
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpontaneous DNA translocation through a van der Waals heterostructure nanopore for single-molecule detection
Yang
Liu†
 a,
Ye
Deng†
a,
Yanmei
Yang
*b,
Yuanyuan
Qu
a,
Chao
Zhang
c,
Yong-Qiang
Li
a,
Mingwen
Zhao
a and
Weifeng
Li
*a
a,
Ye
Deng†
a,
Yanmei
Yang
*b,
Yuanyuan
Qu
a,
Chao
Zhang
c,
Yong-Qiang
Li
a,
Mingwen
Zhao
a and
Weifeng
Li
*a
aSchool of Physics, Shandong University, Jinan, Shandong 250100, China. E-mail: lwf@sdu.edu.cn
bCollege of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Normal University, Jinan, 250014, China. E-mail: yym@sdnu.edu.cn
cCollaborative Innovation Center of Light Manipulations and Applications, School of Physics and Electronics, Shandong Normal University, Jinan 250358, China
First published on 16th August 2021
Abstract
Solid-state nanopore detection and sequencing of a single molecule offers a new paradigm because of its several well-recognized features such as long reads, high throughput, high precision and direct analyses. However, several key technical challenges are yet to be addressed, especially the abilities to control the speed and direct the translocation of the target molecules. In this work, using molecular dynamics (MD) simulations, we found a spontaneous translocation of single-stranded DNA (ssDNA) through a van der Waals (vdW) heterostructure nanopore formed by stacking two graphenic materials, namely those of BC3 and C3N. Our results showed that, without using an external stimulus, ssDNA can be spontaneously transported through such a vdW nanopore from its BC3 side to its C3N side, with the C3N surface demonstrating a stronger capability than the BC3 surface to attract DNA bases. Thus, the distinct binding strengths of BC3 and C3N were concluded to drive the ssDNA translocation. The results indicated the vdW forces playing a leading role during the translocation process. Our simulations also showed, at the edges of the nanopore, a clear energy barrier for nucleotides, resulting in a translocation speed slowed to a value of 0.2 μs per base, i.e., twice as slow as that indicated for the latest published methods. The present findings provide a new architecture for biomolecule detection and sequencing, which may be considered some of the most important functions of nanomaterials in biological and chemical analyses.
1. Introduction
The discovery of DNA sequencing with α-hemolysin1 opens the prelude to single-molecule detection and even sequencing by nanopores. One of the available biological nanopore sequencing methods is to use voltage to drive a DNA molecule to pass through a nanopore in an electronic device where the ion current is modulated in a base-specific manner so that the DNA sequence can be read out.2,3 However, biological nanopores face the drawbacks of low mechanical and chemical stability levels although the sequencing accuracy is high.4 With the rapid development of nanoscience has come the ability to realize a controlled introduction of nano-sized pores into 2D nanomaterials such as graphene, MoS2, and h-BN, for successive DNA sequencing and detection.5–11 However, in most of these cases, the DNA motion through the nanopore is driven by an electric field, where the DNA perforates too rapidly for a high sequencing resolution to be achieved. Furthermore, the electric field-driven movement is only available to molecules with net charges, which most biomolecules do not display.12In the past decade, a great deal of research work has focused on improving the resolution of nanopore sequencing.13,14 Various types of methods have been proposed and widely studied, including change of solvent viscosity, contact friction of the nanopore, ion concentration, charge density of the pore surface, and so on.15–18 On the other hand, efforts have been made to search for a more general stimulus to drive the molecular translocation in a charge-independent manner. As demonstrated in previous studies, biomolecules have different binding affinities for different nano-surfaces.19,20 Thus by designing van der Waals (vdW) heterostructures using two different nano-surfaces, the difference between their binding affinities can drive the translocation of biomolecules between the two components. So far, numerous vdW heterostructures, including graphene/MoS2, graphene/WS2, phosphorene/g-C3N4 and so on, have been synthesized experimentally and used in electronics and optoelectronics.21–25 In a prototype study, Luan et al. carried out a theoretical investigation of spontaneous translocation of DNA and protein molecules through the nanopores in a graphene/MoS2 vdW heterostructure,12,26 and demonstrated the potential use of this heterostructure in molecular sequencing. Their findings have inspired efforts at finding more heterostructure design candidates with outstanding performances. Recently, two new graphene derivatives, namely BC3 and C3N nanomaterials, have been successfully synthesized experimentally.27,28 The significantly high stability, thermal conductivity and carrier mobility levels of BC3 and C3N, and especially their matching lattices,29–31 make them qualified candidates to build vdW heterostructures.29,31–33 A theoretical work has proposed an optoelectronic application of the BC3/C3N vdW heterostructure.34 Due to the different electronegativities of the boron, carbon and nitrogen atoms, there are intrinsic electron transfers in the BC3 and C3N monolayers. This electron redistribution not only benefits sequencing efforts, but also provides distinctive properties that are absent in a pristine graphene surface.
In the present work, we performed molecular dynamics (MD) simulations to study the interactions of ssDNA with a BC3/C3N vdW heterostructure nanopore in order to address the possibility of using the heterostructure nanopore in sequencing applications. Our simulations revealed that both BC3 and C3N attract ssDNA, with ssDNA stably adsorbed on their surfaces without any desorption found. It was particularly interesting to find a spontaneous translocation of ssDNA from the BC3 side of the heterostructure to the C3N side through the nanopores without applying any external force, corresponding to a stronger adsorption of ssDNA onto C3N than onto BC3. A larger free energy barrier at the edges of the nanopore was indicated to effectively reduce the translocation speed of ssDNA to 0.2 μs per base, slower than the previous reported speed through a graphene/MoS2 nanopore.26 In addition, we also found the force driving the translocation of the nucleotides to be mainly dominated by vdW interactions. We believe that the findings of our research will promote the application of vdW heterostructures in DNA sequencing.
2. Simulation methods
A BC3/C3N heterostructure with dimensions of 10.347 nm × 9.264 nm was used in the simulations. The inter-layer separation of the heterostructure was set to 1 nm initially, and optimized to 2.97 Å after a 5 ns equilibration simulation, consistent with the results of functional theory calculations.34 Following previous studies of nanopore sequencing,7,9,35 the boron/nitrogen atoms and carbon atoms were removed in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to generate holes with diameters of about 2 nm as depicted in Fig. 1C. The ssDNA model (sequence: ATCGATCGATCGATCG) was constructed by using Nucleic Acid Builder (NAB) implemented in the Amber Tools package.36
:3 to generate holes with diameters of about 2 nm as depicted in Fig. 1C. The ssDNA model (sequence: ATCGATCGATCGATCG) was constructed by using Nucleic Acid Builder (NAB) implemented in the Amber Tools package.36
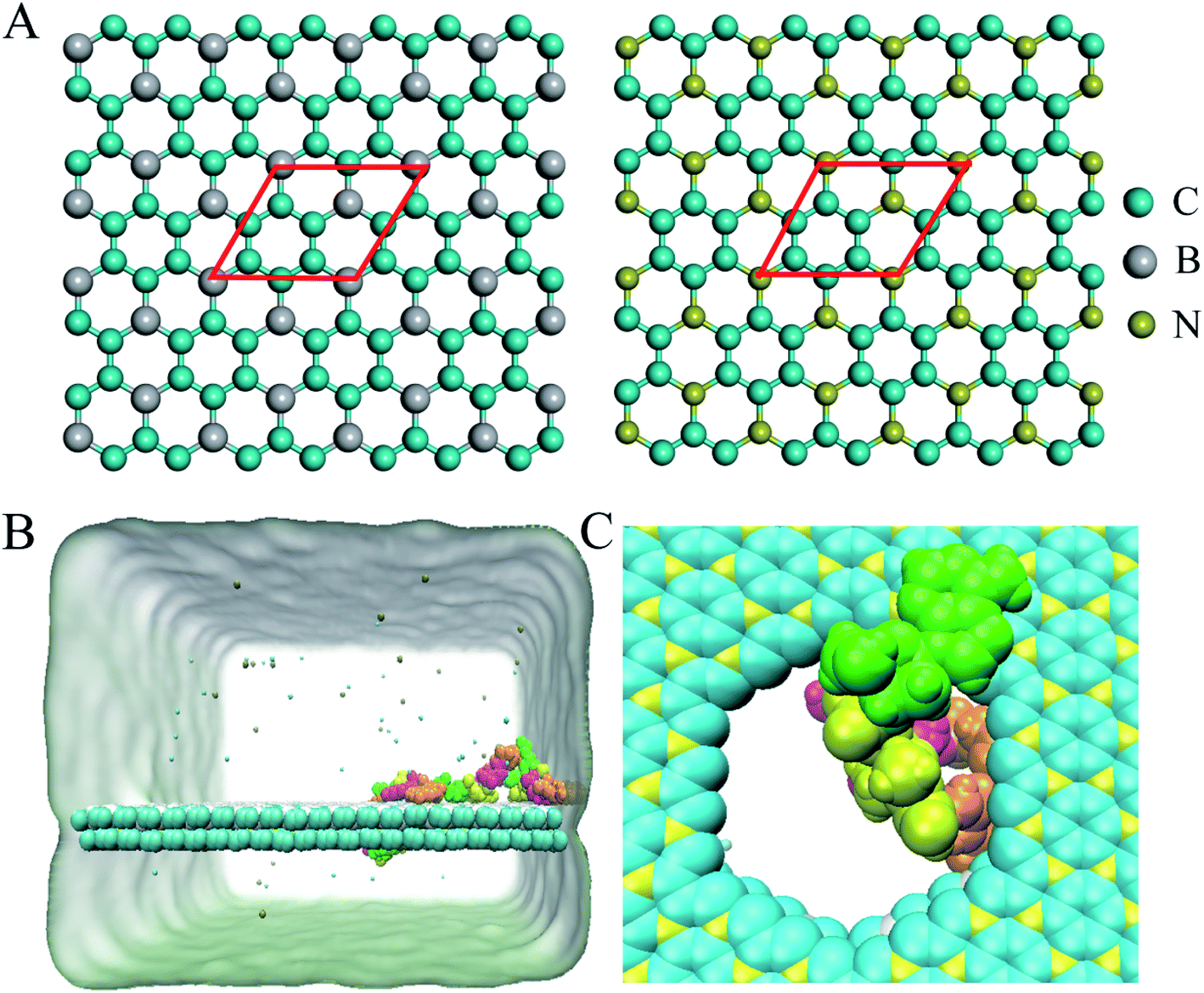 | ||
| Fig. 1 (A) Structures of BC3 and C3N. Atoms in the red rhombuses depict the primitive BC3 and C3N cells. (B) The initial simulated model of ssDNA on a BC3/C3N heterostructure nanopore. Each nucleotide is shown in a different color (A, green; C, magenta; G, orange; T, yellow). K+ and Cl− are shown as cyan and tan spheres. Water molecules are implicitly represented as a light green surface. (C) Local structure of a nucleotide at a nanopore. | ||
All MD simulations were conducted using the GROMACS package.37 The AMBER99sb force field38 was applied for the ssDNAs. The force field for BC3 and C3N was adopted from our previous study.39 The SPC/E water model40 was used in the simulated system. During the simulation, a leap-frog algorithm was used to integrate Newton's equations of motion, and the time step was set to 2 fs. The cut-off for the Lennard-Jones and electrostatic interactions were set to 1.0 nm, and the particle mesh Ewald (PME) method41,42 was used to treat long-range electrostatic interactions. Periodic boundary conditions were applied in all three dimensions. A semi-isotropic pressure coupling at 1 bar was maintained in the z-direction using the Parrinello–Rahman algorithm. After the energy minimization and equilibration, production runs were carried out in the NVT ensemble, where the number of particles, volume and temperature of the simulation systems were constant. The temperature was kept at 300, 350, and 400 K in different MD simulations by using a v-rescale thermostat.43
The potential of mean force (PMF) values of the four types of nucleotide bases on both the BC3 layer and the C3N layer were calculated using the umbrella sampling method.44,45 The distance between the nucleotide base and the nanolayer along the normal direction (equivalently, the z-axis) was chosen as the collective variable (CV). Fifteen sampling windows in total were generated along the selected CV axis to cover the distance from 0.2 nm to 1.5 nm, and each window was sampled for 15 ns. The PMF profiles were calculated based on the weighted histogram analysis method.46 The binding free energy was then defined as the difference between the minimum free energy value of the bound state and that of the fully dissociated state.
3. Results and discussion
3.1 Spontaneous transport of ssDNA through a BC3/C3N nanopore
Following the previous work,7,35,47 the ssDNA was placed at the upper BC3 side of the nanopore at the beginning of the simulation. One terminal base at the 5′ end was placed near the lower C3N side as depicted in Fig. 1B. After equilibration, the nucleotides formed stable complexes with BC3 using π–π stacking interactions, which are commonly found interactions made when DNA binds graphenic nanostructures.48 The local structure of the nanopore is shown in Fig. 1C. Afterward, three production simulations (i.e. simulations to produce data sampling) were carried out at temperatures of 300, 350, and 400 K. Interestingly, in all of the simulations, the ssDNA translocated to the C3N side after a period of simulation time. Fig. 2 depicts a representative translocation process at 350 K.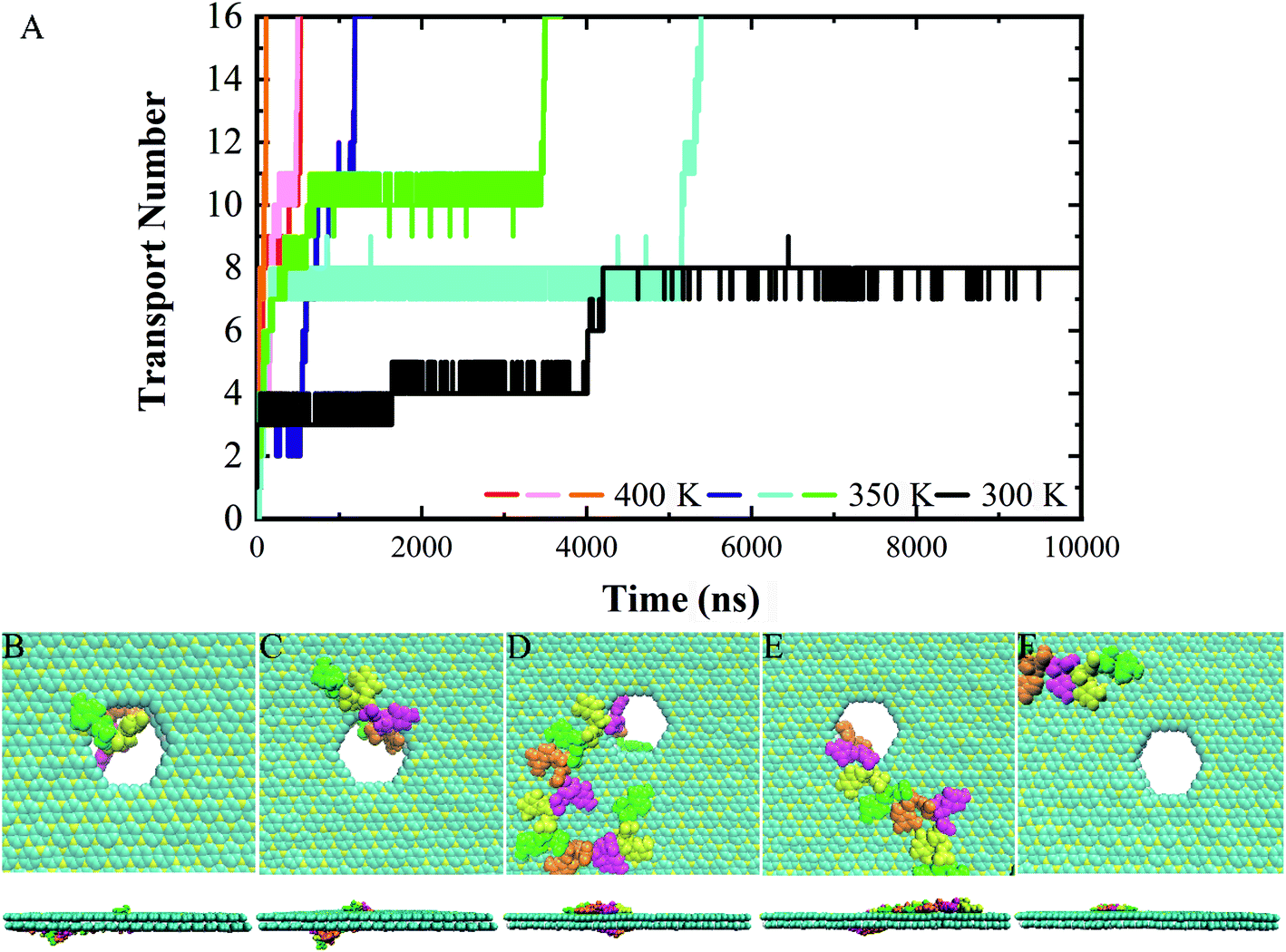 | ||
| Fig. 2 . (A) Time evolution of number of nucleotides transported through a nanopore. (B–F) Representative top- and side-view snapshots of the translocation process in a representative trajectory at (B) 0 ns, (C) 10 ns, (D) 640 ns, (E) 3450 ns, and (F) 3500 ns. | ||
As a quantitative description of the DNA translocation process, Fig. 2A shows the number of nucleotides translocated from BC3 to C3N with respect to simulation time. First, we found the DNA being susceptible to perforating spontaneously at all of the studied temperatures. And consistent with previous research,49,50 our results showed more rapid translocation at higher temperatures, which may provide enough kinetic energy to accelerate the whole process. Specifically, the translocation of ssDNA was calculated to be finished within 1000 ns at 400 K, in contrast to half of the nucleotides of ssDNA still bound to the BC3 side after 10000 ns of simulation at 300 K. Besides, the ssDNA has been shown to perforate in a stepwise manner,51 due to the strong interactions between the nucleotide bases and the nanopore edge, which trap the nucleotide in the pore for certain period of time. Details of the interactions are discussed below.
We chose a representative trajectory at 350 K to further describe the ssDNA translocation process. The simulation began with the adsorption of one nucleotide on the surface of C3N (Fig. 2B). The ssDNA perforated quickly at the initial stage of the simulation: we observed two nucleotides reaching the C3N side at 10 ns (Fig. 2C) and another eight at 640 ns (Fig. 2D). At this time, 11 nucleotides had been adsorbed on the surface of C3N. The translocation then dramatically slowed down, as it took 2810 ns to pass another nucleotide through the nanopore (Fig. 2E). Interestingly, after this nucleotide passed through the nanopore, the translocation became fast again. The last four nucleotides (from the thirteenth to the sixteenth) took only 50 ns to be transferred to the C3N nanosheet (Fig. 2F). Overall, in the simulation, it took about 3500 ns for the entire ssDNA to spontaneously move to the C3N side, at which point the bases of the ssDNA finally formed stable π–π stacking interactions with C3N and diffused freely on the surface.
3.2 The driving force of ssDNA translocation through the nanopore
To understand the dominant force driving the spontaneous movement of the ssDNA, we analyzed the interaction energy (Ep) between the ssDNA and nanopore. As can be seen from Fig. 3A, the Ep gradually decreased along with the translocation of increasingly more nucleotides to the C3N side, suggesting a stronger attraction of the nucleotides for the C3N side of the heterostructure than for the BC3 side. Therefore, a difference between the adsorption strengths of the two sides of the nanopore was concluded to promote the transport of the ssDNA.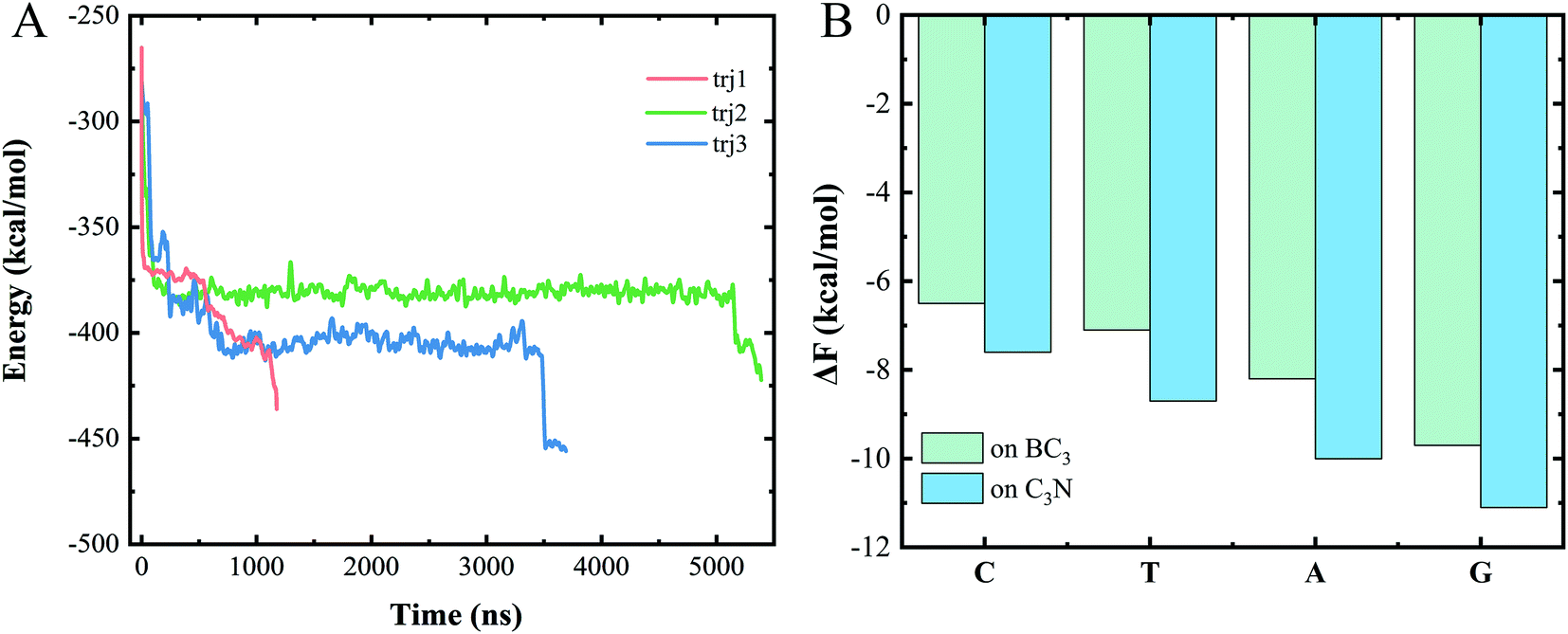 | ||
| Fig. 3 (A) Plots of time evolution of the interaction energy between the ssDNA and BC3/C3N vdW heterojunction for various trajectories. (B) Binding free energy, ΔF, values of the four types of DNA bases on BC3 and C3N, respectively. | ||
To quantitatively assess this binding difference, we calculated the binding affinity (Fb) values of purine (A and G) and pyrimidine (T and C) with BC3 and C3N, respectively. Technically, in the calculations, the base was pulled into the solution from the BC3/C3N surface along the direction normal to the surface. First note that the binding free energies of the four nucleotides, regardless of whether on BC3 or C3N, followed the order G > A > T > C (Fig. 3B). From the perspective of the geometric structures of the bases, it was not surprising to us that the binding energy of a purine, containing both an imidazole and purine ring, was calculated to be greater than that of a pyrimidine, containing only a purine ring. Besides, our results also revealed a greater Ep of adsorption of purine and pyrimidine on C3N than on BC3 (Fig. 3B). This binding affinity difference was concluded to drive the transport of the ssDNA from BC3 to C3N. The transport speed of the ssDNA in the BC3/C3N nanopore was calculated to be 0.2 μs per base, slower than that determined for the heterostructure nanopore composed of graphene and MoS2.26
3.3 The process for transporting the four types of nucleotides
To assess the specific interactions between each type of nucleotide and the vdW heterostructure nanopore and to evaluate the sequencing performance of the nanopore, we established a polymeric model with a 10-unit length for each type of nucleotide, namely polyC, polyT, polyA and polyG. Fig. 5 shows the gradual perforation of each polymer, with each nucleotide staying in the nanopore for a certain period of time. Fig. 4A and B show snapshots highlighting different vdW interactions made between nucleotides and the nanopore, a feature resulting in different residence times for nucleotides in the pore.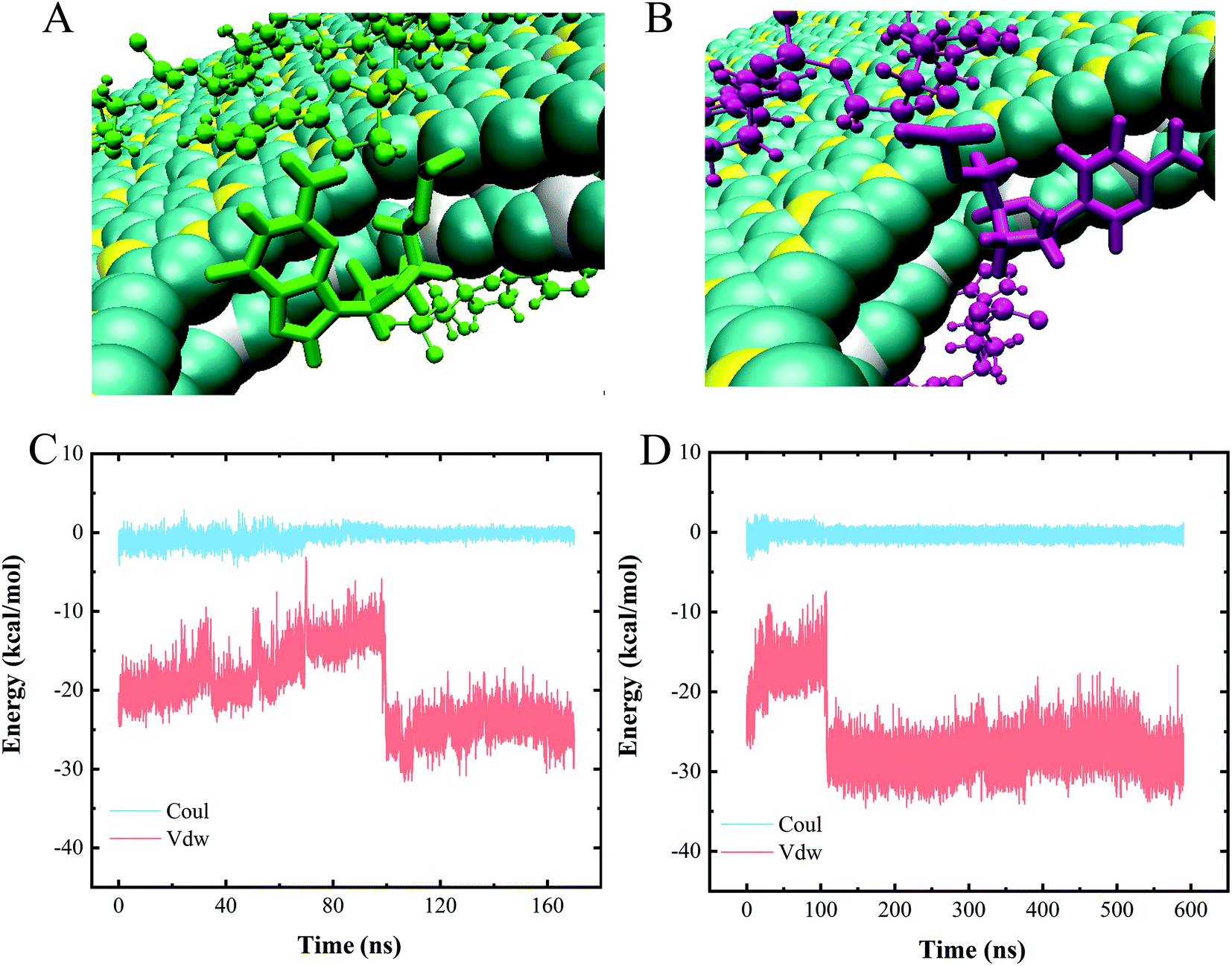 | ||
| Fig. 4 (A and B) Two types of nucleotide stacking interactions inside the heterojunction nanopore. (C and D) Interaction energies of (C) polyC and (D) polyA when translocating through the nanopore. | ||
We also calculated the interaction energies made between one nucleotide of each polymer and the heterojunction nanopore during the entire polymer transport process. Since similar changes with time were observed for the interaction energies of the four types of polymeric models when translocated from BC3 to C3N, only the energy profiles for polyC and polyA are shown as representatives (Fig. 4C and D). Inspection of these plots indicated the entire transport to be a vdW-interaction-dominated process, with only minor changes due to electrostatic interactions. The increases and decreases of the vdW interactions were concluded to correspond to the nucleotide entry into and exit from the nanopore, respectively. Upon the nucleotide reaching the nanopore (Fig. 4A and B), the strength of the π–π stacking bonding between the nucleotide and the surface of the pore was calculated to decrease (Fig. 4C and D), a feature equivalent to a formation of an energy barrier as described previously.52 Such an energy barrier would hinder the translocation of DNA, and may improve the sequencing resolution. Other than that, resistance due to friction from BC3/C3N can also slow down the translocation of DNA, improving the resolution as well. According to our previous study,20 water molecules form a patterned distribution on the surfaces of both BC3 and C3N, resulting in the frictional coefficients of BC3 and C3N being greater than that of graphene, and hence effectively hindering the diffusion of DNA on the surface.
3.4 The speed of nucleotide transported through the nanopore
Measuring ion currents can be used during nanopore sequencing to distinguish the different nucleotide bases. For nanopore sequencing, it is necessary to slow the passage of the DNA through the pore in order to improve resolution. A speed of spontaneous translocation through nanopores of approximately 0.1 μs per base has been previously reported.26 In order to determine the extent to which translocation times differ for the four different nucleotides, we simulated 10 parallel trajectories for each polymer to measure their translocation times.The translocation times of the different polymers are shown in Fig. 5. The translocation times for even one type of polymer were found to differ considerably between the 10 replicates. The average translocation times over 10 replicates were calculated to be 748.6 ± 581.2 ns, 498.3 ± 488.3 ns, 662.0 ± 493.2 ns and 358.1 ± 246.6 ns for polyC, polyT, polyA and polyG, respectively. With such a large uncertainty, however, our data did not provide a meaningful translocation time discrimination. We speculated this wide range of translocation times for any one type of polymer to be due to the two different binding poses of nucleotide when interacting with the nanopore (Fig. 4A and B), i.e., with these two binding poses adopted randomly during the translocation, leading to the large uncertainty of the translocation time. The random translocation of the nucleotides is consistent with a study of DNA and graphene nanopores.53 So translocation time by itself was concluded to not provide sufficient information to sequence DNA.
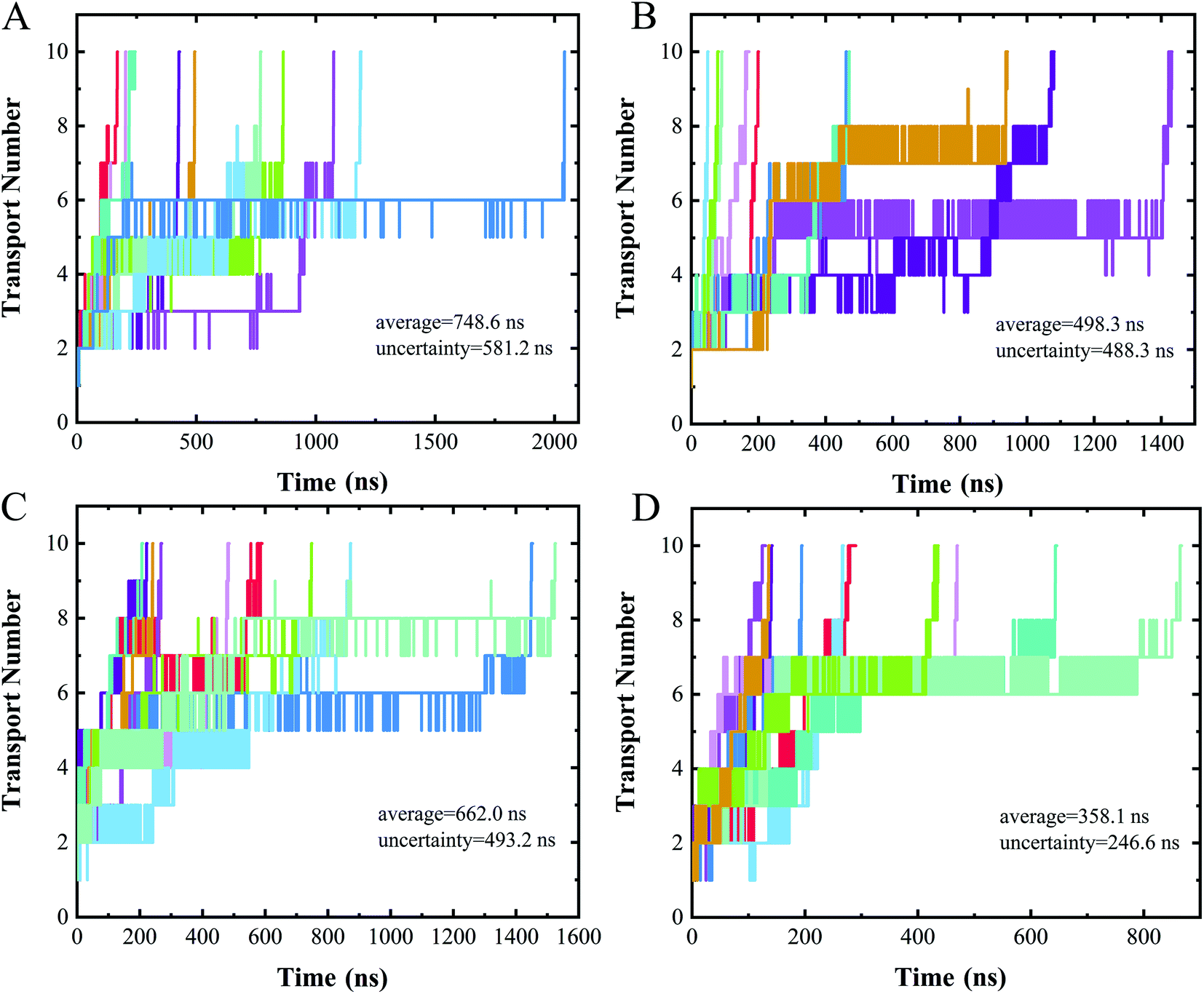 | ||
| Fig. 5 Time evolution of the number of nucleotides transported through a nanopore for (A) polyC, (B) polyT, (C) polyA, (D) polyG. Different colors represent different trajectories of each case. | ||
4. Conclusions
In summary, we used molecular dynamics simulation methods to study the interaction between ssDNA and the BC3/C3N heterojunction structure. The results of the study showed stronger adsorption of ssDNA onto C3N than onto BC3, allowing ssDNA to be spontaneously transported from the BC3 side of the heterojunction structure through the nanopore to the C3N side without an external force. During the entire translocation process, the vdW interaction was found to be predominant. In addition, upon passing through the nanopore, nucleotides were indicated to encounter a relatively large energy barrier, effectively reducing the rate of nucleotide translocation. The translocation speed of ssDNA here was calculated to be 0.2 μs per base, slower than the previously reported transport of ssDNA through the graphene/MoS2 heterojunction nanopore. Therefore, we believe that our research can provide a new architecture for DNA sequencing and promote the application of vdW heterojunctions in DNA sequencing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Natural Science Foundation of Shandong Province (ZR2020MB074, ZR2020JQ04, ZR2020QA049) and National Natural Science Foundation of China (11874238).References
- A. Meller, L. Nivon and D. Branton, Phys. Rev. Lett., 2001, 86, 3435–3438 CrossRef CAS PubMed.
- J. A. Cracknell, D. Japrung and H. Bayley, Nano Lett., 2013, 13, 2500–2505 CrossRef CAS PubMed.
- D. Stoddart, A. J. Heron, J. Klingelhoefer, E. Mikhailova, G. Maglia and H. Bayley, Nano Lett., 2010, 10, 3633–3637 CrossRef CAS PubMed.
- K. Lee, K. B. Park, H. J. Kim, J. S. Yu, H. Chae, H. M. Kim and K. B. Kim, Adv. Mater., 2018, 30, 1704680 CrossRef PubMed.
- N. Yang and X. Jiang, Carbon, 2017, 115, 293–311 CrossRef CAS.
- Z. Zhang, J. Shen, H. Wang, Q. Wang, J. Zhang, L. Liang, H. Agren and Y. Tu, J. Phys. Chem. Lett., 2014, 5, 1602–1607 CrossRef CAS PubMed.
- D. B. Wells, M. Belkin, J. Comer and A. Aksimentiev, Nano Lett., 2012, 12, 4117–4123 CrossRef CAS PubMed.
- S. Banerjee, J. Wilson, J. Shim, M. Shankla, E. A. Corbin, A. Aksimentiev and R. Bashir, Adv. Funct. Mater., 2015, 25, 936–946 CrossRef CAS PubMed.
- J. Feng, K. Liu, R. D. Bulushev, S. Khlybov, D. Dumcenco, A. Kis and A. Radenovic, Nat. Nanotechnol., 2015, 10, 1070–1076 CrossRef CAS PubMed.
- S. Liu, B. Lu, Q. Zhao, J. Li, T. Gao, Y. Chen, Y. Zhang, Z. Liu, Z. Fan, F. Yang, L. You and D. Yu, Adv. Mater., 2013, 25, 4549–4554 CrossRef CAS PubMed.
- G. Danda, P. Masih Das, Y. C. Chou, J. T. Mlack, W. M. Parkin, C. H. Naylor, K. Fujisawa, T. Zhang, L. B. Fulton, M. Terrones, A. T. Johnson and M. Drndic, ACS Nano, 2017, 11, 1937–1945 CrossRef CAS PubMed.
- B. Luan and R. Zhou, J. Phys. Chem. Lett., 2018, 9, 3409–3415 CrossRef CAS PubMed.
- B. Luan, G. Stolovitzky and G. Martyna, Nanoscale, 2012, 4, 1068–1077 RSC.
- B. Luan, D. Wang, R. Zhou, S. Harrer, H. Peng and G. Stolovitzky, Nanotechnology, 2012, 23, 455102 CrossRef PubMed.
- D. Fologea, J. Uplinger, B. Thomas, D. S. McNabb and J. L. Li, Nano Lett., 2005, 5, 1734–1737 CrossRef CAS PubMed.
- U. Mirsaidov, J. Comer, V. Dimitrov, A. Aksimentiev and G. Timp, Nanotechnology, 2010, 21, 395501 CrossRef PubMed.
- B. Luan and A. Aksimentiev, Soft Matter, 2010, 6, 243–246 RSC.
- B. Luan and A. Aksimentiev, J. Phys. Condens. Matter, 2010, 22, 454123 CrossRef PubMed.
- B. Luan and R. Zhou, Nat. Commun., 2019, 10, 4610 CrossRef PubMed.
- Y. Deng, F. Wang, Y. Liu, Y. Yang, Y. Qu, M. Zhao, Y. Mu and W. Li, Nanoscale, 2020, 12, 5217–5226 RSC.
- J. Shim, D.-H. Kang, Y. Kim, H. Kum, W. Kong, S.-H. Bae, I. Almansouri, K. Lee, J.-H. Park and J. Kim, Carbon, 2018, 133, 78–89 CrossRef CAS.
- P. Gnanasekar, D. Periyanagounder and J. Kulandaivel, Nanoscale, 2019, 11, 2439–2446 RSC.
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S. Z. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed.
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 CrossRef CAS PubMed.
- H. Xue, Y. Wang, Y. Dai, W. Kim, H. Jussila, M. Qi, J. Susoma, Z. Ren, Q. Dai, J. Zhao, K. Halonen, H. Lipsanen, X. Wang, X. Gan and Z. Sun, Adv. Funct. Mater., 2018, 28, 1804388 CrossRef.
- B. Luan and R. Zhou, ACS Nano, 2018, 12, 3886–3891 CrossRef CAS PubMed.
- T. C. King, P. D. Matthews, H. Glass, J. A. Cormack, J. P. Holgado, M. Leskes, J. M. Griffin, O. A. Scherman, P. D. Barker, C. P. Grey, S. E. Dutton, R. M. Lambert, G. Tustin, A. Alavi and D. S. Wright, Angew. Chem. Int. Ed., 2015, 54, 5919–5923 CrossRef CAS PubMed.
- J. Mahmood, E. K. Lee, M. Jung, D. Shin, H.-J. Choi, J.-M. Seo, S.-M. Jung, D. Kim, F. Li, M. S. Lah, N. Park, H.-J. Shin, J. H. Oh and J.-B. Baek, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7414–7419 CrossRef CAS PubMed.
- B. Mortazavi, M. Shahrokhi, M. Raeisi, X. Zhuang, L. F. C. Pereira and T. Rabczuk, Carbon, 2019, 149, 733–742 CrossRef CAS.
- B. Mortazavi, Carbon, 2017, 118, 25–34 CrossRef CAS.
- A. H. N. Shirazi, R. Abadi, M. Izadifar, N. Alajlan and T. Rabczuk, Comp. Mater. Sci., 2018, 147, 316–321 CrossRef CAS.
- Y. Tang, H. Chai, H. Zhang, W. Chen, W. Zhang and X. Dai, Phys. Chem. Chem. Phys., 2018, 20, 14040–14052 RSC.
- Y. Liu, V. I. Artyukhov, M. Liu, A. R. Harutyunyan and B. I. Yakobson, J. Phys. Chem. Lett., 2013, 4, 1737–1742 CrossRef CAS PubMed.
- C. Zhang, Y. Jiao, T. He, S. Bottle, T. Frauenheim and A. Du, J. Phys. Chem. Lett., 2018, 9, 858–862 CrossRef CAS PubMed.
- A. B. Farimani, K. Min and N. R. Aluru, ACS Nano, 2014, 8, 7914–7922 CrossRef CAS PubMed.
- W. literature, t. Case, V. Babin, J. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. Cheatham, T. Darden, R. Duke, H. Gohlke, A. Götz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T.-S. Lee and P. A. Kollman, AMBER 14, University of California, San Francisco, 2014 Search PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CrossRef CAS PubMed.
- Y. Deng, F. Wang, Y. Liu, Y. Yang, Y. Qu, M. Zhao, Y. Mu and W. Li, Nanoscale, 2020, 12, 5217–5226 RSC.
- H. Berendsen, J. Grigera and T. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- B. Roux, Comput. Phys. Commun., 1995, 91, 275–282 CrossRef CAS.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1995, 16, 1339–1350 CrossRef CAS.
- A. Barati Farimani, P. Dibaeinia and N. R. Aluru, ACS Appl. Mater. Interfaces, 2016, 9, 92–100 CrossRef PubMed.
- V. A. Karachevtsev, A. M. Plokhotnichenko, M. V. Karachevtsev and V. S. Leontiev, Carbon, 2010, 48, 3682–3691 CrossRef CAS.
- D. Fologea, J. Uplinger, B. Thomas, D. S. Mcnabb and J. Li, Nano Lett., 2005, 5, 1734–1737 CrossRef CAS PubMed.
- M. Belkin and A. Aksimentiev, ACS Appl. Mater. Interfaces, 2016, 8, 12599–12608 CrossRef CAS PubMed.
- H. Qiu, A. Sarathy, J. P. Leburton and K. Schulten, Nano Lett., 2015, 15, 8322–8330 CrossRef CAS PubMed.
- L. Z. Sun, W. P. Cao and M. B. Luo, J. Chem. Phys., 2009, 131, 194904 CrossRef PubMed.
- S. J. Heerema and C. Dekker, Nat. Nanotechnol., 2016, 11, 127–136 CrossRef CAS PubMed.
Footnote |
| † These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2021 |