
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn effective in vivo mitochondria-targeting nanocarrier combined with a π-extended porphyrin-type photosensitizer†
Satrialdi
 ab,
Yuta
Takano
*cd,
Eri
Hirata
e,
Natsumi
Ushijima
e,
Hideyoshi
Harashima
a and
Yuma
Yamada
*a
ab,
Yuta
Takano
*cd,
Eri
Hirata
e,
Natsumi
Ushijima
e,
Hideyoshi
Harashima
a and
Yuma
Yamada
*a
aLaboratory for Molecular Design of Pharmaceutics, Faculty of Pharmaceutical Sciences, Hokkaido University, Kita-12 Nishi-6, Kita-ku, Sapporo 060-0812, Japan. E-mail: u-ma@pharm.hokudai.ac.jp
bPharmaceutics Research Group, School of Pharmacy, Institut Teknologi Bandung, Ganesha 10, Bandung 40132, Indonesia
cResearch Institute for Electronic Science, Hokkaido University, Kita-20 Nishi-10, Kita-ku, Sapporo 001-0020, Japan. E-mail: tak@es.hokudai.ac.jp
dGraduate School of Environmental Science, Hokkaido University, Sapporo 060-0810, Japan
eFaculty of Dental Medicine, Hokkaido University, Sapporo 060-8586, Japan
First published on 21st August 2021
Abstract
A photochemical reaction mediated by light-activated molecules (photosensitizers) in photodynamic therapy (PDT) causes molecular oxygen to be converted into highly reactive oxygen species (ROS) that are beneficial for cancer therapy. As the active oxygen consumer and the primary regulator of apoptosis, mitochondria are known as an important target for optimizing PDT outcomes. However, most of the clinically used photosensitizers exhibited a poor tumor accumulation profile as well as lack of mitochondria targeting ability. Therefore, by applying a nanocarrier platform, mitochondria-specific delivery of photosensitizers can be materialized. The present research develops an effective mitochondria-targeting liposome-based nanocarrier system (MITO-Porter) encapsulating a π-extended porphyrin-type photosensitizer (rTPA), which results in a significant in vivo antitumor activity. A single PDT treatment of the rTPA–MITO-Porter resulted in a dramatic tumor inhibition against both human and murine tumors that had been xenografted in a mouse model. Furthermore, depolarization of the mitochondrial membrane was observed, implying the damage of the mitochondrial membrane due to the photochemical reaction that occurred specifically in the mitochondria of tumor cells. The findings presented herein serve to verify the significance of the mitochondria-targeted nanocarrier system for advancing the in vivo PDT effectivity in cancer therapy regardless of tumor type.
Introduction
During the past century, photodynamic therapy (PDT) has been actively developed as a promising approach for effectively eradicating cancers with minimal effects on healthy cells located adjacent to cancer.1 The PDT effect originates from an energy transfer reaction between light and molecular oxygen, a reaction that is mediated by a photosensitizer, resulting in the production of lethal levels of reactive oxygen species (ROS), mainly singlet oxygen.2,3 The selectivity of this therapy is attributed to the specific accumulation of the non-toxic photosensitizer in the tumor region, accompanied by the precise delivery of harmless visible light to the corresponding area, a process that is known as a dual selectivity concept.4 Singlet oxygen reacts aggressively with a number of vital, biologically relevant molecules, causing irreversible oxidative damage thus exerting a lethal effect on cells.5 However, the reactivity of singlet oxygen is often restricted by its short lifetime and its inability to diffuse deeply into tissue.6 The specific delivery of a photosensitizer and the local production of singlet oxygen at the organelle-level is a promising strategy for overcoming this impediment and therefore producing the maximum benefits of PDT.As an important organelle that has both vital and lethal functions for the cell, mitochondria have been identified as an engaging target for advancing PDT outcomes mainly by activating the intrinsic apoptosis pathway.7–9 A significant increase in ROS concentration inside the mitochondrial compartment during the PDT process leads to destabilization of the mitochondrial membrane. This, in turn, causes the liberation of the proapoptotic proteins that are embedded within the inner mitochondrial membrane, further activating the caspases that are responsible for apoptosis cell death.10 Moreover, the production of excessive levels of mitochondrial ROS generates an inhibitory effect on the electron transport chain, leading to a significant increase in mitochondrial oxygen levels and the effectiveness of the PDT, especially under the low oxygen concentration in many tumors.11,12 However, directing a photosensitizer specifically into mitochondria is a complicated and challenging task, which is due to the double membrane system that prevents undesirable molecules from entering the mitochondria, and this task is more challenging to perform in living organisms.
A number of efforts have been made to design a versatile mitochondria-targeted system for delivering chemotherapeutic agents and photosensitizers for cancer therapy.13 Strategies are developed by exploiting the highly negatively charged and hydrophobic characteristics of the mitochondrial membrane. One of the most popular strategies is to employ delocalized lipophilic cations (DLCs), which contain positive charges combined with lipophilic attributes, for example, triphenyl phosphonium (TPP).14 TPP can be employed either to conjugate the photosensitizer through chemical conjugation15 or to modify the surface of a nanocarrier system containing a photosensitizer.16 Although, TPP shows promising ability for targeting drug molecules into mitochondria, it has been reported that TPP has a negative impact on mitochondrial function, in that it inhibits the electron transport chain.17
Since 2008, our group has been intensively focused on developing a versatile mitochondria-targeted nanodevice using a liposomal platform, namely a MITO-Porter system.18 Nanoparticles that make up the system have a highly fusogenic lipid composition that is combined with an arginine-rich cell-penetrating peptide. This system has been used to carry a wide variety of cargoes, ranging from small molecules to genetic materials, into mitochondria to achieve specific targeting goals.19–21 Moreover, we recently utilized the MITO-Porter system to carry an anthracycline antineoplastic agent into the mitochondria of resistant human renal carcinoma cells, both in vitro and in vivo, resulting in a promising level of inhibition of tumor growth.22,23
Porphyrins, an excellent photosensitizer, have been extensively used in clinical applications due to their ability to induce the formation of singlet oxygen by photoirradiation.24,25 However, most porphyrin compounds have an inadequate capacity for harvesting long-wavelength light, which is of utmost importance for achieving a greater ability to penetrate tissue, which largely consists of water and biomolecules.26,27 Furthermore, the fact that the accumulation is non-specific can result in adverse reactions.28,29 To resolve the problems regarding the light absorption ability and tumor-specific accumulation of photosensitizers, previously, we developed a strategy by utilizing a newly synthesized NIR-activated porphyrin-type photosensitizer (rTPA) encapsulated in a MITO-Porter system as a nanocarrier for mitochondrial targeting.30 This novel mitochondria-targeted PDT system displayed promising in vitro cell-killing ability towards various human cancer cell lines, as indicated by the low minimum concentration needed to eliminate half of the cell population (EC50) in comparison with other reported systems.30 Therefore, evaluating the therapeutic benefits of the rTPA–MITO-Porter becomes relevant and engaging as an integral part of the in vitro-to-in vivo translation process, which is the most significant bottleneck in developing nanoparticle technology as a drug delivery system.
In the present study, we report an in vivo application of our recently developed mitochondria-targeted PDT platform, particularly for treating superficial-type human malignant cells that had been xenografted on a mouse model. We initially optimized the rTPA–MITO-Porter formulation so as to achieve appropriate particle characteristics for an in vivo environment, while still preserving mitochondrial delivery and its photo-induced toxicity. The MITO-Porter system amplifies cytosolic delivery through activation of the macropinocytosis pathway, due to the presence of the arginine-rich cell-penetrating peptide on the surface of the liposome. Electrostatic interactions with the highly negatively charged mitochondrial membrane followed by a membrane fusion process accelerates the mitochondrial delivery of the rTPA molecules. Finally, NIR-light irradiation activates the rTPA molecules to produce lethal levels of singlet oxygen, leading to optimum tumor regression, both in vitro and in vivo (Fig. 1).
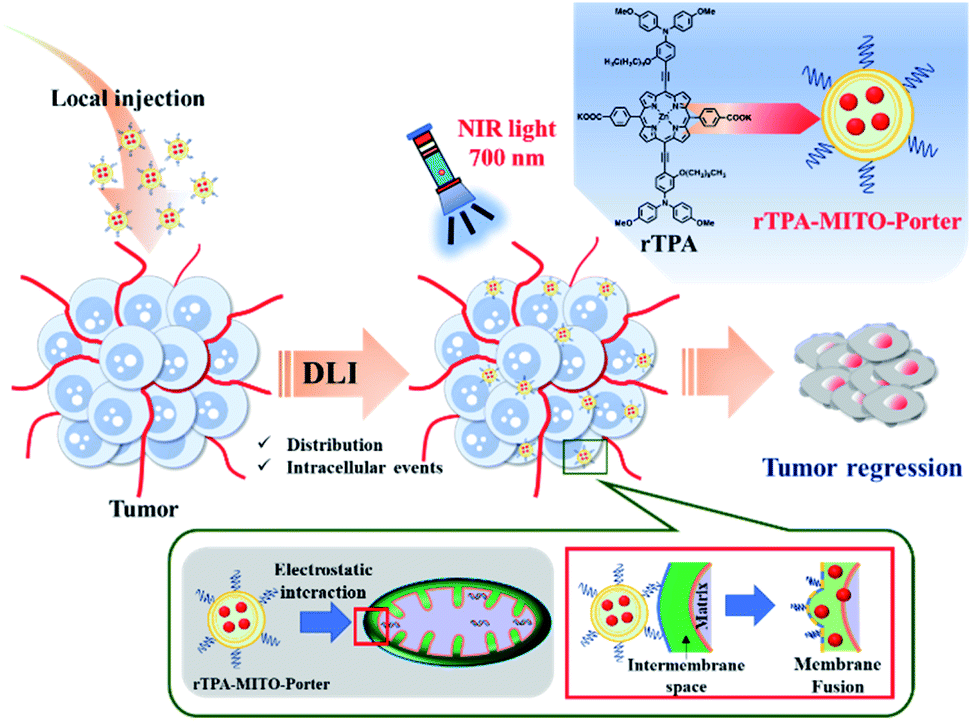 | ||
| Fig. 1 Schematic illustration of the in vivo tumor regression process after the administration of the rTPA–MITO-Porter. The interval between drug administration through a local injection and the light irradiation process (DLI) facilitates particle distribution and intracellular events, including mitochondrial delivery by electrostatic interaction and membrane fusion mechanisms. | ||
Experimental
Materials
Lipid materials consisting of 1,2-dioleoyl-sn-glycero-3-phosphatidyl ethanolamine (DOPE) and cholesterol (Chol) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA) and Sigma Aldrich Corp. (St. Louis, MO, USA), respectively. Stearylated-octaarginine (STR-R8) was acquired from the Toray Research Center, Inc. (Tokyo, Japan). The MitoProbe™ TMRM assay kit was obtained from Thermo Fischer Scientific Inc. (Waltham, MA, USA). SAS cells, a human squamous cell carcinoma cell line of the tongue, were kindly gifted by the National Institutes of Biomedical Innovation, Health, and Nutrition JCRB Cell Bank (Osaka, Japan). rTPA was synthesized and characterized as reported previously.30 All other chemicals and solvents used were purchased as commercially available reagent-grade products.Construction of the rTPA–MITO-Porter
The rTPA–MITO-Porter was prepared using the hydration method, as described in a previous report30 with minor modifications. Briefly, an ethanol solution containing a combination of DOPE and Chol in a molar ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (a total lipid concentration of 2.75 mM) was mixed with 5 mol% of rTPA in chloroform and the solvent was then evaporated to obtain a thin lipid film. The hydration of the lipid film was carried out using 10 mM HEPES buffer containing 290 mM glucose (pH 7.4) for 15 min at room temperature to allow the molecular interactions between the hydration solution and lipid film to be complete, followed by sonication to form spherical-shaped homogeneous particles. The non-encapsulated drugs were precipitated by centrifugation at 20600 × g for 5 min to separate them from the drugs that were encapsulated in the liposomes. Finally, the liposomes were modified with a 10 mol% STR-R8 solution to give the rTPA–MITO-Porter. The particle size and zeta potentials were determined using a dynamic light scattering (DLS) method and the patented phase analysis light scattering technique (M3-PALS technology) (Zetasizer Nano ZS, Malvern Instruments, Worcestershire, UK), respectively. Moreover, the amount of encapsulated rTPA was determined by spectrophotometry (Beckman Coulter DU®730, Brea, CA, USA).
:2 (a total lipid concentration of 2.75 mM) was mixed with 5 mol% of rTPA in chloroform and the solvent was then evaporated to obtain a thin lipid film. The hydration of the lipid film was carried out using 10 mM HEPES buffer containing 290 mM glucose (pH 7.4) for 15 min at room temperature to allow the molecular interactions between the hydration solution and lipid film to be complete, followed by sonication to form spherical-shaped homogeneous particles. The non-encapsulated drugs were precipitated by centrifugation at 20600 × g for 5 min to separate them from the drugs that were encapsulated in the liposomes. Finally, the liposomes were modified with a 10 mol% STR-R8 solution to give the rTPA–MITO-Porter. The particle size and zeta potentials were determined using a dynamic light scattering (DLS) method and the patented phase analysis light scattering technique (M3-PALS technology) (Zetasizer Nano ZS, Malvern Instruments, Worcestershire, UK), respectively. Moreover, the amount of encapsulated rTPA was determined by spectrophotometry (Beckman Coulter DU®730, Brea, CA, USA).
Transmission electron microscopy (TEM) observations
For TEM observations, the specimens were placed on a carbon/formvar film-coated grid, stained with 1% uranyl acetate, dried, and subsequently examined using a TEM (JEM1400, 80 V, JEOL, Tokyo, Japan).Cell culture
SAS cells, human tongue cancer cells, were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Wako, Osaka, Japan) containing 10% (v/v) fetal bovine serum (FBS) (Sigma Aldrich Corp.) and penicillin–streptomycin (Meiji Seika Pharma, Co. Ltd., Tokyo, Japan) under an atmosphere of 5% CO2/air at 37 °C. The cell passage was performed at 90% confluency.Verification of in vitro mitochondrial delivery and phototoxicity
The efficiency of mitochondrial delivery was evaluated using confocal laser scanning microscopy (CLSM) analysis. SAS cells were seeded on a 35 mm glass base dish at a density of 2 × 105 cells followed by a 24 h incubation period. The rTPA encapsulated in the MITO-Porter containing 7-nitrobenz-2-oxa-1,3-diazole dye (NBD-labeled rTPA–MITO-Porter) was transfected into the cells in serum-free medium for 1 h, followed by an additional 2 h in medium containing 10% serum. After the transfection process was completed, the mitochondrial compartment was stained using 0.1 μM MitoTracker™ Deep Red FM (Thermo Fischer Scientific Inc.) for 20 min. The observations were performed using an Olympus FV10i-LIV (Olympus Corporation, Tokyo, Japan) equipped with a water-immersion objective lens (UPlanSApo 60X/NA. 1.2) and a dichroic mirror (DM405/473/559/635). The NBD-labeled rTPA–MITO-Porter and the MitoTracker™ Deep Red FM were excited with 473 nm and 635 nm light, respectively. Two fluorescence detection channels were set using a filter at a bandpass of 490–540 nm to detect the NBD-labeled rTPA–MITO-Porter and 660–710 nm to detect MitoTracker™ Deep Red FM.The verification of the PDT cell-killing ability of the rTPA–MITO-Porter involved the use of SAS cells. Briefly, SAS cells were seeded at a concentration of 5 × 104 cells per mL on a 48-well plate 24 h before the transfection. The cells were transfected with the sample in serum-free medium for 1 h, followed by an additional 2 h incubation in medium containing 10% (v/v) serum. The medium was then replaced with fresh medium prior to the irradiation process using a xenon lamp (MAX-303, Asahi Spectra, Tokyo, Japan) equipped with a bandpass filter (700 ± 6 nm, 68.5 mW cm−2, 5 min). Cell viability was measured using a colorimetric method by measuring the WST-1 absorbance change at 450 nm with the reference at 630 nm using a microplate photometer (EnSpire® Multimode Plate Reader, PerkinElmer, Waltham, MA, USA). The evaluation was independently repeated three times.
Establishment of a tumor-bearing mouse model
Male Balb/c Scl-nu/nu mice (4–6 weeks old) were obtained from Japan SLC Inc. (Shizuoka, Japan). The animals were housed in a specific pathogen-free facility with 12 h light/dark periods and were continuously supplied with food and water. All animal experiments were carried out following protocols that had been previously reviewed and approved by the Institutional Animal Care and Research Advisory Committee at the Faculty of Pharmaceutical Sciences, Hokkaido University, Sapporo, Japan (registration number: 16-0015). To establish a tumor-bearing mouse model, SAS cells with a density of 1 × 106 cells in 80 μL PBS were subcutaneously injected into the right flank of the mouse. The tumor growth was monitored and measured using callipers using the following formula: tumor volume = 0.52 × long axis × short axis2.Evaluation of particle distribution on the tumor tissues
Mice with a tumor volume of around 200 mm3 were intratumorally injected with the rTPA–MITO-Porter containing the fluorescent dye 1,1′-dioctadecyl-3,3,3′,3′-tetra-methylindodicarbocyanine perchlorate (DiD; Invitrogen Corp., Carlsbad, CA, USA). At 6 h or 12 h after liposome administration, the mice were sacrificed, and tumor tissues were collected. The main organs, including the liver, kidneys, lung, spleen, and heart, were also collected. The distribution of the liposomes inside the tumor tissues and several main organs was observed using a FluorVivo™ 300 Small Animal Fluorescence Imaging system (INDEC BioSystems, Los Altos, CA, USA).Evaluation of in vivo PDT antitumor activity
After the tumor volume reached 50 mm3 or six days after tumor inoculation, the SAS cell-bearing mice were randomly divided into five groups and separately intratumorally administered HEPES buffer containing 290 mM glucose pH 7.4 (HBG) with light irradiation (+L), empty MITO-Porter + L, free rTPA + L, rTPA–MITO-Porter, and rTPA–MITO-Porter + L. The rTPA dose was fixed at 8.2 μg per mouse for all treatments containing rTPA. The light irradiation process was performed 12 h after the administration of the solution by means of a xenon lamp with a bandpass filter (700 ± 6 nm, 920 mW cm−2, 20 min). The tumor volume and the bodyweight were monitored at two-day intervals until the endpoint of the experiment (14 days after the first treatment). The relative tumor volume was calculated by comparing the tumor volume with the corresponding initial tumor volume prior to treatment. Furthermore, tumor growth inhibition (%TGI) was calculated using the following equation: %TGI = (1-[Tt/T0/Ct/C0]/1 − [C0/Ct]) × 100, where Tt and Ct are the median tumor volume of the treated and HBG + L group, respectively, at the end of the observation period. In contrast, T0 and C0 represent the median tumor volume of the treated and HBG + L group, respectively, before treatment.31Assessment of mitochondrial membrane potential
The mice bearing SAS cells were intratumorally injected with the rTPA–MITO-Porter (rTPA dose = 8.2 μg per mouse) or an HBG solution (as negative control), followed by irradiation with 700 nm light for 20 min, at 12 h after drug administration. The mice were sacrificed 3 h after the light irradiation process, and tumor tissues were collected. The tumor tissues were incubated with 0.5 μM MitoProbe™ TMRM solution in PBS (−) for 30 min, followed by observation using a CLSM. The Olympus FV10i-LIV is equipped with a water-immersion objective lens (UPlanSApo 60X/NA. 1.2), and a dichroic mirror (DM405/473/559/635) was used to detect the TMRM signal. The tissues were illuminated with 559 nm light to excite the TMRM, and the fluorescence detection channel was set using a filter at a bandpass 570–670 nm to detect the TMRM fluorescence signal. The quantification of the mean fluorescence intensity of the TMRM was then performed from randomly selected CLSM images using the ImageJ software.32Results and discussion
Formulation design and preparation of the rTPA–MITO-Porter for in vivo application
We previously validated that the combination of DOPE and R8 is the key factor for the successful mitochondrial delivery of the MITO-Porter system. At the same time, the helper lipids, such as sphingomyelin, cholesterol, cholesteryl hemisuccinate, and phosphatidic acid, can be replaced or altered without having a significant impact on mitochondrial delivery.33 Furthermore, the addition of cholesterol to the liposomal formulation could improve the packing of phospholipid molecules to result in a more rigid and more stable structure,34,35 which is preferable for maintaining the structure of the liposomes in a harsh in vivo environment. Therefore, we included the cholesterol as the helper lipid in the optimization process, which was then combined with DOPE and R8 as the main building component of the rTPA–MITO-Porter, resulting in a material with a high positive charge with a narrow particle size distribution.The rTPA–MITO-Porter was assembled using the hydration method, as previously reported.30 The hydration method was adopted because it is relatively simple to carry out and allows homogeneous nanoparticles to be produced in a short time. A mixture of DOPE and cholesterol at a total concentration of 2.75 mM was employed to encapsulate the rTPA, followed by surface modification with R8 to produce the rTPA–MITO-Porter. It was found that the use of a high lipid concentration (2.75 mM) allowed us to encapsulate rTPA more efficiently, with an encapsulation rate of 37.6 ± 5.3 μM compared to the previously developed rTPA–MITO-Porter made using sphingomyelin at a low lipid concentration (0.55 mM), which encapsulated only 6.7 ± 2.9 μM of rTPA.30 The high lipid concentration provides more space for the encapsulation of rTPA. This is essential for producing the high dose of the sensitizer needed for in vivo applications as opposed to the in vitro evaluation. DLS measurements revealed that the resulting rTPA–MITO-Porter had an average hydrodynamic diameter of 169 ± 6 nm with homogeneous particle distribution, which is indicated by a small polydispersity index (PDI) of 0.22 ± 0.02 (Fig. 2A). Furthermore, the high positive charge (ζ-potential = 36 ± 1 mV) suggests that the R8 moieties are located on the surface of the rTPA–MITO-Porter (Fig. 2B). The attachment of cationic and hydrophilic R8 peptides to the surface of the MITO-Porter was facilitated by the hydrophobic stearyl chain conjugated to the R8 structure. This resulted in the interaction between stearyl moieties with the hydrophobic region of the phospholipid bilayer of the liposomes, while the hydrophilic R8 would be oriented to the surface of the liposomes. TEM observations confirmed the size distribution seen in the DLS and permitted the morphology of the rTPA–MITO-Porter to be visualized (Fig. 2C), whose featureless round shape is consistent with the formation of spherical multilamellar vesicles.
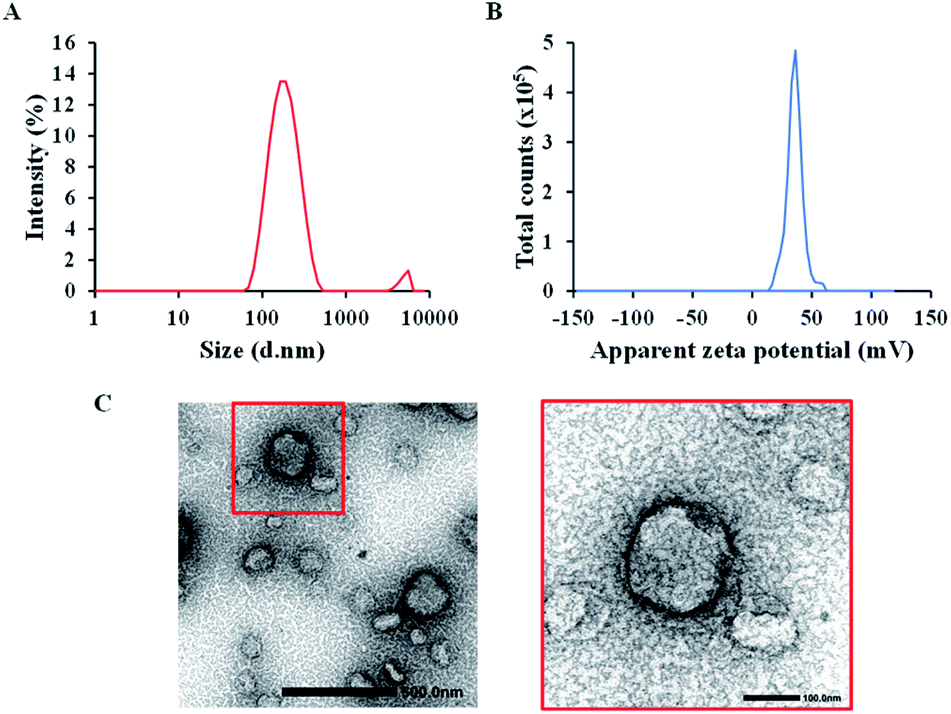 | ||
| Fig. 2 Particle characteristics of the rTPA–MITO-Porter. (A) Particle size distribution with an average diameter of 169 ± 6 nm and a polydispersity index of 0.22 ± 0.02. (B) Zeta potential, indicating the positive charge particle with an average of 36 ± 1 mV. (C) TEM images of the rTPA–MITO-Porter (scale bars: 500 nm) and the magnification image of the particle in the red box (scale bars: 100 nm). | ||
R8, a short peptide sequence containing eight cationic arginine residues, is a member of the arginine-rich cell-penetrating peptide (CPP) group that is extensively used to enhance the cell internalization of nanoparticles. This CPP can interact with cell-surface proteoglycans and activate the macropinocytosis pathway as the primary cellular internalization process.36,37 It also can effectively promote the escape of the nanoparticles from endo-lysosomal vesicles through a membrane fusion process in a pH-independent manner.38 Inside the cytosol, the R8 facilitates the interaction of the MITO-Porter with the mitochondrial membrane via electrostatic interactions. These interactions are accompanied by a membrane fusion process, which is mediated through the highly fusogenic lipid composition of the MITO-Porter system.18,33 Therefore, attaching R8 moieties on the surface of the MITO-Porter system is considered to be the most critical process in constructing the rTPA–MITO-Porter.
Verification of the mitochondrial delivery of the rTPA–MITO-Porter in vitro
SAS cells, which are categorized as squamous carcinoma cells derived from human tongue cancer cells, were transfected with the NBD-labeled rTPA–MITO-Porter for 3 h, as well as the empty MITO-Porter as the control group. The internalization and accumulation of the MITO-Porter in mitochondria were visualized using a CLSM after staining the mitochondrial compartment with the MitoTracker™ Deep Red FM. As indicated in Fig. 3, bright green fluorescence signals of NBD were clearly evident in the interior of the cells that had been treated with both the empty MITO-Porter and the rTPA–MITO-Porter, suggesting that a high-level of particle internalization had occurred. Moreover, most of the nanoparticles were colocalized inside the mitochondrial compartment, as evidenced by the appearance of yellow fluorescence signals, as a result of the overlapping of the green fluorescence signal of the MITO-Porter with the red fluorescence signal of the mitochondria. The level of accumulation in mitochondria was then quantified using Pearson's correlation coefficient (Rr). The results indicated that the presence of rTPA molecules inside the MITO-Porter system had only a negligible effect on the overall accumulation of the MITO-Porter in mitochondria (P = 0.41), with approximately 30% of the particles accumulating in mitochondria. This result indicates the effectivity of the MITO-Porter system in selectively transporting rTPA into the mitochondria of SAS cells.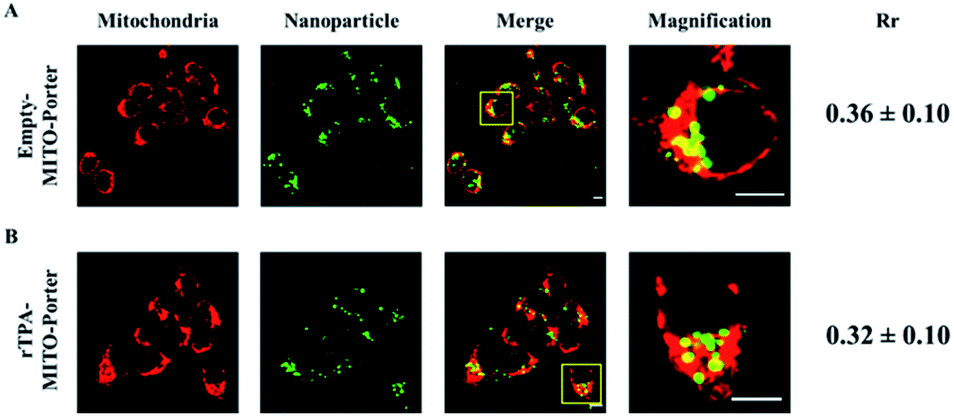 | ||
| Fig. 3 Mitochondrial accumulation profile of (A) the empty-MITO-Porter and (B) the rTPA–MITO-Porter in SAS cells. The level of mitochondrial accumulation was quantified using Pearson's correlation coefficient (Rr). Scale bars: 10 μm. | ||
Validation of in vitro photo-induced cell death by the rTPA–MITO-Porter
The PDT effect is a result of the dynamic interaction between its key elements, specifically the photosensitizer, light, and oxygen molecules. The energy transfer process from visible light to molecular oxygen mediated by a photosensitizer results in the alteration of ground-state oxygen molecules with the formation of reactive and destructive species, mainly singlet oxygen.2,3 The ability of the rTPA–MITO-Porter to induce toxicity against SAS cells was evaluated in the absence or presence of a light irradiation process. As shown in Fig. 4, a dose-dependent toxicity profile was obtained during a 5 min irradiation process by a xenon lamp at 700 ± 6 nm, while negligible toxicity was observed in the absence of irradiation. Furthermore, the EC50 value was determined to be approximately 0.27 ± 0.04 μM. The cell-killing effectivity of the rTPA–MITO-Porter was statistically insignificant (P = 0.268) compared to previously reported results in which a low concentration of sphingomyelin (0.55 mM) was used for preparing a MITO-Porter system,30 indicating that the total lipid concentration and composition have only a minor impact on the PDT effect associated with the rTPA–MITO-Porter. Meanwhile, the robust photo-induced cytotoxicity of the rTPA–MITO-Porter originated from the ability of the rTPA to produce a high-level of singlet oxygen during the NIR-light irradiation process30 in combination with the selective mitochondrial accumulation, as displayed in Fig. 3, which further improves the effectiveness of the PDT.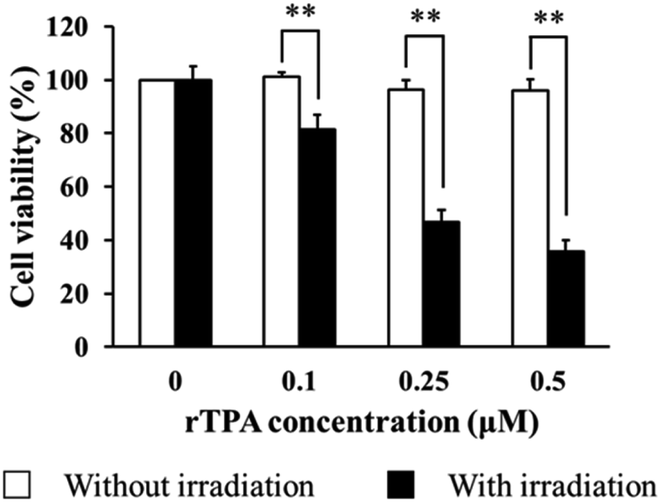 | ||
| Fig. 4 Cytotoxicity profile of the rTPA–MITO-Porter against SAS cells in the presence and absence of light irradiation. The irradiation was performed using 700 ± 6 nm light for 5 minutes with a density of 68.5 mW cm−2. Data represent the average of cell viability with S.D. (n = 3; **p < 0.01 by unpaired T-test). | ||
Evaluation of PDT antitumor activity against the SAS cell-bearing mouse model
Encouraged by the ability to transport rTPA molecules to the mitochondria as well as the ability to induce phototoxicity, we then continued our effort in extending the application of the rTPA–MITO-Porter by evaluating its in vivo antitumor activity. The PDT antitumor activity was evaluated in SAS cells that had been xenografted on a mouse model. SAS cells were selected as the potential target for PDT due to their fatal nature and the fact that the tumor was located on the surface of the body, hence making it easier to treat them by PDT. The cells were subcutaneously inoculated in the right flank of an immunodeficient mouse. When the tumor volume reached 50 mm3, the mice were randomized into five treatment groups, consisting of HBG + L, empty MITO-Porter + L, free rTPA + L, the rTPA–MITO-Porter, and rTPA–MITO-Porter + L. Each group received a specific single treatment with the equivalent of rTPA dose of 8.2 μg per mouse via intratumoral administration followed by a 20 min light irradiation process using a xenon lamp with an optical filter to produce 700 ± 6 nm light. Local injection was chosen since the target tumor is located on the surface of the body, which could be easily accessed by this administration route. Moreover, intratumoral injection provides a relatively lower systemic effect caused by off-target accumulation.Due to the fact that the cell-killing effect of PDT is the result of the interaction of the photosensitizer with light irradiation under an appropriate oxygen concentration, it is important to ensure that the photosensitizer is distributed within the tumor tissues before applying irradiation. The interval between drug administration and the light irradiation process is known as the drug-light interval (DLI). To find the optimum DLI, we performed the biodistribution study by injecting the DiD-labeled rTPA–MITO-Porter subcutaneously followed by the observation of particle accumulation in tumor tissues and several main organs. Based on the biodistribution study, the particles were retained on the tumor tissues for 12 h with negligible distribution to several other main organs, including the liver, spleen, lungs, kidneys, and heart (Fig. S1A†). Moreover, we observed a more extensive particle distribution inside the tumor tissues at 12 h after the injection than after 6 h (Fig. S1B†). Therefore, we proceeded with evaluating the antitumor activity of the rTPA–MITO-Porter using 12 h as the interval between drug administration and the light irradiation process, in order to facilitate the distribution of the particles throughout the tumor tissues, as well as for promoting cellular uptake and the mitochondrial accumulation of rTPA.
As shown in Fig. 5A, B, and S2,† treatment with the rTPA–MITO-Porter without irradiation or the empty MITO-Porter + L resulted in negligible antitumor activity, with a %TGI of 4.63% and 0.94%, respectively. These results indicate that the rTPA–MITO-Porter is biocompatible in the absence of the photoactivation process, and also the empty MITO-Porter with 700 nm light irradiation is biocompatible in the absence of rTPA molecules. In contrast, an efficient and significant inhibition of tumor growth was observed in the group that had been treated with the rTPA–MITO-Porter + L, as evidenced by a %TGI of 80.84%. Interestingly, the treatment with free rTPA + L also showed an inhibition effect on tumor growth, as indicated by a slight decrease in tumor volume at the end of the observation day with a %TGI of 29.07%. This can be attributed to the ability of the rTPA to induce the production of singlet oxygen during the photoirradiation process. However, the effectiveness of the free rTPA in inhibiting tumor growth was significantly lower compared to the rTPA–MITO-Porter, suggesting the importance of the MITO-Porter as a carrier system for optimizing the performance of rTPA. Furthermore, there were no significant changes in the bodyweight of the animals during the treatment process (Fig. 5C), indicating the biocompatibility and biosafety aspects of the treatment. It is also noteworthy that the rTPA–MITO-Porter also exhibited a significant tumor inhibition rate in a simultaneous double implant of the EG7-OVA murine lymphoma mouse model (Fig. S3†). These results suggest the universal use of the rTPA–MITO-Porter as a promising candidate for PDT applications. The significant antitumor activity of the rTPA–MITO-Porter could be associated with the specific localization of the photochemical reaction inside the mitochondrial compartments of tumor cells, regardless of the type of tumor cells.
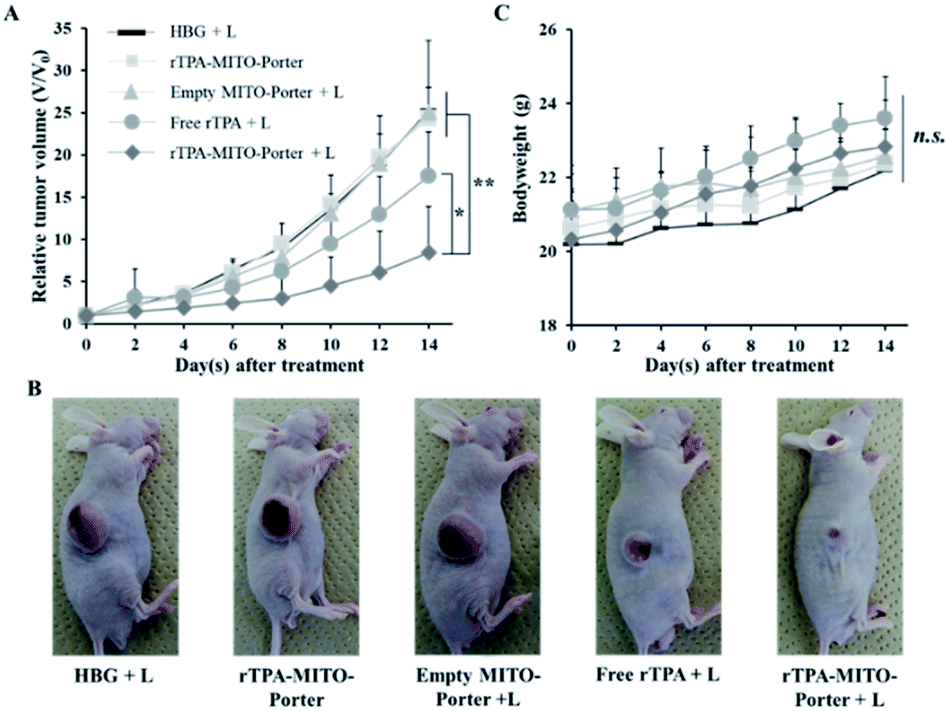 | ||
| Fig. 5 The antitumor evaluation on the SAS cell bearing mice after the single intratumoral administration of HBG, empty MITO-Porter, free rTPA, and rTPA–MITO-Porter. (A) The profile of the relative tumor volume of different treatment groups. (B) The representative photographs of SAS cell bearing mice at the end of the observation period. (C) Changes in body weight during the observation period. The error bars indicate S.D. (n = 4–6; n.s. = not significant; *p < 0.05; **p < 0.01 by ANOVA followed by SNK-test). | ||
Assessment of the mitochondrial membrane potential status after PDT
After obtaining substantial antitumor activity through a single PDT treatment with the rTPA–MITO-Porter, the investigation was continued in an attempt to elucidate the connection between the antitumor activity of the rTPA–MITO-Porter with the state of mitochondria of tumors. We hypothesized that the significant antitumor activity of the rTPA–MITO-Porter during the photoirradiation process would be correlated with the formation of ROS specifically in mitochondria, resulting in an unstable condition inside the mitochondria, which is marked by the status of the mitochondrial membrane potential. Therefore, to verify this hypothesis, we performed an evaluation using tetramethylrhodamine methyl ester (TMRM), a specific fluorescence probe for detecting the mitochondrial membrane potential status. Basically, the TMRM tends to accumulate in healthy mitochondria, resulting in the production of a strong red-orange fluorescence signal upon excitation. When the mitochondrial membrane becomes depolarized, the probe is dispersed in the cytosol, and the fluorescence signal rapidly disappears.In this evaluation, tumor tissues from the rTPA–MITO-Porter and HBG treated mice were collected and stained with a TMRM solution, followed by detecting the TMRM fluorescence signal by CLSM observations. As shown in Fig. 6, an intense red fluorescence signal of TMRM was observed in the negative control group of the HBG treated mice, indicating that the mitochondria were healthy. In contrast, the intensity of the fluorescence signal was dramatically diminished after the PDT treatment with the rTPA–MITO-Porter, suggesting a decrease in mitochondrial membrane potential. The fluorescence intensity was further quantified using the ImageJ software. As a result, the mean fluorescence intensity of TMRM was determined to be 1.9 times lower for the rTPA–MITO-Porter group in comparison to the HBG group. This result suggests that the PDT process of the rTPA–MITO-Porter effectively induced depolarization of the mitochondrial membrane. This result also further confirms that the photochemical reaction was localized inside the mitochondrial compartment of tumors.
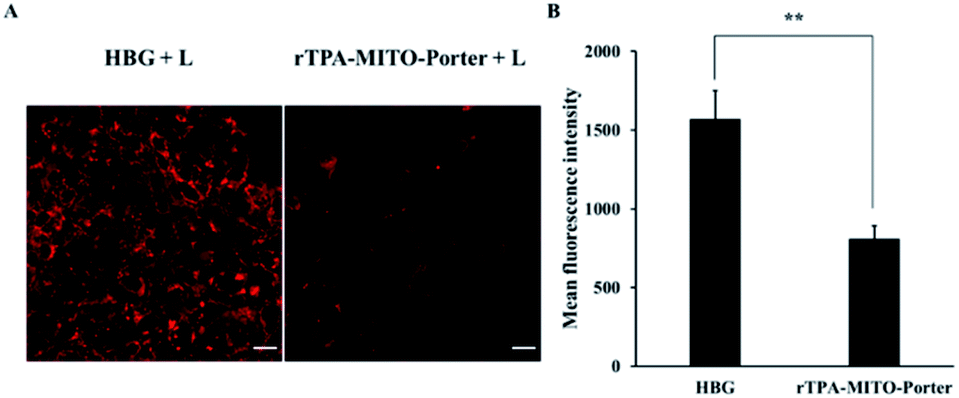 | ||
| Fig. 6 Mitochondrial membrane potential status after PDT treatment, evaluated with TMRM. (A) The representative confocal microscopy images of the excised-tumor tissues from mice treated with HBG and rTPA–MITO-Porter with a 20 min light irradiation process. Scale bars: 20 μm. (B) The mean fluorescence intensity of TMRM, calculated by ImageJ from randomly selected confocal microscopy images. Error bars indicate S.D. (n = 20; **p < 0.01 by unpaired t-test). | ||
Although the PDT process had a high tumor specificity, this therapeutic approach is not very effective against deep-seated tumors. This can be attributed to the difficulty of the photosensitizer to reach and accumulate in such tumors, and the lack of technology for delivering light to the inner part of the body. The application of nanotechnology in the PDT field has opened many innovative issues with the main aim being to improve the tumor accumulation of the photosensitizer,39 particularly in areas that cannot be reached using a conventional drug delivery system. Several approaches have been developed to increase the retention of photosensitizers in tumors, for example, by using an amphiphilic dipeptide or amino acid to form spherical, self-assembled nanosized materials.40,41 Furthermore, the formation of supramolecular metallo-nanophotosensitizers also has promising potential for enhancing antitumor activity with a stimuli–responsive release feature.42 However, the large-scale production of nanoparticles remains challenging, particularly in controlling the quality of nanoparticles, including particle size, homogeneity, and surface characteristics. The application of microfluidic devices has opened new optimism regarding upscaling the production of nanoparticles without essential changes in their physical and functional quality.43 We recently reported the development of a method for the large-scale production of MITO-Porters encapsulating highly hydrophobic substances, such as coenzyme Q10, by applying microfluidic technology and were able to significantly improve intracellular trafficking.44 Therefore, this technology could be further adopted to realize clinical applications of organelle-specific targeting PDT, particularly for the rTPA–MITO-Porter.
Conclusions
In summary, we successfully constructed a novel mitochondrial targeting PDT system, namely the rTPA–MITO-Porter, by incorporating a π-extended porphyrin-type photosensitizer, namely rTPA, into a MITO-Porter system. A notable in vivo antitumor activity was observed by utilizing only a single PDT process of the rTPA–MITO-Porter with minimal systemic toxicity, as indicated by the negligible alteration of the bodyweight of the mice during the treatment. The inhibition of the tumor growth mainly derives from the direct killing effect of the rTPA–MITO-Porter towards the malignant tissues. Additionally, the depolarization of the mitochondrial membrane was detected after the PDT process of the rTPA–MITO-Porter, suggesting the specific localization of the photochemical reaction inside the mitochondrial compartment of tumor tissues as well as the recognition of the mitochondrial delivery of the rTPA compound. Finally, the findings presented in this research serve to verify the considerable functions of the MITO-Porter system as the mitochondrial selective drug delivery system in advancing the in vivo PDT outcomes as well as the achievement in realizing the in vivo applications of the organelle-specific drug delivery technology.Author contributions
Conceptualization: Y. T., H. H., Y. Y. Funding acquisition: Y. T., Y. Y. Investigation: S., Y. T., E. H., N. U. Methodology: S., Y. T., Y. Y. Supervision: Y. T., H. H., Y. Y. Visualization: S., Y. T., Y. Y. Writing – original draft: S., Y. T., Y. Y. Writing – review & editing: S., Y. T., Y. Y.Conflicts of interest
The authors declare no conflict of interest in this work.Acknowledgements
This work was supported, in part, by a Grant-in-Aid for Scientific Research (B) (Grant No. 20H04523 to Y. Y., and 21H0175301 to Y. T.) from the Ministry of Education, Culture, Sports, Science and Technology, the Japanese Government (MEXT), Grant-in-Aid for Scientific Research on Innovative Areas – Platforms for Advanced Technologies and Research Resources “Advanced Bioimaging Support” (JP16H06280), and a grant from the Asahi Glass Foundation (to Y. Y.). This study was also supported by the JSPS Core-to-Core Program, A. Advanced Research Networks, and the Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials. We also thank the Indonesia Endowment Fund for Education (LPDP), Ministry of Finance of the Republic of Indonesia, and Ministry of Education and Culture of the Republic of Indonesia for the scholarship support for Satrialdi. We are grateful to READYFOR Inc., Japan and Shonan Health Innovation Park, Japan for supporting a crowd funding campaign for “Cancer treatment with few side effects by developing technology to deliver drugs to abnormal mitochondria”. A special acknowledgment is addressed to Prof. Mikako Ogawa of Hokkaido University, for providing us with the FluorVivo™ 300 Small Animal Fluorescence Imaging. We also wish to thank Dr Milton Feather for his helpful advice in writing the manuscript.Notes and references
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Photodynamic therapy for cancer, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed
.
- P. Agostinis,
et al. Photodynamic Therapy of cancer: un update, Ca-Cancer J. Clin., 2012, 61, 250–281 CrossRef PubMed
- H. Abrahamse and M. R. Hamblin, New photosensitizers for photodynamic therapy, Biochem. J., 2017, 473, 347–364 CrossRef PubMed
- A. P. Castano, T. N. Demidova and M. Hamblin, Mechanisms in photodynamic therapy: part one, Photodiagn. Photodyn. Ther., 2004, 1, 279–293 CrossRef CAS PubMed
- D. Trachootham, J. Alexandre and P. Huang, Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach?, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed
- J. S. Dysart and M. S. Patterson, Characterization of Photofrin photobleaching for singlet oxygen dose estimation during photodynamic therapy of MLL cells in vitro, Phys. Med. Biol., 2005, 50, 2597–2616 CrossRef CAS PubMed
- F. Guo, M. Yu, J. Wang, F. Tan and N. Li, The mitochondria-targeted and IR780-regulated theranosomes for imaging and enhanced photodynamic/photothermal therapy, RSC Adv., 2016, 6, 11070–11076 RSC
- S. M. Mahalingam, J. D. Ordaz and P. S. Low, Targeting of a Photosensitizer to the Mitochondrion Enhances the Potency of Photodynamic Therapy, ACS Omega, 2018, 3, 6066–6074 CrossRef CAS PubMed
- D. Kessel and Y. Luo, Mitochondrial photodamage and PDT-induced apoptosis, J. Photochem. Photobiol., B, 1998, 42, 89–95 CrossRef CAS
- T. Qi,
et al. A pH-Activatable nanoparticle for dual-stage precisely mitochondria-targeted photodynamic anticancer therapy, Biomaterials, 2019, 213, 119219 CrossRef CAS PubMed
- H. Kurokawa,
et al. High resolution imaging of intracellular oxygen concentration by phosphorescence lifetime, Sci. Rep., 2015, 5, 1–3 Search PubMed
- W. Lv,
et al. A Mitochondria-Targeted Photosensitizer Showing Improved Photodynamic Therapy Effects Under Hypoxia, Angew. Chem., Int. Ed., 2016, 55, 9947–9951 CrossRef CAS PubMed
- Y. Yamada,
et al. Power of mitochondrial drug delivery systems to produce innovative nanomedicines, Adv. Drug Delivery Rev., 2020, 154–155, 187–209 CrossRef CAS PubMed
- J. Zielonka,
et al. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications, Chem. Rev., 2017, 117(15), 10043–10120 CrossRef CAS PubMed
- K. Kirakci,
et al. Cationic octahedral molybdenum cluster complexes functionalized with mitochondria-targeting ligands: Photodynamic anticancer and antibacterial activities, Biomater. Sci., 2019, 7, 1386–1392 RSC
- Z. Yu, Q. Sun, W. Pan, N. Li and B. Tang, A Near-Infrared Triggered Nanophotosensitizer Inducing Domino Effect on Mitochondrial Reactive Oxygen Species Burst for Cancer Therapy, ACS Nano, 2015, 9(11), 11064–11074 CrossRef CAS PubMed
- J. Trnka, M. Elkalaf and M. Andě, Lipophilic triphenylphosphonium cations inhibit mitochondrial electron transport chain and induce mitochondrial proton leak, PLoS One, 2015, 10(4), e0121837 CrossRef PubMed
- Y. Yamada,
et al. MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 423–432 CrossRef CAS PubMed
- Y. Yamada, K. Somiya, A. Miyauchi, H. Osaka and H. Harashima, Validation of a mitochondrial RNA therapeutic strategy using fibroblasts from a Leigh syndrome patient with a mutation in the mitochondrial ND3 gene, Sci. Rep., 2020, 10, 7511 CrossRef CAS PubMed
- E. Kawamura,
et al. Validation of Gene Therapy for Mutant Mitochondria by Delivering Mitochondrial RNA Using a MITO-Porter, Mol. Ther.–Nucleic Acids, 2020, 20, 687–698 CrossRef CAS PubMed
- Y. Yamada,
et al. The use of a MITO-Porter to deliver exogenous therapeutic RNA to a mitochondrial disease's cell with a A1555G mutation in the mitochondrial 12S rRNA gene results in an increase in mitochondrial respiratory activity, Mitochondrion, 2020, 55, 134–144 CrossRef CAS PubMed
- Y. Yamada,
et al. Mitochondrial Delivery of Doxorubicin Using MITO-Porter Kills Drug-Resistant Renal Cancer Cells via Mitochondrial Toxicity, J. Pharm. Sci., 2017, 106, 2428–2437 CrossRef CAS PubMed
- Y. Yamada,
et al. Mitochondrial Delivery
of an Anticancer Drug Via Systemic Administration Using a Mitochondrial Delivery System That Inhibits the Growth of Drug-Resistant Cancer Engrafted on Mice, J. Pharm. Sci., 2020, 109, 2493–2500 CrossRef CAS PubMed
- J. Kou, D. Dou and L. Yang, Porphyrin photosensitizers in photodynamic therapy and its applications, Oncotarget, 2017, 8(46), 81591–81603 CrossRef PubMed
- Q. Zhang,
et al. A promising anticancer drug: A photosensitizer based on the porphyrin skeleton, RSC Med. Chem., 2020, 11, 427–437 RSC
- E. D. Sternberg, D. Dolphin and C. Brückner, Porphyrin-based photosensitizers for use in photodynamic therapy, Tetrahedron, 1998, 54(17), 4151–4202 CrossRef CAS
- J. V. Frangioni, In vivo near-infrared fluorescence imaging, Curr. Opin. Chem. Biol., 2003, 7(5), 626–634 CrossRef CAS PubMed
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Porphyrin and nonporphyrin photosensitizers in oncology: Preclinical and clinical advances in photodynamic therapy, Photochem. Photobiol., 2009, 85, 1053–1074 CrossRef PubMed
- L. b. Li,
et al. Retrospective study of photodynamic therapy vs. photodynamic therapy combined with chemotherapy and chemotherapy alone on advanced esophageal cancer, Photodiagn. Photodyn. Ther., 2010, 7(3), 139–143 CrossRef PubMed
- Satrialdi,
et al. The optimization of cancer photodynamic therapy by utilization of a pi-extended porphyrin-type photosensitizer in combination with MITO-Porter, Chem. Commun., 2020, 56, 1145–1148 RSC
- E. Buck,
et al. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors, Cancer Res., 2008, 68, 8322–8332 CrossRef CAS PubMed
- J. Schindelin,
et al. Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed
- Y. Yamada, Y. Fukuda and H. Harashima, An analysis of membrane fusion between mitochondrial double membranes and MITO-Porter, mitochondrial fusogenic vesicles, Mitochondrion, 2015, 24, 50–55 CrossRef CAS PubMed
- D. Z. Liu, W. Y. Chen, L. M. Tasi and S. P. Yang, Microcalorimetric and shear studies on the effects of cholesterol on the physical stability of lipid vesicles, Colloids Surf., A, 2000, 172, 57–67 CrossRef CAS
- M. L. Briuglia, C. Rotella, A. McFarlane and D. A. Lamprou, Influence of cholesterol on liposome stability and on in vitro drug release, Drug Delivery Transl. Res., 2015, 5, 231–242 CrossRef CAS PubMed
- S. Futaki and I. Nakase, Cell-Surface Interactions on Arginine-Rich Cell-Penetrating Peptides Allow for Multiplex Modes of Internalization, Acc. Chem. Res., 2017, 50, 2449–2456 CrossRef CAS PubMed
- I. A. Khalil, K. Kogure, S. Futaki and H. Harashima, High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression, J. Biol. Chem., 2006, 281, 3544–3551 CrossRef CAS PubMed
- A. El-Sayed, I. A. Khalil, K. Kogure, S. Futaki and H. Harashima, Octaarginine- and octalysine-modified nanoparticles have different modes of endosomal escape, J. Biol. Chem., 2008, 283, 23450–23461 CrossRef CAS PubMed
- C. K. Lim,
et al. Nanophotosensitizers toward advanced photodynamic therapy of Cancer, Cancer Lett., 2013, 334, 176–187 CrossRef CAS PubMed
- K. Liu,
et al. Simple Peptide-Tuned Self-Assembly of Photosensitizers towards Anticancer Photodynamic Therapy, Angew. Chem., Int. Ed., 2016, 55, 3036–3039 CrossRef CAS PubMed
- B. Sun,
et al. Acid-Activatable Transmorphic Peptide-Based Nanomaterials for Photodynamic Therapy, Angew. Chem., Int. Ed., 2020, 59, 20582–20588 CrossRef CAS PubMed
- S. Li,
et al. Smart Peptide-Based Supramolecular Photodynamic Metallo-Nanodrugs Designed by Multicomponent Coordination Self-Assembly, J. Am. Chem. Soc., 2018, 140, 10794–10802 CrossRef CAS PubMed
- N. M. Belliveau,
et al. Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA, Mol. Ther.–Nucleic Acids, 2012, 1(8), e37 CrossRef PubMed
- M. Hibino,
et al. The Use of a Microfluidic Device to Encapsulate a Poorly Water-Soluble Drug CoQ10 in Lipid Nanoparticles and an Attempt to Regulate Intracellular Trafficking to Reach Mitochondria, J. Pharm. Sci., 2019, 108, 2668–2676 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00427a |
| This journal is © The Royal Society of Chemistry 2021 |