
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA platinum nanoparticle doped self-assembled peptide bolaamphiphile hydrogel as an efficient electrocatalyst for the hydrogen evolution reaction†
Deepak K. K.
Kori
,
Rohit G.
Jadhav
,
Likhi
Dhruv
and
Apurba K.
Das
 *
*
Department of Chemistry and Centre for Advanced Electronics (CAE), Indian Institute of Technology Indore, Indore 453552, India. E-mail: apurba.das@iiti.ac.in
First published on 14th September 2021
Abstract
Noble metal-based nanomaterials have shown great potential for catalytic application with higher selectivity and activity. Owing to their self-assembly properties with various molecular interactions, peptides play an essential role in the controlled synthesis of noble metal-based catalysts with high surface area. In this work, a phenylalanine (F) and tyrosine (Y) based peptide bolaamphiphile is prepared by solution-phase peptide synthesis. The peptide bolaamphiphile readily self-assembles into a hydrogel with a cross-linked nanofibrillar network. The platinum nanoparticles (Pt NPs) are in situ generated within the cross-linked nanofibrillar network of the hydrogel matrix of the peptide bolaamphiphile. Benefiting from the synergistic properties of the Pt nanoparticles doped on three-dimensional fibrous networks, Pt6@hydrogel shows efficient catalytic activity for the electrochemical hydrogen evolution reaction (HER) in 0.5 M H2SO4 solution. The Pt6@hydrogel requires an overpotential of 45 mV at −10 mA cm−2 with a Tafel slope of 52 mV dec−1. The Pt6@hydrogel also shows electrocatalytic activity in basic and neutral pH solutions. The excellent activity and stability of Pt6@hydrogel for the HER shows great potential for energy conversion applications.
1. Introduction
With the growing concern about environmental issues and the rise in energy demand, clean and renewable energy sources become inevitable.1–11 In particular, molecular hydrogen (H2) has attracted attention as one of the favourable alternative green energy carriers for diminishing fossil fuels in view of its highly pure products, high gravimetric energy density and environment friendliness.12–15 Despite its application prospects, the exploration of cost-effective and eco-friendly hydrogen production has become a great challenge for scientists in the 21st century.16–18 Among several hydrogen generation technologies, electrochemical water splitting represents a clean and efficient approach to generate molecular hydrogen (H2).19–21 In this regard, an enormous effort has been devoted to developing highly effective and advanced electrocatalysts for hydrogen production.22,23 Although noble metal-free electrocatalytic HER catalysts have been extensively investigated because of the insufficiency and high cost of platinum-based catalysts,16,24–26 the large Tafel slope due to the sluggish mechanism and overpotential with small exchange current density have hampered the widespread application of these noble metal-free catalysts toward practical hydrogen generation.27 Therefore, platinum-based nanomaterials such as Pt nanoparticles (Pt NPs)28 and Pt nanocubes29 are still the most widely used catalysts for the electrocatalytic HER at their lowest overpotential and highest exchange current density.Rational engineering of nanostructures with high surface area, selective activity and long-term durability is an emerging field of interest for developing efficient electrocatalysts.30–32 Supramolecular self-assembling systems with various molecular interactions offer a wide range of nanoscale architectures that mimic natural systems.33 In recent years, biomimetic self-assembled systems have emerged as a sustainable template for the synthesis of inorganic nanomaterials for various applications.34,35 The self-assembled systems offer numerous possibilities for surface functionalization and sustainable conditions for metal ion binding and the formation of inorganic nanomaterials.36,37 The molecular self-assembly offers control over the nanomaterial surface and morphology to tune the catalytic activity of the formed nanomaterials.38–41 Also, the nanomaterials doped in self-assembling systems help to enhance the catalytic activity towards various electrochemical processes.42,43 Peptides with various functionalities can self-assemble into various supramolecular nanostructures with large structural diversity on the micrometric and nanometric scale.44 Peptides with various functional groups as strong metal-binding sites such as –COOH, –OH and –SH and secondary structures lead to stronger interaction between the peptide and inorganic nanomaterial. The synthesis of metal nanoparticles (NPs) in a self-assembled gel phase is an appealing research area. In recent years, several peptides have been engineered to synthesize inorganic nanomaterials without using additional external reducing reagents. Specifically, the synthesis of metallic nanoparticles (NPs) with a controlled arrangement has been achieved using the self-assembly of peptide molecules.45 Zhou et al. used the aniline-GGAAKLVFF peptide to stabilize Pt NPs on amyloid-like fibrils.46 The positive charge on the amyloid-like fibrillar surface offers convenient ways to functionalize the surface with metal ions. Kim et al. introduced an approach for synthesizing programmed superstructures of AuPt NPs on carbon nanotubes using peptide self-assembly.47 The 3D cross-linked networks of swollen gels offer a large open space that serves as a nanoreactor for the growth and nucleation of nanoparticles. The self-assembled gel phase plays a pivotal role in the controlled growth of NPs along with long-term stability and longevity.48,49 Zheng et al. have reported an approach to synthesise self-supported porous monoliths using self-assembly of noble metallic nanospheres and nanowire grafted organic ligands. The self-assembled noble metal nanoparticle-based aerogel displayed more active sites with a low overpotential for the electrochemical HER.48
In this work, we adopt a facile and ingenious approach to prepare ultrafine platinum nanoparticles (Pt NPs) with a particle size of 2.3 nm decorated on the cross-linked nanofibrillar network of a self-assembled peptide bolaamphiphile. The cross-linked nanofibrillar network of peptide bolaamphiphiles is an ideal support for in situ generated Pt NPs. The synthesized Pt NPs on the self-assembled peptide bolaamphiphile network (i.e. Pt@hydrogel) were explored as an active catalyst for the electrochemical HER. The Pt@hydrogel exhibits better hydrogen evolution reaction (HER) performance with low Pt loading than the state-of-the-art commercial Pt/C catalyst. The nanofibrillar network of the peptide bolaamphiphile provides long-term stability to the Pt@hydrogel, which synergistically accelerates HER activity. This research work opens new routes toward the applications of peptide bolaamphiphile hydrogel in the near future.
2. Experimental methods
2.1 Preparation of the peptide bolaamphiphile hydrogel
For the preparation of the hydrogel, 20 mM (15 mg) peptide bolaamphiphile was dispersed in a glass vial having an internal diameter of 10 mm. Then 1 mL phosphate buffer solution (pH 8, 20 mM) was added and sonicated for about 5 minutes. The above solution was allowed to rest, and the self-supporting hydrogel was obtained within 15 minutes. The minimum gelation concentration (MGC) of the hydrogel in phosphate buffer was found to be 10 mM.2.2 Preparation of Pt@hydrogel
In a typical experiment, 15 mg of 20 mM peptide bolaamphiphile was suspended and dispersed in glass vials containing 1 mL sodium phosphate buffer. The pH of the buffer was maintained at 8 throughout the process. Subsequently, 2, 4, 6, and 8 mg of powdered K2PtCl4 salt were added into the phosphate buffer solution containing the pre-mixed peptide bolaamphiphile. The resultant mixtures were vortexed and repeatedly sonicated for the uniform distribution of K2PtCl4 with the peptide bolaamphiphile. Gelation was achieved within 15 min. The synthesis of Pt nanoparticle doped hydrogels, i.e. Pt2@hydrogel, Pt4@hydrogel, Pt6@hydrogel and Pt8@hydrogel, was confirmed visually by the change in colour from light yellow to brown after 36 h (Fig. S1†).2.3 Pt@hydrogel/CP electrode preparations
The catalyst modified bare carbon paper (CP) (1 × 1 cm2) was used as the working electrode. Before each test, bare CP was washed with methanol and 1 M HCl followed by sonication to obtain a clean surface. 3 mg of the lyophilized Pt@hydrogel electrocatalyst was dispersed in 0.5 mL (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v) ethanol/water mixed solvent with gradual addition of 50 μL Nafion. The above mixture was sonicated and vortexed for about 30 min to prepare homogeneously dispersed catalyst ink. 100 μL of ink was drop-cast onto bare CP and kept to dry under high vacuum for 4 h to fabricate Pt@hydrogel/CP. The resulting Pt loading on the working electrode was 0.168 mg cm−2. Similarly, a hydrogel/CP electrode was prepared using a peptide bolaamphiphile xerogel.
:5 v/v) ethanol/water mixed solvent with gradual addition of 50 μL Nafion. The above mixture was sonicated and vortexed for about 30 min to prepare homogeneously dispersed catalyst ink. 100 μL of ink was drop-cast onto bare CP and kept to dry under high vacuum for 4 h to fabricate Pt@hydrogel/CP. The resulting Pt loading on the working electrode was 0.168 mg cm−2. Similarly, a hydrogel/CP electrode was prepared using a peptide bolaamphiphile xerogel.
2.4 Electrochemical measurements
Electrochemical hydrogen evolution reaction (HER) measurements of the catalyst were conducted on Autolab potentiostat PGSTST309 at room temperature with a traditional three-electrode system. A Pt mesh and saturated Ag/AgCl electrode served as the counter and reference electrodes, respectively. Before electrochemical analysis, all electrolytes were purged with H2 for 0.5 h. LSV curves were recorded in saturated H2, 0.5 M H2SO4, 1 M phosphate buffer (pH 7) and 1 M KOH solution at a scan rate of 2 mV s−1. The electrochemical impedance spectroscopy (EIS) experiments were carried out in the frequency range of 0.01 Hz to 105 Hz. The electrochemically active surface area (ECSA) was assessed by the double-layer capacitance (Cdl) quantified from CV curves at variable scan rates (20–200 mV s−1). For 0.5 M H2SO4 and 1 M phosphate buffer electrolyte, the Ag/AgCl electrode (3 M KCl) was used as the reference electrode. However, for 1 M KOH electrolyte, a Hg/HgO reference electrode was used. The Ag/AgCl reference electrode was calibrated in a standard three-electrode cell using a Pt mesh as a counter electrode and Pt wire as a working electrode (Fig. S2†) at different concentrations of H2SO4. All potential values of the HER were calibrated to a reversible hydrogen electrode (RHE) using the equation E(RHE) = E(Ag/AgCl) + 0.51 in 0.5 M H2SO4.3. Results and discussion
3.1 Self-assembly of the peptide bolaamphiphile
Low molecular weight hydrogels (LMWHs) are an interesting field of soft materials and have drawn considerable attention owing to their potential application in diversified areas from biotechnology to advanced materials.50,51 In a previous report, Pt nanoparticles supported on the nanofibrillar network of a peptide bolaamphiphile hydrogel were used as a catalyst for the reduction of p-nitroaniline to p-phenylenediamine.52 In this study, we have synthesised a symmetrical peptide bolaamphiphile, i.e. HO–Y–F–GluA–F–Y–OH (Y: L-tyrosine, F: L-phenylalanine, GluA: glutaric acid), with a central glutaric acid residue by solution-phase peptide synthesis. The synthesis of the peptide bolaamphiphile was confirmed using various spectroscopic methods. The gelation properties of the synthesized peptide bolaamphiphile were studied in phosphate buffer solution at different pHs. The gelator molecules self-assemble at pH 8.0 and form a robust self-supporting hydrogel upon successive sonication under physiological conditions. The formation of a self-supporting hydrogel was confirmed by the vial inversion method (Scheme 1). The phenylalanine and tyrosine based peptide bolaamphiphile offers the necessary hydrophobic and π–π stacking interactions required for the self-organization of gelator molecules.53 In addition, hydrogen bonding interactions that originated from the amide groups of linker amino acids lead to the self-aggregation of peptide bolaamphiphiles. The self-assembled nanostructure of the peptide-based bolaamphiphile was examined by FE-SEM and TEM analysis. The formation of a fibrous morphology was observed by FE-SEM analysis (Fig. 1a) which reveals that the nanofibrillar network structure is responsible for forming the peptide bolaamphiphile based hydrogel. The self-assembled peptide bolaamphiphile nanofibers interconnect to form a highly dense cross-linked nanofibrillar network having an average fiber diameter of 55 nm. The nanoscale fibrous morphology was also observed in the TEM analysis of the diluted solution of the hydrogel, which shows the average widths of the nanofibrils in the range of 70 nm (Fig. 1b).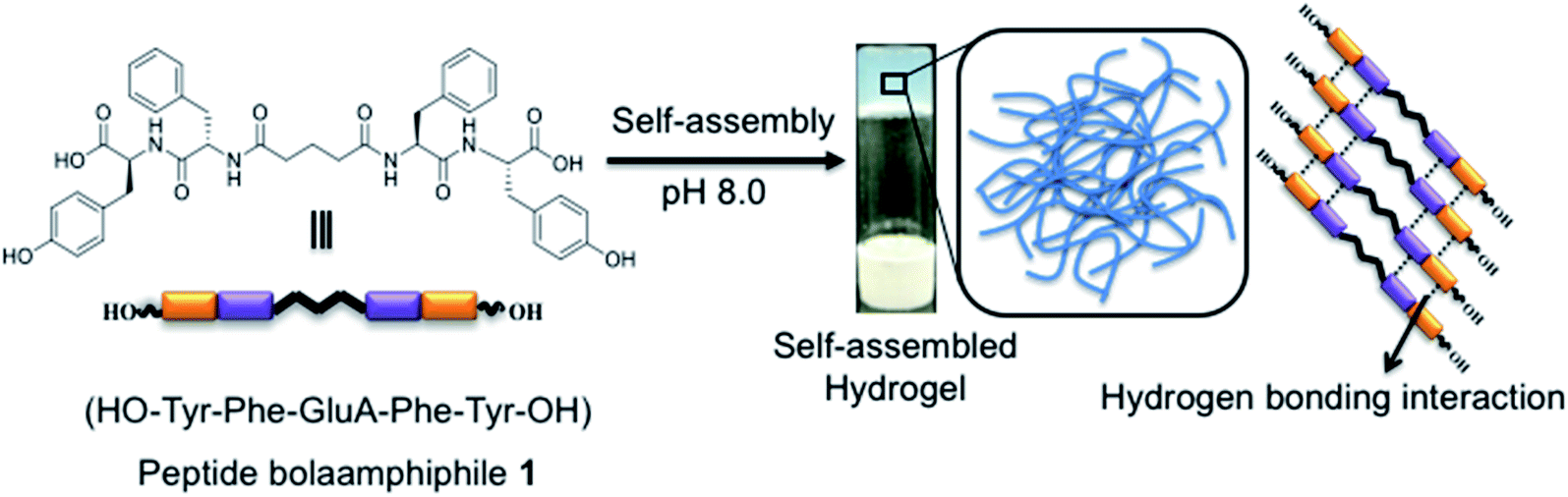 | ||
| Scheme 1 Schematic representation of the formation of the peptide bolaamphiphile based hydrogel. | ||
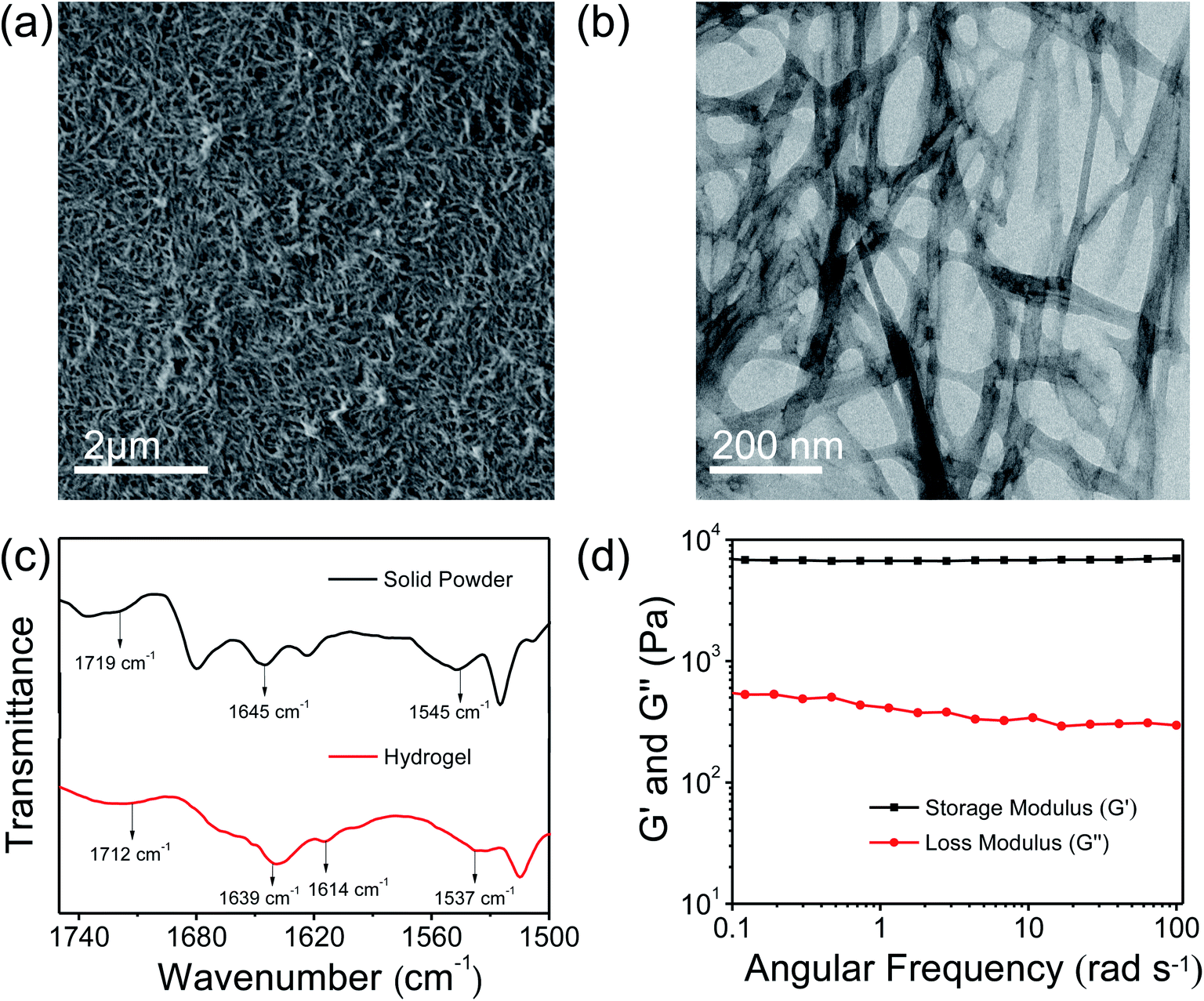 | ||
| Fig. 1 (a) FE-SEM image and (b) TEM image of the peptide bolaamphiphile based hydrogel. (c) FT-IR analysis of the peptide bolaamphiphile based hydrogel and solid powder. (d) Frequency sweep experiment of the peptide bolaamphiphile based hydrogel at a constant strain of 1%. | ||
The self-assembly of peptide bolaamphiphile molecules under optimized conditions using various inter- and intramolecular interactions leads to a self-supporting hydrogel with a cross-linked nanofibrillar network. The peptide-based secondary structures and molecular interactions associated with the formation of the hydrogel were investigated by FT-IR analysis (Fig. 1c). The peak centred at 1719 cm−1 in FT-IR analysis corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the –COOH group of the powdered peptide bolaamphiphile, which shifts at 1712 cm−1 in the gel state. The shift in peak toward lower energy could be ascribed to the involvement of the –COOH group in hydrogen bonding for the self-assembly process. The existence of a prominent amide I band at 1645 cm−1 suggests the disordered arrangement of the peptide bolaamphiphile in a solid powdered state.54 Moreover, the peaks centred at 1639 cm−1 and 1614 cm−1 correspond to the β-sheet arrangement of the self-assembled hydrogel.55 For the solid powder, a peak emerged at 1545 cm−1, which is a representative peak for amide II. The amide II peak was shifted to 1537 cm−1 in the gel state, which indicates the presence of a hydrogen-bonded β-sheet arrangement in the hydrogel. The fluorescence analysis (Fig. S3†) of the hydrogel was performed to investigate the π–π stacking interaction involved during the self-assembly process. The peak at 303 nm in the emission spectrum of the hydrogel suggests the presence of an ordered arrangement of the aromatic side chains of phenylalanine and tyrosine. Additionally, a more prominent peak emerged at 468 nm, which is in accordance with the higher-order self-assembly of peptide bolaamphiphile molecules in the hydrogel.56 The morphological and spectroscopic analysis show that the self-assembly behaviour of the peptide bolaamphiphile is controlled by π–π stacking interactions and hydrogen bonding interactions.
O stretching vibration of the –COOH group of the powdered peptide bolaamphiphile, which shifts at 1712 cm−1 in the gel state. The shift in peak toward lower energy could be ascribed to the involvement of the –COOH group in hydrogen bonding for the self-assembly process. The existence of a prominent amide I band at 1645 cm−1 suggests the disordered arrangement of the peptide bolaamphiphile in a solid powdered state.54 Moreover, the peaks centred at 1639 cm−1 and 1614 cm−1 correspond to the β-sheet arrangement of the self-assembled hydrogel.55 For the solid powder, a peak emerged at 1545 cm−1, which is a representative peak for amide II. The amide II peak was shifted to 1537 cm−1 in the gel state, which indicates the presence of a hydrogen-bonded β-sheet arrangement in the hydrogel. The fluorescence analysis (Fig. S3†) of the hydrogel was performed to investigate the π–π stacking interaction involved during the self-assembly process. The peak at 303 nm in the emission spectrum of the hydrogel suggests the presence of an ordered arrangement of the aromatic side chains of phenylalanine and tyrosine. Additionally, a more prominent peak emerged at 468 nm, which is in accordance with the higher-order self-assembly of peptide bolaamphiphile molecules in the hydrogel.56 The morphological and spectroscopic analysis show that the self-assembly behaviour of the peptide bolaamphiphile is controlled by π–π stacking interactions and hydrogen bonding interactions.
Rheological studies were conducted to determine the mechanical properties of the peptide bolaamphiphile hydrogel. The strain-dependent amplitude sweep measurements confirm the shear-thinning behaviour and anti-shear ability of the hydrogel at a fixed frequency of 10 rad s−1 (Fig. S4†). The amplitude sweep experiment of the hydrogel at a low strain range (0.1 to 10%) maintains its viscoelastic nature because the storage modulus (G′) and loss modulus (G′′) are almost constant at low strain. On the other hand, at higher strain range (>10%), G′ becomes lower than G′′, indicating that the hydrogel loses its viscoelastic nature and turns to the sol state at 99% strain. Frequency sweep analysis (Fig. 1d) was further performed at a constant strain of 1%. The frequency sweep responses reveal that the storage modulus (G′) remained larger than the loss modulus (G′′) throughout the process, indicating the formation of a robust hydrogel. Furthermore, at the low-frequency region G′ is higher than 103 Pa, while the value of storage modulus (G′) is substantially greater than that of the loss modulus (G′′) in the high-frequency region. The chemical surface composition of the peptide bolaamphiphile hydrogel was further investigated by XPS analysis. The high resolution XPS survey profile confirms the existence of C, N, and O elements in the hydrogel (Fig. S5a†). As shown in Fig. S5b,† the C 1s XPS exhibits two characteristic peaks at 284.8 and 284.2 eV attributable to C–C and CC moieties. Similarly, the peaks at 285.5 and 286.2 eV correspond to the presence of C–N and CO groups.57 The high resolution XPS profile of O 1s (Fig. S5c†) displays peaks at 531.5, 532.4 and 533.4 eV which can be ascribed to the C–O, CO and O–C–O bonding interactions.
3.2 Synthesis of Pt@hydrogel
The three dimensional (3D) network offered by supramolecular hydrogels is an important framework for preparing NPs because peptide-based biomaterials are usually biocompatible and nontoxic.49,52 Herein, we present in situ construction of Pt NPs within the matrix of the peptide bolaamphiphile based hydrogel (Scheme 2). The designed tyrosine-based gelator molecules have the unique property of synthesizing nanoparticles within the gel matrix without any external reducing or capping agent. For this purpose, the solid peptide bolaamphiphile was dissolved in sodium phosphate buffer and sonicated for about a minute; then K2PtCl4 salt was added to this solution and again sonicated for 2 min. A transparent self-supporting hydrogel was formed within 15 min upon sonication. The formation of the hydrogel confirms trapping of the Pt precursor within the 3D matrix of the nanofibrillar network. The appearance of a light yellow colour started within 3 h and later, after 24 h, an intense yellow colour was observed. The reduction of Pt(II) to Pt(0) was primarily confirmed by the brown colour appearance of Pt@hydrogel. Several studies report the interaction between surface accumulated –OH groups and Pt(II) ions leading to subsequent reduction of Pt(II) to Pt(0) following the solvolysis reaction pathway.58 The reduction of Pt(II) and subsequent formation of Pt NPs within the cross-linked nanofibrillar network of the peptide bolaamphiphile hydrogel corresponds to the presence of a redox active tyrosine side residue. In the aqueous phosphate buffer solution of gelator peptide and K2PtCl4, the PtCl42− ions undergo the following solvolysis reactions,| PtCl42− + H2O ⇄ PtCl3(H2O)− + Cl− |
| PtCl3(H2O)− + H2O ⇄ PtCl2(H2O)2 + Cl− |
| 2Tyr-OH + PtCl2 → Tyr-O-Tyr + Pt + 2HCl |
 | ||
| Scheme 2 Schematic representation of the synthesis of the Pt nanoparticle doped Pt@hydrogel. | ||
The Pt ions interact with the peptide bolaamphiphile and form a complex by interacting with the hydroxyl groups of tyrosine. The successive proton–electron transfer process leads to the reduction of Pt(II) to Pt(0). As a result, Pt nanoparticles are formed on the peptide fiber surface.
The 3D cross-linked nanofibrillar network allows the nucleation process following the growth of Pt NPs. Recent studies have shown that the mechanical stability of hydrogels can be largely affected by the presence of NPs.59,60 To investigate whether the rheological performance of the hybrid Pt@hydrogel was influenced by the inclusion of Pt nanoparticles, we carried out rheological studies at a given concentration of 20 mM. The oscillatory frequency sweep analysis of Pt@hydrogel (Fig. 2a) reveals that the storage modulus (G′) and loss modulus (G′′) remain almost invariant, and the value of storage modulus (G′) is considerably greater than that of loss modulus (G′′) over the entire experimental frequency limit. This rheological result reveals that the hybrid Pt@hydrogel system also serves as a solid-like gel material. Similarly, upon incorporating Pt NPs the storage modulus (G′) of the hybrid Pt@hydrogel successively increases to G′ = 13557 Pa compared to the native hydrogel, which exhibits a storage modulus (G′) of 6912 Pa. This rheological data imply that the rigidity of the Pt@hydrogel is more than that of the native hydrogel. Furthermore, the synthesis of Pt NPs within the hydrogel matrix was monitored using UV-vis absorption spectroscopy. The UV-vis spectrum (Fig. 2b) of the K2PtCl4 solution shows two absorption peaks of K2PtCl4 salt at 327 nm and 386 nm. However, the UV-vis spectrum of Pt@hydrogel displays the absence of K2PtCl4, suggesting the complete reduction of K2PtCl4 salt to Pt NPs. In addition, the nanofibrillar network of the peptide bolaamphiphile shows outstanding stability towards the nascent Pt nanoparticles for a long time under ambient conditions.
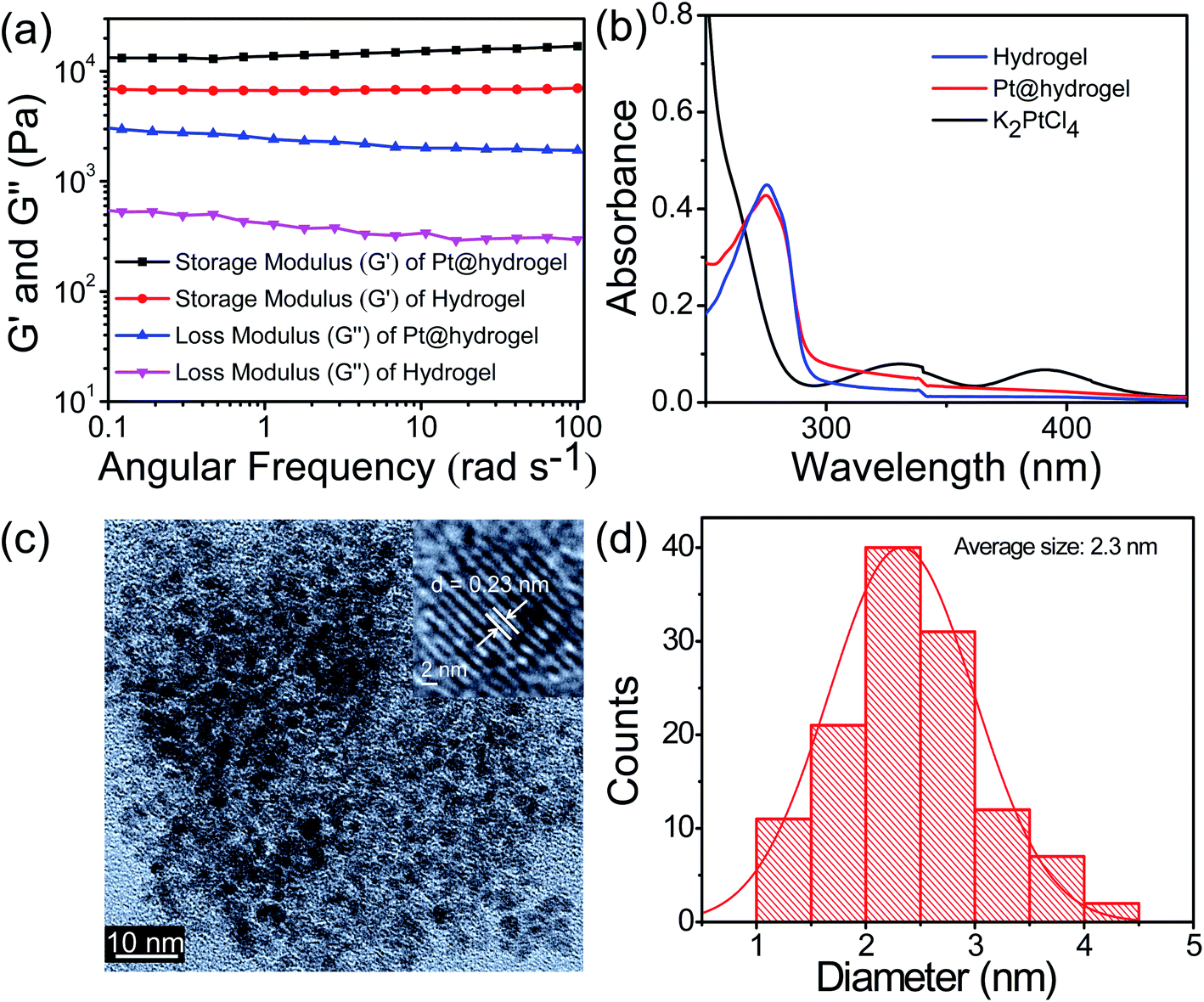 | ||
| Fig. 2 (a) Comparison study of frequency sweep measurements of the peptide bolaamphiphile based hydrogel and Pt@hydrogel at a constant strain of 1%. (b) UV-vis spectra of the hydrogel (blue line), Pt@hydrogel (red line) and K2PtCl4 (black line). (c) HR-TEM image of Pt nanoparticles doped into the peptide bolaamphiphile based hydrogel (the inset shows the lattice fringes of Pt). (d) Histogram showing the size of Pt nanoparticles acquired from the HR-TEM image i.e. (c) (calculated using ImageJ software). | ||
The size distribution and morphology of Pt nanoparticles on peptide bolaamphiphile nanofibers were analysed by TEM. A high-resolution TEM image (Fig. 2c) of Pt6@hydrogel shows uniform and well-defined spherical Pt NPs distributed over the nanofibrillar network of a peptide bolaamphiphile hydrogel. The inset in Fig. 2c displays clearly visible lattice fringes with an interplanar spacing of 0.23 nm obtained from the corresponding HR-TEM image. Similarly, Fig. 2d exhibits the histogram of Pt@hydrogel acquired from the corresponding HR-TEM analysis, which suggests the presence of Pt NPs with an average particle size of 2.3 nm. The peptide bolaamphiphile nanofibers with phenolic residues induce the formation of Pt NPs by the reduction of Pt(II) to Pt(0).
The formation of Pt NPs over peptide bolaamphiphile nanofibers was further confirmed by X-ray photoelectron spectroscopy (XPS). The XPS survey profile of Pt@hydrogel (Fig. 3a) confirms the existence of Pt, C, O and N. The core level XPS profile of C 1s (Fig. 3b) shows the deconvoluted peaks at 284.6 and 284.2 eV attributed to the existence of C–C and CC bonding interactions, respectively.61 The additional peaks at 285.5 and 286.8 eV correspond to C–N and CO bonding interactions present in the peptide bolaamphiphile molecule.61,62 The high-resolution O 1s (Fig. 3c) profile displays the presence of peaks at 532.5, 531.4 and 533.4 eV attributed to the presence of CO, C–O and O–C–O bonding interactions.63 The core level Pt 4f spectrum (Fig. 3d) contains one pair of high-intensity doublets attributable to the Pt 4f7/2 and Pt 4f5/2 splitting. The binding energy at 71.1 eV (for 4f7/2 peak) and 74.3 eV (for 4f5/2 peak) is ascribed to metallic Pt(0),64 suggesting the successful synthesis of Pt nanoparticles on the nanofibrillar network of the peptide bolaamphiphile hydrogel. The XPS analysis confirms the successful conversion of Pt(II) to Pt(0).
 | ||
| Fig. 3 (a) XPS survey profile of Pt@hydrogel with the high-resolution XPS profiles of (b) C 1s, (c) O 1s, and (d) Pt 4f. | ||
3.3 Hydrogen evolution reaction activity of Pt@hydrogel
The catalytic activity for electrochemical hydrogen generation of Pt@hydrogel was studied by several electrochemical techniques such as LSV, Tafel slope analysis and EIS. The effect of Pt amount on electrocatalytic activity was investigated with the synthesis of four Pt doped hydrogels with varying amount of K2PtCl4 during Pt@hydrogel synthesis. The Pt2@hydrogel, Pt4@hydrogel, Pt6@hydrogel and Pt8@hydrogel were synthesized using 2 mg, 4 mg, 6 mg and 8 mg of K2PtCl4, respectively (Table S1†). The iR-corrected LSV curve of Pt6@hydrogel/CP (Fig. S6a†) shows higher catalytic performance for the electrocatalytic HER in 0.5 M H2SO4. The Pt6@hydrogel/CP requires a low overpotential of 45 mV to achieve a current density of −10 mA cm−2. In contrast, Pt2@hydrogel/CP and Pt4@hydrogel/CP exhibited overpotentials of 140 and 133 mV, respectively, to reach a current density of −10 mA cm−2. On the other hand, on further increasing the percentage of Pt loading, a sharp decrease in the performance of Pt8@hydrogel/CP (overpotential of 136 mV to reach −10 mA cm−2 current density) compared to Pt6@hydrogel/CP was observed. This could be due to the agglomeration of Pt NPs upon higher Pt loading in the nanofibrillar network of the peptide bolaamphiphile.65,66 Fig. S6b† compares the Tafel slopes of Pt2@hydrogel/CP, Pt4@hydrogel/CP, Pt6@hydrogel/CP and Pt8@hydrogel/CP derived from the corresponding LSV curves. Pt6@hydrogel/CP shows a smaller Tafel slope (Table S2†) of 52 mV dec−1 than Pt2@hydrogel/CP (81 mV dec−1), Pt4@hydrogel/CP (76 mV dec−1) and Pt8@hydrogel/CP (77 mV dec−1), suggesting the significantly higher HER performance of Pt6@hydrogel/CP. The improved HER activity of the catalysts was also validated through charge transfer resistance (Rct) between the electrode and electrolyte. Nyquist plots (Fig. S6c†) show a dramatically lowered charge transfer resistance (Rct) of 0.68 Ω for Pt6@hydrogel/CP in comparison to that of Pt2@hydrogel/CP (1.9 Ω), Pt4@hydrogel/CP (0.87 Ω) and Pt8@hydrogel/CP (1.76 Ω). The smaller Rct value for Pt6@hydrogel/CP corresponds to the higher HER performance in 0.5 M H2SO4 solution. To further assess the catalytic activity of the as-prepared Pt6@hydrogel/CP, Pt2@hydrogel/CP, Pt4@hydrogel/CP and Pt8@hydrogel/CP, the ECSA was analysed by conducting CV analysis at different scan rates (Fig. S7†). The ECSA was measured using the following equation:7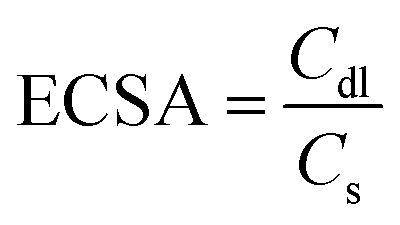 | (1) |
Furthermore, the catalytic activity of carbon paper (CP), bare Pt nanoparticles and commercially available 10% Pt/C towards the HER was also analysed for comparative analysis under identical conditions. In acidic 0.5 M H2SO4 solution, Nafion binder containing Pt6@hydrogel/CP shows stable behaviour and higher conductivity with higher electrochemical catalytic activity. In H2SO4 solution, the iR-corrected LSV curves were recorded (Fig. 4a) at a scan rate of 2 mV s−1. The electrodes prepared with Pt/C and Pt6@hydrogel/CP show higher catalytic performance. However, the conductive CP and hydrogel/CP exhibit poor catalytic activity with low current densities. The 10% Pt/C requires a low overpotential of 37 mV to attain a current density of −10 mA cm−2 (Table S3†). In sharp contrast, Pt6@hydrogel/CP displays outstanding HER activity with the requirement of an overpotential of 45 mV to attain a cathodic current density of −10 mA cm−2 compared to bare Pt nanoparticles (117 mV at a current density of −10 mA cm−2). The high catalytic activity of Pt6@hydrogel/CP relative to bare Pt NPs/CP and hydrogel/CP reflects the synergistic properties associated with the hybrid interface of Pt6@hydrogel/CP nanofibers.
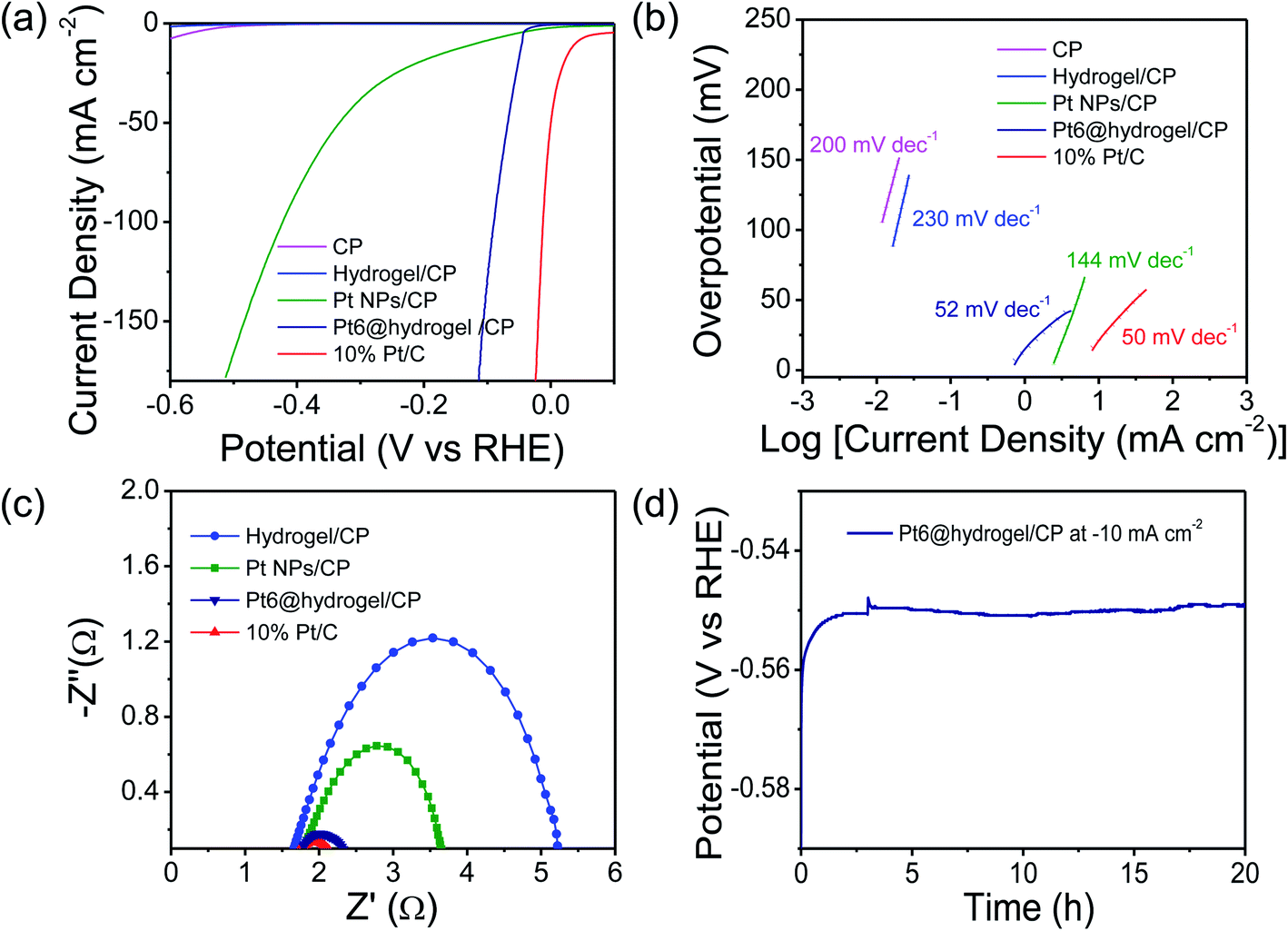 | ||
| Fig. 4 (a) iR-Corrected LSV profiles of CP, hydrogel/CP, Pt NPs/CP, Pt6@hydrogel/CP and 10% Pt/C scanned at 2 mV s−1 in 0.5 M H2SO4 solution; (b) respective Tafel slopes. (c) Nyquist plots of the prepared electrodes measured at a potential of −0.45 V. (d) The chronopotentiometry analysis of Pt6@hydrogel/CP for 20 h in 0.5 M H2SO4. | ||
Insight into the catalytic activity of Pt6@hydrogel/CP was gained using Tafel slopes (Fig. 4b) obtained from LSV profiles. A smaller value of Tafel slope suggests a higher rate of catalytic activity with increasing overpotential. 10% Pt/C shows a Tafel slope of 50 mV dec−1 indicating the highest kinetics for the HER in 0.5 M H2SO4. Moreover, the Pt6@hydrogel/CP shows a smaller Tafel slope of 52 mV dec−1 than bare Pt NPs/CP (144 mV dec−1), implying the faster catalytic pathway for the HER at the electrode–electrolyte interface. The LSV and Tafel slope analysis show that the catalytic activity of the Pt doped hydrogel nanofibers is comparable with that of the reported Pt-based electrocatalysts (Table S4†).4,68–73 The electron transfer kinetics of the prepared electrodes was investigated using EIS analysis. The Nyquist plots (Fig. 4c) of hydrogel/CP, 10% Pt/C, Pt NPs/CP and Pt6@hydrogel/CP were fitted to the equivalent circuit model for the determination of the charge-transfer resistance (Rct) of the electrode. A lower charge transfer resistance at the electrocatalytic interface suggests higher charge transport for electrochemical processes. As expected, hydrogel/CP shows high Rct of 4.8 Ω; however, 10% Pt/C shows lower Rct of 0.59 Ω revealing faster charge transfer and enhanced catalytic activity of Pt/C. Compared to Pt NPs/CP (2 Ω), the Pt6@hydrogel/CP displays Rct of 0.68 Ω at an applied potential of −0.45 V. The low Rct suggests that Pt6@hydrogel/CP attains an advantageous structure for charge transfer and good electrical conductivity during the HER. The chronopotentiometry analysis of Pt6@hydrogel/CP was extended to 20 h (Fig. 4d) at a constant −10 mA cm−2 which further proves the stable behaviour of Pt6@hydrogel/CP over a longer period of time. Furthermore, the iR compensated LSV curve of Pt6@hydrogel/CP after the long-term stability test was also compared with the initial LSV curve obtained before the stability test (Fig. S9†). We observed a slight decrease in the electrocatalytic performance which can be attributed to the leaching of the catalyst due to bubbling of hydrogen gas on the surface of the electrode. The FE-SEM analysis was carried out to investigate the surface morphology of Pt6@hydrogel/CP deposited on CP (working electrode) before and after the stability test. Fig. S10a† displays the FE-SEM micrograph of bare CP having a smooth surface of carbon fibres. Fig. S10b† presents the FE-SEM micrograph of Pt6@hydrogel/CP, which shows the substantial distribution of catalyst nanoparticles on the surface of CP. Compared to bare CP and Pt6@hydrogel/CP (Fig. S10a and b†), the surface of Pt6@hydrogel/CP (Fig. S10c†) is more exposed and porous after the long-term stability test. Similarly, the change in the chemical state of Pt6@hydrogel/CP after the long-term stability test was investigated by XPS. The XPS survey profile of the catalyst (Fig. S11a†) displays the characteristic peaks of C 1s, O 1s, N 1s and Pt 4f which implies the presence of C, O, N and Pt atoms in the Pt6@hydrogel/CP. Fig. S11b† demonstrates the high resolution XPS profile of O 1s which showed the existence of C–O–M bonding interaction after the stability test. Besides that, a change in the high resolution XPS spectrum of C 1s was also seen. Further to this, the high resolution XPS profile of C 1s displays characteristic C–C, CC, C–N, and CO bonding interactions (Fig. S11c†). However, two additional peaks appeared at 291 eV and 292 eV which correspond to the OC–O and CF2 moieties of the Nafion binder.74 Furthermore, no significant change in the high resolution Pt 4f spectrum (Fig. S11d†) of the catalyst after the electrocatalytic performance was observed suggesting the stable behaviour of Pt6@hydrogel/CP on the electrode surface.
We further determined the ECSA to assess the catalytic activity of the catalysts. The Pt6@hydrogel/CP exhibits an ECSA of 0.14 cm2, which is significantly higher than those of hydrogel/CP (ECSA = 0.003 cm2) and Pt NPs/CP (ECSA = 0.005 cm2) but lower than that of Pt/C (ECSA = 0.15 cm2) (Fig. S12 and S13†). The ECSA results reveal the higher HER activity of Pt6@hydrogel/CP, which could be ascribed to the synergistic integration of Pt nanoparticles with the cross-linked nanofibrillar network of the peptide bolaamphiphile hydrogel. The electrocatalytic HER activity of Pt6@hydrogel/CP was also investigated under acidic conditions using different concentrations of H2SO4 electrolyte. Fig. S14a† displays the iR corrected LSV curves of Pt6@hydrogel/CP in 0.1 M H2SO4 and 1 M H2SO4. In 1 M H2SO4, Pt6@hydrogel/CP requires an overpotential of 48 mV (Table S5†) to reach a current density of −10 mA cm−2 which is lower than that in 0.1 M H2SO4 (56 mV). Besides, Pt6@hydrogel/CP exhibits a Tafel slope of 55 mV dec−1 in 1 M H2SO4 which is lower than that in 0.1 M H2SO4 (75 mV dec−1) (Fig. S14b†) suggesting higher HER kinetics in 1 M H2SO4.
The HER performance was also evaluated in alkaline and buffer media because water electrolysis is the most commonly used technique in industry. However, the HER activity in phosphate buffer and alkaline medium is lower than that in acidic medium.12 Therefore, to investigate the HER activity of the as-prepared Pt6@hydrogel in alkaline medium and phosphate buffer simultaneously, all the experiments discussed above were performed in 1 M KOH and 1 M phosphate buffer (pH = 7.0) solutions (Tables S6 and S7†). Fig. S15a† shows the iR-corrected LSV of the HER at 2 mV s−1 in 1 M KOH catalyzed by CP, hydrogel/CP, Pt NPs/CP, Pt6@hydrogel/CP and 10% Pt/C catalysts. The Pt6@hydrogel/CP requires a relatively low overpotential of 140 mV to produce a current density of −10 mA cm−2 which is lower than that of bare Pt NPs/CP (131 mV at a current density of −10 mA cm−2). The 10% Pt/C exhibits the best HER performance with an overpotential of 34 mV to attain a current density of −10 mA cm−2. In addition, CP and hydrogel/CP show poor HER performance, which is consistent with the expected behaviour. Besides, the Tafel slope was found to be 416 mV dec−1 for Pt6@hydrogel/CP, larger than those of bare Pt NPs/CP (101 mV dec−1) and Pt/C (32 mV dec−1), which is consistent with the higher HER activity of bare Pt NPs/CP and 10% Pt/C in 1 M KOH than Pt6@hydrogel/CP (Fig. S15b†). The EIS measurements were further conducted to understand the intrinsic performance of the as-obtained electrodes. The Nyquist plots of CP, hydrogel/CP, Pt NPs/CP, Pt6@hydrogel/CP, and Pt/C are presented in Fig. S15c† to determine the charge transfer resistance (Rct). It is evident from the Nyquist plots that the commercially available 10% Pt/C shows low Rct of 0.6 Ω compared to bare Pt NPs/CP (1.8 Ω) and Pt6@hydrogel/CP (4.2 Ω), respectively, while hydrogel/CP exhibits a maximum value of 13.8 Ω at an applied potential of −0.45 V. These results are consistent with the slower electrocatalytic reaction rate for Pt6@hydrogel/CP than bare Pt NPs/CP and 10% Pt/C in 1 M KOH. The durability of Pt6@hydrogel/CP was studied by conducting a long-term stability test at a fixed current density of −10 mA cm−2 for up to 5 h (Fig. S15d†) without an obvious current drop. Moreover, the LSV curve exhibits a slight decrease in performance after the stability test (Fig. S15d,† inset).
Furthermore, the iR-compensated LSV curves for hydrogel/CP and CP in 1 M phosphate buffer (pH = 7.0) display a straight line with almost negligible current across a tested voltage range indicating no HER activity (Fig. S16a†). Similarly, bare Pt NPs/CP shows a significantly higher overpotential of 491 mV at a current density of −10 mA cm−2 than 10% Pt/C, which requires a greatly reduced overpotential of 418 mV. On the other hand, Pt6@hydrogel/CP requires an overpotential of 424 mV to achieve a current density of −10 mA cm−2 which is comparable to that of the Pt/C. Fig. S16b† shows the corresponding Tafel plots of hydrogel/CP, CP, Pt NPs/CP, Pt6@hydrogel/CP and 10% Pt/C derived from the LSV curves. A Tafel slope of 238 mV dec−1 for Pt6@hydrogel/CP was obtained, which is much higher than those of bare Pt NPs/CP (33 mV dec−1) and 10% Pt/C (29 mV dec−1), respectively. In order to better understand the HER catalytic performance of hydrogel/CP, Pt6@hydrogel/CP, Pt NPs/CP and 10% Pt/C, EIS was carried out (Fig. S16c†). The hydrogel/CP manifests a maximum Rct of 17.2 Ω, while the Rct of Pt6@hydrogel/CP is 9.1 Ω and those of Pt NPs/CP and 10% Pt/C are 4.1 Ω and 3.8 Ω, respectively. These results reveal that the prepared Pt6@hydrogel/CP electrode possesses lower HER activity in 1 M phosphate buffer (pH = 7.0) than bare Pt NPs/CP and Pt/C. The long-term stability is another important parameter for electrocatalysts in the HER process. As shown in Fig. S16d,† Pt6@hydrogel/CP remains stable even after 5 h. In addition, the LSV curves evaluated before and after chronopotentiometry (Fig. S16d,† inset) indicate that Pt6@hydrogel/CP shows long-term electrochemical stability.
The electrocatalytic performance of the catalyst depends upon its composition and surface structure.75 Several studies have shown that short amino acid functionalized peptide nanomaterials offer a novel approach to modulate electrocatalytic HER activity. Peptides have the ability to modify catalytic activity by controlling surface exposure to the solution. Nanoparticles in the 3D matrix of peptides could vary surface exposure by varying the degree of peptide binding to the catalyst surface. The self-assembled peptides can also increase proton transfer kinetics to enhance the kinetics of the HER process. The 3D hydrogel matrix of the bolaamphiphile with various functional groups such as carboxylic acid could act as a proton donor in the near-surface electrode environment and promote proton conduction.76 Aromatic amino acids such as tyrosine and tryptophan peerlessly stimulate proton conductivity and thus have an impact on promoting the HER.77 In this study, the Pt ions interact with the peptide bolaamphiphile and form a complex by interacting with the hydroxyl groups of tyrosine. The successive proton–electron transfer process leads to the reduction of Pt(II) to Pt(0). The Pt(0) formed on the fiber surface forms Pt nanoparticles. The presence of phenylalanine and tyrosine improves the HER by tuning the surface composition and proton conductivity during the electrocatalytic HER process.
4. Conclusion
In summary, we have reported the in situ synthesis of Pt NPs in the 3D cross-linked network of the peptide bolaamphiphile hydrogel based on the molecular self-assembly strategy. These cross-linked nanofibers of the peptide bolaamphiphile were effectively used as a scaffold to synthesize well-dispersed Pt NPs with a particle size of 2.3 nm as supported by the microscopic study. The ultrafine Pt NPs stabilized over a cross-linked nanofibrillar network of the peptide bolaamphiphile have been utilized as an electrocatalyst for the HER. The hybrid interface of Pt6@hydrogel/CP displayed excellent catalytic activity toward the electrochemical HER in 0.5 M H2SO4 solution. The Pt@hydrogel showed an overpotential of 45 mV at −10 mA cm−2 with a Tafel slope of 52 mV dec−1. The Pt6@hydrogel/CP exhibited a low charge transfer resistance of 0.68 Ω and showed outstanding stability for 20 h of reaction time. Similarly, the HER activity of Pt6@hydrogel/CP was also explored in 1 M KOH and 1 M phosphate buffer, which showed relatively lower activity, proving that 0.5 M H2SO4 was optimum for high HER performance. The excellent activity and stability of Pt6@hydrogel/CP for the HER show great potential for energy conversion applications. The current approach could be prolonged for developing diverse nanostructured materials anchored on supramolecular architectures of biomolecules for energy conversion applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Indian Institute of Technology Indore. D. K. K. K., L. D. and R. G. J. thank the University Grants Commission (UGC), Government of India for their doctoral fellowships. The authors thank the Sophisticated Instrumentation Centre (SIC), IIT Indore, for providing all other instrumental facilities. The authors thank SAIF, IIT Bombay for HR-TEM analysis, and IIC, IIT Roorkee and the Central Surface Analytical Facility, IIT Bombay for XPS measurements.References
- Y. Shen, A. C. Lua, J. Xi and X. Qiu, ACS Appl. Mater. Interfaces, 2016, 8, 3464–3472 CrossRef CAS PubMed.
- W. Ren, H. Zhang and C. Cheng, Electrochim. Acta, 2017, 241, 316–322 CrossRef CAS.
- X. Bao, Y. Gong, Y. Chen, H. Zhang, Z. Wang, S. Mao, L. Xie, Z. Jiang and Y. Wang, J. Mater. Chem. A, 2019, 7, 15364–15370 RSC.
- Y. Zhang, J. Yan, X. Ren, L. Pang, H. Chen and S. Liu, Int. J. Hydrogen Energy, 2017, 42, 5472–5477 CrossRef CAS.
- J. Ji, Y. Zhang, L. Tang, C. Liu, X. Gao, M. Sun, J. Zheng, M. Ling, C. Liang and Z. Lin, Nano Energy, 2019, 63, 103849 CrossRef CAS.
- K. Jiang, B. Liu, M. Luo, S. Ning, M. Peng, Y. Zhao, Y. R. Lu, T. S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2019, 10, 1743 CrossRef PubMed.
- R. G. Jadhav, D. Singh, P. V. Krivoshapkin and A. K. Das, Inorg. Chem., 2020, 59, 7469–7478 CrossRef CAS PubMed.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- Z. Xing, C. Han, D. Wang, Q. Li and X. Yang, ACS Catal., 2017, 7, 7131–7135 CrossRef CAS.
- A. Karmakar, K. Karthick, S. Kumaravel, S. S. Sankar and S. Kundu, Inorg. Chem., 2021, 60, 2023–2036 CrossRef CAS PubMed.
- X. Liu, F. Liu, J. Yu, G. Xiong, L. Zhao, Y. Sang, S. Zuo, J. Zhang, H. Liu and W. Zhou, Adv. Sci., 2020, 7, 2001526 CrossRef CAS PubMed.
- G. Sheng, J. Chen, Y. Li, H. Ye, Z. Hu, X. Z. Fu, R. Sun, W. Huang and C. P. Wong, ACS Appl. Mater. Interfaces, 2018, 10, 22248–22256 CrossRef CAS PubMed.
- Z. S. Cai, Y. Shi, S. S. Bao, Y. Shen, X. H. Xia and L. M. Zheng, ACS Catal., 2018, 8, 3895–3902 CrossRef CAS.
- J. Ying, G. Jiang, Z. Paul Cano, L. Han, X. Y. Yang and Z. Chen, Nano Energy, 2017, 40, 88–94 CrossRef CAS.
- S. Kumaravel, K. Karthick, S. S. Sankar, A. Karmakar and S. Kundu, J. Mater. Chem. C, 2021, 9, 1709–1720 RSC.
- A. Guha, T. Veettil Vineesh, A. Sekar, S. Narayanaru, M. Sahoo, S. Nayak, S. Chakraborty and T. N. Narayanan, ACS Catal., 2018, 8, 6636–6644 CrossRef CAS.
- N. Roy, K. T. Leung and D. Pradhan, J. Phys. Chem. C, 2015, 119, 19117–19125 CrossRef CAS.
- X. Li, J. Yu, J. Jia, A. Wang, L. Zhao, T. Xiong, H. Liu and W. Zhou, Nano Energy, 2019, 62, 127–135 CrossRef CAS.
- P. Hota, S. Bose, D. Dinda, P. Das, U. K. Ghorai, S. Bag, S. Mondal and S. K. Saha, ACS Omega, 2018, 3, 17070–17076 CrossRef CAS PubMed.
- M. Sheng, B. Jiang, B. Wu, F. Liao, X. Fan, H. Lin, Y. Li, Y. Lifshitz, S. T. Lee and M. Shao, ACS Nano, 2019, 13, 2786–2794 CrossRef CAS PubMed.
- D. Hou, W. Zhou, X. Liu, K. Zhou, J. Xie, G. Li and S. Chen, Electrochim. Acta, 2015, 166, 26–31 CrossRef CAS.
- W. Lu, W. Li, G. Xiang and L. Wang, Inorg. Chem., 2019, 58, 6529–6533 CrossRef CAS PubMed.
- M. Ogawa, G. Ajayakumar, S. Masaoka, H. B. Kraatz and K. Sakai, Chem.–Eur. J., 2011, 17, 1148–1162 CrossRef CAS PubMed.
- M. Kim, Z. Yang, J. H. Park, S. M. Yoon and B. A. Grzybowski, ACS Appl. Nano Mater., 2019, 2, 2725–2733 CrossRef CAS.
- J. Jia, W. Zhou, G. Li, L. Yang, Z. Wei, L. Cao, Y. Wu, K. Zhou and S. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 8041–8046 CrossRef CAS PubMed.
- Z. Liu, L. Zeng, J. Yu, L. Yang, J. Zhang, X. Zhang, F. Han, L. Zhao, X. Li, H. Liu and W. Zhou, Nano Energy, 2021, 85, 105940 CrossRef CAS.
- Z. Zhao, H. Liu, W. Gao, W. Xue, Z. Liu, J. Huang, X. Pan and Y. Huang, J. Am. Chem. Soc., 2018, 140, 9046–9050 CrossRef CAS PubMed.
- B. Devadas and T. Imae, Electrochem. Commun., 2016, 72, 135–139 CrossRef CAS.
- B. Koo, J. Chu, J. Seo, G. Jung, S. H. Baek, S.-W. Nam, C. Duah, Y. K. Lee, W. Jung and B. Shin, ChemSusChem, 2021, 14, 2585 CrossRef CAS PubMed.
- Y. Wang and J. Zhang, Front. Chem. Sci. Eng., 2018, 12, 838–854 CrossRef CAS.
- Q. Yu, Y. Luo, A. Mahmood, B. Liu and H.-M. Cheng, Electrochem. Energy Rev., 2019, 2, 373–394 CrossRef CAS.
- X. Bao, Y. Gong, X. Zheng, J. Chen, S. Mao and Y. Wang, J. Energy Chem., 2020, 51, 272–279 CrossRef.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- B. A. Krajina, A. C. Proctor, A. P. Schoen, A. J. Spakowitz and S. C. Heilshorn, Prog. Mater. Sci., 2018, 91, 1–23 CrossRef CAS.
- D. Jeon, J. Park, C. Shin, H. Kim, J. W. Jang, D. W. Lee and J. Ryu, Sci. Adv., 2020, 6, eaaz3944 CrossRef CAS PubMed.
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200–5219 RSC.
- S. D. Tillmann, P. Isken and A. Lex-Balducci, J. Phys. Chem. C, 2015, 119, 14873–14878 CrossRef CAS.
- M. Zelzer and R. V. Ulijn, Chem. Soc. Rev., 2010, 39, 3351–3357 RSC.
- D. J. Mikolajczak, J. L. Heier, B. Schade and B. Koksch, Biomacromolecules, 2017, 18, 3557–3562 CrossRef CAS PubMed.
- D. J. Mikolajczak, J. Scholz and B. Koksch, ChemCatChem, 2018, 10, 5665–5668 CrossRef CAS.
- H. Ozay, P. Ilgin, M. K. Sezgintürk and O. Ozay, Renewable Energy, 2020, 155, 500–512 CrossRef CAS.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- L. Leclercq, G. Douyère and V. Nardello-Rataj, Catalysts, 2019, 9, 163 CrossRef.
- A. K. Das and P. K. Gavel, Soft Matter, 2020, 16, 10065–10095 RSC.
- J. Bandak, J. Petzold, H. Hatahet, A. Prager, B. Kersting, C. Elsner and B. Abel, RSC Adv., 2019, 9, 5558–5569 RSC.
- B. Zhou, Z. Sun, D. Li, T. Zhang, L. Deng and Y. N. Liu, Nanoscale, 2013, 5, 2669–2673 RSC.
- E. S. Kang, Y.-T. Kim, Y.-S. Ko, N. H. Kim, G. Cho, Y. H. Huh, J.-H. Kim, J. Nam, T. T. Thach, D. Youn, Y. D. Kim, W. S. Yun, W. F. Degrado, S. Y. Kim, P. T. Hammond, J. Lee, Y.-U. Kwon, D.-H. Ha and Y. H. Kim, ACS Nano, 2018, 12, 6554–6562 CrossRef CAS PubMed.
- Y. Zheng, N. Li, S. Mukherjee, Y. Yang, J. Yan, J. Liu and Y. Fang, ACS Appl. Nano Mater., 2019, 2, 3012–3020 CrossRef CAS.
- B. Adhikari and A. Banerjee, Chem.–Eur. J., 2010, 16, 13698–13705 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- S. Roy and A. Banerjee, Soft Matter, 2011, 7, 5300–5308 RSC.
- I. Maity, D. B. Rasale and A. K. Das, Soft Matter, 2012, 8, 5301–5308 RSC.
- I. Maity, M. K. Manna, D. B. Rasale and A. K. Das, ChemPlusChem, 2014, 79, 413–420 CrossRef CAS PubMed.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS PubMed.
- S. H. Hiew, H. Mohanram, L. Ning, J. Guo, A. Sánchez-Ferrer, X. Shi, K. Pervushin, Y. Mu, R. Mezzenga and A. Miserez, Adv. Sci., 2019, 6, 1901173 CrossRef CAS PubMed.
- I. Maity, D. B. Rasale and A. K. Das, RSC Adv., 2014, 4, 2984–2988 RSC.
- R. G. Jadhav, D. Singh, S. M. Mobin and A. K. Das, Sustainable Energy Fuels, 2020, 4, 1320–1331 RSC.
- D. Varade and K. Haraguchi, Langmuir, 2013, 29, 1977–1984 CrossRef CAS PubMed.
- J. Zaragoza, S. Fukuoka, M. Kraus, J. Thomin and P. Asuri, Nanomaterials, 2018, 8, 882 CrossRef PubMed.
- T. Ghosh, A. Biswas, S. Bhowmik and A. K. Das, Chem.–Asian J., 2021, 16, 215–223 CrossRef CAS PubMed.
- S. S. Jedlicka, J. L. Rickus and D. Y. Zemlyanov, J. Phys. Chem. B, 2007, 111, 11850–11857 CrossRef CAS PubMed.
- M. Vandenbossche, M. Jimenez, A. Dehaese, M. Traisnel and M. Casetta, Green Mater., 2015, 3, 1–9 CrossRef CAS.
- S. Barazzouk and C. Daneault, Nanomaterials, 2012, 2, 187–205 CrossRef CAS PubMed.
- T. Wang, R. Yang, S. Ouyang, H. Shi and S. Wang, RSC Adv., 2015, 5, 48992–48996 RSC.
- M. F. Hossain and J. Y. Park, J. Electrochem. Soc., 2015, 162, B185 CrossRef CAS.
- A. Jindal, K. Tashiro, H. Kotani, T. Takei, S. Reichenberger, G. Marzun, S. Barcikowski, T. Kojima and Y. Yamamoto, ACS Appl. Energy Mater., 2019, 2, 6536–6541 CrossRef CAS.
- M. Li, Y. Zhu, N. Song, C. Wang and X. Lu, J. Colloid Interface Sci., 2018, 514, 199–207 CrossRef CAS PubMed.
- A. Zhang, S. Yu, Y. Jiang, L. Jia, X. Xia, W. Ye and C. Wang, Int. J. Hydrogen Energy, 2015, 40, 16238–16247 CrossRef CAS.
- D. B. Kayan, D. Koçak and M. İlhan, Int. J. Hydrogen Energy, 2016, 41, 10522–10529 CrossRef CAS.
- T. Yang, M. Du, H. Zhu, M. Zhang and M. Zou, Electrochim. Acta, 2015, 167, 48–54 CrossRef CAS.
- Y. Wang, S. Ma, Q. Li, Y. Zhang, X. Wang and X. Han, ACS Sustainable Chem. Eng., 2016, 4, 3773–3779 CrossRef CAS.
- S. Chakrabartty, C. S. Gopinath and C. R. Raj, Int. J. Hydrogen Energy, 2017, 42, 22821–22829 CrossRef CAS.
- Y. Mi, L. Wen, Z. Wang, D. Cao, H. Zhao, Y. Zhou, F. Grote and Y. Lei, Catal. Today, 2016, 262, 141–145 CrossRef CAS.
- M. Meng, H. Yan, Y. Jiao, A. Wu, X. Zhang, R. Wang and C. Tian, RSC Adv., 2016, 6, 29303–29307 RSC.
- W. Wu, Z. Tang, K. Wang, Z. Liu, L. Li and S. Chen, Electrochim. Acta, 2018, 260, 168–176 CrossRef CAS.
- C. N. Loney, S. Maheshwari, N. Pramounmat, M. J. Janik and J. N. Renner, J. Electrochem. Energy Convers. Storage, 2020, 17, 04080 Search PubMed.
- J. L. Yardeni, M. Amit, G. Ashkenasy and N. Ashkenasy, Nanoscale, 2016, 8, 2358–2366 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00439e |
| This journal is © The Royal Society of Chemistry 2021 |