
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural studies into the spin crossover behaviour of Fe(abpt)2(NCS)2 polymorphs B and D†
Helen E.
Mason‡
a,
Jake R. C.
Musselle-Sexton
b,
Judith A. K.
Howard
 a,
Michael R.
Probert
b and
Hazel A.
Sparkes
*c
a,
Michael R.
Probert
b and
Hazel A.
Sparkes
*c
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK
bChemistry, School of Natural and Environmental Sciences, Bedson Building, Newcastle University, Kings Road, NE1 7RU, UK
cDepartment of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: hazel.sparkes@bristol.ac.uk
First published on 12th July 2021
Abstract
The spin-crossover behaviour of [Fe(abpt)2(NCS)2] (abpt = 4-amino-3,5-bis(pyridin-2-yl)-1,2,4-triazole) polymorphs B and D has been studied using single crystal X-ray diffraction to monitor changes in structural features. High pressure single crystal measurements on polymorph B showed that it underwent a monoclinic P21/n (Z′ = 0.5) to triclinic P-1 (Z′ = 2 × 0.5) phase transition between 11.5 and 13.5 kbar, at which point it also starts to undergo a thermally inaccessible spin crossover. In polymorph D which also crystallises in the mononclinic space group P21/n (Z′ = 2 × 0.5) one of the unique Fe centres undergoes a thermal spin transition. It also displays light-induced excited spin-state trapping (LIESST), and a structure has been obtained at 30 K through continuous irradiation with a 670 nm 5 mW CW laser. In addition high pressure single crystal measurements on polymorph D showed a stepped pressure induced spin transition. At ∼9.6 kbar one of the unique Fe centres had undergone a spin transition and by ∼15 kbar both of the unique Fe centres are shown to be essentially low spin, a situation that is thermally inaccessible. Crystallographic data were collected for both polymorphs using variable temperature or high pressure single crystal X-ray diffraction to allow changes in cell parameters, bond lengths and distortion parameters to be monitored for the spin crossover or phase transition.
Introduction
In the 1930s, the first observation of spin crossover was reported.1 Spin crossover compounds display a reversible high spin (HS) to low spin (LS) transition at a metal centre, which can be induced by external stimuli such as temperature, pressure2 or light irradiation3,4 and can occur in both solution and the solid state. The phenomenon is relatively common for 3d4–3d7 metal centres.5 While many of the spin crossover complexes that have been studied are octahedral Fe(II) complexes with nitrogen5–10 ligands, other examples include Fe(III),4,11–14 Co(II)15–17 and Ni(II).18 The change in electronic structure, as a result of the spin crossover, can result in changes in properties including molecular structure, colour and magnetism.19 These property changes give rise to a number of potential applications including molecular switches, data storage or sensors.20–24 There are significant challenges in developing materials for commercial applications as spin crossover is a very complex process. The manner in which the transition occurs e.g. gradual or abrupt, stepped, with or without hysteresis can also affect potential uses. Spin crossover in solution tends to result in a gradual transition as there is no cooperativity between molecules. In the solid state, where cooperativity is possible, a much wider range of transitions are seen. Where strong cooperativity exists between molecules the transitions can be abrupt and show hysteresis or without cooperativity transitions tend to be more gradual and without hysteresis. They can also be full transitions, partial or multi-stepped transitions.25 A large number of factors influence the spin crossover behaviour of a compound e.g. inclusion of solvent in the lattice,8 polymorphism or intermolecular interactions.19 It is therefore important to gain as much insight into the phenomena to allow accurate design and control of potential devices. A number of X-ray diffraction studies have been carried out into the spin crossover behaviour of various compounds to help gain insights into the structure property relationships affecting spin crossover complexes. These have included variable temperature measurements to follow thermally induced structural changes, in situ light irradiation to obtain and characterise Light Induced Excited Spin State Trapping (LIESST) states and high-pressure studies looking at changes upon the application of pressure.3,6,26–30 Alongside magnetic measurements and X-ray diffraction, other techniques31,32 including Mössbauer spectroscopy33 and differential scanning calorimetry34 can also be used to give useful insights into the behaviour of spin crossover complexes.In addition to spin crossover complexes with single metal centres, dimeric10 and polymeric species have also been developed that show spin crossover behaviour. Having more than one metal centre linked can allow for increased cooperativity between the metal centres potentially raising Tc and resulting in hysteresis. Most potential applications require a Tc around room temperature and for applications such as data storage there also needs to be hysteresis. There is therefore much interest in preparing spin-crossover complexes with more than one metal centre. Recently a series of three novel 3D coordination polymers containing Fe(II) metal centres with AgCN linkers have been prepared and found to display spin crossover behaviour.34 These showed thermal spin crossover with Tc ranging from 240–250 K with no hysteresis. The lack of hysteresis was attributed to the relatively long linker distances between the Fe centres having reduced the cooperativity. However, coordination polymers have also been shown to display hysteresis with a particularly exciting set of 1D coordination polymers containing Fe(II), a tetradentate N2O22− and N-(pyrid-4-yl)isocnicotinamide, showing wide, repeatable hysteresis loops, ranging from 46–88 K, centred around room temperature.35
Spin crossover is a complex process so [Fe(abpt)2(NCS)2] (abpt = 4-amino-3,5-bis(pyridin-2-yl)-1,2,4-triazole), which has four reported polymorphs (A–D) all showing slightly different spin crossover behaviour, is a particularly interesting compound to study to try and obtain insights into the phenomenon.36 Three of the polymorphs A,37–39C39,40 and D39,41 undergo at least a partial thermal spin transition at ambient pressure while the fourth polymorph B only undergoes a thermal spin transition at pressures >4.4 kbar.42 Polymorph A has one independent Fe(II) centre (Z′ = 0.5) in the asymmetric unit which undergoes a thermal spin transition without hysteresis, T1/2 was initially reported as 180 K,37 but has been remeasured as T1/2 = 188 K.39A also displays LIESST (light induced excited spin state trapping) TLIESST = 40 K, for which a significant proportion of the HS* state can be maintained for >1000 s up to 30 K. Although the thermally induced spin crossover shows no hysteresis, a light induced thermal hysteresis (LITH) associated with the HS* state has been identified from the photomagnetic data.37 Crystal structures of the high spin (HS), low spin (LS) and LIESST HS* (30 K) have all been determined and changes in crystallographic parameters across the temperature range 375 K to 30 K reported.38 It has also been shown crystallographically that A undergoes a pressure induced spin crossover, as demonstrated by a structure at 5.1(2) kbar. The exact point of the pressure induced spin transition has not been determined as it is challenging to accurately control the pressure increase in the diamond anvil cells used for high pressure crystallography.38 For C which has two independent Fe(II) centres in the asymmetric unit (Z′ = 2 × 0.5), only one of the unique Fe centres undergoes a thermal spin transition T1/2 = 86 K upon cooling.40 The thermal behaviour of C is actually more complicated than a simple thermal spin transition as the method of cooling affects the results obtained: on gradual cooling between 170 and 86 K a tripling of the c axis is associated with the formation of a commensurate modulated structure containing four independent Fe(II) centres in the asymmetric unit (Z′ = 2 × 1 + 2 × 0.5). Further cooling below T1/2 results in the c axis being similar in magnitude to its value above 170 K. Flash cooling of C to 25 K results in the formation of a TIESST (thermal induced excited spin state trapping) state which has a tripling of the c axis and forms a commensurate modulated structure as observed between 170 and 86 K. C also displays LIESST again forming the same commensurate modulated structure just discussed. Polymorph D also contain two unique Fe(II) centres in the asymmetric unit (Z′ = 2 × 0.5), one of which undergoes a gradual partial thermal spin transition, T1/2 = 162 K.39 Above and below the transition the Fe centres are HS/HS and HS/LS respectively and the structures of these have previously been reported.41 As with the other two polymorphs that display a thermal spin transition at ambient pressure, the LS Fe centre in D has also been shown to display LIESST using a 532 nm laser at 20 K.41 The nature of the LIESST HS* structure obtained depends on the laser power used, with a 5 mW cm−2 powered laser the Fe–N bond lengths increase by ∼0.2 Å as commonly observed, however a higher powered 40 mW cm−2 laser more unusually also results in linkage isomerism of the NCS ligands at the Fe centre that undergoes the spin transition. The refined occupancies suggest that 80% of the NCS ligands on this iron centre displayed linkage isomerism, which agreed with that estimated from photomagnetic measurements.41 Such difference in the behaviour under different types of irradiation have the potential to be useful for tuning responses in applications.
It is also worth noting that [Fe(abpt)2(NCSe)2] has been found to have two polymorphs A37,43 and B42 which are isostructural with the respective [Fe(abpt)2(NCS)2] polymorphs. The known spin crossover behaviour is also very similar for the NCS and NCSe polymorphs. In the case of the NCSe polymorph A it is known to display a thermally induced spin crossover T1/2 = 224 K, with a slightly higher than for the NCS polymorph, as well as LIESST. While in both cases for B no thermal spin crossover was observed. High pressure studies have not been carried out for the NCSe polymorphs, so no comparison of the pressure behaviour is possible.
The pressure behaviour of [Fe(abpt)2(NCS)2] B and D at room temperature has not been previously reported and are studied using high pressure single crystal crystallography herein. In addition, the thermal behaviour of D is followed crystallographically from 375 to 30 K, examining changes in the cell parameters, Fe–N bond lengths, distortion parameter (Σ) and the octahedron volume (Vp). These results are compared to those obtained in the high pressure experiments.
Experimental
Synthesis
Synthesis of [Fe(abpt)2(NCS)2] was carried out using a slow diffusion method which was previously reported to produce crystals of the various polymorphs.39 Precursor materials were used without further purification, solvents degassed and all manipulations carried out under a nitrogen atmosphere. FeSO4·7H2O (1 mmol, 0.278 g) and KNCS (2 mmol, 0.194 g) were stirred in MeOH (10 ml) for 15 min. Pale yellow insoluble K2SO4 precipitate was removed by filtration and deionised H2O (10 ml) added to the remaining clear solution. abpt ligand (2 mmol, 0.477 g) was dissolved in MeOH (20 ml) and transferred to a narrow (<5 cm) Schlenk tube. The Fe2+/2(NCS)− solution was carefully injected underneath the abpt solution to form a lower layer. A coloured band containing the target complex immediately formed at the interface between the two layers. Within 1–4 weeks single crystals of B and D suitable for X-ray diffraction studies were formed and separated under the microscope.39X-Ray crystallography
High pressure
For both B and D the following procedure was used for the high pressure single crystal measurements. A single crystal of each polymorph was mounted along with a small ruby chip in a modified Merrill–Bassett type diamond anvil cell (DAC) custom built at Durham University using tungsten carbide backing plates, with an opening angle of 85° and type IA Boehler–Almax diamond anvils with 0.8 mm cutlets. Stainless steel gaskets were pre-indented and drilled to give a gasket chamber of ∼0.3 mm diameter and 0.15 mm depth, with paraffin oil as the pressure transmitting medium. The Ruby R1 fluorescence method was used to determine the pressure in the DAC,53 to ensure that the cell pressure had equilibrated prior to data collection fluorescence measurements were taken before and after each data collection to ensure the pressure had not changed. Exposure of the cell to the laser used for the fluorescence measurements was kept to a minimum as the laser can induce a metastable LIESST spin transition, however since the pressure measurements are almost always taken above TLIESST any structural change would not be long lived so should not affect the results. The pressure in the DAC is increased by tightening screws on the cell, hence it is not possible to accurately control the extent of the pressure increase each time. In the case of B pressure measurements were made at ambient pressure, 9.0(2), 11.5(2), 13.5(2), 16.0(2) and 23.4(2) kbar while for D pressure measurements were made at ambient pressure, 1.8(2), 7.5(2), 9.6(2), 12.0(2) and 15(2) kbar. All measurements were made at room temperature. All data collections were carried out using XIPHOS II,54 part of the XIPHOS diffraction facility.55 This diffractometer, custom built for high pressure studies, is equipped with an Incoatec Ag-Kα (λ = 0.56086 Å) IμS source.56 The data collections were carried out using the Bruker APEX2 software suite,57 integration was performed using SAINT46 and multi-scan absorption corrections were applied to all datasets using SADABS.48 Masking for the diamond anvil cell occluded regions of the data collections was performed within the integration software. The structures were solved using the dual space methods employed within SHELXT and refined by full matrix least squares on F2 in SHELXL49 using the Olex252 graphical interface. All of the non-hydrogen atoms were refined anisotropically and all hydrogen atoms were located geometrically and refined using a riding model with the exception of the hydrogen atoms on N6 which were located in the difference map. Crystal structure and refinement details are given in Tables 2 and 4, Tables S1 and S3 (ESI†).| D–H/Å | H⋯A/Å | D⋯A/Å | D–H⋯A/° | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a 1 − x, 1 − y, 1 − z. b x, 1 − y, −z. c −x, 1 − y, 1 − z. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Polymorph B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N6–H6B⋯N7 | 0.86(2) | 2.45(2) | 2.912(2) | 114.6(17) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C2–H2⋯N4a | 0.93 | 2.83 | 3.629(2) | 144.7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C5–H5⋯N6 | 0.93 | 2.46 | 3.070(2) | 123.7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Polymorph D | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N6–H6B⋯N7 | 0.88(3) | 2.12(2) | 2.862(3) | 141(2) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N13–H13B⋯N14 | 0.80(2) | 2.38(2) | 2.864(3) | 120(2) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C2–H2⋯N4b | 0.93 | 2.81 | 3.605(3) | 144.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C5–H5⋯N6 | 0.93 | 2.48 | 3.090(3) | 122.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C15–H15⋯N11c | 0.93 | 2.77 | 3.582(3) | 145.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C18–H18⋯N13 | 0.93 | 2.43 | 3.042(3) | 123.4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Spin state (temperature/pressure) | HS 300(2) K | HS 100(2) K | HS ambient 296 K | HS 13.5(2) kbar 296 K | LS 23.4(2) kbar 296 K |
|---|---|---|---|---|---|
| Empirical formula | C26 H20 Fe N14 S2 | C26 H20 Fe N14 S2 | C26 H20 Fe N14 S2 | C26 H20 Fe N14 S2 | C26 H20 Fe N14 S2 |
| Formula weight | 648.53 | 648.53 | 648.53 | 648.53 | 648.53 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic | Triclinic |
| Space group | P21/n | P21/n | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a/Å | 11.5730(6) | 11.47261(8) | 11.5739(6) | 9.2535(13) | 8.985(4) |
| b/Å | 9.6589(5) | 9.58685(8) | 9.6588(8) | 11.3483(8) | 11.328(3) |
| c/Å | 12.8541(7) | 12.72647(10) | 12.8455(9) | 12.2374(15) | 12.013(5) |
| α/° | 90 | 90 | 90 | 101.283(7) | 101.89(2) |
| β/° | 101.2740(10) | 100.5225(7) | 101.244(4) | 90.790(8) | 92.74(3) |
| γ/° | 90 | 90 | 90 | 90.653(9) | 91.31(3) |
| Volume/Å3 | 1409.14(13) | 1376.197(18) | 1408.44(17) | 1260.0(3) | 1194.4(8) |
| Z | 2 | 2 | 2 | 2 | 2 |
| ρ calc/g cm−3 | 1.528 | 1.565 | 1.529 | 1.709 | 1.803 |
| μ/mm−1 | 0.730 | 0.747 | 0.384 | 0.429 | 0.453 |
| F(000) | 664.0 | 664.0 | 664.0 | 664.0 | 664.0 |
| Crystal size/mm3 | 0.56 × 0.42 × 0.34 | 0.56 × 0.42 × 0.34 | 0.24 × 0.20 × 0.1 | 0.24 × 0.20 × 0.1 | 0.24 × 0.20 × 0.1 |
| λ/Å | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | AgKα (λ = 0.56086) | AgKα (λ = 0.56086) | AgKα (λ = 0.56086) |
| 2θ range for data collection/° | 4.334 to 52.744 | 5.286 to 52.726 | 3.422 to 47.228 | 2.888 to 45.82 | 2.9 to 47.216 |
| Index ranges | −14 ≤ h ≤ 14, | −14 ≤ h ≤ 14, | −16 ≤ h ≤ 16, | −10 ≤ h ≤ 9, | -9 ≤ h ≤ 9, |
| −11 ≤ k ≤ 12, | −11 ≤ k ≤ 11, | −11 ≤ k ≤ 10, | −15 ≤ k ≤ 15, | −15 ≤ k ≤ 15, | |
| −16 ≤ l ≤ 11 | −15 ≤ l ≤ 15 | −16 ≤ l ≤ 13 | −12 ≤ l ≤ 15 | -12 ≤ l ≤ 12 | |
| Reflections collected | 8218 | 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 396 396 |
20877 |
17608 |
6813 |
| R int/Rsigma | 0.0205/0.0222 | 0.0342/0.0113 | 0.0557/0.0446 | 0.0564/0.0445 | 0.0679/0.0866 |
| Data/restraints/parameters | 2875/0/202 | 2806/0/202 | 2101/0/202 | 1985/346/375 | 1305/346/363 |
| Goodness-of-fit on F2 | 1.029 | 1.073 | 1.081 | 1.060 | 1.075 |
| Final R indexes [I > = 2σ(I)] | R 1 = 0.0283, wR2 = 0.0717 | R 1 = 0.0221, wR2 = 0.0552 | R 1 = 0.0387, wR2 = 0.0924 | R 1 = 0.0399, wR2 = 0.0994 | R 1 = 0.0514, wR2 = 0.1196 |
| Final R indexes [all data] | R 1 = 0.0352, wR2 = 0.0754 | R 1 = 0.0229, wR2 = 0.0556 | R 1 = 0.0800, wR2 = 0.1159 | R 1 = 0.0699, wR2 = 0.1127 | R 1 = 0.0980, |
| wR2 = 0.1373 | |||||
| Largest diff. peak/hole/e Å−3 | 0.19/−0.20 | 0.26/−0.25 | 0.25/−0.25 | 0.20/−0.21 | 0.23/−0.27 |
| 300 K | 100 K | Ambient | 9.0(2) kbar | 11.5(2) kbar | 13.5(2) kbar | 16.0(2) kbar | 23.4(2) kbar | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Σ, the distortion parameter is the sum of the absolute value of the deviation of all 12 cis N–Fe–N angles from 90°. b Vp is the volume of the Fe octahedron calculated in Olex2.52 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe1–N1 (Å) | 2.1310(16) | 2.1309(11) | 2.129(3) | 2.123(3) | 2.107(3) | 2.066(4) | 1.996(4) | 1.956(11) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe1–N2 (Å) | 2.2235(15) | 2.2173(11) | 2.224(3) | 2.203(3) | 2.185(3) | 2.163(12) | 2.096(11) | 2.01(2) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe1–N3 (Å) | 2.1631(13) | 2.1610(10) | 2.166(2) | 2.146(2) | 2.122(2) | 2.098(5) | 2.034(4) | 1.984(7) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe1′–N1′ (Å) | — | — | — | — | — | 2.026(12) | 1.975(10) | 1.93(2) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe1′–N2′ (Å) | — | — | — | — | — | 2.116(5) | 2.043(5) | 2.029(11) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe1′–N3′ (Å) | — | — | — | — | — | 2.050(5) | 1.989(5) | 1.940(11) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe1 Σa (°) | 76.5(4) | 77.5(3) | 77.7(8) | 84.3(8) | 82.2(7) | 82(2) | 71(2) | 60(4) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe1 Vpb (Å3) | 13.168(6) | 13.137(4) | 13.168(11) | 12.896(9) | 12.601(9) | 12.10(3) | 11.09(2) | 10.25(4) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe1′ Σa (°) | — | — | — | — | — | 70(2) | 66(2) | 55(4) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe1′ Vpb (Å3) | — | — | — | — | — | 11.43(2) | 10.49(2) | 9.96(5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Spin state (Fe1/Fe2) (temperature/pressure) | HS HS 300 K | HS/LS 30 K | HS/HS* LIESST 30 K | HS/HS ambient, 296 K | Mainly HS/LS 9.6(2) kbar, 296 K | LS/LS 15.0(2) kbar, 296 K |
|---|---|---|---|---|---|---|
| Empirical formula | C26 H20 Fe N14 S2 | C26 H20 Fe N14 S2 | C26 H20 Fe N14 S2 | C26 H20 Fe N14 S2 | C26 H20 Fe N14 S2 | C26 H20 Fe N14 S2 |
| Formula weight | 648.53 | 648.53 | 648.53 | 648.53 | 648.53 | 648.53 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/c | P21/c | P21/c | P21/c |
| a/Å | 10.8097(3) | 10.8082(4) | 10.7016(4) | 10.7819(15) | 10.6923(8) | 10.4607(17) |
| b/Å | 15.9326(4) | 15.6979(5) | 15.9017(6) | 15.870(4) | 15.3683(18) | 15.203(4) |
| c/Å | 17.4617(5) | 16.9850(6) | 17.1557(6) | 17.415(3) | 16.4865(12) | 16.348(3) |
| β/° | 106.8470(10) | 107.6180(10) | 106.0400(10) | 106.875(10) | 107.682(4) | 105.886(11) |
| Volume/Å3 | 2878.30(14) | 2746.61(17) | 2805.79(18) | 2851.7(9) | 2581.1(4) | 2500.7(9) |
| Z | 4 | 4 | 4 | 4 | 4 | 4 |
| ρ calc/g cm−3 | 1.497 | 1.568 | 1.535 | 1.511 | 1.669 | 1.723 |
| μ/mm−1 | 0.714 | 0.749 | 0.733 | 0.377 | 0.416 | 0.430 |
| F(000) | 1328.0 | 1328.0 | 1328.0 | 1328.0 | 1328.0 | 1328.0 |
| Crystal size/mm3 | 0.4 × 0.24 × 0.16 | 0.4 × 0.24 × 0.16 | 0.4 × 0.24 × 0.16 | 0.18 × 0.14 × 0.12 | 0.18 × 0.14 × 0.12 | 0.18 × 0.14 × 0.12 |
| λ/Å | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | AgKα (λ = 0.56086) | AgKα (λ = 0.56086) | AgKα (λ = 0.56086) |
| 2θ range for data collection/° | 3.532 to 52.744 | 3.614 to 52.734 | 3.558 to 52.742 | 2.796 to 39.206 | 2.926 to 39.064 | 2.94 to 39.182 |
| Index ranges | −13 ≤ h ≤ 13, | −13 ≤ h ≤ 13, | −12 ≤ h ≤ 13, | −12 ≤ h ≤ 12, | −12 ≤ h ≤ 12, | −12 ≤ h ≤ 12, |
| −19 ≤ k ≤ 19, −21 ≤ l ≤ 21 | −19 ≤ k ≤ 19, −21 ≤ l ≤ 21 | −19 ≤ k ≤ 19, −21 ≤ l ≤ 21 | −14 ≤ k ≤ 15, −19 ≤ l ≤ 19 | −14 ≤ k ≤ 14, −17 ≤ l ≤ 19 | −14 ≤ k ≤ 14, −16 ≤ l ≤ 16 | |
| Reflections collected | 23130 |
31139 |
22558 |
15847 |
22491 |
13610 |
| R int/Rsigma | 0.0340/0.0298 | 0.0363/0.0245 | 0.0357/0.0312 | 0.0858/0.0948 | 0.0599/0.0403 | 0.0727/0.0687 |
| Data/restraints/parameters | 5880/1/407 | 5621/0/403 | 5744/0/403 | 2524/346/403 | 2669/346/403 | 2354/346/393 |
| Goodness-of-fit on F2 | 1.018 | 1.039 | 1.029 | 1.047 | 1.084 | 1.085 |
| Final R indexes [I > = 2σ(I)] | R 1 = 0.0340, wR2 = 0.0792 | R 1 = 0.0279, wR2 = 0.0646 | R 1 = 0.0293, wR2 = 0.0682 | R 1 = 0.0821, wR2 = 0.1916 | R 1 = 0.0498, wR2 = 0.1220 | R 1 = 0.0735, wR2 = 0.1762 |
| Final R indexes [all data] | R 1 = 0.0505, wR2 = 0.0864 | R 1 = 0.0336, wR2 = 0.0673 | R 1 = 0.0379, wR2 = 0.0719 | R 1 = 0.1318, wR2 = 0.2231 | R 1 = 0.0705, wR2 = 0.1343 | R 1 = 0.1088, wR2 = 0.1986 |
| Largest diff. peak/hole/e Å−3 | 0.20/−0.28 | 0.34/−0.45 | 0.36/−0.38 | 0.48/−0.42 | 0.31/−0.34 | 0.46/−0.52 |
Results
Polymorph B and D structural features
The structures obtained here at 300 K are consistent with the previously published structures for the polymorphs. Polymorph, B, crystallises in the monoclinic space group P21/n with half a molecule in the asymmetric unit (Z′ = 0.5) due to the central Fe sitting on an inversion centre, Fig. 1. While Polymorph D crystallises in the monoclinic space group P21/c and has 2 × 0.5 molecules in the asymmetric unit so there are two unique Fe centres, see Fig. 2. However, the main structural features of the two polymorphs are very similar. The Fe(II) centres are in approximately octahedral Fe–N6 coordination geometry, with the six coordinated nitrogen atoms: one from each NCS group and two from each abpt ligand (one pyridyl nitrogen and one triazole nitrogen). The structures contain intramolecular N–H···N hydrogen bonding interactions existing between the NH2 group attached to the triazole and the N of the free pyridyl, and intramolecular C–H···N interactions between a pyridyl C–H and the N of the NH2 group attached to the triazole and a pyridyl C–H and the uncoordinated N on the triazole (Table 1). In addition, there are weak π–π interactions existing between pairs of counterpart abpt pyridyl groups on adjacent molecules. For Polymorph B centroid (N2, C2–C6) to centroid (N7#1, C9#1–C13#1, #1 = ½ + x, 3/2 − y, ½ + z) distance ∼3.7 Å with an offset of ∼1.3 Å at 300(2) K, while for Polymorph D only Fe2 is involved in weak π–π interactions with the centroid (N9, C15–C19) to centroid (N14#2, C22#2–C26#2, #2 = −x, ½ + y, ½ − z) distance ∼3.8 Å, offset distance ∼2.0 Å at 300(2) K. At 300(2) K the Fe–N bond lengths, distortion parameter and octahedron volume are consistent with those associated with a HS Fe centre for both polymorphs. | ||
| Fig. 1 Structure of B with the atomic numbering scheme depicted at 100(2) K. Thermal ellipsoids depicted at the 50% probability level. Symmetry code i = 1 − x, 1 − y, 1 − z. | ||
 | ||
| Fig. 2 Structure of D with the atomic numbering scheme depicted at 100(2) K. Thermal ellipsoids depicted at the 50% probability level. Symmetry codes I = −x, 1 − y, 1 − z, ii = −x, 1 − y, −z. | ||
Polymorph B thermal behaviour
In line with the magnetic data for Polymorph B no thermal spin transition was observed upon cooling from 300(2) to 100(2) K, at ambient pressure, and the structures obtained at these temperatures were essentially identical to the published structure reported at 293(2) K.42 It is however known that B does undergo a thermal spin transition above 4.4 kbar.42Polymorph B pressure induced spin crossover
The study carried out here monitored the structure of polymorph B upon the application of pressure at room temperature, with structures reported between ambient pressure and 23.4(2) kbar. The unit cell parameters are provided in Table 2, Table S1 and Fig. S1 (ESI†).Up to a pressure of 11.5(2) kbar reductions in the length of all of the cell axes are observed which results in an ∼8.8% decrease in the cell volume. However, the key structural features for B up to this pressure are essentially consistent with those already described at 300(2) K and the parameters associated with the Fe centre indicate that it is HS, Table 3 and Table S1 (ESI†).
Somewhere between 11.5(2) and 13.5(2) kbar the crystal undergoes a phase transition from monoclinic to triclinic (P). The cell axes at 13.5(2) kbar are similar in magnitude to those obtained at ambient pressure (with a and b switched in line with cell conventions) however the initial monoclinic α and γ cell angles (β and γ respectively in the triclinic cell) have distorted significantly away from 90° hence the triclinic cell. The phase transition results in an increase in the number of molecules in the asymmetric unit from Z′ = 0.5 (ambient pressure) to Z′ = 2 × 0.5 (13.5(2) kbar). The phase transition occurs within a single crystal so despite the reduction in symmetry it is not surprising that the overall structure is still very similar to the 300(2) K structure discussed previously. However, with the phase change is the start of a spin crossover with a reduction in the Fe–N bond lengths for the system with one of the Fe centres undergoing a significantly larger change than the other at this stage, 0.02–0.04 Å (Fe1) and 0.07–0.08 Å (Fe1′). Alongside the change in the Fe–N bond length there is also a reduction in the distortion parameter (Σ) and octahedron volume (Vp) which is commonly observed for spin crossover compounds. As expected larger reductions are observed for Fe1′ than Fe1, due to the spin crossover having progressed further, see Fig. 3, Table 3 and Table S1 (ESI†).
 | ||
| Fig. 3 Change in Fe–N bond length for B as a function of pressure. At 13.5 kbar and above there were two unique Fe centres, the second of which has been given the solid filled markers. | ||
Increasing the pressure in the DAC further to 23.4(2) kbar resulted in further reductions in the length of the cell axes and increases in all of the cell angles which results in the cell volume being ∼15% smaller than at ambient pressure. The structure is still very similar to that observed at ambient pressure but it was noted that there were differences in the two unique molecules. The angle calculated between planes through the two 6 membered rings on the same ligand changed from 35.1(1)° at ambient pressure to 38.3(5)° (Fe1) and 32.0(6)° (Fe1′). Nonetheless both of the unique Fe centres appear to have undergone a spin transition from HS towards LS, with the Fe–N bond lengths now ranging from 0.14–0.18 Å shorter than at ambient pressure. Usually a reduction of ∼0.2 Å is observed for a complete transition so it is possible that the transition is not quite complete. The distortion parameter and octahedron volume have also decreased significantly towards those expected for a LS Fe centre. This indicates that B has undergone a thermally inaccessible spin transition (Table 3). Given the isostructural nature with the [Fe(abpt)2(NCSe)2] polymorph B,42 it would be interesting to establish whether [Fe(abpt)2(NCSe)2] polymorph B also undergoes a pressure induced spin crossover.
Polymorph D thermal spin crossover and LIESST
Upon cooling one of the unique Fe centres, Fe2, undergoes a spin transition, T1/2 = 162 K,39 and the HS/HS (room temperature) and HS/LS (90 K and 20 K) structures have previously been reported.41 The LIESST HS* structure for polymorph D has also been previously reported using both a 532 nm, 5 mW laser and a 532 nm, 40 mW laser at 20 K.41In the study reported herein, the structural parameter changes upon cooling are monitored upon cooling from 300 to 30 K. Whilst the a axis length does not change significantly as a result of the spin crossover, upon cooling there are small decreases in the b and c axes lengths and a small increase in the β angle, which results in an ∼4.5% decrease in the cell volume across the temperature range, see Table 4, Table S2 and Fig. S2 (ESI†). Typically, the Fe centre (Fe2) which undergoes the spin crossover showed a decrease of ∼0.18 Å in the Fe2–N bond lengths and a reduction in the distortion parameter (Σ) of ∼15° and in the octahedron volume (Vp) of ∼2.8 Å3 (Fig. 4 and Table 5). These values are in line with those seen for Polymorph A38 or other Fe–N spin crossover compounds.19 No significant changes were observed in the parameters associated with Fe1 which does not undergo a spin transition upon cooling.
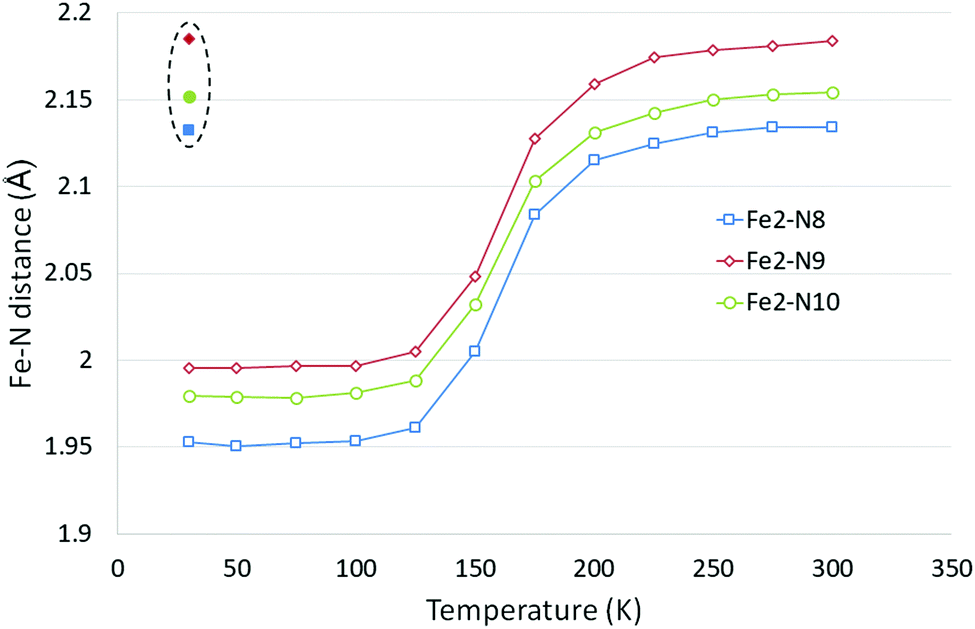 | ||
| Fig. 4 Fe2–N bond length changes in D as a function of temperature, LIESST values are contained within the dashed oval. Error bars are not included as they are essentially obscured by the data markers. | ||
| 300(2) K | 275(2) K | 250(2) K | 225(2) K | 200(2) K | 175(2) K | 150(2) K | 125(2) K | 100(2) K | 75(2) K | |
|---|---|---|---|---|---|---|---|---|---|---|
| Fe1–N1 (Å) | 2.1146(18) | 2.1179(19) | 2.1153(18) | 2.1167(17) | 2.1170(17) | 2.1172(16) | 2.1145(14) | 2.1141(16) | 2.1140(16) | 2.1152(15) |
| Fe1–N2 (Å) | 2.2243(16) | 2.2209(17) | 2.2224(16) | 2.2207(15) | 2.2199(16) | 2.2179(15) | 2.2184(13) | 2.2153(15) | 2.2148(13) | 2.2132(15) |
| Fe1–N3 (Å) | 2.1530(16) | 2.1508(16) | 2.1505(16) | 2.1490(15) | 2.1490(15) | 2.1500(15) | 2.1529(13) | 2.1500(15) | 2.1497(13) | 2.1487(14) |
| Fe2–N8 (Å) | 2.134(2) | 2.134(2) | 2.1313(19) | 2.1247(18) | 2.1151(20) | 2.0836(18) | 2.0052(15) | 1.9613(16) | 1.9536(13) | 1.9521(15) |
| Fe2–N9 (Å) | 2.1838(16) | 2.1812(16) | 2.1786(16) | 2.1743(15) | 2.1589(16) | 2.1278(16) | 2.0484(14) | 2.0053(15) | 1.9969(13) | 1.9970(14) |
| Fe2–N10 (Å) | 2.1546(15) | 2.1531(16) | 2.1503(15) | 2.1424(15) | 2.1315(16) | 2.1036(15) | 2.0322(14) | 1.9884(15) | 1.9812(13) | 1.9784(15) |
| Σ a Fe1 (°) | 73.1(5) | 73(5) | 72.7(5) | 72.4(4) | 72.1(5) | 72.1(5) | 71(4) | 70.1(4) | 70.4(3) | 70.2(4) |
| V p b Fe1 (Å3) | 13.024(6) | 13.012(7) | 13.006(6) | 13.001(6) | 12.998(6) | 12.998(6) | 12.996(5) | 12.957(6) | 12.951(5) | 12.950(6) |
| Σ a Fe2 (°) | 64.8(5) | 64.3(4) | 64.2(5) | 63.5(4) | 63.2(4) | 60.8(4) | 54(4) | 49.5(3) | 49.5(3) | 49.6(4) |
| V p b Fe2 (Å3) | 12.965(6) | 12.942(7) | 12.901(6) | 12.799(6) | 12.599(6) | 12.109(6) | 10.917(5) | 10.267(5) | 10.152(4) | 10.128(5) |
| 50(2) K | 30(2) K | 30(2) K LIESST | Ambient | 1.8(2) kbar | 7.5(2) kbar | 9.6(2) kbar | 12(2) kbar | 15(2) kbar | Ambient pressure released | |
|---|---|---|---|---|---|---|---|---|---|---|
| Fe1–N1 (Å) | 2.1165(15) | 2.1166(14) | 2.1232(14) | 2.118(5) | 2.116(4) | 2.100(5) | 2.069(5) | 2.045(5) | 1.984(9) | 2.106(10) |
| Fe1–N2 (Å) | 2.2126(15) | 2.2115(14) | 2.2203(14) | 2.212(4) | 2.210(4) | 2.179(5) | 2.144(6) | 2.117(5) | 2.012(10) | 2.203(11) |
| Fe1–N3 (Å) | 2.1475(15) | 2.1503(14) | 2.1434(14) | 2.150(4) | 2.149(3) | 2.135(4) | 2.110(5) | 2.084(4) | 1.988(8) | 2.147(8) |
| Fe2–N8 (Å) | 1.9504(15) | 1.9527(14) | 2.1320(15) | 2.129(5) | 2.127(4) | 1.985(4) | 1.956(5) | 1.949(4) | 1.947(8) | 2.144(11) |
| Fe2–N9 (Å) | 1.9958(15) | 1.9958(13) | 2.1848(14) | 2.180(5) | 2.176(5) | 2.026(6) | 1.993(6) | 1.984(5) | 1.969(10) | 2.188(13) |
| Fe2–N10 (Å) | 1.9792(14) | 1.9798(13) | 2.1519(14) | 2.161(4) | 2.147(4) | 2.007(5) | 1.983(5) | 1.976(5) | 1.983(10) | 2.165(10) |
| Σ a Fe1 (°) | 70.1(4) | 70.4(3) | 74.1(4) | 72(1) | 71(1) | 70(1) | 68(2) | 67(1) | 56(3) | 73(3) |
| V p b Fe1 (Å3) | 12.948(6) | 12.958(5) | 13.016(5) | 12.954(15) | 12.953(15) | 12.61(2) | 12.127(6) | 11.707(19) | 10.40(3) | 12.82(4) |
| Σ a Fe2 (°) | 50.0(4) | 49.7(4) | 66.1(4) | 65(1) | 63(1) | 52(2) | 49(2) | 50(1) | 52(3) | 61(3) |
| V p b Fe2 (Å3) | 10.118(5) | 10.134(4) | 12.954(6) | 12.952(18) | 12.881(15) | 10.594(17) | 10.174(17) | 10.056(16) | 10.00(3) | 13.16(4) |
The LIESST HS* structure was also obtained during our study using a 670 nm, 5 mW laser at 30(2) K, with no evidence of linkage isomerism. This was as expected since a previous study41 reporting the LIESST structure for D found no linkage isomerism when a low powered laser was used, as is the case here, but did find linkage isomerism when using a high powered laser. Changes were observed in the cell parameters of the 30(2) K LIESST HS* structure relative to the 30(2) K ground state the a axis decreases slightly along with the β angle, while the b and c axes increase slightly resulting in a 2% increase in the cell volume. As expected, the Fe2–N distances, distortion parameter and octahedron volume parameters also increase and are consistent with those associated with a HS Fe centre.
Polymorph D pressure induced spin crossover
The effect of pressure on the spin transition behaviour of polymorph D was examined by collecting datasets using a DAC at six different pressures between ambient and 15.0(2) kbar. For all structures, the crystal system and spacegroup remained as monoclinic P21/c, however significant structural changes were observed, see Fig. 5 and Table 5. | ||
| Fig. 5 Fe–N bond length changes in D as a function of pressure. Error bars are not included as they are essentially obscured by the data markers. | ||
At ambient and 1.8(2) kbar the structure obtained was consistent with the room temperature structure i.e. both Fe centres were in the HS state. At 7.5(2) kbar the Fe2–N bond lengths, Σ and Vp had reduced significantly indicating that Fe2 had mostly undergone a spin crossover, which appeared to be essentially complete at 9.6(2) kbar. The structure at 7.5(2) kbar is therefore very similar to the low temperature structure obtained for D. What is noticeable at 9.6(2) kbar is that the Fe1–N bond lengths, Σ and Vp have started to reduce significantly, and this continues until the 15.0(2) kbar dataset where the transition appears to be nearly but probably not quite complete as the change in Fe–N bond lengths (∼0.13–0.2 Å) is slightly less than the expected ∼0.2 Å reduction. It is possible looking at the plot of bond length versus pressure for Fe1 that the spin transition has begun at 7.5(2) kbar however this is not totally clear. Alongside the changes in structural parameters relating to the Fe centres there was a small reduction of ∼0.1 Å in the a axis length and a slightly larger decrease in the b and c axes of ∼0.6 Å and ∼1 Å respectively. The β angle initially increased by ∼1° as the pressure increased up to 9.6(2) kbar before dropping to ∼1° below the value seen at ambient pressure. The cell volume showed an ∼12.5% reduction between ambient pressure and 15.0(2) kbar.
After reaching 17.0 kbar, at which pressure the data quality were poor, the pressure in the DAC was released and another dataset collected at ambient pressure. This showed that the transition was reversible with both Fe centres having reverted back to the HS state with the cell parameters and Fe parameters consistent with those at the start of the experiment (Table 5 and Table S3, ESI†). It was noted that the quality of the crystal had reduced as a result of the change in pressure.
This result is particularly interesting as applying pressure to D has resulted in the occurrence of a spin transition at both Fe1 and Fe2, a situation that is inaccessible thermally.
Summary of the four [Fe(abpt)2(NCS)2] polymorphs
As discussed in the introduction, the four reported polymorphs of [Fe(abpt)2(NCS)2] are particularly interesting as they show quite different spin crossover behaviour despite having very similar structures. Some of these properties along with selected structural parameters are detailed in Table 6.| Polymorph | A | B | C | D | ||
|---|---|---|---|---|---|---|
| Fe1 | Fe2 | Fe1 | Fe2 | |||
| Z′ | 0.5 | 0.5 | 2 × 0.5 | 2 × 0.5 | ||
| Thermal spin crossover | Yes | No | Yes | No | No | Yes |
| T 1/2 (K) | 188 | — | 86 | 162 | ||
| LIESST | Yes | — | Yes commensurate modulated structure | — | — | Yes |
| Pressure induced spin crossover | Yes | Yes by 23.6(2) kbar, after phase transition (11.5(2)–13.5(2) kbar) giving Z′ = 2 × 0.5 | Unknown | Yes > ∼15.0(2) kbar | Yes > ∼9.6(2) kbar | |
Examining an overlay of the unique molecules shows that the molecular confirmations are relatively similar, as would be expected. The largest differences are seen in the relative orientations of the two pyridyl rings on the abpt ligand for B (green) and D Fe2 (grey), see Fig. 6. These happen to be two of the Fe centres that don’t undergo a thermal spin crossover, however this may well just be coincidental for these structures and it is most likely that it is a function of the resultant packing. It is widely recognised that intermolecular interactions play a significant role in the spin crossover behaviour of compounds.19,58Table 7 summarises the reported spin crossover behaviour of Fe(abpt)2X2 compounds found in the Cambridge Structural Database, along with details of π–π interactions. The main points to note are that all of the Fe centres that do undergo at least a partial thermal spin crossover display π–π interactions between their adpt ligands, while and all of the Fe centres for which the attached abpt ligands are not involved in π–π interactions are not reported to undergo a thermal spin crossover. There are of course a couple of anomalies, in addition to B discussed herein, there are also two complexes that are not reported to undergo a thermal spin crossover that do have π–π interactions Fe(abpt)2[N(CN)2]259 and Fe(abpt)2(NCSe)2 polymorph B,42 the latter of which is isostructural with B reported herein. As discussed herein B does undergo a pressure induced spin crossover and also a thermal spin crossover at pressure above 4.4 kbar.42
 | ||
| Fig. 6 Overlay of each of the unique Fe centres at room temperature, constructed in Mercury by selecting the Fe1/Fe2, N1/N8, N2/N9 and N3/N10 and calculating the overlay. Two different views of overlay shown (cyan) A, (green) B, (red) C Fe1, (blue) C Fe2, (yellow) D Fe1, (grey) D Fe2. | ||
| Structurea | Temperature | Thermal spin crossover? | T 1/2 | π–πb | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a If multiple structures are reported for a compound only one structure has been selected for each unique polymorph to avoid duplication. b Both ends means that both the pyridyl rings on the abpt are involved in π–π interactions with the two pyridyl rings on an abpt on another molecule while one end means only one pyridyl ring on an abpt interacts with a pyridyl on an abpt in another molecule. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| A37–39 | 275 | Y | 188 | Both ends | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| B42 | 300 | N | — | One end | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C39,40 | RT | Fe1 | Y | 86 | Both ends | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe2 | N | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| D39,41 | 300 | Fe1 | N | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe2 | Y | 162 | One end | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe(abpt)2(C(CN)3)258 | RT | Y | 336 | Both ends | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe(abpt)2((NC)2CC(OCH3)C(CN)2)258 | RT | N | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe(abpt)2((NC)2CC(OC2H5)C(CN)2)258 | RT | Y: magnetic data indicates ∼60% spin crossover | 377 | Both ends | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe(abpt)2((NC)2CC(OC2H5)C(CN)2)258 | RT | Fe1 | Y: magnetic data indicates ∼60% crossover | 383 | Both ends | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe2 | Both ends | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe(abpt)2[N(CN)2]260 | RT | N: redetermined see paper59 | — | Both ends | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe(abpt)2[N(CN)2]259 | RT | Fe1 | Y | ∼86 stepped Fe1 then Fe2 | Both ends | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe2 | Both ends | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe(abpt)2(C16SO3)261 | 92 | Y | Unknown | One end | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe(abpt)2Cl262 | 85 | N | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe(abpt)2(NCSe)2 A37,43 | 375 | Y | Both ends | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe(abpt)2(NCSe)2 B42 | RT | N | — | One end | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Conclusions
In summary, the spin crossover behaviour of [Fe(abpt)2(NCS)2] polymorph B and D have been structurally characterised. In the case of B, it does not undergo a thermal spin crossover but was found to undergo a pressure induced spin crossover. Somewhere between 11.5(2) and 13.5(2) kbar it undergoes a phase transition from a monoclinic to triclinic crystal system with Z′ increasing from 0.5 to 2 × 0.5. At the same point the Fe centres start to display a spin crossover which by 23.4(2) kbar is approximately complete.Polymorph D has previously been reported to undergo a thermal spin crossover in one of the two unique Fe centres, the second Fe centre remains HS upon cooling. Herein the cell parameters and structural parameters were monitored from 300(2) to 30(2) K. A LIESST structure was also obtained at 30(2) K. In addition, a high pressure single crystal study showed that D underwent a stepped pressure induced spin crossover. The Fe centre that undergoes a thermal spin transition underwent a pressure induced spin crossover by ∼9.6(2) kbar while the second Fe centre began to undergo the spin crossover at a higher pressure and was essentially LS by ∼15.0(2) kbar.
In all cases the spin crossover upon cooling or increasing pressure resulted in the expected reduction in the Fe–N bond lengths, Σ and Vp for the Fe centre that underwent the spin crossover.
The ability to undergo a spin crossover for general Fe(abpt)2X2 complexes could be linked to the presence of intermolecular π–π interactions. All of the complexes that undergo at least a partial thermal spin crossover displayed π–π interactions between pyridyl rings on abpt ligands and all of the complexes that did not have π–π interactions did not undergo a thermal spin crossover. There were three complexes identified with π–π interactions but that do not undergo a thermal spin crossover, however B does undergo a pressure induced spin transition.
It was particularly interesting to note within this study that high pressure was able to induce a thermally inaccessible spin crossover at Fe centres in both B and D.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. E. M. was grateful to Professor Jonathan W. Steed (Durham University) for useful discussions. She was also grateful to the EPSRC and Durham University for financial support.References
- L. Cambi and L. Szegö, Ber. Dtsch. Chem. Ges., 1931, 64(10), 2591–2598 CrossRef.
- P. Gütlich, V. Ksenofontov and A. B. Gaspar, Coord. Chem. Rev., 2005, 249, 1811–1829 CrossRef.
- A. Hauser, Light-induced spin crossover and the high-spin → low-spin relaxation. Spin crossover in transition metal compounds II, Top. Curr. Chem., 2004, 234, 155–198 CrossRef CAS.
- M. Nakaya, R. Ohtani, L. F. Lindoy and S. Hayami, Inorg. Chem. Front., 2021, 8, 484–498 RSC.
- P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342–391 CrossRef PubMed.
- S. De, L. M. Chamoreau, H. El Said, Y. L. Li, A. Flambard, M. L. Boillot, S. Tewary, G. Rajaraman and R. Lescouezec, Front. Chem., 2018, 6, 326 CrossRef PubMed.
- T. Matsuyama, K. Nakata, H. Hagiwara and T. Udagawa, Crystals, 2019, 9, 276 CrossRef CAS.
- D. Sertphon, P. Harding, K. S. Murray, B. Moubaraki, S. M. Neville, L. Liu, S. G. Telfer and D. J. Harding, Crystals, 2019, 9, 116 CrossRef.
- T. Houari, E. Cuza, D. Pinkowicz, M. Marchivie, S. Yefsah and S. Triki, Crystals, 2018, 8, 401 CrossRef.
- A. R. Craze, N. F. Sciortino, M. M. Badbhade, C. J. Kepert, C. E. Marjo and F. Li, Inorganics, 2017, 5, 62 CrossRef.
- M. Nihei, T. Shiga, Y. Maeda and H. Oshio, Coord. Chem. Rev., 2007, 251, 2606–2621 CrossRef CAS.
- K. Takahashi, K. Yamamoto, T. Yamamoto, Y. Einaga, Y. Shiota, K. Yoshizawa and H. Mori, Crystals, 2019, 9, 81 CrossRef.
- W. Phonsri, L. C. Darveniza, S. R. Batten and K. S. Murray, Inorganics, 2017, 5, 51 CrossRef.
- T. Nakanishi, A. Okazawa and O. Sato, Inorganics, 2017, 5, 53 CrossRef.
- A. B. Gaspar, M. C. Muñoz, V. Niel and J. A. Real, Inorg. Chem., 2001, 40, 9–10 CrossRef CAS PubMed.
- P. Nielsen, H. Toftlund, A. D. Bond, J. F. Boas, J. R. Pilbrow, G. R. Hanson, C. Noble, M. J. Riley, S. M. Neville, B. Moubaraki and K. S. Murray, Inorg. Chem., 2009, 48, 7033–7047 CrossRef CAS PubMed.
- A. Ondo and T. Ishida, Crystals, 2018, 8, 155 CrossRef.
- O. A. Qamar, C. Cong and H. B. Ma, Dalton Trans., 2020, 49, 17106–17114 RSC.
- P. Guionneau, Dalton Trans., 2014, 43, 382–393 RSC.
- K. S. Kumar and M. Ruben, Angew. Chem., Int. Ed., 2021, 60, 7502–7521 CrossRef CAS PubMed.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- J. Linares, E. Codjovi and Y. Garcia, Sensors, 2012, 12, 4479 CrossRef CAS PubMed.
- Spin Crossover in Transition Metal Compounds I, II and III, ed. P. Gütlich and H. A. Goodwin, Springer-Verlag, Berlin, Heidelberg, New York, 2004 Search PubMed.
- Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Ltd, Chichester, UK, 2013 Search PubMed.
- M. Griffin, S. Shakespeare, H. J. Shepherd, C. J. Harding, J.-F. Létard, C. Desplanches, A. E. Goeta, J. A. K. Howard, A. K. Powell, V. Mereacre, Y. Garcia, A. D. Naik, H. Mueller-Bunz and G. G. Morgan, Angew. Chem., Int. Ed., 2011, 50, 896–900 CrossRef CAS PubMed.
- J. A. Real, B. Gallois, T. Granier, F. Suezpanama and J. Zarembowitch, Inorg. Chem., 1992, 31, 4972–4979 CrossRef CAS.
- T. Granier, B. Gallois, J. Gaultier, J. A. Real and J. Zarembowitch, Inorg. Chem., 1993, 32, 5305–5312 CrossRef CAS.
- A. B. Gaspar, G. Molnar, A. Rotaru and H. J. Shepherd, C. R. Chim, 2018, 21, 1095–1120 CrossRef CAS.
- Z. Yan, L. F. Zhu, L. W. Zhu, Y. Meng, M. N. Hoque, J. L. Liu, Y. C. Chen, Z. P. Ni and M. L. Tong, Inorg. Chem. Front., 2017, 4, 921–926 RSC.
- J.-F. Létard, P. Guionneau, L. Rabardel, J. A. K. Howard, A. E. Goeta, D. Chasseau and O. Kahn, Inorg. Chem., 1998, 37, 4432–4441 CrossRef PubMed.
- E. Collet and P. Guionneau, C. R. Chim, 2018, 21, 1133–1151 CrossRef CAS.
- J. A. Wolny, V. Schunemann, Z. Nemeth and G. Vanko, C. R. Chim, 2018, 21, 1152–1169 CrossRef CAS.
- S. Chorazy, T. Charytanowicz, D. Pinkowicz, J. H. Wang, K. Nakabayashi, S. Klimke, F. Renz, S. Ohkoshi and B. Sieklucka, Angew. Chem., Int. Ed., 2020, 59, 15741–15749 CrossRef CAS PubMed.
- T. Kosone, S. Okuda, M. Kawata, S. Arai, R. Kosuge and T. Kawasaki, ACS Omega, 2021, 6, 12187–12193 CrossRef CAS PubMed.
- C. Lochenie, W. Bauer, A. P. Railliet, S. Schlamp, Y. Garcia and B. Weber, Inorg. Chem., 2014, 53, 11563–11572 CrossRef CAS PubMed.
- J. Tao, R.-J. Wei, R.-B. Huang and L.-S. Zheng, Chem. Soc. Rev., 2012, 41, 703–737 RSC.
- N. Moliner, M. C. Muñoz, S. Létard, J.-F. Létard, X. Solans, R. Burriel, M. Castro, O. Kahn and J. A. Real, Inorg. Chim. Acta, 1999, 291, 279–288 CrossRef CAS.
- H. E. Mason, W. Li, M. A. Carpenter, M. L. Hamilton, J. A. K. Howard and H. A. Sparkes, New J. Chem., 2016, 40, 2466–2478 RSC.
- C.-F. Sheu, S.-M. Chen, S.-C. Wang, G.-H. Lee, Y.-H. Liu and Y. Wang, Chem. Commun., 2009, 7512–7514 RSC.
- C.-H. Shih, C.-F. Sheu, K. Kato, K. Sugimoto, J. Kim, Y. Wang and M. Takata, Dalton Trans., 2010, 39, 9794–9800 RSC.
- C.-F. Sheu, C.-H. Shih, K. Sugimoto, B.-M. Cheng, M. Takata and Y. Wang, Chem. Commun., 2012, 48, 5715–5717 RSC.
- A. B. Gaspar, M. C. Mũnoz, N. Moliner, V. Ksenofontov, G. Levchenko, P. Gütlich and J. A. Real, Monatsh. Chem., 2003, 134, 285–294 CrossRef CAS.
- H. E. Mason, M. L. Hamilton, J. A. K. Howard and H. A. Sparkes, New J. Chem., 2018, 42, 18028–18037 RSC.
- A. E. Goeta, L. K. Thompson, C. L. Sheppard, S. S. Tandon, C. W. Lehmann, J. Cosier, C. Webster and J. A. K. Howard, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, C55, 1243–1246 CrossRef CAS.
- Bruker, SMART-NT, Data Collection Software, Version 5.63, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 2000 Search PubMed.
- Bruker, SAINT+ Integration Engine, Data Reduction Software, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 2007 Search PubMed.
- Bruker, SAINT-NT, Data Reduction Software Ver. 6.14, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 2000 Search PubMed.
- Bruker, SADAB, Bruker AXS area detector scaling and absorption correction, Bruker Analytical X-ray Instruments Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- L. Palatinus, S. J. Prathapa and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46, 2774–2780 CrossRef CAS.
- M. R. Probert, J. A. Coome, A. E. Goeta and J. A. K. Howard, Acta Crystallogr., Sect. A: Found. Crystallogr., 2011, 67(Supplement), C528 Search PubMed.
- M. R. Probert, C. M. Robertson, J. A. Coome, J. A. K. Howard, B. C. Michell and A. E. Goeta, J. Appl. Crystallogr., 2010, 43, 1415–1418 CrossRef CAS.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
- Bruker, APEX2, Data Collection Software, Version 1.08, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 2004 Search PubMed.
- G. Dupouy, M. Marchivie, S. Triki, J. Sala-Pala, J. Y. Salaun, C. J. Gomez-Garcia and P. Guionneau, Inorg. Chem., 2008, 47, 8921–8931 CrossRef CAS PubMed.
- C. F. Sheu, S. Pillet, Y. C. Lin, S. M. Chen, I. J. Hsu, C. Lecomte and Y. Wang, Inorg. Chem., 2008, 47, 10866–10874 CrossRef CAS PubMed.
- N. Moliner, A. B. Gaspar, M. C. Munoz, V. Niel, J. Cano and J. A. Real, Inorg. Chem., 2001, 40, 3986–3991 CrossRef CAS PubMed.
- N. G. White, H. L. C. Feltham, C. Gandolfi, M. Albrecht and S. Brooker, Dalton Trans., 2010, 39, 3751–3758 RSC.
- J. A. Kitchen, A. Noble, C. D. Brandt, B. Moubaraki, K. S. Murray and S. Brooker, Inorg. Chem., 2008, 47, 9450–9458 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2086314–2086340. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1nj02607k |
| ‡ Died December 2019. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |