
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInduced pluripotent stem cell-derived vascular networks to screen nano–bio interactions†
Luís
Estronca
a,
Vitor
Francisco
 b,
Patrícia
Pitrez
b,
Inês
Honório
b,
Lara
Carvalho
c,
Helena
Vazão
b,
Josephine
Blersch
b,
Akhilesh
Rai
a,
Xavier
Nissan
d,
Ulrich
Simon
e,
Mário
Grãos
b,
Leonor
Saúde
c and
Lino
Ferreira
*a
b,
Patrícia
Pitrez
b,
Inês
Honório
b,
Lara
Carvalho
c,
Helena
Vazão
b,
Josephine
Blersch
b,
Akhilesh
Rai
a,
Xavier
Nissan
d,
Ulrich
Simon
e,
Mário
Grãos
b,
Leonor
Saúde
c and
Lino
Ferreira
*a
aFaculty of Medicine, University of Coimbra, 3000-548, Coimbra, Portugal. E-mail: lino@uc-biotech.pt
bCenter for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal
cInstituto de Medicina Molecular e Instituto de Histologia e Biologia do Desenvolvimento, Faculdade de Medicina da Universidade de Lisboa, 1649-028, Lisboa, Portugal
dCECS, I-STEM, AFM, Institute for Stem Cell Therapy and Exploration of Monogenic diseases, Evry cedex, France
eInstitute of Inorganic Chemistry, RWTH Aachen University, Germany
First published on 12th February 2021
Abstract
The vascular bioactivity/safety of nanomaterials is typically evaluated by animal testing, which is of low throughput and does not account for biological differences between animals and humans such as ageing, metabolism and disease profiles. The development of personalized human in vitro platforms to evaluate the interaction of nanomaterials with the vascular system would be important for both therapeutic and regenerative medicine. A library of 30 nanoparticle (NP) formulations, in use in imaging, antimicrobial and pharmaceutical applications, was evaluated in a reporter zebrafish model of vasculogenesis and then tested in personalized humanized models composed of human-induced pluripotent stem cell (hiPSC)-derived endothelial cells (ECs) with “young” and “aged” phenotypes in 3 vascular network formats: 2D (in polystyrene dish), 3D (in Matrigel) and in a blood vessel on a chip. As a proof of concept, vascular toxicity was used as the main readout. The results show that the toxicity profile of NPs to hiPSC-ECs was dependent on the “age” of the endothelial cells and vascular network format. hiPSC-ECs were less susceptible to the cytotoxicity effect of NPs when cultured in flow than in static conditions, the protective effect being mediated, at least in part, by glycocalyx. Overall, the results presented here highlight the relevance of in vitro hiPSC-derived vascular systems to screen vascular nanomaterial interactions.
New conceptsThe exposure of humans to environments with increased levels of nanoparticles in the air as well as to pharmaceutical nanoformulations for regenerative and therapeutic medicine requires a better knowledge of their bioactivity/safety. In the past, these tests were performed in low throughput in vivo tests (e.g. mice) that did not account for differences with the human system and in non-personalized human cells, i.e., in cells that did not have patient-specific information and ageing signatures. In this study, we have developed personalized vascular networks with variable complexity (2D, 3D, and a blood vessel on a chip) formed by human induced pluripotent stem cell-derived endothelial cells with a “young” or “aged” phenotype, to screen vascular–nanomaterial interactions. We have used a library of 30 nanomaterials with different physico-chemical properties and relevance for clinical molecular imaging, protective formulations, antimicrobial coatings, catalysis and pharmaceutical applications. The complexity of the vascular network, and particularly its ability to express glycocalyx, as well as the age of the cells influenced largely their sensitivity to the nanomaterials. The platform presented in this study is very promising for high-throughput screening of nano–bio interactions and for the identification of nanomaterials able to eliminate more specifically aged vascular cells. |
1. Introduction
Nanomaterials and nanomaterial-based technologies enable the development of new materials and applications across disciplines, including mechanical and electrical engineering, agriculture, energy generation and medicine.1–3 The interactions of these nanomaterials with the human body are only partially known.4–6 The development of new cell-based platforms for the rapid profiling of nanomaterials in terms of bioactivity, toxicity, and biodegradation, among other aspects, is in great need.7,8 The majority of the nanomaterials that enter the human body, independent of the route of entry, will certainly circulate and be transported in the vascular system through the blood vessels and therefore it is critical to study their impact in individuals with differences in their vascular biology because of their genetic background or pathologies. The effects of nanomaterials in the disruption of the integrity of endothelial cell–cell communication9 and in the induction of endothelial10 and smooth muscle11 cell toxicity have been identified.Standard protocols for assessing the effect of nanomaterials on the vascular system involve testing in animals.5,10 Unfortunately, these tests are of low-throughput, and expensive, yield limited mechanistic information, do not address the current policies of regulatory agencies to use alternatives to animal testing, and do not account for differences between species. Moreover, with the advent of personalized medicine, new cell technologies are required to provide patient-specific information about nanomaterial bioactivity/safety. In this respect, human-induced pluripotent stem cells (hiPSCs) represent a potential source of endothelial cells (ECs) and smooth muscle cells (SMCs).12–14 These humanized systems are, in some aspects, superior to animal models because they recapitulate human ageing, metabolism and disease profiles. Unfortunately, hiPSC-derived ECs and SMCs have not been used to study the bioactivity/safety of nanomaterials among different individuals. These cells may be cultured under flow shear conditions in microfluidic systems to better mimic the in vivo conditions.
In this work, the impact of nanomaterials in iPSC-derived vascular networks was evaluated in 2D, 3D and blood vessel on a chip in vitro models. While the first two models run in static conditions, the blood vessel on a chip model runs in flow conditions, similar to in vivo conditions, which allows the formation of a functional glycocalyx layer on top of endothelial cells. Thirty nanomaterials that are normally used for different applications such as clinical molecular imaging, protective formulations, antimicrobial coatings, pharmaceutical formulations and catalysts have been selected (Fig. 1). The nanomaterials with a metallic or polymeric composition and sizes ranging between 1.4 and 400 nm were initially tested in the zebrafish embryo vasculogenesis/angiogenesis to investigate their in vivo effect. Subsequently, the nanomaterials were tested in the in vitro human iPSC-derived vascular networks from two individuals in the same timeframe as the zebrafish tests (i.e. 24 h) to evaluate their acute toxicity. One of the iPSC lines was derived from fetal cells (cord blood of a healthy newborn), while the other was generated from fibroblasts of a 14 year-old female patient with Hutchinson–Gilford progeria syndrome (HGPS).15 HGPS is a rare and fatal disease caused by a single point mutation of the LMNA gene leading to the accumulation of an abnormally truncated lamin A protein called progerin, which in turn leads to accelerated ageing.16 The accumulation of progerin is also observed during physiological ageing, although at much lower levels.17 The motivation here was to investigate differences in the interaction of nanomaterials with vascular cells with a “young” and “aged” phenotype. As a proof of concept, we have selected vascular toxicity as the main readout because it is relatively easy to quantify; however, cellular NP internalization was also quantified in some experiments. We have used monoculture of both iPSC-derived ECs either in polystyrene culture dishes (2D) or in Matrigel (3D) to screen, in static conditions. Some of the toxic formulations were further tested in a blood vessel on a chip formed by a co-culture of iPSC-ECs and iPSC-SMCs.
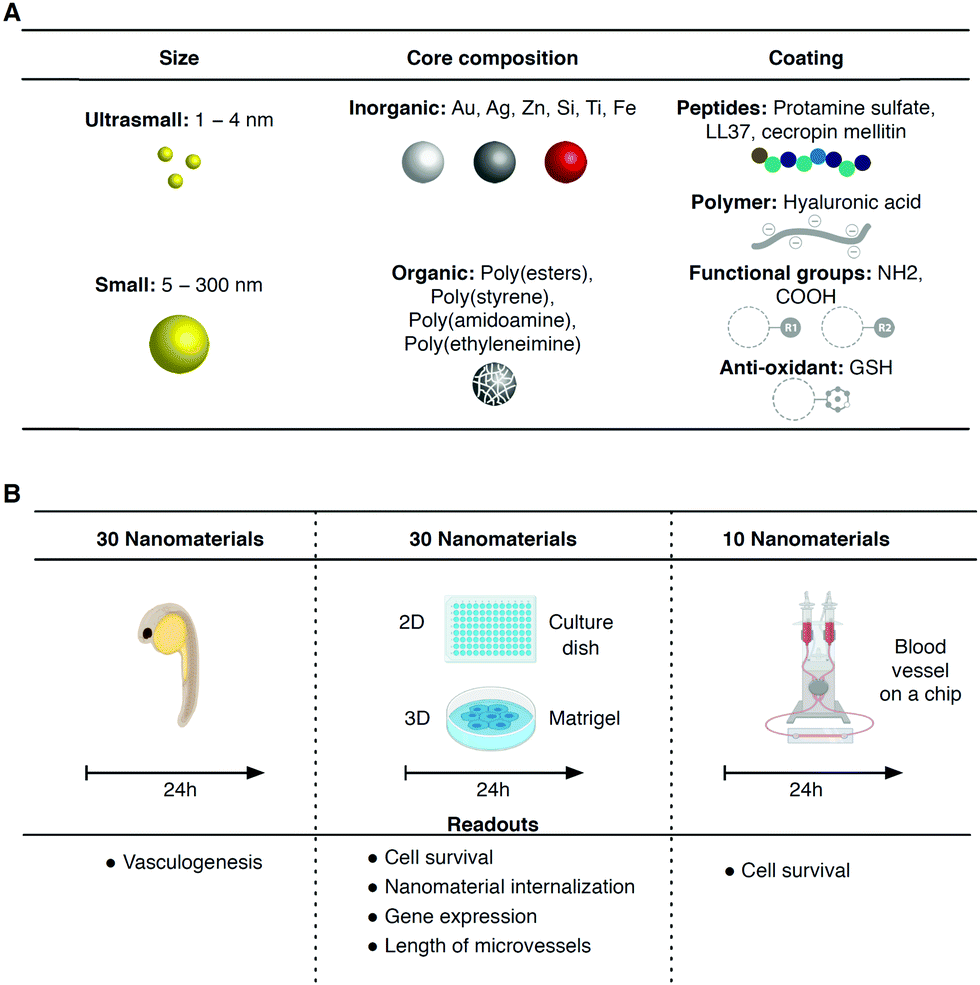 | ||
| Fig. 1 NP library and in vivo/in vitro models to screen their toxicity. (A) Properties of the NPs. The NPs can be categorized based on their size as: ultrasmall (3 NPs) or small (27 NPs); based on their composition as: inorganic (15 NPs) or organic (15 NPs); and by the type of the coating: peptides (3), polymers (2), functional groups (4) or anti-oxidants (2). (B) In vivo and in vitro models to screen NP toxicity. In vivo model: 30 NPs were tested at 1 μg mL−1 in 4 hpf zebrafish embryos (manually dechorionated) for 24 h. The readout was vasculogenesis. 2D model (in culture plates, static conditions): 30 NPs were incubated at 4 different concentrations (6, 12.5, 25 and 50 μg mL−1) for 24 h with N-iPSC ECs or HGPS-iPSC ECs. The readouts were: (i) cell survival and (ii) nanomaterial internalization. 3D model (in Matrigel, static conditions): 30 NPs were incubated at 2 concentrations (12.5 and 25 μg mL−1) together with iPSC ECs in culture plates with Matrigel for 24 h. The readouts were: (i) length of microvessels formed, (ii) cell survival and (iii) gene expression. Blood vessel on a chip (microfluidic system; flow conditions): 10 NPs were tested at 50 μg mL−1 in arterial flow conditions in a co-culture of N-iPSC SMCs and N-iPSC ECs for 24 h. The readout was cell survival. | ||
2. Results
Characterization of nanomaterials
We have selected 30 nanomaterials, both organic and inorganic, with different sizes and zeta potentials to evaluate in our vascular screening platform (Table 1). We focused on nanomaterials used for: (i) molecular imaging (NP4), (ii) sunscreen (NP15 and NP16), (iii) antimicrobial applications (NP29, NP918 or NP1219), (iv) pharmaceutical applications (NP20 and NP21,20 NP24,21 and light-triggerable formulations recently synthesized by us such as NP30, NP23, NP26, NP27, NP18 (all described in reference ref. 22), NP22,23 NP14,23 and NP2524) and (v) reaction catalysis or biomedical applications (NP1, NP2, NP3, NP5, NP6, NP7, NP8, or silica-based NPs such as NP13 and NP28). Many of these NPs may get inside of the human body and interact with blood vessels in the circulation. Dynamic light scattering (DLS) analyses showed that the average sizes of the inorganic nanomaterials suspended in PBS varied between 1 (NP1) and 360 nm (NP29), while those for organic nanomaterials varied between 32 (NP10) and 427 nm (NP30) (Fig. 2A.1 and A.2). Most of the nanomaterials maintained their size after 24 h in suspension with a few exceptions (NP8 and NP16 slightly increased their diameters while NP28 decreased significantly their diameters, likely due to a poor initial dispersion of the NPs as shown by TEM analyses, see below). The hydrodynamic diameter of the nanomaterials ranged between 1.1 and 427 nm. Transmission electron microscopy (TEM) analyses were performed to confirm the sizes and shapes of some of the nanomaterials (Fig. S1, ESI†).| NP number | Core | Ligand | Size (nm) | Zeta (mV) | Source/ref. | NP number | Core | Ligand | Size (nm) | Zeta (mV) | Source/ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Determined in water by DLS (n = 3). Ligands: MS – sodium 3-(diphenylphosphino)benzene sulfonate; GSH – glutathione; HA – hyaluronic acid; DEX – dextran; LL37 – antimicrobial peptide; CM – cecropin melittin. | |||||||||||
| NP1 | Au | MS | 1.4 | — | 25 | NP16 | TiO2 | — | 10–30 | — | http://ssnano.com |
| NP2 | Au | GSH | 1.5 | — | 25 | NP17 | PS | COOH | 100 | — | http://micromod.de |
| NP3 | Au | HA | 5 ± 1a | −25 ± 2a | N.A. | NP18 | C11 | — | 65 ± 5 | 14 ± 1 | 22 |
| NP4 | SPION | DEX | 40 | 31 | http://biopal.com | NP19 | PS | NH2 | 100 | — | http://micromod.de |
| NP5 | Au | MS | 13.9 | — | NP20 | PLGA | — | 170 ± 7 | −9 ± 3 | 20 | |
| NP6 | Au | GSH | 12 | — | NP21 | PLGA–PS | — | 218 ± 9 | 7 ± 2 | 20 | |
| NP7 | Au | MS | 15 ± 1.5 | — | 25 | NP22 | P1C5 | — | 350 ± 35 | 21 ± 2 | 23 |
| NP8 | Au | HA | 20 ± 3a | −26 ± 3a | N.A. | NP23 | A9 | — | 265 ± 23 | 21 ± 2 | 22 |
| NP9 | Au | LL37 | 21 ± 8 | 15 ± 2 | 18 | NP24 | PEI:DS | — | 91 ± 6 | 15 ± 3 | 21 |
| NP10 | PM | COOH | 25 | — | http://micromod.de | NP25 | PEI–DMNC:DS | — | 108 ± 10 | 27 ± 2 | 24 |
| NP11 | PM | NH2 | 25 | — | http://micromod.de | NP26 | E1 | — | 330 ± 24 | 7 ± 1 | 22 |
| NP12 | Au | CM | 14 ± 1 | 28 ± 2 | 19 | NP27 | E2 | — | 260 ± 18 | −2 ± 2 | 22 |
| NP13 | SiO2 | — | 7–14 | — | http://plasmachem.com | NP28 | SiO2 | — | 10–20 | — | http://ssnano.com |
| NP14 | P1C7 | — | 74 ± 10 | 19 ± 1 | 23 | NP29 | Ag | — | <6000 | — | http://sciessent.com |
| NP15 | ZnO | — | 10–30 | — | http://ssnano.com | NP30 | A1 | — | 420 ± 14 | −1 ± 1 | 22 |
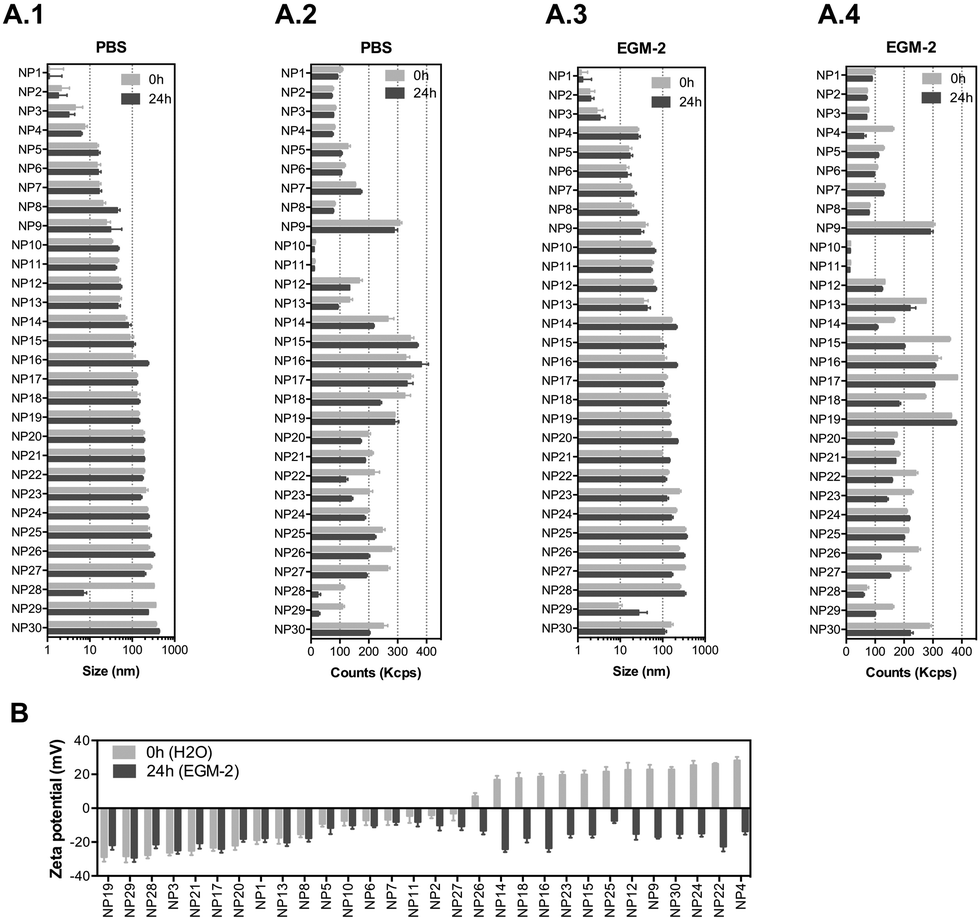 | ||
| Fig. 2 Physicochemical characterization of the NP library. (A) NP sizes and counts measured by dynamic light scattering at 0 and 24 h in PBS (A.1 and A.2) and EGM-2 medium (A.3 and A.4). (B) Zeta potentials of NP library at 0 h (in molecular biology grade water) and 24 h (in EGM-2 medium). After 24 h in EGM-2, the NPs were centrifuged and resuspended in H2O for zeta potential measurements. In A and B, results are expressed as mean ± SEM (n = 3). | ||
To study the effect of salts and proteins on NP size and stability, the hydrodynamic diameters of NPs (suspended at a concentration of 50 μg mL−1) was evaluated by DLS in a cell culture medium with serum (2% fetal bovine serum (FBS); concentration of serum used to culture ECs) for 24 h (duration of most in vitro experiments) (Fig. 2A.3 and A.4). In the cell culture medium, with some exceptions (NP12, NP14, NP18, NP29 and NP30 increased their diameters, while NP16 and NP17 slightly decreased their diameters), the NPs maintained their diameters for at least 24 h. The hydrodynamic diameters of the NPs ranged between 1.1 and 357 nm. All the NPs showed a negative zeta potential in cell culture medium, which indicates that proteins and salts have adsorbed on their surfaces (Fig. 2B). The most negatively charged NP was NP29. Overall, we have selected a library of NPs that is relevant for biomedical and environmental applications. These NPs maintained a reasonable level of stability in the cell culture medium for at least 24 h.
Nanomaterial screening in zebrafish
We evaluated the effect of the nanomaterials in vasculogenesis and angiogenesis using the zebrafish transgenic Tg(fli1a:EGFP)y1 line.26 In this transgenic line, the promoter of the endothelial marker fli1 drives the expression of EGFP in blood vessels, thus allowing the in vivo analysis of the vasculature. In zebrafish embryos, the circulation begins after 24 h-post-fertilization (hpf) in a single circulatory loop.27 Vasculogenesis of the dorsal aorta, cardinal vein and a primitive cranial vasculature occurs at this stage.28 After the formation of this primitive vasculature, the formation of new blood vessels occurs via angiogenesis, the intersegmental vessels (ISVs) of the trunk being among the first angiogenic vessels to be formed.29 The ISVs sprout from the dorsal aorta along the somite boundaries, which ultimately interconnect between each other to form the dorsal longitudinal anastomotic vessel (DLAV) (Fig. S2A, ESI†). Having this in mind, we evaluated the effect of nanomaterials in the vascular development of ISVs during the first 24–28 hpf. Fertilized embryos were selected and manually dechorionated to avoid any interference of the chorion in the internalization of the NPs. Indeed, previous studies have demonstrated that the presence of the chorion may affect the toxicity of some NPs.30 For example, Ag NPs caused higher mortality and malformations in embryos without chorion as compared with embryos with chorion at the same concentrations.30 In our study, the dechorionation caused higher sensitivity of the embryos to their environment, so we used NPs at a concentration of 1 μg mL−1. The number of ISVs, the average length of the first 10 ISVs and the total lengths of the ISVs were evaluated (Fig. 3 and Fig. S2B, ESI†). Our results showed that some NPs had a negative impact on angiogenesis by decreasing the length of ISVs (NP1, NP3, NP4, NP7, NP8, NP14, NP17, NP18, NP21, NP24, NP25 and NP26) whereas one nanomaterial (NP9) had a positive impact on angiogenesis by increasing the length and number of ISVs (although not statistically significant relative to control). The pro-angiogenic activity of LL37–Au NPs is aligned with our previous results showing that these NPs promoted neovascularization in a wound healing animal model.18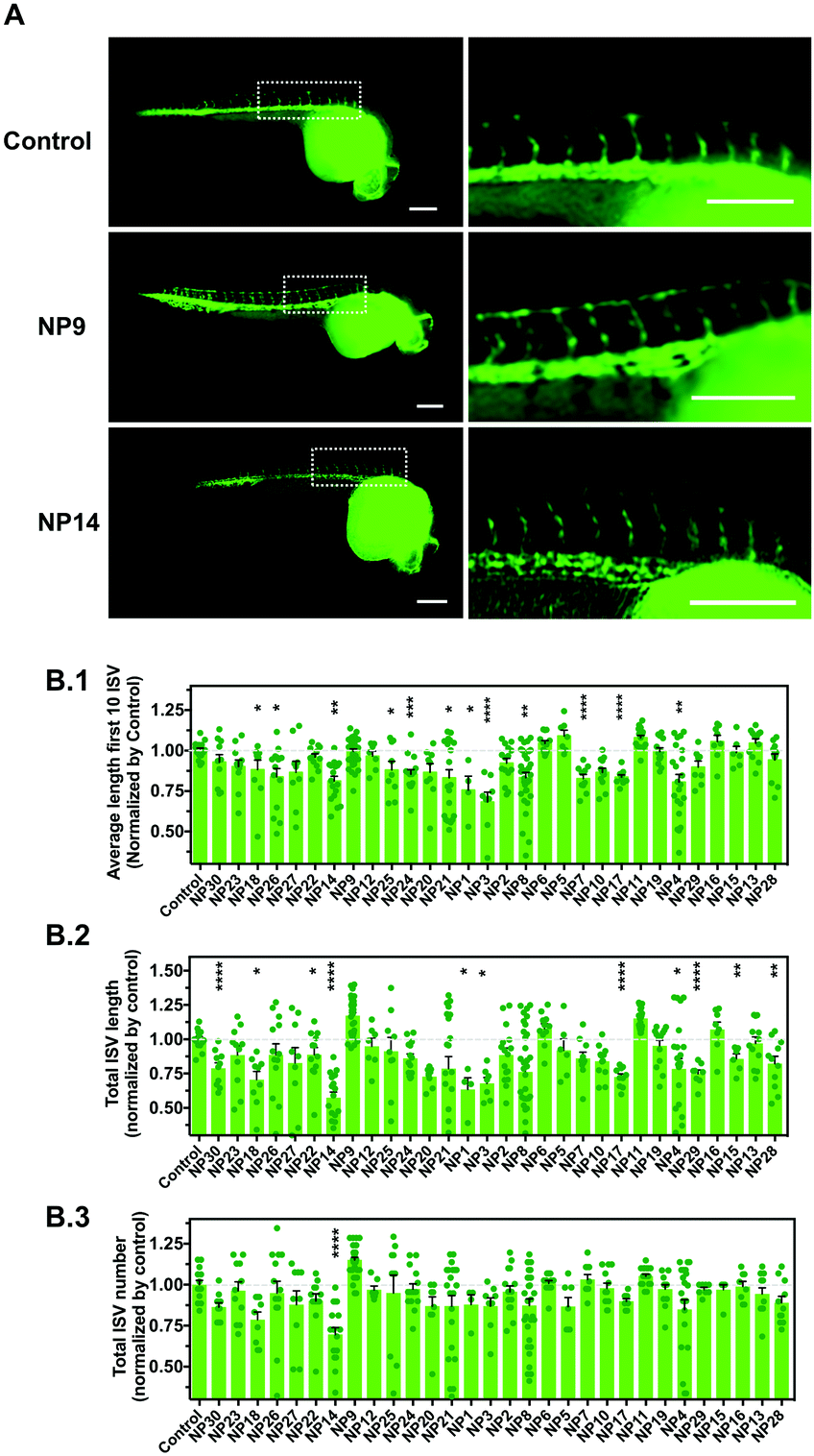 | ||
| Fig. 3 Impact of nanomaterials on the embryonic vascular development of zebrafish embryos. (A) Representative fluorescence microscopy images (all embryos and magnification of the first 10 ISVs) of zebrafish embryos with ∼28 hpf in control conditions (vehicle) and after 24 h treatment with NP9 and NP14. Scale bar is 200 μm. (B.1) Impact of 24 h incubation of nanomaterials on the average length of the first 10 ISVs of zebrafish embryos with ∼28 hpf. (B.2) Impact of 24 h incubation of nanomaterials on the total ISV length of zebrafish embryos with ∼28 hpf. (B.3). Impact of 24 h incubation of nanomaterials on the total number of ISVs of zebrafish embryos with ∼28 hpf. In B.1, B.2 and B.3, results are mean ± SEM (n = 6–24) and statistical analyses were performed by an unpaired t-test. *, **, and **** denote statistical significance (P < 0.05; P < 0.01; P < 0.0001) relative to control. | ||
Development of a vascular screening platform based on iPSC-derived vascular cells: derivation of endothelial cells from hiPSCs
iPSC lines were differentiated into ECs according to a protocol previously reported by us in ref. 13 (Fig. 4A). Undifferentiated cells were cultured in chemically defined medium supplemented with BMP4 and FGF-basic for 5 days to generate mesoderm progenitor cells followed by their differentiation into endothelial progenitor cells (PECAM1+ cells) in a medium containing VEGF and Tβ4 for an additional 5 days. PECAM1+ cells were then selected using magnetism-activated cell sorting and cultured for up to 7 passages in an endothelial medium supplemented with VEGF165 and TGF-β inhibitor (SB431542). Gene expression analysis in cells differentiated for 7 passages (between 24 and 28 days after cell seeding) indicated that both N-iPSC ECs (from non-disease cord blood cells) and HGPS-iPSC ECs expressed EC markers such as PECAM1, CDH5, KDR and vWF as human umbilical artery ECs (HUAECs), although with some fluctuations in the expression of these markers (Fig. 4B). In general, the mRNA levels of the EC markers were higher in HGPS-iPSC ECs than in N-iPSC ECs. N-iPSC-derived or HGPS-iPSC-derived PECAM1+ cells cultured for 7 passages expressed high levels of EC markers such as PECAM1 [by immunocytochemistry (Fig. 4C) and flow cytometry (Fig. S3A, ESI†)], VE-cadherin and ZO-1 at cell–cell contacts (Fig. 4C) as well as endoglin (CD105). In contrast to N-iPSC ECs, HGPS-iPSC ECs expressed progerin, the truncated form of lamin A. Indeed, 61 ± 18% (n = 3) of HGPS-iPSC ECs had accumulation of progerin, as quantified by immunofluorescence. The presence of nuclear blebs (characteristics of progeria cells) was also observed by immunostaining of lamin A/C (inset in Fig. 4C). At the functional level, both N-iPSC ECs13 and HGPS-iPSC ECs responded to pro-inflammatory stimuli such as TNF-α for 24 h leading to an increase in the expression of adhesion molecules, which then mediated the adhesion of monocytes (Fig. S3C, ESI†), and hiPSC lines. The derived cells expressed EC markers at the gene and protein levels and were functional.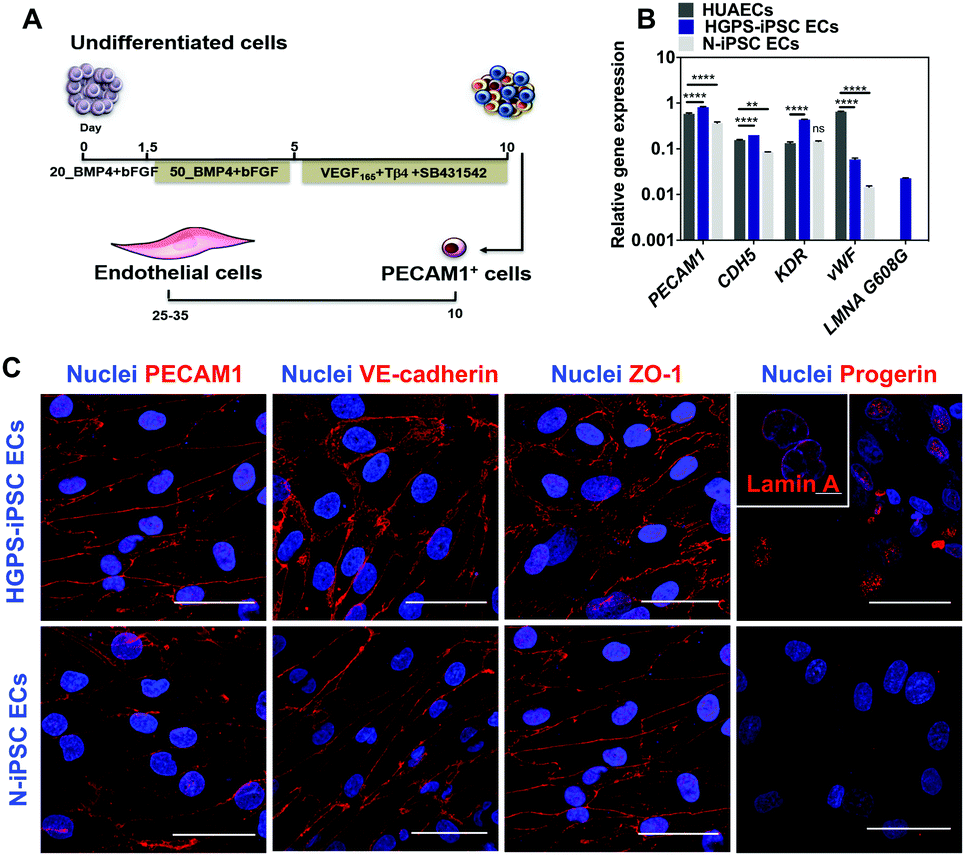 | ||
| Fig. 4 Characterization of iPSC-ECs. (A) Protocol for the differentiation of N-iPSCs and HGPS-iPSCs into EC cells. Undifferentiated iPSCs were transferred to Petri dishes coated with fibronectin and were cultured in a differentiation medium for a period of 10 days (see Methods section for more details), after which the PECAM1+ cells were selected by magnetism-activated cell sorting and cultured with full culture medium supplemented with VEGF165 and SB431542 for 7 passages (∼25 days) with the medium changed every 2–3 days. (B) qRT-PCR analyses for the expression of endothelial and progeria genes. Expression of EC (PECAM1, CDH5, KDR and vWF) and progeria (progerin, which is encoded by LMNA G608G gene) markers in HGPS-iPSC and N-iPSCs at passage 7. Human umbilical aortic endothelial cells (HUAECs) were used as controls. Results are mean ± SEM (n = 3). **, and **** denote statistical significance (P < 0.01, and P < 0.0001) normalized by the housekeeping gene (GAPDH). Statistical analyses were performed by an unpaired t-test. (C) Immunofluorescence analysis performed on HGPS-iPSC ECs and N-iPSC ECs at passage 7 for ECs (PECAM1, VE-cadherin, and ZO-1), progeria (progerin) and lamin A/C (inset) markers. Cell nuclei were labelled with DAPI (blue). Scale bar is 50 μm, except in the inset where scale bar is 10 μm. | ||
Vascular screening of NPs in a 2D model
The impact of the nanomaterial library was evaluated in ECs derived from N-iPSCs or HGPS-iPSCs cultured in static conditions. Cells were cultured for 24 h, washed and then exposed for 24 h to different concentrations of the nanomaterials (6, 12.5, 25 and 50 μg mL−1) (Fig. 5A). The effect of the NPs was quantified by two tests: (i) cell metabolism through the measurement of mitochondrial activity by a PrestoBlue assay and (ii) cell necrosis by counting cell nuclei using a Hoechst 33342/propidium iodide (PI) staining (Hoechst to quantify the total number of cells; PI to quantify the dead cells). Because some NPs may interfere with the PrestoBlue assay,31 cells after being incubated with NPs for 24 h were washed 3 times with PBS before incubating with PrestoBlue. These washing steps (which were not necessary and thus not performed in the Hoechst 33342/PI assay) likely induced higher cell detachment/loss, especially when NPs cause some toxicity, which may lead to an apparent low mitochondrial activity. This might be the reason why the PrestoBlue results appeared to show more toxic effect for some NPs than the corresponding Hoechst 33342/PI assay. Therefore, primarily, we have used Hoechst/PI staining to compare all the experimental groups. From the thirty nanomaterials tested, 7 induced significant toxicity (>60% at 50 μg mL−1; based on PI staining) towards both hPSC-derived ECs tested (i.e., NP1, NP3, NP14, NP15, NP18, NP22 and NP30) whereas 2 nanomaterials (NP25 and NP28) induced significant toxicity only towards N-iPS ECs (Fig. 5B–E). The high toxicity observed for NP1 and NP3 NPs (i.e., ultrasmall Au NPs) is in agreement with previous results obtained in HeLa cells, with a reported IC50 of about 50 μM.25,32 The toxicity of NP15 (ZnO) is in line with the toxic profile observed previously in human aortic ECs (>50% for 4 to 24 h incubation at 50 μg mL−1),33,34 human cardiac microvascular ECs (>60% for 12 to 24 h incubation at 50 μg mL−1)35 and in human umbilical vein ECs (HUVECs) (>75% for 24 h incubation at 32 μg mL−1).36,37 Regarding NP28, the results are also in accordance with those reported in the literature for HUVECs exposed for 24 h, with toxicities ranging from 20 to 60% for 50 and 75 μg mL−1 SiO2 concentrations, respectively.38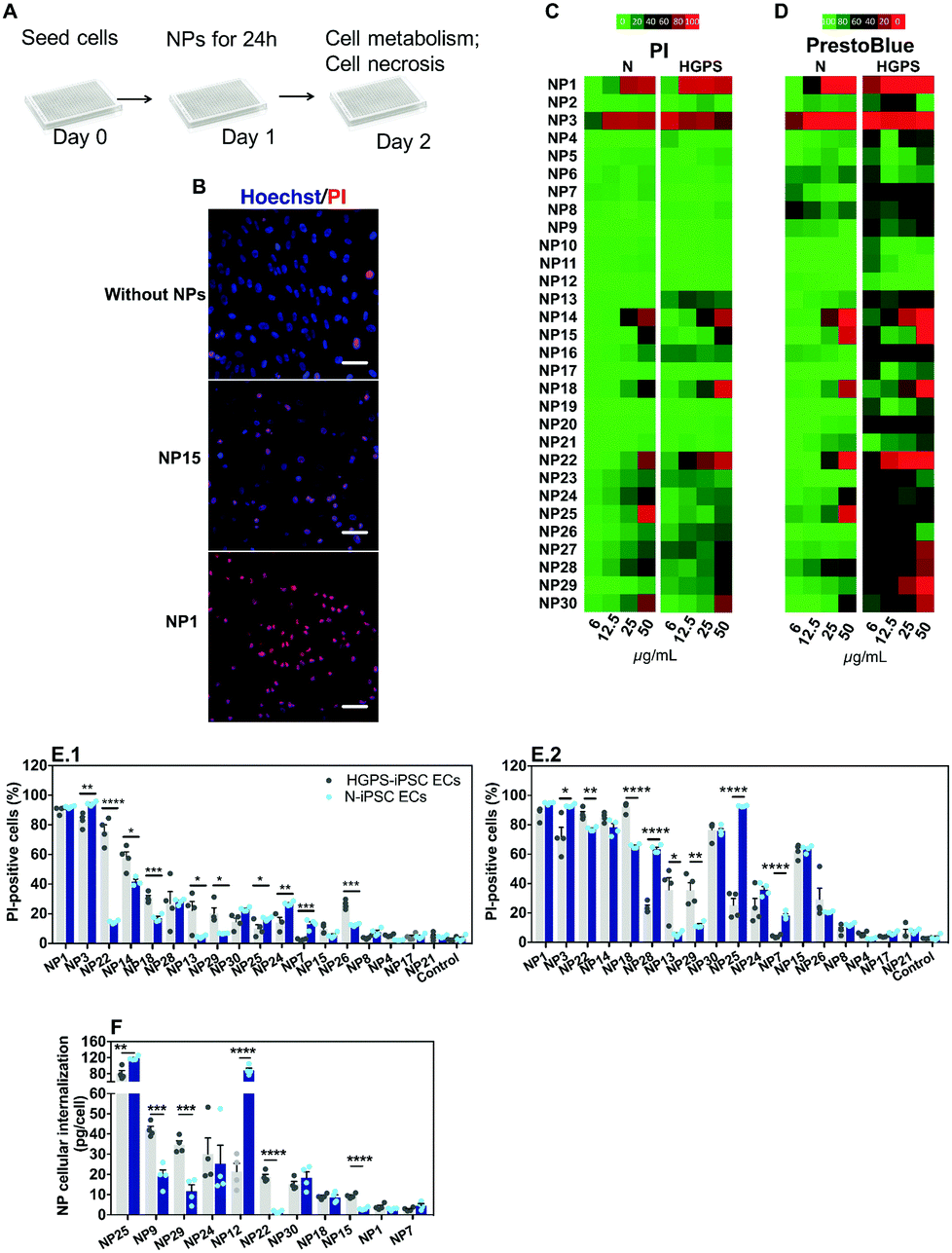 | ||
| Fig. 5 Impact of nanomaterials in iPSC-ECs cultured in 2D. (A) Schematic representation of the protocol used. Cells were seeded at density of 1 × 105 cells per cm2 and were allowed to rest for 24 h, after which NPs were added in fresh medium at concentrations between 6 and 50 μg mL−1 for a 24 h period. Cell viability was evaluated by Hoechst 33342/PI staining, while cell metabolism was quantified by a PrestoBlue assay. (B) Representative images of Hoechst 33342/PI staining for N-iPSC ECs with (NP1 or NP15, both at 50 μg mL−1) or without exposure to NPs for 24 h. Scale bar is 50 μm. (C and D) Heat maps of NP toxicity based on Hoechst 33342/PI staining (C) and PrestoBlue assay (D; normalized by control cells, without NPs). (E) Toxicity observed by PI staining in N-iPSC ECs and HGPS-iPSC ECs exposed to NPs at 25 μg mL−1 (E.1) and 50 μg mL−1 (E.2). Results are mean ± SEM (n =4). *, **, and **** denote statistical significance (P < 0.05, P < 0.01, and P < 0.0001). (F) ICP-MS analyses in N-iPSC ECs and HGPS-iPSC ECs after being exposed to NPs for 24 h (except for NP1, which was 4 h incubation). Results are mean ± SEM (n =4). **, ***, and **** denote statistical significance (P < 0.01, P < 0.001, and P < 0.0001). In E and F, the statistical analyses were performed by an unpaired t-test. | ||
NPs showed a differential toxic effect against both types of ECs. HGPS-iPSC ECs were more sensitive to NP13, NP18, NP22, NP26 and NP29 NPs as compared to N-iPSC ECs (Fig. 5B–E). However, NP3, NP7, NP24, NP25, and NP28 were more toxic for N-iPSC-derived ECs than for HGPS-iPSC ECs.
To investigate whether the nanomaterial toxicity correlates with its capacity to be internalized by ECs, we performed inductively coupled plasma mass spectrometry measurements (ICP-MS) on N-iPSC ECs and HGPS-iPSC ECs exposed to 11 different nanomaterials (25 μg mL−1) for 24 h (Fig. 5F). The concentration of NPs was selected based on a compromise between cell death and NP quantification before cell death. For one of the nanomaterials tested (NP1), the accumulation was evaluated after 4 h of incubation with cells, because of the high toxicity observed after 24 h incubation. Overall, our results indicate that the toxicity of the NPs was not dependent on the concentration internalized by the cells. For example, NP24, NP25 and NP30 had similar ranges of cytotoxicity (Fig. 5E.1) against N-iPSC ECs but significant differences in NP uptake (Fig. 5F). On the other hand, the cellular uptake of NP1 and NP7 was very low compared to that of other NP formulations; however, they exerted a higher cytotoxic effect against ECs. In addition, one (NP12) showed high cellular internalization (Fig. 5F), mostly by N-iPSC ECs, without showing measurable toxicity (Fig. 5C). Interestingly, some nanomaterials were more internalized by HGPS-iPSC ECs than by N-iPSC ECs and this is surprising taking into account that HGPS-iPSC ECs showed lower cell doublings than N-iPSC ECs (Fig. S3B, ESI†).
Overall, our results indicate that: (i) NPs showed a differential toxic effect against both types of ECs and (ii) there is not a direct correlation between the concentration of NPs internalized by cells and the observed toxicity in vitro under static conditions. Regarding this last point, some NPs showed high internalization efficiency without causing any toxic effect on cells, whereas others caused high toxicity with very low internalization efficiency.
Vascular screening of NPs in a 3D model
The vascular impact of the NPs was then screened by a Matrigel assay39 comprising the formation of microvessels with a patent lumen, as previously demonstrated by us in ref. 12. The optimization of the assay (e.g. cell exposure time to nanomaterials, concentration of nanomaterials, and type of readouts) was carried out with somatic ECs (human umbilical arterial ECs) (Fig. S4, ESI†). Cells were cultured on top of Matrigel in the presence of NPs for 24 or 48 h after which EC viability and tube length was measured. The option of mixing the NPs during the formation of microvessels, and not after, was inspired by the in vivo strategy described below in zebrafish embryos in which the effect of the NPs was evaluated during, and not after, embryonic vasculature formation. Our results indicate that cells exposed to NP1 at 1 μg mL−1 for 24 h had no significant effect, while at 12.5 μg mL−1 showed approximately 12% and 14% of apoptosis and necrosis, respectively, and were unable to form vascular microvessels in Matrigel (cells showed clear altered morphology) (Fig. S4, ESI†). Therefore, for the screening assay in Matrigel we have used N-iPSC-ECs, at a concentration of nanomaterials of 12.5 μg mL−1 and we evaluated the length of the microvessels after 24 h (Fig. 6A). Interestingly, only three formulations could negatively affect microvessel formation, specifically NP1, NP3 and NP29 that reduced or even abolished the formation of microvessels (Fig. 6B.1). Interestingly, NP9 enhanced angiogenesis, as LL37 peptide is a recognized pro-angiogenic peptide.18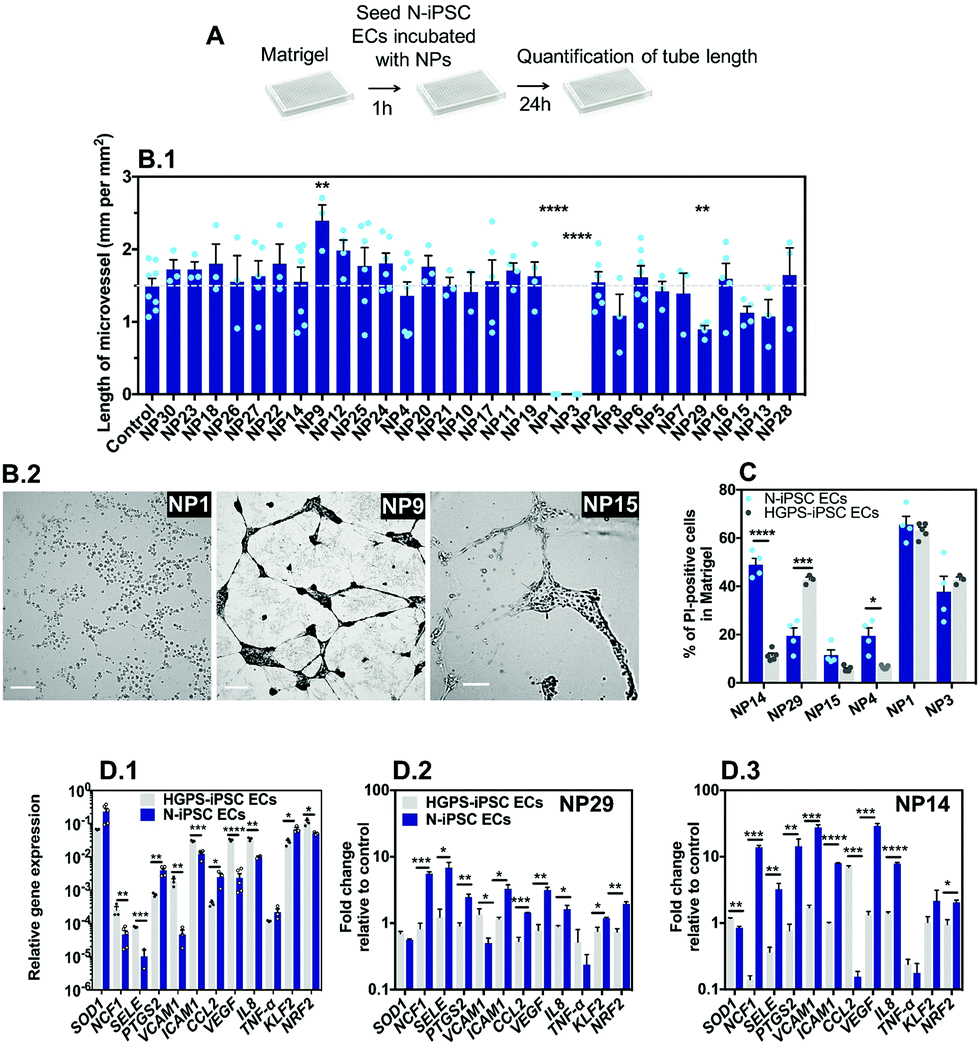 | ||
| Fig. 6 Impact of nanomaterials on iPSC-ECs cultured in 3D. (A) Schematic representation of the protocol to evaluate the effect of NPs in the formation of capillary-like networks by iPSC-ECs on Matrigel. Cells were seeded on top of Matrigel at a density of 2 × 103 cells per well together with NPs (12.5 μg mL−1) for 24 h, after which cell viability and tube formation was analyzed. (B.1) Impact of NP library on the length of microvessels formed by N-iPSC ECs. Results are average ± SEM (n = 4). **, and **** denote statistical significance (P < 0.01, and P < 0.0001, respectively) relative to control. (B.2) Representative images of capillary-like networks formed in Matrigel by N-iPSC ECs treated for 24 h with 12.5 μg mL−1 of NP1, NP9 or without NPs (control). Scale bar is 200 μm. (C) Toxicity was measured by Hoechst 33342/PI staining at 24 h, in HGPS-iPSC ECs or N-iPSC ECs cultured in the presence of NP1, NP3, NP4, NP14, NP15 and NP29 in Matrigel at 25 μg mL−1. *, ***, and **** denote statistical significance (P < 0.05, P < 0.001, and P < 0.0001). (D) Relative gene expression of genes involved in oxidative stress (NRF2), inflammation (ICAM1, SELE, VCAM1 and IL8) and endothelial homeostasis (KLF2, and VEGF) in N-iPSC ECs and HGPS-iPSC ECs before (D.1) and after exposure to 25 μg mL−1 of NP29 (D.2) or NP14 (D.3). *, **, and *** denote statistical significance (P < 0.05, P < 0.01, and P < 0.001). In B.1, C and D, the statistical analyses were performed by an unpaired t-test. | ||
Next, to compare the vascular response in 3D versus 2D (see section above), we have mixed NP formulations (at a concentration of 25 μg mL−1) with N-iPSC ECs or HGPS-iPSC ECs, cultured the cells in Matrigel for 24 h and measured cell toxicity by Hoechst/PI staining. We have selected 6 NP formulations for these analyses including NP1, NP3, NP4, NP14, NP15 and NP29 since they showed cytotoxicity in a 2D model (Fig. 5C). The toxicity levels (based on PI staining) of NP1 and NP3 were lower than the ones observed in a 2D model (NP1: 60 vs. 90%; NP3: 40 vs. 90%, all based in N-iPSC ECs); however, it was higher for NP29 (N-iPSC ECs: 20 vs. 10%; HGPS-iPSC ECs: 40 vs. 20%). Our results further showed differences in NP toxicity against N-iPSC ECs and HGPS-iPSC ECs (Fig. 6C). For example, NP4 and NP14 induced higher toxicity towards N-iPSC ECs than HGPS-iPSC ECs. In contrast, NP29 induced higher toxicity towards HGPS-iPSC ECs than N-iPSC ECs, similar to the results obtained with the 2D model (Fig. 5).
Previous studies have shown that the accumulation of progerin in ECs induced a pro-inflammatory program of EC activation characterized by an overexpression of leukocyte adhesion molecules (VCAM1, E-selectin)40 and pro-inflammatory cytokines (IL8)40 and a downregulation of the transcription factor Kruppel-like factor (KLF2),40 which regulates EC pro-inflammatory activation, but also a decrease in the anti-oxidative stress response program characterized by an impaired transcriptional activity of the transcription factor NRF2.41 Indeed, HGPS-iPSC ECs have higher reactive oxidative species levels than N-iPSC ECs (Fig. S5, ESI†). To identify the mechanism behind the differential toxicity of NPs against N-iPSC ECs and HGPS-iPSC ECs, we evaluated the expression of genes involved in oxidative stress (NRF2), inflammation (SELE, ICAM1, VCAM1 and IL-8) and endothelial homeostasis (KLF2, and VEGF) in cells exposed to NP14 and NP29 (both at 25 μg mL−1) for 24 h (Fig. 6D). In the absence of NPs, HGPS-iPSC ECs showed higher expression of inflammatory and oxidative stress but lower KLF2 mRNA transcripts than N-iPSC ECs. After exposure to NPs, the inflammatory response of both cells was significantly different: only N-iPSC ECs showed a significant up-regulation of pro-inflammatory mRNA and the response seemed higher for NP14 than for NP29. Moreover, N-iPSC ECs, but not HGPS-iPSC ECs, showed an upregulation of mRNA of the NRF2 gene that encodes nuclear factor erythroid 2-related factor involved in the cellular antioxidant response. Altogether, our results indicate that: (i) a small number of NPs (3 out of 30) interfered with microvessel formation in Matrigel at the concentration tested (12.5 μg mL−1); (ii) the Matrigel assay showed differences in the vascular toxicity profile (evaluated by PI staining) of NPs relative to the 2D model, but in both cases the importance of EC background was critical for the biological response; (iii) the differential biological response of N-iPSC ECs and HGPS-iPSC ECs to NPs involved differential regulation of genes related with inflammation and oxidative stress.
Screening of NPs in a blood vessel on a chip
The vascular models described above have several limitations, including the fact that they are not perfused and do not take into account the heterotypic interactions between ECs and SMCs. Cells exposed to flow change their morphology and more importantly they express a layer of proteins, glycoproteins, glycolipids, proteoglycans and glycosaminoglycans called glycocalyx.42 The glycocalyx is involved in several functions, such as regulation of vascular permeability, acting as a selective barrier to macromolecules (based on size and charge), as well as a mechanosensor of fluid shear stress. Thus, we decided to prepare a blood vessel on a chip to screen nano–bio interactions. In general, these chips have only one layer of endothelial cells to evaluate nano–bio interactions.43 Here, we have successfully developed a chip with two-cell monolayers of SMCs and ECs. The vessel was perfused with medium, at shear rates similar to in vivo, and the EC monolayer was able to secrete a functional glycocalyx layer on top. For this purpose, SMCs (for initial studies we have used somatic cells) were cultured in a microfluidic chamber for 12 h to form a monolayer followed by the plating of ECs on top of SMCs for 4 h and finally by the perfusion of both cells for 5 days (Fig. S6, ESI†). Under these conditions, SMCs were in the outer part of the vessel, while ECs remained in the luminal side (Fig. S6A and B, ESI†). Interestingly, ECs co-cultured with SMCs under flow conditions showed higher levels of glycocalyx than ECs alone cultured under flow conditions (Fig. S7A, ESI†). To verify that the glycocalyx was indeed located in the luminal side of ECs, we have used GFP-expressing ECs, which were co-cultured with SMCs for 5 days under flow as previously described (Fig. S7B and C, ESI†). As expected, the layer of glycocalyx was located on top of the EC layer in the luminal side, showing the polarization of the glycocalyx. The formation of the glycocalyx was also demonstrated by an EC uptake assay of DiI-LDL (Fig. S7D, ESI†). In this case, cells cultured under flow conditions for 5 days had lower uptake of Dil-LDL than cells cultured for 5 days and exposed to heparinase III to remove the heparin sulfate in glycocalyx.In order to verify the effect of flux in the formation of glycocalyx, SMCs and ECs were co-cultured as previously described and the heparan sulfate intensity was quantified at different time points (Fig. S8A, ESI†). The amount of heparan sulfate increased more than 2.5-fold up to 5 days in flow, at 20 dyn cm−2 (Fig. S8B and C, ESI†). The recovery of heparan sulfate was also evaluated. Cells cultured under flow for 5 days were treated with heparinase III to degrade the heparan sulfate, and its recovery was followed over time (Fig. S8D, ESI†). The kinetics of the recovery was similar to the one observed for the heparan sulfate formation induced by the shear stress. Therefore, for subsequent experiments with iPSC-derived cells, they were co-cultured for 5 days at 20 dyn cm−2. We performed experiments with N-iPSC SMCs and N-iPSC ECs (Fig. 7A) but not with HGPS-iPSC-derived cells because the HGPS-iPSC SMCs were sensitive to flow and detached overtime.44 N-iPSC SMCs obtained using protocols previously published by us in ref. 45 were cultured in a microfluidic chamber followed by the culture of N-iPSC ECs on top of the SMCs (Fig. 7B). Cells were then cultured under flow conditions (20 dyn cm−2) for 5 days. Under these conditions, N-iPSC SMCs were located in the outer part of the blood vessel and positively stained for α-smooth muscle actin (α-SMA) whereas the iPSC-ECs, which were located in the inner part of the blood vessel, were positively stained for VE-cadherin (Fig. 7B.1). As expected, ECs expressed glycocalyx, the expression being higher in flow than in static conditions (Fig. 7B.2) and in co-culture with SMCs versus ECs alone (Fig. 7C). Moreover, the enzymatic removal of heparan sulfate by heparinase in vascular cells cultured under flow conditions for 5 days allowed higher accumulation of Dil LDL (Fig. 7D) than cells without enzymatic treatment. Altogether, we have developed a blood vessel on a chip from ECs and SMCs derived from iPSCs. ECs co-cultured with SMCs under flow conditions expressed higher levels of heparan sulfate than ECs cultured in static conditions and thus represent a better match to the in vivo blood vessel physiology.
 | ||
| Fig. 7 Impact of nanomaterials in a blood vessel on a chip composed by iPSC-ECs and iPSC-SMCs. (A) Protocol to generate a blood vessel on a chip. A suspension of SMCs (4.1 × 104 cells per cm2) was applied in each channel of an Ibidi μ-Slide VI0,4 plate. SMCs were maintained in static conditions for 12 h, after which a suspension of ECs (12.5 × 104 cells per cm2) was applied on top of SMCs and the co-culture was maintained in static conditions for 4 h, after which cells were perfused with EGM-2 medium at 20 dyn cm−2 for 5 days. (B) Immunofluorescence analyses performed on a co-culture of N-iPSC SMCs and N-iPSC ECs for: SMC (α-SMA), EC (VE-cadherin) (B1) and glycocalyx (heparan sulfate) (B2) markers. Scale bar is 50 μm. (C) Heparan sulfate intensity in a co-culture of N-iPSC SMC and N-iPSC EC cells vs. monoculture of N-iPSC ECs after 5 days under arterial flow conditions (20 dyn cm−2). **** denotes statistical significance (P < 0.0001). (D) Effect of glycocalyx impairment (by heparinase III treatment) on the uptake of DiI-LDL by N-iPSC ECs after 5 days in co-culture with N-iPSC SMCs under arterial flow conditions (20 dyn cm−2). **** denotes statistical significance (P < 0.0001). In C and D, the statistical analyses were performed by an unpaired t-test. (E) Schematic representation of the protocol. Cells (co-culture of N-iPSC SMCs and N-iPSC ECs) were cultured for 5 days in arterial flow conditions (20 dyn cm−2), after which NPs were incubated for 24 h and characterized for cell metabolism and death. (F) Cell viability after 24 h incubation in flow conditions measured by Hoechst 33342/PI staining. Arrows show the difference in toxicity between static (monoculture of N-iPSC ECs) and flow conditions (co-culture N-iPSC SMCs and N-iPSC ECs) for the same NPs. Results are mean ± SEM (n = 4–6). *, and **** denote statistical significance (P < 0.05, and P < 0.0001) against control (cells without NPs). (G) Effect of the removal of endothelial glycocalyx, by heparinase III treatment, on the metabolism of N-iPSC ECs and N-iPSC SMCs cultured in the microfluidic chip for 24 h with NP1 or NP2. ** denotes statistical significance (P < 0.01) against control (cells without NPs). (H.1) Cell metabolism of N-iPSC ECs after 24 h incubation with NP1 or NP2 under arterial flow conditions. **** denotes statistical significance (P < 0.0001) against control (cells without NPs). (H.2) Cell metabolism of N-iPSC SMCs after 24 h incubation with NP1 or NP2 under arterial flow conditions. In F, G and H, results are mean ± SEM (n = 3) and statistical analyses were performed by an unpaired t-test. | ||
Next, we have tested the 10 NPs identified in previous vascular models with higher cytotoxicity effects in a blood vessel on a chip (Fig. 7E). In this model, we used ECs and SMCs derived from N-iPSCs. Ten NPs were exposed for 24 h under flow conditions to the blood vessel on a chip. For two of the formulations (NP15 and NP25) we have also quantified the cellular internalization of the NPs by ICP-MS analyses. The results show that NP internalization was 34.6 pg per cell and 1.1 pg per cell (assuming that all the NPs were taken up by ECs alone) of NP15 and NP25, respectively, which means approximately 3-fold lower than in static conditions (Fig. S9, ESI†). Interestingly, all the NPs tested showed lower toxicity than in static conditions against N-iPSC ECs (Fig. 7F). The decrease in toxicity may be explained by the protective role of glycocalyx expressed in the blood vessel on a chip. To demonstrate this hypothesis, the toxicities of NP1 and NP2 were evaluated in 3 different models: (i) a co-culture of N-iPSC SMCs and N-iPSC ECs (Fig. 7G), (ii) a monoculture of N-iPSC ECs (Fig. 7H.1) and (iii) a monoculture of N-iPSC SMCs (Fig. 7H.2). As shown before (Fig. 7C and Fig. S7A, ESI†), a co-culture of SMCs and ECs led to an increase of the production of the glycocalyx layer shown by a significant increase in intensity of heparan sulfate, one important and main component of the glycocalyx. Our results showed that when ECs had this protective glycocalyx layer the toxicity of NP1 was considerably lower than in cells without this layer (Fig. 7G). In addition, when the glycocalyx layer was impaired, either by enzymatic degradation (Fig. 7G) or by using the monoculture cell model of ECs (that produces less glycocalyx; Fig. 7H.1), the toxicity was higher relative to cells with a glycocalyx layer. iPS-SMCs showed no toxicity towards these NPs at the concentrations tested (Fig. 7H.2). Therefore, the presence of a glycocalyx layer influenced the impact of NPs on ECs and might explain differences in the toxicities of nanomaterials in static vs. flow conditions.
Overall, our results indicate that most of the nanomaterials that had a negative impact on the angiogenesis of blood vessels in the transgenic zebrafish model showed vascular toxicity in the 2D and 3D models but with differences according to the type of vascular cells and their organizational complexity. In addition, the toxicities of the tested NPs were lower in the blood vessel on a chip than in the 2D model. The results further indicate that the decrease in the NP toxicity, at least in part, was mediated by the expression of a protective glycocalyx layer in ECs cultured in the blood vessel on a chip.
3. Discussion
The current work describes a platform to study the bioactivity/toxicity of nanomaterials based on vascular cells derived from hiPSCs, which were cultured in 3 different platforms with increasing level of complexity: (i) monoculture in poly(styrene) (named “2D”); (ii) monoculture in Matrigel (named “3D”) and (iii) co-culture (both ECs and SMCs) under flow conditions (named “blood vessel on a chip”). These 3 platforms captured different aspects of the impact of the nanomaterials and the results were validated in zebrafish embryos, more specifically, in the capacity of the NPs to interfere with embryonic vasculogenesis. The 2D model is the most suitable model for high-throughput screening; however, it does not recapitulate the three-dimensional organization of the blood vessel as well as the dynamic environment of the bloodstream, which may induce phenotype alterations in ECs (e.g. absence of glycocalyx expression) and NPs may deposit at higher levels onto the cells, which increases NP uptake. The 3D model is suitable to reproduce the geometry of the blood vessel in vitro since ECs associate into 3D tubes with lumen formation;12,39 however, as a 2D model, it does not take into account the complexity of the blood vessel or the effect of flow. The blood vessel on a chip is suitable to reproduce the complexity (as it includes ECs and SMCs) and flow culture conditions as observed in the in vivo blood vessels; however, it is complex for use in high-throughput screening. Our results indicate that the three platforms were able to identify, to different extents, the NPs that showed toxicity against zebrafish embryos. From the 18 NP formulations that interfered with the vasculogenesis process in zebrafish embryos (Fig. 3), 15 NP formulations have been identified by the in vitro models (2D and 3D models; the number of PI+ cells was above 20%), their toxicity level being dependent on the hiPSC-EC type. Some of the formulations were also tested in the blood vessel on a chip and the results further confirmed their toxicity; however, in general, at lower intensity. Table S1 (ESI†) summarizes the main findings obtained among the four systems as well as rodent data found in the literature.Previous studies have used 2D models based on somatic ECs, but not on ECs derived from hiPSCs generated from different individuals, to screen multiple NP formulations.46 In addition, hiPSC-ECs have been used to screen small molecules12 but not nanomaterials. The use of hiPSCs enables the in vitro modelling of different individuals with or without diseases. The development of iPSC technology allows the access of a virtually unlimited number of cells from any individual, which can then be differentiated into any kind of cells and be used for high-throughput screening studies. This technology has allowed, for instance, the screening of large libraries of small compounds to find hits that rescue genetic diseases47,48 or interfere with human development.12 We have used hiPSCs as a source of ECs to study nanomaterial interactions, which have not yet been investigated. We have studied the impact of nanomaterials on hiPSC-ECs having embryonic (N-iPSC ECs)12 and aged phenotypes (HGPS-iPSC ECs), as confirmed by the accumulation of progerin and the existence of dysmorphic nuclei in the latter. The selection of HGPS-iPSC ECs in the context of this work was to investigate the biological response of aged cells to NPs, which is a topic that remains largely unexplored.
Our results indicate that HGPS-iPSC ECs and N-iPSC ECs had differential toxicity profiles against NPs, both in 2D and 3D models. The ultrasmall NP1 and NP3 were shown to be the most toxic of all NPs of the library for both types of ECs, even for concentrations at 6 μg mL−1 (2D model); five NPs (NP14, NP15, NP18, NP22 and NP30) showed toxicity levels higher than 60% (measured by a PI assay) for both EC-derived cells; three NPs (2D system: NP25 and NP28; 3D system: NP14) showed higher toxicities against N-iPSC ECs than HGPS-iPSC ECs; one NP formulation (NP29) showed higher toxicity (3-fold) for HGPS-iPSC ECs than for N-iPSC ECs. The vascular toxicities observed for some NPs in the 2D model are in good accordance with those reported in the literature using somatic cells. For example, NP1 and NP3 showed high toxicities against HeLa cells.32 Zinc oxide NPs were observed to be toxic (>50%) at 50 μg mL−1 in human aortic ECs,34 human cardiac microvascular ECs35 and in HUVECs;36,37 all these results are in good accordance with the 60% toxicity observed in our study for ECs derived from iPSCs. SiO2 NPs have also been reported to induce toxicities from 20 to 60% in HUVECs for NP concentrations between 50 and 75 μg mL−1,38 while Ag NPs have been reported to induce toxicity in rat brain microvessel ECs49 and to demonstrate anti-angiogenic properties.50 It is possible that the higher toxicity of Ag NPs against HGPS-iPSC ECs, as compared to that of N-iPSC ECs, is related to a decrease in the antioxidative stress response program in HGPS-iPSC ECs. It is known that Ag NPs induce cytotoxicity through oxidative stress, leading to the production of reactive oxygen species (ROS) in their surface or though the direct interaction with cell mitochondria.51 On the other hand, the nuclear accumulation of progerin impairs the transcriptional activation of NRF2, due to the physical interaction of both proteins, which reduces the anti-oxidative stress program of HGPS cells.41 Therefore, it is expected that HGPS-iPSC ECs are more sensitive to NPs that induce toxicity by oxidative stress such as Ag NPs. In contrast, the higher toxicity of NP14 against N-iPSC ECs as compared to HGPS-iPSC ECs may be related to the differential inflammatory program between cells. Our results show that HGPS-iPSCs have a pro-inflammatory program before NP exposure, characterized by higher levels of VCAM1, SELE, IL8 and ICAM1 mRNA transcripts as compared to N-iPSC ECs. It is possible that the pro-inflammatory status of HGPS-iPSC ECs makes these cells less sensitive to inflammatory cytokines than N-iPSC ECs. Indeed, the pro-inflammatory program of HGPS-iPSC ECs is not significantly affected after exposure to NPs, while the opposite was observed in N-iPSC ECs.
The EC response to NPs is largely influenced by the flow shear stress and the interaction with SMCs. EC genotype/phenotype as well as endocytic capacity are influenced by flow shear stress. For example, ECs cultured under increased shear stress have decreased oxidative stress and inflammation52 and showed differential NP uptake as compared to ECs cultured under static conditions.43,53 Importantly, EC response to flow shear stress is influenced by the neighbouring SMCs.54,55 Although in vitro co-culture systems of ECs and SMCs under flow conditions have been established for somatic ECs and SMCs,54,55 the generation of co-culture systems with hiPSC-derived vascular cells remains unexplored. The model developed in the current work allows the performance of assays at arterial conditions by submitting cells to shear stresses of 20 dyn cm−2. In this model, cells aligned parallel to the flow direction, and ECs produced a glycocalyx (a glycoprotein–polysaccharide meshwork) layer in the apical region as it occurs in vivo. Previous studies have shown that glycocalyx influences NP uptake by somatic ECs.56,57 Our results showed that NP toxicity against ECs is significantly lower in cells cultured under flow conditions and in co-culture with SMCs. Our results further showed that glycocalyx mediated, at least in part, the reduction in NP internalization, since the degradation of glycocalyx by heparinase significantly reduced cell metabolism/viability.
In many cases, the impact of nanomaterials is screened in rodents;4–6 however, these experiments are expensive, time consuming and low throughput. Zebrafish embryos can be used to investigate vascular toxicity of NPs because they are relatively cheap, the developmental processes are well studied and the toxicity of the NPs can be monitored in real time using transgenic lines with fluorescent markers.58,59 Therefore, in the current work, zebrafish embryos were used to validate the toxicity profiles of the NP library evaluated in the in vitro tests. Because some NPs are trapped by the chorion of zebrafish,60 we have removed it to perform the screening. Our results showed that 18 NP formulations had an impact in the angiogenesis of the intersegmental vessels of zebrafish embryos. Most of the formulations (15 out 18) were confirmed by the in vitro tests.
The iPSC-derived vascular networks described here might be useful to screen other types of nano–bio interactions. For example, recent studies have shown that some NPs can disrupt the VE–cadherin interactions between endothelial cells (called nanoEL effect), negatively charged NPs being the ones with the highest impact.9,61 For this purpose, we have tested the effect of silica NPs (formulation NP13) in a monolayer of N-iPSC ECs for 1 h after which we have evaluated its paracellular permeability against FITC–dextran and VE–cadherin interaction between cells by confocal microscopy (Fig. S10, ESI†). Our results clearly show the sensitiveness of our iPSC-derived vascular network to the nanoEL effect.
4. Conclusions
We have developed personalized human in vitro platforms with a variable level of complexity (2D, 3D and blood vessel on a chip formats) and ageing phenotype to evaluate the interaction of nanomaterials with the vascular system. Taking into account the toxicity as the main readout, the NP toxicity profile was dependent on the age and vascular network format, the NPs being less cytotoxic in the blood vessel on a chip format due to the protective effect of glycocalyx. The platform presented here is very promising for high-throughput screening of nano–bio interactions in the context of regenerative and therapeutic medicine.5. Experimental section
Nanomaterial library: characterization
The diameter and morphology of some of the nanomaterials were characterized using a PHILIPS CM-12 transmission electron microscope at 100 kV. A few microliters of an aqueous suspension of nanomaterials was placed on a 200-mesh copper grid coated with a Formvar film and then wiped off by filter paper. The dried grid was then examined under an electron microscope. Additionally, the nanomaterials were analysed by photon correlation spectroscopy (PCS) using quasi-elastic light scattering equipment (Zeta-Pals™ Zeta Potential Analyzer, Brookhaven Instruments Corp., Holtsville, NY) and the ZetaPlus™ Particle Sizing Software (version 4.03). The nanomaterial suspension (2 mL, 50 μg mL−1 in PBS or EGM-2 medium containing FBS) was added to a cuvette and allowed to stabilize for 10 min and then analysed (3 times) at room temperature. Nanomaterials suspended in EGM-2 medium were then centrifuged (8000g, 8 min) and resuspended in 1 mM KCl (2 mL, 50 μg mL−1), at 25 °C. The nanomaterial suspension was finally characterized by zeta potential analyses. All data were recorded with at least 5 runs (in triplicate) with a relative residual value (measure of data fit quality) of 0.03.In vivo studies
Zebrafish (Danio rerio) were maintained in accordance with Institutional and National Animal Care protocols, in a re-circulating system at 28 °C on a 14 h-light, 10 h-dark cycle, fed twice daily. Transgenic adult zebrafish [Tg(fli1a:EGFP)y1]26 expressing enhanced green fluorescent protein (EGFP) under control of the fli1 promotor were used in this work. Genders were housed separately until the day before breeding, then placed in tanks at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 male:female ratio. Fish were left undisturbed overnight and fertilized embryos were collected 4 hours after the light was turned on the next morning. For each condition, a minimum of 20 embryos within 4 hours-post-fertilization (hpf) were manually dechorionated and randomly placed in glass Petri dishes containing 10 mL of embryo medium. NPs at 1 μg mL−1 were added to the embryo medium and incubated for 24 hours, after which the embryos were fixed with 4% PFA at 4 °C overnight. Embryos were then washed twice with PBS. As a control, the same amount of vehicle (water) was used. For each experimental group a minimum of eight embryos were analysed for the number of ISVs along the anterior–posterior axis, the average length of the first 10 ISVs and the total length of ISVs. Different batches of embryos and pools of embryos from different mating pairs were used among different experiments. Images of the embryos embedded in 3% carboxymethyl cellulose were obtained using a Zeiss/P.A.L.M. laser dissecting microscope equipped with a Fluar 5×/0.25 M27 objective. The number and length of ISVs were directly measured in each image by the same user using ImageJ software.
:1 male:female ratio. Fish were left undisturbed overnight and fertilized embryos were collected 4 hours after the light was turned on the next morning. For each condition, a minimum of 20 embryos within 4 hours-post-fertilization (hpf) were manually dechorionated and randomly placed in glass Petri dishes containing 10 mL of embryo medium. NPs at 1 μg mL−1 were added to the embryo medium and incubated for 24 hours, after which the embryos were fixed with 4% PFA at 4 °C overnight. Embryos were then washed twice with PBS. As a control, the same amount of vehicle (water) was used. For each experimental group a minimum of eight embryos were analysed for the number of ISVs along the anterior–posterior axis, the average length of the first 10 ISVs and the total length of ISVs. Different batches of embryos and pools of embryos from different mating pairs were used among different experiments. Images of the embryos embedded in 3% carboxymethyl cellulose were obtained using a Zeiss/P.A.L.M. laser dissecting microscope equipped with a Fluar 5×/0.25 M27 objective. The number and length of ISVs were directly measured in each image by the same user using ImageJ software.
iPSC culture
HGPS-iPS cells were generated from skin fibroblasts of an HGPS patient and were kindly donated by Xavier Nissan.15 N-iPSCs were generated from cord blood (hiPSCs K2, passages 32–35)62 and were kindly donated by Ulrich Martin. Cells at passage 35–45 have been used for the differentiation studies. Both iPSCs were cultured on inactivated mouse embryonic fibroblasts (MEFs), as previously described.12,13,45iPSCs: differentiation into SMCs
The differentiation of iPSCs into SMCs was performed through an intermediary embryoid body (EB) stage. Undifferentiated iPSCs were treated with type IV collagenase (2 mg mL−1, Invitrogen) for 2 h and then transferred (2:1) to low attachment plates (Corning) containing a differentiation medium [10 mL, 80% KO-DMEM, 20% FBS, 0.5% L-glutamine, 0.2% β-mercaptoethanol, 1% nonessential amino acids and 50 U mL−1:50 mg mL−1 penicillin–streptomycin solution] to form EBs. EBs were cultured for 10 days at 37 °C, 5% CO2 in a humidified atmosphere, with medium changes every 3–4 days. CD34+ cells were isolated from EBs at day 10 using magnetism-activated cell sorting (MACS). Isolated cells were grown on 24-well plates coated with 0.1% gelatin and containing endothelial growth medium-2 (EGM-2) supplemented with retinoic acid (1 mM, Sigma).
iPSCs: differentiation into ECs
Undifferentiated iPSCs were treated with type IV collagenase (2 mg mL−1, Invitrogen) for 1 h and then transferred to Petri dishes coated with fibronectin (1 μg cm−2, Calbiochem) and containing a differentiation medium [10 mL of 50% Iscove's modified Dulbecco's medium (IMDM, Gibco), 50% F12 (Gibco), 5 mg mL−1 BSA (Sigma), 0.2% β-mercaptoethanol, 15 mg mL−1 transferrin (Sigma), 7 mg mL−1 Insulin (Sigma) and 50 U mL−1 penicillin–streptomycin (Lonza)]. The differentiation medium was supplemented with BMP4 (10 ng mL−1, Peprotech) and FGF-basic (20 ng mL−1, Peprotech). After 1.5 days, the medium was replaced with fresh differentiation medium supplemented with BMP4 (50 ng mL−1) and FGF-basic (20 ng mL−1). After 5 days, the medium was further replaced by fresh differentiation medium supplemented with VEGF165 (50 ng mL−1, Peprotech), Tβ4 (100 ng mL−1, Caslo) and SB431542 (10 μM, Tocris). At the end of day 10, the PECAM1+ cells were selected by magnetic labelling, plated in 0.1% gelatin-coated dishes and cultured with EGM-2 medium supplemented with VEGF165 (50 ng mL−1) and SB431542 (10 μM). The medium was changed every 2–3 days.Primary vascular cell culture
Human vascular smooth muscle cells (Lonza, CC-2579) were cultured in MCDB 131 medium (Gibco) supplemented with FBS (20% v/v, Gibco), L-glutamine (Gibco, 1 mM) and penicillin–streptomycin (50 U mL−1, Lonza). Human umbilical artery endothelial cells (HUAECs) and human umbilical vein endothelial cells (HUVECs) were cultured in EGM-2 (Lonza). Cell cultures were maintained at 37 °C, and 5% CO2 in a humidified atmosphere, with media changed every 2 days.Cell characterization: immunofluorescence analyses
Cells were washed with PBS, fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 15 min at room temperature and washed again with PBS. Cells were permeabilized (when necessary) with 0.1% Triton for 10 min, blocked with 1% (w/v) BSA for 30 min and incubated for 1 h with specific primary antibodies: mouse anti-human heparan sulfate (10E4 epitope, US Biological), rabbit anti-human smooth muscle α-actin (Abcam), mouse anti-human calponin (calponin1, Santa Cruz Biotechnology), mouse anti-human PECAM1 (clone JC70A, Dako), mouse anti-human VE-Cadherin (Santa Cruz Biotechnology), rabbit lamin A/C (H-110, Santa Cruz Biotechnology) and rabbit anti-human ZO-1 (Life Technologies). The binding of primary antibodies was detected with: goat anti-rabbit IgG Alexa 488, goat anti-mouse IgG Alexa 488 or goat anti-mouse IgG Alexa 555 (Life Technologies). Cell nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI, Sigma) and the slides were examined using an LSM 710 confocal microscope (Zeiss) or a high-content fluorescence microscope IN Cell 2200 (GE Healthcare). In the case of heparan sulfate immunostaining, the imageJ software was used to quantify the overall intensity of heparan sulfate normalized by the area of the image in, at least, 8 images per condition.Cell characterization: flow cytometry analyses
Cells were dissociated with non-enzymatic cell dissociation buffer (Gibco) for 10 min, followed by gentle pipetting and washes in PBS with 5% FBS. Single cells were aliquoted in PBS with 5% FBS (between 100000 and 150000 cells were used per condition) and stained with either isotype controls or antigen-specific fluorescent-conjugated antibodies for 30 min at 4 °C. The following antibodies have been used: PECAM1 (eBioscience, 5 μL per 100 μL of cell suspension), CD105 (Miltenyi Biotec, 10 μL per 100 μL of cell suspension) and VEGFR2 (R&D Systems, 10 μL per 100 μL of cell suspension). The flow cytometry analyses were performed in a BD Accuri C6 and data analysis was performed with the FlowJo_V10. Ten thousand events were collected in each run. The percentages shown in dot plots were calculated based on the isotype controls represented by light blue. Isotype controls had 1% overlap with the protein of interest.
Cell characterization: ROS levels
N-iPSC ECs and HGPS-iPS ECs were plated in a 96-well plate at a density of 2 × 104 cells per well and cultured for 24 h before incubation with 5 μM CellROX® Deep Red (Invitrogen) for 2 h. Cells were then washed, fixed with 3.7% formaldehyde for 15 min at room temperature and washed two times with PBS. Images were taken from each well (9 images per well) using the IN cell Analyzer 2200 (IN Cell 2200, GE Healthcare) and analysed using the Analyzer Workstation software. For oxidative stress measurements the mean intensity of CellROX® Deep Red was registered in both the cell nucleus and cytoplasm. The total value of the measured intensity was used and normalized according to cell area. Assays were performed in triplicate.Cell characterization: quantitative real-time polymerase chain reaction (qRT-PCR) analyses. Total RNA was extracted with an RNeasy Micro Kit (Qiagen) and immediately stored at −80 °C. RNA was quantified using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Inc., USA) at 260 nm. The cDNA was reverse transcribed from 1 μg of total RNA using a TaqMan reverse transcription reagents kit (Invitrogen) according to the manufacturer's instructions. The cDNA obtained was stored at −20 °C until further analysis by real-time PCR was performed. Real-time PCR analyses were performed using the fluorescent dye SYBR Green (Applied Biosystems) or TaqMan technology (Life Technologies) and the 7500 Fast Real Time PCR System (Applied Biosystems). Specific set of Taqman MGB probe (Life Technologies) and primers (designed by Sigma) used in this work (Tables 2 and 3).
| Probe | Sense | Antisense |
|---|---|---|
| TaqMan PCR conditions: initial step at 50 °C for 2 min; after that another step at 95 °C for 10 min; 45 cycles at 95 °C for 15 s and at 60 °C for 1 min. After amplification, melting curves were acquired and used to determine the specificity of PCR products. | ||
| CGCTGAGTACAACCT | ACTGCAGCAGCTCGGGG | TCTGGGGGCTCTGGGC |
| Sense | Antisense | |
|---|---|---|
| SYBR Green PCR conditions: initial denaturation step at 94 °C for 5 min; 40 cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 33 s and extension at 72 °C for 30 s. At the end was performed a final 7 min extension at 72 °C. After amplification, melting curves were acquired and used to determine the specificity of PCR products. | ||
| PECAM1 | AGATACTCTAGAACGGAAGG | CAGAGGTCTTGAAATACAGG |
| CDH5 | ACGGGATGACCAAGTACAGC | ACACACTTTGGGCTGGTAGG |
| SELE | AGCTTCCCATGGAACACAAC | CTGGGCTCCCATTAGTTCAA |
| GAPDH | AGCCACATCGCTCAGACACC | GTACTCAGCGCCAGCATCG |
| ICAM1 | CAAGGCCTCAGTCAGTGTGA | CCTCTGGCTTCGTCAGAATC |
| IL8 | CCTTGGCAAAACTGCACCTT | CTGGCCGTGGCTCTCTTG |
| KDR | GTACATAGTTGTCGTTGTAGG | TCAATCCCCACATTTAGTTC |
| KLF2 | CGTTTGTAGATGACAATGA | AGAAGTTTCAGGTGACTGA |
| NRF2 | AAGGGCCTTAATTTGTACT | ATAAAAACGAACCAGGTAG |
| VCAM1 | ACTTGATGTTCAAGGAAGAG | TCCAGTTGAACATATCAAGC |
| VEGF | AGAAGGAGGAGGGCAGAATC | ACACAGGATGGCTTGAAGATG |
| vWF | TGTATCTAGAAACTGAGGCTG | CCTTCTTGGGTCATAAAGTC |
Cell characterization: monocyte-EC adhesion assay
THP1 (1 × 106 cells per mL) was incubated with CFSE (5 μM, Molecular Probes) for 15 min at 37 °C and washed three times in the culture medium to remove the unbound dye at the ratio of 1:2 for 30 min. iPSC-derived ECs were cultured in 24-well plates until confluence. The cells were then treated with 10 nM TNF-α (Peprotech), a pro-inflammatory cytokine, for 6 h. Next, the cells were washed 3 times in the culture medium and co-cultured with CSFE-labeled THP1 monocytes. Then, the cells were washed 3 times with PBS to remove unbound THP1 cells, the nuclei were stained with Hoechst (Molecular Probes), and the images were acquired using an InCell Analyser HCA System (GE Healthcare) and analyzed by the corresponding software. For non-treated and TNF-α-treated cells the percentage of THP1 cells was calculated relative to the total number of nuclei counted for each field. Each condition was performed in triplicate and a minimum of 9 fields were acquired for each well; on average 500 cells were counted per field.
Culture of vascular cells in flow conditions
A suspension of SMCs (derived from N-iPSCs or somatic human SMCs, 30 μL in MCDB 131 medium, in order to have 4.1 × 104 cells per cm2) was applied in each channel of an Ibidi plate (μ-Slide VI0,4 Luer, Ibidi) and allowed to flow inside by capillary force. After 1 h, each channel was filled with MCDB 131 medium. The cells were maintained in static conditions at 37 °C, and 5% CO2 in a humidified atmosphere for 12 h, after which a suspension of ECs (N-iPSC ECs or HUAECs, and 30 μL of EGM-2 medium, in order to have of 12.5 × 104 ECs per cm2) was applied on top of SMCs. After 1 h, each channel was filled with EGM-2 medium, and the cells were maintained at 37 °C, and 5% CO2 in a humidified atmosphere for 4 h in static conditions. After 4 h, the cells were perfused with EGM-2 medium at the physiological flow rate (20 dyn cm−2) using an Ibidi pump system.Cell characterization: heparan sulfate analyses
ECs co-cultured with SMCs in a microfluidic system for 5 days were characterized for the expression of heparan sulfate. Cells were stained with heparan sulfate (10E4 Epitope, USBiological) and the overall intensity of heparan sulfate of each image was quantified using ImageJ software. In a parallel experiment, cells were treated with heparinase III (Flavobacterium heparinum 0.5 U mL−1, Sigma) for 30 min and washed with EGM-2 medium. Cells treated with heparinase were then exposed to low density lipoprotein from human plasma complexed with DiI (DiI LDL, Invitrogen) at 20 μg mL−1 at 37 °C for 4 h. Cells were then washed and maintained with medium. Images were acquired using an InCell Analyser HCA system and analysed using the corresponding software.Nanomaterials library: preparation
The following nanomaterials have been used: zinc oxide (Product Number: 8410DL, SkySpring NanoMaterials), Agion® silver antimicrobial type AJ (Product Number: AJ10D, Sciessent LLC), silicon oxide (Product Number: 6807NM, SkySpring Nanomaterials, Inc.), silicon dioxide (Product Number: PL-SiOF, Plasmachem), titanium oxide (Product Number: 7910DL, SkySpring Nanomaterials, Inc.), Molday ION MI-750 (Product Number:CL-50Q01-6A-53, BioPAL), micromer®-greenF 25 nm-NH2 (Product Number: 29-01-251), micromer®-greenF 25 nm-COOH (Product Number: 29-02-251), micromer®-greenF, 100 nm-NH2 (Product Number: 29-01-102) and micromer®-greenF 100 nm-COOH (Product Number: 29-02-102, Micromod Partikeltechnologie GmbH). The nanomaterials including 1.4MS–Au NPs, 1.5GSH–Au NPs, 12GSH–Au NPs, 13.9MS–Au NPs and 15MS–Au NPs were provided by the Ulrich Simon group and the synthesis and characterization have been reported elsewhere.25,63,64 PLGA,20 PLGA–PS,20 CM–AuNPs,19 LL37–AuNPs,18 PEI:DS,21 PEI–DMNC,24 A1,22 A9,22 C11,22 E1,22 E2,22 P1C5,23 and P1C723 have been synthesized by us and the synthesis protocols were previously published. 5HA–Au NPs were obtained by ligand exchange of 1.4MS–Au NPs with thiol end-modified HA. 20HA–Au NPs were obtained by ligand exchange of citrate–Au NPs (10 nm) with thiol end-modified HA. Thiol end-modified HA was obtained by reductive amination. Briefly, HA (MW 8–15 kDa, 100 mg) and cystamine dihydrochloride (60 mg) were dissolved in 0.1 M borate buffer (10 mL, pH 8.5) with 0.4 M NaCl and stirred for 2 h. NaBH3CN was added to the solution at a final concentration of 200 mM and reacted at 40 °C for 5 days. The reaction mixture was incubated with 100 mM DTT for 12 h to introduce a free thiol group. The mixture was dialyzed (MWCO: 2 kDa) during 4 days (2 days against 100 mM of NaCl, 25% ethanol for 1 day and pure water for 1 day). The purified thiol end-modified HA was freeze-dried for 2 days and characterized by 1H NMR and Ellman's assay. The formation of HA–Au NPs was assessed by UV-vis spectra, TEM, elemental analysis and DLS.Toxicity of nanomaterials in iPSC-ECs cultured in 2D
N-iPSC ECs were seeded the day before the experiment at a density of 1 × 105 cells per cm2 into 96 or 384 well plates. Nanomaterials suspended in water at 1 mg mL−1 were added to cells at a concentration between 6 and 50 μg mL−1 for 24 h. In the control, the same amount of water without NPs was added to the cells. After 24 h incubation, cell viability was assessed by PrestoBlue® assay and apoptosis levels were monitored by propidium iodide (PI)/Hoechst 33342 staining. PrestoBlue assay is based on a resazurin-based solution that indicates the reducing power of living cells and therefore measures indirectly their number. Hoechst 33342, a blue-fluorescence dye (excitation/emission maxima at 350/461 nm, when bound to DNA), stains the condensed chromatin in apoptotic cells more brightly than the chromatin in normal cells and PI is a red-fluorescence dye (excitation/emission maxima ∼535/617 nm when bound to DNA), and permeant only to dead cells. For each measurement, the cell medium was removed, PI/Hoechst (10 μM) was added in EGM-2 medium and incubated for 10 min, after which the apoptosis levels were checked by microscopy by verifying the co-localization of the PI and Hoechst signals. Cells were then washed twice with PBS and a PrestoBlue solution (10% v/v, in EGM-2 medium) was added for 2 h at 37 °C, upon which the fluorescence was measured using a synergy H1 multi-mode reader (BioTek) at 590 nm with excitation at 560 nm.ICP-MS analyses
NP internalization was monitored by ICP-MS. In this case, the intracellular levels of Zn (in ZnO, PEI-DS, PEI-DMNC, A1, C11 and P1C5 NPs), Ag (in Ag NPs) or Au (in LL37–Au, CM–Au and 1.4MS–Au NPs) were measured after N-iPSC EC and HGPS-iPSC EC exposure to NPs for 24 h (with the exception of 1.4MS–Au NPs, which were only incubated for 4 h; longer incubation times would result in cell death) at a density of 1 × 105 cells per cm2 in 96 or 384-well plates. After incubation, NPs that were not internalized by the cells were washed (three times with PBS) and the cells were then lysed, collected and lyophilized. The samples were analysed by ICP-MS for the concentration of intracellular levels of Zn, Ag or Au, depending on the NP composition. To convert the concentration of each element into a concentration of NP, a suspension of NPs (not exposed to cells) was also quantified by ICP-MS. Finally, the concentration of NP per cell was calculated.Toxicity of nanomaterials in iPSC-ECs cultured in 3D
A 96 multiwell plate was coated with Matrigel (BD) (50 μL per well – Matrigel was thawed at 4 °C and both tips and plates were kept cold during the procedure) and placed at 37 °C for 30 min. Cells were seeded on top of the polymerized Matrigel at a density of 2 × 103 cells per well together with nanoparticles (12.5 μg mL−1) in EGM-2 medium (100 μL). After 24 h, bright field images were acquired in an InCell Analyser HCA System, and capillary-like networks analyzed using the Ibidi ACAS image software.Toxicity of nanomaterials in a blood vessel on a chip
The effect of NPs was evaluated under flow conditions. N-iPSC ECs and N-iPSC SMCs were co-cultured as described in the “Culture of vascular cells in flow conditions” section for 5 days in a microfluidic system. Cells were then washed and perfused with NPs at 50 μg mL−1 in EGM-2 medium at the physiological flow rate (20 dyn cm−2) for 24 h. Cell viability was assessed as described above for static conditions.Statistical analyses
An unpaired t test or one-way ANOVA analysis of variance with a Bonferroni post-test was performed for statistical tests. Results were considered statistically different when P < 0.05. Data are shown as mean ± SEM.Author contributions
Experiments were designed by LE, VF, PP, IH, HV, MG and LF. Nanoparticle preparations were performed by LE, VF, JB, AR and US. HGPS iPS were prepared by XN. Cellular experiments were performed by LE, PP and IH. Analysis of results was performed by LE, VF, PP, IH and LF. Zebrafish work was performed by LE, LC and LS. The manuscript was written by LE, VF and LF.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by FEDER through the Program COMPETE and by Portuguese fund through FCT in the context of the projects PTDC/SAU-TOX/121887/2010, POCI-01-0145-FEDER-029229, POCI-01-0145-FEDER-016390 and POCI-01-0145-FEDER-029414, as well as the European project ERAatUC (ref. 669088). PP wishes to thank FCT for a BD fellowship (SFRH/BD/71042/2010). The iMM Fish Facility was supported by CONGENTO LISBOA-01-0145-FEDER-022170, co-financed by FCT (Portugal) and Lisboa 2020, under the PORTUGAL 2020 agreement (European Regional Development Fund). The authors have no conflict of interest to disclose.References
- D. M. Smith, J. K. Simon and J. R. Baker, Jr., Nat. Rev. Immunol., 2013, 13, 592–605 CrossRef CAS.
- G. V. Lowry, A. Avellan and L. M. Gilbertson, Nat. Nanotechnol., 2019, 14, 517–522 CrossRef CAS.
- Z. Zhang, X. Li, J. Yin, Y. Xu, W. Fei, M. Xue, Q. Wang, J. Zhou and W. Guo, Nat. Nanotechnol., 2018, 13, 1109–1119 CrossRef CAS.
- S. J. Hawkins, L. A. Crompton, A. Sood, M. Saunders, N. T. Boyle, A. Buckley, A. M. Minogue, S. F. McComish, N. Jimenez-Moreno, O. Cordero-Llana, P. Stathakos, C. E. Gilmore, S. Kelly, J. D. Lane, C. P. Case and M. A. Caldwell, Nat. Nanotechnol., 2018, 13, 427–433 CrossRef CAS.
- L. Ye, K. T. Yong, L. Liu, I. Roy, R. Hu, J. Zhu, H. Cai, W. C. Law, J. Liu, K. Wang, J. Liu, Y. Liu, Y. Hu, X. Zhang, M. T. Swihart and P. N. Prasad, Nat. Nanotechnol., 2012, 7, 453–458 CrossRef CAS.
- K. Yamashita, Y. Yoshioka, K. Higashisaka, K. Mimura, Y. Morishita, M. Nozaki, T. Yoshida, T. Ogura, H. Nabeshi, K. Nagano, Y. Abe, H. Kamada, Y. Monobe, T. Imazawa, H. Aoshima, K. Shishido, Y. Kawai, T. Mayumi, S. Tsunoda, N. Itoh, T. Yoshikawa, I. Yanagihara, S. Saito and Y. Tsutsumi, Nat. Nanotechnol., 2011, 6, 321–328 CrossRef CAS.
- M. Faria, M. Bjornmalm, K. J. Thurecht, S. J. Kent, R. G. Parton, M. Kavallaris, A. P. R. Johnston, J. J. Gooding, S. R. Corrie, B. J. Boyd, P. Thordarson, A. K. Whittaker, M. M. Stevens, C. A. Prestidge, C. J. H. Porter, W. J. Parak, T. P. Davis, E. J. Crampin and F. Caruso, Nat. Nanotechnol., 2018, 13, 777–785 CrossRef CAS.
- B. Fadeel, L. Farcal, B. Hardy, S. Vazquez-Campos, D. Hristozov, A. Marcomini, I. Lynch, E. Valsami-Jones, H. Alenius and K. Savolainen, Nat. Nanotechnol., 2018, 13, 537–543 CrossRef CAS.
- M. I. Setyawati, C. Y. Tay, S. L. Chia, S. L. Goh, W. Fang, M. J. Neo, H. C. Chong, S. M. Tan, S. C. Loo, K. W. Ng, J. P. Xie, C. N. Ong, N. S. Tan and D. T. Leong, Nat. Commun., 2013, 4, 1673 CrossRef CAS.
- N. Bayat, V. R. Lopes, J. Scholermann, L. D. Jensen and S. Cristobal, Biomaterials, 2015, 63, 1–13 CrossRef CAS.
- A. P. Sommer, Circ. Res., 2010, 106, e10 CrossRef CAS.
- H. Vazao, S. Rosa, T. Barata, R. Costa, P. R. Pitrez, I. Honorio, M. R. de Vries, D. Papatsenko, R. Benedito, D. Saris, A. Khademhosseini, P. H. Quax, C. F. Pereira, N. Mercader, H. Fernandes and L. Ferreira, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3022–E3031 CrossRef CAS.
- S. Rosa, C. Praca, P. R. Pitrez, P. J. Gouveia, X. L. Aranguren, L. Ricotti and L. S. Ferreira, Sci. Rep., 2019, 9, 3826 CrossRef CAS.
- J. Ribas, Y. S. Zhang, P. R. Pitrez, J. Leijten, M. Miscuglio, J. Rouwkema, M. R. Dokmeci, X. Nissan, L. Ferreira and A. Khademhosseini, Small, 2017, 13, 1603737 CrossRef.
- X. Nissan, S. Blondel, C. Navarro, Y. Maury, C. Denis, M. Girard, C. Martinat, A. De Sandre-Giovannoli, N. Levy and M. Peschanski, Cell Rep., 2012, 2, 1–9 CrossRef CAS.
- A. De Sandre-Giovannoli, R. Bernard, P. Cau, C. Navarro, J. Amiel, I. Boccaccio, S. Lyonnet, C. L. Stewart, A. Munnich, M. Le Merrer and N. Levy, Science, 2003, 300, 2055 CrossRef CAS.
- K. Cao, C. D. Blair, D. A. Faddah, J. E. Kieckhaefer, M. Olive, M. R. Erdos, E. G. Nabel and F. S. Collins, J. Clin. Invest., 2011, 121, 2833–2844 CrossRef CAS.
- M. Comune, A. Rai, K. K. Chereddy, S. Pinto, S. Aday, A. F. Ferreira, A. Zonari, J. Blersch, R. Cunha, R. Rodrigues, J. Lerma, P. N. Simoes, V. Preat and L. Ferreira, J. Controlled Release, 2017, 262, 58–71 CrossRef CAS.
- A. Rai, S. Pinto, T. R. Velho, A. F. Ferreira, C. Moita, U. Trivedi, M. Evangelista, M. Comune, K. P. Rumbaugh, P. N. Simoes, L. Moita and L. Ferreira, Biomaterials, 2016, 85, 99–110 CrossRef CAS.
- R. S. Gomes, R. P. das Neves, L. Cochlin, A. Lima, R. Carvalho, P. Korpisalo, G. Dragneva, M. Turunen, T. Liimatainen, K. Clarke, S. Yla-Herttuala, C. Carr and L. Ferreira, ACS Nano, 2013, 7, 3362–3372 CrossRef CAS.
- J. Maia, T. Santos, S. Aday, F. Agasse, L. Cortes, J. O. Malva, L. Bernardino and L. Ferreira, ACS Nano, 2011, 5, 97–106 CrossRef CAS.
- J. Blersch, V. Francisco, C. Rebelo, A. Jimenez-Balsa, H. Antunes, S. Pinto, S. Simoes, A. Rai and L. Ferreira, Nanoscale, 2020, 12, 9935–9942 RSC.
- J. Blersch, V. Francisco, C. Rebelo, A. Jimenez-Balsa, H. Antunes, C. Gonzato, S. Pinto, S. Simoes, K. Liedl, K. Haupt and L. Ferreira, Angew. Chem., Int. Ed., 2020, 59, 1985–1991 CrossRef CAS.
- C. Boto, E. Quartin, Y. Cai, A. Martin-Lorenzo, M. B. G. Cenador, S. Pinto, R. Gupta, T. Enver, I. Sanchez-Garcia, D. Hong, R. Pires das Neves and L. Ferreira, Nat. Commun., 2017, 8, 15204 CrossRef.
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941–1949 CrossRef CAS.
- N. D. Lawson and B. M. Weinstein, Dev. Biol., 2002, 248, 307–318 CrossRef CAS.
- S. Isogai, M. Horiguchi and B. M. Weinstein, Dev. Biol., 2001, 230, 278–301 CrossRef CAS.
- B. L. Roman, V. N. Pham, N. D. Lawson, M. Kulik, S. Childs, A. C. Lekven, D. M. Garrity, R. T. Moon, M. C. Fishman, R. J. Lechleider and B. M. Weinstein, Development, 2002, 129, 3009–3019 CAS.
- A. V. Gore, K. Monzo, Y. R. Cha, W. Pan and B. M. Weinstein, Cold Spring Harbor Perspect. Med., 2012, 2, a006684 Search PubMed.
- K. T. Kim and R. L. Tanguay, Environ. Health Toxicol., 2014, 29, e2014021 CrossRef.
- K. J. Ong, T. J. MacCormack, R. J. Clark, J. D. Ede, V. A. Ortega, L. C. Felix, M. K. Dang, G. Ma, H. Fenniri, J. G. Veinot and G. G. Goss, PLoS One, 2014, 9, e90650 CrossRef.
- Y. Pan, A. Leifert, D. Ruau, S. Neuss, J. Bornemann, G. Schmid, W. Brandau, U. Simon and W. Jahnen-Dechent, Small, 2009, 5, 2067–2076 CrossRef CAS.
- A. Gojova, B. Guo, R. S. Kota, J. C. Rutledge, I. M. Kennedy and A. I. Barakat, Environ. Health Perspect., 2007, 115, 403–409 CrossRef CAS.
- S. Liang, K. Sun, Y. Wang, S. Dong, C. Wang, L. Liu and Y. Wu, Chem.-Biol. Interact., 2016, 258, 40–51 CrossRef CAS.
- J. Sun, S. Wang, D. Zhao, F. H. Hun, L. Weng and H. Liu, Cell Biol. Toxicol., 2011, 27, 333–342 CrossRef CAS.
- Y. Gong, Y. Ji, F. Liu, J. Li and Y. Cao, J. Appl. Toxicol., 2017, 37, 895–901 CrossRef CAS.
- Y. Gu, S. Cheng, G. Chen, Y. Shen, X. Li, Q. Jiang, J. Li and Y. Cao, Toxicol. Mech. Methods, 2017, 27, 191–200 CrossRef CAS.
- M. Filipova, O. K. Elhelu, S. H. De Paoli, Z. Fremuntova, T. Mosko, D. Cmarko, J. Simak and K. Holada, PLoS One, 2018, 13, e0206557 CrossRef.
- P. Nowak-Sliwinska, K. Alitalo, E. Allen, A. Anisimov, A. C. Aplin, R. Auerbach, H. G. Augustin, D. O. Bates, J. R. van Beijnum, R. H. F. Bender, G. Bergers, A. Bikfalvi, J. Bischoff, B. C. Bock, P. C. Brooks, F. Bussolino, B. Cakir, P. Carmeliet, D. Castranova, A. M. Cimpean, O. Cleaver, G. Coukos, G. E. Davis, M. De Palma, A. Dimberg, R. P. M. Dings, V. Djonov, A. C. Dudley, N. P. Dufton, S. M. Fendt, N. Ferrara, M. Fruttiger, D. Fukumura, B. Ghesquiere, Y. Gong, R. J. Griffin, A. L. Harris, C. C. W. Hughes, N. W. Hultgren, M. L. Iruela-Arispe, M. Irving, R. K. Jain, R. Kalluri, J. Kalucka, R. S. Kerbel, J. Kitajewski, I. Klaassen, H. K. Kleinmann, P. Koolwijk, E. Kuczynski, B. R. Kwak, K. Marien, J. M. Melero-Martin, L. L. Munn, R. F. Nicosia, A. Noel, J. Nurro, A. K. Olsson, T. V. Petrova, K. Pietras, R. Pili, J. W. Pollard, M. J. Post, P. H. A. Quax, G. A. Rabinovich, M. Raica, A. M. Randi, D. Ribatti, C. Ruegg, R. O. Schlingemann, S. Schulte-Merker, L. E. H. Smith, J. W. Song, S. A. Stacker, J. Stalin, A. N. Stratman, M. Van de Velde, V. W. M. van Hinsbergh, P. B. Vermeulen, J. Waltenberger, B. M. Weinstein, H. Xin, B. Yetkin-Arik, S. Yla-Herttuala, M. C. Yoder and A. W. Griffioen, Angiogenesis, 2018, 21, 425–532 CrossRef.
- B. Yap, G. Garcia-Cardena and M. A. Gimbrone, FASEB J., 2008, 22, 471.11 Search PubMed.
- N. Kubben, W. Zhang, L. Wang, T. C. Voss, J. Yang, J. Qu, G. H. Liu and T. Misteli, Cell, 2016, 165, 1361–1374 CrossRef CAS.
- J. M. Tarbell and L. M. Cancel, J. Intern. Med., 2016, 280, 97–113 CrossRef CAS.
- J. Han, B. J. Zern, V. V. Shuvaev, P. F. Davies, S. Muro and V. Muzykantov, ACS Nano, 2012, 6, 8824–8836 CrossRef CAS.
- P. R. Pitrez, L. Estronca, L. M. Monteiro, G. Colell, H. Vazão, D. Santinha, K. Harhouri, D. Thornton, C. Navarro, A.-L. Egesipe, T. Carvalho, R. L. Dos Santos, N. Lévy, J. C. Smith, J. P. de Magalhães, A. Ori, A. Bernardo, A. De Sandre-Giovannoli, X. Nissan, A. Rosell and L. Ferreira, Nat. Commun., 2020, 11, 4110 CrossRef CAS.
- H. Vazao, R. P. das Neves, M. Graos and L. Ferreira, PLoS One, 2011, 6, e17771 CrossRef CAS.
- S. Y. Shaw, E. C. Westly, M. J. Pittet, A. Subramanian, S. L. Schreiber and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7387–7392 CrossRef CAS.
- G. Lee, C. N. Ramirez, H. Kim, N. Zeltner, B. Liu, C. Radu, B. Bhinder, Y. J. Kim, I. Y. Choi, B. Mukherjee-Clavin, H. Djaballah and L. Studer, Nat. Biotechnol., 2012, 30, 1244–1248 CrossRef CAS.
- S. Blondel, A. L. Egesipe, P. Picardi, A. L. Jaskowiak, M. Notarnicola, J. Ragot, J. Tournois, A. Le Corf, B. Brinon, P. Poydenot, P. Georges, C. Navarro, P. R. Pitrez, L. Ferreira, G. Bollot, C. Bauvais, D. Laustriat, A. Mejat, A. De Sandre-Giovannoli, N. Levy, M. Bifulco, M. Peschanski and X. Nissan, Cell Death Dis., 2016, 7, e2105 CrossRef CAS.
- W. J. Trickler, S. M. Lantz, R. C. Murdock, A. M. Schrand, B. L. Robinson, G. D. Newport, J. J. Schlager, S. J. Oldenburg, M. G. Paule, W. Slikker, Jr., S. M. Hussain and S. F. Ali, Toxicol. Sci., 2010, 118, 160–170 CrossRef CAS.
- S. Gurunathan, K. J. Lee, K. Kalishwaralal, S. Sheikpranbabu, R. Vaidyanathan and S. H. Eom, Biomaterials, 2009, 30, 6341–6350 CrossRef CAS.
- C. Gonzalez, H. Rosas-Hernandez, M. A. Ramirez-Lee, S. Salazar-Garcia and S. F. Ali, Arch. Toxicol., 2016, 90, 493–511 CrossRef CAS.
- K. Okahara, B. Sun and J. Kambayashi, Arterioscler., Thromb., Vasc. Biol., 1998, 18, 1922–1926 CrossRef CAS.
- J. Han, V. V. Shuvaev, P. F. Davies, D. M. Eckmann, S. Muro and V. R. Muzykantov, J. Controlled Release, 2015, 210, 39–47 CrossRef CAS.
- M. Balcells, J. Martorell, C. Olive, M. Santacana, V. Chitalia, A. A. Cardoso and E. R. Edelman, Circulation, 2010, 121, 2192–2199 CrossRef.
- T. Ziegler, R. W. Alexander and R. M. Nerem, Ann. Biomed. Eng., 1995, 23, 216–225 CrossRef CAS.
- B. Uhl, S. Hirn, R. Immler, K. Mildner, L. Mockl, M. Sperandio, C. Brauchle, C. A. Reichel, D. Zeuschner and F. Krombach, ACS Nano, 2017, 11, 1498–1508 CrossRef CAS.
- M. J. Cheng, R. Kumar, S. Sridhar, T. J. Webster and E. E. Ebong, Int. J. Nanomed., 2016, 11, 3305–3315 CrossRef CAS.
- J. Gao, C. T. Mahapatra, C. D. Mapes, M. Khlebnikova, A. Wei and M. S. Sepulveda, Nanotoxicology, 2016, 10, 1363–1372 CrossRef CAS.
- C. Bai and M. Tang, J. Appl. Toxicol., 2020, 40, 37–63 CrossRef CAS.
- S. Vranic, Y. Shimada, S. Ichihara, M. Kimata, W. Wu, T. Tanaka, S. Boland, L. Tran and G. Ichihara, Int. J. Mol. Sci., 2019, 20, 882 CrossRef CAS.
- J. Wang, L. Zhang, F. Peng, X. Shi and D. Tai Leong, Chem. Mater., 2018, 30, 9 Search PubMed.
- A. Haase, R. Olmer, K. Schwanke, S. Wunderlich, S. Merkert, C. Hess, R. Zweigerdt, I. Gruh, J. Meyer, S. Wagner, L. S. Maier, D. W. Han, S. Glage, K. Miller, P. Fischer, H. R. Scholer and U. Martin, Cell Stem Cell, 2009, 5, 434–441 CrossRef CAS.
- A. Leifert, Y. Pan, A. Kinkeldey, F. Schiefer, J. Setzler, O. Scheel, H. Lichtenbeld, G. Schmid, W. Wenzel, W. Jahnen-Dechent and U. Simon, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8004–8009 CrossRef CAS.
- J. Broda, J. Setzler, A. Leifert, J. Steitz, R. Benz, U. Simon and W. Wenzel, Nanomedicine, 2016, 12, 1409–1419 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00550a |
| This journal is © The Royal Society of Chemistry 2021 |