
In vitro and in vivo models for anti-amyloidosis nanomedicines
Aleksandr
Kakinen
 *a,
Ibrahim
Javed
a,
Thomas P.
Davis
*ab and
Pu Chun
Ke
*abc
*a,
Ibrahim
Javed
a,
Thomas P.
Davis
*ab and
Pu Chun
Ke
*abc
aAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: a.kakinen@uq.edu.au; t.davis@uq.edu.au
bARC Centre of Excellence in Convergent Bio-Nano Science and Technology, Monash Institute of Pharmaceutical Sciences, Monash University, 381 Royal Parade, Parkville, VIC 3052, Australia. E-mail: pu-chun.ke@monash.edu
cZhongshan Hospital, Fudan University, 111 Yixueyuan Rd, Xuhui District, Shanghai, 200032, China
First published on 5th January 2021
Abstract
Amyloid diseases are global epidemics characterized by the accumulative deposits of cross-beta amyloid fibrils and plaques. Despite decades of intensive research, few solutions are available for the diagnosis, treatment, and prevention of these debilitating diseases. Since the early work on the interaction of human β2-microglobulin and nanoparticles by Linse et al. in 2007, the field of amyloidosis inhibition has gradually evolved into a new frontier in nanomedicine offering numerous interdisciplinary research opportunities, especially for materials, chemistry and biophysics. In this review we summarise, for the first time, the in vitro and in vivo models employed thus far in the field of anti-amyloidosis nanomedicines. Based on this systematic summary, we bring forth the notion that, due to the complex and often overlapping physiopathologies of amyloid diseases, there is a crucial need for the appropriate use of in vitro and in vivo models for validating novel anti-amyloidosis nanomedicines, and there is a crucial need for the development of new animal models that reflect the behavioural, symptomatic and cross-talk hallmarks of amyloid diseases such as Alzheimer's (AD), Parkinson's (PD) diseases and type 2 diabetes (T2DM).
 Aleksandr Kakinen | Aleksandr Kakinen is a research fellow at Australian Institute for Bioengineering and Nanotechnology (AIBN), the University of Queensland, Australia. He obtained his PhD degree (2014) at Tallinn University of Technology, Estonia, and worked as a postdoctoral fellow (2016–2020) at Monash Institute of Pharmaceutical Sciences, Monash University, Australia. His research interests range from nanomedicine and amyloids diseases to structural biology and nanotoxicology. |
 Ibrahim Javed | Ibrahim Javed is a research fellow at AIBN, the University of Queensland (UQ), Australia. He obtained his PhD degree (2019) from Monash Institute of Pharmaceutical Sciences at Monash University. His research concerns the in vivo disease modelling of amyloidogenesis, the pharmacology and biological interaction of nanomedicines, and the gut–brain axis of neurological disorders. |
 Thomas P. Davis | Thomas P. Davis is the Director of ARC Centre of Excellence in Convergent Bio-Nano Science and Technology (CBNS). He is a Chair Professor of Precision Medicine at the University of Queensland and a Professor at Monash University. Prior to his appointment at the University of Queensland he spent 21 years as a senior academic at the University of New South Wales in Sydney. His research focuses on the application of polymer science and nanotechnology to therapeutic applications. |
 Pu Chun Ke | Pu Chun Ke is a Professor at Zhongshan Hospital of Fudan University, a Signature Project Leader at CBNS, Monash University, and a Principal Reseach Fellow at the University of Queensland. He was a Distinguished Visiting Scientist at CSIRO, Australia (2014) and Assistant and tenured Associate Professor at Clemson University, USA (2003–2013). His postdoctoral research was performed at the University of California, San Diego (2001–2003) supported by a LJIS Postdoctoral Fellowship in Biophysics (Burroughs Wellcome Fund). His research concerns the biophysical mechanisms and mitigation of brain and metabolic diseases, the protein corona and environmental science. |
1. Introduction
Amyloid diseases refer to a wide range of human conditions characterized by extracellular plaques and intracellular lesions present in the central nervous system (CNS), pancreatic islets, bloodstream, cerebrospinal fluid (CSF), and/or other bodily organs. Accumulation of these proteinaceous substances, whose atomic structures are underlined by a ubiquitous cross-β peptide backbone,1 has been hypothesized as causative to neuronal or β-cell degeneration, loss in cognitive function, impaired motor skills, among other histological and behaviour markers of dementia and metabolic disorders.2 Together, amyloid diseases account for over 400 million in their global burden and, despite intensive research in the past decades, have found no cure except selective symptomatic treatments.Amyloidosis describes the dynamic process of molecular self-assembly, where amyloid proteins/peptides stagger in a linear fashion towards fibrillar structures of high plasticity, triggered by dysregulated homeostasis in the local pH, ionic strength and chaperones and incited by misfolding events in association with cell membranes (Fig. 1a).3,4 Among the miscellaneous by-products of amyloidosis, the oligomeric and protofibrillar species have been implicated as the most cytotoxic,5,6 and hence serve as major targets for AD (nano)medicines. Clinical evidence supports the notion that the aetiologies of amyloid diseases are considerably more complex than amyloidosis, which are also undermined by disrupted autophagy, heightened or impaired immune response, as well as familial and other environmental factors.7,8
 | ||
| Fig. 1 Amyloidogenesis and application of in vitro models for validating anti-amyloid nanomedicines. (a) Schematic of the self-assembly of amyloidogenic peptides and proteins, going through the states of monomers, oligomers, protofibrils and amyloid fibrils via the kinetic processes of peptide nucleation, elongation and saturation. (b) Applications of in vitro models for validating anti-amyloid nanomedicines. (c) Anti-amyloid mechanisms with nanomaterials. (d) Major modes of interaction involved in the anti-amyloid activity of nanomaterials. | ||
In recent years, the use of nanomaterials has emerged as a new frontier in the development of therapeutic solutions to amyloid diseases, alternative to the use of small molecules, monoclonal antibodies and peptidomimetics that are often stigmatized due to their countless failures in the clinic.9–11 This trend is well justified by the advantages of nanomaterials in their diverse physicochemical properties, ease for functionalisation and drug loading, tuneable biocompatibility and robust capacities in anti-inflammation, biodistribution and blood–brain barrier (BBB) translocation. For these applications, cell lines and animal models are typically employed to test the efficiency of a nanomaterial inhibitor against amyloidosis (membrane damage, cell viability, reactive oxygen species/ROS production), cellular/tissue inflammation (cytokine secretion, endoplasmic reticulum/ER stress, microglial activity), plaque accumulation (immunohistochemical/birefringence quantifications ex vivo or in vivo) as well as animal behaviour (swimming/walking trajectories, memory, etc.) that are symptomatic to the diseases.
As the field of amyloid diseases is vast, while the need for developing facile, economic and reliable models alternative to lab rats is pressing,12,13 in this review we focus on the state-of-the-art of in vitro and in vivo models employed in the rapidly expanding literature for testing nanomaterials against amyloidosis. This effort, first of its kind to our knowledge, serves two purposes: one is to highlight the progress of this new frontier in nanomedicine,14–17 and the other is to facilitate the development of materials research and systematic testing of nanomedicines against a range of debilitating human amyloid diseases.
2. In vitro models for validating anti-amyloid nanomedicines
Animal cell and tissue cultures have become indispensable tools for research in biology and medicine in the recent decades.18,19 Several in vitro approaches have been developed to understand the etiology and pathogenesis of a broad range of amyloid diseases. The most common types of cell cultures are tumour-derived cell lines, immortalised cell lines, primary cells and ex vivo organotypic models.20 Tissue explants and organotypic slice cultures faithfully represent organ architecture and may have advantages over cell lines, but they are more difficult to prepare and maintain in a viable state. Moreover, their inherent variability leads to poor reproducibility in experiments.21 Primary cells are obtained directly from animals or humans. These cells are typically slow in growth and carry features of the tissues of their origin.20 Usually, primary cells have a finite lifespan, whereas immortalised and cancerous cells give rise to continuous cell lines. On the other hand, while cancer cell lines are established from tumours, immortalised cell lines are produced through transformation or genetic alteration of primary cells.20 However, despite the fact that the development of immortalised cell lines removed the need to use tissue as a source for in vitro experiments, such cell lines often present genetic and metabolic abnormalities compared to normal animal cells.22The in vitro models have enjoyed wide applications in the field of drug development and assessment of their efficacy and toxicity (Fig. 1b).23–25 Particularly, cell cultures have been actively utilised in the development of nanomedicines to combat amyloid diseases such as Alzheimer's disease (AD), Parkinson's disease (PD) and type 2 diabetes mellitus (T2DM).17
Anti-amyloid nanomaterials may act as medicines themselves or be used as nanocarriers to deliver drugs to specific parts of the body. Anti-amyloid agents can shift the balance from peptide–peptide interaction towards peptide–nanoparticle interaction and suppress peptide fibrillization. It has been shown that ceria nanocrystals, ZnO nanoparticles (NPs), carbon nanotubes (CNTs), graphene oxide (GO) and graphene quantum dots (GQDs) can sequester toxic amyloid species, reduce their concentration in solution and, as a result, reduce toxicity (Fig. 1c).17 However, it has been noted that inhibition of amyloid aggregation may not always prevent cell degeneration. Counterintuitively, accelerated aggregation of amyloidogenic peptides could be used as an alternative to eliminate the toxic oligomeric species (Fig. 1c). For example, (2-hydroxyl ethyl acrylate) star-shaped polymeric NPs can accelerated the fibrillization of human islet amyloid polypeptide (IAPP) through the formation of polymer–peptide complexes, thereby reducing IAPP-elicited toxicity in pancreatic beta cells and islets.26 Depending on the structure and surface chemistry, NPs can disrupt the self-assembly of unfolded peptides/proteins via hydrophobic interactions, aromatic stacking, hydrogen bonding, and salt-bridging, which can serve as a mechanistic basis for the design of anti-amyloid nanomedicines (Fig. 1d).17,27 In addition, the physicochemical properties of NPs affect corona formation in a biological milieu.28 For example, it has been shown that the anti-amyloid properties of NPs strongly depend on their pre-formed corona composition.29,30 Furthermore, since each disease is characterised by different plasma proteomes, the same nanomaterial incubated with the plasma of patients with different pathologies may acquire disease-specific and personalised protein corona31,32 to impact the effectiveness of treatment strategies.
Despite the wide use of in vitro models for the development of nanotechnologies in biomedical applications and for anti-amyloid nanomedicine in particular, a notable gap remains between bench discoveries and clinical translation of NPs. An often overlooked key factor which affects the biological fate of NPs is the considerable functional variations between cell sexes.33 For example, it has been recently demonstrated that cell sex led to differences in NP uptake between male and female human amniotic stem cells, with greater uptake elicited by female cells.34 Similarly, AD is known to be more prevalent among women than men.35 Thus, considering sex as a biological variable in amyloidosis and its treatment is a topic to be exploited.
2.1 Cancer and tumour cell models
Current efforts in the development of nano-based strategies against amyloidosis have been mainly focused on age-related neurological diseases, such as AD and PD. Traditional in vitro cell culture techniques usually involve cultivation of 2D monolayers of cells. One of the most frequently used in vitro models for amyloid-related neuroscience is the SH-SY5Y cell line (Table 1). SH-SY5Y neuroblastoma-derived cells are often employed as a model of neuronal function and differentiation.36 The common use of this neuronal cell model takes advantage of its human origin, neuronal properties and ease of maintenance.36 In terms of application, SH-SY5Y cells have been utilised for testing the cytotoxicity and anti-amyloidogenic activities of nanomaterials involving a variety of cell viability assays such as 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT), propidium iodide (PI) and lactate dehydrogenase (LDH) assays (Table 1). In addition, Chen et al. used SH-SY5Y cells as a tauopathy cell model,37 demonstrating that nanocomposites fabricated by controlled assembly of ultrasmall ceria nanocrystals (CeNCs) and iron oxide nanocrystals (IONCs) on the surface of mesoporous silica nanoparticles (MSNs) relieved the AD symptoms by mitigating mitochondrial oxidative stress, suppressing tau hyperphosphorylation, and preventing neuronal death both in vitro and in vivo (Fig. 2a). SH-SY5Y cells have shown variable responses to amyloid-β (Aβ) peptide associated with the pathology of AD. Particularly, SH-SY5Y cells displayed viability of 65% and 56% with 1 μM38 and 20 μM of Aβ, respectively.39 SH-SY5Y cells have also been utilised as a cell uptake40 and proliferation model.41 In a recent study by Liu et al.40 on the effect of ultrasound-mediated nanomedicine delivery through the BBB, SH-SY5Y was employed for the assessment of cellular uptake, cytotoxicity and endoplasmic reticulum stress of the nanomedicine. Quercetin-modified sulphur NPs (Qc@SNPs) embedded in the shell of microbubbles (MB) combined with ultrasound pulses (US) improved cell uptake by up to 5 times via the sonoporation effect, compared to the endocytosis pathway (Fig. 2b and c).40 Despite its wide use, the SH-SY5Y cell line lacks many of the features that define neurons, including neuronal morphology, inhibited cell division, and expression of neuron-specific markers.42 Moreover, the lack of a standardised protocol to maintain this cell line in culture leads to variable cell growth, differentiation into various neuronal cell types and inconsistent experimental outcomes.43| Cell origin | Description | Disease | NPs applied | Application/method applied | Ref. |
|---|---|---|---|---|---|
| Human | |||||
| SH-SY5Y | Neuroblastoma cell line. Widely used as a neuronal cell model in neuroscience, including AD and PD research. | ND | AGuIX® (Gd3+) + KLVFF/LPFFD + PEG + Cyanine 5.5 | Cytotoxicity (MTT) | 62 |
| AD | Cur–SPIO + PEG–pLacA-CoP + PVP | Cytotoxicity (MTT) | 63 | ||
| βCas AuNPs | Cytotoxicity (PI) | 39 | |||
| PA-LIP, CL-LIP | Cell proliferation, cytotoxicity (MTT) | 41 | |||
| RI-OR2-TAT NLIPs | Cell viability (MTS, LDH) | 64 | |||
| PPI-MalGD | Cytotoxicity (MTT) | 38 | |||
| Qc@SNPs-MB | Cell uptake, cytotoxicity (MTT), ER stress assay (thapsigargin (Tg) induced) | 40 | |||
| CeNC/IONC/MSN-T807-MB | Cell apoptosis (Annexin V-FITC kit), western blotting, RT-PCR, mitochondrial ROS scavenging activity (MitoSOX reagent), cellular uptake, intracellular Tau aggregation | 37 | |||
| PMA + LND/MND | Cytotoxicity (MTT) | 65 | |||
| CLPFFD-AuNR | Cell viability (MTS), cell uptake | 66 | |||
| FeO + DSPE–PEG–NHS + CR + rutin | Cytotoxicity (MTT) | 67 | |||
| PD | Fullerenols | Cytotoxicity (PI) | 68 | ||
| Caco-2 | Heterogeneous human epithelial colorectal adenocarcinoma cells. The most commonly used intestinal permeability model and a model for epithelial-like phenotypes. | AD | CS-SLN + BACE1 siRNA + CPP | Cell permeability | 69 |
| BE(2)-C | Human neuroblastoma cell line from a metastatic site (bone marrow). | AD | COOH–AuNPs | Cytotoxicity (MTT) | 46 |
| GI-2 | Established from a human brain tumour gliosarcoma. | AD | PLGA–Cur NPs | Cell uptake | 47 |
| HeLa | Human cervical cancer cell line. The oldest and most commonly used human cell line. | AD | nDa + PEG-MNSs + AβO-Ab | Toxicity (ViaCount) | 48 |
| HEPG3 | Human hepatocellular carcinoma. HepG2-derived stable cell line containing an integrated wild-type duck hepatitis B virus (DHBV) head-to-tail unit-length genomic DNA dimer. | AD | nDa + PEG-MNSs + AβO-Ab | Toxicity (ViaCount) | 48 |
| Mouse | |||||
| N2a | Mouse neuroblastoma cell line. Has a neuronal and amoeboid stem cell morphology, allowing it to differentiate in response to environmental factors. | AD | CPDs | Cytotoxicity (MTT) | 50 |
| USPIO NPs + Aβ1–42 | Neurotoxicity | 51 | |||
| RAW264.7 | Mouse leukemic monocyte macrophage cell line. Established from an ascites of a tumour induced in a male mouse by intraperitoneal injection of Abselon Leukaemia Virus (A-MuLV). Suitable transfection host. | AD | nDa + PEG-MNSs + AβO-Ab | Toxicity (ViaCount) | 48 |
| βTC-6 | Insulin-secreting cell line derived from a pancreatic tumour (insulinoma) arising in a transgenic mouse expressing the large T-antigen of simian virus 40 (SV40) in pancreatic β-cells. | T2DM | GQDs | Cytotoxicity (Alamar Blue) | 27 |
| CRSi NRs | Cytotoxicity (PI) | 70 | |||
| βLg AuNPs | Cytotoxicity (PI) | 45 | |||
| c-AuNPs, PEG–AuNPs | Cytotoxicity (PI) | 39 | |||
| Starshaped PHEA | Cytotoxicity (PI) | 26 | |||
| Rat | |||||
| PC12 | Rat pheochromocytoma derived and widely used in neuroscience. Synthesizes and stores dopamine. | AD | SiNCs-scFvs | Cytotoxicity (MTT) | 44 |
| MSN-CQ-IgG | Cytotoxicity (MTT) | 57 | |||
| MeSMC | Cytotoxicity (MTT) | 55 | |||
| BP-BTA | Cytotoxicity (MTT) | 56 | |||
| NC-KLVFF | Cell adhesion, cytotoxicity (CCK-8) | 71 | |||
| aAβmAb-MagNPs | Cytotoxicity (XTT) | 59 | |||
| UCNP-C60-KLVFF | Cytotoxicity (DCFH-DA) | 60 | |||
| PEG–PLA + TGN and QSH | Cytotoxicity (MTT) | 72 | |||
 | ||
| Fig. 2 Nanoconstructs for anti-amyloid cellular studies. (a) Proposed mechanism of CeNC/IONC/MSN nanocomposite efficiency in the amelioration of cognitive impairment in AD based on the results observed using SH-SY5Y as a neuronal cell model. Reproduced with the permission from ref. 37. Copyright 2018 American Chemical Society. (b) Ultrasound pulses (US) improved cellular uptake of Qc@SNPs embedded in the shells of microbubbles (Qc@SNPs-MB) (labelled with ruthenium) by up to 5 times in SH-SY5Y cells, compared to non-US conditions. (c) TEM images of Qc@SNPs-MB distribution in SH-SY5Y cells. The bottom images are the enlargements of the white squares above, and the white arrows indicate locations of Qc@SNPs-MB + US. Panels (b and c) reproduced with the permission from ref. 40. Copyright 2020 The Royal Society of Chemistry. (d) Scanning electron microscopy (SEM) image of Aβ nanodepletors (porous silica nanostructures) and (e) alleviation of Aβ-induced neurotoxicity by Aβ nanodepletors in PC12 cell line. Panels (d and e) reproduced with the permission from ref. 44. Copyright 2020 Wiley. (f) TEM image of β-lactoglobulin (bLg)-coated AuNPs. (g) Co-fibrillization of pathogenic islet amyloid polypeptide (IAPP) and functional beta lactoglobulin (bLg) via gold nanoparticles (AuNPs) and (h) elimination of IAPP toxicity using βTC-6 pancreatic cell line. Panels (f–h) reproduced with the permission from ref. 45. Copyright 2017 American Chemical Society. | ||
BE(2)-C is another human neuroblastoma cell line that has been employed as a neuronal cytotoxicity model in anti-AD research. For example, Liao et al. used BE(2)-C cells to investigate the effect of gold nanoparticles (AuNPs) on Aβ aggregation and toxicity.46 They demonstrated that co-incubation of Aβ with negatively charged AuNPs reduced Aβ-associated cytotoxicity, which could be used as a potential nano-chaperone to inhibit and redirect Aβ aggregation. Other human tumour-derived in vitro models used to validate anti-amyloid nanomaterials include gliosarcoma cell line Gi-247 and hepatocellular carcinoma cell line HEPG3.48 Gi-2 was employed to investigate the cell uptake of brain-targeting (with Tet-1 peptide) curcumin–PLGA NPs as a potential AD nanomedicine.47 In the study by Viola et al., the cytotoxicity of magnetic nanostructures for non-invasive diagnostic imaging of early-stage AD was characterised using four different cell lines, including HEPG3 and HeLa cells.48
Alongside human-originated models, mouse- and rat-derived cell lines have found wide applications for validating anti-amyloid nanomedicines. The mouse neuroblastoma cell line N2a has a neuronal and amoeboid stem cell morphology, allowing it to differentiate in response to environmental factors.49 N2a cells have been utilised in AD research, particularly in the evaluation of various nanomaterials on Aβ-induced neurotoxicity (Table 1).50,51 This cell line has been shown to be sensitive to Aβ, displaying 70% viability at 10 μM Aβ42.51
Another cancer-derived in vitro cytotoxicity model that has been widely used for validating anti-amyloid properties of nanomaterials is rat pheochromocytoma-derived PC12 cell line.52 As these cells can reversibly respond to the administration of the nerve growth factor (NGF) and express a sympathetic neuronal phenotype, PC12 represents a model for neuronal differentiation53 and has been utilised for the development of AD nanomedicines (Table 1). PC12 is a homogeneous and easy-to-handle cell line that synthesises, releases and stores catecholamines (e.g. dopamine).54 The capacity of organic and inorganic NPs to ameliorate Aβ aggregation and toxicity has been evaluated using several cell viability assays such as MTT,44,55–57 LDH,58 sodium 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)-carbonyl]-2H-tetrazolium (XTT)59 and dichloro-dihydro-fluorescein diacetate (DCFH-DA).60 The viability of PC12 cells varied between 70% for 5 μM of Aβ57 to 55% for 50 μM of Aβ.56 PC12 has also been often used as an in vitro platform to investigate the anti-amyloid efficacy of NPs prior to in vivo assays. For instance, Jung et al. have observed that highly porous silica nanostructures (Fig. 2d) can attenuate Aβ-induced neurotoxicity (Fig. 2e) using PC12 cells before introducing the nanomaterial to mice.44
In addition to AD and PD, anti-amyloid nanomedicines have been also explored for applications against T2DM-associated pathogenic protein misfolding. βTC-6, for example, is an insulin-secreting mouse pancreatic beta-cell line derived from insulinoma.61 The inhibitory effect of polymeric and metallic nanomaterials on the aggregation and toxicity of IAPP was determined with βTC-6 cells.26,39,45 In particular, Javed et al. inhibited IAPP toxicity by constructing an IAPP and β-lactoglobulin (bLg) double “corona” on AuNPs via β-sheet stacking (Fig. 2f–h).45
2.2 Immortalised cell models
In contrast to tumour-derived cell lines, immortalised cell lines have been used for a greater range of purposes, including validating anti-amyloid nanomedicines (Fig. 1b and Table 2). One of such applications is brain drug delivery. A major challenge in treating brain-associated amyloid disorders is to deliver therapeutics to the brain by crossing the BBB.24 The BBB comprises specialised microvascular endothelial cells, pericytes, astrocytes and neurons that couple local neuronal functions to local cerebral blood flow and regulate the transport of blood components and molecules in and out of the CNS.73 Under certain pathological conditions of diabetes, PD and AD, for example, the BBB permeability is increased.73 Nanotechnology-based approaches can lead to improved non-invasive delivery of therapeutic agents across the BBB. Among human-derived immortalised cell models, human brain capillary endothelial cell line hCMEC/D3 has been most frequently employed for the studies of anti-amyloid nanomedicines (Table 2). Since hCMEC/D3 is derived from human temporal lobe microvessels, it has been used for cytotoxicity41 and cell uptake studies,74 in addition to serving as a BBB model system.75,76 For instance, the BBB uptake of theranostic nanovehicles (TNVs) that were capable of targeting cerebrovascular amyloid and treating cerebrovascular inflammation resulting from cerebral amyloid angiopathy (CAA) led to a 30-fold increase in the internalisation of TNVs by hCMEC/D3 cells pre-treated with Aβ40 compared to the uptake of Alexa Fluor 647-labelled anti-amyloid antibody (IgG4.1).75| Cell origin | Description | Disease | NPs applied | Application/method applied | Ref. |
|---|---|---|---|---|---|
| Human | |||||
| hCMEC/D3 | Immortalised human brain capillary endothelial cells. Derived from human temporal lobe microvessels. Immortalised by lentiviral transduction of the catalytic subunit of human telomerase and SV40-T antigen. Used as a human BBB model in drug transport, neurotoxicity and neuroscience. | AD | Lipid–PEG2000–Cur LIPs | Cell uptake | 74 |
| PA-LIP, CL-LIP | Cell proliferation, cytotoxicity (MTT) | 41 | |||
| mApoE-PA-LIP | BBB transwell system. Cytotoxicity (MTT) | 76 | |||
| RI-OR2-TAT NLIPs | Uptake and transcytosis, cytotoxicity | 64 | |||
| CAA | Gd-DTPA + IgG-anti-Aβ + CS + 125I + CTX | Uptake by BBB cells | 75 | ||
| HEK293 | Human embryonic kidney cells generated by transfection of cultures of normal human embryonic kidney cells with sheared adenovirus 5 DNA. One of the most common cell lines used for research purposes – widely used in cell biology and biotechnology as hosts for gene expression and transfection. | T2DM | c-AgNPs, PEI–AgNPs, PEG-IONPs, PC-IONPs | Toxicity (Calcein-AM) | 78 |
| hA53T α-syn HEK293 | HEK293T transfected cells with pCMV5-myc-A53T αS. Over-expressing αS. | PD | GQD | Immunostaining | 79 |
| Jurkat human T cells | Immortalised T lymphocytes first derived from the peripheral blood of a child suffering from leukemia. Most often used as a prototypical T cell line to study multiple events in T cell biology. | T2DM | c-AuNPs, PEG–AuNPs | Immune response (LSPR immunoassay) | 39 |
| Mouse | |||||
| bEnd.3 | BBB model by mouse brain cell line transformed with the polyoma virus middle T-antigen derived from BALB/c mice. Widely used for blood vascular research. | AD | Qc@SNPs-MB | Cytotoxicity (MTT) | 40 |
| PEG-PLA + TGN and QSH | Cell uptake, cytotoxicity (MTT) | 72 | |||
| BV-2 | Retroviral-immortalised microglial cells derived from C57BL/6 mice and transformed by recombinant retrovirus J2. Most frequently used substitute for primary cultures of microglia. Have similar functions as primary microglia. Have morphological, phenotypical and functional markers of macrophages. | AD | KLVFF-NC | Microglia phagocytosis | 71 |
| Qc@SNPs-MB | Anti-inflammatory activity (cytokines production (ELISA)) | 40 | |||
| LAG | Mouse fibroblast-like spontaneously immortalised cell line derived from connective tissue of the C3H strain. | AD | PLGA–Cur NPs | Cytotoxicity (Alamar Blue, MTT) | 47 |
| NIH/3T3s | Mouse embryonic fibroblast line. Established from a NIH Swiss mouse embryo. Proven useful in DNA transfection studies. | AD | nDa + PEG-MNSs + AβO-Ab | Toxicity (ViaCount) | 48 |
| Others | |||||
| B14 | Immortalised Chinese hamster fibroblast | AD | GATG-Mor | Cytotoxicity (MTT) | 82 |
| MDCK | Madin–Darby canine kidney epithelial cells. Widely utilised to investigate the processing of Aβ precursor protein (APP), as well as the sorting of its proteolytic products. | AD | Cur–SPIO + PEG–pLacA-CoP + PVP | Monolayer permeability | 63 |
Human embryonic kidney HEK293 cells are one of the most commonly employed cell lines for nanomedicine,77 including for validating the anti-amyloid potential of AuNPs against T2DM (Table 2).78 HEK293 cells are popular for their ease of growth and transfection. Its transfected α-synuclein (αS) over-expressing strain (hA53T αS HEK293) was a host to GQDs against α-synucleinopathy, where GQDs penetrated through the BBB to protect against dopamine neuron loss induced by preformed αS fibrils (Fig. 3a–c).79 However, while HEK263 cell line yields homogenous populations and is suitable for large-scale experiments, high passage numbers can lead to genetic and epigenetic alterations.80 Among the non-human cancer-derived cell lines, the mouse brain bEnd.3 cell line40,72 and Madin–Darby canine kidney epithelial cells (MDCK)63,81 are also common BBB model systems for evaluating anti-amyloid nanomedicines.
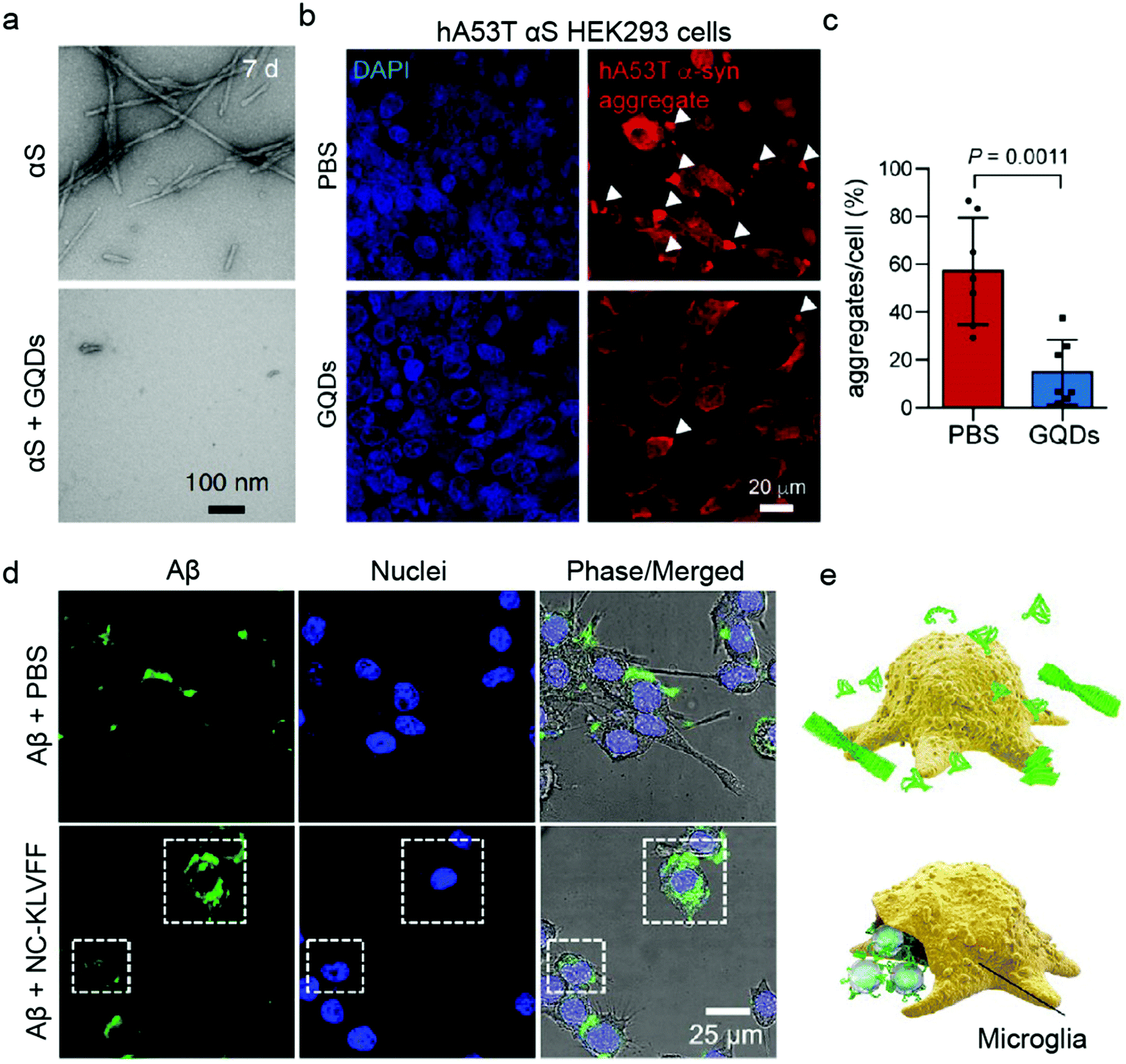 | ||
| Fig. 3 Use of immortalised cell models for validating anti-amyloid nanomedicines. (a) TEM images of preformed α-synuclein (αS) fibrils in the absence (top) and presence (bottom) of GQDs. (b and c) GQDs treatment reduced αS aggregates induced by overexpression of human A53T αS in HEK293 cells. Panels (a–c) reproduced with the permission from ref. 79. Copyright 2018 Springer Nature. (d) Fluorescence images and (e) schematic showing Aβ elimination by BV-2 microglia cells treated with PBS and NC-KLVFF. Panels (d and e) reproduced with the permission from ref. 71. Copyright 2018 American Chemical Society. | ||
The application of immortalised cell lines in anti-amyloid research is however not limited to cell uptake and BBB models. The immune response to IAPP with AuNPs inhibitors has been studied using Jurkat human T cells, which are immortalised T lymphocytes.39 In a recent study, Zhao et al. determined the potential of nanocomposites to regain endocranial microglia's potency in phagocytosing Aβ using a retroviral-immortalised microglial cell line BV-2 (Fig. 3d and e).71 Moreover, the anti-inflammatory activity of quercetin-modified embedded nano-sulphur microbubbles has been assessed by BV-2 cells.40 A mouse-derived fibroblast-like spontaneously immortalised cell line derived from connective tissue (LAG)47 and an embryonic fibroblast line NIH/3T3s48 were used for AD research, where the cytotoxicities of polymeric and silica NPs were determined. Similarly, immortalised Chinese hamster fibroblast cell line B14 was utilised to study the anti-Aβ aggregation property of dendrimers.82
2.3 Primary cell cultures
While primary cultures have the potential to overcome many of the difficulties inherent to cell lines, isolating and culturing primary dopaminergic neurons from post-mortem human brains remains difficult tasks. Therefore, primary dopaminergic neurons are usually obtained from embryonic murine brain tissues. For example, Kim et al. used primary neurons derived from C57BL/6 mice to validate the protective potential of GQDs against dopamine neuron loss induced by αS fibrils.79 Apart from immortalised microglial cells, primary microglia have been utilised in neurodegenerative research. For instance, Daria et al. demonstrated that functional impairment of aged microglial cells in amyloid plaque-bearing tissues can be reversed through factors secreted by young microglia isolated from postnatal day 5 WT mouse brains.83 In another study, AD-associated proteomic signatures of APPPS1 and APP-KI-derived microglia have been compared.84 The kinetic differences in proteomic profiles correlated with the presence of fibrillar Aβ, suggesting that fibrillar Aβ may trigger AD-associated microglial phenotypes and their corresponding functional decline.In addition to immortalised cell lines, primary cell cultures have found ample applications as BBB models in nanomedicine (Table 3). Specifically, human85 and non-human75,86 derived brain microvascular endothelial cell lines have been applied to assess the transport of anti-amyloid nanomedicines across the BBB. For instance, the uptake and potential cytotoxicity of curcumin-containing nanoclusters was investigated in human brain capillary endothelial cells.85 Loureiro et al. used porcine brain capillary endothelial cells for the brain delivery of pegylated liposomes functionalized with an anti-transferrin receptor monoclonal antibody (OX26MAb) and an anti-Aβ peptide antibody (19B8Mab) for AD therapy.86 Recently, the same group developed an in vitro BBB model by culturing endothelial cells (ECs) derived from hematopoietic stem cells isolated from umbilical cord blood and pericytes.87 This transwell BBB system exhibited low permeability to non-permeable markers (such as sucrose, fluorescein sodium) and a high transendothelial electrical resistance (TEER), and hence was considered a human brain-like endothelial system. Similarly, Kim et al. combined primary mouse astrocytes and primary mouse brain microvascular endothelial cells (BMEC) in an in vitro BBB permeability experiment.79 The astrocytes were seeded onto the underside of the transwell inserts and BMEC were cultivated on top of the inserts. Furthermore, Agyare et al. utilised bovine brain microvascular endothelial cells to validate the cellular uptake and ability of theranostic nanovehicles to inhibit the secretion of pro-inflammatory cytokines.75 In addition to BBB permeability studies, primary cells have been also used to evaluate the cytotoxicity of anti-amyloid AD and PD nanomedicines.79,85
| Cell origin | Description | Disease | NPs applied | Application/method applied | Ref. |
|---|---|---|---|---|---|
| Human | |||||
| BCECs | BBB model by brain capillary endothelial cells | AD | Cur-loaded Lf-NLC | Cytotoxicity (MTT), cellular uptake, intracellular stability | 85 |
| HBLECs | Human brain-like endothelial cells (HBLECs). BBB model by co-culture of endothelial cells (ECs) derived from hematopoietic stem cells isolated from umbilical cord blood on the upper side of a filter insert and pericytes at the bottom of the well. | AD | OX26 mAb-SLN | Permeability assay (14C-sucrose), cellular accumulation | 87 |
| Others | |||||
| PBCEC | BBB model by porcine brain capillary endothelial cells. | AD | PEG–DSPS–LIPs + OX26MAb and 19B8MAb | Cellular uptake | 86 |
| Primary mouse neurons | Isolated from C57BL/6 mice. | PD | GQD | Cell viability (TUNEL, LDH, neurite outgrowth assays), mitochondrial dysfunction (8-OHG), Lewy body pathology (western blot) | 79 |
| Primary mouse astrocytes | Isolated from C57BL/6 mice. | PD | GQD | BBB permeability | 79 |
| BMEC | Mouse primary brain microvascular endothelial cells. Isolated from C57BL/6 mice. | PD | GQD | BBB permeability | 79 |
| BBMVEC | BBB model by bovine brain microvascular endothelial cells. | CAA | Gd-DTPA + IgG-anti-Aβ + CS + 125I + CTX | Cellular uptake, cytokines inhibition | 75 |
Traditional 2D cell culture systems have improved our understanding of human amyloid diseases, but these models have limited value as disease models because they do not recreate authentic interactions between cells. However, models based on recent technological innovations in three-dimensional (3D) culture systems can open new frontiers in the characterisation of pathological mechanisms and the feasibility of high-throughput drug screening. The first 3D culture model suitable for studying amyloid diseases was a human neural stem-cell-derived 3D culture system expressing familial AD (FAD) mutations (APP (K670N/M671L and V717I) and PSEN1 (1E9) genes).88 These cells were found to accumulate both senile Aβ plaques and neurotrophic factors, two hallmarks of AD not previously observed in 2D cultures or most animal models. Human brain organoids have recently emerged as new invaluable tools facilitating a range of research applications in neurodegenerative and amyloid diseases, including disease mechanisms and progression, drug discovery and development.89,90 However, to our knowledge, these 3D model systems have yet to be applied to validating anti-amyloid nanomedicines.
2.4 Ex vivo models
While in vitro and ex vivo testing methods appear similar, they entail significant differences. Both approaches involve experiments on a biological matter, conducted outside of a living organism and in a laboratory setting.91 However, in contrast to in vitro, the living tissues for ex vivo experiments are not created or cultivated artificially but taken directly from a living organism. Although in vitro primary cultures have provided important knowledge about the mechanisms of neurogenerative and brain-associated amyloid diseases as well as potential drug targets, they do not fully mimic the organisation of cells in the brain and the extracellular matrix within the CNS. In general, ex vivo models (Table 4) are more complex in terms of cell diversity and, therefore, closer to in vivo conditions.91 For example, isolated rat brains and mouse brain slices have been used to validate MRI contrast probes for non-invasive diagnosis of early-stage AD (Fig. 4a).48,59 In another study, mouse brain sections have been used to validate multifunctional Gd-based NPs for AD diagnostics.62 It was shown that ultra-small GdNPs grafted with two Aβ42 targeting peptides (KLVFF and LPFFD) selectively bound Aβ amyloid plaques in ex vivo AD mouse hippocampus (Fig. 4b). Furthermore, Fülöp et al. used mouse hippocampal slices for ex vivo electrophysiological experiments.92 Specifically, they synthesised foldamer-dendrimer constructed of ordered recognition segments (helical β-peptide foldamers) conjugated to disordered linker regions (G0-PAMAM dendrimer) (Fig. 4c). This arrangement afforded wrapping of the Aβ42 oligomers through the repeating binding sites displayed over the oligomeric surface (Fig. 4d) that rescued the long-term potentiation (LTP) of the toxic Aβ oligomers in ex vivo mouse hippocampal slices (Fig. 4e).| Cell origin | Description | Disease | NPs applied | Application/method applied | Ref. |
|---|---|---|---|---|---|
| Mouse | |||||
| Mouse brain slices | 400 μm thick, from eight-month-old 5×FAD and WT mice. APP/PS1 transgenic mice that co-express five familial AD (FAD) mutations. Develop cerebral amyloid plaques at 2 months of age. | AD | nDa + PEG-MNSs + AβO-Ab | MRI | 48 |
| APP/PS1/TTR mouse brain slices | 10 μm thick slices. Triple genetically modified AD mouse model. Generated by crossing APP/PS1 mice with TTRnull mice (TTR−/−). Display amyloid plaques in the cortex and the hippocampus | AD | AGuIX® (Gd3+) + KLVFF/LPFFD + PEG + Cyanine 5.5 | Immunohistochemical analyses of brain sections | 62 |
| Mouse hippocampal slices | 350 μm-thick, from 7-months-old mice (CD1) | AD | G0-PAMAM + MMP + helical b-peptide foldamer | Electrophysiology (a hippocampus slice long-term potentiation model) | 92 |
| Mouse islets | Isolated from C57BL/6 mice | T2DM | Starshaped PHEA | Ex vivo viability (PI) | 26 |
| Rat | |||||
| Rat brains | Brains extracted from 2-day old Sprague-Dawley rats | AD | aAβmAb-MagNPs | Ex vivo brains incubated with NPs. MRI | 59 |
| Rat hippocampal slices | Prepared from 6- to 8-day-old male Wistar rats. Transverse hippocampal slices (400 μm thick) | AD | IndOH-LNCs | Ex vivo Aβ neurotoxicity | 95 |
| Rat hippocampal tissue | From embryonic day-12 Wistar rat embryos. Free-floating spherical clusters of neural stem cells (NSC) formed in the presence of specific mitotic growth factors | AD | Cur–PLGA-NPs | NSC proliferation and neuronal differentiation | 93 |
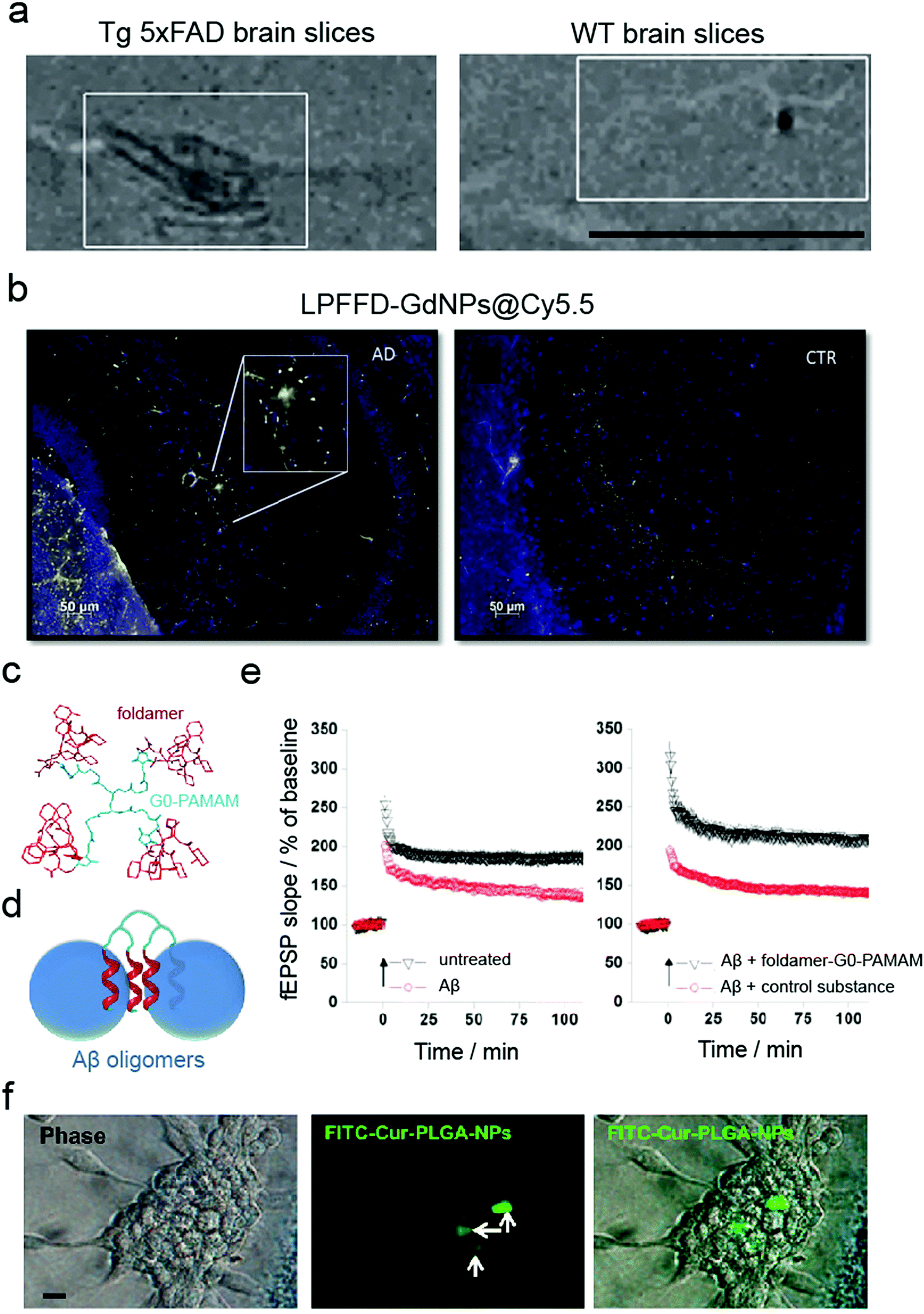 | ||
| Fig. 4 Use of ex vivo models for validating anti-amyloid nanomedicines. (a) MRI imaging of brain sections from 5×FAD (left) and WT (right) eight-month-old mice probed with magnetic nanostructures (MNS) with Aβ oligomer-specific antibodies, demonstrating binding of MNS to the Aβ deposits in 5×FAD slices but not in the control. Panel (a) reproduced with the permission from ref. 48. Copyright 2015 Springer Nature. (b) Binding of LPFFD-grafted GdNPs to Aβ amyloid plaques on the brain slices of APPswe/PS1A246/TTR transgenic mice (left) but not on control wild-type (WT) brain slices (right). Panel (b) reproduced from ref. 62, an open-source article from Springer Nature. (c–e) Ex vivo electrophysiological experiments in mouse hippocampal slices: (c) design and (d) principle of action of the foldamer-dendrimer conjugates. (e) Foldamer-G0-PAMAM protected against the synaptic plasticity damage caused by Aβ42 oligomers by using a hippocampus slice long-term potentiation model. Panels (c–e) reproduced from ref. 92, an open-source article from Plos. (f) Cellular uptake of FITC–Cur–PLGA-NPs: phase contrast and fluorescent images of the neurospheres formed by hippocampus-derived neural stem cells (from Wistar rats). Panel (f) reproduced with the permission from ref. 93. Copyright 2014 American Chemical Society. | ||
Rat hippocampal slices derived from 6- to 8-day old male Wistar rats have been used as an ex vivo Aβ neurotoxicity model. Tiwari et al. acquired hippocampal-derived neuronal stem cells (NSC) from embryonic day-12 rat embryos to study the effect of curcumin-encapsulated PLGA NPs on neurogenesis (Fig. 4f).93 It has been proposed that the induction of neurogenesis could be a therapeutic approach against AD by influencing the brain self-regenerative capacity. In addition, mouse islets have been employed as an ex vivo toxicity model in T2DM research. Specifically, Pilkington et al. demonstrated that OH-terminated star-shaped polymers promoted IAPP amyloid aggregation while mitigated the peptide-induced cytotoxicity.26
Despite several advantages, ex vivo models are difficult to reproduce and the preparations are expensive and time-consuming requiring significant expertise. Therefore, ex vivo models are generally unsuitable for large-scale studies.94 Moreover, these models originate from animals with significant differences in physiology, gene expression patterns and drug metabolism compared to humans.
3. In vivo models for validating anti-amyloid nanomedicines
Preclinical studies with animal models play a crucial role to extrapolate the therapeutic relevance, safety and efficacy of nanomedicines to human. It has been highly desirable to select or develop an animal model which closely mimics the pathophysiology of a human disease. However, there have always been limitations in employing higher animal models that have more human relevance, e.g., non-human primates. Such constraints include ethical checks, limitation on the number of animals available for statistical significance, availability of these sophisticated animal models and ease of experimentation. Therefore, small rodents like rats and mice as well as simplified but still comprehensive animal models like zebrafish and Caenorhabditis elegans have been extensively employed in the literature for pre-clinical studies of nanomedicines. These animal models provide relevance to human physiology, genetic make-up, pathologies and organic distribution to provide valuable pre-clinical data for validating the pharmacology, pharmacokinetics, therapeutic efficacy and safety of nanomedicines. In this section, the wild type or genetically modified mice and rats (as mammalian models), zebrafish as well as C. elegans models will be discussed as pre-clinical models for anti-amyloid nanomedicine (Fig. 5). | ||
| Fig. 5 In vivo models for anti-amyloid nanomedicine. (a) In vivo models and delivery methods on anti-amyloid nanomedicines (NPs). (b) Applications of in vivo models for validating anti-amyloid nanomedicines. | ||
3.1 Mammalian models
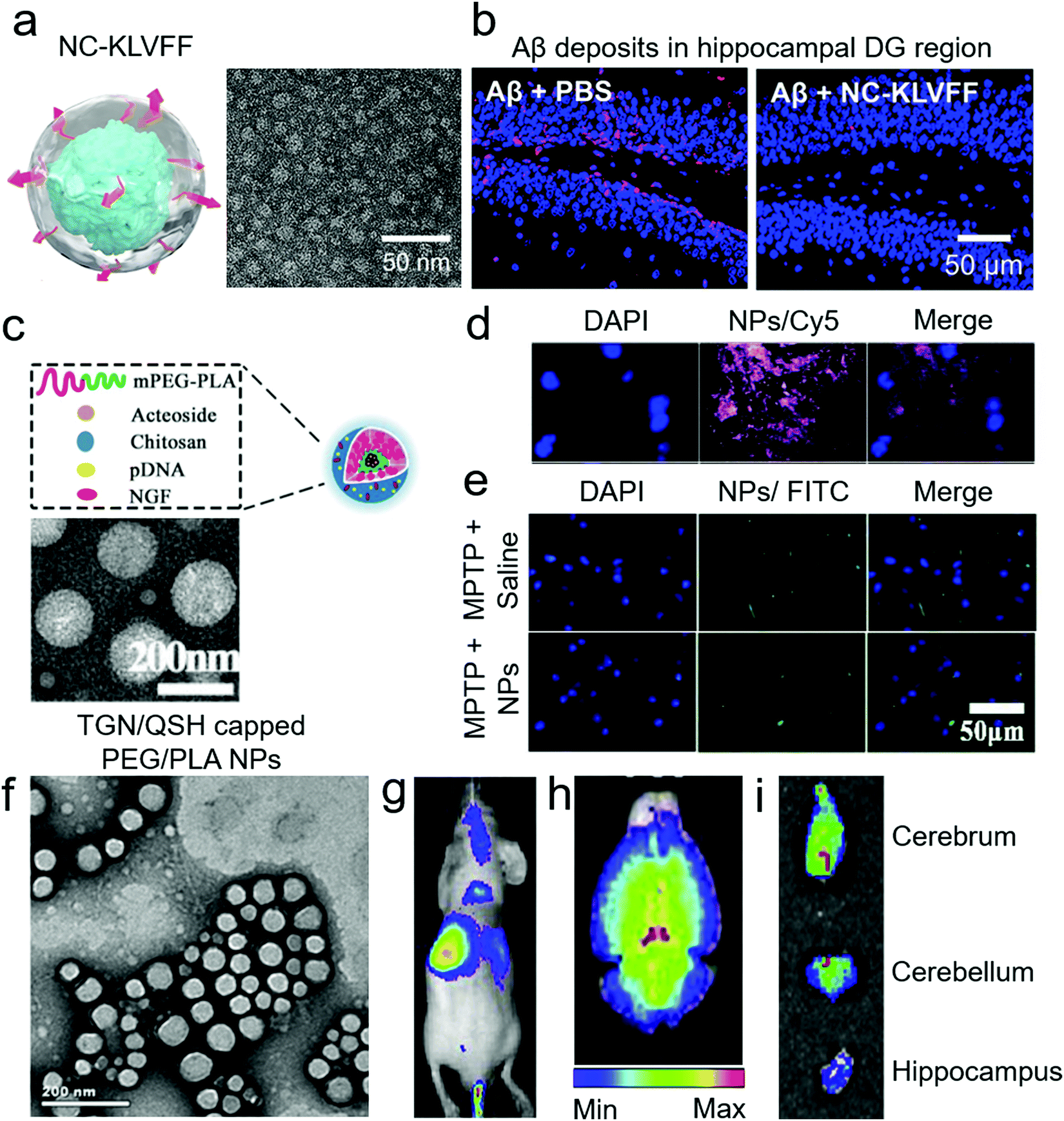 | ||
| Fig. 6 Use of non-transgenic mouse models for anti-amyloid nanomedicines. (a) Schematic and TEM image of KLVFF peptide-capped nanocomposites (NC-KLVFF). The nanocomposites alleviated Aβ toxicity from ICR-Swiss WT mice. (b) Deposition of Aβ in the hippocampal dentate gyrus (DG) region of mice and this deposition was prevented by intravenous injection of nanocomposites. Panels (a and b) reproduced with the permission from ref. 71. Copyright 2019 American Chemical Society. (c) Schematic and TEM image of acetoside-loaded chitosan NPs. (d) The NPs distributed to the substantia nigra of C57 WT mice, with peak concentration showing 2 h after intraperitoneal injection. (e) In the MPTP-induced PD model, the NPs inhibited αS fibrillization. Panels (c–e) reproduced with the permission from ref. 105. Copyrights 2020 Elsevier. (f) TEM image of TGN/QSH-capped PEG/PLA NPs. These NPs were loaded with coumarin-6, coumarin-7 and 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl indotricarbocyanine iodide dye. Panels (g and h) present in vivo and ex vivo brain distributions of the NPs in nude mice after tail vein injection. (i) Ex vivo distribution of the NPs in different regions of the brain in Aβ-injected ICR WT mice. Panels (f–i) reproduced with the permission from ref. 72. Copyright 2014 Elsevier. | ||
Another major application of Aβ-injected WT mouse model is for the imaging of neuronal plaques (Table 5). Zhang et al. used nude WT mice for near-infrared imaging of Aβ plaques.72 Preformed Aβ fibrils were injected bilaterally to the hippocampus and traced by intravenously injected NPs one week after the Aβ treatment. Polylactic acid/PEG NPs were equipped with targeting capacity via TGN/QSH peptides and loaded with a DiR probe (Fig. 6f). By optimising the targeting peptide ratios, NPs were able to label Aβ deposits in vivo more effectively. Although Aβ was injected bilaterally to the hippocampus, the NPs were able to tag both sides of the hippocampus in ex vivo imaging (Fig. 6g–i). Similarly, Hartig et al. used C57B6 WT mice to deliver the amyloid-specific thioflavin T (ThT) dye to the brain.106 Polymeric NPs with a shell of butyl-cyanoacrylate and core of ThT/polystyrene were directly injected to the hippocampus of mice, and the brain was fixed and sliced three days post-treatment. The NPs were found to be localized in microglia and cytoplasm of granule cells. However, the ability of these NPs to target amyloid-plaques in vivo and their BBB permeability are yet to be explored.
| Animal | Description | Disease | NPs | Applications and methods | Ref. |
|---|---|---|---|---|---|
| WT mice | The phenotype with respect to a given inherited characteristic that is considered to be the “normal” type commonly found in natural populations. | AD | nDa + PEG-MNSs + AβO-Ab | Intranasal delivery. MRI detection of early AD. | 48 |
| Nude mice | Mutants with a spontaneous deletion in the FOXN1 gene. Deteriorated or absent thymus, resulting in an inhibited immune system due to reduced number of T cells. The phenotype is lack of body hair and hence “nude”. Applications in imaging and tumour research. | AD | PPEG–PLA + TGN and QSH | Tail vein injection. Dissected brain imaging. | 72 |
| ICR-Swiss mice | Albino mice originating from Switzerland (SWISS) and outbred by the Institute of Cancer Research (ICR) in the USA. General use in oncological and pharmaceutical research. | AD | KLVFF-NCs | Aβ intracerebroventricular injection, NP intravenous administration daily for 3 weeks. Biodistribution, hippocampus neuronal morphology and Aβ levels | 71 |
| PEG–PLA + TGN and QSH | Bilateral Aβ injection into hippocampus. Brain imaging. | 72 | |||
| C57BL/6 mice | Inbred strain. The most widely used “genetic background” for genetically modified mice for use as models of human diseases. | AD | RI-OR2-TAT NLIPs | Intraperitoneal injection. Biodistribution | 64 |
| ThT-LPs (HL-23) | Intrahippocampal injection. Dentate gyrus vibratome sectioning and EM analysis. | 106 | |||
| C57BL/6J mice | C57BL/6J is the parental sub-strain of C57BL/6; “J” is the laboratory code for The Jackson Laboratory. Is the most widely used inbred strain. A general-purpose strain and background strain for spontaneous and induced mutations. | AD | USPIO NPs + Aβ1–42 | Femoral intravenous injection. μMRI. | 110 |
| C57BL/6J mice PD model | PD model induced by stereotaxical injection of sonicated preformed αS fibrils (PFFs) into the striatum. Induces endogenous αS aggregation and subsequent nigral dopaminergic neuron degeneration. | PD | GQD | PD model – injection of a-syn PFFs. Behavioural tests (cylinder and pole) | 79 |
| B6SJLF1/J mice | An inbred mouse. A cross between C57BL/6J females (B6) and SJL/J males (SJL) mice. Often used in the production of transgenic mice. Genetically and phenotypically uniform. | CAA | Gd-DTPA + IgG-anti-Aβ + CS + 125I + CTX | Plasma pharmacokinetics and tissue distribution. SPECT/CT and MR imaging. | 75 |
WT mice have also been used as a background control in parallel to transgenic mice of amyloid diseases. Viola et al. used B6SJLF1 WT mice together with 5×FAD transgenic mice to study the colocalization, distribution and MRI imaging of Aβ plaques.48 5×FAD possesses five AD-related mutations: Swedish (K670N/M671L), Florida (I716V) and London (V717I) mutations in APP, and M146L and L286V mutations in PSEN1.107 Magnetic NPs were capped with Aβ oligomer-specific antibodies that directed the NPs towards Aβ sites in the brain. The NPs were administered via the intranasal route to bypass the BBB. NPs accumulated and yielded MRI signals in the temporal or hippocampal regions of transgenic mice only, while WT mice served as a negative background control. A study with a similar cohort used FVB/N WT mice as a negative control for IAPP induced diabetes.108 IAPP expressed by WT mice did not show the propensity to fibrillate due to the proline substitution in IAPP20–29.109 Geisler et al. demonstrated that estrogen (E2) reversed the diabetic symptoms in human-IAPP transgenic mice, while using lean FVB/N WT mice as a negative control.108
| Animal | Description | Disease | NPs | Animal application and methods | Ref. |
|---|---|---|---|---|---|
| APP/PS1 mouse | Transgenic AD mouse model with two mutations: Swedish and PSEN1. Amyloid plaque deposition starts at ∼6 months of age. Main use in neurobiology research and AD. | AD | USPIO NPs + Aβ | Femoral intravenous injection. μMRI. | 110 |
| PA-LIP, CL-LIP | Repeated intraperitoneal injection. Sink-effect. | 41 | |||
| Qc@SNPs-MB | Tail vein injection twice a week for 5 weeks. In vivo fluorescence images, learning and cognitive abilities (Morris water maze test). | 40 | |||
| mApoE-PA-LIPs | Intraperitoneal injection. Sink effect. | 76 | |||
| PPI-MalGD | Intraperitoneal injection, short term intranasal and long-term intranasal administrations. Memory test. Brain tissue analysis (immonohistochemistry, FTIR microscopy). | 38 | |||
| anti-Aβ-SPIONs | Tail vein injection. MRI, brain histology. | 119 | |||
| FeO + DSPE–PEG–NHS + CR + rutin | Intravenous injection. MRI. | 67 | |||
| Aβ1–40-Gd, Aβ-MION | Femoral intravenous injection. In vivo and ex vivo MRI brain imaging, brain histology. | 110 | |||
| 5 × FAD mouse | APP/PS1 transgenic mice that co-express five familial AD (FAD) mutations ([APP K670N/M671L (Swedish) + I716V (Florida) + V717I (London) and PS1 M146L + L286V]). Develop cerebral amyloid plaques and gliosis at 2 months of age, achieve massive Aβ burdens and memory impairment. | AD | SiNCs–scFvs | Stereotaxic injection. Ex vivo brain imaging. | 44 |
| nDa/PEG-MNCs + AβO-Ab | Intranasal delivery. MRI detection of early AD. | 48 | |||
| Tg2576 mouse | AD transgenic mice – carries the Swedish double mutation (K670N/M671L). Express mutant human APP at about 5.5 times the level of endogenous murine APP in the brain. AD characteristics: Aβ plaques (11–13 months), dystrophic neuritis, astrogliosis, microgliosis. | AD | (P(HDCA-co-MePEGCA)) + Aβ1–42-mAb | Intravenous injection. Behavioural and memory tests, Aβ levels in brain and plasma. | 120 |
| Cur-SPIO + PEG–pLacA-CoP + PVP | Femoral vein injection. Immunohistochemical examination of the mouse brains. Ex vivo MRI brain imaging. | 63 | |||
| RI-OR2-TAT NLIPs | Intraperitoneal injection. Biodistribution, memory test (NOR test). | 64 | |||
| αS A53T mouse | Overexpress human αS in the brain at levels about 6× higher than endogenous mouse αS with a PD-associated mutation (A53T). Develop severe progressive motor impairments around one year of age. | PD | GQD | Intraperitoneal injection. Behaviour analysis (hindlimb clasping function). | 79 |
 | ||
| Fig. 7 Use of transgenic mouse models for anti-amyloid nanomedicines. (a) TEM image of curcumin-capped Fe NPs. When injected intravenously to Tg2576 mice, the NPs were distributed to the brain and were able to label Aβ plaques. (b) Triple-labelled Aβ plaques in mice brain section; amyloid plaques (red), curcumin (yellow) and iron (blue) co-localized in some plaques. Panels (a and b) reproduced with the permission from ref. 63. Copyrights 2015 Elsevier. (c) Difference in Aβ depositions, both neuronal and angiopathic, among transgenic mice and humans. The Aβ deposition in humans was intense and involved vascular amyloid-deposits. (d) Microglia were highly active and were embedded inside the Aβ plaques in humans, while they were weak and limited to the surroundings in APP23 transgenic mice. Similarly, Aβ plaques in transgenic mice displaced neurons, glia and cellular processes but not in humans. Panels (c and d) reproduced with the permission from ref. 118. Copyright 2004 Elsevier. (e) APP/PS1 transgenic mice produced from traditional B6 mice displayed higher Aβ plaques but APP/PS1 transgenic mice derived from new wild-types mice (CAST, WSB and PWK) displayed higher Aβ production, lower Aβ plaques and higher Aβ oligomers. (f) WSB.APP/PS1 presented Aβ deposits around the cerebral blood vessels (traced by X34; a Congo-red derivative) and fibrin staining outside the vessels, indicating compromised vascular integrity. Panels (e and f) reproduced from ref. 96, an open-source article from PLoS. | ||
Despite all these transgenic efforts to introduce AD pathologies and Aβ overexpression in mice, there is still a gap between transgenic mice models and the actual AD pathologies in human subjects. This gap may be responsible for the failure of AD therapies in clinical trials that had shown promising results in transgenic mice models. Schwab et al. has demonstrated the limitations of transgenic mice by comparing Swedish double mutation KM670/671NL AD mice and actual human samples (Fig. 7c and d).118 Both samples presented Aβ lesions, ApoE inclusion and astrocytes activation. However, the mouse samples displayed astrocytes at the periphery of Aβ lesions but human samples showed activated astrocytes in the core. The complement receptor CD11b was highly active in human samples while mouse samples had weak activation of CD11b. Furthermore, there were significantly higher tau tangles in the human samples, compared to the mice samples. To address these limitations, Onos et al. has recently introduced another cohort of transgenic mice derived from WT mice that can represent AD pathologies with better human relevance.96 Two widely used AD transgenes of APPswe and PS1de9 (APP/PS1) were cross-introduced into three WT mice strains CAST/EiJ, WSB/EiJ and PWK/PhJ that represented natural genetic variations suitable for modelling AD pathogenesis. The resulting transgenic models presented cognitive loss, neurodegeneration in cortical and hippocampus regions, expression of toxic Aβ oligomers rather than plaques (Fig. 7e), mixed AD pathologies of vascular Aβ deposition (Fig. 7f), and transcriptional profiles. However, further research is needed to validate these novel mouse models.
| Animal | Description | Disease | NPs | Animal application and methods | Ref. |
|---|---|---|---|---|---|
| Wistar rat | An outbred albino rat – one of the most popular rats used for laboratory research. Used in all fields of medical and biological research. Easy to handle. Its longevity and high rate of spontaneous tumours make it an ideal choice for ageing studies. | AD | Cur–PLGA-NPs | Stereotaxic injection into hippocampus. Biodistribution and pharmacokinetic. Learning and memory impairments. | 93 |
| IndOH-LNCs | Intracerebroventricular injection. In vivo neurotoxicity, behavioural test (Y-maze test). | 95 | |||
| PS-80-PIP-SLN | AD model by ibotenic acid-stimulated lesions of basalis magnacellularis. Brain histopathological analysis. | 130 | |||
| Sprague-Dawley rat | An outbred multipurpose breed of albino rat used extensively in medical research. Was developed from Wistar rat. One of the two most frequently used rat models. Life span is 2.5–3.5 years. Calm and easy to handle. | AD | TFB–PLGA NPs, TFB-SLNs | Intranasal administration. Drug biodistribution, plasma and brain pharmacokinetics. | 131 |
| Cur loaded Lf-NLC | AD model by intraperitoneal injection of d-gal and bilateral injection in the dorsal hippocampus of Aβ. Intravenous administration of NPs. Biodistribution (brain cryotome sectioning). | 85 | |||
| CN-SLN | Intracerebroventricular injections. Behavioural studies (open field test, novel object recognition, radial arm maze training, water maze and Y-maze tests). | 132 | |||
| CeNC/IONC/MSN-T807 | Tauopathy rat model by okadaic acid microinjection into right the hippocampus. Learning and memory impairments (Morris water maze test). | 37 | |||
In terms of PD rat models. Paumier et al. injected αS monomers and preformed fibrils to the WT Sprague-Dawley rats via the intrastriatal route. It resulted in significant nigrostriatal degeneration, fibrillization of endogenous αS and formation of Lewy bodies in the frontal and insular cortices, amygdala and the substantia nigra pars compacta.127 Another approach is the lentiviral-based induction of αS overexpression. Bianco et al. injected human αS overexpressing lentivirus into the substantia nigra of Wistar rats.128 This resulted in a selective loss of dopaminergic neurons in nigral regions and an abundance of αS-positive inclusions. However, no behavioural or cognitive decline was examined in this study. Rat models have also been developed for IAPP overexpression and diabetes. The IAPP from rats and mice lack the propensity to aggregate or to form toxic oligomers, as observed in human. A fusion DNA of human IAPP was injected to Sprague-Dawley rat eggs and the born rats were studied for 20 months.129 The human IAPP transgenic rats presented significant weight-loss, high blood-glucose and impaired insulin response. Their mean β-cells mass was decreased at the 20 month time-point and IAPP oligomers were more toxic to replicating β-cells compared to static β-cells.
3.2 Zebrafish
The pathological progression of amyloid diseases is complex and age dependant. In human, there is a simultaneous involvement of multiple pathogenic and etiological pathways that are difficult to model in rodent models. Although rodents, especially the mouse models, are widely used for modelling amyloid diseases, the focus has been on engineering the genetic-overexpression of amyloid proteins or amyloid precursor proteins, as with AD.133 These models have provided tissue degeneration and amyloid-plaque build-ups. However, therapeutic strategies succeeded in cleaning these plaques in rodents, failed in human clinical trials.134 The reason lies in the etiological complexity of these diseases, lack of genetic diversity in rodent models and their natural propensity to forgo amyloid diseases. The search for new lab-rats led to the use of zebrafish (Danio rerio) for the modelling of amyloid diseases, particularly AD (Table 8). Although zebrafish do not naturally develop amyloid diseases, they provide an alternative to mice for preclinical screening of nanomedicines. The zebrafish models can address the internal/external validity of animal modelling, provide a checklist for quality control for replication of therapeutic results at different sites and enable high-throughput screening.111 Furthermore, zebrafish possess all the protein machinery required to study AD pathologies, i.e., APPa and APPb, Aβ production, γ-secretase enzyme family and presenilin 1, 2 and 3. The neuro-regenerative ability allows zebrafish to act as a suitable animal model for neurodegenerative amyloid diseases.135 The ex-utero development of zebrafish larvae also provides genetic-modulation opportunities. The physiological function of Aβ to modulate cardiovascular development was demonstrated for the first time in zebrafish.136 Zebrafish embryos were injected with morpholino to induce Aβ deficiency. The larvae developed cardiovascular defects that were reversible by treating with Aβ. This indicates that AD therapies should target Aβ aggregation but not production. Alongside proteomics, zebrafish also display AD-relevant neurotransmission, i.e., γ-amino butyric acid, cholinergic and glutaminergic transmission.137 The complexity of amyloid diseases can be modelled in zebrafish by manipulation of multiple genes in embryos, a characteristic limitation of rodents.135,138 Recently, Javed et al. have demonstrated the use of embryonic, larval and adult zebrafish models for mimicking the different pathological manifestations of amyloid diseases.39,139,140 Embryonic zebrafish provide an advanced quasi-in vivo model system with an opportunity to study the toxicity of amyloid proteins. Specifically, zebrafish embryos were exposed to Aβ and IAPP via direct injection to perivitelline and immersion in amyloid protein solutions directly or after removal of the chorionic membrane.140 Direct injection of amyloid proteins to the perivitelline space was highly effective as it required the least amount of the protein (fM concentrations in nL injection volume) (Fig. 8a and b). This optically transparent model also allowed imaging of amyloid proteins interacting with embryonic cell membranes and direct observations of IAPP toxicity and ROS via fluorescence microscopy. Co-injection of amyloid proteins with different nanomaterials, i.e., CNT functionalized with the fragments of β-lactoglobulin amyloids (Fig. 8c), GQDs and chiral silica nano-ribbons, mitigated the toxicity of amyloid proteins in zebrafish embryos.27,70,140,141 To further develop this model, Javed et al. employed larval and adult zebrafish to study the clinical and histological pathologies of AD by using direct injection, as discussed for WT mice and rats in above sections. Aβ was injected directly to the interventricular space of zebrafish larvae and adults.39 In the case of zebrafish larvae, fibrillization of Aβ was imaged in vivo by co-injection the peptide with Congo-red dye (Fig. 8d). The total swimming distance and movement frequency were decreased for larval zebrafish. The pathologies were rescued by systemic injection of β-casein-capped AuNPs, which sequestered toxic Aβ species (Fig. 8e). Injection of Aβ to adult zebrafish resulted in a cognitive decline (Fig. 8f). The adult zebrafish were trained to avoid swimming in the red-labelled part of the tank, by using an electric shock (12 V) as a penalty. The healthy control fish learned to avoid swimming into the red-labelled area of the tank while Aβ-treated fish were unable to differentiate between the normal and red-labelled areas of the tank. The training time was 20 min and observation time was 1 min. The immunohistochemistry on the brain sections of the fish displayed neuronal deposition of Aβ, degeneration of synapses (Fig. 8g), DNA damage, increased acetylcholine esterase activity, enhanced glutamate levels and ROS production in the brain. The symptoms in adult zebrafish were also prevented by systemic injection of β-casein AuNPs. This in vivo model was further used to study the acceleratory impact of Pseudomonas aeruginosa FapC amyloid seeds on the fibrillization and pathological manifestations of Aβ.139| Animal | Description | Disease | NPs | Animal application and methods | Ref. |
|---|---|---|---|---|---|
| Zebrafish | Danio rerio – has become a leading vertebrate model for studying maternal germ cell specification due to large embryo clutch sizes, genetic tractability, optical clarity, and rapid germ line formation. Display neuropathological and behavioural phenotypes. Suitable for neuroscience research because of their complex behaviours, high genetic and physiological homology to humans. | AD | βCas-AuNPs | Injecting Aβ into the cerebroventricular space of larvae. Zebrafish larvae behavioural pathology. Behavioural pathology and cognitive function test of adult zebrafish. | 39 |
| AD, T2DM | Amf-MWCNTs | Microinjection into the embryos. ROS assay, toxicity, hatching. | 140 | ||
| T2DM | GQD | Microinjected into the yolk of the embryos. ROS assay, toxicity. | 27 | ||
| CRSi NRs | Microinjected into the yolk of the embryos. ROS assay, toxicity. | 70 | |||
| IAPP, 19–29 S20G, 8–20 fibrils | Microinjected into the yolk of the embryos. Toxicity (hatching, abnormality). | 141 | |||
| C. elegans CL2006 | Expresses human Aβ42 under the control of the muscle-specific promoter unc-54, leading to progressive adult onset paralysis. Temperature sensitive. | AD | UCNP-C60-KLVFF | Paralysis and motility impairments. Immunofluorescent staining experiments. | 60 |
| BP-BTA | Paralysis and motility impairments. Fluorescent microscopy. | 56 |
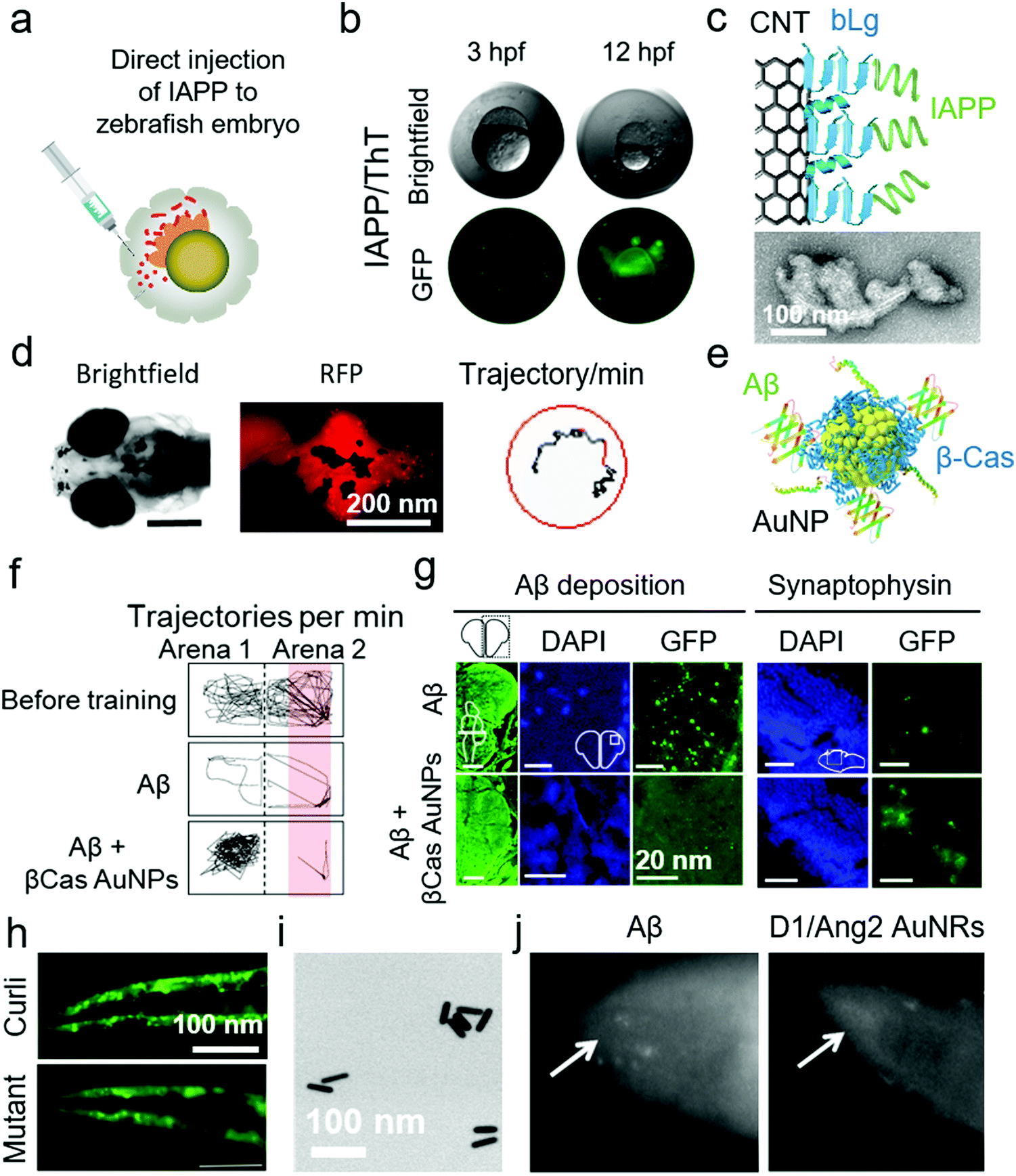 | ||
| Fig. 8 Use of zebrafish and C. elegans for anti-amyloid nanomedicine research. (a) Development of zebrafish embryos as a toxicity model for human IAPP by direct injection to the perivitelline space. (b) IAPP-injected embryos were dead 12 h post fertilization (hfp). The interaction of IAPP with embryonic cells was traceable by ThT dye. (c) Use of β-lactoglobulin (bLg) amyloid-fragments coated CNTs to sequester IAPP, a scheme that protected zebrafish embryos against IAPP toxicity. Panels (a–c) reproduced from ref. 140. Copyright 2018 American Chemical Society. (d) Zebrafish larvae were injected with Aβ in the intraventricular space. Fibrillization of injected Aβ was traceable by Congo-red injections via the same route. Aβ injection to zebrafish larvae suppressed their swimming behaviour. (e) β-Casein-capped AuNPs were used to scavenge toxic Aβ species, demonstrating efficacy in vitro and in vivo. (f) Microinjection of Aβ to adult zebrafish induced cognitive dysfunction, (g) cerebral Aβ deposition and synaptic degeneration. (h) C. elegans that express αS, displayed an overproduction of αS plaques in the muscle cells when exposed to curli-producing E. coli. However, normal αS deposition was observed upon exposure to non-curli producing mutant E. coli. Panels (d–h) reproduced from ref. 39 and 148. Copyright 2019 and 2016 Springer Nature. Dopamine receptor 1 and Angiopep2 peptide capped Au nanorods (AuNRs) (TEM image in panel (i)) rescued C. elegans from producing Aβ deposits. (j) Fluorescence microscopy images of Aβ-producing C. elegans with and without AuNRs treatment. Reproduced with the permission from ref. 150. Copyright 2017 Elsevier. | ||
3.3 Caenorhabditis elegans
The quest for increasing the lifespan in human has led to the use of Caenorhabditis elegans as animal models. C. elegans age and die in a few weeks that is 2000-fold less than human, however, the genetic makeups for aging pathways display some similarities between the two species. Modulation of insulin and insulin-like growth factors, Toll signalling, adenosine monophosphate kinases, telomeres and reduced ROS increase the lifespan in C. elegans.142 These genetic traits of aging allowed the introduction of C. elegans to amyloid research (Table 8). Initially, C. elegans were genetically engineered to express human Aβ in their muscle cells by incorporating human cDNA clones in muscle-specific unc-54 promoter/enhancer of C. elegans.143 It resulted in the production of immunoreactive and thioflavin S binding amyloid plaques in the muscles of C. elegans. The Aβ expression further induced ROS generation, muscle paralysis and reduction in the chemotactic movement that was specific to the oligomeric not the fibrillar state of Aβ.144,145 Like zebrafish larvae, C. elegans also provide an opportunity for live imaging of amyloidosis with amyloid-specific dyes.146 Similarly, αS-expressing transgenic C. elegans strains were produced that displayed αS deposits in the peripheral muscle cells, decreased locomotion and body-bend movements as well as loss of dopaminergic neurons.147 These αS-expressing C. elegans were also used to study the acceleratory effect of orally-fed curli-producing Escherichia coli (Fig. 8h).148 In terms of nanomedicine research, Yin et al. used Aβ-expressing C. elegans to evaluate the efficacy of resveratrol NPs.149 Resveratrol, a lipophilic antioxidant, was encapsulated inside PEG/polycaprolactone NPs. These NPs relieved Aβ-induced ROS and muscle paralysis. Similarly, Zavala et al. synthesized dopamine receptor 1 and Angiopep 2-capped Au nanorods (NRs) that protected Aβ toxicity in C. elegans by inhibiting Aβ fibrillization (Fig. 8i and j).1504. Conclusions and outlook
This review has presented the state-of-the-art concerning the use of in vitro and in vivo models for validating anti-amyloidosis nanomedicines. These model systems are both an essential passage and a roadblock for the mitigation of a wide range of debilitating human amyloid diseases, from AD, PD and T2DM to systemic amyloidosis, and much more. In the light of the countless failures that have shrouded and stigmatised research on dementia, it is apparent that each animal model has its unique strengths as well as deficiencies in accurately representing the pertinent human disease pathologies. Needless to say, a main contributor to this dilemma arises from our still evolving knowledge of amyloidogenesis in vivo, further complicated by our limited understanding of its multifactorial manifestations such as inflammation, autophagy, protein “corona”, as well as crosstalk between the pathologies.9,29,30,151,152 For the laboratory investigators, i.e., chemists, materials scientists, biomedical engineers, nanotechnologists, pharmacists and structural biophysicists, the choice of a specific cell line or an animal model often evokes a debate and then a compromise between the extent of scientific discovery, quality of data and the practicalities of cost, ethics, accessibility, time and available expertise associated with the models. It should be noted that current research in the field of anti-amyloidosis nanomedicine is still nascent, as reflected by the prevalence of test tube-only data, although the use of a cell line, often in conjunction with an animal model, has become an increasingly new trend. On the other hand, while beyond the scope of this review, in silico methodologies for simulating the structure and kinetics of amyloid protein aggregation as well as for capturing single-molecule/particle interaction between amyloid proteins and their nanoparticle inhibitors, should be acknowledged as indispensable tools for enhancing laboratory research in revealing the inner workings of complex nano–bio systems.17,153–159 It is our hope that this review will play a role in informing our peers of the current development, availability as well as the pros and cons of existing in vitro and in vivo models employed in anti-amyloidosis nanomedicine. Continued improvement and smart use of such model systems will facilitate our quest for deciphering the origin of amyloid diseases and finding a cure towards their elimination.160Abbreviations
| Ab | Antibody |
| AD | Alzheimer's disease |
| Amf | Amyloid fragment |
| AuNPs | Gold nanoparticles |
| AuNR | Gold nanorod |
| Aβ | Amyloid-beta |
| BACE1 | Beta-secretase responsible for amyloid-(A) generation in the brain |
| BBB | Blood brain barrier |
| bLg | Beta lactoglobulin |
| BP | Black phosphorus |
| bPEI | Polyethyleneimine |
| BTA | Thioflavin-T derivative |
| c-AgNPs | Citrate-coated AgNPs |
| CAA | Cerebral amyloid angiopathy |
| CL | Cardiolipin |
| CN | Chrysin |
| CNS | Central nervous system |
| CNTs | Carbon nanotubes |
| CPDs | Cationic phosphorus dendrimers |
| CPP | Cell penetrating peptide |
| CQ | Clioquinol |
| CR | Congo Red |
| CRSi NRs | Chiral silica nanoribbons |
| CS | Chitosan |
| CSF | Cerebrospinal fluid |
| CTX | Cyclophosphamide |
| Cur | Curcumin |
| DA | Dopamine |
| DCFH-DA | Dichloro-dihydro-fluorescein diacetate |
| DSPC | 1,2-Distearoyl-sn-glycero-3-phosphocholine |
| DSPE | 1,2-Dioleoyl-sn-glycero-3-phosphoethanolaminen |
| ECs | Endothelial cells |
| EGCG | Epigallocatechin gallate |
| ELISA | Enzyme-linked immunosorbent assay |
| FAD | Familial AD mutations |
| G0-PAMAM | Generation 0 poly(amidoamine) dendrimer |
| GATG | Gallic acid-triethylene glycol dendrimer |
| Gd-DTPA | Gadopentetate dimeglumine |
| GH | Galantamine hydrobromide |
| (GH-SLNs)NPs | Galantamine hydrobromide-loaded solid–lipid NPs |
| GO | Graphene oxide |
| GQD | Graphene quatum dots |
| IAPP | Islet amyloid poppypeptide |
| IndOH | Indomethacin |
| IONC | Iron oxide nanocrystals |
| LDH | Lactate dehydrogenase |
| Lf | Lactoferrin |
| LIPs | Liposomes |
| LNCs | Lipid-core nanocapsules |
| LND | Lipid-nanodiscs |
| LPs | Core–shell latex particles (HL-23) |
| mAb | Monoclonal antibody |
| MAG | Maghemite |
| MagNPs | Maghemite nanoparticles |
| MalGD | Maltose glycodendrimers |
| mApoE | ApoE-derived peptide |
| MB | Microbubbles |
| MeSMC | Metallosupramolecular complexes |
| MION | Monocrystalline iron oxide nanoparticles |
| MMP | Tetra-maleimidopropionlyl |
| MNCs | Magnetic nanoclusters |
| MND | Macro-nanodiscs |
| Mor | Morpholine groups |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MSNs | Mesoporous silica nanoparticles |
| MTT | 3-(4,5-Dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide |
| MWCNT | Multi-wall carbon nanotubes |
| NC | Nanoclusters |
| ND | Neurodegeneration |
| nDA | Nitro-dopamine |
| NGF | Nerve growth factor |
| NHS | N-Hydroxysuccinimide |
| NLC | Nanostructured lipid carrier |
| NLIPs | Nanolipopsomes |
| NPs | Nanoparticles |
| OL | Odorranalectin |
| OX26MAb | Anti-transferrin receptor monoclonal antibody |
| PA | Phosphatidic acid |
| PBCA | Poly(n-butylcyanoacrylate) |
| PBS | Phosphate buffer saline |
| PC | Phosphorylcholine |
| PD | Parkinson's disease |
| PEG | Polyethylene glycol |
| pLacA-CoP | Polylactic acid block copolymer |
| PEI | Polyethylenimine |
| PHEA | Poly(2-hydroxyethyl acrylate) |
| P(HDCA-co-MePEGCA) | PEG–poly[hexadecyl cyanoacrylate-co-methoxypoly(ethylene glycol)cyanoacrylate] |
| PI | Propidium iodide |
| PIP | Piperine |
| PLA | Poly(lactic acid) |
| PLGA | Poly(lactide-co-glycolide) |
| PMA | Polymethacrylate-copolymer |
| PPI | Poly(propylene imine) |
| PS-80 | Polysorbate-80 |
| PVP | Polyvinylpyrrolidone |
| Qc@SNPs | Quercetin-modified sulfur nanoparticles |
| QSH | D-Enantiomeric peptide QSHYRHISPAQV |
| RHT | Rivastigmine |
| RI-OR2-TAT | Retro-inverso peptide |
| ROS | Reactive oxygen species |
| scFvs | Anti-Aβ single-chain variable fragments |
| SiNC | Porous silica nanostructures |
| SLN | Solid lipid nanoparticles |
| SWCNT | Single-wall carbon nanotubes |
| T2DM | Type 2 diabetes mellitus |
| TEER | Transendothelial electrical resistance |
| TFB | Tarenflurbil |
| TGN | TGNYKALHPHNG ligand |
| ThT | Thioflavin-T |
| TNVs | Theranostic nanovehicles |
| UCNP | Upconversion nanoparticle |
| US | Ultrasound |
| USPIO | Ultrasmall superparamagnetic iron oxide |
| WT | Wild-type |
| XTT | Sodium 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)-carbonyl]-2H-tetrazolium |
| αS | Alpha-synuclein |
| βCas | Beta casein |
| 19–29 S20G | 19–29 amino acids IAPP fragment with S20G mutation |
| 19B8MAb | Anti-Aβ peptide antibody |
| 8–20 | 8–20 amino acids IAPP fragment |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by ARC Project No. CE140100036.References
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. Ø. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453–457 CrossRef CAS.
- R. M. Koffie, M. Meyer-Luehmann, T. Hashimoto, K. W. Adams, M. L. Mielke, M. Garcia-Alloza, K. D. Micheva, S. J. Smith, M. L. Kim, V. M. Lee, B. T. Hyman and T. L. Spires-Jones, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4012–4017 CrossRef CAS.
- M. G. Iadanza, M. P. Jackson, E. W. Hewitt, N. A. Ranson and S. E. Radford, Nat. Rev. Mol. Cell Biol., 2018, 19, 755–773 CrossRef CAS.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS.
- S. T. Ferreira and W. L. Klein, Neurobiol. Learn. Mem., 2011, 96, 529–543 CrossRef CAS.
- H. A. Lashuel, D. Hartley, B. M. Petre, T. Walz and P. T. Lansbury, Nature, 2002, 418, 291 CrossRef CAS.
- P. C. Ke, M.-A. Sani, F. Ding, A. Kakinen, I. Javed, F. Separovic, T. P. Davis and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 6492–6531 RSC.
- A. Webers, M. T. Heneka and P. A. Gleeson, Immunol. Cell Biol., 2020, 98, 28–41 CrossRef.
- P. C. Ke, R. Zhou, L. C. Serpell, R. Riek, T. P. J. Knowles, H. A. Lashuel, E. Gazit, I. W. Hamley, T. P. Davis, M. Fändrich, D. E. Otzen, M. R. Chapman, C. M. Dobson, D. S. Eisenberg and R. Mezzenga, Chem. Soc. Rev., 2020, 49, 5473–5509 RSC.
- P. Chen, F. Ding, R. Cai, I. Javed, W. Yang, Z. Zhang, Y. Li, T. P. Davis, P. C. Ke and C. Chen, Nano Today, 2020, 35, 100937 CrossRef CAS.
- N. Andrikopoulos, Y. Li, L. Cecchetto, A. Nandakumar, T. Da Ros, T. P. Davis, K. Velonia and P. C. Ke, Nanoscale, 2020, 12, 14422–14440 RSC.
- Editorial, Alzheimer’s disease: expedition into the unknown, The Lancet, 2016, 388, 2713, DOI:10.1016/S0140-6736(16)32457-6.
- S. Reardon, Nature, 2018, 563, 611–612 CrossRef CAS.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, J. Am. Chem. Soc., 2008, 130, 15437–15443 CrossRef CAS.
- B. R. Sahoo, T. Genjo, T. W. Nakayama, A. K. Stoddard, T. Ando, K. Yasuhara, C. A. Fierke and A. Ramamoorthy, Chem. Sci., 2019, 10, 3976–3986 RSC.
- T. John, A. Gladytz, C. Kubeil, L. L. Martin, H. J. Risselada and B. Abel, Nanoscale, 2018, 10, 20894–20913 RSC.
- P. C. Ke, E. H. Pilkington, Y. Sun, I. Javed, A. Kakinen, G. Peng, F. Ding and T. P. Davis, Adv. Mater., 2020, 32, 1901690 CrossRef CAS.
- X. Sun, N. Ni, Y. Ma, Y. Wang and D. T. Leong, Small, 2020, 16, 2003000 CrossRef CAS.
- X. Ding, F. Peng, J. Zhou, W. Gong, G. Slaven, K. P. Loh, C. T. Lim and D. T. Leong, Nat. Commun., 2019, 10, 41 CrossRef CAS.
- S. Bhatia, T. Naved and S. Sardana, Introduction to Pharmaceutical Biotechnology, 2019, vol. 3 DOI:10.1088/2053-2563/aafac0ch1.
- K. Walsh, J. Megyesi and R. Hammond, Neurobiol. Dis., 2005, 18, 2–18 CrossRef CAS.
- K. Gordon, T. Clouaire, X. X. Bao, S. E. Kemp, M. Xenophontos, J. I. de Las Heras and I. Stancheva, Nucleic Acids Res., 2013, 42, 3529–3541 CrossRef.
- H. P. Modarres, M. Janmaleki, M. Novin, J. Saliba, F. El-Hajj, M. RezayatiCharan, A. Seyfoori, H. Sadabadi, M. Vandal, M. D. Nguyen, A. Hasan and A. Sanati-Nezhad, J. Controlled Release, 2018, 273, 108–130 CrossRef CAS.
- L. Cucullo, B. Aumayr, E. Rapp and D. Janigro, Curr. Opin. Drug Discovery Dev., 2005, 8, 89–99 CAS.
- K.-T. Rim, Toxicol. Environ. Health Sci., 2019, 11, 94–103 CrossRef.
- E. H. Pilkington, M. Lai, X. Ge, W. J. Stanley, B. Wang, M. Wang, A. Kakinen, M.-A. Sani, M. R. Whittaker, E. N. Gurzov, F. Ding, J. F. Quinn, T. P. Davis and P. C. Ke, Biomacromolecules, 2017, 18, 4249–4260 CrossRef CAS.
- M. Wang, Y. Sun, X. Cao, G. Peng, I. Javed, A. Kakinen, T. P. Davis, S. Lin, J. Liu, F. Ding and P. C. Ke, Nanoscale, 2018, 10, 19995–20006 RSC.
- P. C. Ke, S. Lin, W. J. Parak, T. P. Davis and F. Caruso, ACS Nano, 2017, 11, 11773–11776 CrossRef CAS.
- S. Mirsadeghi, R. Dinarvand, M. H. Ghahremani, M. R. Hormozi-Nezhad, Z. Mahmoudi, M. J. Hajipour, F. Atyabi, M. Ghavami and M. Mahmoudi, Nanoscale, 2015, 7, 5004–5013 RSC.
- M. Mahmoudi, O. Akhavan, M. Ghavami, F. Rezaee and S. M. Ghiasi, Nanoscale, 2012, 4, 7322–7325 RSC.
- C. Corbo, R. Molinaro, M. Tabatabaei, O. C. Farokhzad and M. Mahmoudi, Biomater. Sci., 2017, 5, 378–387 RSC.
- J. Ren, R. Cai, J. Wang, M. Daniyal, D. Baimanov, Y. Liu, D. Yin, Y. Liu, Q. Miao, Y. Zhao and C. Chen, Nano Lett., 2019, 19, 4692–4701 CrossRef CAS.
- K. Shah, C. E. McCormack and N. A. Bradbury, Am. J. Physiol.: Cell Physiol., 2014, 306, C3–C18 CrossRef CAS.
- V. Serpooshan, S. Sheibani, P. Pushparaj, M. Wojcik, A. Y. Jang, M. R. Santoso, J. H. Jang, H. Huang, R. Safavi-Sohi, N. Haghjoo, H. Nejadnik, H. Aghaverdi, H. Vali, J. M. Kinsella, J. Presley, K. Xu, P. C.-M. Yang and M. Mahmoudi, ACS Nano, 2018, 12, 2253–2266 CrossRef CAS.
- J. L. Podcasy and C. N. Epperson, Dialogues Clin. Neurosci., 2016, 18, 437–446 Search PubMed.
- H. Xicoy, B. Wieringa and G. J. M. Martens, Mol. Neurodegener., 2017, 12, 10 CrossRef.
- Q. Chen, Y. Du, K. Zhang, Z. Liang, J. Li, H. Yu, R. Ren, J. Feng, Z. Jin, F. Li, J. Sun, M. Zhou, Q. He, X. Sun, H. Zhang, M. Tian and D. Ling, ACS Nano, 2018, 12, 1321–1338 CrossRef CAS.
- O. Klementieva, E. Aso, D. Filippini, N. Benseny-Cases, M. Carmona, S. Juvés, D. Appelhans, J. Cladera and I. Ferrer, Biomacromolecules, 2013, 14, 3570–3580 CrossRef CAS.
- I. Javed, J. He, A. Kakinen, A. Faridi, W. Yang, T. P. Davis, P. C. Ke and P. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 10462–10471 CrossRef CAS.
- Y. Liu, Y. Gong, W. Xie, A. Huang, X. Yuan, H. Zhou, X. Zhu, X. Chen, J. Liu, J. Liu and X. Qin, Nanoscale, 2020, 12, 6498–6511 RSC.
- L. Ordóñez-Gutiérrez, F. Re, E. Bereczki, E. Ioja, M. Gregori, A. J. Andersen, M. Antón, S. M. Moghimi, J.-J. Pei, M. Masserini and F. Wandosell, Nanomedicine, 2015, 11, 421–430 CrossRef.
- L. Agholme, T. Lindström, K. Kågedal, J. Marcusson and M. Hallbeck, J. Alzheimer's Dis., 2010, 20, 1069–1082 CAS.
- M. Buttiglione, F. Vitiello, E. Sardella, L. Petrone, M. Nardulli, P. Favia, R. d’Agostino and R. Gristina, Biomaterials, 2007, 28, 2932–2945 CrossRef CAS.
- H. Jung, Y. J. Chung, R. Wilton, C. H. Lee, B. I. Lee, J. Lim, H. Lee, J.-H. Choi, H. Kang, B. Lee, E. A. Rozhkova, C. B. Park and J. Lee, Adv. Funct. Mater., 2020, 30, 1910475 CrossRef CAS.
- I. Javed, Y. Sun, J. Adamcik, B. Wang, A. Kakinen, E. H. Pilkington, F. Ding, R. Mezzenga, T. P. Davis and P. C. Ke, Biomacromolecules, 2017, 18, 4316–4322 CrossRef CAS.
- Y. H. Liao, Y. J. Chang, Y. Yoshiike, Y. C. Chang and Y. R. Chen, Small, 2012, 8, 3631–3639 CrossRef CAS.
- A. Mathew, T. Fukuda, Y. Nagaoka, T. Hasumura, H. Morimoto, Y. Yoshida, T. Maekawa, K. Venugopal and D. S. Kumar, PLoS One, 2012, 7, e32616 CrossRef CAS.
- K. L. Viola, J. Sbarboro, R. Sureka, M. De, M. A. Bicca, J. Wang, S. Vasavada, S. Satpathy, S. Wu, H. Joshi, P. T. Velasco, K. MacRenaris, E. A. Waters, C. Lu, J. Phan, P. Lacor, P. Prasad, V. P. Dravid and W. L. Klein, Nat. Nanotechnol., 2015, 10, 91–98 CrossRef CAS.
- R. Salto, J. D. Vílchez, M. D. Girón, E. Cabrera, N. Campos, M. Manzano, R. Rueda and J. M. López-Pedrosa, PLoS One, 2015, 10, e0135614 CrossRef.
- T. Wasiak, M. Ionov, K. Nieznanski, H. Nieznanska, O. Klementieva, M. Granell, J. Cladera, J.-P. Majoral, A. M. Caminade and B. Klajnert, Mol. Pharmaceutics, 2012, 9, 458–469 CrossRef CAS.
- J. Yang, Y. Z. Wadghiri, D. M. Hoang, W. Tsui, Y. Sun, E. Chung, Y. Li, A. Wang, M. de Leon and T. Wisniewski, NeuroImage, 2011, 55, 1600–1609 CrossRef.
- R. H. S. Westerink and A. G. Ewing, Acta Physiol., 2008, 192, 273–285 CrossRef CAS.
- L. A. Greene and A. S. Tischler, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 2424–2428 CrossRef CAS.
- L. Smirnova, G. Harris, J. Delp, M. Valadares, D. Pamies, H. T. Hogberg, T. Waldmann, M. Leist and T. Hartung, Arch. Toxicol., 2016, 90, 2725–2743 CrossRef CAS.
- M. Li, S. E. Howson, K. Dong, N. Gao, J. Ren, P. Scott and X. Qu, J. Am. Chem. Soc., 2014, 136, 11655–11663 CrossRef CAS.
- Y. Li, Z. Du, X. Liu, M. Ma, D. Yu, Y. Lu, J. Ren and X. Qu, Small, 2019, 15, 1901116 CrossRef.
- J. Geng, M. Li, L. Wu, C. Chen and X. Qu, Adv. Healthcare Mater., 2012, 1, 332–336 CrossRef CAS.
- W. Li, Y. Zhou, N. Zhao, B. Hao, X. Wang and P. Kong, Environ. Toxicol. Pharmacol., 2012, 34, 272–279 CrossRef CAS.
- H. Skaat, E. Corem-Slakmon, I. Grinberg, D. Last, D. Goez, Y. Mardor and S. Margel, Int. J. Nanomed., 2013, 8, 4063–4076 Search PubMed.
- Z. Du, N. Gao, X. Wang, J. Ren and X. Qu, Small, 2018, 14, 1801852 CrossRef.
- V. Poitout, L. E. Stout, M. B. Armstrong, T. F. Walseth, R. L. Sorenson and R. P. Robertson, Diabetes, 1995, 44, 306–313 CrossRef CAS.
- M. Plissonneau, J. Pansieri, L. Heinrich-Balard, J.-F. Morfin, N. Stransky-Heilkron, P. Rivory, P. Mowat, M. Dumoulin, R. Cohen, É. Allémann, É. Tóth, M. J. Saraiva, C. Louis, O. Tillement, V. Forge, F. Lux and C. Marquette, J. Nanobiotechnol., 2016, 14, 60 CrossRef.
- K. K. Cheng, P. S. Chan, S. Fan, S. M. Kwan, K. L. Yeung, Y. X. Wáng, A. H. Chow, E. X. Wu and L. Baum, Biomaterials, 2015, 44, 155–172 CrossRef CAS.
- M. Gregori, M. Taylor, E. Salvati, F. Re, S. Mancini, C. Balducci, G. Forloni, V. Zambelli, S. Sesana, M. Michael, C. Michail, C. Tinker-Mill, O. Kolosov, M. Sherer, S. Harris, N. J. Fullwood, M. Masserini and D. Allsop, Nanomedicine, 2017, 13, 723–732 CrossRef CAS.
- B. R. Sahoo, T. Genjo, M. Bekier, S. J. Cox, A. K. Stoddard, M. Ivanova, K. Yasuhara, C. A. Fierke, Y. Wang and A. Ramamoorthy, Chem. Commun., 2018, 54, 12883–12886 RSC.
- C. Adura, S. Guerrero, E. Salas, L. Medel, A. Riveros, J. Mena, J. Arbiol, F. Albericio, E. Giralt and M. J. Kogan, ACS Appl. Mater. Interfaces, 2013, 5, 4076–4085 CrossRef CAS.
- B. Hu, F. Dai, Z. Fan, G. Ma, Q. Tang and X. Zhang, Adv. Mater., 2015, 27, 5499–5505 CrossRef CAS.
- Y. Sun, A. Kakinen, C. Zhang, Y. Yang, A. Faridi, T. P. Davis, W. Cao, P. C. Ke and F. Ding, Nanoscale, 2019, 11, 11933–11945 RSC.
- G. Rassu, E. Soddu, A. M. Posadino, G. Pintus, B. Sarmento, P. Giunchedi and E. Gavini, Colloids Surf., B, 2017, 152, 296–301 CrossRef CAS.
- A. Faridi, Y. Sun, Y. Okazaki, G. Peng, J. Gao, A. Kakinen, P. Faridi, M. Zhao, I. Javed, A. W. Purcell, T. P. Davis, S. Lin, R. Oda, F. Ding and P. C. Ke, Small, 2018, 14, 1802825 CrossRef.
- Y. Zhao, J. Cai, Z. Liu, Y. Li, C. Zheng, Y. Zheng, Q. Chen, H. Chen, F. Ma, Y. An, L. Xiao, C. Jiang, L. Shi, C. Kang and Y. Liu, Nano Lett., 2019, 19, 674–683 CrossRef.
- C. Zhang, X. Wan, X. Zheng, X. Shao, Q. Liu, Q. Zhang and Y. Qian, Biomaterials, 2014, 35, 456–465 CrossRef CAS.
- I. Wilhelm, C. Fazakas and I. A. Krizbai, Acta Neurobiol. Exp., 2011, 71, 113–128 Search PubMed.
- S. Mourtas, A. N. Lazar, E. Markoutsa, C. Duyckaerts and S. G. Antimisiaris, Eur. J. Med. Chem., 2014, 80, 175–183 CrossRef CAS.
- E. K. Agyare, K. M. Jaruszewski, G. L. Curran, J. T. Rosenberg, S. C. Grant, V. J. Lowe, S. Ramakrishnan, A. K. Paravastu, J. F. Poduslo and K. K. Kandimalla, J. Controlled Release, 2014, 185, 121–129 CrossRef CAS.
- S. Mancini, S. Minniti, M. Gregori, G. Sancini, A. Cagnotto, P. O. Couraud, L. Ordóñez-Gutiérrez, F. Wandosell, M. Salmona and F. Re, Nanomedicine, 2016, 12, 43–52 CrossRef CAS.
- A. A. Stepanenko and V. V. Dmitrenko, Gene, 2015, 569, 182–190 CrossRef CAS.
- M. Wang, A. Kakinen, E. H. Pilkington, T. P. Davis and P. C. Ke, Biomater. Sci., 2017, 5, 485–493 RSC.
- D. Kim, J. M. Yoo, H. Hwang, J. Lee, S. H. Lee, S. P. Yun, M. J. Park, M. Lee, S. Choi, S. H. Kwon, S. Lee, S.-H. Kwon, S. Kim, Y. J. Park, M. Kinoshita, Y.-H. Lee, S. Shin, S. R. Paik, S. J. Lee, S. Lee, B. H. Hong and H. S. Ko, Nat. Nanotechnol., 2018, 13, 812–818 CrossRef CAS.
- B. H. Falkenburger and J. B. Schulz, Parkinson's Dis. Relat. Disord., 2006, 261–268 CAS.
- A. Trapani, E. De Giglio, D. Cafagna, N. Denora, G. Agrimi, T. Cassano, S. Gaetani, V. Cuomo and G. Trapani, Int. J. Pharm., 2011, 419, 296–307 CrossRef CAS.
- B. Klajnert, T. Wasiak, M. Ionov, M. Fernandez-Villamarin, A. Sousa-Herves, J. Correa, R. Riguera and E. Fernandez-Megia, Nanomedicine, 2012, 8, 1372–1378 CrossRef CAS.
- A. Daria, A. Colombo, G. Llovera, H. Hampel, M. Willem, A. Liesz, C. Haass and S. Tahirovic, EMBO J., 2017, 36, 583–603 CrossRef CAS.
- L. Sebastian Monasor, S. A. Müller, A. V. Colombo, G. Tanrioever, J. König, S. Roth, A. Liesz, A. Berghofer, A. Piechotta, M. Prestel, T. Saito, T. C. Saido, J. Herms, M. Willem, C. Haass, S. F. Lichtenthaler and S. Tahirovic, eLife, 2020, 9, e54083 CrossRef.
- F. Meng, S. Asghar, S. Gao, Z. Su, J. Song, M. Huo, W. Meng, Q. Ping and Y. Xiao, Colloids Surf., B, 2015, 134, 88–97 CrossRef CAS.
- J. A. Loureiro, B. Gomes, G. Fricker, I. Cardoso, C. A. Ribeiro, C. Gaiteiro, M. A. Coelho, C. Pereira Mdo and S. Rocha, Colloids Surf., B, 2015, 134, 213–219 CrossRef CAS.
- J. A. Loureiro, S. Andrade, A. Duarte, A. R. Neves, J. F. Queiroz, C. Nunes, E. Sevin, L. Fenart, F. Gosselet, M. A. Coelho and M. C. Pereira, Molecules, 2017, 22, 277 CrossRef.
- S. H. Choi, Y. H. Kim, M. Hebisch, C. Sliwinski, S. Lee, C. D’Avanzo, H. Chen, B. Hooli, C. Asselin, J. Muffat, J. B. Klee, C. Zhang, B. J. Wainger, M. Peitz, D. M. Kovacs, C. J. Woolf, S. L. Wagner, R. E. Tanzi and D. Y. Kim, Nature, 2014, 515, 274–278 CrossRef CAS.
- H. Wang, Front. Synaptic Neurosci., 2018, 10, 15 CrossRef.
- S. Logan, T. Arzua, S. Canfield, E. Seminary, S. Sison, A. Ebert and X. Bai, Compr. Physiol., 2019, 14, 565–611 Search PubMed.
- L. Lossi and A. Merighi, Front. Vet. Sci., 2018, 5, 164 CrossRef.
- L. Fülöp, I. M. Mándity, G. Juhász, V. Szegedi, A. Hetényi, E. Wéber, Z. Bozsó, D. Simon, M. Benkő, Z. Király and T. A. Martinek, PLoS One, 2012, 7, e39485 CrossRef.
- S. K. Tiwari, S. Agarwal, B. Seth, A. Yadav, S. Nair, P. Bhatnagar, M. Karmakar, M. Kumari, L. K. Chauhan, D. K. Patel, V. Srivastava, D. Singh, S. K. Gupta, A. Tripathi, R. K. Chaturvedi and K. C. Gupta, ACS Nano, 2014, 8, 76–103 CrossRef CAS.
- F. M. Lopes, I. J. Bristot, L. L. da Motta, R. B. Parsons and F. Klamt, NeuroMol. Med., 2017, 19, 241–255 CrossRef CAS.
- A. Bernardi, R. L. Frozza, A. Meneghetti, J. B. Hoppe, A. M. O. Battastini, A. R. Pohlmann, S. S. Guterres and C. G. Salbego, Int. J. Nanomed., 2012, 7, 4927–4942 CrossRef CAS.
- K. D. Onos, A. Uyar, K. J. Keezer, H. M. Jackson, C. Preuss, C. J. Acklin, R. O’Rourke, R. Buchanan, T. L. Cossette and S. J. S. Rizzo, PLoS Genet., 2019, 15, e1008155 CrossRef CAS.
- K. Nishitomi, G. Sakaguchi, Y. Horikoshi, A. J. Gray, M. Maeda, C. Hirata-Fukae, A. G. Becker, M. Hosono, I. Sakaguchi and S. S. Minami, J. Neurochem., 2006, 99, 1555–1563 CrossRef CAS.
- C. G. Specht and R. Schoepfer, BMC Neurosci., 2001, 2, 11 CrossRef CAS.
- V. Ilievski, P. K. Zuchowska, S. J. Green, P. T. Toth, M. E. Ragozzino, K. Le, H. W. Aljewari, N. M. O’Brien-Simpson, E. C. Reynolds and K. Watanabe, PLoS One, 2018, 13, e0204941 CrossRef.
- Y. Nisemblat, H. Belinson, I. Dolev and D. M. Michaelson, Neurodegener. Dis., 2008, 5, 166–169 CrossRef CAS.
- M. Asai, C. Hattori, N. Iwata, T. C. Saido, N. Sasagawa, B. Szabó, Y. Hashimoto, K. Maruyama, S. I. Tanuma and Y. Kiso, J. Neurochem., 2006, 96, 533–540 CrossRef CAS.
- S. H. Kim, E. M. Knight, E. L. Saunders, A. K. Cuevas, M. Popovech, L.-C. Chen and S. Gandy, F1000Research, 2012, 1, 70 Search PubMed.
- T. Ali, M. J. Kim, S. U. Rehman, A. Ahmad and M. O. Kim, Mol. Neurobiol., 2017, 54, 6490–6506 CrossRef CAS.
- S. Niu, L.-K. Zhang, L. Zhang, S. Zhuang, X. Zhan, W.-Y. Chen, S. Du, L. Yin, R. You and C.-H. Li, Theranostics, 2017, 7, 344 CrossRef CAS.
- Y. Xue, N. Wang, Z. Zeng, J. Huang, Z. Xiang and Y.-Q. Guan, J. Mater. Sci. Technol., 2020, 43, 197–207 CrossRef.
- W. Härtig, B. R. Paulke, C. Varga, J. Seeger, T. Harkany and J. Kacza, Neurosci. Lett., 2003, 338, 174–176 CrossRef.
- T. Urano and C. Tohda, Phytother. Res., 2010, 24, 1658–1663 CrossRef CAS.
- J. G. Geisler, W. Zawalich, K. Zawalich, J. R. Lakey, H. Stukenbrok, A. J. Milici and W. C. Soeller, Diabetes, 2002, 51, 2158–2169 CrossRef CAS.
- T. Gurlo, S. Ryazantsev, C.-J. Huang, M. W. Yeh, H. A. Reber, O. J. Hines, T. D. O’Brien, C. G. Glabe and P. C. Butler, Am. J. Pathol., 2010, 176, 861–869 CrossRef CAS.
- Y. Z. Wadghiri, E. M. Sigurdsson, M. Sadowski, J. I. Elliott, Y. Li, H. Scholtzova, C. Y. Tang, G. Aguinaldo, M. Pappolla, K. Duff, T. Wisniewski and D. H. Turnbull, Magn. Reson. Med., 2003, 50, 293–302 CrossRef CAS.
- M. Jucker, Nat. Med., 2010, 16, 1210–1214 CrossRef CAS.
- J. Sweatt, Mechanisms of Memory, 2003, p. 337 Search PubMed.
- K. K. Cheng, C. F. Yeung, S. W. Ho, S. F. Chow, A. H. Chow and L. Baum, AAPS J., 2013, 15, 324–336 CrossRef CAS.
- D. Carradori, C. Balducci, F. Re, D. Brambilla, B. Le Droumaguet, O. Flores, A. Gaudin, S. Mura, G. Forloni and L. Ordoñez-Gutierrez, Nanomedicine, 2018, 14, 609–618 CrossRef CAS.
- D. R. Borchelt, T. Ratovitski, J. Van Lare, M. K. Lee, V. Gonzales, N. A. Jenkins, N. G. Copeland, D. L. Price and S. S. Sisodia, Neuron, 1997, 19, 939–945 CrossRef CAS.
- J. L. Jankowsky, D. J. Fadale, J. Anderson, G. M. Xu, V. Gonzales, N. A. Jenkins, N. G. Copeland, M. K. Lee, L. H. Younkin and S. L. Wagner, Hum. Mol. Genet., 2004, 13, 159–170 CrossRef CAS.
- A. Savonenko, G. M. Xu, T. Melnikova, J. L. Morton, V. Gonzales, M. P. Wong, D. L. Price, F. Tang, A. L. Markowska and D. R. Borchelt, Neurobiol. Dis., 2005, 18, 602–617 CrossRef CAS.
- C. Schwab, M. Hosokawa and P. L. McGeer, Exp. Neurol., 2004, 188, 52–64 CrossRef CAS.
- L. O. Sillerud, N. O. Solberg, R. Chamberlain, R. A. Orlando, J. E. Heidrich, D. C. Brown, C. I. Brady, T. A. Vander Jagt, M. Garwood and D. L. Vander Jagt, J. Alzheimer's Dis., 2013, 34, 349–365 CAS.
- D. Carradori, C. Balducci, F. Re, D. Brambilla, B. Le Droumaguet, O. Flores, A. Gaudin, S. Mura, G. Forloni, L. Ordoñez-Gutierrez, F. Wandosell, M. Masserini, P. Couvreur, J. Nicolas and K. Andrieux, Nanomedicine, 2018, 14, 609–618 CrossRef CAS.
- M. Sanati, F. Khodagholi, S. Aminyavari, F. Ghasemi, M. Gholami, A. Kebriaeezadeh, O. Sabzevari, M. J. Hajipour, M. Imani and M. Mahmoudi, ACS Chem. Neurosci., 2019, 10, 2299–2309 CrossRef CAS.
- Y. Wang, J. Liu, Z. Zhang, P. Bi, Z. Qi and C. Zhang, Neurosci. Lett., 2011, 487, 70–72 CrossRef CAS.
- N. A. Singh, V. Bhardwaj, C. Ravi, N. Ramesh, A. K. A. Mandal and Z. A. Khan, Front. Aging Neurosci., 2018, 10, 244 CrossRef.
- A. Levit, A. M. Regis, A. Gibson, O. H. Hough, S. Maheshwari, Y. Agca, C. Agca, V. Hachinski, B. L. Allman and S. N. Whitehead, Brain, Behav., Immun., 2019, 80, 25–34 CrossRef CAS.
- C. Agca, J. J. Fritz, L. C. Walker, A. I. Levey, A. W. Chan, J. J. Lah and Y. Agca, BMC Neurosci., 2008, 9, 1–13 CrossRef.
- E. N. Wilson, A. R. Abela, S. Do Carmo, S. Allard, A. R. Marks, L. A. Welikovitch, A. Ducatenzeiler, Y. Chudasama and A. C. Cuello, Cereb. Cortex, 2016, 27, bhv332 CrossRef.
- K. L. Paumier, K. C. Luk, F. P. Manfredsson, N. M. Kanaan, J. W. Lipton, T. J. Collier, K. Steece-Collier, C. J. Kemp, S. Celano and E. Schulz, Neurobiol. Dis., 2015, 82, 185–199 CrossRef CAS.
- C. L. Bianco, J. Ridet, B. Schneider, N. Deglon and P. Aebischer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10813–10818 CrossRef.
- A. E. Butler, J. Jang, T. Gurlo, M. D. Carty, W. C. Soeller and P. C. Butler, Diabetes, 2004, 53, 1509–1516 CrossRef CAS.
- M. Yusuf, M. Khan, R. A. Khan and B. Ahmed, J. Drug Targeting, 2013, 21, 300–311 CrossRef CAS.
- E. Muntimadugu, R. Dhommati, A. Jain, V. G. S. Challa, M. Shaheen and W. Khan, Eur. J. Pharm. Sci., 2016, 92, 224–234 CrossRef CAS.
- A. Vedagiri and S. Thangarajan, Neuropeptides, 2016, 58, 111–125 CrossRef CAS.
- J. Götz, L. G. Bodea and M. Goedert, Nat. Rev. Neurosci., 2018, 19, 583–598 CrossRef.
- S. Reardon, Nature, 2018, 563, 611–613 CrossRef CAS.
- M. Newman, E. Ebrahimie and M. Lardelli, Front. Genet., 2014, 5, 189 Search PubMed.
- S. Luna, D. J. Cameron and D. W. Ethell, PLoS One, 2013, 8, e75052 CrossRef CAS.
- S. Santana, E. P. Rico and J. S. Burgos, Am. J. Neurodegener. Dis., 2012, 1, 32 Search PubMed.
- M. Newman, S. Nornes, R. N. Martins and M. T. Lardelli, Neurosci. Lett., 2012, 519, 14–19 CrossRef CAS.
- I. Javed, Z. Zhang, J. Adamcik, N. Andrikopoulos, Y. Li, D. E. Otzen, S. Lin, R. Mezzenga, T. P. Davis and F. Ding, Adv. Sci., 2020, 2001299 CrossRef.
- I. Javed, T. Yu, G. Peng, A. Sánchez-Ferrer, A. Faridi, A. Kakinen, M. Zhao, R. Mezzenga, T. P. Davis, S. Lin and P. C. Ke, Nano Lett., 2018, 18, 5797–5804 CrossRef CAS.
- A. Kakinen, Y. Xing, N. Hegoda Arachchi, I. Javed, L. Feng, A. Faridi, A. M. Douek, Y. Sun, J. Kaslin, T. P. Davis, M. J. Higgins, F. Ding and P. C. Ke, Nano Lett., 2019, 19, 6535–6546 CrossRef CAS.
- C. J. Kenyon, Nature, 2010, 464, 504–512 CrossRef CAS.
- C. D. Link, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9368–9372 CrossRef CAS.
- J. Drake, C. D. Link and D. A. Butterfield, Neurobiol. Aging, 2003, 24, 415–420 CrossRef CAS.
- Y. Wu, Z. Wu, P. Butko, Y. Christen, M. P. Lambert, W. L. Klein, C. D. Link and Y. Luo, J. Neurosci., 2006, 26, 13102–13113 CrossRef CAS.
- C. D. Link, C. J. Johnson, V. Fonte, M.-C. Paupard, D. H. Hall, S. Styren, C. A. Mathis and W. E. Klunk, Neurobiol. Aging, 2001, 22, 217–226 CrossRef CAS.
- P. Cao, Y. Yuan, E. A. Pehek, A. R. Moise, Y. Huang, K. Palczewski and Z. Feng, PLoS One, 2010, 5, e9312 CrossRef.
- S. G. Chen, V. Stribinskis, M. J. Rane, D. R. Demuth, E. Gozal, A. M. Roberts, R. Jagadapillai, R. Liu, K. Choe and B. Shivakumar, Sci. Rep., 2016, 6, 1–10 CrossRef CAS.
- H. Yin, J. Si, H. Xu, J. Dong, D. Zheng, X. Lu and X. Li, J. Biomed. Nanotechnol., 2014, 10, 1536–1544 CrossRef CAS.
- F. Morales-Zavala, H. Arriagada, N. Hassan, C. Velasco, A. Riveros, A. R. Álvarez, A. N. Minniti, X. Rojas-Silva, L. L. Muñoz and R. Vasquez, Nanomedicine, 2017, 13, 2341–2350 CrossRef CAS.
- A. Nandakumar, Y. Xing, R. R. Aranha, A. Faridi, A. Kakinen, I. Javed, K. Koppel, E. H. Pilkington, A. W. Purcell, T. P. Davis, P. Faridi, F. Ding and P. C. Ke, Biomacromolecules, 2020, 21, 988–998 CrossRef CAS.
- E. H. Pilkington, O. J. R. Gustafsson, Y. Xing, J. Hernandez-Fernaud, C. Zampronio, A. Kakinen, A. Faridi, F. Ding, P. Wilson, P. C. Ke and T. P. Davis, ACS Nano, 2018, 12, 6066–6078 CrossRef CAS.
- K. Koppel, H. Tang, I. Javed, M. Parsa, M. Mortimer, T. P. Davis, S. Lin, A. L. Chaffee, F. Ding and P. C. Ke, Nanoscale, 2020, 12, 12317–12328 RSC.
- Y. Sun, A. Kakinen, Y. Xing, P. Faridi, A. Nandakumar, A. W. Purcell, T. P. Davis, P. C. Ke and F. Ding, Small, 2019, 15, 1805166 CrossRef.
- A. Kakinen, Y. Sun, I. Javed, A. Faridi, E. H. Pilkington, P. Faridi, A. W. Purcell, R. Zhou, F. Ding, S. Lin, P. C. Ke and T. P. Davis, Sci. Bull., 2019, 64, 26–35 CrossRef CAS.
- P. Nedumpully-Govindan, E. N. Gurzov, P. Chen, E. H. Pilkington, W. J. Stanley, S. A. Litwak, T. P. Davis, P. C. Ke and F. Ding, Phys. Chem. Chem. Phys., 2016, 18, 94–100 RSC.
- C. Bai, Z. Lao, Y. Chen, Y. Tang and G. Wei, Front. Chem., 2020, 8, 51 CrossRef CAS.
- L. Xie, Y. Luo, D. Lin, W. Xi, X. Yang and G. Wei, Nanoscale, 2014, 6, 9752–9762 RSC.
- Z. Yang, C. Ge, J. Liu, Y. Chong, Z. Gu, C. A. Jimenez-Cruz, Z. Chai and R. Zhou, Nanoscale, 2015, 7, 18725–18737 RSC.
- J. M. Long and D. M. Holtzman, Cell, 2019, 179, 312–339 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |