solution in ethanol (31–35%) and bromotrimethylsilane (97%) were purchased from Sigma-Aldrich. Methacrylic acid (≥99%) was obtained from Merck. Aluminium oxide 90 active neutral (70–230 mesh ASTM) was received from Merck Millipore. 2,2′-Bipyridine (>99%) was purchased from TCI Chemicals Germany. Ethyl 2-bromoisobutyrate = EBiB (98%) and hydrazine monohydrate (98%) were obtained from Alfa Aesar. Sodium bicarbonate (>99%) was received from Fluka. Hydrochloric acid (37%) was purchased from Bernd Kraft GmbH. Dry dichloromethane (99,8%) and methanol (99,8%) were purchased from Acros Organics. Pre-wetted cellulose regenerated tubing membranes with a 3500 D MWCO were purchased from Fisher Scientific. CuBr was synthesized by reduction of CuBr2 with equimolar amounts of NaSO3 in distilled water at room temperature. The precipitating colorless solid was stirred for 30 min in glacial acetic acid and then washed with methanol (×3) and diethyl ether (×3). The obtained solid was dried under vacuum and stored under argon atmosphere. The hydroquinone monomethyl ether stabilizer of DEGMA was removed by passing it through active neutral alumina prior to use. All other chemicals were used without further purification.Characterization
Fourier transform infrared (FTIR) spectra were recorded with a Bruker Vertex 70 spectrometer under ambient air (16 scans at a resolution of 4 cm−1) in attenuated total reflectance (ATR) mode. Differential scanning calorimetry (DSC) measurements were performed using a Netzsch DSC 204 F1 Phoenix. Samples were prepared in aluminum crucibles with pierced lids and heated under nitrogen at a rate of 5 K min−1, 10 K min−1 or 20 K min−1. Solution NMR spectra were recorded with a Bruker Advance III HD 300/400 spectrometer at 25 °C (1H at 300/400 MHz, 13C at 75/101 MHz, 31P at 121/162 MHz) using CDCl3, D2O and the residual protons of solvent and carbon as reference. Elemental analysis was performed with a Leco 900 CHN analyzer. A Netzsch Iris TG 209C was used for thermogravimetric analysis (TGA). Measurements were conducted in aluminum oxide crucibles heating from room temperature to 880 °C under nitrogen atmosphere, followed by heating to 1000 °C under a mixture of nitrogen and oxygen (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with a rate of 10 K min−1. TGA and CHN results were used to determine the molar amount of coupling molecules per gram of particles. Transmission electron micrographs were measured with a JEOL JEM-2010 with 200 kV used for electron acceleration. Samples were prepared on Plano S160-3 carbon coated copper grids.
:1) with a rate of 10 K min−1. TGA and CHN results were used to determine the molar amount of coupling molecules per gram of particles. Transmission electron micrographs were measured with a JEOL JEM-2010 with 200 kV used for electron acceleration. Samples were prepared on Plano S160-3 carbon coated copper grids.
Powder X-ray diffraction (PXRD) patterns were recorded with a Bruker D8 Advance diffractometer (Bruker, Karlsruhe, Germany) in a Bragg–Brentano θ–θ-geometry (goniometer radius 280 mm). A 2θ range from 7 to 120° (step size 0.013°) was recorded in 1 h scan time. Cu-Kα radiation (λ = 154.0596 pm, 40 kV, 40 mA) was used with a 12 μm Ni foil to reduce Kβ radiation. A variable divergence slit was mounted at the primary beam side (irradiated sample area: 10 × 7 mm). A LYNXEYE 1D detector was used at the secondary beam side. Background caused by white radiation and sample fluorescence was reduced by limiting the energy range of the detection. Interpretation of the XRD data was performed via Rietveld analysis using TOPAS 5. Crystallographic structure and micro-structure were refined, while instrumental line broadening was included in a fundamental parameters approach. The mean crystallite size 〈L〉 was calculated at the mean volume weighted column height derived from the integral breadth. The background of standard measurements was fitted by a Chebyshev polynomial function of 15th degree.
Zeta potential measurements were performed with a Zetasizer Nano ZSP (Malvern Panalytical, Nürnberg Germany). For the measurement, the solid particle sample was dispersed in deionized water under ultra-sonication for 15 minutes. Sample concentration was ∼0.05 wt%. Titration was performed from the initial pH, once to basic and once to acidic conditions. At every pH value three measure points were taken each consisting of 20 separate measurements.
Dynamic light scattering (DLS) studies were performed on an ALV/CGS-3 compact goniometer system with an ALV/LSE-5003 correlator at a 90° goniometer angle, using noninvasive backscattering (λ = 632.8 nm). Samples from reaction solution were diluted, homogenized in an ultrasound bath, and equilibrated for about five minutes before measurement.
Microscope images were recorded under polarized light using an Olympus BX60 microscope equipped with a Sony CCD-Iris color camera.
To generate the external alternating magnetic fields either an high frequency generator 1997 1G 5/3000 or a TruHeat HF 5010, both from TRUMPF Hüttinger Elektronik GmbH + Co. KG (Freiburg, Germany) were used. Measurements were carried out with a 3-winded copper coil (d = 50 mm) with water cooling at a constant frequency of 1.95 MHz up to 3000 W generator power for the first system. For the TruHeat system a 5-winded copper coil (d = 40 mm) with water cooling was used. Measurements were performed at a constant frequency of 313 kHz and a generator power up to 5000 W. Temperatures in the high frequency oven were measured with a Fotemp1 from Optocon (Dresden Germany) with a TS2 fiber optic temperature sensor. For temperature measurements the samples are pricked with a needle. The tip of the sensor is placed into the resulting hole and the polymer is pressed together at slightly elevated temperature to assure best possible contact. The whole sample is placed into the induction coil.
Syntheses
FTIR, 1H and 13C NMR spectra of the literature known compounds, as well as the IR spectra of the synthesized copolymers and composites are shown in the ESI (Fig. S1–S17†).Diethyl(6-bromohexyl) phosphonate. 4.40 g (100 mmol) of 1,6-dibromohexane were heated to 150 °C. 16.60 g (100 mmol) of triethyl phosphite were added dropwise within one hour. The reaction mixture was kept at 150 °C for further 5 h. After cooling to room temperature, the volatile side products were removed applying a rotational evaporator. The crude product was fractionally distilled at reduced pressure (Bpt0.08 mbar: 123 °C). Yield: 19.97 g, 66 mmol, 66%, colorless oil.
1H NMR δ (CDCl3): 3.99–4.12 (4H, O–CH2–CH3), 3.34–3.43 (2H, CH2–Br), 1.34–1.92 (10H, CH2), 1.24–1.33 (6H, O–CH2–CH3) ppm. 13C NMR δ (CDCl3): 61.25 d (O–CH2), 33.60 (CH2–Br), 32.28 (CH2–CH2), 29.50 d, 27.47, 25.41 d (CH2–P), 22.13 (CH2–CH2), 32.68 (O–CH2–CH3) ppm. 31P NMR δ (CDCl3): 32.14 ppm. FTIR: 2982 (νas C–H2), 2933 (νs C–H2), 2864 (νs C–H2), 1439 (δ C–H2), 1394, 1242 (ν P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1167, 1101, 1024, 955 (ν P–OH), 787, 644 cm−1.
O), 1167, 1101, 1024, 955 (ν P–OH), 787, 644 cm−1.
6-(Diethoxyphosphoryl)-N,N,N-trimethylhexan-1-aminium bromide. 4.20 g (14 mmol) of diethyl-(6-bromohexyl)phosphonate and 10 mL (42 mmol) of trimethylamine (33% in ethanol) were stirred for 24 h at room temperature. The crude product was filtered, and the filtrate was freed from the solvent with a rotary evaporator. Yield: 3.82 g, 11 mmol, 76%, white solid.
1H NMR δ (CDCl3) = 4.00–4.12 m (4H, O–CH2–CH3), 3.59–3.71 m (2H, CH2–Br), 3.45 s (9H, CH3), 1.36–1.82 m (10H, CH2), 1.27–1.33 t (6H, O–CH2) ppm. 13C NMR δ (CDCl3): 66.61 (N–CH3), 61.45 d (O–CH2), 53.32 (N–CH3), 29.84 d (CH2–P), 25.61, 25.27 d (CH2P), 22.84, 22.14 (CH2–CH2), 16.43 (O–CH2–CH3) ppm. 31P NMR δ (CDCl3): 32.14 ppm. FTIR: 2983 (νas C–H2), 2941 (νas C–H2), 2858 (νs C–H2), 1488, 1394 (δ C–H2), 1389, 1246 (ν PO), 1163, 1095, 1026, 953 (ν P–OH), 792, 521 cm−1.
N,N,N-Trimethyl-6-phosphonhexan-1-aminium bromide. 2.35 g (6 mmol) of 6-(diethoxyphosphoryl)-N,N,N-trimethylhexane-1-ammonium bromide were dissolved in 10 mL dry dichloromethane at room temperature under argon atmosphere. 1.5 mL (12 mmol) of bromotrimethylsilane were added dropwise over an hour. The mixture was stirred at room temperature for further 24 h until the conversion to the silyl ester was completed (studied by 1H NMR spectroscopy). The solvent was then removed applying a rotary evaporator. Subsequently 10 mL of a methanol
:water mixture (3:2, v/v) were added and the solution was stirred for another for 24 h. Methanol was removed with a rotary evaporator and the product was dried in vacuo. The resulting solid was recrystallized from 2-propanol. Yield: 1.60 g, 5 mmol, 88%, white solid.
1H NMR δ (D2O) = 3.24–3.31 m (2H, N–CH2), 3.07 s (9H, N(CH3)3), 1.28–1.83 m (10H, CH2) ppm. 13C NMR δ (D2O): 66.61 (N–CH2), 52.71 (N–CH3), 29.13 d (CH2–P), 27.18, 25.84, 24.86, 21.97 (CH2–CH2) ppm. 31P NMR δ (D2O): 30.04 ppm. FTIR: 2943 (νas C–H2), 2933 (νas C–H2), 2858 (νs C–H2), 2156, 1637 (H2O) 1481 (δ C–H2), 1217 (ν PO), 1105, 1055 (ν C–N), 989, 970, 933 (ν P–OH), 914, 768, 717, 660 cm−1. CHNtheo (C9H23BrNO3P): C: 35.54; H: 7.62; N: 4.61, CHNexp: C: 33.90, H: 7,07, N: 4,48.
CHNexp: C: 9.09, H: 1.71, N: 4.48.
:ethanol (1:2, v/v) was chosen to ensure a stable dispersion over the entire reaction process. The exchange took place within 48 h at room temperature. The particles were then decanted magnetically. If necessary, the separation was supported by adding a small amount of hexane. Excess of phosphonic acid was removed by washing with an ethanol:hexane mixture (v/v = 1:1, 3 × 10 mL).
FTIR: 3334 (ν O–H), 2925 (νas C–H2), 2852 (νs C–H2), 1637 (CO), 1481 (δ C–H2), 964 (ν PO), 536 (ν Fe–O) cm−1.
Sodium 4-(methacryloyloxy)butan-1-sulfonate. Sodium methacrylate was prepared freshly before synthesis by stirring 10.20 g (119 mmol) methacrylic acid and 9.9 g (118 mmol) sodium bicarbonate at room temperature. Completion of the reaction was determined with 1H NMR spectroscopy. The obtained white solid was dried under vacuum and used without further purification. 3.60 g (33 mmol) sodium methacrylate were dissolved in 35 mL ethanol and heated to reflux. Subsequently 5.00 g (37 mmol) 1,4-butane sultone were added dropwise. Heating was continued for 8 h. After cooling to room temperature, the solvent was removed under vacuum. The obtained white solid was washed with small amounts of xylene (3×) and ethanol (3×) and dried under vacuum. Yield: 5.43 g, 22 mmol, 67%, white solid.
1H NMR δ (D2O) = 6.08 s (1H, =C–H), 5.63–5.67 m (1H, =C–H), 4.14–4.21 m (2H, O–CH2), 2.88–2.95 m (2H, S–CH2), 1.88 s (3H, CH3), 1.78–1.82 m (4H, CH2) ppm. 13C NMR δ (D2O): 170.07 (CO), 136.06 (CCH2), 126.49 (CH2C), 64.94 (C–O), 50.50 (C–S), 26.68, 20.88, 17.33 (CH2–CH2, CH3), ppm. FTIR: 3535 (νas CO), 3493 (νas CO), 2956 (νas C–H2), 2931 (νas C–H3), 2860 (νas C–H3), 1716 (νas CO), 1630, 1570 (νas CC), 1450, 1416 (δ CC), 1348, 1327, 1296, 1165 (νas C–O), 1049, 906 (δ CH2), 779 cm−1. CHNtheo (C8H13NaO5S): C: 39.34; H: 5.37, CHNexp: C: 37.24; H: 5.65.
Synthesis of P(DEGMA). The synthesis was carried out via an ATRP reaction. For this purpose, 3.76 g (20 mmol) of DEGMA were dissolved in 35 mL of dry toluene. The solution was degassed with three freeze–pump–thaw cycles. In a second flask, 12.8 mL of dry methanol were placed and degassed with three freeze–pump–thaw cycles. 28.69 mg (0.2 mmol) of CuBr and 62.79 mg (0.4 mmol) of 2,2′-bipyridine were added to the frozen methanol. Two further vacuum argon cycles were then carried out. The two solutions were combined and finally 29.35 μL (0.2 mmol) degassed EBiB were added, and the mixture was stirred at 70 °C, for 24 h. The solid obtained was washed several times with water, as a result of which the copper complex contained was separated off. The sample was dried under vacuum at 50 °C. Yield: 2.95 g, 78%, colorless solid.
1H NMR δ (CDCl3) = 4.04 s (2H, COOCH2), 3.46–3.67 m (6H, O–CH2), 3.33 s (3H, O–CH3), 1.70–1.91 m (2H, CH2), 0.73–1.03 m (3H, CH3) ppm. FTIR: 3463 (νas CO), 2931 (νas C–H2), 2873 (νas C–H3), 1735 (νas CO), 1475, 1259, 1101 (νas C–O), 971, 856, 740 cm−1. CHNtheo: C: 57.43; H: 8.57, CHNexp: C: 55.37; H: 57.43.
Synthesis of the P(sodium-4-(methacryloyloxy)butan-1-sulfonate). The synthesis was carried out under argon atmosphere. 2 g (8.19 mmol) of sodium 4-(methacryloyloxy)butanesulfonate were dissolved in 14 mL water. The pH was adjusted to 9 with a 1 M NaOH solution. The solution was degassed with 3 freeze–pump–thaw cycles. In a second flask, 4.6 mL methanol were also degassed with 3 freeze–pump–thaw cycles. Then 11.76 mg (0.082 mmol) of CuBr and 25.77 mg (0.165 mmol) 2,2′-bipyridine were added. The solutions were combined and 12 μL (0.082 mmol) degassed EBiB were added. The reaction solution was stirred at 70 °C for 12 h. A solid precipitated out during the reaction. By adding a further 15 mL water (previously degassed), the polymer was redissolved and 25.77 mg 2,2′-bipyridine as well as 9 μL hydrazine monohydrate were added. The mixture was stirred again at 70 °C for 12 h. The product obtained was dialyzed against water. The copper complex was removed by dialysis against diluted HCl. The sample was dried under vacuum at 60 °C. Yield: 1.07 g, 53% pale yellow solid.
1H NMR δ (D2O) = 3.99 s (2H, COOCH2), 2.90 s (2H, S–CH2), 1.77 s (6H, CH2), 0.82–1.00 m (3H, CH3) ppm. FTIR: 3449 (ν O–H, water), 2945 (νas C–H2), 2887 (νas C–H3), 1706 (νas CO), 1619 (νas SO), 1475, 1158 (νas C–O), 1058, 956, 726 cm−1. CHNtheo: C: 39.34; H: 5.37, CHNexp: C: 39.38; H: 5.49.
Synthesis of the P(DEGMA-co-sodium-4-(methacryloyloxy)butan-1-sulfonate). Various amounts of sodium 4-(methacryloyloxy)butan-1-sulfonate (1.02–3.41 mmol) and 1.93 g (10.24 mmol) DEGMA were dissolved in 18 mL water. The pH was adjusted to 9 with a 1 M NaOH solution. The solution was degassed with 3 freeze–pump–thaw cycles. In a second flask, 6.5 mL methanol were also degassed with 3 freeze–pump–thaw cycles. Then 14.86 mg (0.102 mmol) CuBr and 32.00 mg (0.205 mmol) 2,2′-bipyridine were added. The solutions were combined and 15.03 μL degassed EBiB (0.1024 mmol) were added. The reaction solution was stirred at 70 °C for 12 h. Then 32.00 mg of 2,2′-bipyridine and 23 μL of hydrazine monohydrate were added and the mixture was heated for a further 12 h. The crude product obtained was concentrated and dialyzed against water for 3 days. Similar to the homopolymer, the sample was not completely decolorized, and the copper was removed by dialyzing against diluted HCl. The product was freed from the solvent. Mean yield 70% (±7%).
1H NMR δ (D2O) = 4.13 s (20H, COOCH2/DEGMA), 3.99 s (2H, COOCH2/anionic), 3.59–3.73 m (66H, CH2–CH2/DEGMA), 3.36 s (34H, O–CH3), 2.85 s (2H, S–CH2), 1.75–1.99 m (24H, CH2), 0.84–1.09 m (34H, CH3) ppm. FTIR: 3595 (νas CO), 3440 (νas CO), 2990 (νas C–H2), 2934 (νas C–H3), 2871, 2821 (νas C–H3), 1725 (νas CO), 1458, 1402, 1353, 1233, 1099 (νas C–O), 1043, 952, 861, 741, 601, 530 cm−1.
:1) under ultrasonication. The appropriate amount of functionalized nanoparticles (2 wt%, 5 wt%, 10 wt% and 20 wt%) is added. The solvent is slowly removed under stirring.
Polymer and composite films were prepared by compression molding at 80 °C for 24 h in a Teflon form, which was held using a vice.
Self-healing experiments were performed by cutting the samples through half thickness. Afterwards the edges were gently pressed together at room temperature. Then the samples are heated to 80 °C for 24 h. Afterwards the samples were put back into the Teflon molds and heated to 80 °C for another 24 h. For the self-healing by induction heating the generator power was adjusted to obtain a macroscopic temperature of 58 °C. Healing was performed in two segments of 24 h duration.
FTIR: 3580 (νas CO), 3433 (νas CO), 2998 (νas C–H2), 2934 (νas C–H3), 2885, 2821 (νas C–H3), 1725 (νas CO), 1458, 1395, 1353, 1240, 1107 (νas C–O), 1043, 966, 861, 587 (Fe–O–Fe) cm−1.
Results and discussion
General procedure for the composite synthesis and investigation of self-healing
The schematic synthetic route for the nanocomposites is shown in Scheme 1. First oleic acid coated iron oxide nanoparticles were synthesized. N,N,N-Trimethyl-6-phosphonhexan-1-aminium bromide was used to introduce positive surface charges. As the counterpart anionic copolymers were synthesized via ATRP and mixed with the functionalized particles. To check the self-healing ability composite samples were cut and then cured thermally or with an alternating magnetic field.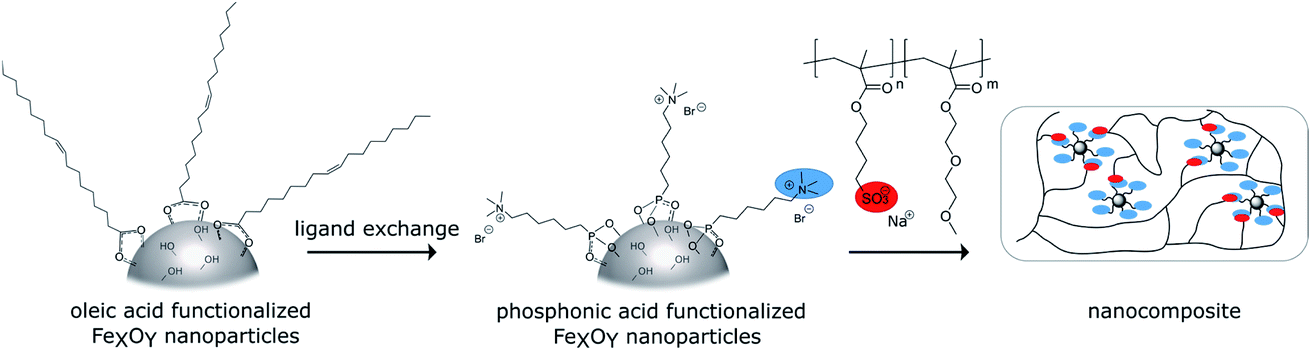 | ||
| Scheme 1 Syntheses of the self-healing nanocomposites. | ||
Synthesis of the cationic phosphonic acid
Organophosphorus molecules bearing functional groups are ideal molecules for the surface-functionalization of transition metal oxide systems, due to their strong bonding to the surface, their high chemical stability and the variety of functions that can be incorporated in the molecules.43 Goal of this study was the synthesis of iron oxide nanoparticles with a permanent cationic or anionic surface functionalization that is only slightly affected by environmental changes. Therefore, carboxylic acid or primary amine functions were not good choices because environmental pH would have a large effect on their protonation causing pH changes to have a large effect on surface charge. Excellent groups for inducing permanent negative and positive charges are sulfonate and alkylated ammonium ions, respectively. Preliminary experiments showed that surface functionalization with sulfonate group-modified organophosphorus molecules can lead to possible interactions of the anionic function with the iron oxide surface: thus, they act as bridging agents, leading to extended particle agglomeration. For our approach, we therefore used coupling agents with permanent positive charges. We chose organic ammonium ions due to their high charge stability independent of the chemical environment. These coupling reagents were synthesized in a three-step approach (Scheme 2).26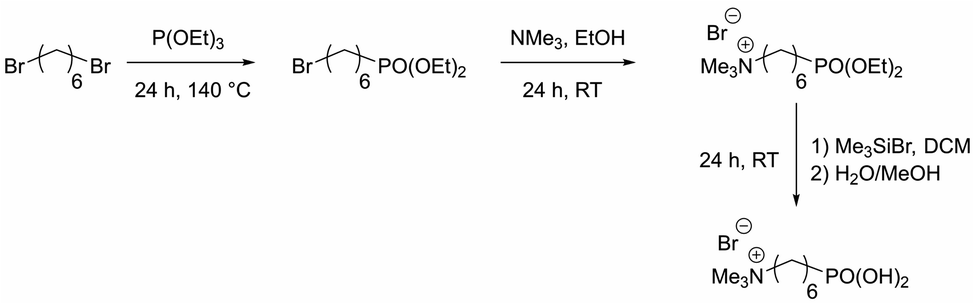 | ||
| Scheme 2 Synthesis route for N,N,N-trimethyl-6-phosphonhexan-1-aminium bromide. | ||
The molecules used in our study consisted of the phosphonic acid group, a C6-spacer and the cationic functionality. The C6-chain was chosen because shorter spacer lengths often lead to agglomeration during the particle functionalization process even in the presence of the electrostatic repulsion of cationic groups. In addition, the corresponding dibromohexane, which is required as a starting compound in the synthesis, is unlike the longer-chain derivatives easily available. Synthesis starts with a Michaelis–Arbuzov reaction, resulting in a ω-bromophosphonic acid ester. Cationic functionalities are introduced via a nucleophilic substitution with trimethylamine. In the last step the phosphonic acid functionality is obtained by transesterification with bromotrimethylsilane followed by hydrolysis. CHN analysis as well as NMR and FTIR spectroscopy confirmed the product formation with an overall yield of 44%.
Particle synthesis and functionalization
Oleic acid (OA)-functionalized iron oxide nanoparticles were synthesized by the thermal decomposition of a Fe(acac)3 precursor.41 X-ray diffraction (XRD) analysis confirmed the formation of nanoparticles in a cubic spinel structure (space group: Fd3m). The mean crystallite size was determined with Rietveld analysis as 6.19 nm (see Fig. 1).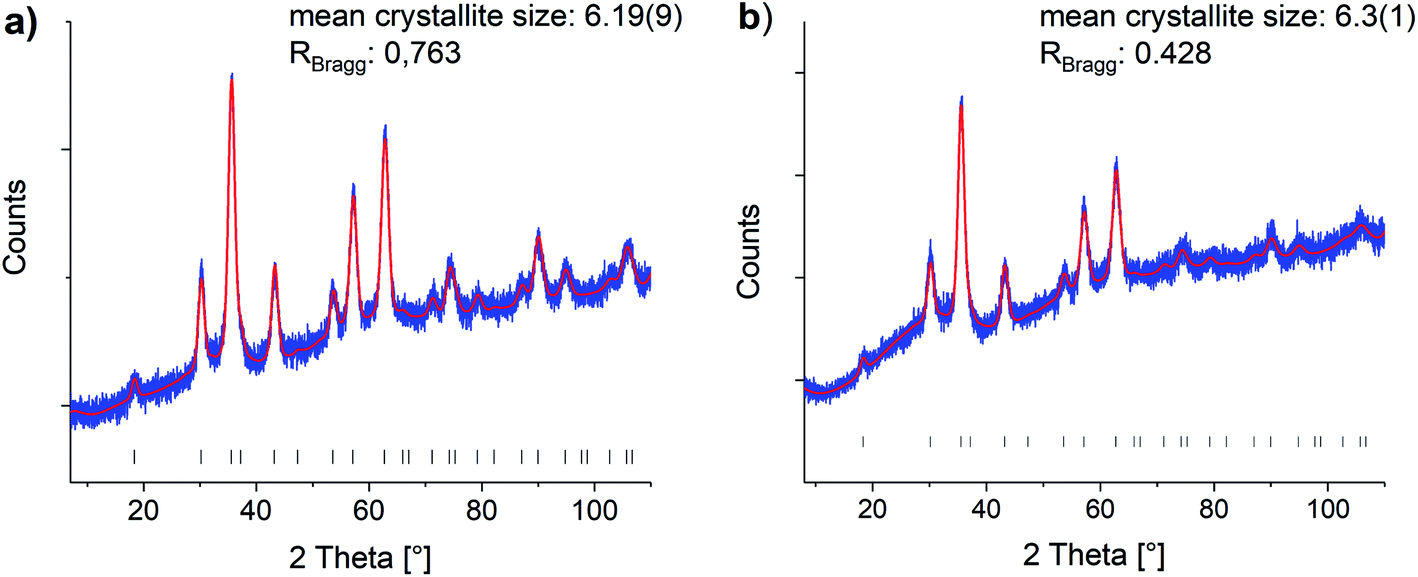 | ||
| Fig. 1 Rietveld refinements of the (a) oleic acid functionalized and (b) phosphonic acid functionalized particles (0.200P@FexOy). Blue: experimental data, red: Rietveld refinement, black hkl-values. Reference taken from COD1513301.44 | ||
Attachment of the oleic acid groups on the particle surface was confirmed with IR spectroscopy showing the characteristic vibrations of COO− bonded to Fe–O at 1695–1200 cm−1. The organic content determined by TGA was 25% varying within a 6% range between different oleic acid functionalized particle batches. TEM measurements confirmed the formation of nanoparticles with an approximate diameter of 7 nm, which fits well with the determined crystallite size from XRD analysis, verifying a high crystallinity of the particles. The small particle core size indicates that the particles are superparamagnetic, as iron oxide nanoparticles in this size range exhibit superparamagnetic behavior.45,46 DLS experiments in hexane revealed a hydrodynamic diameter of 8 nm, which again is in good agreement with TEM and XRD data. IR, as well as DLS and TGA results are shown in the ESI (Fig. S18–S20†). An exchange protocol for the oleic acid molecules on the surface against N,N,N-trimethyl-6-phosphonohexane-1-ammonium bromide was developed. The exchange with the organophosphorus coupling reagent was optimized to produce the maximum occupancy for monolayer modification. Since increasing electrostatic repulsion between the cationic groups and thus more difficult attachment can be expected as modification of the particle surface progresses, low degrees of functionalization from 0.025 to 0.6 mmol per 150 mg nanoparticles (samples 0.025P@FexOy–0.600P@FexOy) were particularly investigated. Oleic acid functionalized particles are dispersible in n-hexane, while the ionically functionalized particles are better dispersible in polar solvents such as ethanol or water. The polarity of the particles increases significantly with increasing degree of functionalization. A solvent ratio of hexane:ethanol of 1:2 (v/v) was chosen for the exchange reaction, since this ensures a stable dispersion over a wide concentration range and the organophosphorus molecules and oleic acid are soluble in it. Fig. 2 illustrates the stability of 0.002P@FexOy and OA@FexOy particle dispersions in different solvents.
 | ||
| Fig. 2 Stability of the particle dispersions (a) OA@FexOy and (b) 0.002P@FexOy in hexane and (c) 0.002P@FexOy and (d) OA@FexOy in hexane:ethanol (1:2). | ||
FTIR spectroscopy was used to investigate the particle functionalization (see Fig. 3). All spectra were normalized to the Fe–O stretching vibration at 536 cm−1. The main characteristic of the modified particles is the intense vibration band at 964 cm−1, which proves the bond formation between the phosphonic acid groups and the iron oxide surface. The intensity of the signal increases with progressive functionalization.47–49 In addition the IR spectra reveal the increase of C–H stretching and deformation vibrations at 2945–2813 cm−1 and 1645–1402 cm−1. The CO stretching vibration at 1637 cm−1, which is present in all samples, shows a non-quantitative exchange of the oleic acid. The signal intensity suggests a significantly larger amount of remaining oleic acid compared to similar exchange protocols of DA and PEG phosphonic acid derivatives reported in literature.23,50 As mentioned above, this effect can be assigned to the increasing electrostatic repulsion between the cationic groups with increasing surface coverage. Remaining oleic acid molecules on the surface seem to act as a spacer between the cationic charges. 31P NMR spectroscopy was conducted on the supernatant solutions of the functionalization reaction mixtures, showing residual unreacted phosphonic acid starting from sample 0.100P@FexOy (see Fig. S21†).
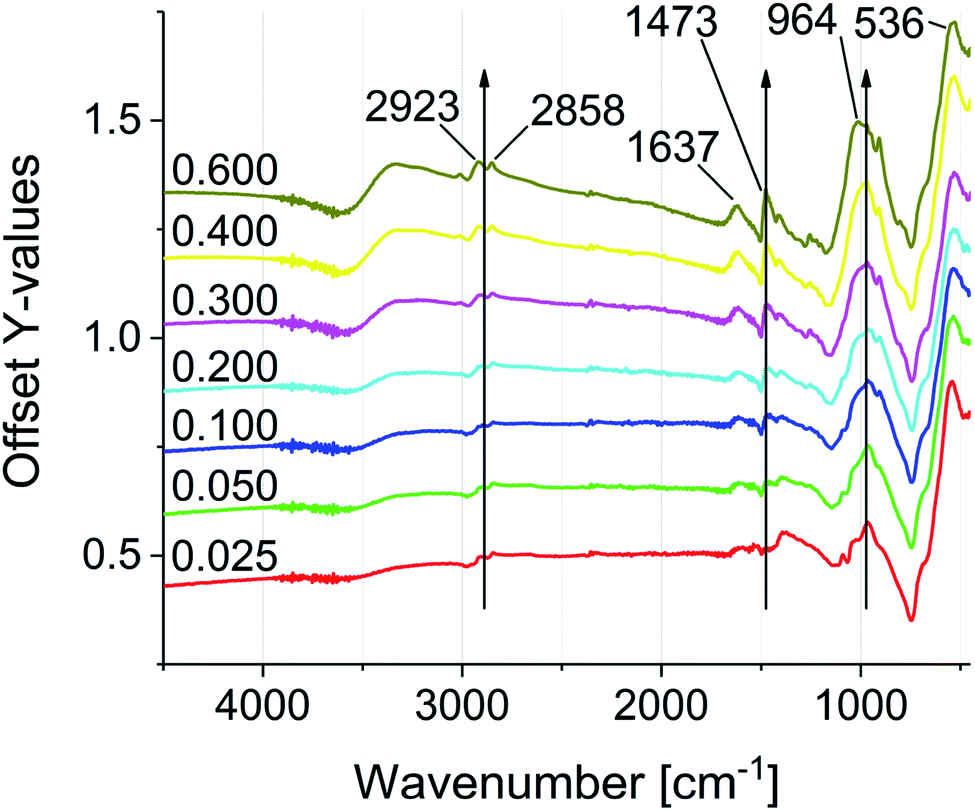 | ||
| Fig. 3 FTIR spectra of the phosphonic acid functionalized particles (0.025P@FexOy–0.600P@FexOy). | ||
Surface coverage was studied using TGA and CHN analysis. TGA measurements were performed after a 10 min vacuum step at 100 °C to remove any remaining solvent adsorbed on the particle surface (Fig. 4). Every measurement consists of two steps. Initially, samples were heated to 880 °C under nitrogen. In this first segment the cationic particles show a mass loss at 50 °C to 200 °C. This is caused by solvent desorption, which could not be removed even in the preheating step, due to strong interaction with the positively charged particle surface. The phosphonic acid decomposes in several steps, starting at about 230 °C for all samples. The mass loss for all samples increases compared to the oleic acid functionalized particles, indicating an increase of organic material bound to the surface. As expected, the mass loss increases from sample 0.025P@FexOy to 0.600P@FexOy. The organic content, obtained at the end of the N2 segment (char yield) varies between 22% and 34%. In the second step, samples are heated to 1000 °C under synthetic air. For all samples, an increase in mass is observed in the air segment, due to oxidation of the particles. While the mass increase is quite small (∼3%) for the oleic acid functionalized particles, the phosphonic acid functionalized particles show a larger increase of 7% to 12%. This indicates an additional oxidation mechanism. While for the oleic acid a complete desorption is expected during heating, the anchoring group of phosphonic acid “PO3H2” is reported to remain on the particle surface even at higher temperatures.26 By switching to oxygen-containing atmosphere the phosphonate is oxidized to the phosphate in addition to particle oxidation. As a result, the mass increase is magnified with phosphonic acid functionalization. A summary of the TGA data is presented in the ESI (Table S1†).
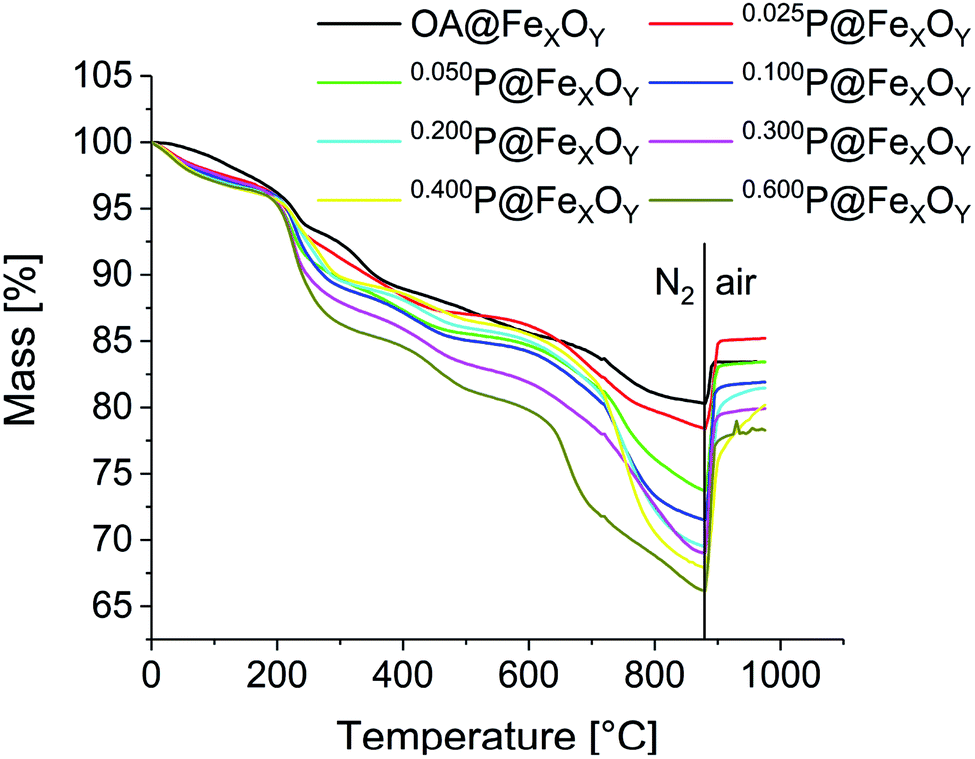 | ||
| Fig. 4 TG curves of oleic acid and the phosphonic acid functionalized nanoparticles, heating from room temperature to 880 °C under nitrogen atmosphere, followed by heating to 1000 °C under a mixture of nitrogen and oxygen (4:1) with a rate of 10 K min−1. | ||
In combination with the TGA results, CHN analysis was used to calculate the surface coverage. Because of the previously discussed oxidation occurring during TGA at elevated temperatures, when using synthetic air, the residual mass at the end of the N2 segment (carbon/hydrogen content below CHN detection limit) was used to calculate the surface coverage. The following equation was applied:
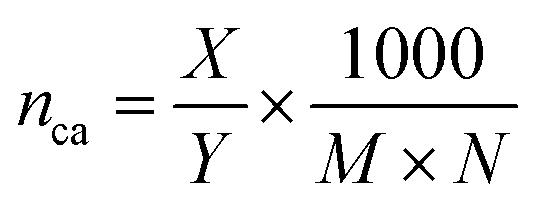 :HOA = 6.31). This indicates the presence of additional organic material on the particle surface, for example adsorbed solvent molecules that could not be removed during the drying process (C:Hethanol = 3.97). As a result an exact quantification of the remaining oleic acid was not possible. The phosphonic acid corrected carbon and hydrogen values are shown in Table S2.†
:HOA = 6.31). This indicates the presence of additional organic material on the particle surface, for example adsorbed solvent molecules that could not be removed during the drying process (C:Hethanol = 3.97). As a result an exact quantification of the remaining oleic acid was not possible. The phosphonic acid corrected carbon and hydrogen values are shown in Table S2.†
To investigate the influence of the functionalization on the particles XRD, TEM and DLS measurements were also conducted for the functionalized particles. The results are presented exemplary for 0.200P@FexOy in comparison with the oleic acid functionalized particles. While the DLS measurements show a slight increase in the hydrodynamic radius upon functionalization there is no change in the crystallite size determined by XRD, nor the particle size determined by TEM within the measuring accuracy (see Fig. 5). Furthermore, no change in color was observed during functionalization indicating no major changes in the iron oxide composition.
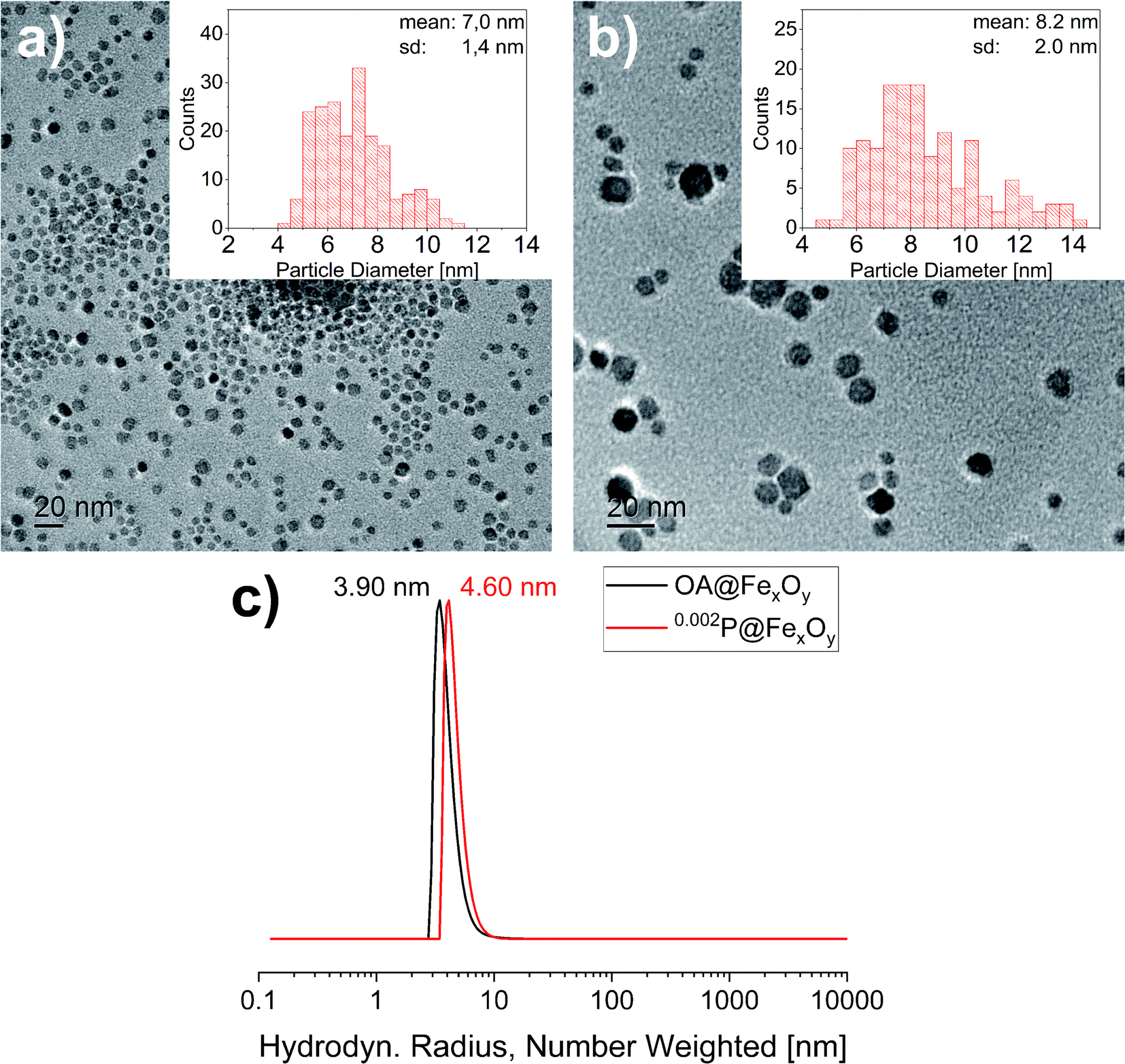 | ||
| Fig. 5 Transmission electron micrographs of (a) OA@FexOy and (b) 0.002P@FexOy. Histograms: statistical size distribution of particles (N = 200). (c) DLS measurement of OA@FexOy and 0.002P@FexOy. | ||
Zeta-potential measurements were performed to determine the isoelectric point as well as surface charge dependent of pH value (Fig. 6).
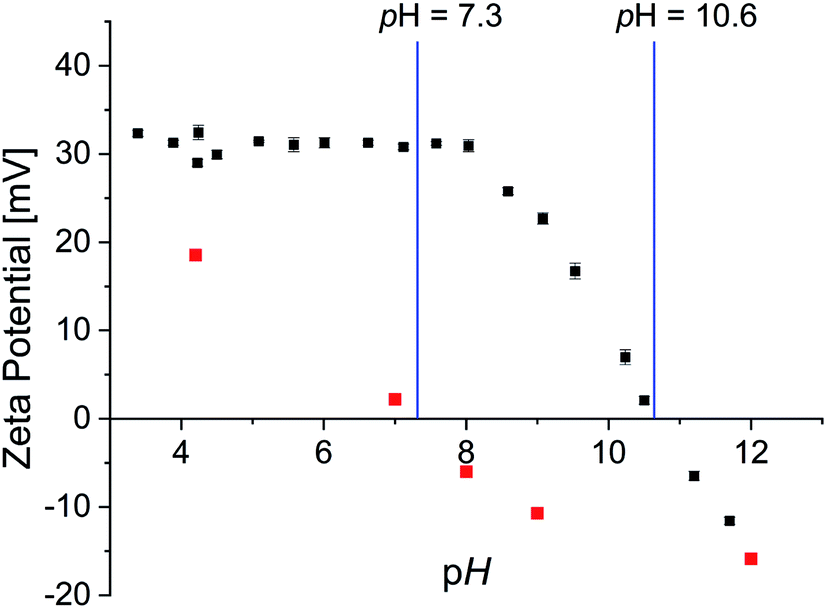 | ||
| Fig. 6 Zeta potential of 0.002P@FexOy in water (black) and OA@FexOy in ethanol (red) as a function of pH. | ||
The surface charge of the phosphonic acid modified particles starts at a high positive value of 30 mV and remains constant up to pH 8. Thereafter a linear decrease of the surface charge is observed. The isoelectric point (IEP) was determined at a pH of 10.6 and is thus significantly higher than described for non-ionic functionalized iron oxide nanoparticle surfaces of 7.9.51,52 Nonetheless considering the expected permanent positive charge the quaternary ammonium functionalities should provide, the decrease at high pH values is rather surprising. CHN analysis of the particles confirmed the absence of nitrogen containing groups at pH 11.5 (Table S3†). As the IR data suggest that the organic molecules remaining on the surface at pH 11.5 are oleic acid, analogous experiments were performed with oleic acid functionalized iron oxide nanoparticles. Zeta potential measurements showed a linear decrease of surface charge with increasing pH with an IEP of 7.3 (Fig. 6). CHN analysis displays only a partial removal of the oleic acid upon higher pH values, supporting the presumption of oleic acid in the particle surface after the functionalization. In order to further examine the absence of nitrogen content on the particle surface at high pH values, the supernatant solution of the functionalized particles was analyzed. Both 1H and 31P NMR spectroscopy, showed the intact phosphonic acid, confirming a desorption mechanism rather than a decomposition process (Fig. S22†). This is quite surprising, as the binding of a vast variety of phosphonic acid derivates to metal oxide surfaces is described as very stable.26,50,53
Polymer synthesis
Electrostatic self-healing nanocomposites, require beside charged nano building blocks charged polymer chains. For our methyl ammonium ion charged iron oxide nanoparticles we selected sulfonic acid groups in the polymer.54,55 Self-healing nanocomposites require a polymer matrix with a low Tg to grant a high chain mobility.21 Since typical anionic polyelectrolytes, like poly(acrylic acid), tend to have high Tg values (>100 °C)56 it is often necessary to use random copolymers in which a second monomer delivers a lower glass transition temperature. Furthermore, the second monomer must be hydrophilic to guarantee a good miscibility with the polar sulfonic acid derivate. Hence for a self-healing approach based on electrostatic interactions a tailoring of the matrix polymer is necessary. We have chosen methyl methacrylates as the polymerizable groups, because they show high reactivity in the polymerization process largely independent of the functionalized side chains. Based on previous arguments we used sodium-4-(methacryloyloxy)butan-1-sulfonate (SMBS) and di(ethylene glycol) methyl ether methacrylate as monomers. All synthesized polymers were prepared via activators regenerated by electron transfer atom transfer radical polymerization (AGET ATRP), using hydrazine as the reducing agent. Literature known procedures were used, applying a Cu(I) catalyst with 2,2′-bipyridine as ligand.21 In a first approach, we synthesized the corresponding homopolymers from the selected monomers. Applying standard ATRP conditions lead to low conversions and yields particularly in aqueous reaction mixtures. The reason for that seems to be the lability of the [CuBr(bipy2)] complex.57,58 By adding hydrazine after 12 h reaction time, yields could be improved up to 53% for the ionic homopolymer and over 60% for P(DEGMA) and the synthesized copolymers. 1H NMR confirmed product formation and the removal of unreacted monomer during work up (see Fig. S23†). Removal of the copper complex was achieved by dialysis against diluted HCl. Commercially available poly(styrene sulfonate) (P(SS), 70000 Da) was used as an anionic reference system.
For both synthesized homopolymers, DSC measurements were conducted to determine Tg values (see Fig. S25†). P(DEGMA) shows a very low Tg of about −23 °C, which is in good agreement with literature reports.49 Contrary the Tg of the anionic homopolymer is much higher at 166 °C. In order to assess the variability of the Tg by adjusting the polymer composition copolymers with monomer ratios of 1:10, 1:8, 1:5 and 1:3 (SMBS:DEGMA) were synthesized by weighing the respective amounts of monomer (10Pol, 8Pol, 5Pol, 3Pol). Fig. 7 shows the 1H NMR spectra of the respective polymers. The spectra were normalized to the COO–CH2 signal at 4.11 ppm.
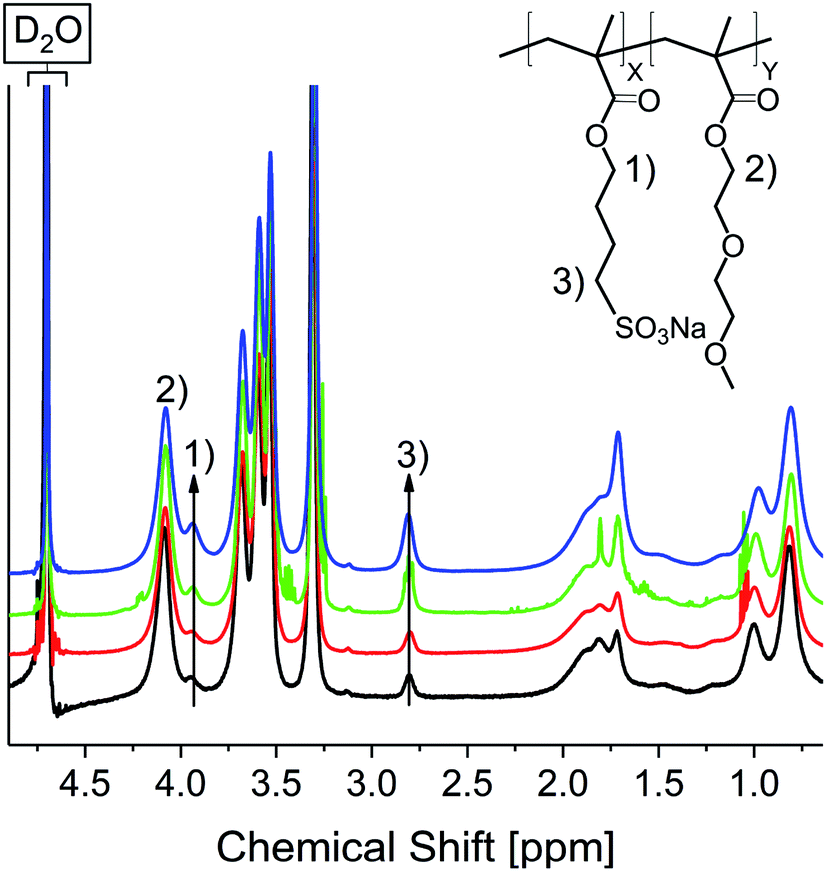 | ||
| Fig. 7 1H NMR spectra of 10Pol (black), 8Pol (red), 5Pol (green), 3Pol (blue). | ||
It is clearly visible that the signals of the ionic polymer increase with the increasing ionic proportion in the monomer composition. The polymer composition determined by integration of 1H NMR signals matches the weighted monomer ratios (see Table 1).
| Sample | DEGMAtheo | SMBStheo | DEGMAfound | SMBSfound | T g [°C] |
|---|---|---|---|---|---|
| P(DEGMA) | n | — | n | — | −23 |
| P(SMBS) | — | n | — | n | 126 |
| 10Pol | 10n | n | 11.0n | n | −18 |
| 8Pol | 8n | n | 8.1n | n | −16 |
| 5Pol | 5n | n | 5.2n | n | 26 |
| 3Pol | 3n | n | 4.0n | n | 50 |
The obtained polymer samples are hydrophilic and slowly adsorb water from the ambient air. This water affinity of polyelectrolytes is well known and studied extensively, as the water content governs a plethora of properties such as mechanical strength, rigidity and ion conductivity.59–62 Therefore, storage conditions such as humidity and temperature have to be controlled.
Fig. 8 shows the measured DSC curves in the area of the glass transition temperature for the obtained polymer samples. Comparing the glass transition temperatures of the synthesized polymers, it is apparent the increase of the ionic species elevates the glass transition temperature.
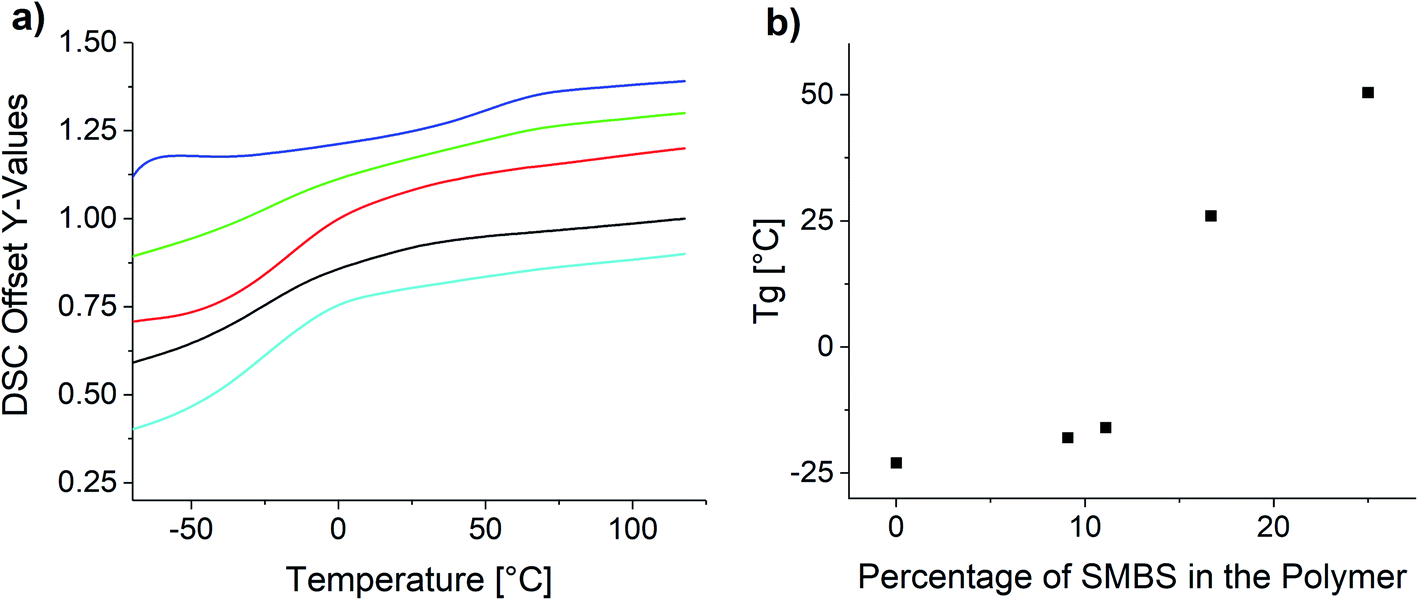 | ||
| Fig. 8 (a) DSC curves of the synthesized copolymers and (b) influence of the polymer composition on the glass transition temperature. | ||
The observed Tg increase with increasing proportion of ionic species is below the rule of mixture. This is in agreement with reported copolymers and empiric models like the Gordon–Taylor equation.63,64 Especially in the lower molar percentages the Tg only increases very slowly, whereas at the higher molar concentrations the increase is significantly higher. P(SS) has a much higher crystallinity and shows a distinct melting peak at 235 °C (see Fig. S26†).
To assess the thermal stability of the used polymers TGA measurements were performed. The results are shown in Fig. 9.
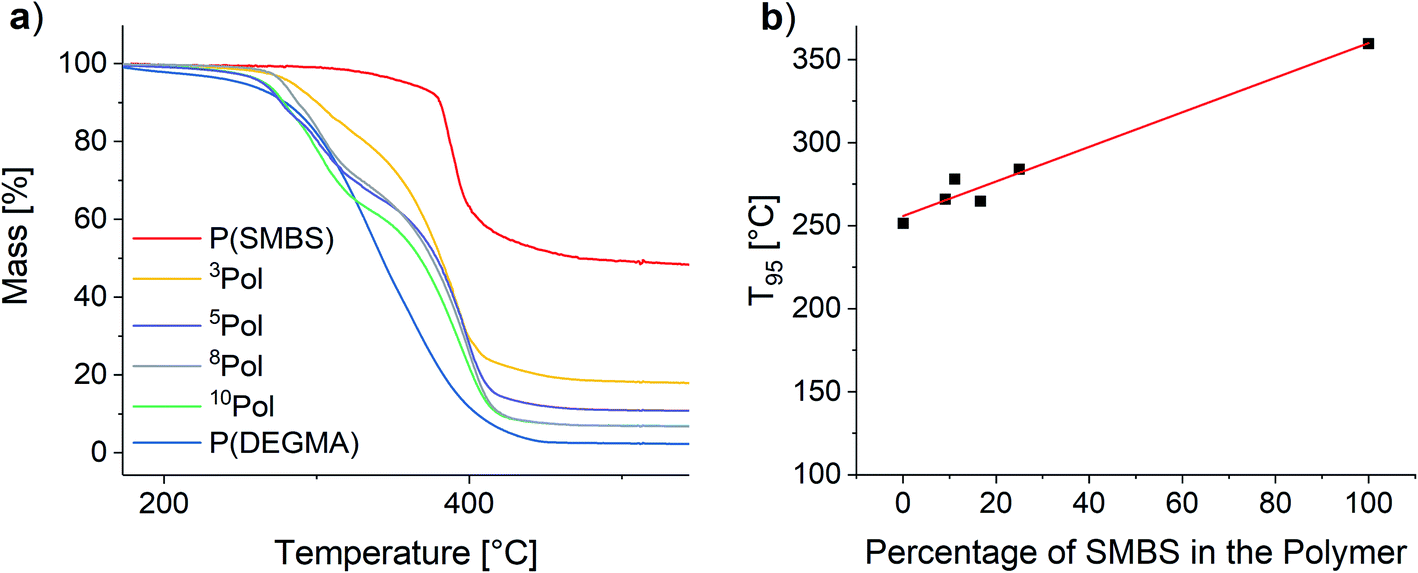 | ||
| Fig. 9 (a) TGA of the synthesized polymers and (b) influence of the polymer composition on the T95 values. | ||
For all polymers, the decomposition starts at temperatures above 250 °C. The onset for the matrix polymer is at 274 °C. In contrast, the temperature stability of the anionic polymer is much higher, showing decomposition at 375 °C. As a result, the synthesized copolymers show two distinct decomposition steps at the respective decomposition temperatures of the homopolymers. T95 values (temperature at 5% mass loss) of the polymers were considered to assess the polymer stability. By increasing the proportion of SMBS in the polymers an almost linear increase of the T95 values from 251 °C for P(DEGMA) to 360 °C for P(SMBS) is observed (see Fig. 9). P(SS) shows an even higher T95 of 447 °C (see Fig. S27†). Furthermore, P(DEGMA) shows an almost complete decomposition (char yield: 2%), whereas the ionic P(SMBS) and P(SS) reveal residual masses of 48% and 40%, respectively.
Composite synthesis
Nanocomposites were obtained by mixing the synthesized copolymers with the respective amount of cationic functionalized nanoparticles in a water/THF solvent mixture (1:1, v/v). Particle contents between 1 and 20 wt% were used. P(SS) was mixed with the filler as a control experiment. Solvents were evaporated at room temperature under vigorous stirring. The solid polymers were pressed into a Teflon mold using a vice at 80 °C. The compositions of all synthesized composites are shown in Table 2. Using P(SS) as the polymer matrix, extremely brittle composites were obtained which could not be shaped by compression molding.
| 10Pol | 8Pol | 5Pol | 3Pol | P(SS) | |
|---|---|---|---|---|---|
| 1 wt% 0.200P@FexOy | C11 | C21 | C31 | C41 | CK1 |
| 2 wt% 0.200P@FexOy | C12 | C22 | C32 | C42 | CK2 |
| 5 wt% 0.200P@FexOy | C13 | C23 | C33 | C43 | CK3 |
| 20 wt% 0.200P@FexOy | C14 | C24 | C34 | C44 | CK4 |
Black, solid, and highly magnetic samples were acquired. All specimens show no macroscopic agglomeration of the particles within the polymer (see Fig. 10).
 | ||
| Fig. 10 Image of the 3Pol based nanocomposites C41, C42, C43 and C41 (1 wt%, 2 wt%, 5 wt% and 20 wt% particle content). Dimensions: 0.5 cm × 0.5 cm × 1 mm and investigation of the magnetic properties of C41. | ||
Analogously to the polymer samples, TGA measurements were performed with the composite materials. The results for the 10Pol und 3Pol based systems are shown exemplarily in Fig. 11. In contrast to the polymer composition, the particle content shows only a small influence on the material's thermal stability. While slight increases of the thermal stability and thus the T95 values are observed for all composites the influence seems to decrease with increasing proportion of the ionic component in the polymer. For the 10Pol based systems decomposition is shifted from 266 °C to 275 °C by increasing the particle content from 0 wt% to 20 wt%. The shift is only about 5 °C for the 3Pol system for the same particle contents. As already observed for the polymer systems the char yields of the 3Pol based composites is much larger than the 10Pol based systems. The observed increase of char yields of the composites compared to the respective polymers almost linearly correlates with the particle content. Upon introducing 20 wt% particles char yields increase from 6% to 24% for the 10Pol and from 16% to 26% for the 3Pol based systems. All recorded TGA data is included in the ESI (Fig. S28†).
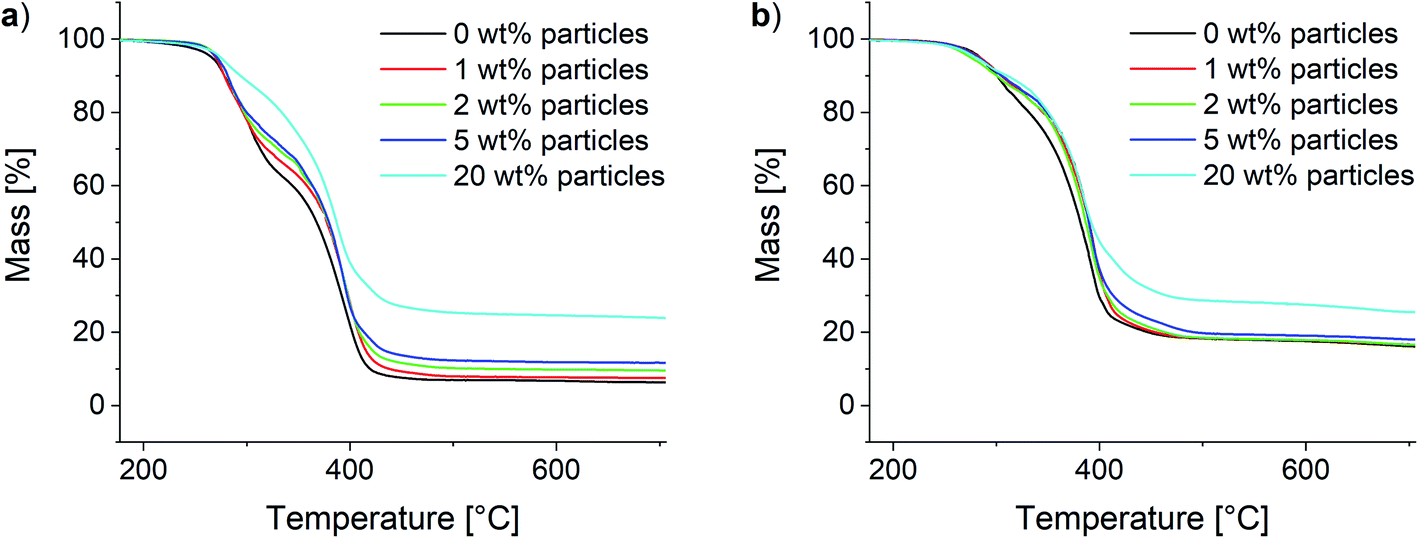 | ||
| Fig. 11 Thermogravimetric analysis of the synthesized composites. (a) 10Pol and (b) 3Pol based systems. | ||
Two different effects were observed for composites with low and high SMBS contents in DSC analysis. Fig. 12 shows the obtained curves for the 10Pol and 3Pol based systems as representatives of systems with a low and high ionic character, respectively. The DSC data of all samples are displayed in the ESI (see Fig. S29†). DSC measurements revealed no changes of the Tgs for 10Pol and 8Pol based composites in comparison to the pristine copolymers. These results are quite surprising because we expected that due to the strong ionic interactions between particles and polymer matrix the chain mobility is reduced and thereby the Tg is increased. Moll and Kumar explain the almost complete absence of Tg shift described for many strongly interacting composite systems by the fact that an almost irreversible connection between polymer and filler occurs. As a result, the layer is dynamically decoupled from the rest of the “free polymer”. Furthermore, they postulate this bonded layer does not relax in the time scale of the DSC measurements, whereby only the bulk polymer is recorded.65 For the composites with higher P(SMBS) content a decrease in Tg from 26 °C to −10 °C for 5Pol based systems respectively 50 °C to −15 °C for 3Pol based systems was observed. These results can be attributed to comprised solvent,66,67 which could not be removed completely, especially for the composites containing more polar polymer. Independent of the polymer composition no deviations in the Tg were observed between the composites containing 1 wt%, 2 wt%, 5 wt% and 20 wt% particles.
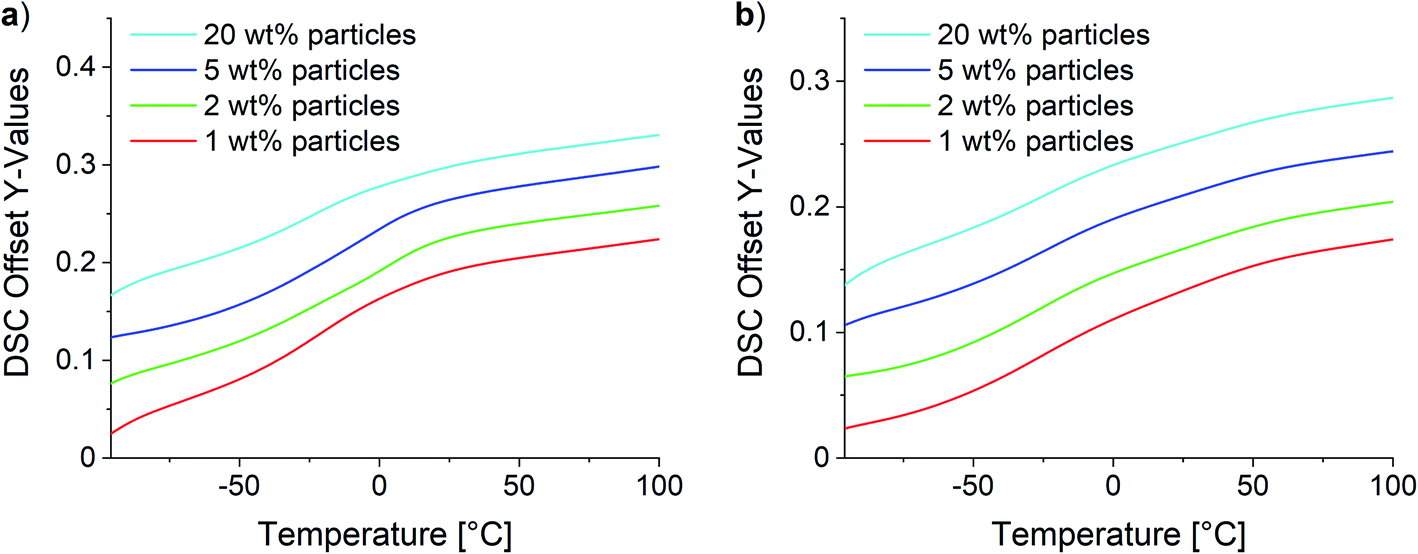 | ||
| Fig. 12 DSC of the synthesized composites. (a) 10Pol and (b) 3Pol based systems. | ||
Self-healing experiments
Self-healing studies were performed on 1 cm × 1 cm × 0.2 cm specimens. Two types of self-healing experiments were performed, varying in the type of heat supply necessary for the healing process. Firstly, regular heating in an oven was applied. For the self-healing experiments the composite samples were cut through half thickness. The separated ends were gently pressed together and heated to 80 °C for 24 h. Hereafter, the samples were put back into the Teflon molds and heated for another 24 h. After every step microscope images were taken. The results are shown exemplarily for the C44 system in Fig. 13. | ||
| Fig. 13 Microscope images of (a) C44, (b) cut through half thickness, (c) gently pressed together at room temperature then heated to 80 °C for 24 h and (d) another 24 h at 80 °C in Teflon mold. | ||
Within the first 24 h an almost complete recombination of the surfaces was observed. After another 24 h the crack completely disappeared for all samples. In general, the healing is faster for samples with lower SMBS content, due to the higher chain mobility in low Tg systems. For further investigations, the 10Pol based systems were excluded, as they rather showed melting and convergence upon heating, which did not allow the investigation of the actual healing process based on the ionic interactions.
In preparation for the self-healing experiments in alternating electromagnetic fields, heating studies were performed. Fig. 14 shows exemplarily the determined temperature at varying generator outputs over time for composites C41–C44. As expected, the heating efficiency increases with increasing particle content. The slight decrease in temperature in the first segment can be attributed to the water cooling of the HF coil. Starting from that point with every increase of the applied power a sharp increase in temperature is observed. Within five minutes an equilibrium is reached, and the temperature stabilizes. For C41, with the lowest particle content of 1 wt%, a maximum of 58 °C is achievable. With a particle content of 2 wt% the maximum temperature slightly increases to 63 °C. For C43 and C44 temperatures above 80 °C (temperature used in oven self-healing experiments) are possible.
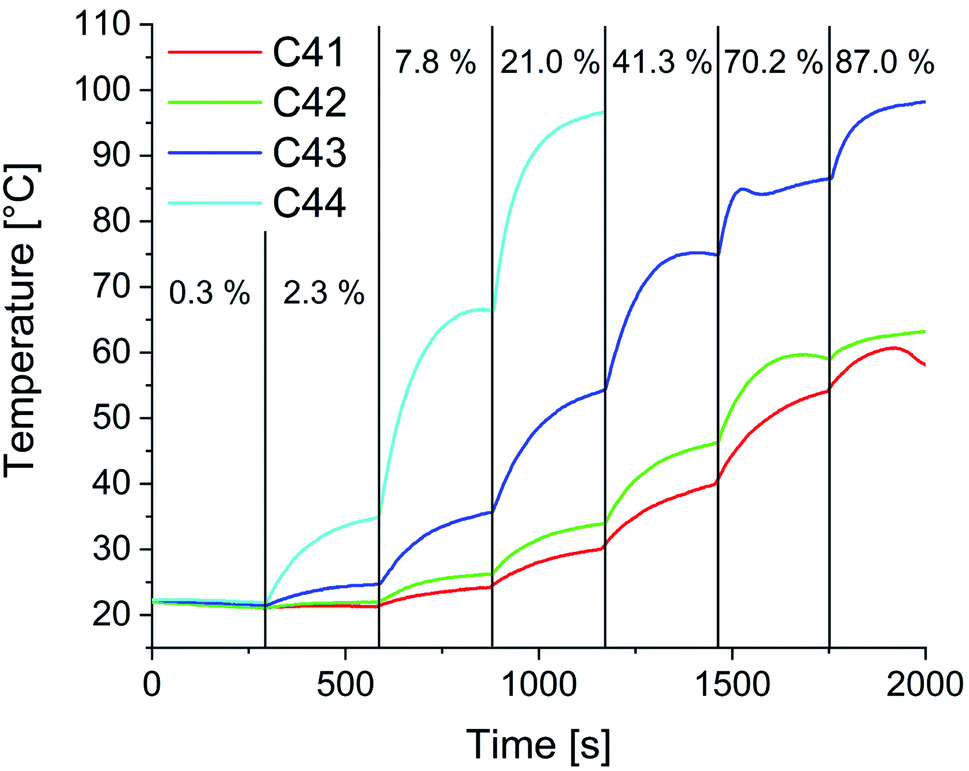 | ||
| Fig. 14 Heating studies for composites C41–C44 in alternating magnetic fields. Frequency: 1.95 MHz, field strength: 0.5–1.1 kA m−1, percentage values refer to the applied part of the total generator power of 3000 W for the respective heating segment. | ||
The influence of the electromagnetic field on the 3Pol polymer sample was also examined as a control experiment without particles. Even at full power, no significant heating of the sample was observed (see Fig. S30†). At this point the heating mechanism for the composites is not fully clarified yet, as for the used small particles a lower heating efficiency due to Néel and Brown relaxation can be expected. At higher frequencies heating might also occur due to Eddy currents.
In the next step induction heating was used as a stimulus to initiate the healing process. Field strengths were adjusted to reach a macroscopic temperature of 55 °C, thus lying below the healing experiments in the regular oven. Analogously to the healing experiments in the regular oven, microscope images were recorded after pressing the edges together, 24 h and 48 h healing time (Fig. 15).
 | ||
| Fig. 15 Microscope images of the (a) untreated sample C44, (b) the cut sample and the healed sample after (c) 24 h and (d) 48 h at 58 °C in the induction furnace. | ||
As shown for sample C44 in Fig. 15 the cut healed completely. A control experiment was performed with the polymer sample 3Pol, where no crack healing was observed (see Fig. S31†). Due to the absence of the particles, the alternating magnetic field does not induce heat in the sample, which is necessary for the healing process. Furthermore, the removal of the ionic interactions, which occur between the particles and the polymer in the composites, hampers the self-healing. Comparing the conventional oven heating and heating in an alternating magnetic field shows, that the induction heating allows faster and more efficient healing at lower macroscopic temperatures.
Conclusions
Novel functional nanocomposite materials based on anionic methacrylate-based copolymers and iron oxide nanoparticles were synthesized. Linear, flexible polymer chains were obtained by ATRP synthesis of DEGMA and SMBS at 70 °C. 1H NMR, FTIR and DSC were used for characterization. ATRP showed high flexibility towards tailoring the polymer composition and hence the polymers Tg. For these polyelectrolytes as well as the later synthesized composites the control of storage conditions, such as humidity and temperature are necessary, as these have decisive influence on the water content of the samples and thus their mechanical properties. Iron oxide nanoparticles were synthesized and functionalized with phosphonic acid derivatives bearing quaternary ammonium moieties in ω-position. Zeta potential measurements confirmed the positive surface charge necessary for a uniform dispersion in the polymer system at moderate pH-values. On top of that, self-healing properties are achieved. Particle contents of 1–20 wt% were investigated, all showing promising self-healing in the μm length scale. Introduction of superparamagnetic iron oxide nanoparticles introduced magnetic properties and the opportunity to induce heat with an external alternating magnetic field. Experiments in an alternating magnetic field revealed the possibility of spatially resolved healing at lower macroscopic temperatures necessary for conventional healing. The self-healable nanocomposites lay a basis for the development of advanced nanocomposites possibly combining the self-assembly by ionic interactions with DA-healing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Instrumentation and technical assistance for this work were provided by the Service Center X-ray Diffraction, with financial support from Saarland University and the German Science Foundation (project number INST 256/349-1). The authors thank Dr R. Haberkorn and Dr D. Becker for the support in collection of the X-ray diffraction data presented in this paper. We also thank Susanne Harling for CHN analysis and Sarah Schumacher for zeta potential measurements.References
- T. Engel and G. Kickelbick, Furan-Modified Spherosilicates as Building Blocks for Self-Healing Materials, Eur. J. Inorg. Chem., 2015, 1226–1232 CrossRef CAS.
- T. Osada, K. Kamoda, M. Mitome, T. Hara, T. Abe, Y. Tamagawa, W. Nakao and T. Ohmura, A Novel Design Approach for Self-Crack-Healing Structural Ceramics with 3D Networks of Healing Activator, Sci. Rep., 2017, 7, 1–2 CrossRef CAS.
- B. Grabowski and C. Tasan, Self-Healing Metals, in Advances in Polymer Science, 2015, vol. 273, pp. 387–407 Search PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, A Thermally Re-Mendable Cross-Linked Polymeric Material, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Self-Healing and Thermoreversible Rubber from Supramolecular Assembly, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, High-Water-Content Mouldable Hydrogels by Mixing Clay and a Dendritic Molecular Binder, Nature, 2010, 463, 339–343 CrossRef CAS.
- Y. L. Rao, A. Chortos, R. Pfattner, F. Lissel, Y. C. Chiu, V. Feig, J. Xu, T. Kurosawa, X. Gu, C. Wang, M. He, J. W. Chung and Z. Bao, Stretchable Self-Healing Polymeric Dielectrics Cross-Linked through Metal-Ligand Coordination, J. Am. Chem. Soc., 2016, 138, 6020–6027 CrossRef CAS PubMed.
- R. S. Chen, N. A. Mohd Amran and S. Ahmad, Reinforcement Effect of Nanocomposites with Single/Hybrid Graphene Nanoplatelets and Magnesium Hydroxide: Thermal Stability, Flame Retardancy and Mechanical Performance, J. Therm. Anal. Calorim., 2019, 137, 79–92 CrossRef CAS.
- L. Zhou, Y. Zhou, Y. Shi, T. Chen, T. Zou, D. Zhou and Q. Fu, Enhancing Thermal Stability of P(VDF-HFP) Based Nanocomposites with Core-Shell Fillers for Energy Storage Applications, Compos. Sci. Technol., 2020, 186, 107934 CrossRef CAS.
- Y. Ul-haq, I. Murtaza, S. Mazhar, R. Ullah and M. Iqbal, Dielectric, Thermal and Mechanical Properties of Hybrid PMMA/RGO/Fe2O3 Nanocomposites Fabricated by In Situ Polymerization, Ceram. Int., 2019, 46, 5828–5840 CrossRef.
- M. G. Mohamed and S. W. Kuo, Functional Silica and Carbon Nanocomposites Based on Polybenzoxazines, Macromol. Chem. Phys., 2019, 220, 1–13 CrossRef.
- M. G. Mohamed and S. W. Kuo, Functional Polyimide/Polyhedral Oligomeric Silsesquioxane Nanocomposites, Polymers, 2019, 11, 26 CrossRef.
- L. Han, X. Lu, M. Wang, D. Gan, W. Deng, K. Wang, L. Fang, K. Liu, C. W. Chan, Y. Tang, L. T. Weng and H. Yuan, A Mussel-Inspired Conductive, Self-Adhesive, and Self-Healable Tough Hydrogel as Cell Stimulators and Implantable Bioelectronics, Small, 2017, 13, 1–9 Search PubMed.
- J. Li, Q. Liu, D. Ho, S. Zhao, S. Wu, L. Ling, F. Han, X. Wu, G. Zhang, R. Sun and C. P. Wong, Three-Dimensional Graphene Structure for Healable Flexible Electronics Based on Diels-Alder Chemistry, ACS Appl. Mater. Interfaces, 2018, 10, 9727–9735 CrossRef CAS PubMed.
- A. Gantar, N. Drnovšek, P. Casuso, A. Pérez-San Vicente, J. Rodriguez, D. Dupin, S. Novak and I. Loinaz, Injectable and Self-Healing Dynamic Hydrogel Containing Bioactive Glass Nanoparticles as a Potential Biomaterial for Bone Regeneration, RSC Adv., 2016, 6, 69156–69166 RSC.
- A. J. R. Amaral, M. Emamzadeh and G. Pasparakis, Transiently Malleable Multi-Healable Hydrogel Nanocomposites Based on Responsive Boronic Acid Copolymers, Polym. Chem., 2018, 9, 525–537 RSC.
- B. C. K. Tee, C. Wang, R. Allen and Z. Bao, An Electrically and Mechanically Self-Healing Composite with Pressure- and Flexion-Sensitive Properties for Electronic Skin Applications, Nat. Nanotechnol., 2012, 7, 825–832 CrossRef CAS PubMed.
- S. Dai, X. Zhou, S. Wang, J. Ding and N. Yuan, A Self-Healing Conductive and Stretchable Aligned Carbon Nanotube/Hydrogel Composite with a Sandwich Structure, Nanoscale, 2018, 10, 19360–19366 RSC.
- S. Schäfer and G. Kickelbick, Self-Healing Polymer Nanocomposites Based on Diels-Alder-Reactions with Silica Nanoparticles: The Role of the Polymer Matrix, Polymer, 2015, 69, 357–368 CrossRef.
- T. Engel and G. Kickelbick, Self-Healing Nanocomposites from Silica-Polymer Core-Shell Nanoparticles, Polym. Int., 2014, 63, 915–923 CrossRef CAS.
- S. Schäfer and G. Kickelbick, Double Reversible Networks: Improvement of Self-Healing in Hybrid Materials via Combination of Diels-Alder Cross-Linking and Hydrogen Bonds, Macromolecules, 2018, 51, 6099–6110 CrossRef.
- T. Engel and G. Kickelbick, Thermoreversible Reactions on Inorganic Nanoparticle Surfaces: Diels-Alder Reactions on Sterically Crowded Surfaces, Chem. Mater., 2013, 25, 149–157 CrossRef CAS.
- S. Schäfer and G. Kickelbick, Diels–Alder Reactions on Surface-Modified Magnetite/Maghemite Nanoparticles: Application in Self-Healing Nanocomposites, ACS Appl. Nano Mater., 2018, 1, 2640–2652 CrossRef.
- M. Zhong, Y. T. Liu and X. M. Xie, Self-Healable, Super Tough Graphene Oxide-Poly(Acrylic Acid) Nanocomposite Hydrogels Facilitated by Dual Cross-Linking Effects through Dynamic Ionic Interactions, J. Mater. Chem. B, 2015, 3, 4001–4008 RSC.
- J. Odent, J. M. Raquez, P. Dubois and E. P. Giannelis, Ultra-Stretchable Ionic Nanocomposites: From Dynamic Bonding to Multi-Responsive Behavior, J. Mater. Chem. A, 2017, 5, 13357–13363 RSC.
- C. Heinrich, L. Niedner, B. Oberhausen and G. Kickelbick, Surface-Charged Zirconia Nanoparticles Prepared by Organophosphorus Surface Functionalization with Ammonium or Sulfonate Groups, Langmuir, 2019, 35, 11369–11379 CrossRef CAS PubMed.
- Z. Guo, K. Lei, Y. Li, H. W. Ng, S. Prikhodko and H. T. Hahn, Fabrication and Characterization of Iron Oxide Nanoparticles Reinforced Vinyl-Ester Resin Nanocomposites, Compos. Sci. Technol., 2008, 68, 1513–1520 CrossRef CAS.
- J. Santoyo Salazar, L. Perez, O. De Abril, L. Truong Phuoc, D. Ihiawakrim, M. Vazquez, J. M. Greneche, S. Begin-Colin and G. Pourroy, Magnetic Iron Oxide Nanoparticles in 10–40 nm Range: Composition in Terms of Magnetite/Maghemite Ratio and Effect on the Magnetic Properties, Chem. Mater., 2011, 23, 1379–1386 CrossRef CAS.
- L. Qiao, Z. Fu, J. Li, J. Ghosen, M. Zeng, J. Stebbins, P. N. Prasad and M. T. Swihart, Standardizing Size- and Shape-Controlled Synthesis of Monodisperse Magnetite (Fe3O4) Nanocrystals by Identifying and Exploiting Effects of Organic Impurities, ACS Nano, 2017, 11, 6370–6381 CrossRef CAS.
- E. C. Vreeland, J. Watt, G. B. Schober, B. G. Hance, M. J. Austin, A. D. Price, B. D. Fellows, T. C. Monson, N. S. Hudak, L. Maldonado-Camargo, A. C. Bohorquez, C. Rinaldi and D. L. Huber, Enhanced Nanoparticle Size Control by Extending LaMer's Mechanism, Chem. Mater., 2015, 27, 6059–6066 CrossRef CAS.
- R. Gupta, K. Pancholi, R. De Sa, D. Murray, D. Huo, G. Droubi, M. White and J. Njuguna, Effect of Oleic Acid Coating of Iron Oxide Nanoparticles on Properties of Magnetic Polyamide-6 Nanocomposite, JOM, 2019, 71, 3119–3128 CrossRef CAS.
- S. Ali, S. A. Khan, Z. H. Yamani, M. T. Qamar, M. A. Morsy and S. Sarfraz, Shape- and Size-Controlled Superparamagnetic Iron Oxide Nanoparticles Using Various Reducing Agents and Their Relaxometric Properties by Xigo Acorn Area, Appl. Nanosci., 2019, 9, 479–489 CrossRef CAS.
- M. Torres-Lugo and C. Rinaldi, Thermal Potentiation of Chemotherapy by Magnetic Nanoparticles, Nanomedicine, 2013, 8, 1689–1707 CrossRef CAS.
- C. C. Corten, M. W. Urban and F. Shelby, Repairing Polymers Using an Oscillating Magnetic Field, Adv. Mater., 2009, 21, 5011–5015 CrossRef CAS.
- M. Yoonessi, B. A. Lerch, J. A. Peck, R. B. Rogers, F. J. Solá-Lopez and M. A. Meador, Self-Healing of Core-Shell Magnetic Polystyrene Nanocomposites, ACS Appl. Mater. Interfaces, 2015, 7, 16932–16937 CrossRef CAS PubMed.
- A. S. Ahmed and R. V. Ramanujan, Magnetic Field Triggered Multicycle Damage Sensing and Self Healing, Sci. Rep., 2015, 5, 1–10 Search PubMed.
- Y. Yang, J. He, Q. Li, L. Gao, J. Hu, R. Zeng, J. Qin, S. X. Wang and Q. Wang, Self-Healing of Electrical Damage in Polymers Using Superparamagnetic Nanoparticles, Nat. Nanotechnol., 2019, 14, 151–155 CrossRef CAS.
- N. Hohlbein, A. Shaaban and A. M. Schmidt, Remote-Controlled Activation of Self-Healing Behavior in Magneto-Responsive Ionomeric Composites, Polymer, 2015, 69, 301–309 CrossRef CAS.
- Y. Zhang, Y. Wang, Y. Wen, Q. Zhong and Y. Zhao, Self-Healable Magnetic Structural Color Hydrogels, ACS Appl. Mater. Interfaces, 2020, 12, 7486–7493 CrossRef CAS PubMed.
- M. T. Rahman, M. Asadul Hoque, G. T. Rahman, M. A. Gafur, R. A. Khan and M. K. Hossain, Study on the Mechanical, Electrical and Optical Properties of Metal-Oxide Nanoparticles Dispersed Unsaturated Polyester Resin Nanocomposites, Results Phys., 2019, 13, 102264 CrossRef.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, Monodisperse MFe2O4 (M = Fe, Co, Mn) Nanoparticles, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed.
- W. D. Niederhauser, E. Broderick and F. F. Owings, Method for Preparing Salts of Sulfoalkyl Methacrylates, US Pat., 2964557A, 1960 Search PubMed.
- C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Surface Modification Using Phosphonic Acids and Esters, Chem. Rev., 2012, 112, 3777–3807 CrossRef.
- F. F. Ferreira, E. Granado, W. Carvalho, S. W. Kycia, D. Bruno and R. Droppa, X-Ray Powder Diffraction Beamline at D10B of LNLS: Application to the Ba2FeReO6 Double Perovskite, J. Synchrotron Radiat., 2006, 13, 46–53 CrossRef CAS PubMed.
- S. R. Cooper, L. K. Plummer, A. G. Cosby, P. Lenox, A. Jander, P. Dhagat and J. E. Hutchison, Insights into the Magnetic Properties of Sub-10 nm Iron Oxide Nanocrystals through the Use of a Continuous Growth Synthesis, Chem. Mater., 2018, 30, 6053–6062 CrossRef CAS.
- J. Mohapatra, F. Zeng, K. Elkins, M. Xing, M. Ghimire, S. Yoon, S. R. Mishra and J. P. Liu, Size-Dependent Magnetic and Inductive Heating Properties of Fe3O4 Nanoparticles: Scaling Laws across the Superparamagnetic Size, Phys. Chem. Chem. Phys., 2018, 20, 12879–12887 RSC.
- Y. Okada, H. Asama, N. Koike, S. Yamashita, N. Maeta, A. Uesaka and H. Kamiya, Direct Ordering of Anchoring Events at the Surface of Iron Oxide Nanoparticles Enabled by A Stepwise Phase-Transfer Strategy, ChemistrySelect, 2018, 3, 8458–8461 CrossRef CAS.
- F. Brodard-Severac, G. Guerrero, J. Maquet, P. Florian, C. Gervais and P. H. Mutin, High-Field 17O MAS NMR Investigation of Phosphonic Acid Monolayers on Titania, Chem. Mater., 2008, 20, 5191–5196 CrossRef CAS.
- S. Han, M. Hagiwara and T. Ishizone, Synthesis of Thermally Sensitive Water-Soluble Polymethacrylates by Living Anionic Polymerizations of Oligo(Ethylene Glycol) Methyl Ether Methacrylates, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- K. Davis, B. Qi, M. Witmer, C. L. Kitchens, B. A. Powell and O. T. Mefford, Quantitative Measurement of Ligand Exchange on Iron Oxides via Radiolabeled Oleic Acid, Langmuir, 2014, 30, 10918–10925 CrossRef CAS.
- M. Baalousha, Aggregation and Disaggregation of Iron Oxide Nanoparticles: Influence of Particle Concentration, PH and Natural Organic Matter, Sci. Total Environ., 2009, 407, 2093–2101 CrossRef CAS PubMed.
- E. Tombácz, K. Farkas, I. Földesi, M. Szekeres, E. Illés, I. Y. Tóth, D. Nesztor and T. Szabó, Polyelectrolyte Coating on Superparamagnetic Iron Oxide Nanoparticles as Interface between Magnetic Core and Biorelevant Media, Interface Focus, 2016, 6, 1–8 CrossRef.
- M. Raza, A. Bachinger, N. Zahn, G. Kickelbick, I. Solid and S. Chemistry, Interaction and UV-Stability of Various Organic Capping Agents on the Surface of Anatase Nanoparticles, Materials, 2014, 2890–2912 CrossRef.
- D. J. Pietrzyk and J. Belisle, Acidity of Aromatic Sulfonic Acids and Their Use as Titrants in Nonaqueous Solvents, Anal. Chem., 1966, 38, 969–973 CrossRef CAS.
- X. Li, Y. Wan and C. Sun, Covalent Modification of a Glassy Carbon Surface by Electrochemical Oxidation of R-Aminobenzene Sulfonic Acid in Aqueous Solution, J. Electroanal. Chem., 2004, 569, 79–87 CrossRef CAS.
- Y. Kawaguchi, Y. Itamura, K. Onimura and T. Oishi, Effects of the Chemical Structure on the Heat Resistance of Thermoplastic Expandable Microspheres, J. Appl. Polym. Sci., 2005, 96, 1306–1312 CrossRef CAS.
- S. Smolne and M. Buback, Kinetic Investigations of Cu-Mediated ATRP in Aqueous Solution, Macromol. Chem. Phys., 2015, 216, 894–902 CrossRef CAS.
- M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Understanding the Fundamentals of Aqueous ATRP and Defining Conditions for Better Control, Macromolecules, 2015, 48, 6862–6875 CrossRef CAS.
- J. Kittle, J. Levin and N. Levin, Water Content of Polyelectrolyte Multilayer Films Measured by Quartz Crystal Microbalance and Deuterium Oxide Exchange, Sensors, 2021, 21, 771–782 CrossRef CAS PubMed.
- T. Liu, Q. An, Q. Zhao, J. Wu, Y. Song, B. Zhu and C. Gao, Synergistic Strenghening of Polyelectrolyte Complex Membranes by Functionalized Carbon Nanotubes and Metal Ions, Sci. Rep., 2015, 5, 7782 CrossRef CAS PubMed.
- H. H. Hariri, A. M. Lehaf and J. B. Schleno, Mechanical Properties of Osmotically Stressed Polyelectrolyte Complexes and Multilayers: Water as a Plasticizer, Macromolecules, 2012, 45, 9364–9372 CrossRef CAS.
- A. Ostendorf, M. Scho and C. Cramer, Ionic Conductivity of Solid Polyelectrolyte Complexes with Varying Water Content: Application of the Dynamic Structure Model, Phys. Chem. Chem. Phys., 2019, 21, 7321–7329 RSC.
- F. N. Kelley and F. Bueche, Viscosity and Glass Temperature Relations for Polymer-Diluent Systems, J. Polym. Sci., 1961, 50, 549–556 CrossRef CAS.
- M. Gordon and J. S. Taylor, Ideal Copolymers and the Second-Order Transitions of Synthetic Rubbers. I. Noncrystalline Copolymers, Rubber Chem. Technol., 1953, 26, 323–335 CrossRef CAS.
- J. Moll and S. K. Kumar, Glass Transitions in Highly Attractive Highly Filled Polymer Nanocomposites, Macromolecules, 2012, 45, 1131–1135 CrossRef CAS.
- M. Eriksson, H. Goossens and T. Peijs, Influence of Drying Procedure on Glass Transition Temperature of PMMA Based Nanocomposites, Nanocomposites, 2015, 1, 36–45 CrossRef CAS.
- B. J. Ash, R. W. Siegel and L. S. Schadler, Glass-Transition Temperature Behavior of Alumina/PMMA Nanocomposites, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 4371–4383 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00417d |
| This journal is © The Royal Society of Chemistry 2021 |