
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanomaterials for the electrochemical nitrogen reduction reaction under ambient conditions
Juan
Wen
a,
Linqing
Zuo
a,
Haodong
Sun
a,
Xiongwei
Wu
 b,
Ting
Huang
a,
Zaichun
Liu
a,
Jing
Wang
*a,
Lili
Liu
*a,
Yuping
Wu
*a,
Xiang
Liu
a and
Teunis
van Ree
c
b,
Ting
Huang
a,
Zaichun
Liu
a,
Jing
Wang
*a,
Lili
Liu
*a,
Yuping
Wu
*a,
Xiang
Liu
a and
Teunis
van Ree
c
aState Key Laboratory of Materials-oriented Chemical Engineering, School of Energy Science and Engineering, Institute for Advanced Materials, Nanjing Tech University, Nanjing 211816, China. E-mail: wuyp@njtech.edu.cn; wjx2015@njtech.edu.cn
bCollege of Chemistry and Materials, Hunan Agriculture University, Changsha, Hunan 410128, China
cDepartment of Chemistry, University of Venda, Thohoyandou 0950, South Africa
First published on 4th August 2021
Abstract
As an important chemical product and carbon-free energy carrier, ammonia has a wide range of daily applications in several related fields. Although the industrial synthesis method using the Haber–Bosch process could meet production demands, its huge energy consumption and gas emission limit its long-time development. Therefore, the clean and sustainable electrocatalytic N2 reduction reaction (NRR) operating under conditions have attracted great attention in recent years. However, the chemical inertness of N2 molecules makes it difficult for this reaction to proceed. Therefore, rationally designed catalysts need to be introduced to activate N2 molecules. Here, we summarize the recent progress in low-dimensional nanocatalyst development, including the relationship between the structure and NRR performance from both the theoretical and experimental perspectives. Some insights into the development of NRR electrocatalysts from electronic control aspects are provided. In addition, the theoretical mechanisms, reaction pathways and credibility studies of the NRR are discussed. Some challenges and future prospects of the NRR are also pointed out.
 Yuping Wu | Yuping Wu, FRSC, Distinguished Professor of Nanjing Tech University, received his Ph. D. degree from the Institute of Chemistry, CAS, in 1997. In 2003, he was promoted to full professor in Fudan University, China. In 2015 he moved to Nanjing Tech University. He has published over 350 papers in peer-reviewed journals with H-index > 80, was named in the World's Most Influential Minds (2015) list by Thomson Reuters, won the Albert Nelson Marquis Lifetime Achievement Award (2019), and was listed as one of the Highly Cited Researchers all over the world. He conducted pioneering research work on ARLBs, gel-type and pore-free separators for lithium batteries with high safety, and nitrogen fixation. |
1 Introduction
Against the background of increasing population and global energy consumption connected with burning fossil fuels and worldwide environmental issues, creating an energy system based on efficient, clean and sustainable resources is particularly significant to secure our energy supply.1,2 Ammonia (NH3) as one of the most widely produced industrial chemicals has found wide applications in the manufacture of fertilizers (at least 80% of total ammonia annually) and pharmaceuticals, as well as energy conversion.3 Particularly, compared to other energy carriers, NH3 is recognized as an ideal storage medium for hydrogen (H2), because it has a relatively high hydrogen content (17.6% by mass) and can exist easily in liquid form.4–8 Besides, nitrogen is an indispensable element of life and a crucial component of various bio-macromolecules (amino acids, nucleotides and the like). However, since the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond has a high bond energy of 945 kJ mol−1 at 298 K, it is difficult to crack and hydrogenate.9 Even though the atmospheric nitrogen (N2) content is 78%, organisms normally obtain N in the form of NH3 or nitrate (NO3−) instead of directly using N2 molecules. In nature, NH3 can be obtained by fixing N2 either by lightning or through the action of microbial nitrogenases.10 Generally, bacteria of the genus Azotobacter use adenosine triphosphate (ATP) as an energy transporter, and an iron–molybdenum–sulphur (FeMoS) core cluster as the main N2 binding site to achieve the conversion of N2 into NH3 through migrating multiple protons and electrons under ambient conditions.11–13 However, microorganisms containing nitrogenase in nature alone cannot sufficiently match the demand for fertilizer use. Industrially, the Haber–Bosch process with Fe-based14,15 or Ru-based16,17 catalysts has been the main NH3 production method over the last 100 years, and is operated under harsh conditions (around 400 °C, 50 bar), thus suffering from high energy consumption and severe equipment requirements.18,19 In addition, H2 is generally produced by the steam reformation of CH4, which also depletes fossil fuels immensely and causes excessive CO2 emissions. The social cost of CO2 emissions and the impact of global fuel prices on geopolitics also limit the long-term development of conventional NH3 processes.20 Therefore, NH3 synthesized from water and atmospheric N2 using electric power is a promising method to convert unstably stored electricity into chemical energy. Moreover, feedstocks like water and N2 are abundant and eco-friendly, acting as a proton source and N source, respectively. Since Davy's group developed the N2 reduction reaction (NRR) method in 1807, researchers around the world have used various catalysts with different structures and compositions to activate N2.21 Attracted by the attention of various countries and the huge prospects of electrocatalysis, the NRR field has developed tremendously over the past five years (Fig. 1a). So far, various active centre nanomaterials such as precious metals (Au, Ag, Ru, Pd, etc.), non-precious metals (Fe, Mo, Co, Ti, etc.), and metal-free (B, C, etc.) (Fig. 1c) have been investigated to match some suitable low-dimensional substrates to improve the catalytic performance of NH3 yield and faradaic efficiency (FE). However, in some representative studies (Fig. 1b), around 70% of the catalysts appear in the low performance zone of low NH3 yield or low FE, which is not conducive to the industrialization of the production process. According to S. Giddey's suggestion, the commercial process needs to achieve an NH3 yield of 6.5 × 10−7 mol cm−2 s−1 or 3978 μg mgcat.−1 h−1 (mass loading: 0.1 mg cm−1), and a FE of about 50%, with a current density above 0.25 A cm−2.22 Therefore, these issues should be solved before attempting to produce NH3 on a large scale: firstly, the NN bond is hard to activate – the high NN bond energy makes it difficult for the N2 activation reaction to proceed. Therefore, some key strategies to improve the kinetics of the NRR need to be developed. Secondly, the competing hydrogen evolution reaction (HER) should be considered. Since the NRR and HER have similar reaction potentials, choosing a catalyst with high selectivity that can inhibit the HER is the key to finding an efficient and sustainable NH3 synthesis route.23 Moreover, a rational material morphology should be designed because of the significant difference in the reaction rates of materials on different exposed surfaces with different morphologies. Therefore, it is necessary to design a series of advanced catalysts to realize revolutionary progress in the NRR field.
N triple bond has a high bond energy of 945 kJ mol−1 at 298 K, it is difficult to crack and hydrogenate.9 Even though the atmospheric nitrogen (N2) content is 78%, organisms normally obtain N in the form of NH3 or nitrate (NO3−) instead of directly using N2 molecules. In nature, NH3 can be obtained by fixing N2 either by lightning or through the action of microbial nitrogenases.10 Generally, bacteria of the genus Azotobacter use adenosine triphosphate (ATP) as an energy transporter, and an iron–molybdenum–sulphur (FeMoS) core cluster as the main N2 binding site to achieve the conversion of N2 into NH3 through migrating multiple protons and electrons under ambient conditions.11–13 However, microorganisms containing nitrogenase in nature alone cannot sufficiently match the demand for fertilizer use. Industrially, the Haber–Bosch process with Fe-based14,15 or Ru-based16,17 catalysts has been the main NH3 production method over the last 100 years, and is operated under harsh conditions (around 400 °C, 50 bar), thus suffering from high energy consumption and severe equipment requirements.18,19 In addition, H2 is generally produced by the steam reformation of CH4, which also depletes fossil fuels immensely and causes excessive CO2 emissions. The social cost of CO2 emissions and the impact of global fuel prices on geopolitics also limit the long-term development of conventional NH3 processes.20 Therefore, NH3 synthesized from water and atmospheric N2 using electric power is a promising method to convert unstably stored electricity into chemical energy. Moreover, feedstocks like water and N2 are abundant and eco-friendly, acting as a proton source and N source, respectively. Since Davy's group developed the N2 reduction reaction (NRR) method in 1807, researchers around the world have used various catalysts with different structures and compositions to activate N2.21 Attracted by the attention of various countries and the huge prospects of electrocatalysis, the NRR field has developed tremendously over the past five years (Fig. 1a). So far, various active centre nanomaterials such as precious metals (Au, Ag, Ru, Pd, etc.), non-precious metals (Fe, Mo, Co, Ti, etc.), and metal-free (B, C, etc.) (Fig. 1c) have been investigated to match some suitable low-dimensional substrates to improve the catalytic performance of NH3 yield and faradaic efficiency (FE). However, in some representative studies (Fig. 1b), around 70% of the catalysts appear in the low performance zone of low NH3 yield or low FE, which is not conducive to the industrialization of the production process. According to S. Giddey's suggestion, the commercial process needs to achieve an NH3 yield of 6.5 × 10−7 mol cm−2 s−1 or 3978 μg mgcat.−1 h−1 (mass loading: 0.1 mg cm−1), and a FE of about 50%, with a current density above 0.25 A cm−2.22 Therefore, these issues should be solved before attempting to produce NH3 on a large scale: firstly, the NN bond is hard to activate – the high NN bond energy makes it difficult for the N2 activation reaction to proceed. Therefore, some key strategies to improve the kinetics of the NRR need to be developed. Secondly, the competing hydrogen evolution reaction (HER) should be considered. Since the NRR and HER have similar reaction potentials, choosing a catalyst with high selectivity that can inhibit the HER is the key to finding an efficient and sustainable NH3 synthesis route.23 Moreover, a rational material morphology should be designed because of the significant difference in the reaction rates of materials on different exposed surfaces with different morphologies. Therefore, it is necessary to design a series of advanced catalysts to realize revolutionary progress in the NRR field.
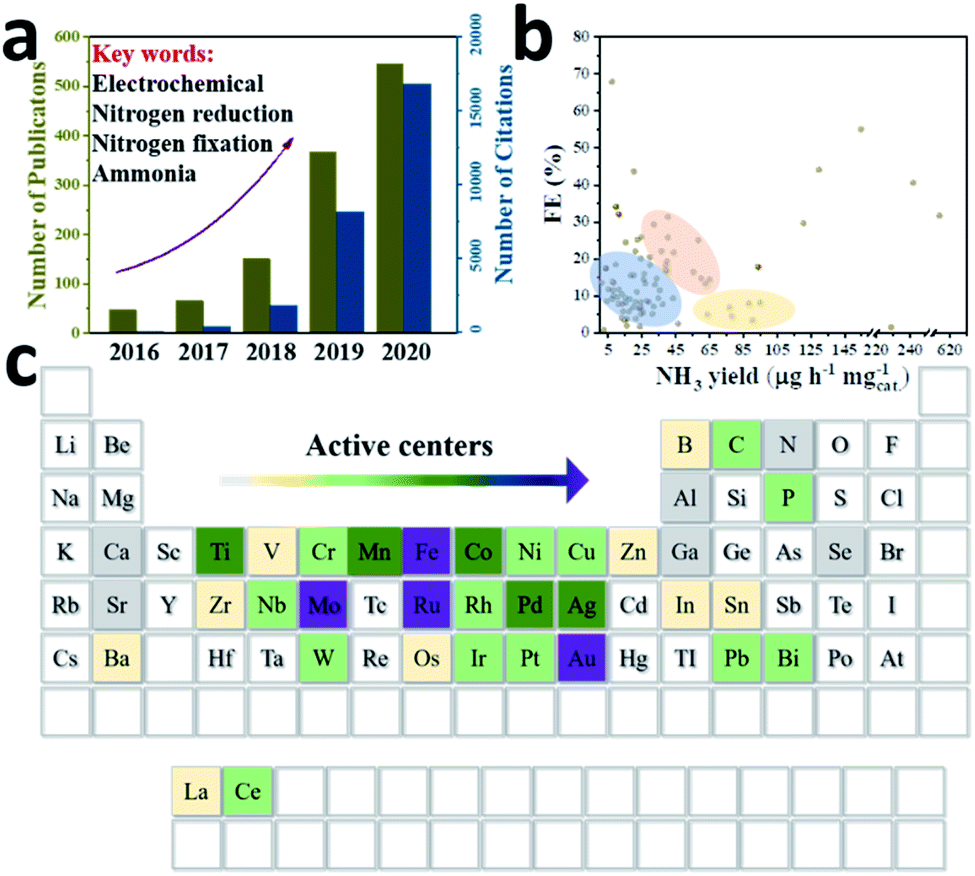 | ||
| Fig. 1 (a) The number of publications and citations of NRR articles in the Web of Science in the last 5 years. (b) Reported NH3 yield rate and FE of some representative articles. (c) The types of NRR electrocatalyst active centre published to date. The direction of the arrow represents the number of reports. | ||
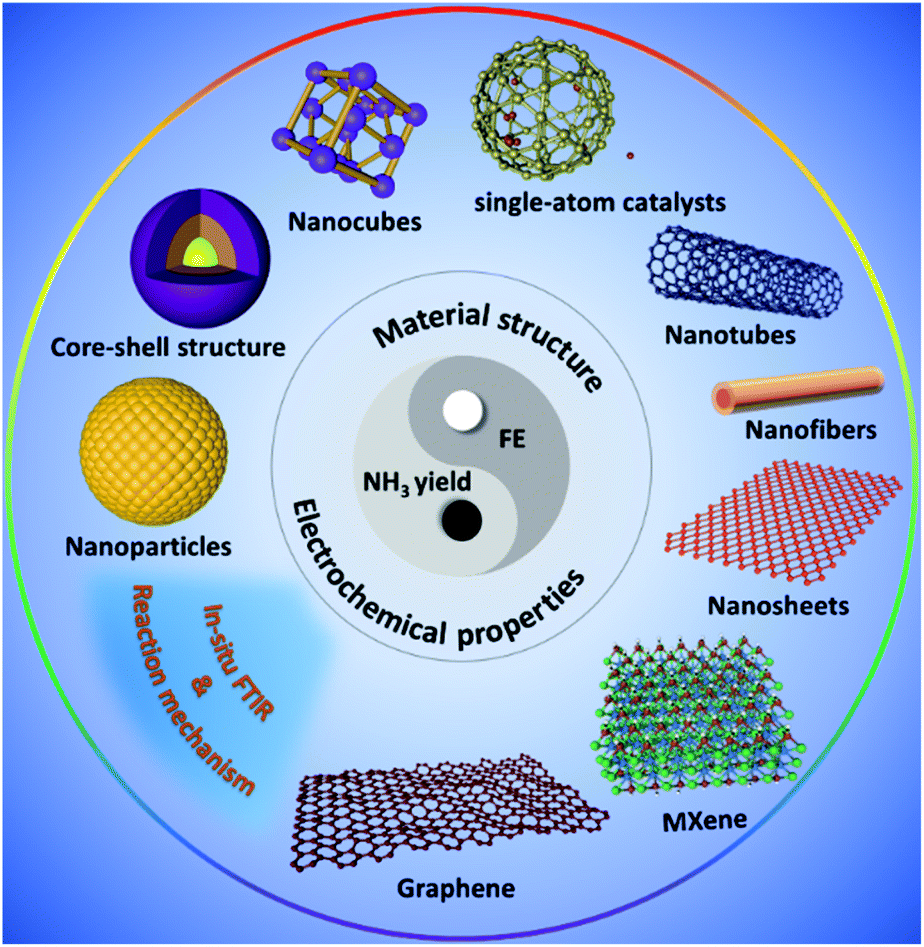 | ||
| Fig. 2 Schematic overview of the NRR materials covered in this section. | ||
In this review, we will mainly summarize the recent progress of some advanced low-dimensional nanomaterials in N2 fixation under ambient conditions from experimental and theoretical perspectives, aiming at inspiring new perceptions for the future in this field. Firstly, we discuss the NRR mechanisms, reaction intermediates and credibility studies of the NRR. Then, we summarize some typical low-dimensional nanomaterials, discussing the relationship between the structure and NRR performance from both the theoretical and experimental perspectives. Finally, we provide some insights into the development of NRR electrocatalysts from the perspective of electronic control.
2 Reaction mechanisms
2.1 Theoretical basis
The electrochemical NRR is generally completed in an H-type cell. The overall reaction equation is shown in eqn (1):| N2 + H2O → NH3 + O2 | (1) |
The anodic reaction is shown in eqn (2) and (3):
| Acidic electrolyte: 3H2O → 3/2O2 + 6H+ + 6e− | (2) |
Alkaline or neutral electrolyte:
| 6OH− → 3/2O2 + 3H2O + 6e− | (3) |
The cathodic reaction is shown in eqn (4) and (5):
| Acidic electrolyte: N2 + 6H+ + 6e− → 2NH3 | (4) |
Alkaline or neutral electrolyte:
| N2 + 6H2O + 6e− → 2NH3 + 6OH− | (5) |
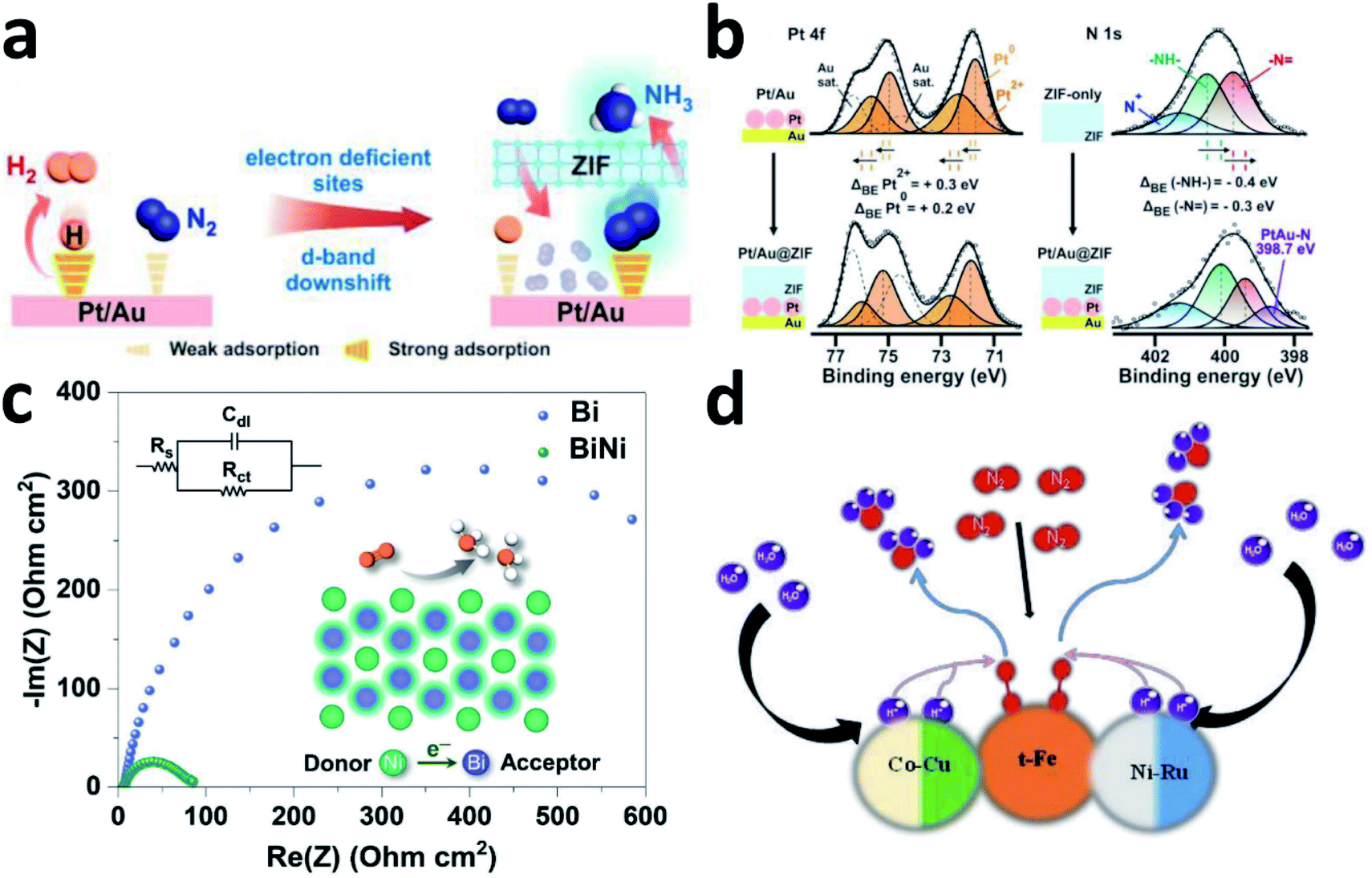
The first important step of the NRR is the adsorption and activation of N2, which is highly related to the electronic state of the electrocatalyst. For the ground state of N2, the bonding HOMO is 3σg, and the antibonding LUMO is 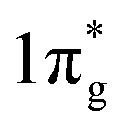 . Taking transition metals as an example, N2 donates electrons to the empty d orbital from its HOMO and accepts electrons to its LUMO, thereby being adsorbed on the catalyst and forming bonds.24 This electron donation and acceptance process is beneficial to adsorbing N2 and weakening the NN bonds.25 The NRR is accompanied by a series of hydrogenation steps, including the breaking of NN bonds and the formation of N–H bonds. Generally speaking, N2 hydrogenation has two main mechanisms, the dissociative mechanism and the associative mechanism (Fig. 3).26,27 In the dissociative mechanism, the NN bond is broken before hydrogenation. Then, each N atom is hydrogenated separately until two NH3 molecules are formed. This approach requires a high activation energy to overcome the high energy barrier during the direct cleavage of NN bonds. In the associative mechanism, one N atom in the N2 molecule is first adsorbed on the surface of the electrocatalyst, and then gradually hydrogenated. Generally, the mainstream pathways in the associative mechanism for converting N2 to NH3 can be classified into two hydrogenation routes, distal hydrogenation and alternating hydrogenation. In the distal hydrogenation path, H is added one by one to the N atom farthest away from the catalyst surface. When this N atom is converted to NH3, the N–N bond is broken. Then, the other N atom adsorbed on the surface of the catalyst is converted into NH3 in the same way. In the alternating hydrogenation route, hydrogenation occurs alternately on the two N atoms. After the first N atom is converted to NH3, the remaining –NH2 will absorb another proton to form a second NH3 molecule. The enzymatic pathways belonging to the associative pathway exhibit the unique characteristics of lateral adsorption, rather than terminal adsorption in the alternating and distal pathways. The current research work shows that the N2 fixation in the Haber–Bosch process is mainly caused by the dissociative mechanism.28 Therefore, high-temperature and high-pressure reaction conditions are necessary for the cleavage of NN bonds. However, cleavage of the NN bond in the associative mechanism is not necessary, which significantly reduces the energy input during the N2 activation process, allowing the reaction to proceed under relatively mild conditions. Therefore, the separate or mixed partial radical steps of the distal, alternating, and enzymatic pathways dominate in the NRR.
. Taking transition metals as an example, N2 donates electrons to the empty d orbital from its HOMO and accepts electrons to its LUMO, thereby being adsorbed on the catalyst and forming bonds.24 This electron donation and acceptance process is beneficial to adsorbing N2 and weakening the NN bonds.25 The NRR is accompanied by a series of hydrogenation steps, including the breaking of NN bonds and the formation of N–H bonds. Generally speaking, N2 hydrogenation has two main mechanisms, the dissociative mechanism and the associative mechanism (Fig. 3).26,27 In the dissociative mechanism, the NN bond is broken before hydrogenation. Then, each N atom is hydrogenated separately until two NH3 molecules are formed. This approach requires a high activation energy to overcome the high energy barrier during the direct cleavage of NN bonds. In the associative mechanism, one N atom in the N2 molecule is first adsorbed on the surface of the electrocatalyst, and then gradually hydrogenated. Generally, the mainstream pathways in the associative mechanism for converting N2 to NH3 can be classified into two hydrogenation routes, distal hydrogenation and alternating hydrogenation. In the distal hydrogenation path, H is added one by one to the N atom farthest away from the catalyst surface. When this N atom is converted to NH3, the N–N bond is broken. Then, the other N atom adsorbed on the surface of the catalyst is converted into NH3 in the same way. In the alternating hydrogenation route, hydrogenation occurs alternately on the two N atoms. After the first N atom is converted to NH3, the remaining –NH2 will absorb another proton to form a second NH3 molecule. The enzymatic pathways belonging to the associative pathway exhibit the unique characteristics of lateral adsorption, rather than terminal adsorption in the alternating and distal pathways. The current research work shows that the N2 fixation in the Haber–Bosch process is mainly caused by the dissociative mechanism.28 Therefore, high-temperature and high-pressure reaction conditions are necessary for the cleavage of NN bonds. However, cleavage of the NN bond in the associative mechanism is not necessary, which significantly reduces the energy input during the N2 activation process, allowing the reaction to proceed under relatively mild conditions. Therefore, the separate or mixed partial radical steps of the distal, alternating, and enzymatic pathways dominate in the NRR.
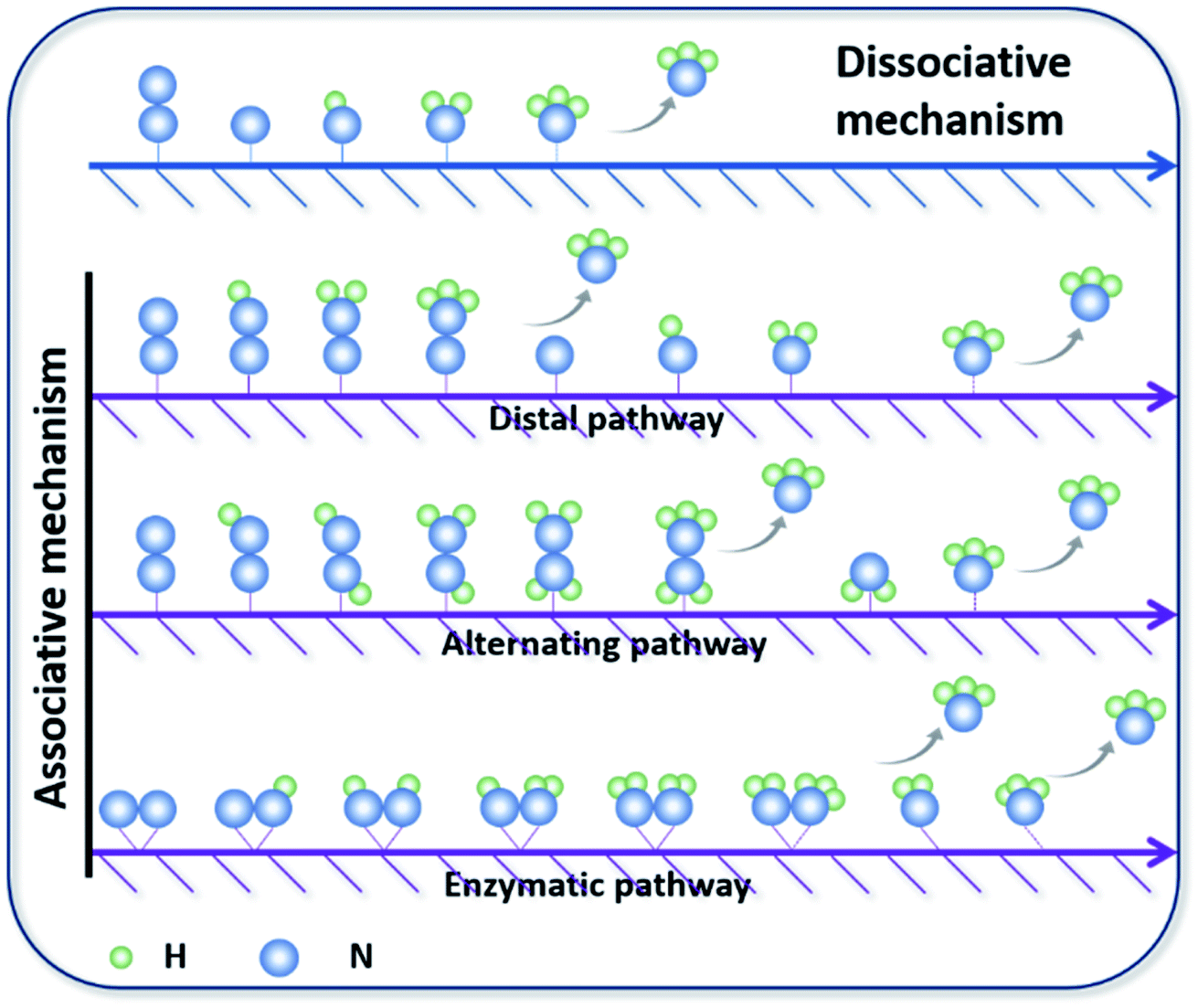 | ||
| Fig. 3 Mainstream mechanisms for N2 reduction to NH3 on catalysts. Copyright 2016, Elsevier B.V.26 Copyright 2016, American Chemical Society.27 | ||
2.2 Research on intermediate products
Although it is important to determine the NRR mechanism to further improve the absorbance and activation of N2 on the surface of the catalyst, few experimental studies have been published on the significant effects of the NRR. In situ Fourier transform infrared spectroscopy (in situ FTIR) technology based on electrochemistry has become a new method to explore the N2 reduction pathways, employed to detect the change of functional groups (like *NNH, *HNH, and *NH3) on the electrocatalyst surface during the NRR. By using in situ FTIR, key information about the real-time adsorption state of reactants and the structural transformation of intermediates on the catalyst surface could be obtained during the reaction process.29 Shao's group investigated the composition and content of Pt and Au surface functional groups at different potentials by surface-enhanced infrared absorption spectroscopy (SEIRAS).30Fig. 4a shows the change in functional groups at the Au surface in N2-saturated solution under the first CV segment. It can be seen clearly that the intensity of δ(H–N–H) and δ(N–N) gradually increases as the potential grows more negative in the N2-saturated solution. At the Pt surface, the characteristic peaks between 1300 and 1500 cm−1 are not observed (Fig. 4b), due to the much faster kinetics of the HER. Based on this analysis, it was believed that there was an associative reaction mechanism on the Au surfaces. Furthermore, they deduced a new possible NRR pathway on Rh surfaces by SEIRAS and differential electrochemical mass spectrometry (DEMS), that is, first reduction to N2H2 on the Rh surface, and then chemical dissociation in the electrolyte to form NH3.31 The optimal potential and materials have also been analysed by Yan.32 Combining LSV (Fig. 4c), in situ FTIR (Fig. 4d) and in situ Raman (Fig. 4e), they studied the catalyst reaction interface. Compared to the BCP (left) and PVDF@BCP (right), the in situ FTIR (Fig. 4d) of ECOF@BCP (middle) additionally displayed obvious ν(N–H) with increasing intensity as the potential became more negative, which is consistent with LSV. According to the position of the cathodic peak in the LSV, the potential of −0.3 V vs. RHE was selected for in situ Raman spectroscopy. Fig. 4e shows the gradual formation of NH3 on the surface of ECOF@BCP.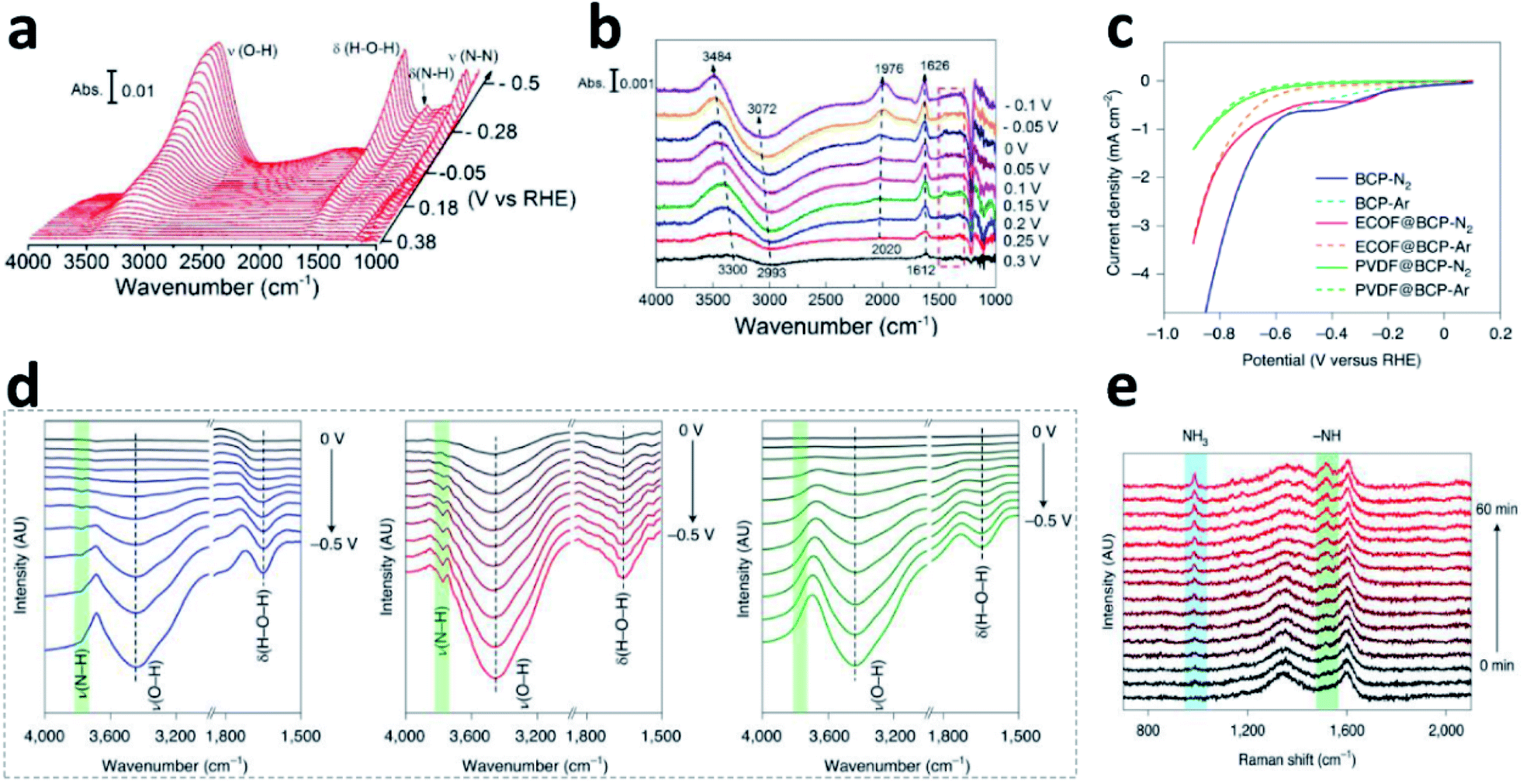 | ||
| Fig. 4 FTIR spectra during the CV on the Au (a)/Pt (b) film in N2-saturated solution. Copyright 2018, American Chemical Society.30 (c) LSV of different electrodes in feed gas. (d) In situ FTIR spectra of BCP (left), ECOF@BCP (middle) and PVDF@BCP (right) under feed gas. (e) In situ Raman spectra of ECOF@BCP at −0.3 V vs. RHE. Copyright 2021, Springer Nature.32 | ||
2.3 Credibility studies
The fundamental problem with the NRR is that the amount of NH3 produced is very small, yet NH3 is a common contaminant in the laboratory environment, which has led to a large number of rigorous experiments being carried out to verify the reliability of the NRR activity of the material. The reasons for the discrepancies in the experimental data can be broadly classified as follows: contamination of the raw material (water, catalyst, catalyst carrier, electrolyte, and feed gas), air contamination, deterioration of the colour developer and decomposition of the catalyst. For feedstock contamination, eight sets of comparison experiments using the controlled variable method for NRR systems can be verified: (1) non-catalyst-Ar-open circuit voltage (OCV), (2) non-catalyst-N2-OCV, (3) non-catalyst-Ar-working voltage (WV), (4) non-catalyst-N2-WV, (5) catalyst-Ar-OCV, (6) catalyst-N2-WV, (7) catalyst-Ar-WV, (8) catalyst-N2-WV. For air contamination, the same volume of electrolyte placed daily on the operating table allows for monitoring of NH3 contamination in the laboratory. For colour reagent deterioration, the standard curve should be calibrated weekly and all batches of electrolyte are coloured for the same time. For catalysts containing N, 15N NMR is necessary to ensure that the source of NH3 is N2 and not the decomposition of the catalyst.3 Nanomaterial NRR electrocatalysts
For electrocatalytic N2 fixation, nanomaterials have a uniform exposed lattice and can realize more exposed active sites under a certain mass loading compared with conventional bulk catalysts. Nanomaterials encompass a broad spectrum of tunable physical and chemical properties including a high surface to volume ratio and abundant active sites, making them immensely useful for a wide range of catalytic reactions. Low-dimensional nanocatalysts are highly designable because their surface structures and electronic properties are very sensitive to particle size and morphology. In this section, we will briefly summarize the synthetic methods and corresponding unique properties of these advanced nanomaterials, which are grouped into four categories based on material morphology (Fig. 2): zero-dimensional (0D) nanomaterials, including nanoparticles (NPs), nanospheres, quantum dots (QDs) and single-atom catalysts (SACs); one-dimensional (1D) nanomaterials such as nanofibers (NFs), nanowires (NWs), nanorods (NRs), and nanotubes (NTs); and two-dimensional (2D) nanomaterials such as nanosheets (NSs), graphene (G), C3N4, MXene, and other non-layered carbon compounds.3.1 Zero-dimensional (0D) nanomaterials
0D nanomaterials have ultra-small sizes, high specific surface area and huge numbers of active edge sites. The most typical 0D materials are NPs, which can be crystalline or amorphous, monometallic, multicomponent, or metal-free materials. In catalysis, NPs are the most common nanomaterials due to the relatively easier synthetic procedures. Over the past few decades, a rich variety of chemical methods (like chemical exfoliation, sonication-assisted exfoliation, chemical vapor deposition (CVD), solvothermal processing, cautery technique, and laser ablation technique) have been developed to fabricate 0D NPs with well-controlled sizes and shapes.33 In this section, we will briefly review the synthetic strategies for various 0D NRR catalysts. The performance of some 0D electrocatalysts is summarized in Table 1.| Catalystref. | Electrolyte | NH3 yield (μg h−1 mgcat.−1) | NH3 yield (×10−10 mol s−1 cm−2) | FE (%) | Potential (V vs. RHE) |
|---|---|---|---|---|---|
| MV-MoN@NC34 | 0.1 M HCl | ≈76.90 | 6.90 | −0.20 | |
| AuCu/ZIF-8 (ref. 35) | 0.1 M HCl | 63.90 | — | 5.00 | −0.20 |
| Pt/Au@ZIF36 | THF + CF3LIO3S + 1% ethanol | >130 | — | 44.00 | — |
| Au NPs37 | 0.1 M Na2SO4 | 17.49 | — | 5.79 | −0.14 |
| Au1Cu1 (ref. 38) | 0.05 M H2SO4 | 154.91 | — | 54.96 | −0.2 |
| Fe2(MoO4)3 (ref. 39) | 0.1 M Na2SO4 | 18.16 | — | 9.10 | −0.60 |
| Mn3O4 NPs40 | 0.1 M Na2SO4 | 25.95 | — | 5.51 | −0.50 |
| ZrO2 NPs41 | 0.1 M HCl | 24.74 | — | 5.00 | −0.45 |
| C/Y stabilized ZrO2 (ref. 42) | 0.1 M Na2SO4 | 24.60 | — | 8.20 | −0.50 |
| np-PdH0.43 (ref. 43) | 0.1 M PBS | 20.40 | — | 43.60 | −0.15 |
| Body-centered cubic PdCu NPs44 | LiCl | 35.70 | — | 11.50 | −0.10 |
| B and N dual-doped C nanospheres45 | 0.05 M H2SO4 | 15.70 | — | 8.10 | −0.04 |
| Defect-rich MoS2 nanoflowers46 | 0.1 M Na2SO4 | 29.28 | — | 8.34 | −0.40 |
| BP QDs/MnO2 (ref. 47) | 0.1 M Na2SO4 | 25.30 | — | 6.70 | −0.50 |
| BP QDs@SnO2−x (ref. 48) | 0.1 M Na2SO4 | 29.22 | — | 18.00 | −0.40 |
| Ru SAs/N–C49 | 0.05 M H2SO4 | 120.90 | — | 29.60 | −0.20 |
| Fe-(O–C2)4 (ref. 50) | 0.1 M KOH | 32.10 | — | 29.30 | −0.10 |
| SACs-MoS2-Fe51 | 0.1 M KCl | 613.70 | 15.93 | 31.60 | −0.20 |
| Nix-N-C-700-3h52 | 0.5 M Li2SO4 | — | 18.79 | 21.00 | — |
| SA-Mo/NPC53 | 0.1 M KOH | 34.00 | — | 14.60 | −0.30 |
The NPs were fabricated applying a series of solvothermal strategies using various precursors. The general approach for preparing NPs is using a solvothermal process to prepare precursors followed by annealing treatment, or using sacrificial templates. Some pioneering researches on TiO2 as the NRR nanocatalyst were done by Sun's group.54,55 They fabricated C-doped TiO2 NPs by dispersing titanium butoxide in water/ethanol (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30) solution, followed by hydrothermal and annealing treatment. A yield of 16.22 μg h−1 mgcat.−1 NH3 was obtained with a 1.84% FE at −0.7 V vs. RHE in Na2SO4. However, the amorphous carbon may cover the active sites of the catalyst during the synthesis process, resulting in a relatively low yield of NH3. Therefore, modulating the vacancy concentration and defect states may increase the active site density effectively. Recently, Cu ions with mixed valences of +1 and +2 were also introduced into TiO2 NPs by hydrothermal reaction to modulate the oxygen vacancy concentration and Ti3+ defect states (Fig. 5a).55 Due to the chemical compensation effect, mixed-valent Cu can induce different electronic defect states in TiO2, thereby spontaneously adjusting the oxygen vacancies (OVs) concentration. Introduction of mixed valence Cu modulates the surface of TiO2 (101) to generate OVs with different Ti3+ 3d1 defect states below the Fermi energy. By adjusting the coordination structure, the activation and adsorption of N2 can be enhanced effectively. Moreover, the catalytic performance is related to both Ti3+ and Cu contents. When the content of Cu was 6.9%, the Cu-doped TiO2 NPs attained a high FE of 21.99% with 21.31 μg h−1 mgcat.−1 NH3 yield at −0.55 V vs. RHE in 0.5 M LiClO4. Indeed, semiconductor materials with OVs can contribute to a certain NRR activity.56 If combined with other NRR active components like noble Au, the catalytic performance may also improve synergistically. Due to the easy Ce3+/Ce4+ redox cycle and abundant OVs, CeO2 has been widely investigated as a catalyst.57 Zhang's group reported a core–shell structure Au@CeO2 with a small core layer of Au NPs (<10 nm).58 The combination of abundant OVs in the shell layer of CeO2 NPs and small-sized Au NPs in the core layer enhances the NRR performance synergistically. The same group also developed core–shell Au/C NPs (Fig. 5b) by laser ablation, which consists of a spherical Au core and 1–2-layer graphite carbon shell with a large number of defects.59 The ultrathin carbon layer inhibits the aggregation of Au nanospheres effectively during the electrochemical reaction. Alternatively, there are a large number of carbon vacancies in the ultrathin carbon layer, which can provide additional catalytically active sites for the NRR. Hence, the core–shell Au/C NPs can afford an excellent performance (NH3: 241.9 μg h−1 mgcat.−1, FE: 40.5%) at −0.45 V vs. RHE in neutral solution. Apparently, the synergistic effect between the carbon vacancies in the ultrathin graphitic carbon layer and the inner active Au nanospheres enhanced the NRR activity.
:30) solution, followed by hydrothermal and annealing treatment. A yield of 16.22 μg h−1 mgcat.−1 NH3 was obtained with a 1.84% FE at −0.7 V vs. RHE in Na2SO4. However, the amorphous carbon may cover the active sites of the catalyst during the synthesis process, resulting in a relatively low yield of NH3. Therefore, modulating the vacancy concentration and defect states may increase the active site density effectively. Recently, Cu ions with mixed valences of +1 and +2 were also introduced into TiO2 NPs by hydrothermal reaction to modulate the oxygen vacancy concentration and Ti3+ defect states (Fig. 5a).55 Due to the chemical compensation effect, mixed-valent Cu can induce different electronic defect states in TiO2, thereby spontaneously adjusting the oxygen vacancies (OVs) concentration. Introduction of mixed valence Cu modulates the surface of TiO2 (101) to generate OVs with different Ti3+ 3d1 defect states below the Fermi energy. By adjusting the coordination structure, the activation and adsorption of N2 can be enhanced effectively. Moreover, the catalytic performance is related to both Ti3+ and Cu contents. When the content of Cu was 6.9%, the Cu-doped TiO2 NPs attained a high FE of 21.99% with 21.31 μg h−1 mgcat.−1 NH3 yield at −0.55 V vs. RHE in 0.5 M LiClO4. Indeed, semiconductor materials with OVs can contribute to a certain NRR activity.56 If combined with other NRR active components like noble Au, the catalytic performance may also improve synergistically. Due to the easy Ce3+/Ce4+ redox cycle and abundant OVs, CeO2 has been widely investigated as a catalyst.57 Zhang's group reported a core–shell structure Au@CeO2 with a small core layer of Au NPs (<10 nm).58 The combination of abundant OVs in the shell layer of CeO2 NPs and small-sized Au NPs in the core layer enhances the NRR performance synergistically. The same group also developed core–shell Au/C NPs (Fig. 5b) by laser ablation, which consists of a spherical Au core and 1–2-layer graphite carbon shell with a large number of defects.59 The ultrathin carbon layer inhibits the aggregation of Au nanospheres effectively during the electrochemical reaction. Alternatively, there are a large number of carbon vacancies in the ultrathin carbon layer, which can provide additional catalytically active sites for the NRR. Hence, the core–shell Au/C NPs can afford an excellent performance (NH3: 241.9 μg h−1 mgcat.−1, FE: 40.5%) at −0.45 V vs. RHE in neutral solution. Apparently, the synergistic effect between the carbon vacancies in the ultrathin graphitic carbon layer and the inner active Au nanospheres enhanced the NRR activity.
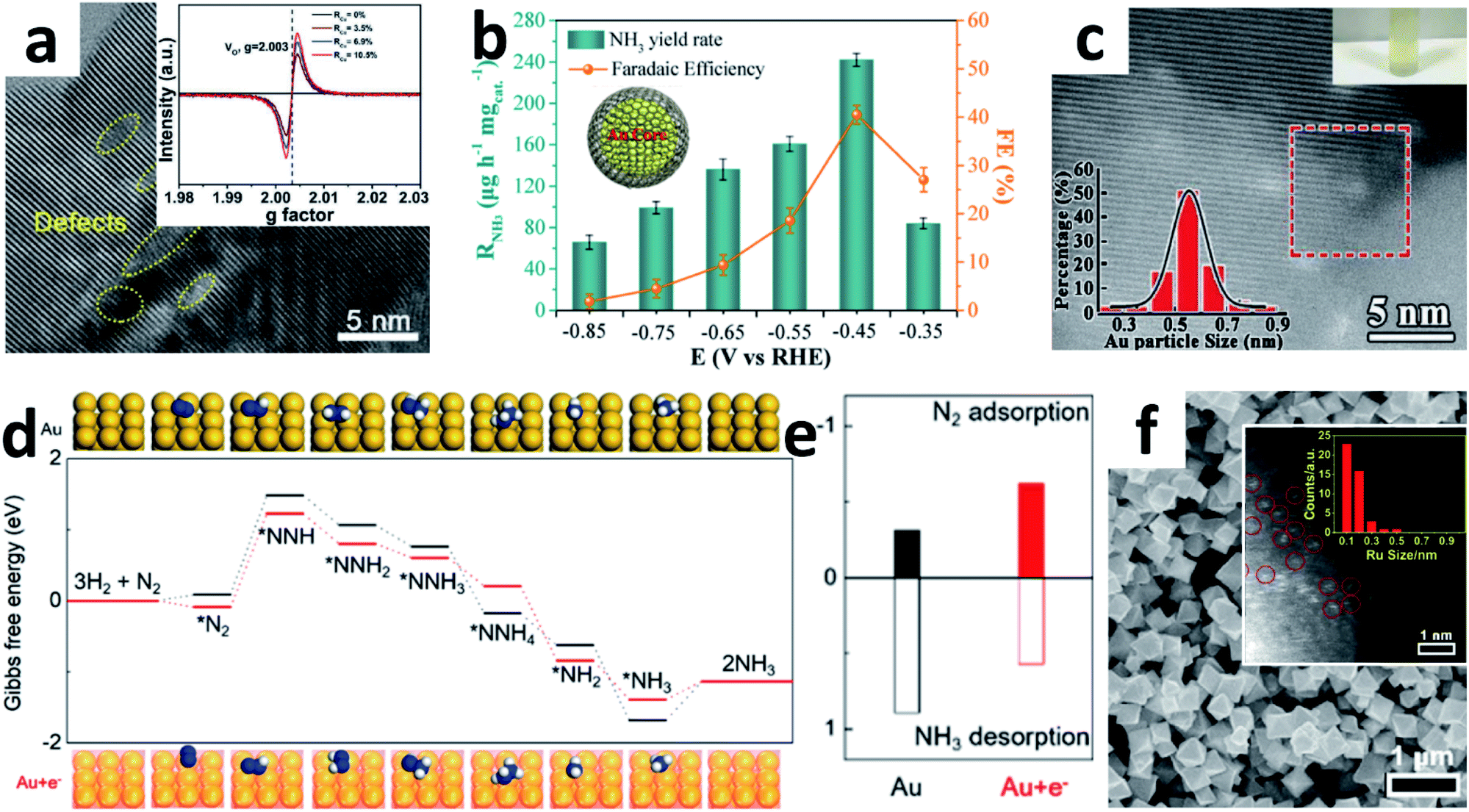 | ||
| Fig. 5 (a) HRTEM image of Cu–TiO2 NPs (inset: EPR spectra of Cu–TiO2 with different Cu contents). Copyright 2020, Wiley-VCH.55 (b) Performance of Au@C at different applied potentials (inset: the structural diagram of Au@C). Copyright 2019, American Chemical Society.59 (c) HAADF-STEM image of the TA-reduced Au/TiO2 (top inset: the photograph of the catalyst suspended in water; bottom inset: Au particle size distribution). Copyright 2017, Wiley-VCH.60 (d) Gibbs free energy diagram of the NRR for Au and Au + e− (insets: the corresponding optimized adsorption structures). (e) The calculated adsorption energies and desorption energies for adsorption onto Au and Au + e−.61 (f) SEM image of Ru@ZrO2/NC (inset: the size-distribution histogram of Ru). Copyright 2019, Elsevier Inc.69 | ||
To utilize Au effectively, Jiang's group embedded Au sub-nanoclusters onto the TiO2 surface and stabilized them with lattice oxygen to form Au–O–Ti bonds. The positively charged Au centre in the Au–O–Ti bond is the active site for forming the chemically adsorbed Au–N2 bond. These small Au sub-nanoclusters (≈0.5 nm) with few coordination sites as isolated precious metal active centres dispersed on the TiO2 support lead to effective and stable NRR performance (Fig. 5c).60 Xue and co-workers constructed inorganic donor−acceptor metal couples to increase electron density.61 The highly coupled Au6/Ni NPs exhibited a high FE of 67.8% at −0.14 V vs. RHE. The author suggested that Ni provides numerous electrons for Au, which boosts the NRR activity of electron-rich Au. DFT calculations further proved that both the Gibbs free energy (ΔG) of N2 dissociation and the desorption energy of NH3 molecules on the electron-rich Au surface were reduced (Fig. 5d and e). Quantum dots (QDs) are nanoclusters that behave like atoms, which can significantly affect the chemical and physical properties of nanomaterials. Taking BP QDs as an example, the ultrafine BP QDs anchored on either MnO2 NSs47 or SnO2−x NTs48 could synergistically promote the NRR. Shao's group developed a 1T-MoS2/g-C3N4 with ultra-small sized MoS2 QDs for the NRR.62 The metallic 1T-MoS2 nanodots anchored on g-C3N4 NSs have abundant active sites and excellent conductivity, thereby achieving a comparable FE of 20.48% under demanding HER conditions.
Single-atom catalysts (SACs) are the ultimate form of 0D NPs, where all the atoms are surface active sites. Due to the homogeneity of the catalytically active sites, low coordination environment and maximal utilization efficiency, SACs have high catalytic activity, stability and selectivity for a series of electrochemical processes.63–66 Some reports have explored the application of SACs anchored on N-doped porous carbon for the NRR.53,67,68 In 2018, Zeng's group prepared single Ru atoms distributed on N-doped C by pyrolyzing a Ru-containing derivative of ZIF-8.49 The isolated Ru atom was dispersed on the whole Ru SAs/N–C and without Ru–Ru bond contribution. The Ru SAs/N–C reached a high NH3 production rate (120.9 μg h−1 mgcat.−1) and FE (29.6%) at −0.2 V vs. RHE. In particular, the FE of Ru SAs/N–C was 2.1 times higher than that of Ru NPs/N–C. Reducing the size of Ru particles to the atomic level should obviously improve efficiency. In addition, Tao et al. developed a thermal pyrolysis method anchoring single Ru atoms onto a Zr-based MOF to suppress the HER.69 They claimed that control of the Ru size by the choice of ligand and the inhibitory effect of ZrO2 on the HER facilitate the dissociation of N2. DFT also showed that the Ru sites with OVs were major active centres for enhancing N2 fixation. Due to these remarkable properties, the Ru@ZrO2/NC catalysts (Fig. 5f) achieved a significant NH3 production rate of 3.665 mg h−1 mgRu−1 at −0.21 V vs. RHE. Moreover, the FEs of Ru@ZrO2/C and Ru@ZrO2/NC were significantly higher than those of Ru/C and Ru/NC, indicating that ZrO2 could probably suppress the HER during the NRR.
3.2 One-dimensional (1D) nanomaterials
As an emerging class of nanomaterials, 1D materials exhibit excellent catalytic activity due to their fast charge transport, large active surface area and rapid mass transport.70–72 Compared with their bulk, 0D and 2D counterparts, the 1D structure has the following characteristics: first, the diameter of the material is limited to the nanoscale, so it shows high atom efficiency while preserving the relatively stable structure. Then, the large number of open spaces between adjacent 1D nanostructures facilitate both chemical accessibility and mass transport of electrolyte deep into the electrode.73 Moreover, for electrocatalysis, the surface of the 1D nanomaterials can greatly facilitate the formation and release of bubbles, thus preventing them from occupying the active sites and hindering subsequent reactions.74–76 The synthetic methods for 1D nanomaterials include electrospinning, electrodeposition, solvothermal reactions, template-assisted methods, and mediated growth.70,71 Thus, advanced 1D nanomaterials including NFs, NWs, NTs and NRs will be briefly reviewed in this section. The performance of some 1D electrocatalysts is summarized in Table 2.| Catalystref. | Electrolyte | NH3 yield (μg h−1 mgcat.−1) | NH3 yield (×10−10 mol s−1 cm−2) | FE (%) | Potential (V vs. RHE) |
|---|---|---|---|---|---|
| Nb2O5 NF77 | 0.1 M HCl | 43.6 | — | 9.26 | −0.55 |
| AuPdP NWs78 | 0.1 M Na2SO4 | 18.78 | 1.23 | 15.44 | −0.30 |
| CBC/Vr-ReSe2@CBC/CBC79 | 0.1 M Na2SO4 | — | 4.62 | 42.5 | −0.25 |
| Ti3+ self-doped TiO2−x NWs80 | 0.1 M Na2SO4 | — | 0.35 | 14.62 | −0.55 |
| C@CoS@TiO2 NFs81 | 0.1 M Na2SO4 | — | 8.09 | 28.60 | −0.55 |
| Fe-doped W18O49 NWs82 | 0.25 M LiClO4 | 24.70 | — | 20.0 | −0.15 |
| Fe–MnO2 (ref. 83) | 0.1 M Na2SO4 | 39.20 | — | 16.80 | −0.29 |
| α-Fe2O3@mTiO2 (ref. 84) | 0.1 M Na2SO4 | 27.20 | — | 13.30 | −0.50 |
| Fe–N/C-CNTs85 | 0.1 M KOH | 34.83 | — | 9.28 | −0.20 |
| CoVP@NiFeV-LDH HHNTs86 | 0.05 M H2SO4 | — | 4.44 | 13.80 | −0.30 |
| Mo2C NRs87 | 0.1 M HCl | 95.10 | — | 8.13 | −0.30 |
| Bi4O11/CeO2 (ref. 88) | 0.1 M HCl | 23.21 | — | 10.16 | −0.20 |
| CNT@C3N4–Fe & Cu89 | 0.25 M LiClO4 | 9.86 | — | 34.00 (−0.80 V) | −1.20 |
| Surface-rough Rh2Sb NRs75 | 0.5 M Na2SO4 | 228.85 | — | 1.50 | −0.45 |
| IrP2@PNPC-NF90 | 0.05 M H2SO4 | 94.00 | — | 17.80 | −0.20 |
The electrospinning method is one of the most convenient methods for constructing complex 1D nanomaterials. Up to now, various 1D NWs and NTs have been synthesized by a spinneret electrospinning process for research on batteries and electrocatalysis.70,91 Yu's group designed an amorphous Bi4V2O11/CeO2 hybrid obtained by electrospinning.88 The OVs and defective sites in Bi4V2O11/CeO2 are able to trap electrons in the metastable state, so that the electrons are injected into the antibonding orbital of the adsorbed N2, thereby weakening the NN triple bond and promoting catalytic activity. Similarly, Sun's group also reported a novel metal oxide synthesized by this method.77 The Nb2O5 NFs were shown to be a superior NRR catalyst with a high NH3 yield (43.60 μg h−1 mgcat.−1 at −0.55 V vs. RHE) and FE (9.26%). DFT calculations show that the electrocatalytic reaction takes place on the active sites on the surface (181), and the key energy barrier could be eliminated by applying a potential of −0.56 V. The same group also reported the Ti3+ self-doped TiO2−x NWs supported on Ti mesh.80 Introducing Ti3+ into the TiO2 surface could decrease the energy barrier of electron transfer and increase the number of active sites to obtain a higher NH3 yield (3.51 × 10−11 mol s−1 cm−2) with 14.62% FE at −0.55 V vs. RHE, compared to TiO2/TM (1.89 × 10−11 mol s−1 cm−2, 6.49%). Wang et al. synthesized AuPdP NWs by simultaneously incorporating Pd and P into a controlled morphology Au particle (Fig. 6a).78 There is a strong interaction between Au, Pd and P in the NWs. Pd, which has a stronger adsorption effect on H, improves the H adsorption on the Au surface;92 and the incorporation of P provides more valence electrons, which is beneficial to the selective hydrogenation of N2 to NH3. Thus, AuPdP NWs achieved an NH3 yield of 18.78 μg h−1 mgcat.−1 and FE of 15.44% at −0.3 V vs. RHE. Although defect engineering has been repeatedly proven as a rational strategy to enhance electrocatalytic performance, it has poor selectivity between the NRR and HER. The side-effects of the HER mean that the NRR is still far from commercialization. Recently, Liu et al. reported a novel “sandwich” structure, CBC/Vr-ReSe2@CBC/CBC (CBC = carbonized bacterial cellulose, Vr-ReSe2 = vacancy-rich ReSe2).79 The surface of the hydrophobic CBC layer resists the intimate contact between water molecules and Vr-ReSe2@CBC NFs, and slightly increases the adhesive force to N2 bubbles, so the FE is boosted by 32% at −0.25 V vs. RHE compared with Vr-ReSe2@CBC without the CBC sandwich structure.
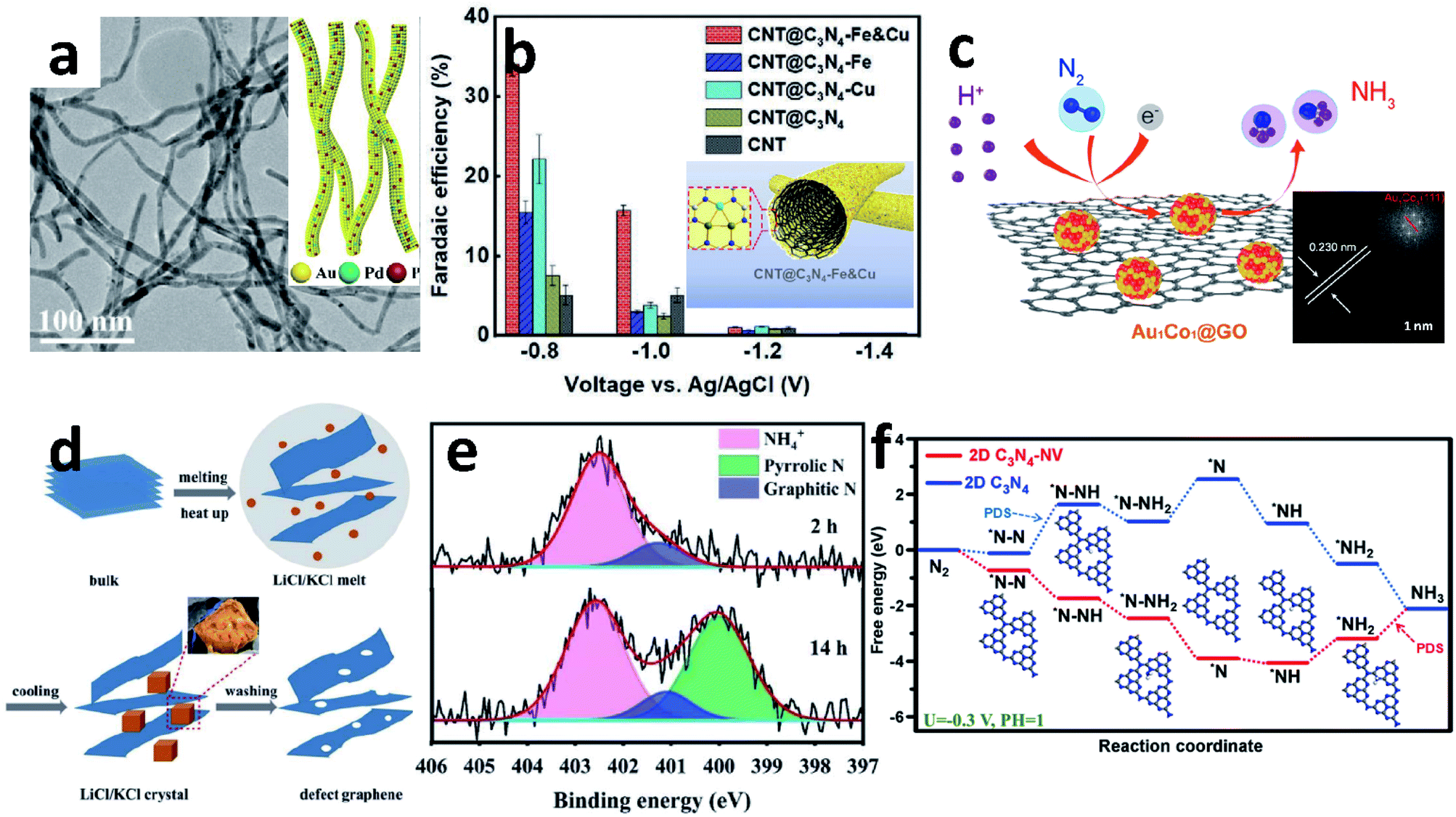 | ||
| Fig. 6 (a) TEM image of AuPdP NWs (inset: schematic diagram of AuPdP NWs). Copyright 2019, American Chemical Society.78 (b) The FE of different materials at different potentials (inset: schematic diagram of CNT@C3N4-Fe & Cu). Copyright 2020, Wiley-VCH.89 (c) Schematic diagram of the NRR of Au1Co1@GO (inset: HRTEM image of Au1Co1@GO). Copyright 2019, American Chemical Society.117 (d) Schematic diagram of defect graphene (DG) synthesis. (e) N 1s spectra of DG-800 after reaction. Copyright 2020, The Royal Society of Chemistry.131 (f) Gibbs free energy of C3N4 and C3N4-NV for the NRR. Copyright 2021, Elsevier B.V.138 | ||
NRs are the most common 1D morphology because they contain exposed stepped faces, remarkably boosting their catalytic performance.93–96 In 2017, Ren et al. demonstrated Mo2N NRs as an efficient NRR electrocatalyst.97 According to the DFT calculations, after nitrogenation, the ΔG of the potential determining step of the NRR on MoO2 decreased dramatically. Later, this group developed MoC NRs based on Mo2N, which was synthesized by pyrolyzing a Mo3O10(C6H8N)2·2H2O precursor in an Ar atmosphere. The FE of MoC is 1.8-fold that of Mo2N.87 Porous FeVO4 NRs have been reported as an effective NRR catalyst contributing to the dual active centres.98 The dual active centres of the Fe2c-V2c dimer can jointly back-donate their 3d-electrons into the antibonding 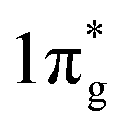 orbital of *N2, and this process strongly polarizes and activates N2, increasing the NH3 yield up to 52.8 μg h−1 mg−1. Moreover, coordination of Fe and Cu also enhanced the NRR. From Chu's report, the ordered sub-nano space in the surface cavities of CNT@g-C3N4 can accommodate several Fe and Cu atoms, forming a sub-nano reactor with tight coordination between Fe and Cu.89 Compared to their single-metal counterparts, the coordination between Cu and Fe can accelerate the adsorption of N2 synergistically to optimize the reaction path and lower the energy barrier, thereby improving the FE to up to 34% (Fig. 6b).
orbital of *N2, and this process strongly polarizes and activates N2, increasing the NH3 yield up to 52.8 μg h−1 mg−1. Moreover, coordination of Fe and Cu also enhanced the NRR. From Chu's report, the ordered sub-nano space in the surface cavities of CNT@g-C3N4 can accommodate several Fe and Cu atoms, forming a sub-nano reactor with tight coordination between Fe and Cu.89 Compared to their single-metal counterparts, the coordination between Cu and Fe can accelerate the adsorption of N2 synergistically to optimize the reaction path and lower the energy barrier, thereby improving the FE to up to 34% (Fig. 6b).
3.3 Two-dimensional (2D) nanomaterials
2D materials have a single and fully exposed crystal surface and unique electronic state, arousing enormous interest over a wide field of energy and storage.99,100 Compared with their bulk, 0D and 1D counterparts, the 2D structure has the following characteristics: first, the 2D nanomaterials have exceptional flexibility and mechanical strength due to nanoscale thickness and strong in-plane covalent bonding. Then, thanks to their ultra-thin thickness and large transverse dimensions, 2D materials have a high specific surface area and surface atomic exposure. Moreover, for surfaces that expose a large number of atoms, researchers can design 2D nanomaterials for surface modification, elemental doping or vacancy engineering at the atomic level. Especially in recent years, various 2D nanomaterials (such as NSs, MXene, G, and C3N4) have been developed and utilized in the NRR electrocatalysis. The synthetic methods for 2D nanomaterials are mainly solvothermal reactions, CVD, template-assisted methods, topochemical transformations, salt-assisted synthesis, and self-assembly methods.100–103 The performance of some 2D electrocatalysts is summarized in Table 3.| Catalystref. | Electrolyte | NH3 yield (μg h−1 mgcat.−1) | NH3 yield (×10−10 mol s−1 cm−2) | FE (%) | Potential (V vs. RHE) |
|---|---|---|---|---|---|
| Co-FePS3 NSs118 | 0.1 M KOH | 90.60 | — | 3.38 | −0.40 |
| Sn/SnS2 (ref. 119) | 0.1 M PBS | 23.8 | — | 3.40 | −0.80 |
| B4C NSs120 | 0.1 M Na2SO4 | 26.57 | — | 15.95 | −0.75 |
| Bi NS105 | 0.1 M Na2SO4 | 13.23 | 0.42 | 10.46 | −0.80 |
| FeS2-Mo17.3 NSs121 | 0.1 M KOH | 26.15 | — | 14.41 | −0.20 |
| Few-layer black phosphorus NSs122 | 0.01 M HCl | 31.37 | — | 3.09 | −0.70 |
| Crystalline red phosphorus nanoribbons109 | 0.1 M Na2SO4 | 15.40 | — | ≈3.00 | −0.40 |
| IrP2@PNPC nanofilm90 | 0.05 M H2SO4 | 94.00 | ≈10.00 | −0.20 | |
| 1T-MoS2/g-C3N4 (ref. 62) | 0.1 M HCl | 29.97 | — | 20.48 | −0.30 |
| Hydroxyl-rich Ti3C2Tx QDs123 | 0.1 M HCl | 62.94 | — | 13.30 | −0.50 |
| Au/Ti3C2 (ref. 124) | 0.1 M HCl | 30.06 | — | 18.34 | −0.20 |
| Rh2P@NPC125 | 0.1 M Na2SO4 | 37.60 | — | 7.64 | −0.25 |
| Au1Co1@GO117 | 0.5 M K2SO4 | 36.82 | — | 22.03 | −0.20 |
| CoO QD/RGO111 | 0.1 M Na2SO4 | 21.50 | — | 8.30 | −0.60 |
| CoS2/NS-G126 | 0.05 M H2SO4 | 25.00 | — | ≈6.00 | −0.20 |
| rGO/Fe@Fe3O4/CP127 | 0.2 M NaHCO3 | — | 1.30 | 6.25 | −0.30 |
| Boron-doped graphene (BG)128 | 0.05 M H2SO4 | — | 1.60 | 10.80 | −0.50 |
Nørskov et al. suggested using a less reactive main group element, semiconductor or thin insulator, to suppress the HER process and enhance the FE.104 Qiao's group reported a semiconducting Bi NS with effective p-orbital electron delocalization and abundant exposure of edge sites, which exhibits a high NRR electrocatalytic activity.105 Notably, materials with HER characteristics also promote the NRR. Wang's group developed ultrathin carbon NSs with Mo2C DQs, which have high reactivity for both the NRR and HER.106 Unlike the back-donation process of the transition metal complex, the B atoms have the characteristics of monovalent borylene, and the N2 unit can be used as a Lewis s-donor and p-receptor.107 In 2019, Zhang et al. developed a novel boron NS (BNS), whose B atoms were oxidized and H deactivated as a NRR electrocatalyst with an NH3 production rate of 13.22 μg h−1 mgcat.−1.108 Compared with traditional NSs, 2D nanoribbons have stronger size confinement effects. Recently, Liu et al. reported triclinic crystalline red phosphorus (cRP) nanoribbons as a large-scale synthetic nanocatalyst for the NRR in 0.1 M Na2SO4.109
As an ideal substrate to load electrocatalysts, graphene (G) has a large specific surface area and abundant material attachment sites, which can make the material grow evenly on the G surface to avoid agglomeration. Many G-based materials have been reported for the NRR.110–114 Recent work by Chu et al. developed an advanced electrocatalyst, CuO/RGO, which was synthesized by a microwave-assisted solvothermal method.115 The CuO/RGO exhibited a high NH3 yield of 1.8 × 10−10 mol s−1 cm−2 and FE of 3.9% at −0.75 V vs. RHE in 0.1 M Na2SO4, far outperforming the bare CuO or RGO alone. The highly stable and efficient mode for the N2 adsorption activity of CuO was attributed to the (111) facet of surface end groups. Similarly, a-Au/CeOx-RGO achieved a comparable NH3 performance due to the amorphous Au, which has more structural distortion and a higher concentration of catalytic NRR active sites.116 The alloyed Au1Co1 NPs dispersed on GO can also synergistically boost the activation of the NN bond, and its synergistic effect is mainly reflected in Au1Co1 (111) (Fig. 6c).117
G-Based materials are always modified by the introduction of defects129 or heteroatoms130 to improve their usage. Defective G disrupts the original electronic arrangement and improves the transfer of electrons from the electrocatalyst to the chemisorbed molecules. He's group prepared an ultrathin porous dopant-free defect G using the molten salt method (Fig. 6d).131 It is noteworthy that the defect G utilizes the product NH3 from the NRR instead of N2 as the N source for N doping (Fig. 6e). G can also be activated by lone pairs of electrons in electron-rich dopants, or by empty orbitals in electron-deficient dopants, thereby destroying the integrity of π-conjugation.132,133 Boron (B) is an effective doping example, which can cause electron deficiency in G and thus increase electrocatalytic activity.134,135 The local electron-deficient environment at the B-doped site provides a powerful binding site for the Lewis base, leading to improved electrocatalytic activity. Recently, Zheng's group reported B-doped G. In this work, the distribution structures and contents of B have distinct effects on NH3 yield and FE. Among the structure of BC3, BC2O and BCO2, the G-like BC3-type bond plays a key role in enhancing N2 fixation. At a doping level of 6.2%, the B-doped G achieves an NH3 production rate of 9.8 μg h−1 cm−2 and a remarkable FE of 10.8% at −0.5 V vs. RHE in aqueous solutions. B doping in the G framework leads to the redistribution of electron density, where the electron-deficient boron sites provide enhanced binding capability to N2 molecules.128 Wang's group developed B, N co-doped G with abundant defects by thermal treatment of GO and boric acid under an NH3 atmosphere followed by annealing. Edge carbon atoms occurring near B–N pairs were found to be the active sites for the NRR. Thus, the NRR with a high NH3 production rate (7.75 μg h−1 mgcat.−1) and excellent FE (13.79%) was achieved at −0.3 V vs. RHE.136
Analogous to G, the crystal structure of graphitic carbon nitride (C3N4) may be described as a hexagonal carbon framework with N-substituted carbon by the sp2 hybridization of C and N atoms. C3N4 and related materials possess evenly distributed holes, a tunable electronic structure and uniform pyridinic-N coordinators rich in lone electron pairs able to capture metal ions in the ligands. C3N4 has a more negative conduction position relative to the reduction potential of N2/NH3, which favours the activation of adsorbed N2.137 The 2D C3N4-NV, which has a larger specific surface area and abundant NVs, has a lower reaction ΔG (Fig. 6f).138 This is because NVs can enrich electrons and transfer them to the 2π* orbital of N2, thereby enhancing the adsorption effect.
MXenes have been widely reported in the electrocatalysis field due to their diverse metal active centres and abundant surface end groups.139–141 For example, Ti3C2Tx (T = F, OH),142 Ti3C2Tx (T = O, OH),143 MXene/TiFeOx-700,144 and Ti3C2OH QDs123 achieved good NRR activities by modifying the surface end groups. Li and co-workers developed small sized fluorine-free Ti3C2Tx (T = O, OH) NSs for the NRR, synthesized by TMAOH intercalation and delamination.143 The size effect and fluorine-free characteristics result in an NH3 yield 1.8 times that of fluorine-based treatment. Tan's group reported a SAC Ru doped Mo2CTx NS material for the NRR in 0.5 M K2SO4, which increased the FE to 25.77%.145 Among them, the SAC Ru is the main electron back-donation centre for the absorbance and activation of N2 molecules.
4 Modification of nanomaterials
4.1 Forming alloy centres
N2 activation is a complicated process. Although the single active site can activate N to a certain extent, it cannot balance well the adsorption energy of multiple reaction intermediate species. Extending the single active site to dual or multiple sites may further improve the overall NRR performance of the catalyst.Bimetallic nanocatalysts have been introduced into NRR systems to enhance selectivity for N2. In 2001, Jens K. Nørskov and co-workers reported a bimetallic ammonia synthesis strategy that used interpolation to find a rational catalyst in the periodic table.146 They combined metals with high and low N2 interaction energies to obtain the required interaction strength for preparation of NH3 synthesis catalysts. By the introduction of N atoms to order the binary alloy, bimetallic catalysis could be achieved. Yu's group developed an amorphous BiNi alloy for the NRR, which was designed to activate N2 molecules by optimizing the adsorption energy and adjusting the crystallinity.147 The BiNi alloy has a significantly smaller area-specific impedance than Bi (Fig. 7c), indicating that the 3D nanoporous alloy framework promotes charge transport, thereby enhancing the NRR activity.
 | ||
| Fig. 7 (a) Schematic depicting the functions of Pt/Au@ZIF. (b) XPS investigations on the Pt/Au@ZIF platform. Copyright 2020, Wiley-VCH.36 (c) EIS spectra of Bi and BiNi. Copyright 2020, Wiley-VCH.147 (d) Schematic illustration of a possible mechanism of RuFeCoNiCu NPs. Copyright 2020, Wiley-VCH.148 | ||
For metal–carbon-based materials, bimetallic alloying has been employed to alter such weak metal–carbon interactions into synergistic metal–metal interactions, thereby optimizing the electronic structure of monometals and accordingly, promoting the catalytic activity. Taking Pt/Au@ZIF as an example, ZIF electronically modified the d-band structure of the Pt/Au electrocatalyst and created electron deficient sites to achieve more favourable N adsorption of H (Fig. 7a).36 According to the XPS spectra, after encapsulating the ZIF, the binding energy of Pt 4f shifts left and that of N 1s shifts right, clearly highlighting the electron density transfer from Pt to the ZIF framework by the Pt–N ZIF interaction (Fig. 7b). This electron migration from Pt to ZIF will form electron-deficient sites on the Pt surface, thereby forming N2 adsorption sites. Moreover, the hydrophobic ZIF pores act as a barrier that prevents trace amounts of water from reaching the electrocatalytic sites, thereby inhibiting the HER. As a result, Pt/Au@ZIF has a FE >44 times that of Pt/Au (≈1%), and reaches a high NH3 production rate of >161 μg h−1 mgcat.−1 under ambient conditions. AuCu/ZIF-8 was also reported for the NRR due to the large active surface areas and conductive carbon frameworks of the ZIF-8 precursor. The highly dispersed bimetallic active sites achieved an unprecedented NH3 yield of 23.3 μg h−1 mgcat.−1.35
Due to the unique characteristics such as corrosion resistance under harsh conditions and adjustable performance (shorter distance between active sites facilitates component optimization), high-entropy alloys (HEAs) have also been applied in the NRR. The HEAs are synthesized by controlling the coordination entropy within 5 or more metal elements. Wang's group reported HEA RuFeCoNiCu NPs as a pH-universal NRR catalyst. When multiple H atoms are adsorbed, they are mainly distributed over the hollow positions, and the adsorption energy decreases rapidly with the H coverage.148 Therefore, the surface H adsorption will affect the Co–Ni–Ni sites, but the optimal site of the NRR, t-Fe, is not at a hollow position and will not be affected significantly. Clearly, different metals may play different roles and work in a synergetic way to provide outstanding NRR performance (Fig. 7d). This catalyst has a surprising NH3 yield of 57.1 μg h−1 mgcat.−1 at 0.05 V vs. RHE in 0.1 M KOH, and the corresponding FE is 38.5%. It also has excellent NRR activity in 0.1 M Li2SO4, 0.1 M Na2SO4, and 0.1 M HCl electrolytes.
4.2 Optimizing the valence state
N2 activation is essentially an electron donating–accepting process between the catalyst surface and N2 molecules. The different valence states for the active centre atoms will affect the energy barrier of the activation of N2 molecules. Although both NbO2 and Nb2O5 have similar octahedral structures, the catalytic properties differ significantly due to the different oxidation states of Nb (Fig. 8a). According to the report by Zheng's group, the FE of NbO2 is almost 3.5 times higher than that of Nb2O5. For Nb2O5, the charge exchange and transfer mainly occur between the Nb atoms and N2, and the electrons on the adjacent Nb atoms are transferred back to weaken the NN bond while strengthening the metal–N bond.107 For NbO2, the Jahn–Teller distortion destroys degeneracy, causing the t2g level to split into partially occupied dxy orbitals and partially unoccupied dyz and dxz orbitals. Therefore, when N2 molecules are adsorbed on NbO2, they not only bind electrons from its 3σg orbital to the empty orbital of Nb, but also accept d-electrons of Nb and back-donate to its anti-bonding 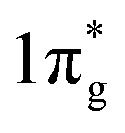 orbital (Fig. 8b). This back-donation can effectively weaken the NN bond and activate the adsorbed N2 molecules. For Nb4+ with an electron occupying a 4d orbital, it should be easier to form such a back donor than Nb5+ without 4d electrons, so it exhibits better N2 fixation activity.24 Moreover, introducing mixed valences into nanomaterials to modulate the vacancy concentration and metal defect states could also induce the NRR process. Sun's group introduced Cu ions with mixed valences of +1 and +2 to modulate the defect states in TiO2. Theoretical calculations suggest that when the Ti3+ 3d1 defect states present the splitting of eg and t2g orbitals, the adsorption and activation of N2 enhanced strongly.55
orbital (Fig. 8b). This back-donation can effectively weaken the NN bond and activate the adsorbed N2 molecules. For Nb4+ with an electron occupying a 4d orbital, it should be easier to form such a back donor than Nb5+ without 4d electrons, so it exhibits better N2 fixation activity.24 Moreover, introducing mixed valences into nanomaterials to modulate the vacancy concentration and metal defect states could also induce the NRR process. Sun's group introduced Cu ions with mixed valences of +1 and +2 to modulate the defect states in TiO2. Theoretical calculations suggest that when the Ti3+ 3d1 defect states present the splitting of eg and t2g orbitals, the adsorption and activation of N2 enhanced strongly.55
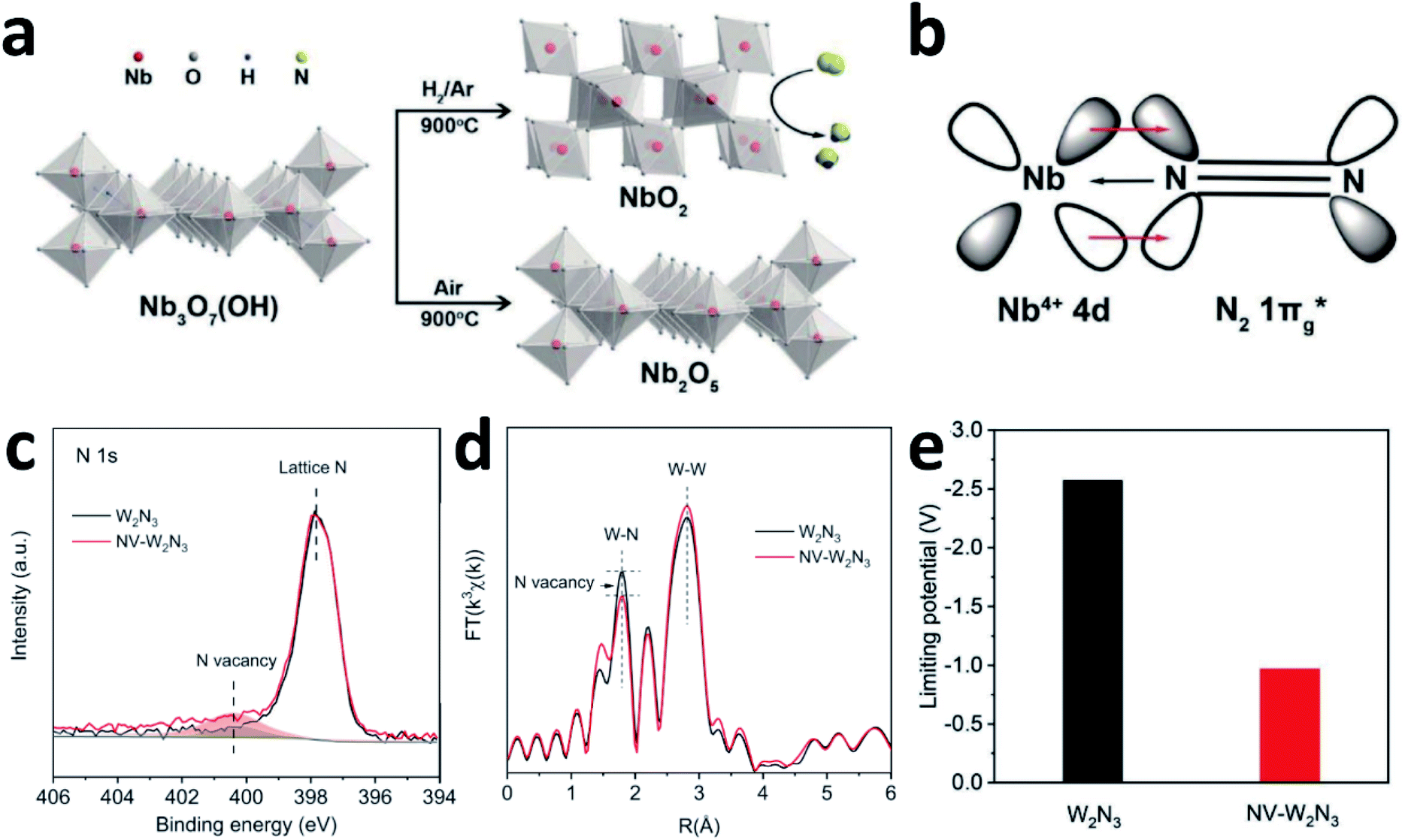 | ||
| Fig. 8 (a) Synthetic schematic of NbO2 and Nb2O5. (b) The interaction between Nb4+ and N2. Copyright 2018, Wiley-VCH.107 (c) N 1s spectra and (d) FT-EXAFS plots of the pristine W2N3 and NV-W2N3. (e) Magnitudes of the theoretical limiting potentials of NH3 production on W2N3 and NV-W2N3. Copyright 2019, Wiley-VCH.159 | ||
4.3 Introducing vacancies
For a long time, surface chemical modifications like defect engineering and heteroatom doping have been regarded as efficient methods to boost NRR activity, due to the electronic structures of the electrochemical catalytic surfaces resulting in the modulation of adsorption properties.149,150 Some introduced vacancy nanomaterials are summarized in Table 4.| Catalystref. | Electrolyte | NH3 yield (μg h−1 mgcat.−1) | NH3 yield (×10−10 mol s−1 cm−2) | FE (%) | Potential (V vs. RHE) |
|---|---|---|---|---|---|
| OVs-Bi-VO4 (ref. 151) | 0.2 M Na2SO4 | 8.60 | — | 10.04 | −0.50 |
| OVs-MoO2 (ref. 152) | 0.1 M HCl | ∼12.20 | — | 8.20 | −0.15 |
| Bo-OV-MnO2 (ref. 153) | 0.5 M LiClO4 | 54.2 | — | 16.80 (−0.20 V) | −0.40 |
| Au@CeO2 (ref. 58) | 0.01 M H2SO4 | 10.60 | 4.61 | 9.50 | −0.40 |
| Al-doped Co3O4 (ref. 154) | 0.1 M KOH | — | 0.65 | 6.25 | −0.20 |
| OVs-CeO2 (ref. 155) | 0.1 M Na2SO4 | 16.4 | — | 3.70 (−0.40 V) | −0.50 |
| Polymeric CN-NVx (ref. 156) | 0.1 M HCl | 8.09 | — | 11.59 | −0.20 |
| NVs-CN/C600 (ref. 157) | 0.1 M HCl | 2.9 | — | 16.8 | −0.30 |
| S-NVs-C3N4 (ref. 158) | 0.5 M LiClO4 | 32.7 | — | 14.1 | −0.40 |
| NVs-W2N3 NSs159 | 0.1 M KOH | 11.66 | 0.38 | 11.67 | −0.20 |
| C3N4-NVs138 | 0.1 M HCl | 17.85 | — | 10.96 | −0.30 |
| Vs-FePS3 NSs160 | 0.1 M HCl | 38.8 | 0.63 | 12.36 (−0.20 V) | −0.25 |
| B-doped VS2 (ref. 161) | 0.5 M LiClO4 | 55.7 | — | 16.40 (−0.20 V) | −0.40 |
| Mo–SnS2-SVs162 | 0.5 M LiClO4 | 41.3 | — | 20.80 (−0.40 V) | −0.50 |
| MoS3-SVs163 | 0.5 M LiClO4 | 51.7 | — | 12.8 | −0.30 |
| MoS2-SVs164 | 0.1 M HCl | 23.38 | — | 17.9 | −0.35 |
| Sb2S3-SVs165 | 0.1 M Na2SO4 | 10.85 | — | 3.75 (−0.30 V) | −0.40 |
| N-doped MoS2 (ref. 166) | 0.1 M Na2SO4 | 69.82 | — | 9.14 | −0.30 |
| CoS1−x (ref. 167) | 0.05 M H12SO4 | ∼12.1 | — | ∼16.5 | −0.15 |
| SV-1T-MoS2@MoO3 (ref. 168) | 0.05 M H2SO4 | 116.1 | — | 18.9 | −0.20 |
| SVs-ZnS-rGO169 | 0.1 M HCl | 51.2 | — | 28.2 (−0.10 V) | −0.15 |
In contrast, Zhang's work anchored Fe atoms into OVs and doped Fe on the lattice, enhancing the adsorption and activation of N2 on SnO2 and improving the electrical conductivity of SnO2, respectively.170 This Fe–SnO2 catalyst had a unique surface structure. In state of metallic Fe NPs or clusters, the elemental Fe in Fe–SnO2 is composed of oxygen vacancy-anchored SAC Fe and lattice doped Fe, which largely lowered the energy barrier for activating chemically inert N2 molecules. The electron paramagnetic resonance (EPR) spectra confirmed that many OVs existed within SnO2 and Fe–SnO2. However, the peak intensities of O 1s in the XPS and EPR spectra clearly decreased after Fe doping, which indicates that Fe may have been anchored by OVs. The extended X-ray absorption fine structure (EXAFS) of the catalyst showed a first-shell Fe–O contribution with a coordination number of about 3.6 at 2.01 Å, and a first-shell Fe–Sn contribution with a coordination number of about 0.4 at 3.12 Å, corresponding to lattice doped Fe. Moreover, the first-shell Fe–Sn contribution with a coordination number of about 4.4 at 3.87 Å can be ascribed to Fe anchored at OVs, forming a Sn–Fe–Sn structure. Therefore, the elemental Fe in Fe–SnO2 can be categorized into isolated single-atom Fe on the surface with OVs, and lattice doped Fe. Consequently, the Fe–SnO2 catalyst exhibited superior catalytic performance with a high NH3 yield of 82.7 μg h−1 mgcat.−1 and a high FE of 20.4%.
Qiao's group experimentally and theoretically studied the NVs on W2N3, which acts as an efficient and stable NRR catalyst.159 NV-W2N3 NSs were prepared by annealing under an NH3 atmosphere, and the NVs were generated by further annealing (5% H2/Ar at 500 °C for 3 h). According to the XPS N 1s spectra and EXAFS (Fig. 8c and d), compared to the pristine W2N3 NSs, the intensity of the peak (NVs) in NV-W2N3 increased and the coordination number of the W–N bond decreased, proving that vacancies were created. The authors also claimed that the high valence state of W and the surface distortion induced by the 2D morphology contribute to the good stability of NV-W2N3. Based on the pristine W2N3 theoretical model, they investigated the effect of introducing NVs on N2 activation. When N vacancies are involved, significant charge transfer is observed between W2N3 and the adsorbed N2 molecule. More importantly, inducing NVs decreases the theoretical limiting potentials of NH3 production significantly (Fig. 8e), which is convenient for the NRR.
N bond.
5 Summary and outlook
Due to the urgent need to solve energy problems and the great attraction of finding new energy materials, the NRR is considered to be the most effective strategy for industrial production of NH3 under mild conditions. Over the past five years, researchers from various countries have explored some effective and promising active centres to boost the adsorption and activation of N2, combined with some low-dimensional nanomaterial substrates with high specific surface area, abundant exposed active sites, and unique electronic morphology to improve the performance of the NRR. In this review, we have briefly discussed some advanced low-dimensional nanomaterials with various structures in the development of the NRR, and also mentioned approaches to catalyst design and characteristics. In pursuit of more practical NRR catalysts with higher activity and selectivity, we provide some prospects for future research.(1) Combination of theoretical calculations and some advanced characterization techniques (like NMR, XAFS, etc.) with experiments. A series of catalysts has been obtained using the results of theoretical calculations. Moreover, the adsorption energy of different sites on the catalyst surface could be explored by atomic-scale NMR technology. Using these two methods to design NRR catalysts in a targeted manner can save researchers time in screening catalysts.
(2) Improvement of the material preparation method. Constructing small-sized catalysts with diatomic or polyatomic active centres and combining them with low-dimensional nano-matrices. The diatomic or polyatomic active centres can reasonably regulate the adsorption process of each intermediate product in the N2 activation process. Compounding with low-dimensional nanosized substrates can effectively adjust the exposed crystal faces of the catalyst, which is convenient for researchers to screen for active crystal faces. High-porosity 3D nanomaterials also have comparable potential as suitable substrate supported catalysts.
(3) Some elements with mixed valences are selected as the components of the catalyst to adjust the defect concentration and metal defect valences. N2 activation is essentially an electron donor–acceptor process between the catalyst surface and N2 molecules. The different valences of the active centre atoms will affect the energy barrier for activation of N2 molecules.
(4) Designing a strategy that combines defect engineering and heteroatom doping. Defect engineering and heteroatom doping can change the electronic structure of the electrochemical catalyst surface, enhance the electron cloud density of the active site, and promote the adsorption and activation of N2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation Committee of China (Distinguished Youth Scientists Project of 51026004), Research Foundation of State Key Lab (ZK201805 and ZK201717), Jiangsu Distinguished Professorship Program (2016) and Sanyo Chemical Co. Ltd is gratefully acknowledged.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- G. Soloveichik, M. Acharya, H. Cheeseman, D. Wicks and D. Tew, Renewable Energy to Fuels through Utilization of Energy-Dense Liquids (REFUEL), U.S. DOE, 2016 Search PubMed.
- V. Rosca, M. Duca, M. T. d. Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1055–1074 CrossRef CAS.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- R. Schlgl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- B. E. Smith, R. L. Richards and W. E. Newton, Catalysts for Citrogen Fixation, 2004 Search PubMed.
- J. A. Pool, E. Lobkovsky and P. J. Chirik, Nature, 2004, 427, 527–530 CrossRef CAS PubMed.
- Y. Wang, M. Craven, X. Yu, J. Ding, P. Bryant, J. Huang and X. Tu, ACS Catal., 2019, 9, 10780–10793 CrossRef CAS PubMed.
- H. Wang, H. Li, M. Zhang, Y. Song, J. Huang, H. Huang, M. Shao, Y. Liu and Z. Kang, ACS Appl. Mater. Interfaces, 2018, 10, 16308–16314 CrossRef CAS PubMed.
- D. L. Nelson and M. M. Cox, Chem. Educ., 2001, 6, 69–70 CrossRef.
- B. A. MacKay and M. D. Fryzuk, Chem. Rev., 2004, 104, 385–402 CrossRef CAS PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
- J. Chatt, A. J. Pearman and R. L. Richards, Nature, 1975, 253, 39–40 CrossRef CAS.
- V. A. A. Sclafani and M. Schiavello, J. Electrochem. Soc., 1982, 130, 735–737 Search PubMed.
- M. Kitano, J. Kujirai, K. Ogasawara, S. Matsuishi, T. Tada, H. Abe, Y. Niwa and H. Hosono, J. Am. Chem. Soc., 2019, 141, 20344–20353 CrossRef CAS PubMed.
- M. Hattori, S. Iijima, T. Nakao, H. Hosono and M. Hara, Nat. Commun., 2020, 11, 2001 CrossRef CAS PubMed.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- C. J. van der Ham, M. T. Koper and D. G. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- G. Hochman, A. S. Goldman, F. A. Felder, J. M. Mayer, A. J. M. Miller, P. L. Holland, L. A. Goldman, P. Manocha, Z. Song and S. Aleti, ACS Sustainable Chem. Eng., 2020, 8, 8938–8948 CrossRef CAS.
- H. Davy, Philos. Trans. R. Soc. London, 1807, 97, 1–56 Search PubMed.
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS.
- R. Silverstein and W. A. Bulen, Biochemistry, 1970, 9, 3809–3815 CrossRef CAS PubMed.
- L. Huang, J. Wu, P. Han, A. M. Al-Enizi, T. M. Almutairi, L. Zhang and G. Zheng, Small Methods, 2019, 3, 1800386 CrossRef.
- J. Feng and H. Pan, J. Mater. Chem. A, 2020, 8, 13896–13915 RSC.
- X. F. Li, Q. K. Li, J. Cheng, L. Liu, Q. Yan, Y. Wu, X. H. Zhang, Z. Y. Wang, Q. Qiu and Y. Luo, J. Am. Chem. Soc., 2016, 138, 8706–8709 CrossRef CAS PubMed.
- M. A. Shipman and M. D. Symes, Catal. Today, 2016, 286, 57–68 CrossRef.
- J. Humphreys, R. Lan and S. Tao, Advanced Energy and Sustainability Research, 2020, 2, 2000043 CrossRef.
- H. Zhou, B. Xiong, L. Chen and J. Shi, J. Mater. Chem. A, 2020, 8, 20286–20293 RSC.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, Angew. Chem., Int. Ed., 2020, 59, 10479–10483 CrossRef CAS PubMed.
- S. Liu, T. Qian, M. Wang, H. Ji, X. Shen, C. Wang and C. Yan, Nat. Catal., 2021, 4, 322–331 CrossRef CAS.
- B. L. Li, M. I. Setyawati, H. L. Zou, J. X. Dong, H. Q. Luo, N. B. Li and D. T. Leong, Small, 2017, 13, 1700527 CrossRef PubMed.
- X. Yang, F. Ling, J. Su, X. Zi, H. Zhang, H. Zhang, J. Li, M. Zhou and Y. Wang, Appl. Catal., B, 2020, 264, 118477 CrossRef CAS.
- X. Lv, L. Wang, G. Wang, R. Hao, J. Ren, X. Liu, P. N. Duchesne, Y. Liu, W. Li, Z. Yuan and G. A. Ozin, J. Mater. Chem. A, 2020, 8, 8868–8874 RSC.
- H. Y. F. Sim, J. R. T. Chen, C. S. L. Koh, H. K. Lee, X. Han, G. C. Phan-Quang, J. Y. Pang, C. L. Lay, S. Pedireddy, I. Y. Phang, E. K. L. Yeow and X. Y. Ling, Angew. Chem., Int. Ed., 2020, 59, 16997–17003 CrossRef CAS PubMed.
- C. Chen, C. Liang, J. Xu, J. Wei, X. Li, Y. Zheng, J. Li, H. Tang and J. Li, Electrochim. Acta, 2020, 335, 135708 CrossRef CAS.
- Y. Liu, L. Huang, X. Zhu, Y. Fang and S. Dong, Nanoscale, 2020, 12, 1811–1816 RSC.
- H. Xian, H. Guo, Z. Chen, G. Yu, A. A. Alshehri, K. A. Alzahrani, F. Hao, R. Song and T. Li, ACS Appl. Mater. Interfaces, 2020, 12, 2445–2451 CrossRef CAS PubMed.
- C. Wang, X. Zhu and P. Zuo, Chem. Eng. J., 2020, 396, 125163 CrossRef CAS.
- T. Xu, D. Ma, C. Li, Q. Liu, S. Lu, A. M. Asiri, C. Yang and X. Sun, Chem. Commun., 2020, 56, 3673–3676 RSC.
- S. Luo, X. Li, M. Wang, X. Zhang, W. Gao, S. Su, G. Liu and M. Luo, J. Mater. Chem. A, 2020, 8, 5647–5654 RSC.
- W. Xu, G. Fan, J. Chen, J. Li, L. Zhang, S. Zhu, X. Su, F. Cheng and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 3511–3516 CrossRef CAS PubMed.
- W. Tong, B. Huang, P. Wang, L. Li, Q. Shao and X. Huang, Angew. Chem., Int. Ed., 2020, 59, 2649–2653 CrossRef CAS PubMed.
- S. Xiao, F. Luo, H. Hu and Z. Yang, Chem. Commun., 2020, 56, 446–449 RSC.
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Energy Mater., 2018, 8, 1801357 CrossRef.
- C. Wang, J. Gao, J. Zhao, D. Yan and X. Zhu, Small, 2020, 16, 1907091 CrossRef CAS PubMed.
- Y. T. Liu, D. Li, J. Yu and B. Ding, Angew. Chem., Int. Ed., 2019, 58, 16439–16444 CrossRef CAS PubMed.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, e1803498 CrossRef PubMed.
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. R. Zheng, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2020, 59, 13423–13429 CrossRef CAS PubMed.
- J. Li, S. Chen, F. Quan, G. Zhan, F. Jia, Z. Ai and L. Zhang, Chem, 2020, 6, 885–901 CAS.
- S. Mukherjee, X. Yang, W. Shan, W. Samarakoon, S. Karakalos, D. A. Cullen, K. More, M. Wang, Z. Feng, G. Wang and G. Wu, Small Methods, 2020, 4, 1900821 CrossRef CAS.
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lu, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. R. Adzic and H. L. Xin, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS PubMed.
- K. Jia, Y. Wang, Q. Pan, B. Zhong, Y. Luo, G. Cui, X. Guo and X. Sun, Nanoscale Adv., 2019, 1, 961–964 RSC.
- T. Wu, H. Zhao, X. Zhu, Z. Xing, Q. Liu, T. Liu, S. Gao, S. Lu, G. Chen, A. M. Asiri, Y. Zhang and X. Sun, Adv. Mater., 2020, 32, e2000299 CrossRef PubMed.
- J. Dong, J. Han, Y. Liu, A. Nakajima, S. Matsushita, S. Wei and W. Gao, ACS Appl. Mater. Interfaces, 2014, 6, 1385–1388 CrossRef CAS PubMed.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- G. Liu, Z. Cui, M. Han, S. Zhang, C. Zhao, C. Chen, G. Wang and H. Zhang, Chem.–Eur. J., 2019, 25, 5904–5911 CrossRef CAS PubMed.
- W. Li, C. Zhang, M. Han, Y. Ye, S. Zhang, Y. Liu, G. Wang, C. Liang and H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 44186–44195 CrossRef CAS PubMed.
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed.
- Z. H. Xue, S. N. Zhang, Y. X. Lin, H. Su, G. Y. Zhai, J. T. Han, Q. Y. Yu, X. H. Li, M. Antonietti and J. S. Chen, J. Am. Chem. Soc., 2019, 141, 14976–14980 CrossRef CAS PubMed.
- X. Xu, X. Tian, B. Sun, Z. Liang, H. Cui, J. Tian and M. Shao, Appl. Catal., B, 2020, 272, 118984 CrossRef CAS.
- Z. Liu, T. Huang, H. Chang, F. Wang, J. Wen, H. Sun, M. Hossain, Q. Xie, Y. Zhao and Y. Wu, Energy Environ. Mater., 2020, 4, 255–262 CrossRef.
- Y. Zhai, Z. Zhu, C. Zhu, K. Chen, X. Zhang, J. Tang and J. Chen, Mater. Today, 2020, 38, 99–113 CrossRef CAS.
- Y. Qiu, X. Peng, F. Lü, Y. Mi, L. Zhuo, J. Ren, X. Liu and J. Luo, Chem.–Asian J., 2019, 14, 2770–2779 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- Q. Qin, T. Heil, M. Antonietti and M. Oschatz, Small Methods, 2018, 2, 1800202 CrossRef.
- X. W. Guo, S. M. Chen, H. J. Wang, Z. M. Zhang, H. Lin, L. Song and T. B. Lu, J. Mater. Chem. A, 2019, 7, 19831–19837 RSC.
- H. Tao, C. Choi, L. X. Ding, Z. Jiang, Z. Han, M. Jia, Q. Fan, Y. Gao, H. Wang, A. W. Robertson, S. Hong, Y. Jung, S. Liu and Z. Sun, Chem, 2019, 5, 204–214 CAS.
- Q. Wei, F. Xiong, S. Tan, L. Huang, E. H. Lan, B. Dunn and L. Mai, Adv. Mater., 2017, 29, 1602300 CrossRef PubMed.
- Q. Zhao, M. Zhao, J. Qiu, W. Y. Lai, H. Pang and W. Huang, Small, 2017, 13, 1701091 CrossRef PubMed.
- L. Zhang, P. Lu, Y. Luo, J. Y. Zheng, W. Ma, L. Ding and H. Wang, J. Mater. Chem. A, 2021, 9, 9609–9615 RSC.
- X. Li and J. Wang, InfoMat, 2019, 2, 3–32 CrossRef.
- J. Wen, H. Chang, T. Huang, M. Hossain, Z. Liu, H. Sun, Y. Zhu, Y. Chen, Q. Huang and Y. Wu, Electrochim. Acta, 2020, 367, 137421 CrossRef.
- N. Zhang, L. Li, J. Wang, Z. Hu, Q. Shao, X. Xiao and X. Huang, Angew. Chem., Int. Ed., 2020, 59, 8066–8071 CrossRef CAS PubMed.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed.
- J. Han, Z. Liu, Y. Ma, G. Cui, F. Xie, F. Wang, Y. Wu, S. Gao, Y. Xu and X. Sun, Nano Energy, 2018, 52, 264–270 CrossRef CAS.
- H. Wang, D. Yang, S. Liu, S. Yin, Y. Xu, X. Li, Z. Wang and L. Wang, ACS Sustainable Chem. Eng., 2019, 7, 15772–15777 CrossRef CAS.
- F. Lai, W. Zong, G. He, Y. Xu, H. Huang, B. Weng, D. Rao, J. A. Martens, J. Hofkens, I. P. Parkin and T. Liu, Angew. Chem., Int. Ed., 2020, 59, 13320–13327 CrossRef CAS PubMed.
- B. Li, X. Zhu, J. Wang, R. Xing, Q. Liu, X. Shi, Y. Luo, S. Liu, X. Niu and X. Sun, Chem. Commun., 2020, 56, 1074–1077 RSC.
- Y. T. Liu, X. Chen, J. Yu and B. Ding, Angew. Chem., Int. Ed., 2019, 58, 18903–18907 CrossRef CAS PubMed.
- Y. Tong, H. Guo, D. Liu, J. Liang, X. Yan, P. Su, S. Zhou, J. Liu, G. Q. M. Lu and S. X. Dou, Angew. Chem., Int. Ed., 2020, 59, 7356–7361 CrossRef CAS PubMed.
- T. Huang, Z. Liu, Y. Zhang, F. Wang, J. Wen, C. Wang, M. Hossain, Q. Xie, S. Yao and Y. Wu, J. Mater. Chem. A, 2020, 8, 13679–13684 RSC.
- W. Q. Qiu, Y. L. Luo, R. L. Liang and J. D. Qiu, Chemistry, 2020, 26, 10226–10229 CrossRef CAS PubMed.
- Y. Wang, X. Cui, J. Zhao, G. Jia, L. Gu, Q. Zhang, L. Meng, Z. Shi, L. Zheng, C. Wang, Z. Zhang and W. Zheng, ACS Catal., 2018, 9, 336–344 CrossRef.
- M. Arif, G. Yasin, L. Luo, W. Ye, M. A. Mushtaq, X. Fang, X. Xiang, S. Ji and D. Yan, Appl. Catal., B, 2020, 265, 118559 CrossRef CAS.
- X. Ren, J. Zhao, Q. Wei, Y. Ma, H. Guo, Q. Liu, Y. Wang, G. Cui, A. M. Asiri, B. Li, B. Tang and X. Sun, ACS Cent. Sci., 2019, 5, 116–121 CrossRef CAS PubMed.
- C. Lv, C. Yan, G. Chen, Y. Ding, J. Sun, Y. Zhou and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 6073–6076 CrossRef CAS PubMed.
- X. Wang, S. Qiu, J. Feng, Y. Tong, F. Zhou, Q. Li, L. Song, S. Chen, K. H. Wu, P. Su, S. Ye, F. Hou, S. X. Dou, H. K. Liu, G. Q. Max Lu, C. Sun, J. Liu and J. Liang, Adv. Mater., 2020, 32, e2004382 CrossRef PubMed.
- X. Yang, F. Ling, X. Zi, Y. Wang, H. Zhang, H. Zhang, M. Zhou, Z. Guo and Y. Wang, Small, 2020, 16, e2000421 CrossRef PubMed.
- Y. Zhao, J. Liang, C. Wang, J. Ma and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1702524 CrossRef.
- C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- Q. Liu, X. Zhang, B. Zhang, Y. Luo, G. Cui, F. Xie and X. Sun, Nanoscale, 2018, 10, 14386–14389 RSC.
- X. Xiang, Z. Wang, X. Shi, M. Fan and X. Sun, ChemCatChem, 2018, 10, 4530–4535 CrossRef CAS.
- X. Zhu, Z. Liu, Q. Liu, Y. Luo, X. Shi, A. M. Asiri, Y. Wu and X. Sun, Chem. Commun., 2018, 54, 11332–11335 RSC.
- Z. Wang, K. Zheng, S. Liu, Z. Dai, Y. Xu, X. Li, H. Wang and L. Wang, ACS Sustainable Chem. Eng., 2019, 7, 11754–11759 CrossRef CAS.
- X. Ren, G. Cui, L. Chen, F. Xie, Q. Wei, Z. Tian and X. Sun, Chem. Commun., 2018, 54, 8474–8477 RSC.
- P. Shen, Y. Liu, Q. Li and K. Chu, Chem. Commun., 2020, 56, 10505–10508 RSC.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- C. Chen, N. W. Li, B. Wang, S. Yuan and L. Yu, Nanoscale Adv., 2020, 2, 5496–5503 RSC.
- L. Huang, Z. Hu, H. Jin, J. Wu, K. Liu, Z. Xu, J. Wan, H. Zhou, J. Duan, B. Hu and J. Zhou, Adv. Funct. Mater., 2020, 30, 1908486 CrossRef CAS.
- X. Xiao, H. Wang, P. Urbankowski and Y. Gogotsi, Chem. Soc. Rev., 2018, 47, 8744–8765 RSC.
- M. Och, M. B. Martin, B. Dlubak, P. Seneor and C. Mattevi, Nanoscale, 2021, 13, 2157–2180 RSC.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2016, 7, 706–709 CrossRef.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S. Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- H. Cheng, L. X. Ding, G. F. Chen, L. Zhang, J. Xue and H. Wang, Adv. Mater., 2018, 30, e1803694 CrossRef PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- X. Zhang, T. Wu, H. Wang, R. Zhao, H. Chen, T. Wang, P. Wei, Y. Luo, Y. Zhang and X. Sun, ACS Catal., 2019, 9, 4609–4615 CrossRef CAS.
- Q. Liu, X. Zhang, J. Wang, Y. Zhang, S. Bian, Z. Cheng, N. Kang, H. Huang, S. Gu, Y. Wang, D. Liu, P. K. Chu and X. F. Yu, Angew. Chem., Int. Ed., 2020, 59, 14383–14387 CrossRef CAS PubMed.
- M. M. Shi, D. Bao, S. J. Li, B. R. Wulan, J. M. Yan and Q. Jiang, Adv. Energy Mater., 2018, 8, 1800124 CrossRef.
- K. Chu, Y. Liu, Y. Li, H. Zhang and Y. Tian, J. Mater. Chem. A, 2019, 7, 4389–4394 RSC.
- H. Y. Zhou, J. C. Li, Z. Wen and Q. Jiang, Phys. Chem. Chem. Phys., 2019, 21, 14583–14588 RSC.
- R. Zhao, C. Liu, X. Zhang, X. Zhu, P. Wei, L. Ji, Y. Guo, S. Gao, Y. Luo, Z. Wang and X. Sun, J. Mater. Chem. A, 2020, 8, 77–81 RSC.
- X. Zhang, Q. Liu, X. Shi, A. M. Asiri, Y. Luo, X. Sun and T. Li, J. Mater. Chem. A, 2018, 6, 17303–17306 RSC.
- F. Wang, Y. p. Liu, H. Zhang and K. Chu, ChemCatChem, 2019, 11, 1441–1447 CrossRef CAS.
- S. J. Li, D. Bao, M. M. Shi, B. R. Wulan, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1700001 CrossRef PubMed.
- Q. Wang, G. Zheng, S. Hao, X. Liu, J. Zheng, Y. Wang, Z. Su, N. Xu, Y. He, L. Lei and X. Zhang, ACS Sustainable Chem. Eng., 2019, 8, 44–49 CrossRef.
- H. Huang, F. Li, Q. Xue, Y. Zhang, S. Yin and Y. Chen, Small, 2019, 15, 1903500 CrossRef CAS PubMed.
- P. Li, W. Fu, P. Zhuang, Y. Cao, C. Tang, A. B. Watson, P. Dong, J. Shen and M. Ye, Small, 2019, 15, e1902535 CrossRef PubMed.
- W. Qiu, X. Y. Xie, J. Qiu, W. H. Fang, R. Liang, X. Ren, X. Ji, G. Cui, A. M. Asiri, G. Cui, B. Tang and X. Sun, Nat. Commun., 2018, 9, 3485 CrossRef PubMed.
- H. Wang, J. Wang, R. Zhang, C. Cheng, K. Qiu, Y. Yang, J. Mao, H. Liu, M. Du, C. Dong and X. Du, ACS Catal., 2020, 10, 4914–4921 CrossRef CAS.
- L. Zhang, L. X. Ding, G. F. Chen, X. Yang and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 2612–2616 CrossRef CAS PubMed.
- Z. Jin, C. Liu, Z. Liu, J. Han, Y. Fang, Y. Han, Y. Niu, Y. Wu, C. Sun and Y. Xu, Adv. Energy Mater., 2020, 10, 2000797 CrossRef CAS.
- D. Liu, G. Zhang, Q. Ji, Y. Zhang and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 25758–25765 CrossRef CAS PubMed.
- J. Su, H. Zhao, W. Fu, W. Tian, X. Yang, H. Zhang, F. ling and Y. Wang, Appl. Catal., B, 2020, 265, 118589 CrossRef.
- P. Chen, N. Zhang, S. Wang, T. Zhou, Y. Tong, C. Ao, W. Yan, L. Zhang, W. Chu, C. Wu and Y. Xie, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6635–6640 CrossRef CAS PubMed.
- C. Li, Y. Fu, Z. Wu, J. Xia and X. Wang, Nanoscale, 2019, 11, 12997–13006 RSC.
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS.
- Y. Jiang, L. Yang, T. Sun, J. Zhao, Z. Lyu, O. Zhuo, X. Wang, Q. Wu, J. Ma and Z. Hu, ACS Catal., 2015, 5, 6707–6712 CrossRef CAS.
- W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Mullen, Angew. Chem., Int. Ed., 2014, 53, 1570–1574 CrossRef CAS PubMed.
- Y. Du, C. Jiang, W. Xia, L. Song, P. Li, B. Gao, C. Wu, L. Sheng, J. Ye, T. Wang and J. He, J. Mater. Chem. A, 2020, 8, 55–61 RSC.
- Y. Li, Z. Zhou, P. Shen and Z. Chen, ACA Nano, 2009, 3, 1952–1958 CrossRef CAS PubMed.
- S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J. B. Baek and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110–3116 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- C. Chen, D. Yan, Y. Wang, Y. Zhou, Y. Zou, Y. Li and S. Wang, Small, 2019, 15, e1805029 CrossRef PubMed.
- S. Wang, D. Guo, M. Zong, C. Fan, X. Jun and D.-H. Wang, Appl. Catal., A, 2021, 617, 118112 CrossRef CAS.
- Z. Zhao, Y. Long, S. Luo, Y. Luo, M. Chen and J. Ma, J. Energy Chem., 2021, 60, 546–555 CrossRef.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- X. Guo, S. Lin, J. Gu, S. Zhang, Z. Chen and S. Huang, Adv. Funct. Mater., 2020, 31, 2008056 CrossRef.
- Y. Luo, G. Chen, L. Ding, X. Chen, L. Ding and H. Wang, Joule, 2019, 3, 279–289 CrossRef CAS.
- J. Zhao, L. Zhang, X. Y. Xie, X. Li, Y. Ma, Q. Liu, W. H. Fang, X. Shi, G. Cui and X. Sun, J. Mater. Chem. A, 2018, 6, 24031–24035 RSC.
- T. Li, X. Yan, L. Huang, J. Li, L. Yao, Q. Zhu, W. Wang, W. Abbas, R. Naz, J. Gu, Q. Liu, W. Zhang and D. Zhang, J. Mater. Chem. A, 2019, 7, 14462–14465 RSC.
- Y. Guo, T. Wang, Q. Yang, X. Li, H. Li, Y. Wang, T. Jiao, Z. Huang, B. Dong, W. Zhang, J. Fan and C. Zhi, ACS Nano, 2020, 14, 9089–9097 CrossRef CAS PubMed.
- W. Peng, M. Luo, X. Xu, K. Jiang, M. Peng, D. Chen, T. S. Chan and Y. Tan, Adv. Energy Mater., 2020, 10, 2001364 CrossRef CAS.
- S. D. Claus, J. H. Jacobsen, B. S. Clausen, S. Bahn, A. Logadottir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404–8405 CrossRef PubMed.
- Z. Fang, P. Wu, Y. Qian and G. Yu, Angew. Chem., Int. Ed., 2021, 60, 4275–4281 CrossRef CAS PubMed.
- D. Zhang, H. Zhao, X. Wu, Y. Deng, Z. Wang, Y. Han, H. Li, Y. Shi, X. Chen, S. Li, J. Lai, B. Huang and L. Wang, Adv. Funct. Mater., 2020, 31, 2006939 CrossRef.
- Z. Xiao, C. Xie, Y. Wang, R. Chen and S. Wang, J. Energy Chem., 2021, 53, 208–225 CrossRef.
- G. F. Chen, X. Cao, S. Wu, X. Zeng, L. X. Ding, M. Zhu and H. Wang, J. Am. Chem. Soc., 2017, 139, 9771–9774 CrossRef CAS PubMed.
- J. Yao, D. Bao, Q. Zhang, M. Shi, Y. Wang, R. Gao, J. Yan and Q. Jiang, Small Methods, 2019, 3, 1800333 CrossRef.
- G. Zhang, Q. Ji, K. Zhang, Y. Chen, Z. Li, H. Liu, J. Li and J. Qu, Nano Energy, 2019, 59, 10–16 CrossRef CAS.
- K. Chu, Y. Liu, Y. Cheng and Q. Li, J. Mater. Chem. A, 2020, 8, 5200–5208 RSC.
- X. W. Lv, Y. Liu, R. Hao, W. Tian and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2020, 12, 17502–17508 CrossRef CAS PubMed.
- B. Xu, L. Xia, F. Zhou, R. Zhao, H. Chen, T. Wang, Q. Zhou, Q. Liu, G. Cui, X. Xiong, F. Gong and X. Sun, ACS Sustainable Chem. Eng., 2019, 7, 2889–2893 CrossRef CAS.
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246–10250 CrossRef CAS PubMed.
- G. Peng, J. Wu, M. Wang, J. Niklas, H. Zhou and C. Liu, Nano Lett., 2020, 20, 2879–2885 CrossRef CAS PubMed.
- K. Chu, Q. Li, Y. Liu, J. Wang and Y. Cheng, Appl. Catal., B, 2020, 267, 118693 CrossRef CAS.
- H. Jin, L. Li, X. Liu, C. Tang, W. Xu, S. Chen, L. Song, Y. Zheng and S. Z. Qiao, Adv. Mater., 2019, 31, e1902709 CrossRef PubMed.
- H. Wang, Z. Li, Y. Li, B. Yang, J. Chen, L. Lei, S. Wang and Y. Hou, Nano Energy, 2021, 81, 105613 CrossRef CAS.
- Q. Li, Y. Guo, Y. Tian, W. Liu and K. Chu, J. Mater. Chem. A, 2020, 8, 16195–16202 RSC.
- K. Chu, J. Wang, Y. Liu, Q. Li and Y. Guo, J. Mater. Chem. A, 2020, 8, 7117–7124 RSC.
- K. Chu, H. Nan, Q. Li, Y. Guo, Y. Tian and W. Liu, J. Energy Chem., 2021, 53, 132–138 CrossRef.
- M. You, S. Yi, X. Hou, Z. Wang, H. Ji, L. Zhang, Y. Wang, Z. Zhang and D. Chen, J. Colloid Interface Sci., 2021, 599, 849–856 CrossRef CAS PubMed.
- X. Wang, J. Bai, Y. Wang, X. Lu, Z. Zou, J. Huang and C. Xu, Green Energy Environ., 2020 DOI:10.1016/j.gee.2020.11.016.
- L. Zeng, S. Chen, J. van der Zalm, X. Li and A. Chen, Chem. Commun., 2019, 55, 7386–7389 RSC.
- C. Li, R. Xu, S. Ma, Y. Xie, K. Qu, H. Bao, W. Cai and Z. Yang, Chem. Eng. J., 2021, 415, 128958 CrossRef.
- X. Zi, J. Wan, X. Yang, W. Tian, H. Zhang and Y. Wang, Appl. Catal., B, 2021, 286, 119870 CrossRef CAS.
- J. Zhao, X. Liu, X. Ren, X. Sun, D. Tian, Q. Wei and D. Wu, Appl. Catal., B, 2021, 284, 119746 CrossRef CAS.
- L. Zhang, M. Cong, X. Ding, Y. Jin, F. Xu, Y. Wang, L. Chen and L. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10888–10893 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |