
rGO–diaminobutane surfaces with optimized N doping and hydrodynamics as dual proton–electron conductors and carbon photocatalysts
Mohammad Razaul
Karim
 *,
Mohammed M.
Rahman
* and
Abdullah M.
Asiri
*,
Mohammed M.
Rahman
* and
Abdullah M.
Asiri
Department of Chemistry, King Abdulaziz University, Jeddah 21589, Saudi Arabia. E-mail: krazaul@yahoo.com; mmrahman@kau.edu.sa
First published on 17th November 2020
Abstract
The hybrids of reduced graphene oxide (rGO) and diaminoalkane (DAA) including rGO–DAP, rGO–DAB, and rGO–DAPN (DAP = 1,4-diaminopropane, DAB = 1,4-diaminobutane and DAPN = 1,4-diaminopantane) were synthesized by solventless vapor phase reduction of the respective GO-hybrids, namely, GO–DAP, GO–DAB, and GO–DAPN. Within this rGO–DAA series, the optimization of N-doping, electronic conductivity (EC), and water dynamics (evaluated by the measurement of the proton conductivity value) is observed in rGO–DAB. The EC of rGO–DAB is 1.72 μA V−1, which is higher than the EC of reported rGO–alkylamine (rGO–AA) hybrids. The PC of rGO–DAB is ≈10−3–10−4 S cm−1, which is slightly less than that of pristine GO. The extent of N doping at the graphitic carbon skeleton is 6.7%, which is higher than that of all the reported rGO–AA hybrids. The rGO–DAB could afford photocatalytic water splitting (WS) and dye degradation (DD). The efficiency toward WS is 18.48 μmol h−1 g−1, whereas the efficiency toward the degradation of methylene blue (MB) revealed a kLH value of 0.015 min−1. Both these values are higher than those of the reported rGO–AA hybrids. The DAP precursor contributes to N doping in rGO. The hybrids thus function as bandgap semiconductors and aid light-harvesting photocatalysis. The high EC and super-fast hydrodynamics supported the electron mediation and propagation of water/MB molecules to the N-doped center during reactions. Therefore, in addition to the generation of electron/hole pairs, the materials can trigger the mechanistic pathway of photocatalysis. These findings suggest that the catalytic activity of rGO-based materials can be improved not only by engineering the surface and bandgap property but also by improving the electronic conductivity and hydrodynamics within the staked layers of rGO.
Introduction
The ionic conductivity of materials, in particular the proton conductivity (PC), is an essential property for devising solid electrolytes for fuel cells, sensors, steam electrolysis cells, and so on.1 Graphene oxide (GO)-based hybrid materials also ensure numerous device applications.2–4 In recent years, we have found that the photocatalytic activity of layered GO-based hybrids in the aqueous system have a close relationship with their PC.5 The inherent fact is that the high surface area of GO-based photocatalysts can be completely involved in catalysis only if the water/reactant molecules can move within the interlayer space (IS) of the materials. Otherwise, even the surface area of a GO or reduced graphene oxide (rGO) hybrid is very high, and the staked material functions as a bulk particle with low catalytic efficiency.6 Thus, GO/rGO-based photocatalytic materials must allow water dynamics within the layers. The PC value signifies the extent of such water dynamics at the IS.7 Thus, water permission justified by measuring the PC values of the hybrids by solid-state impedance analysis at different temperatures and relative humidity conditions is considered for understanding the mechanistic pathway of photocatalysis. However, devising GO-based hybrid photocatalysts is stipulated by some other necessary conditions, including the opening of graphene's bandgap to endow the light-harvesting property.8 The N doping in rGO is a common strategy for such kind and we observed that intercalating alkylamine (AA) can contribute both N doping and flexible IS in rGO.5The present work aims to develop organic photocatalysts by forming hybrids with rGO and diaminoalkane (DAA) precursors. The rGO component was selected for its excellent two-dimensional surface property, electronic conductivity and scaffolding capability. The DAA precursor was selected for its polar amino terminal, N-doping capability in rGO, and possible functions as a better spacer group. We expected that due to the presence of diamino groups, compared with AA, the DAA precursor will result in higher N-doping and flexibility of IS in the hybrids. Both these facts are reported conditions for devising rGO-based organic photocatalysts. In practice, we found that the high PC and electronic conductivity (EC) of rGO–DAA hybrids revealed excellent photocatalytic activity towards both the water splitting (WS) and dye degradation (DD) processes.
Proton conductors transport protons instead of hydrogen atoms to the oxygen compartment in a hydrogen fuel cell, and thus, the cell operates without the risk of explosion.9 The commonest proton conductors employed in devices are Nafion, phosphoric acid, MOFs and sulfonic acid derivatives.10–12 Nowadays, carbon-based proton conductors for battery applications attract worldwide attention due to their cheap and non-toxic nature and easy processability.13 Majorly, GO-based materials belong to this class. GO displays proton conductivity for its flat-shaped structure and hydrophilic oxygenous functional groups.14 Intercalating some hydrophilic materials including dicarboxylic acids, amines, and sulfonic acids can amplify the PC of GO.15 In GO or rGO, the intercalation of other ingredients is considered to develop other functionalities including magnetism, semi-conductivity, catalysis, electrolysis, and sensing performance. However, the PC of rGO lowered for some inorganic intercalation, as in such cases the hydrophilic sites are blocked. In rGO, the generation of sp2 carbon sites increases the EC. The EC of rGO is significant to devise catalysts, electrodes, sensors and so on. The rGO can transport or mediate electrons in addition to its function as a template for scaffolding active functional ingredients. Materials showing dual PC and EC are highly fascinating as the two conduction phenomena depending on two different carriers usually perturb each other and are rarely observed.
The PC of any carbon material is assisted by the presence of hydrophilic sites and C–C sp3 sites. The hydrophilic sites can adsorb water films, which allows for the proton transference by the reformation of hydrogen bonds. The PC hence can be improved by increasing hydrophilicity. In reverse, the EC is supported by the hydrophobic domain of the materials.16 In GO, the hydrophobic domain is associated with π networked C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C sp2 sites. Thus, GO exhibits PC and rGO exhibits EC.17 Thus, the control over GO's PC and EC can be achieved by regulating the extent of GO reduction, single/double-bonded carbon domain generation, oxygenated functionalization or intercalation of a second material.18 In GO/rGO hybrids, the nature of guest materials that are inserted at the GO interlayer is a major issue. As GO is an insulator, in many applications, reduction of GO is needed to recover the EC. However, the transformation of GO into rGO destroys or blocks the water adsorbing sites.19 Moreover, the difficulties toward the hydronium ion's bond reformation process arise due to the shrinking of GO's IS during reduction. Previously, the AA precursors were intercalated in rGO with subsequent solid-state reduction. The resulting rGO–AA hybrids were sustained with both the PC and EC, which confirmed the formation of N-doped rGO. Some rGO–AA hybrids were successfully utilized for the photocatalytic degradation of water and methylene blue (MB) dye.5,6 However, there exists the possibility of increasing the catalytic efficiency by improving the water dynamics at the IS and increasing the extent of N doping. Based on these issues, in his work, we considered the insertion of DAA precursors including 1,3-diaminopropane (DAP), 1,4-diaminobutane (DAB) and 1,5-diaminopentane (DAPN) at the rGO interlayer to obtain rGO–DAP, rGO–DAB and rGO–DAPN hybrids (Fig. 1). We considered the doping of N atoms at the graphitic carbon skeleton to develop organic-only photocatalysts. Originally, G is a zero bandgap conductor. rGO possesses almost similar conduction properties to G. N doping can open G's bandgap. The bandgap opening can adopt rGO for the light-harvesting photocatalytic activity. Our hypothesis also includes that the double amine groups will increase the N doping, hydrophilicity, and stability of the hybrids. Hence, it might function for increasing the PC to some moderate values. Besides, the chain length possessing 3–5 carbon atoms will barely damage the polarity due to the presence of two amine groups. Moreover, the bulky size of diaminoalkane would be capable to generate enough IS for supporting hydrodynamics-oriented catalytic performance.
C sp2 sites. Thus, GO exhibits PC and rGO exhibits EC.17 Thus, the control over GO's PC and EC can be achieved by regulating the extent of GO reduction, single/double-bonded carbon domain generation, oxygenated functionalization or intercalation of a second material.18 In GO/rGO hybrids, the nature of guest materials that are inserted at the GO interlayer is a major issue. As GO is an insulator, in many applications, reduction of GO is needed to recover the EC. However, the transformation of GO into rGO destroys or blocks the water adsorbing sites.19 Moreover, the difficulties toward the hydronium ion's bond reformation process arise due to the shrinking of GO's IS during reduction. Previously, the AA precursors were intercalated in rGO with subsequent solid-state reduction. The resulting rGO–AA hybrids were sustained with both the PC and EC, which confirmed the formation of N-doped rGO. Some rGO–AA hybrids were successfully utilized for the photocatalytic degradation of water and methylene blue (MB) dye.5,6 However, there exists the possibility of increasing the catalytic efficiency by improving the water dynamics at the IS and increasing the extent of N doping. Based on these issues, in his work, we considered the insertion of DAA precursors including 1,3-diaminopropane (DAP), 1,4-diaminobutane (DAB) and 1,5-diaminopentane (DAPN) at the rGO interlayer to obtain rGO–DAP, rGO–DAB and rGO–DAPN hybrids (Fig. 1). We considered the doping of N atoms at the graphitic carbon skeleton to develop organic-only photocatalysts. Originally, G is a zero bandgap conductor. rGO possesses almost similar conduction properties to G. N doping can open G's bandgap. The bandgap opening can adopt rGO for the light-harvesting photocatalytic activity. Our hypothesis also includes that the double amine groups will increase the N doping, hydrophilicity, and stability of the hybrids. Hence, it might function for increasing the PC to some moderate values. Besides, the chain length possessing 3–5 carbon atoms will barely damage the polarity due to the presence of two amine groups. Moreover, the bulky size of diaminoalkane would be capable to generate enough IS for supporting hydrodynamics-oriented catalytic performance.
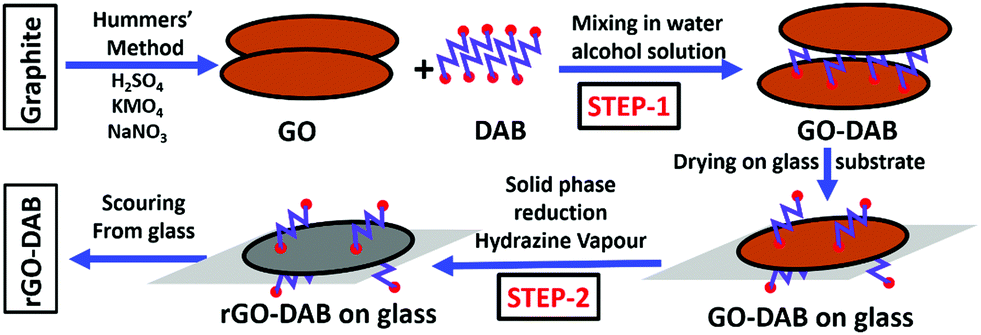 | ||
| Fig. 1 Scheme for the synthesis of rGO–DAA hybrids. Two major steps are involved in the synthetic route. In step 1, GO synthesized from graphite powder by Hummers’ method is mixed with DAB in a water–alcohol mixture. In step 2, the GO–DAB film on the glass sheet is exposed to hydrazine vapor in a closed flask to obtain rGO–DAB. Finally, black rGO–DAB powder is scoured from the glass substrate. | ||
In practice, rGO–DAB supported both the high PC and EC. The solid-state electrochemical investigation shows that compared with GO, though the PC of rGO–DAB is slightly less, it is almost double the PC values that we have previously observed in IMBA–rGO hybrids. The EC of rGO–DAB was increased almost 106 times, which is also higher than that for the previously devised rGO–AA hybrids. The material thus exhibited excellent photocatalytic activity toward WS and MB degradation. Though the efficiency of WS and DD is much lower than that of inorganic hybrids, it is significantly higher than that of pristine rGO and previously reported rGO–AA organic hybrids.
Experimental procedure
All the materials were purchased from Wako Pure Chemicals, Japan.Choice of material
rGO was selected for its function as a conductive template. The DAB precursor was selected for its amine functional groups and butyl alkyl chain. The amine functional groups can afford to open the graphene's bandgap by N doping in rGO. The double alkyl group of DAB was expected to function as a spacer group and scaffold two neighboring rGO nanosheets for resulting in an optimum ID. Moreover, DAB being polar was expected to recover the hydrophilic property in the hybrid. DAP and DAPN were also selected based on a similar hypothesis. The scheme for the synthesis of the hybrids is presented in Fig. 1.Preparation of GO
Modified Hummers' method was followed to synthesize GO from graphite powder.20 Graphite powder (1.5 g), H2SO4 (75 ml, 97%), and finely meshed NaNO3 (1.5 g) were mixed with continuous stirring in an ice bath for 30 min. Then, 4.5 g KMnO4 powder was added with stirring to keep the temperature below 20 °C for 1.5 h. The mixture was heated to 35 °C for 40 min. Following this, 270 ml water was added slowly increasing the temperature to 95 °C for 2 h and 600 ml water and 18 ml H2O2 solution (30%) were added. The mixture was centrifuged at 4500 rpm for 15 min, and the residue was washed with 5% HCl solution (×1) and water (×3). The precipitate was dried at 60 °C for 20 h.Preparation of rGO–diaminoalkane
First, 20 mg GO was dispersed in 20 ml H2O + 10 ml EtOH by sonication for 2 h. The solution was centrifuged at 4500 rpm for 1.5 h to discard heavier particles. The GO–DAB solution was prepared by dissolving 8.8 mg DAB in 20 ml ethanol. This DAB–alcohol solution was added to the as-prepared GO solution with vigorous stirring for 12 h under the ambient condition to obtain a GO–DAB mixture. Part of the GO–DAB mixture was then poured on the surface of a 5 × 9 cm glass sheet and dried under the ambient condition. This process was repeated several times to dry the entire GO–DAB mixture on the glass. Thus, the glass-supported GO–DAB film was generated. The glass sheet was then inclined inside the glass beaker, the bottom of which was filled partly with a hydrazine solution. The beaker was then covered by a glass sheet for 5 days and was kept under the ambient condition. In 5 days, the grey GO–DAB film was converted into a black rGO–DAB film. The film was scratched out from the glass surface and cleaned with H2O and CH2Cl2 to remove the unreacted materials. The black powdered form of rGO–DAB was thus obtained. rGO–DAP and rGO–DAPN were prepared similarly by taking 7.4 and 10.2 g of DAP and DAP, respectively. IMBA–rGO was prepared according to the process described in our previous report.5Characterization
The morphology of the sample's surface was analyzed by field-emission SEM (Hitachi High-Tech, SU-8000). Raman spectroscopy was studied using an NRS-3100 micro Raman spectrometer of JASCO, Japan, functioning with a 532 nm excitation source at room temperature. A Rigaku X-ray diffractometer RAD-2A equipped with a 2.0 kW Cu Kα X-ray source was employed for powder X-ray diffraction (PXRD) studies. The samples for PXRD measurements were collected from scratching the surface of pellets incubated for 30 min under 40 Pascal pressure. The chemical structure was justified using an XPS instrument of Thermo Scientific, Sigma Probe. A monochromatized X-ray source (Al Kα, hν = 1486.6 eV) and a discharge source (He I, hν = 21.2 eV) were employed. The systems were ensured with vacuums better than 10−7 Pa.Conductivity measurements
According to our previous report, proton conductivities of pellet samples were measured by a four-probe impedance/gain phase analyzing system using a Solartron 1260/1296 in the frequency range from 1 to 106 Hz.21 First, 100 mg GO sample was mashed into a powder and mechanically pressed under 40 pascal for 30 min to obtain coin-shaped pellets. Both faces of the pellets were connected to gold wires (50 μm diameter) using gold paste. Much care was taken to avoid the connection between the two faces. The PC values (σ) were calculated as follows: σ = d/RA, where d is the thickness, R is the resistance obtained from the radius of the semicircular curve of the Nyquist plot and A is the area of the surface of the sample pellet's face. ‘d’ and ‘A’ for all the samples had fixed values as 0.1 cm and 1 cm2, respectively. In Nyquist plots, the semicircular curves are fitted from the real (Z′) and imaginary parts (Z′′) of the Cole–Cole plot of impedance analysis. A series of measurements were accomplished by modulating the temperature and relative humidity (RH). The RH and temperature were controlled using an incubator (SH-221, ESPEC). A 1 h incubation time was maintained before every measurement. The electronic conductivities of the pellets were measured using a two-electrode system (Ivium Compactstat), where the electrodes are placed 2 cm apart.Photocatalytic performance
Photocatalytic H2 gas production by water splitting was accomplished in a 120 ml quartz pot. First, 10 mg of the catalyst in the pot was mixed with 120 ml ultrapure water followed by N2 bubbling for 25 min. Then, the solution was stirred continuously and irradiated using a Xe lamp (500 W) for 36 h. The H2 gas produced was estimated by gas chromatography (Shimadzu, GC-8A) assembled directly with the reactor. The data were collected once every 3 h. The photocatalytic performance of the samples toward the degradation of MB was investigated by monitoring the degree of dye degradation with the time of light irradiation. The light source for the experiment was a 500 W Tungsten filament lamp. In a 200 ml conical flask (Pyrex glass), 60 mg sample was dispersed in 120 ml 1 × l0−5 M MB solution. Before running the experiment, the flask was placed in a dark place (with sonication) for 35 min to avail the adsorption–desorption equilibrium state. Then, the solution was irradiated for 280 min. The sample solution from the reactor was collected in every 40 min. The catalytic particle was removed by centrifugation and filtration, while the concentration of MB in the solution was monitored by observing the absorbance (at 664 nm). A PerkinElmer (LAMBDA-35) UV-vis spectrophotometer was employed for this purpose.Results and discussion
Fig. 2(a–c) presents the optical photographs of the change in the glass-supported film color. The brown GO–DAB solution (Fig. 2a) on glass changes to grey after drying (Fig. 2b). After incubation for five days in hydrazine vapor, the rGO–DAB film finally becomes black (Fig. 2c). The SEM image of rGO–DAB presented in Fig. 2d and e depicts the layered structure of the hybrid. In Fig. 2d, the folds indicate that the sheet-like nature sustains even after reduction. The cross-sectional image (Fig. 2e) shows the deposition of the hybrids in layers. The other samples displayed an almost similar morphology. The IS of the materials was justified by PXRD analysis. Fig. 2f represents the IS of graphite, GO, rGO, and rGO–DAB. Graphite shows a typical semi-crystalline structure. The GO displays a peak at 2θ ≈ 11.08, which equals an IS of 7.98 Å.22 In rGO, the IS decreased to 3.96 Å (2θ = 22.43°). The IS of rGO–DAB has an intermediate value of 5.13 Å (17.26°). The position of PXRD peaks for rGO–DAPN, rGO–DAB, and rGO–DAP are 18.37, 18.45, and 21.22° with the corresponding IS values as 4.82, 4.81, and 4.18 Å, respectively. The quantification of sp3 or sp2 domain was accomplished by Raman spectroscopy. In Fig. 2g, the Raman spectra of GO, graphite, rGO–DAP, rGO–DAB and rGO–DAPN exhibit typical Raman shifts and peaks (D-band and G-band) for carbon materials. The D band near 1350 cm−1 associated with the breathing mode appears for the A1g symmetry in graphite. The G band around 1580 cm−1 appears due to the bond stretching motion of carbon atom's in-plane E2g pairs.23 Graphite shows its G band at ∼1577 cm−1, which shifts to ∼1590 cm−1 after oxidation to GO. This G band shifts to ∼1594, 1590, and 1601 cm−1 in rGO–DAP, rGO–DAB, and rGO–DAPN, respectively. This shifting of the G band position in the high-wavenumber region indicates the change in the electronic structure of the hybrid. The ID/IG value, which is the ratio of D and G band intensity, was found to be 0.655 for graphite. This value changes to 0.934, 0.917, 0.876, and 0.894 in GO, rGO–DAP, rGO–DAB, and rGO–DAPN, respectively. The ID/IG value signifies the ratio of sp2 and sp3 carbon atoms. Therefore, GO materials having a higher EC usually have a lower ID/IG value. For the oxidation of G into GO, some epoxy sites are generated accompanying the conversion of graphitic sp2 CC bonds into sp3 C–C. The conductive π-network in G is thus broken. Then, N-doping resulting in some resonance between the electron pairs of N atoms and π electrons of GO increases the π-networks. Hence, the EC is recovered in rGO–DAB.24
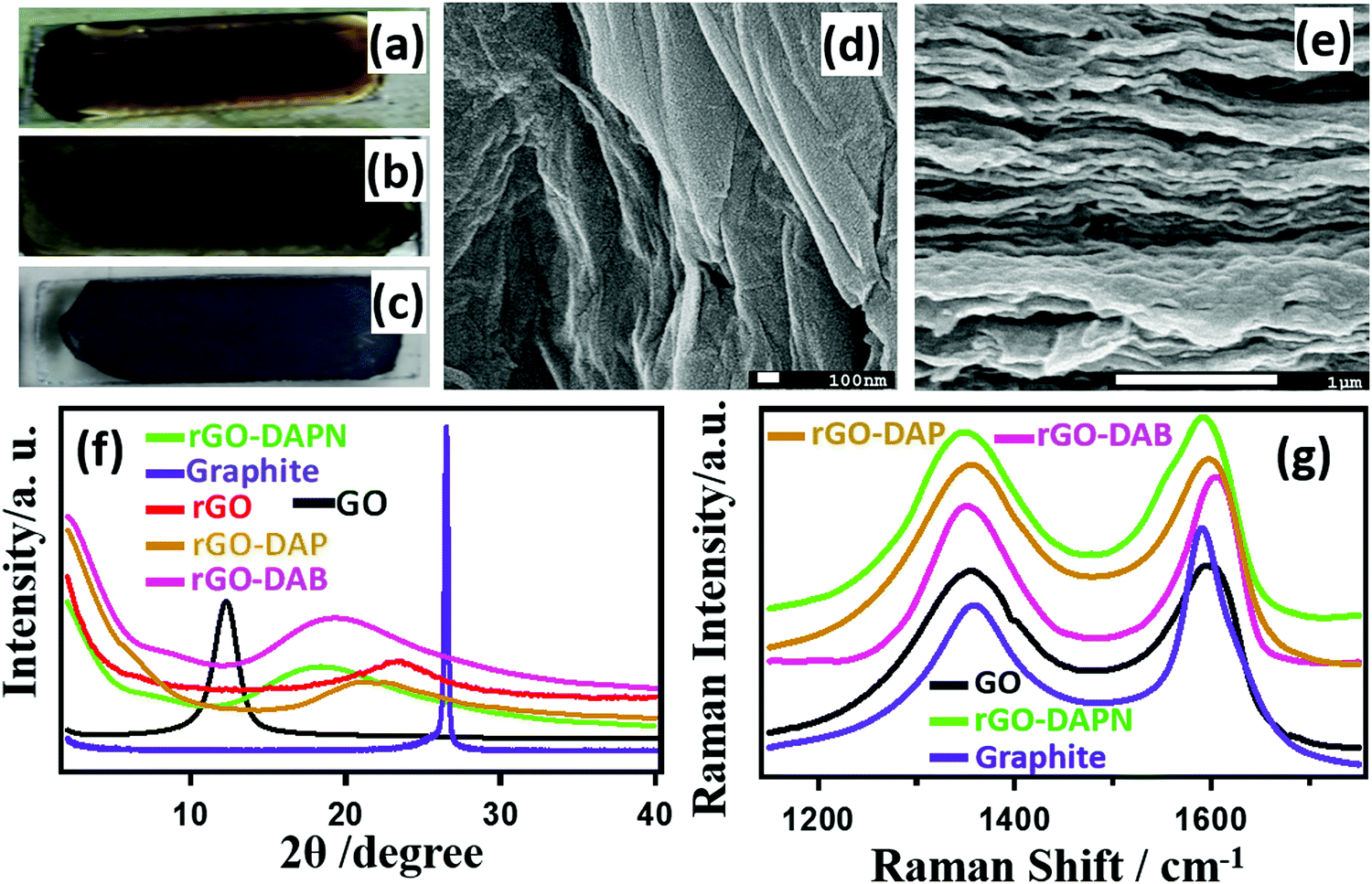 | ||
| Fig. 2 Characterization of reduced rGO–DAB hybrid. Optical images of glass-supported GO–DAB solution (a), GO–DAB film (b), and rGO–DAB film (c). SEM image of the rGO–DAB film surface (d) and cross-section (e). PXRD patterns of rGO–DAB and other samples (f). Raman spectra of rGO–DAB and other samples (g). | ||
The nature of functional groups in the materials was studied by IR and XPS analysis. The IR spectra in Fig. 3a indicate the formation of rGO–DAB hybrids. The presence of amine groups in rGO–DAB matches with the previous report.25 The IR peaks at 3100–3300 cm−1, 3300–3500 cm−1 and 1080–1360 cm−1 arise from N–H, O–H and C–N bonds. GO exhibit typical IR spectra. In rGO–DAB, peaks for GO's oxygenated sites are decreased. Peaks at 1180, 1640 and 1730 cm−1 arise from the stretching vibration of –C–O–C–, C–OH and –CO (carboxylic, and carbonyl) bonds. Other materials show almost similar IR spectra. The XPS spectra in Fig. 3(b–d) confirms the presence of oxygenated sites and/or amine precursors in GO and rGO–DAB. The epoxy (C–O–C) groups at the interior of the GO nanosheet generate peaks at 286.8–287.0 eV. The edge carbonyl (–CO) and carboxylic (–COOH) groups show XPS peaks at 287.8–288.0 and 289.0–289.3 eV, respectively.26 The major chemical changes during GO–DAB → rGO–DAB conversion include the decomposition of C–O–C groups and the formation of C–N functional groups. The C–O–C and C–N functional group contents of 33.7% and 0.7% in GO respectively are changed into 11.3% and 6.7% in rGO–DAB. Even after reduction, there exist some residual polar sites in the form of edge oxygenated sites in rGO–DAB. The number of epoxy carbon sites is reduced for both the decomposition process and the formation of some C–N bonds. The type of C–N bonds in rGO–DAB is shown in the N 1s XPS spectra (Fig. 3d). Several N 1s components for the materials appearing in different positions confirm the existence of different types of bonded nitrogen. However, three major peaks including protonated, graphitic and pyrrolic N, respectively appeared at 401.7, 401.1 and 399.8 eV.27
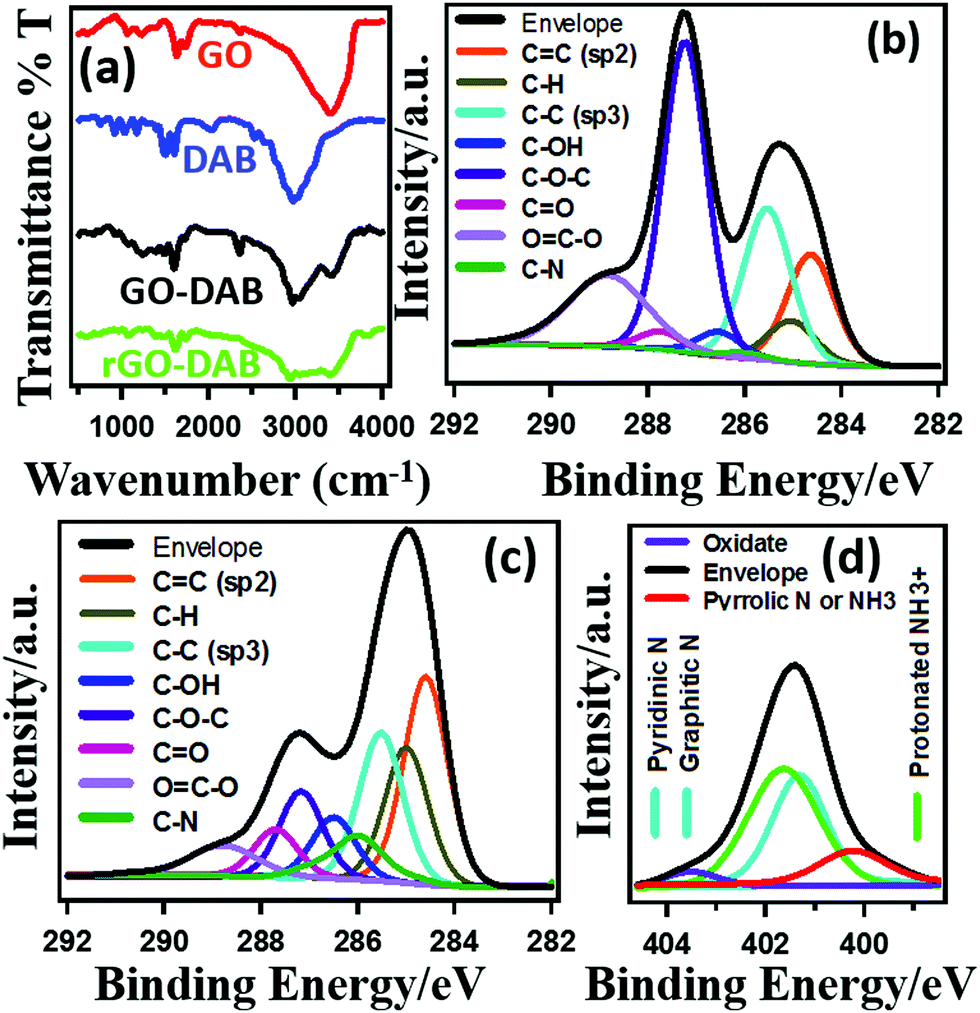 | ||
| Fig. 3 Chemical structure of the rGO–DAB hybrid. IR spectra of rGO–DAB, GO–DAB, rGO and GO (a). C 1s spectra of GO (b), rGO–DAB (c), and N 1s spectra of rGO–DAB (d). | ||
The results for PC measurement of the samples are displayed in Fig. 4. As rGO–DAB displayed the optimized conductivity and photocatalytic efficiency, here, we presented the dataset associated with rGO–DAB only. rGO–DAP and rGO–DAPN displayed significantly lower PC values. Fig. 4a shows typical Cole–Cole plots obtained during the impedance measurements for GO and rGO–DAB at different RH values and temperatures. The semicircle-shaped curves existing with a second semicircle primarily indicate the proton-driven conductivity. However, PC was confirmed from the isotope effect, where the D2O humidified sample displayed a lower PC than the H2O humidified sample. The RH-dependent PC values in Fig. 4b indicate that rGO–DAB has slightly lower (less than one order) PC values than that for GO. At 30 °C, the conductivities of GO and rGO–DAB increase from 3.3 × 10−4 and 2.7 × 10−4 S cm−1, respectively at 40% RH to 6.4 × 10−3 and 6.1 × 10−3 S cm−1, respectively at 90% RH. Fig. 4c shows that the PC value also rises with the temperature. At 40% RH and 30 °C, the σ value for GO and rGO–DAB are 3.3 × 10−4 and 2.7 × 10−4 S cm−1, respectively. These values reach 1.3 × 10−3 and 1.2 × 10−3 S cm−1, respectively at 90 °C. The Arrhenius plots of ln(σT) versus T−1 in Fig. 4d ravels the activation energies (Ea) for proton conduction in GO and GO–BA as 0.324 and 0.291 eV, respectively. As the proton conduction value is related to the hydrophobicity and water adsorbing capacity of a material, we also accomplished the water adsorbing capacity of the samples by thermogravimetric (TGA) analysis (Fig. 4e). Before the thermal analysis, the samples were incubated for 6 h at r.t. and 90%. The weight losses follow the trend as GO > GO–DAB > rGO–DAB. rGO–DAB possessing the lowest value for weight losses implies its lowest water adsorbing capacity and hydrophilicity. The weight losses up to 100 °C represent the existence of moisture at the GO interlayer. The decomposition of functional groups is indicative of the weight losses at 100–200 °C. The H+ ion is transported by the support of H2O molecules. The H+ ion alone cannot propagate without the formation of H3O+ ion. A continuous reformation of hydronium ions takes place during the propagation of the H+ ion. The PC value of rGO–DAB is slightly less than that of pristine GO. In general, PC is supported by hydrophilic ID. GO possessing about 30% oxygen content is hydrophilic enough to support faster PC. Due to the solid-state reduction of GO–DAB, part of GO's polarity is lost in rGO–DAB, though the DAB precursor contributes some hydrophilicity that is not sufficient enough to recover the loss that resulted from the reduction. The PC in rGO–DAB is thus reduced. The loss of hydrophilicity of GO is also clear from the TGA analysis in Fig. 4e.
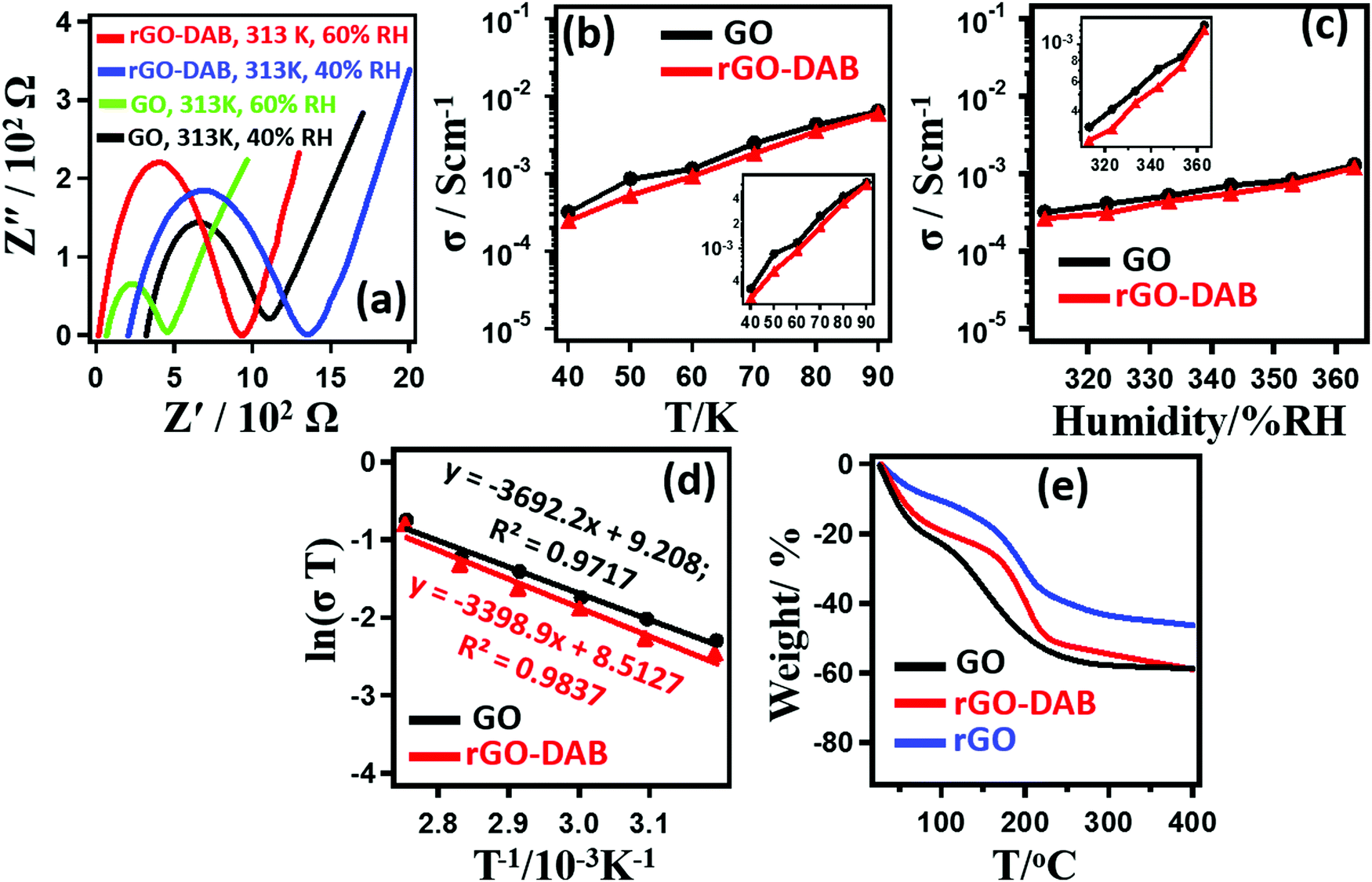 | ||
| Fig. 4 Proton conduction behaviour of GO and rGO–DAB. Typical Nyquist plots from impedance analysis of rGO–DAB at different relative humidity values and temperatures (a). Temperature-dependent proton conductivity values (fitted and magnified plot in the inset) (b). RH-dependent proton conductivity values (c). Arrhenius plots of temperature-dependent proton conductivity show perfect linear lines (d). Result for the thermogravimetric analysis of GO, rGO, and rGO–DAB reveals the presence of water adsorbing capacity (e). | ||
The performance toward photocatalytic WS and DD with associated electron conduction property is displayed in Fig. 5. In Fig. 5a, the amount of generated H2 gas (per gram of catalyst) in the WS process in the presence of RTiO2, rGO–DAA, rGO, and previously reported isomethylbutylamine–rGO hybrids (IMBA–rGO) is presented.5 The experimental duration was 36 h, maintaining the data collection frequency as once every 3 h. Pristine rGO cannot afford the generation of H2. However, the rest samples exhibit a gradual increase in the amount of photocatalytically generated H2 gas with the photo-irradiation time. In 36 h rGO–DAB, IMBA–rGO, rGO–DAP, RTiO2, and rGO–DAPN could generate 665.13, 593.75, 472.47, 432.55, and 360.27 μmol g−1 H2 gas, respectively. Fig. 5b represents the amount of H2 gas generated by the hybrids after 12, 24, and 36 h of light irradiation. Calculated H2 gas-producing efficiencies in Fig. 5c show the trend as rGO–DAB (18.48 μmol h−1 g−1) > IMBA–rGO (16.49 μmol h−1 g−1) > rGO–DAP (13.14 μmol h−1 g−1) > RTiO2 (12.02 μmol h−1 g−1) > rGO–DAPN (10.01 μmol h−1 g−1). The results for the study of DD activities for MB degradation are exhibited in Fig. 5(d–g). The absorption spectra for MB solutions shown in Fig. 5d exhibit typical peaks around 664 nm. The topmost line with the highest absorption peak intensity is observed for 1 × 10−4 MB solution in water. The gradual decrease in the peak intensity during light irradiation indicates the successful degradation of MB solution. The initial dye concentration (C0) displays the absorbance as A0, whereas the absorbance (A) of the solution after a certain time is proportional to the concentration (C) at that time. Thus, the ratio C/C0 was calculated using the variable (A/A0) values. Fig. 5(e) represents the photo-irradiation time-dependent C/C0 values. Pure rGO does not show any significant photocatalytic activity. However, the C/C0 value associated with the DD activity of rGO–DAB increases dramatically and displays a higher value than that for the previously reported IMBA–rGO. The relative trends can be represented as rGO–DAB > IMBA–rGO > rGO–DAP. For control experiments, the MB degradation in the absence of rGO–DAB and presence of rGO was justified with the light irradiation. However, insignificant DD efficiencies indicated that the photocatalytic activity is not assisted by the photolysis of MB. The DD efficiencies (η) can be calculated as follows: η = (1 − C/C0) × 100. The respective η values for rGO–DAB, IMBA–rGO, and rGO–DAP were calculated as 98.68, 91.72, and 81.78% after 280 min light irradiation. Fig. 5(f) displays the photoirradiation time-dependent ln(C0/C) values for the DD activity of the materials. The linear patterns imply that ‘ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C0/C’ values are directly proportional to the time of photo-irradiation. Thus, the plot indicates the presence of the proportionality constant, namely, kLH. In fact, the equation ‘ln C0/C = kLH·t′ signifies the Langmuir–Hinshelwood-type unimolecular reaction model in case of heterogeneous catalysis. The high regression coefficient (R2) values as 0.99, 0.99, and 0.98 for rGO–DAB, IMBA–rGO and, rGO–DAP indicate excellent linearity within the variables accompanying the pseudo-first-order-type reaction kinetic model. The kLH values were calculated to be 0.015, 0.013, and 0.006 min−1 for rGO–DAB, IMBA–rGO, and rGO–DAP, respectively. The maximum DD efficiency having k = 0.015 min−1, for rGO–DAB is lower than those values for some reported nitrogen-doped rGO–inorganic hybrids. However, it is higher than previously reported rGO–AA hybrids and some other G-based hybrid photocatalysts including graphene–C3N4–CdS (0.012 min−1), graphene-C3N4 (0.004 min−1), and pristine TiO2 (0.009 min−1). The kLH values exhibited by ZnO, graphene–TiO2, and graphene–ZnO as 0.022, 0.098, and 0.248 min−1 are higher than that of rGO–DAB.28–31Table 1 represents the MB degradation efficiency of rGO–DAB and reported organic hybrids. Compared with inorganic materials, though rGO–DAB displays a lower catalytic efficiency, this novel material representing entirely of organic-only origin indicates the possibility of devising hybrid photocatalysts with higher efficiency in the near future.
C0/C’ values are directly proportional to the time of photo-irradiation. Thus, the plot indicates the presence of the proportionality constant, namely, kLH. In fact, the equation ‘ln C0/C = kLH·t′ signifies the Langmuir–Hinshelwood-type unimolecular reaction model in case of heterogeneous catalysis. The high regression coefficient (R2) values as 0.99, 0.99, and 0.98 for rGO–DAB, IMBA–rGO and, rGO–DAP indicate excellent linearity within the variables accompanying the pseudo-first-order-type reaction kinetic model. The kLH values were calculated to be 0.015, 0.013, and 0.006 min−1 for rGO–DAB, IMBA–rGO, and rGO–DAP, respectively. The maximum DD efficiency having k = 0.015 min−1, for rGO–DAB is lower than those values for some reported nitrogen-doped rGO–inorganic hybrids. However, it is higher than previously reported rGO–AA hybrids and some other G-based hybrid photocatalysts including graphene–C3N4–CdS (0.012 min−1), graphene-C3N4 (0.004 min−1), and pristine TiO2 (0.009 min−1). The kLH values exhibited by ZnO, graphene–TiO2, and graphene–ZnO as 0.022, 0.098, and 0.248 min−1 are higher than that of rGO–DAB.28–31Table 1 represents the MB degradation efficiency of rGO–DAB and reported organic hybrids. Compared with inorganic materials, though rGO–DAB displays a lower catalytic efficiency, this novel material representing entirely of organic-only origin indicates the possibility of devising hybrid photocatalysts with higher efficiency in the near future.
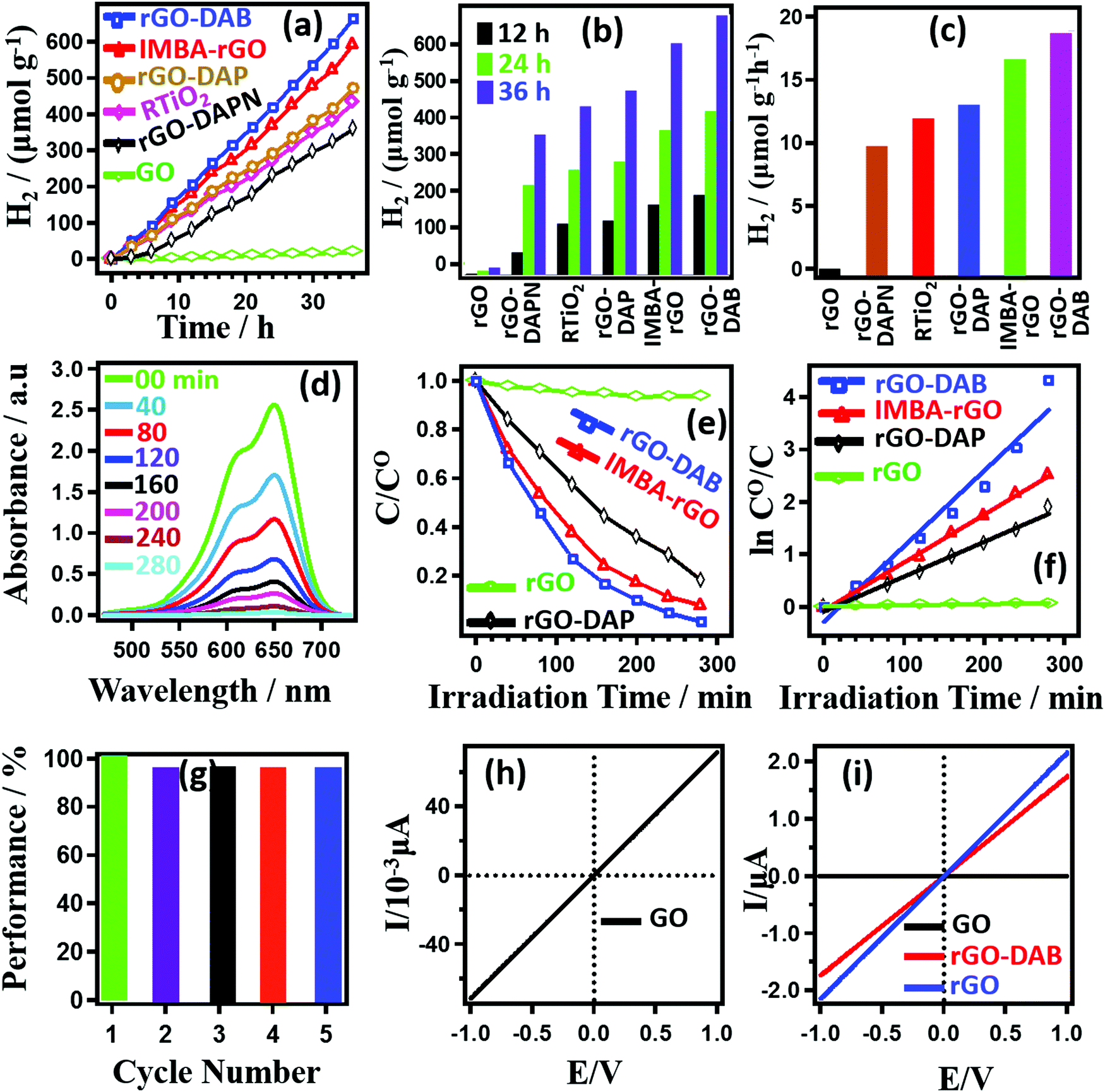 | ||
| Fig. 5 Photocatalytic performance of rGO–DAB and other hybrids: the amount of H2 generated by rGO–DAB, IMBA–rGO, rGO–DAP, RTiO2 and rGO–DAPN in 36 h photoirradiation time (a). Quantity of H2 gas generated every 12 h (b). H2 gas production efficiencies (c). Photo-irradiation time-dependent UV-Vis absorption spectra of MB solution undergoing the dye-degradation process in the presence of rGO–DAB (d). Photoirradiation time-dependent C/C0 profile (e) and ln(C0/C) values (f) during the DD experiment. Catalytic recyclability of rGO–DAB in terms of cycle number-dependent MB degradation efficiency (g). Electronic conductivity (EC) of GO (h) and comparative EC of GO, rGO, and rGO–DAB (i). | ||
| Materials | MB degradation efficiency (%) | k LH values | Ref. |
|---|---|---|---|
| rGO–DAB | 98.68 | 0.015 | — |
| rGO–methylpropylamine | 98.48 | 0.013 | 6 |
| rGO–isomethylbutylamine | 91.72 | 0.013 | 5 |
| Graphene–C3N4–CdS | — | 0.012 | 29 |
| rGO–propylamine | 88.14 | 0.008 | 6 |
| rGO–methylamine | 85.24 | 0.007 | 5 |
| rGO–ethylamine | 81.76 | 0.006 | 6 |
| rGO–DAP | 81.78 | 0.006 | — |
| Graphene–C3N4 | — | 0.004 | 29 |
Comparatively, inorganic photocatalysts are more effective than the rGO–DAB hybrid. The bandgap of inorganic hybrids can be tuned in a wider range due to huge structural variation. The properties of an inorganic semiconductor depend on its atomic component. Thus, the photocatalytic activity of inorganic materials can be modulated in a better way by choosing a wide range of nanoparticles including metallic oxide and sulfide. In contrast, the semiconductor behavior in rGO arises majorly for N-doping.
We further justified the catalytic efficiency of rGO–DAB in terms of recyclability towards the DD process. rGO–DAB displaying the maximum efficiency toward both the WS and DD process was considered only for the study of recyclability. Five cycles of DD experiment were accomplished. In Fig. 5(f), the DD performance of rGO–DAB against the cycle number is displayed. After performing the DD experiment in each cycle, powdered rGO–DAB was separated by filtration and washing using water and acetone. Finally, the material was dried at 50 °C for 24 h. The material thus was used consecutively in repeated runs. The result thus obtained shows a slight lowering of MB degradation performance in the second run. The efficiency reduces from 96.82% to 91.74% after running the first cycle of 280 min. The efficiency remains almost the same in the next three runs with respective values as 91.52, 91.38, and 91.35%. The photocatalytic degradation of MB in aqueous systems is a well-reported phenomenon. The removal of MB's color takes place due to the degradation of the dye molecules. The dye molecules become oxidized with the removal of carbon, nitrogen, and sulfur atoms in the form of CO2, NH4+, NO3−, and SO42−, respectively. Successive hydroxylation reactions of MB molecules result in the aromatic ring opening to form these end products. Related detailed degradation pathway has been reported by A. Houas et al.29Fig. 5(h and i) represents the EC of GO, rGO, and rGO–DAB. GO is almost insulator with the EC value ≈10−3 μA V−1. The conductivity significantly increases in rGO and rGO–DAB (≈103 times) with respective values around 2.14 and 1.72 μA V−1. The EC of GO materials depends on the extent of epoxy group decomposition. Epoxy groups contain sp3 carbon centers, which are nonconductive. Staying at the interior of GO, the epoxy group causes breakthrough within the conductive sp2 carbon network of graphite. Thus, less epoxy content in rGO favors a higher EC. Compared with rGO–AA, the extent of decomposition of epoxy groups in rGO–DAB is higher. Thus, the EC of rGO–DAB is also higher. rGO–DAB exists with a more flexible ID. It seems that the reducing agents can easily pass through the more flexible IS in GO–DAB and reduces the epoxy sites by a higher degree.
Here, rGO–DAB functions as a heterogeneous catalyst. The hybrid was synthesized by the novel vapor phase reduction of GO–DAB. This strategy was considered for optimizing N doping, hydrodynamics and catalytic efficiency. Previously, we have observed that hybrids of rGO and AA could function as organic photocatalysts.5,6 In such hybrids, N-doped rGO could function as light-harvesting materials to adopt light-driven photocatalytic behavior. The AA precursor endows the hybrid with flexible IS and hydrophilic properties. The mechanistic pathway of photocatalysis thus becomes easier. In rGO–DAB, we could increase the extent of N doping, hydrophilicity and ID. The rGO–DAB thus displayed the optimized photocatalytic activity toward both the WS and DD. The increased N doping and hydrophilicity were obtained due to the presence of diamine groups in DAB. The optimized IS was availed due to the size of the butyl group and affinity of DAB precursor to scaffold two oppositely faced rGO precursors by its two edge amine groups. We propose that an optimized separation of adjacent rGO nanosheet is afforded by double amine-headed DAB units. As in the previous report, we observed that a too flexible IS does not support faster hydrodynamics, and the butyl group seems to function for optimizing the IS and photocatalytic performance. GO-based materials transport proton by the reformation of hydrogen bonds in the hydronium ions (H3O+) through the adsorbed water film at the IS (Fig. 5a).32 Though materials showing proton conductivity are desired in hydrogen fuel cells, in recent reports we have depicted the association of hydronium ion's passivity at the rGO interlayer with the photocatalytic mechanism. GO-based proton conductors can allow the permission of hydrogen ions in fuel cells. Similarly, the GO-based photocatalyst needs to allow water molecules through the IS (Fig. 6a). In our previous reports, rGO–AA revealed dual proton–electronic conductivity and photocatalytic ability. The materials displayed the properties of N-doped rGO, fast electron mediation affinity and facile support of hydrodynamics at the interlayer. These issues simultaneously afforded the PC and photocatalysis. In the present manuscript, the PC is increased significantly. The extent of the N content in the hybrids also increased as the DAB precursor contains double amine groups. Both the water adsorptivity (Fig. 4e) and IS (Fig. 2f) are increased in rGO–DAB. The hydration dynamic thus radically increased, for which the photocatalytic performance was improved. In rGO–DAB, the DAB molecules having two amine terminals can firmly be attached with two opposite rGO nanosheets. Thus, a highly flexible IS is obtained. In contrast, the AA can tie the rGO nanosheet at a single terminal. Thus, the IS is less flexible as the other terminal of AA can be bent (Fig. 6b). Thus, we see that even BA and DAB have a similar number of alkyl carbons, and the PC, IS, and catalytic ability of rGO–DAB are comparatively higher. The photocatalytic ability of the rGO-based material is supported by graphene's N doping, bandgap opening, EC and PC. The optimization of all these parameters made rGO–DAB an excellent multifunctional hybrid. The material is a perfect organic-only composite. The layered pattern of the material was sustained due to mild solid-phase reduction. The light-harvesting property resulted from the N-doping and bandgap opening. Though rGO and G possess almost zero bandgap property, the N-doping became a facile way to widen the bandgap. The reasons behind fast proton hopping in thin layered materials were described by Maier et al.33 Maier correlated the charged center's randomization, degree of structural defects, and space charge domain with proton hopping. Oxidized carbon materials possessing flexible IS and extended hydrophilic groups can accommodate the moisture film, which supports the PC. Though GO and some of its hybrids can support high PC, due to the lack of mobile electron the EC remains low. However, by reducing GO in rGO, though the EC is increased, the decay of hydrophilic sites and shrinking of IS result in a lower PC. Herein, we considered the insertion of DAB at rGO for optimizing not only the EC and PC but also the photocatalytic efficiency. In Fig. 2, the IR and XPS spectra indicate that the DAB groups introduce polar amine groups and doped N. The reduction in vapor phase seems to be efficient to convert GO–DAB into rGO–DAB with ensuring the existence of a wider IS and 8.2% residual oxygenated functional sites. However, the Raman shifts imply the recovery of graphene's conductive π-electron network in rGO–DAB. Both the EC and PC in the materials thus sustain simultaneously. The PXRD data confirm that the DAB precursor function as a spacer group to widen the IS from 3.96 Å in rGO to 5.13 Å in rGO–DAB. Moreover, it is confirmed that DAB can result in an intermediate IS (compared with rGO–DAP and rGO–DAPN). The presence of DAB at the interlayer of rGO has a significant effect on the PC, as the wider IS can support the facile reformation of hydronium ions during proton conduction. However, a too large or too short IS does not necessarily favor the PC.7 Thus, even rGO–DAP has a slightly lower IS value than rGO–DAPN, and the PC of rGO–DAP is much higher. Moreover, the carbon chain length is an influential factor for PC. The DAPN chain with five carbon attributes lower the hydrophobicity in rGO–DAPN, for which the PC is lower. This observation concluded that DAB having four carbon-based linear alkyl chains is the best candidate for decorating dual EC and PC. From the Raman and XPS study, the formation of the C–N chemical bond between DAB and rGO is confirmed. Thus, the hybrid gain stability and washing the hybrids with several nonpolar and polar solvents do not reduce the PC value. The reduction of GO into rGO results in the loss of sp3 electron insulative carbon centers and the gaining of the conductive p–π domain. The electrons hence flow along the rGO nanosheet surface. At the same time, the protons can migrate through the lamellar water attached to the doped-N, residual polar groups, and N-terminals of the intercalated DAB molecules. The PC and EC thus are reverse in directions (Fig. 6c).
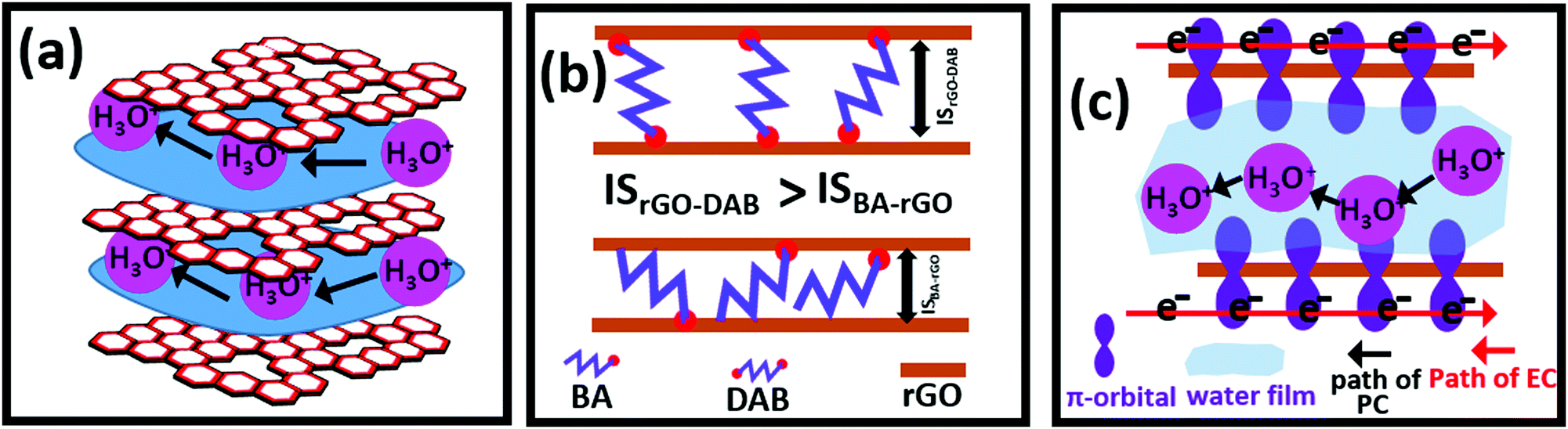 | ||
| Fig. 6 Scheme for the component's stacking pattern and conduction phenomena in rGO–DAB. PC is supported along the humidified interlayer space by the reformation of hydronium ions (a). DAB can function as a better spacer group than BA, as the double polar terminal of DAB can scaffold the neighbouring rGO nanosheet (b). EC is supported by the π-electron networks, whereas PC is supported by the humidified interlayer space (c). | ||
The rGO–DAB contains 6.7% C–N content, which is higher than the other hybrids. The TGA profile also reveals the excellent water adsorbing capacity of rGO–DAB. The WS and DD affinity of the hybrid is the consequence of this C–N content, water permission process, flat sheet structure, surface property, and energy harvesting capability. The EC of semiconductor material can be tuned in wide ranges. Usually, semiconductor composites with low current conduction majorly are used for electrochemical sensing.34–40 However, herein, the high current conduction and performance of photocatalysis is triggered by some scientific facts including the photo-generation of electron/hole pairs; the migration of electron/holes at the catalyst surface; the chemical reaction between electrons/holes with reactant water molecules/MB dye and the charge recombination process (Fig. 7).41–43 Taking these facts under consideration, the photocatalytic action of some metallic nanoparticles and nitrogen-doped rGO/G was reported before.44,45 The photocatalytic activity of rGO–DAB can also be explained considering these similar issues.
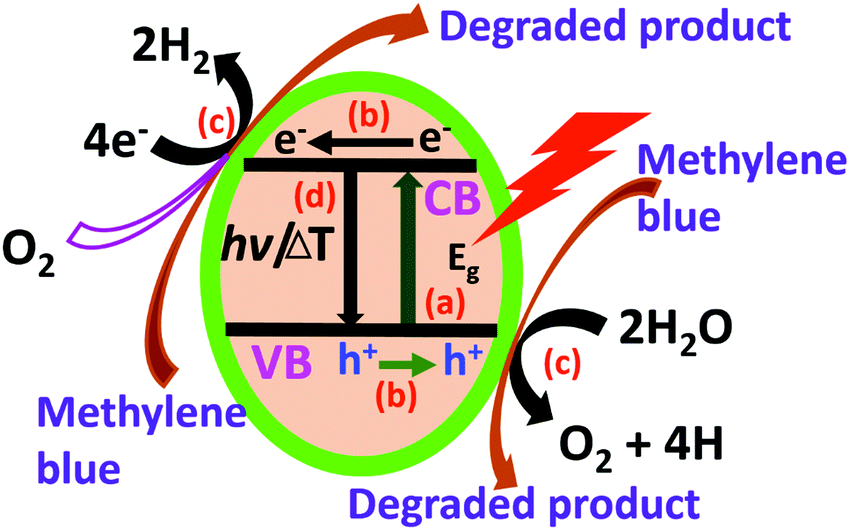 | ||
| Fig. 7 Scheme for the mechanistic pathway of rGO–DAB toward WS and DD. The water splitting and degradation of methylene blue dye involve several mechanistic steps including electron–hole pair generation by the absorption of photons (a); propagation of electrons and holes along the surface of the catalyst particles (b); the accomplishment of chemical reactions associated with the capture of electrons and holes (c) and finally, the recombination of charges (d). | ||
The N atom from DAB molecules substitutes some C atoms from the GO honeycomb structure. This N-doped rGO behaves like a bandgap semiconductor and can produce electron/hole pairs when exposed to light. The possible forms of doped N revealed from XPS analysis include pyrrolic, graphitic, pyridinic, and oxidative N (Fig. 8a). N is pentavalent, while the carbon atom that is replaced is tetravalent. Thus, dopant N endows rGO–DAB with the N-type semiconductor nature in addition to the opening of graphene's bandgap (Fig. 8b). Originally, conductive G turns to insulator GO. The rGO is conductive but can exhibit light-harvesting property. However, due to N doping, the linear combination of N and C atom orbitals generates LCOA orbitals of different energy states. This rearrangement can enhance the light-harvesting property. Hence, in the initiation step of photocatalysis rGO–DAB can introduce electron/hole pairs. In the next step, the electron/hole pair migrates through the conductive rGO hybrids. The rGO nanosheet possesses almost a two-dimensional structure, and they are separated from each other by the DAB units. Therefore, the composite possesses an entire sheet-like structure, and there is no bulk mass. Thus, the full surface exposures for the electron/hole movement sustain, which is not possible even in the spherical TiO2 nanoparticle. The photocatalytic efficiency of rGO–DAB is thus higher than the bulk-sized TiO2 nanoparticles. In the third step, the electron/hole pair migrates around the water/MB molecules, which can enter the IS. The reactant molecules can reach around the entire surface of the rGO nanosheets as the IS is sufficiently flexible. It seems that the whole mass of rGO–DAB behaves like a giant flat sheet. The photocatalytic reaction thus takes place through optimized efforts. The evidence of easy permission of reactant molecules is not only the significant water loss in the TGA profile but also the high PC values. The persistent catalytic efficiency with the cycle number indicates the possible application of the material in real uses. In the starting cycle, we propose that the adsorbed and untied intercalated materials including DAB become washed and result in a slight lowering of catalytic performance. There also exists another option of dye degraded products to be adsorbed on the catalyst surface. However, the PXRD pattern recorded after the starting run shows that the IS remains unchanged. This observation implies that no structural or morphological change of rGO–DAB hybrid happens when part of the degraded product is adsorbed onto the surface. The unchanged DD efficiencies in the rest cycles imply that the washing of adsorbed or unreacted materials is almost finished during the first run.
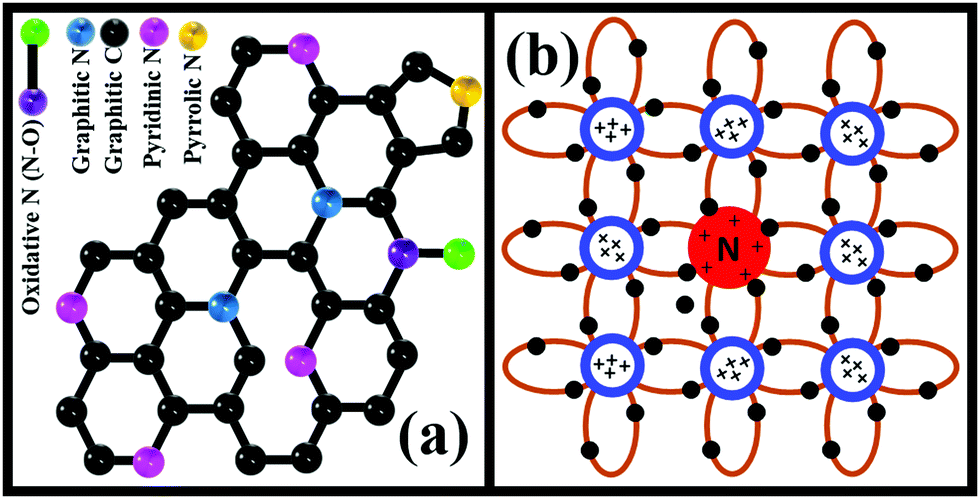 | ||
| Fig. 8 Scheme for the changes in the graphitic skeleton of rGO in rGO–DAA due to the N doping afforded by DAA precursors. The types of nitrogenous functional groups in rGO–DAB hybrids include pyrrolic, graphitic, oxidative and pyridinic N (a). Nitrogen atoms with valency 5 can populate additional electrons and holes when it replaces carbon atoms from the graphitic skeleton (b). | ||
Conclusions
A new strategy of engineering the semiconductor bandgap in rGO by inserting a series of organic molecules including 1,4-diaminoprobane (DAP), 1,4-diaminobutane (DAB) and 1,4-diaminopantane (DAPN) is presented. We found that the intercalation of the diaminoalkane (DAA) precursors into rGO by solid-phase reduction process results in the formation of highly stable rGO–DAA-type organic-only hybrids. Within the series, the rGO–DAB hybrids exhibit a high PC value (≈10−3–10−4 S cm−1) that is slightly less than pristine rGO. The EC value was observed as high as 1.72 μA V−1. The materials could afford photocatalytic performances when justified by DD (toward the degradation of MB) and WS process. The DD performances of rGO–DAB were maximum within the series. The kLH value of 0.015 min−1 is higher than that for rGO–DAP (0.006 min−1) and previously reported IMBA–rGO (0.013 min−1). The WS capability follows the trend as rGO–DAB (18.48 μmol h−1 g−1) > IMBA–rGO (16.49 μmol h−1 g−1) > rGO–DAP (13.14 μmol h−1 g−1) > RTiO2 (12.02 μmol h−1 g−1) > rGO–DAPN (10.01 μmol h−1 g−1). Surface morphology, spectroscopic investigation, and weight loss profile were employed to characterize the samples. The rGO–DAB hybrid exists with the optimized amount (6.7%) of doped N. The DAB could generate a highly flexible IS and exhibit excellent PC, EC, N-doping, and hydrophilicity. The strategy depicted herein can be considered for the further development of rGO-based organic photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under grant no. G: 575-130-1441. The authors, therefore, acknowledge with thanks DSR for technical and financial support.References
- Y. Meng, J. Gao, Z. Zhao, J. Amoroso, J. Tong and K. S. Brinkman, J. Mater. Sci., 2019, 54, 9291–9312 CrossRef CAS.
- P. T. Yin, S. Shah, M. Chhowalla and K.-B. Lee, Chem. Rev., 2015, 115, 2483–2531 CrossRef CAS.
- Y. Murashima, M. R. Karim, R. Furue, T. Matsui, H. Takehira, K. Wakata, K. Toda, R. Ohtani, M. Nakamura and S. Hayami, Inorg. Chem. Front., 2016, 3, 842–848 RSC.
- M. R. Karim, H. Shinoda, M. Nakai, K. Hatakeyama, H. Kamihata, T. Matsui, T. Taniguchi, M. Koinuma, K. Kuroiwa, M. Kurmoo, Y. Matsumoto and S. Hayami, Adv. Funct. Mater., 2013, 23, 323–332 CrossRef CAS.
- M. R. Karim, M. M. Rahman, A. M. Asiri and S. Hayami, ACS Appl. Mater. Interfaces, 2020, 12, 10829–10838 CrossRef.
- M. R. Karim, M. M. Rahman and A. M. Asiri, J. Taiwan Inst. Chem. Eng., 2020, 122, 87–96 CrossRef.
- M. R. Karim, M. S. Islam, K. Hatakeyama, M. Nakamura, R. Ohtani, M. Koinuma and S. Hayami, J. Phys. Chem. C, 2016, 120, 21976–21982 CrossRef CAS.
- Y. Ikeda, M. R. Karim, H. Takehira, K. Hatakeyama, T. Matsui, T. Taniguchi, M. Koinuma, Y. Matsumoto and S. Hayami, Chem. Lett., 2014, 43, 486–488 CrossRef CAS.
- K.-D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- S. Bose, T. Kuila, T. X. H. Nguyen, N. H. Kim, K.-T. Lau and J. H. Lee, Prog. Polym. Sci., 2011, 36, 813–843 CrossRef CAS.
- L. Vilčiauskas, M. E. Tuckerman, G. Bester, S. J. Paddison and K. D. Kreuer, Nat. Chem., 2012, 4, 461–466 CrossRef.
- L. Yang, J. Tang, L. Li, X. Chen, F. Ai, W. Z. Yuan, L. Wang and Y. Zhang, RSC Adv., 2012, 2, 5950–5953 RSC.
- Y. Shudo, M. S. Islam, M. R. Karim, N. N. Rabin, K. Wakata, R. Ohtani, M. Nakamura, L. F. Lindoy and S. Hayami, Global Challenges, Consequences, and Prospects, 2017, 1, 1700054 CrossRef.
- M. R. Karim, K. Hatakeyama, M. Koinuma and S. Hayami, J. Mater. Chem. A, 2017, 5, 7243–7256 RSC.
- K. Hatakeyama, M. R. Karim, C. Ogata, H. Tateishi, A. Funatsu, T. Taniguchi, M. Koinuma, S. Hayami and Y. Matsumoto, Angew. Chem., Int. Ed., 2014, 53, 6997–7000 CrossRef CAS.
- M. R. Karim, M. M. Rahman and A. M. Asiri, ACS Appl. Electron. Mater., 2020, 2, 1304–1312 CrossRef CAS.
- M. R. Karim, M. S. Islam, N. N. Rabin, R. Ohtani, M. Nakamura, M. Koinuma and S. Hayami, Aust. J. Chem., 2017, 70, 642–645 CrossRef CAS.
- Y. Shudo, M. R. Karim, R. Ohtani, M. Nakamura and S. Hayami, ChemElectroChem, 2018, 5, 238–241 CrossRef CAS.
- Y. Ikeda, M. R. Karim, H. Takehira, K. Hatakeyama, T. Taniguchi, M. Koinuma, Y. Matsumoto and S. Hayami, Bull. Chem. Soc. Jpn., 2014, 87, 639–641 CrossRef CAS.
- K. Hatakeyama, M. R. Karim, C. Ogata, H. Tateishi, T. Taniguchi, M. Koinuma, S. Hayami and Y. Matsumoto, Chem. Commun., 2014, 50, 14527–14530 RSC.
- M. R. Karim, K. Hatakeyama, T. Matsui, H. Takehira, T. Taniguchi, M. Koinuma, Y. Matsumoto, T. Akutagawa, T. Nakamura, S. Noro, T. Yamada, H. Kitagawa and S. Hayami, J. Am. Chem. Soc., 2013, 135, 8097–8100 CrossRef CAS.
- M. R. Karim, M. S. Islam, N. N. Rabin, H. Takehira, K. Wakata, M. Nakamura, R. Ohtani, K. Toda and S. Hayami, ChemistrySelect, 2017, 2, 4248–4254 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- M. C. Hsiao, S. H. Liao, M. Y. Yen, P. I. Liu, N. W. Pu, C. A. Wang and C. C. Ma, ACS Appl. Mater. Interfaces, 2010, 2, 3092–3099 CrossRef CAS.
- Y. Ikeda, M. R. Karim, H. Takehira, T. Matsui, T. Taniguchi, M. Koinuma, Y. Matsumoto and S. Hayami, Chem. Lett., 2013, 42, 1412–1414 CrossRef CAS.
- M. R. Karim, H. Takehira, T. Matsui, Y. Murashima, R. Ohtani, M. Nakamura and S. Hayami, Curr. Inorg. Chem., 2014, 4, 191–219 CrossRef CAS.
- J. A. Hong, E. D. Jung, J. C. Yu, D. W. Kim, Y. S. Nam, I. Oh, E. Lee, J.-W. Yoo, S. Cho and M. H. Song, ACS Appl. Mater. Interfaces, 2020, 12, 2417–2423 CrossRef CAS.
- K. Ravichandran, N. Chidhambaram, T. Arun, S. Velmathid and S. Gobalakrishnane, RSC Adv., 2016, 6, 67575–67585 RSC.
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J. M. Herrmann, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- H. Fan, X. Zhao, J. Yang, X. Shan, L. Yang, Y. Zhang, X. Li and M. Gao, Catal. Commun., 2012, 29, 29–34 CrossRef CAS.
- Y. Wang, R. Shi, J. Lin and Y. Zhu, Appl. Catal., B, 2010, 100, 179–183 CrossRef CAS.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- J. Maier, Solid State Ionics, 1987, 23, 59–67 CrossRef CAS.
- M. M. Rahman, A. Wahid, A. M. Asiri, M. R. Awual and M. R. Karim, New J. Chem., 2020, 44, 4952–4959 RSC.
- M. M. Rahman, M. M. Hussain, M. N. Arshad, M. R. Awual and A. M. Asiri, New J. Chem., 2019, 43, 9066–9075 RSC.
- M. M. Alam, A. M. Asiri, M. T. Uddin, M. A. Islam, M. R. Awual and M. M. Rahman, New J. Chem., 2019, 43, 8651–8659 RSC.
- M. M. Alam, A. M. Asiri, M. T. Uddin, Inamuddin, M. A. Islam, M. R. Awual and M. M. Rahman, New J. Chem., 2019, 43, 4849–4858 RSC.
- M. M. Rahman, T. A. Sheikh, A. M. Asiri and M. R. Awual, New J. Chem., 2019, 43, 4620–4632 RSC.
- M. M. Rahman, M. M. Alam, A. M. Asiri and M. R. Awual, New J. Chem., 2017, 41, 9159–9169 RSC.
- M. M. Alam, M. T. Uddin, A. M. Asiri, M. R. Awual, M. A. Fazal, M. M. Rahman and M. A. Islam, Mater. Chem. Phys., 2020, 251, 123029 CrossRef CAS.
- H. Ohmagari, M. R. Karim, Y. Shudo, S. Ida, R. Ohtani and S. Hayami, MRS Adv., 2018, 47–48, 2847–2854 CrossRef.
- Y. Shudo, M. R. Karim, K. Wakata, H. Ohmagari, N. Kameda and S. Hayami, J. Inclusion Phenom. Macrocyclic Chem., 2019, 94, 283–286 CrossRef CAS.
- N. N. Rabin, H. Ohmagari, M. S. Islam, M. R. Karim and S. Hayami, J. Inclusion Phenom. Macrocyclic Chem., 2019, 94, 263–281 CrossRef CAS.
- H. Takehira, M. S. Islam, M. R. Karim, Y. Shudo, R. Ohtani, L. F. Lindoy, T. Taniguchi, M. Osada and S. Hayami, ChemistrySelect, 2017, 2, 6941–6944 CrossRef CAS.
- T. Mayu, M. S. Islam, M. R. Karim, N. N. Rabin, R. Ohtani, M. Nakamura, L. F. Lindoy and S. Hayami, Bull. Chem. Soc. Jpn., 2017, 90, 950–954 CrossRef.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |