
Cytotoxic and antitumor peptides as novel chemotherapeutics
Xin
Luan†
a,
Ye
Wu†
a,
Yi-Wen
Shen
a,
Hong
Zhang
a,
Yu-Dong
Zhou
ab,
Hong-Zhuan
Chen
a,
Dale G.
Nagle
*ac and
Wei-Dong
Zhang
 *ad
*ad
aInstitute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China
bDepartment of Chemistry and Biochemistry, College of Liberal Arts, University of Mississippi, University-1848, MS 38677, USA
cDepartment of BioMolecular Sciences and Research Institute of Pharmaceutical Sciences, School of Pharmacy, University of Mississippi, University-1848, MS 38677, USA. E-mail: dnagle@olemiss.edu
dSchool of Pharmacy, Second Military Medical University, Shanghai, 200433, China. E-mail: wdzhangy@hotmail.com
First published on 10th August 2020
Abstract
Covering: up to 2020
Treatment resistance and drug-induced refractory malignancies pose significant challenges for current chemotherapy drugs. There have been increasing research efforts aimed at developing novel chemotherapeutics, especially from natural products and related derivatives. Natural cytotoxic peptides, an emerging source of chemotherapeutics, have exhibited the advantage of overcoming drug resistance and displayed broad-spectrum antitumor activities in the clinic. This highlight examines the increasingly popular cytotoxic peptides from isolated natural products. In-depth review of several peptides provides examples for how this novel strategy can lead to the improved anti-tumor effects. The mechanisms and current application of representative natural cytotoxic peptides (NCPs) have also been discussed, with a particular focus on future directions for interdisciplinary research.
 Xin Luan | Associate Prof. Xin Luan received his BSc in Pharmaceutics from China Pharmaceutical University (2011). He was awarded his PhD degree in Pharmacology at Shanghai Jiao Tong University (2016), under the supervision of Prof. Hong-Zhuan Chen. He subsequently worked for two years at the University of Michigan under the guidance of Prof. Duxin Sun. His current appointment is Associate Professor at the Shanghai University of Traditional Chinese Medicine. He is currently working on anti-tumor pharmacology and delivery systems of natural products. |
 Ye Wu | Ye Wu obtained his BSc in Pharmaceutics (2013) and Master degree in Pathology and Pathophysiology (2017) at Chengdu Medical College. He is currently pursuing his PhD studies at Shanghai University of Traditional Chinese Medicine, under the supervision of Prof. Wei-Dong Zhang and Associate Prof. Xin Luan. His research mainly focuses on the conformational constraint and targeted delivery of natural cytotoxic peptides. |
 Dale G. Nagle | Prof. Dale G. Nagle obtained his PhD in Pharmacy/Marine Natural Products from Oregon State University in 1995 with Prof. William H. Gerwick. Following postdoctoral work in marine chemical ecology and small-molecule regulation of transcription, respectively, he joined the University of Mississippi and is a Professor in the Department of BioMolecular Sciences, an Associate Member of the Molecular Cancer Therapeutics Program in the UM Medical Center's Cancer Institute, and an International Professor at the Shanghai University of Traditional Chinese Medicine. His primary areas of research are mitochondria-targeted natural products, traditional Chinese medicines, natural products that regulate hypoxic signaling, and drug discovery for metastatic breast cancer. |
 Wei-Dong Zhang | Prof. Wei-Dong Zhang obtained his BSc and Master degree in Natural Medicinal Chemistry from the Second Military Medical University in 1988 and 1991, respectively. He received his PhD in Natural Medicinal Chemistry from the Shanghai Institute of Pharmaceutical Industry (1998), under the supervision of Professor Hui-Ting Li. He is currently a professor of the Shanghai University of Traditional Chinese Medicine and Second Military Medical University. His research mainly focuses on Chinese medicine formulas, isolation, structural identification and modification, total synthesis, and structure–activity relationships of bioactive natural products. |
1 Introduction
Cancer is a major public health problem and has become the second leading cause of death worldwide, after cardiovascular and cerebrovascular diseases. Despite considerable progress in the treatment of various forms of cancer, each of the current therapies still has its own limitation(s). For early stage cancer, surgery and radiation are effective approaches to remove primary tumors. However, local treatments cannot prevent tumor invasion/metastasis, and the metastatic spread of tumor cells is the primary cause of cancer-related mortality.1 Immunotherapy is revolutionizing the treatment of cancer by checkpoint blockade of the cytotoxic T lymphocyte-associated protein 4 (CTLA-4) or the programmed cell death 1 (PD-1) pathway. However, only a relatively small group of patients responds to immune-checkpoint blockade therapy and considerable uncertainty remains in the application of cancer immunotherapy. In contrast, chemotherapeutics, including anthracyclines and taxanes, are still listed as first-line drugs for the systemic treatment of advanced and metastatic cancers, despite the acute nonspecific cytotoxicity against normal tissues and the risk of developing multi-drug resistance.2 Over the last two decades, cytotoxic peptides have emerged as promising chemotherapeutics to combat cancer and multidrug resistant infections.As natural defences, peptide-based toxins and skin secretions from animals have been used to cope with external threats (e.g., predators). At the same time, peptidyl components from endogenic immune/defence systems have also shown potential anti-bacteria and antitumor effects. Because of their unique structural and mechanistic diversity, these natural cytotoxic peptides (NCPs) provide an unparalleled source of lead compounds for chemotherapeutic development, and continue to play an important role in antitumor drug discovery and development.
In comparison to ‘traditional’ small-molecule chemotherapeutics that often non-selectively inhibit cellular growth in both normal and neoplastic tissues,3 NCPs have exhibited several highly appealing properties: (1) they typically range from 5 to 50 amino acid residues in length, making it easy for chemical synthesis and modification; (2) some relatively selective NCPs exhibit minimal off-target effects; (3) the most common multiple-targeted mechanisms of cytotoxic peptides, especially those selective for tumor-specific cell membrane lysis, are believed to overcome certain forms of drug resistance; and (4) their molecular scaffolds are suitable to modulate relatively undruggable targets and/or protein–protein interactions.4 After comprehensively considering the species source (animal, plant and microorganism), structural diversity (α-helix, β-sheets, loop alone or in combination) and significance of NCPs, we chose a series of representative NCPs to highlight their well-studied therapeutic targets and latest applications.
However, it is worth noting that the successful translation of NCPs still has some challenges, including suboptimal therapeutic activity, structural instability, potential off-target effects, and restricted membrane-permeability. To address those shortcomings, we also proposed the potential solutions and future directions to advance NCPs-based chemotherapeutic drug discovery.
2 Antitumor targets of NCPs
2.1 Cell membrane
There are two types of mechanisms involved in the membrane disruption/permeation effects of NCPs: “carpet-like” and “toroidal pore” models. In the “carpet-like” model, the NCPs are placed in parallel to the cell membrane, like a carpet within a structure.5 Aurein 1.2 and citropin 1.1 (Fig. 1 and Table 1) are a family of peptides (13–25 residues) that were originally isolated from the defensive skin secretions of the closely related Australian tree frogs Litoria aurea and Litoria citropa, respectively.6,7 As surface-acting membrane disrupting peptides, they both display a broad-spectrum inhibitory effects on over 90% of the tumor cell lines from the US National Cancer Institute's 60-cell line panel via the “carpet-like” mechanism [Fig. 2(1)].8 The C-terminal amide group has been shown to be critical for structural stabilization, charge, and bioactivity.8 In the “toroidal model”, the NCPs with lipid headgroups are vertically embedded in the plasma membrane bilayer. For example, magainin 2 (Fig. 1 and Table 1), a class of α-helical antimicrobial peptides (AMPs) from the skin of the African clawed frog, Xenopus laevis, can irreversibly lyse tumor cells by inducing transmembrane pores like a typical “toroidal model” agent [Fig. 2(1)], and have a relatively low toxicity towards normal cells.9,10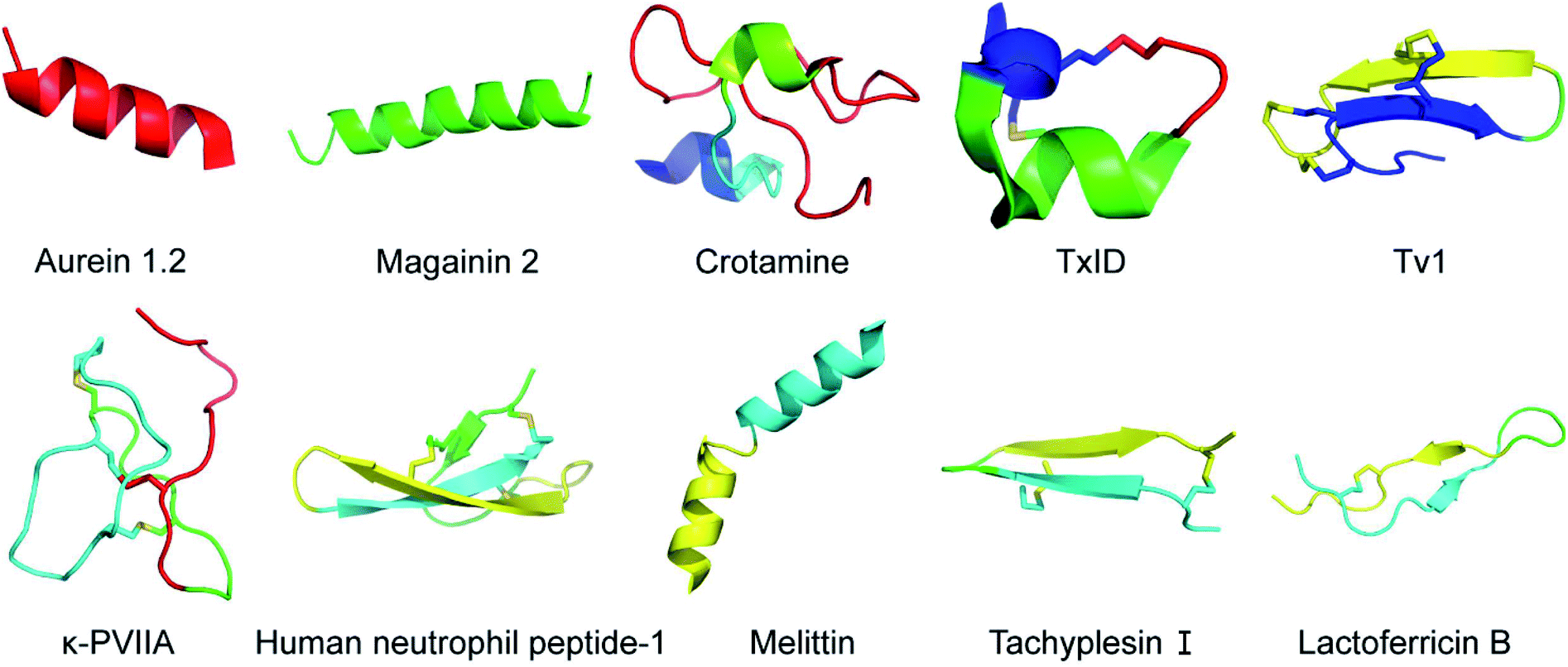 | ||
Fig. 1 Structural diagram of representative NCPs generated using PyMOL. Aurein 1.2 (Protein Data Bank ID: 1VM5), magainin 2 (Protein Data Bank ID: 2MAG), crotamine (Protein Data Bank ID: 1Z99), TxID (Protein Data Bank ID![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2M3I), Tv1 (Protein Data Bank ID: 2MIX), κ-PVIIA (Protein Data Bank ID: 2N8E), human neutrophil peptide-1 (Protein Data Bank ID: 3GNY), melittin (Protein Data Bank ID: 2MLT), tachyplesin I (Protein Data Bank ID: 1WO1), lactoferricin B (Protein Data Bank ID: 1LFC). :2M3I), Tv1 (Protein Data Bank ID: 2MIX), κ-PVIIA (Protein Data Bank ID: 2N8E), human neutrophil peptide-1 (Protein Data Bank ID: 3GNY), melittin (Protein Data Bank ID: 2MLT), tachyplesin I (Protein Data Bank ID: 1WO1), lactoferricin B (Protein Data Bank ID: 1LFC). | ||
| Target(s) | Species | Name | Sequence | Ref. |
|---|---|---|---|---|
| Cell membrane | Litoria aurea | Aurein 1.2 | GLFDIIKKIAESF–NH2 | 6 |
| Litoria citropa | Citropin 1.1 | GLFDVIKKVASVIGGL–NH2 | 7 | |
| Xenopus laevis | Magainin 2 | GIGKFLHSAKKFGKAFVGEIMNS | 36 | |
| Anoplius samariensis | Anoplin | GLLKRIKTLL–NH2 | 15 | |
| Ion channel | Conus textile | TxID | GC1C2SHPVC1SAMSPIC2–NH2 | 19 |
| Terebra variegata | Tv1 | TRIC1C2GC3YWNGSKDVC3SQSC1C2 | 23 | |
| Conus purpurascens | κ-PVIIA | C1RIPNQKC2FQHLDDC3C2SRKC1NRFNKC3V | 25 | |
| Cytoskeleton | Symploca sp. VP642 | Dolastatin 10 | Refer to Fig. 3 | 29 |
| Organelles | Crotalus durissus terrificus | Crotamine | YKQC1HKKGGHC2FPKEKIC3LPPSSDFGKMDC2RWRWKC1C3KKGSG | 37 |
| Cell cycle and apoptosis | Trididemnum solidum | Didemnin B | Refer to Fig. 3 | 38 |
| Aplidium albicans | Plitidepsin | Refer to Fig. 3 | 39 | |
| Tumor angiogenesis | Homo sapiens | Human neutrophil peptide-1 | AC1YC2RIPAC3IAGERRYGTC2IYQGRLWAFC3C1 | 40 |
| Multi-target synergistic effect targets | Apis mellifera | Melittin | GIGAVLKVLTTGLPALISWIKRKRQQ–NH2 | 41 |
| Tachypleus tridentatus | Tachyplesin I | KWC1FRVC2YRGIC2YRRC1R–NH2 | 42 | |
| Bos taurus | Lactoferricin B | FKCRRWQWRMKKLGAPSITCVRRAF | 43 | |
| Glycine max | Lunasin | SKWQHQQDSCRKQLQGVNLTPCEKHIMEKIQGRGDDDDDDDDDN | 44 | |
| Homo sapiens | LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 45 |
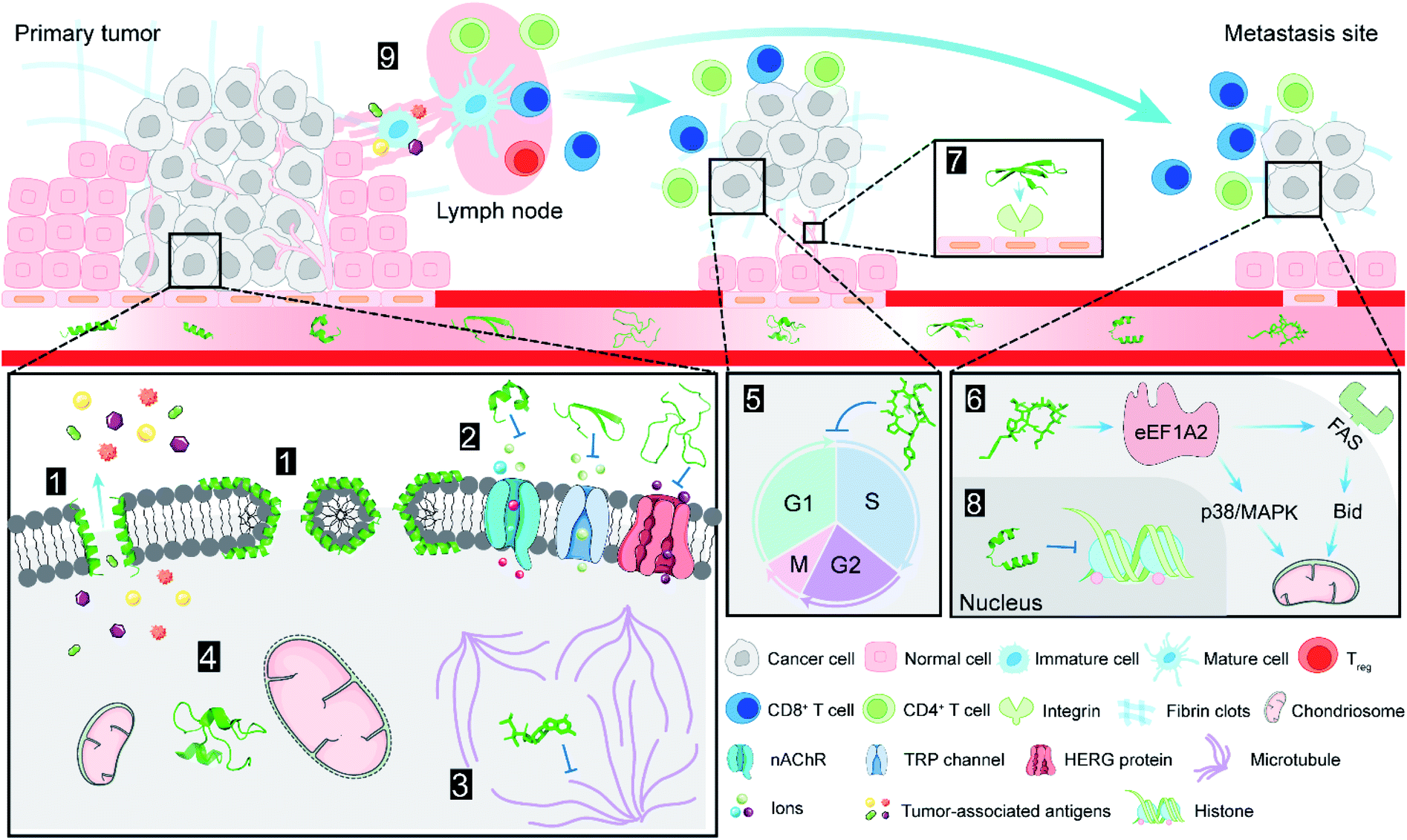 | ||
| Fig. 2 Antitumor targets of NCPs. (1) NCPs lyse cell membrane by the “carpet-like” and “toroidal pore” models; (2) NCPs block various ion channels that are overexpressed on cell membrane; (3) microtubules are inhibited by dolastatin 10; (4) crotamine causes depolarization of mitochondrial membrane and induces tumour cell death; (5) didemnin B displays antitumor activity through the inhibition of cell cycle; (6) plitidepsin interacts with eEF1A2 which involved in the oxidative stress/p38-MAPK pathway and FAS/CD95 pathway, and subsequent apoptotic program; (7) human neutrophil peptide-1 specifically binds to integrin and suppresses angiogenesis; (8) lunasin inhibits acetylation of core histones H3 and H4; (9) tumor-associated antigens induced by lactoferricin B treatment are released into the tumor microenvironment (TME), and cause systemic immune response. NCPs are shown as green ribbon or stick and ball models. | ||
The tumor cell selectivity of NCPs may result from the physiological and biochemical differences between tumor and normal cells. For example, the abundance of anionic substances (i.e., phosphatidylserine and heparan sulfate) in tumor cells increase the negative charge on cell membrane.11 The surface of normal cell membrane is mainly composed of neutral phospholipids and sterols, and the relatively abundant membrane-bound cholesterol can also protect normal cells from NCPs by altering membrane fluidity.12 Cationic NCPs possess the ability to form membrane potential-mediated electrostatic interactions, thereby displaying a selectivity not achievable with ‘traditional’ chemotherapeutic drugs.13 Tumor cells distinctly have more microvilli than normal cells, and these microvilli can increase the surface area for increased NCP binding.14 These factors are all significant contributors to the tumor cell selectivity of NCPs. Anoplin (Table 1), the shortest peptide from wasp venom, is one of the representative NCPs with high selectivity towards tumor cells. It exerts lytic activity on tumor cells without detectable haemolytic activity in red blood cells.15
The membrane-lytic mechanism of NCPs offers an attractive solution for cancer related multidrug-resistance (MDR). In general, the response of cancer cells to chemotherapy correlates with the intracellular concentration of chemotherapeutic agents, that is determined by the balance between drug influx and efflux.16 Drug efflux pumps (e.g. P-glycoprotein and multidrug-resistance protein-mediated) function as vital MDR factors that contribute to the accelerated efflux of chemotherapeutics, and eventually lead to the reduced sensitivity to chemotherapy.17 In this context, cell membrane lysis by NCPs has a much lower tendency to induce MDR when compared with conventional chemotherapeutic agent.
2.2 Ion channel
Ion channels are a class of membrane proteins that are often overexpressed in many forms of cancer, and the selective blockade of ion channels is envisioned to have potential therapeutic effects on cancer.18 Among these ion channels, nicotinic acetylcholine receptors (nAChRs) play a pivotal role in cancer development. Members of a ligand-gated ion channel family, nAChRs are composed of five distinct subunits (i.e., α, β, γ, δ and ε) that are further divided into different subtypes. The Conus textile α-conotoxin TxID (Fig. 1 and Table 1) is an α3β4 nAChR selective antagonist with the potential for development as a novel anticancer drug.19 The synthetic TxID has been shown to inhibit the proliferation of cervical20 and lung cancer cells,21 mediated through the α3β4 nAChRs which provide a potentially novel basis for chemotherapeutic intervention [Fig. 2(2)].Transient receptor potential (TRP) channels, members of the Ca2+ channel family, are intimately associated with cancer initiation and metastasis by modulating intracellular Ca2+ concentrations.22 Toxin Tv1 (Fig. 1 and Table 1), a bioactive disulfide-rich peptide isolated from predatory marine snail Terebra variegate, has a unique structural scaffold in comparison to other snail venom peptides.23 Results from in vitro and in vivo studies on chemically synthesized Tv1 revealed that Tv1 selectively inhibited hepatocellular carcinoma (HCC) cells and significantly reduced tumor size in a syngeneic mouse model, through the modification of TRPC6 and/or TRPV6 channels [Fig. 2(2)] that are overexpressed in the HCC models.24 Similarly, κ-conotoxins-PVIIA (κ-PVIIA) (Fig. 1 and Table 1), a 27 amino acid peptide from Conus purpurascens venom,25 binds to the potassium channel protein HERG, which is overexpressed on the plasma membrane of many tumor cells [Fig. 2(2)].26
2.3 Cytoskeleton
Microtubules [Fig. 2(3)] are a critical component of the eukaryotic cytoskeleton, which not only maintains cell morphology and intra-cellular structures, but also protects the integrity of cell division, genome replication, and other vital functions.27 Compared with normal cells, tumor cells have underdeveloped cytoskeletons with slower and less effective self-repair due to insufficient microfilament content, resulting in tumor cell death following exposure to NCPs.28 Dolastatin 10 (Fig. 3 and Table 1) is an anti-microtubule pentapeptide originally isolated from the sea hare Dolabella auricularia, and subsequently found to be produced by the marine cyanobacterium Symploca sp.29 Dolastatin 10 and analogues (i.e., dolastatin 15 and symplostatin 1) demonstrated sub-nanomolar anti-proliferative activity in vitro towards various cancer cell lines, but their unfavourable pharmacokinetic profiles limited their further clinical development.30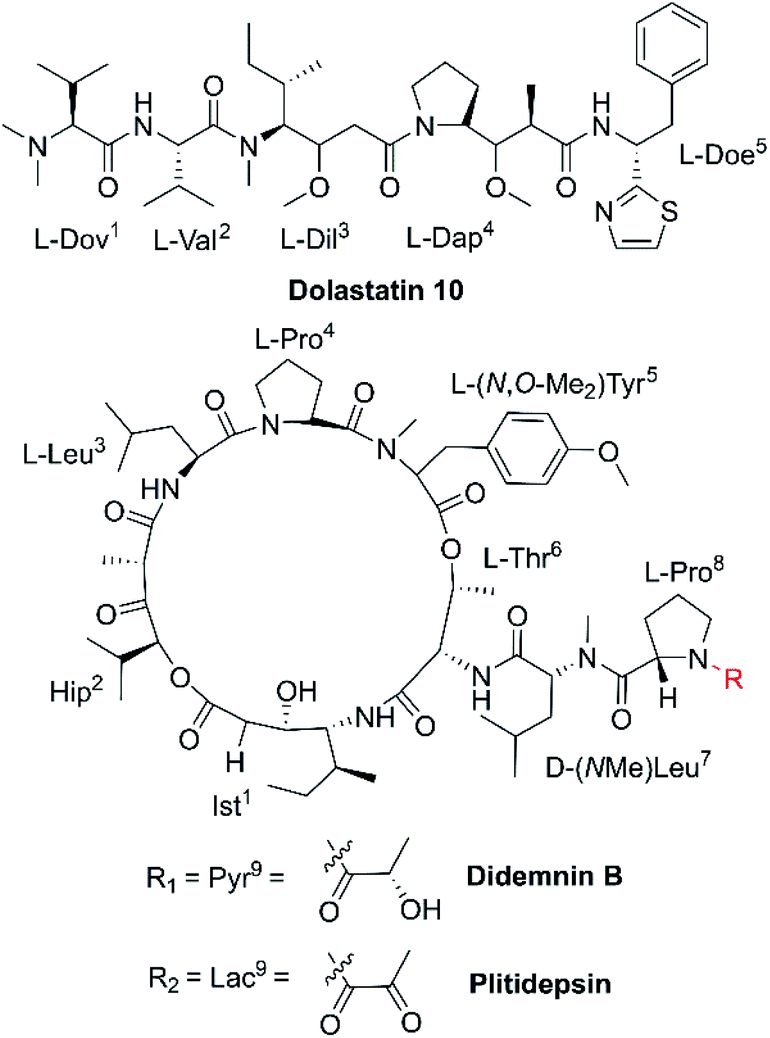 | ||
| Fig. 3 Chemical structures of dolastatin 10, didemnin B and plitidepsin. | ||
2.4 Organelles
In addition to the well-recognized role as the primary cellular energy powerhouse, mitochondria are also the central regulators of cell death pathways.31 Crotamine (Fig. 1 and Table 1), the major component of venom from the South American rattlesnake Crotalus durissus terrificus, is the first venom peptide classified as natural cell penetrating and antimicrobial defensin-like peptide with enhanced antiproliferative activity.32 Its structure encompasses a short N-terminal α-helix and anti-parallel triple-stranded β-sheets arranged in an αβ1β2β3 topology, stabilized by three disulfide bridges.33 Electrostatic interactions are induced between crotamine and the negatively charged cardiolipins within mitochondrial membranes. While inducing a rapid intracellular Ca2+ overload, crotamine can cause depolarization of the inner mitochondrial membrane in B16F10 cells and selectively induce tumor cell death [Fig. 2(4)].34 Notably, the fluorescent derivative of crotamine is also a potent theranostic tool for targeted delivery.352.5 Cell cycle and apoptosis
The cell cycle is mainly regulated by various cyclins and cyclin-dependent protein kinases (CDKs). Drugs that inhibit the activity of these enzymes and/or their gene expression can block the progression of the cell cycle and exert favourable antitumor effects.46 The non-ribosomal peptide didemnin B (Fig. 3 and Table 1), a marine tunicate (ascidian or sea-squirt)-derived cyclic depsipeptide isolated from Trididemnum solidum, has displayed promising antitumor effects through the inhibition of DNA, RNA and protein synthesis, initiation of apoptosis and G1 to S phase blockade [Fig. 2(5)].38 The unique structural features of didemnin B are the D-configuration of its N-methyl Leu, two specific chemical units (isostatine (Ist) and α-(α-hydroxyisovaleryl) propionyl (HIP)), and two nonproteinogenic L-amino acids (N,O-dimethyl Tyr and pyruvyl Pro).47 As the first marine-derived peptide in clinical trial, the continuous clinical development of didemnin B was hindered by the unsatisfactory response and significant toxicity risk, which could be potentially solved by the development of derivatives.48Plitidepsin (Fig. 3, Tables 1 and 2), originally isolated from the Mediterranean tunicate Aplidium albicans in 1990, has been manufactured through total synthesis and commercialized as Aplidin®.39 The first-in-class peptide drug approved for the treatment of relapsed or refractory multiple myeloma (MM), leukemia, and lymphoma,49 plitidepsin is only slightly different from the previously mentioned didemnin B, where a pyruvoyl-proline residue is replaced by a lactyl-proline residue. This minor structural modification causes a striking disparity in the anticancer mechanisms. Plitidepsin can interact with the overexpressed eukaryotic elongation factor 1A2 (eEF1A2) which is involved in the oxidative stress/p38-MAPK pathway, FAS/CD95 pathway, and subsequently induces a rapid apoptotic response [Fig. 2(6)].50
| Name | Source | Developer | Clinical stage | Disease area | Ref. |
|---|---|---|---|---|---|
| Plitidepsin (Aplidin®) | Plitidepsin | PharmaMar | Approved | Multiple myeloma, leukemia, lymphoma | 39 |
| Enfortumab vedotin-ejfv (PADCEV™) | Dolastatin 10 | Astellas Pharma & Seattle Genetics | Approved | Metastatic urothelial cancer | 64 |
| Polatuzumab vedotin (Polivy™) | Dolastatin 10 | Roche/Genetech | Approved | Non-Hodgkin lymphoma, chronic lymphocytic leukemia, lymphoma, B-cell and follicular lymphoma | 65 |
| Brentuximab vedotin (Adcetris®) | Dolastatin 10 | Seattle Genetics | Approved | Anaplastic large T-cell systemic malignant lymphoma, Hodgkin's disease | 66 |
| Balixafortide | Polyphemusin | Polyphor | Phase III | Metastatic breast cancer, locally recurrent breast cancer | 67 |
| LTX-315 | Lactoferricin B | Lytix Biopharma AS | Phase II | Soft tissue sarcoma | 68 |
2.6 Tumor angiogenesis
Angiogenesis, the formation of new blood vessels from existing vasculature, is required for tumor growth and metastasis.51 Vascular endothelial growth factor (VEGF) and its receptors play an important role in the regulation of the angiogenesis process.52 Chavakis and co-workers found that human neutrophil peptide-1 (HNP-1) (Fig. 1 and Table 1), that belongs to a peptide series known as α-defensins, specifically inhibited VEGF-dependent endothelial cell adhesion, migration, and proliferation by forming a ternary complex with fibronectin and α5β1 integrin, and thus suppressed VEGF-induced angiogenesis in vivo [Fig. 2(7)].40 These findings provide new insights into the potential of HNPs as angiogenesis inhibitor. However, the structural complexity of HNP-1, especially the three intramolecular disulfide bridges, increases its synthetic complexity.532.7 Multi-target synergistic effect
Melittin (Fig. 1 and Table 1), the predominant pharmacologically active component of honeybee venom, is a 26-amino acid, amphipathic, cell-penetrating, α-helical peptide. The helix-interrupting Pro14 (along with Gly12) facilitates the formation of a kinked bend in its structure which could create a donut-shaped toroidal pore within the cell membrane.54 Melittin produces a multi-targeted antitumor effect, that consists of cytolytic activity, antiproliferative, anti-invasion and anti-metastatic effects in vivo.41 However, its relatively low selectivity and hemolytic activity need to be addressed before melittin can be brought into the clinic.55Tachyplesin I (Fig. 1 and Table 1) was extracted from the blood lymphatic granulosa cells of the limulus horseshoe crab, Tachypleus tridentatus. This peptide is stabilized by two disulfide bonds (Cys3–Cys16, Cys7–Cys12) that form a uniquely rigid and stable β-hairpin with three tandem tetrapeptide repeats.42 In human gastric adenocarcinoma BGC-823 cells, tachyplesin I promoted cell cycle arrest at the G0/G1 phase while decreasing the percentage of cells at S phase, and changed the expression of correlative oncogene and tumor suppressor gene.56 In addition, the activation of C4b and C3b subcomponents of complement was observed in TSU human bladder cancer cells treated with tachyplesin I, suggesting the activation of classic complement cascade.57
Lactoferricin B (LfcinB) (Fig. 1 and Table 1) is composed of an 18 amino acid loop formed through disulfide bonding from the bactericidal domain of bovine lactoferrin. It exhibits an α-helical structure in lactoferrin, and becomes an amphipathic structure dominated by β-sheets upon dissociation.43 Both LfcinB and its synthetic analogues have exhibited cytotoxicity against numerous cancer cells at low concentrations with minimal toxicity to normal cells.58,59 Mechanisms responsible for this tumor cell-selective cytotoxicity are multi-faceted, including cell cycle arrest, initiation of apoptosis, induction of tumor necrosis, anti-angiogenesis, inhibition of metastasis, and immune modulation [Fig. 2(9)].60
The soybean metabolite lunasin (Table 1) is a 44 amino acid peptide component of the 2S albumin seed protein with three proposed functional domains that include an aspartic acid tail, an RGD domain, and a chromatin-binding helical domain.44 Its RGD domain is critical for cellular internalization and activity against cancer-stem cells through interaction with the αvβ3 integrin via the FAK/ERK/NF-κB signalling pathway.61 The aspartic acid tail inhibited the acetylation of core histones H3 and H4 and the phosphorylation of retinoblastoma protein in a concentration-dependent manner (Fig. 2).62
The peptide LL-37 (Table 1) is the only member of the cathelicidin family isolated from the innate human immune system, and is produced through the selective cleavage of its precursor hCAP-18 by serine protease 3.45 Studies have demonstrated that LL-37 can effectively inhibit tumorigenesis through multiple regulatory effects on tumor cell apoptosis, the cell cycle, immunocyte activation, and autophagy.63
3 Currently approved anticancer applications
3.1 Antibody drug conjugates
Although the application of dolastatin 10 as a single agent requires further investigation, dolastatin 10 and its derivatives monomethyl auristatin E and F (Fig. 4 and Table 2), have been chemically connected with monoclonal antibodies via environmental responsive linker forming antibody drug conjugates (ADCs).69 These dolastatin 10 ADCs and other derivatives have been approved by the FDA or are in phase I to III clinical trials. Dolastatin 10-derived ADCs have become an important part of the marine-derived cancer chemotherapeutic pipeline.70 This successful targeted strategy offers innovative anticancer agents for treating otherwise intractable cancers.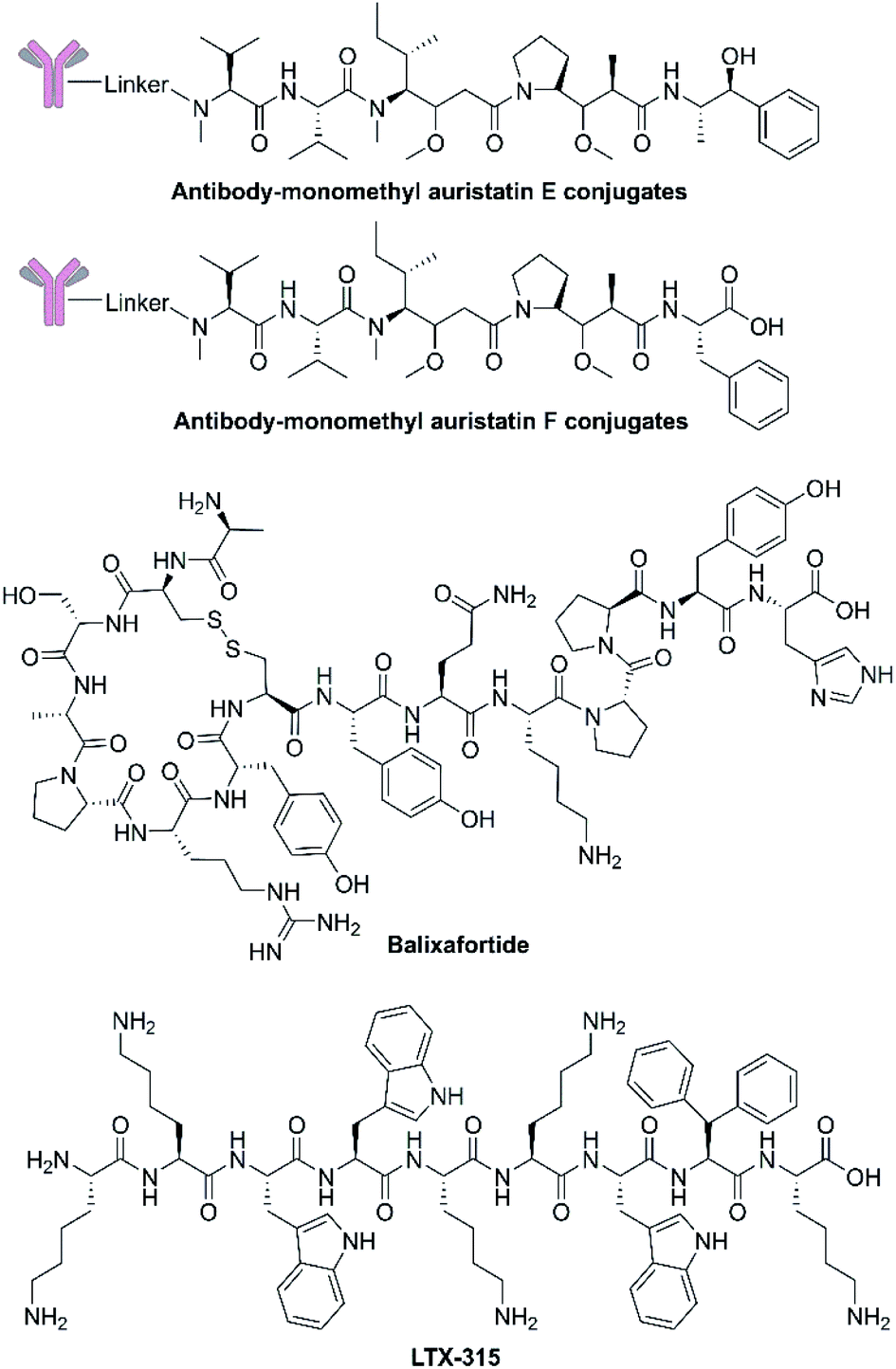 | ||
| Fig. 4 Chemical structures of antibody drug conjugates based on dolastatin 10, balixafortide and LTX-315. | ||
3.2 Chemical structural modification
Extensive structure–activity relationship research indicates that modification, addition, and elimination of structural components of NCPs can effectively enhance their antitumor activity and stability while reducing toxicity. The best example is balixafortide (POL6326) (Fig. 4 and Table 2), a potent, selective antagonist of C-X-C chemokine receptor type 4 (CXCR4) which is primarily expressed by hematopoietic stem cells and leukocytes and overexpressed in numerous human tumor cell lines. Through the binding with its ligand, stromal cell-derived factor-1α (SDF-1), CXCR4 can promote tumor immune escape and create a favourable niche for tumor growth and metastasis.71 The design of balixafortide is based on polyphemusin, a natural peptide from the horseshoe crab, Limulus polyphemus. Blockade of the CXCR4 pathway by balixafortide plays a key role in the suppression of metastatic spread, sensitization of tumor cells to chemotherapy, and activation of the immune system in the tumor microenvironment (TME).72 The completed results of phase I and II trials revealed that balixafortide plus eribulin enhanced antitumor effects in patients with HER2-negative metastatic breast cancer and improved tolerance relative to eribulin alone, while producing no dose-limiting toxicities.67 Therefore, balixafortide shows great promise for its forthcoming phase III trial (Clinical Trials.gov Identifier: NCT03786094).3.3 Oncolytic immunotherapies
As the first-in-class oncolytic peptide, LTX-315 (Fig. 4 and Table 2), a derivative of lactoferricin, can effectively lyse cancer cell membranes while reprograming the TME. Intratumoral treatment with LTX-315 result in rapid cell lysis (necrosis) through membrane destabilization followed by the subsequent release of Danger-Associated Molecular Patterns (DAMPs) (such as calreticulin, ATP and high mobility group box protein 1) and tumor-associated antigens (TAAs) into the TME.68 Those intracellular content will initiate the recruitment and maturation of dendritic cells (DCs) into the TME. Mature DCs are then primed for engulfment of whole-cell tumor antigens and subsequent presentation to T cells, creating tumor-specific cytotoxic CD8+ T lymphocytes that are capable of eradicating residual cancer cells.68 The LTX-315 peptide produced a complete tumor regression and systemic immune response in a rat fibrosarcoma model, and overcame the resistance to immunotherapy when combined with CTLA-4 checkpoint blockade therapy.73 These findings suggest its potential as a novel cancer immunotherapeutic agent and an ideal combination choice for other immunotherapies.74 Currently, an Open Label Phase II Single Centre Study is recruiting subjects to determine the safety and effectiveness of LTX-315 to induce T-cell infiltration prior to tumor infiltrating lymphocyte expansion in patients with soft tissue sarcoma (Clinical Trials.gov Identifier: NCT03725605).3.4 Targeted co-delivery systems
For some NCPs, such as melittin, clinical application is limited by nonspecific cytotoxicity, hemolytic effect, or rapid degradation. Recent research efforts in the field of nanotechnology hold promise in overcoming these bottlenecks that hinder the clinical development of certain NCPs. Aronson et al.75 successfully integrated such a cytolytic NCP payload into the lipid nanoparticles, and this strategy facilitated the selective destruction of cancer cells while circumventing off-target toxicity towards healthy tissues. Another self-assembling lipid nanoparticle (α-melittin-NP) can also shield cytotoxicity of melittin to antigen presenting cells (APCs), but maintain the its ability to promote whole tumor antigen release in situ and result in the activation of APCs in lymph nodes (LNs). This nanoparticle makes melittin an ideal LN-targeted whole-cell nanovaccine for cancer immunotherapy in a synergistic manner.76 Use of the chemotherapeutic agent, doxorubicin (DOX), is associated with a number of serious side effects, including dose-dependent cardiomyopathy, gastrointestinal toxicity, bone marrow suppression and potential drug resistance.77 The combination of NCPs and DOX via biocompatible nanomaterials may provide an attractive strategy to overcome the potential toxicities of both agents, while reducing DOX-associated drug resistance. For example, a melittin–RADA32 hybrid peptide hydrogel that was loaded with DOX has been developed. It also regulates the TME for melanoma chemoimmunotherapy. This peptide hydrogel dramatically reduced the side effects of melittin and DOX, while significantly enhanced antitumor efficacy and innate immune response.78 These co-delivery systems have demonstrated the synergistic potential of NCPs and conventional chemotherapeutic drugs to elicit sustained antitumor effects and, thus, provide novel drug discovery paradigms.4 The potential development of NCPs
Although the translation of NCPs into the clinic has made considerable progress, obstacles still remain in many aspects of drug discovery from NCP candidates. For example, the high proteolytic susceptibility of natural peptides leads to their short half-life and low bioavailability in vivo. The inherent conformational flexibility of NCPs impedes the efficacy in complex physiological environments, and the low membrane permeability limits the interaction with intracellular targets for some NCPs.4.1 Conformational constraints
To increase stability and improve physicochemical properties of peptides, structurally constrained peptide technologies have been widely adopted in both basic research and industrial production. The constrained/stapled peptide technologies were firstly developed to mimic key binding epitopes of proteins, including α-helix and β-hairpin motifs, for the regulation of intracellular protein–protein interactions. Studies have demonstrated that these constrained peptides possess increased cell permeability and improved stability, exhibit increased binding affinity and target protein specificity. In general, conformational helix constraint is fulfilled by linking one of the termini to a side-chain crosslink, or cross-linking two side-chain residues through structurally diverse chemical bonds.79 Among these, the all-hydrocarbon stapled peptide technology is considered as one of the most successful and promising stapling strategies.80 The peptide, ALRN-6924 (Fig. 5), is an optimized ATSP-7041 derivative that was the first example of an all-hydrocarbon stapled-peptide drug to be evaluated in ongoing clinical trials for advanced solid tumors and lymphomas (Clinical Trials.gov Identifier: NCT02264613).81 We have applied this stapling strategy to design and synthesize a series of novel hydrocarbon-stapled melittin analogues, and these staple-modified melittin peptides exhibited enhanced anti-hepatoma activity and improved protease resistance relative to native melittin peptides.55 Further, these stapled peptides retained their important peripheral residues that enhance both bioactivity and membrane permeability.82 With the success of these stapling strategies, we anticipate that such stapled peptide technologies will be widely applied in structural modification of other α-helical NCPs and their derivatives.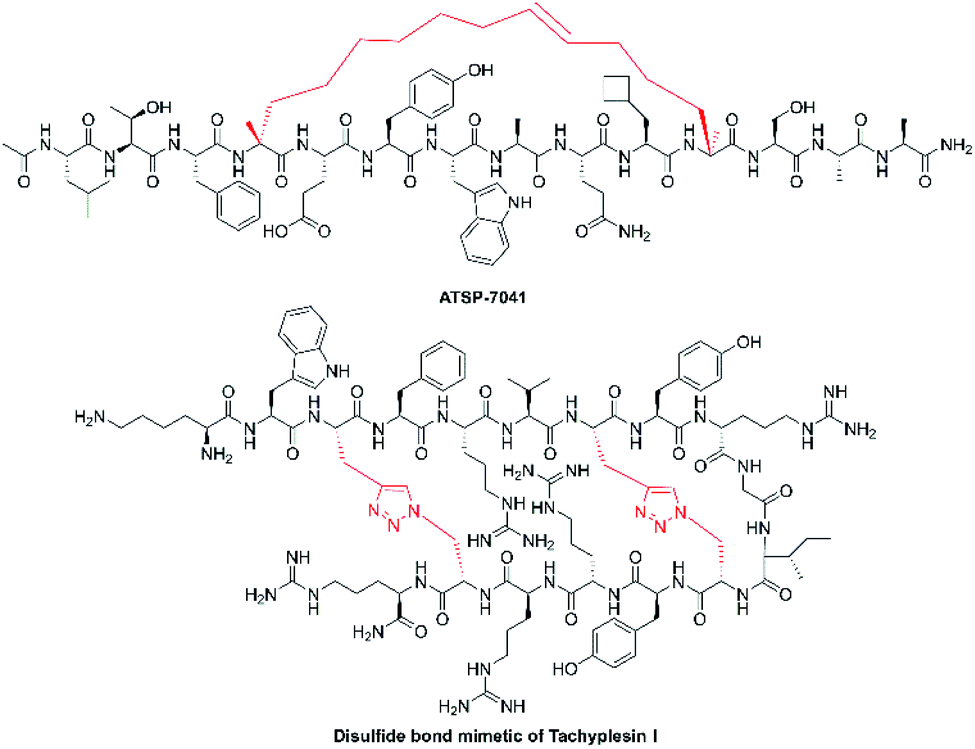 | ||
| Fig. 5 Chemical structures of ATSP-7041 and disulfide bond mimetic of tachyplesin I. | ||
Disulfide-rich scaffolds confer peptides with increased stability and rigidity. However, disulfide isomerases, as well as reductive glutathione in vivo, can affect or dissociate the disulfide bonds, and lead to structural rearrangement with potential loss of activity.83 Hence, the strategy of developing disulfide surrogates is to enhance redox stability and conformation rigidity of NCP analogues. The Brust group84 and Meldal et al.85 used triazole as a disulfide bond mimetic to lock the conformational structure of conotoxin MrIA and tachyplesin I (Fig. 5) without losing biological activities, respectively. Liu et al.83 also developed diaminodiacid-strategy, using C–C and S–C bonds to maintain the β-hairpin ribbon structure of tachyplesin I. It is anticipated that these methods will provide a highly efficient and practically useful strategy to constrain analogues of disulfide bond-containing NCPs for their translational therapeutic development.
4.2 Peptide–drug conjugates
Like most small-molecule drugs, some NCPs are not sufficiently selective for tumor cells over normal cells, resulting in significant clinical toxicity and limited therapeutic index.75 To minimize off-target effects and generalized toxicities, peptide–drug conjugates (PDCs) have been developed. The two component conjugates typically contain a peptide and a specific high affinity molecule which is designed to bind to specific binding sites on tumor cells.86 The difunctional PDC, BT1718, is composed of a constrained bicyclic peptide derived from membrane type 1-matrix metalloprotease (MT1-MMP) using a proprietary phage display peptide technology, coupled to mertansine (a thiol-containing maytansinoid-based small molecule toxin). Pre-clinical evaluations have demonstrated that BT1718 can target MT1-MMP which is expressed in a variety of advanced solid tumors, including triple-negative breast cancer (TNBC), non-small cell lung cancer, and various sarcomas. Phase I trials with BT1718 are underway (Clinical Trials. gov Identifier: NCT03486730). Inspired by BT1718, we hypothesize a possible way to add a trans-membrane molecule or functional molecule onto NCPs by using an elaborate linker. For example, cell penetrating peptides (CPPs), such as TAT and poly-Arg or poly-Lys, have been broadly used.87 These CPPs may increase the cell membrane permeability when linked to NCPs, achieving a reasonable level of intracellular target engagement for antitumor activity. Another ingenious design is covering up the cationic charge of NCPs by an anionic sequence via a cleavable linker that is sensitive to the TME. Li and co-workers developed activatable cell lytic peptides targeting TNBC, and these TME-responsive NCPs showed specific targeting in vivo upon systematic administration and displayed no nonspecific toxicity.88 Alternatively, cell targeting peptides, such as RGD211 and NGR212, provide the shortest specific sequences with high affinity to their targets. For example, synthetic RGD-tachyplesin exhibited a doubly targeted inhibitory effect on proliferating endothelial cells and B16 melanoma cells, both in vitro and in vivo.89 Such difunctional or multifunctional conjugates may show promise to greatly accelerate the discovery, optimization, and preclinical evaluation of NCPs and ultimately facilitate the clinical translation of NCPs.5 Concluding remarks
Solid tumor-forming cancers continue to pose a severe threat to human health worldwide through their uncontrolled growth and ability to form distant metastases. Further, cancer patients are especially susceptible to opportunistic microbial infections due to their weakened immune systems which are caused by conventional chemotherapies.90 Compared with traditional chemotherapeutics, NCPs derived from innate immune systems also play a pivotal role in preventing pathogens through their antimicrobial activity.91 Evidence suggests that bacteria exist in symbiotic relationships with certain tumors.92 In such cases, NCPs with both antimicrobial and anticancer activity may have a distinct advantage. Multi-mechanism NCPs can potentially act in a synergistic manner to increase the antitumor efficacy of conventional single-target chemotherapeutics. Multi-drug resistance (MDR) efflux has become a nearly insurmountable bottleneck for the use of certain small-molecule chemotherapeutic agents93 and NCPs offer the potential as new classes of drug candidates with enormous promise to meet the increasing therapeutic needs. The development of NCP-based chemotherapeutics is still in its infancy, and just as with traditional small-molecule based drugs, numerous challenges exist that can hinder their translation into the clinic (e.g., suboptimal therapeutic activity, potential side effects, low stability, and poor membrane-permeability). Recently developed technical innovations and novel strategies in peptide chemistry show potential to address these shortcomings, and improve the pharmacokinetic properties of peptides while reducing associated undesired effects. Taken together, NCP-derived agents and their synthetic analogues hold great promise for the future of novel chemotherapeutic drug discovery.6 Conflicts of interest
There are no conflicts to declare.7 Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81903654), Program for Professor of Special Appointment (Young Eastern Scholar) at Shanghai Institutions of Higher Learning (QD2018035), Shanghai “Chenguang Program” of Education Commission of Shanghai Municipality (No. 18CG46), Shanghai Sailing Program (No. 19YF1449400), Shanghai Engineering Research Centre for the Preparation of Bioactive Natural Products (16DZ2280200), the National Key Research and Development Program of China (2017YFC1700200), Graduate Innovation Training research project of the Shanghai University of Traditional Chinese Medicine (No. Y2019064), and the U.S. NIH grant CA199016.8 References
- M. Mohme, S. Riethdorf and K. Pantel, Nat. Rev. Clin. Oncol., 2017, 14, 155–167 CrossRef CAS.
- K. D. Miller, L. Nogueira, A. B. Mariotto, J. H. Rowland, K. R. Yabroff, C. M. Alfano, A. Jemal, J. L. Kramer and R. L. Siegel, Ca-Cancer J. Clin., 2019, 69, 363–385 CrossRef.
- C. Lengauer, L. J. Diaz and S. Saha, Nat. Rev. Drug Discovery, 2005, 4, 375–380 CrossRef CAS.
- M. R. Naylor, A. T. Bockus, M. J. Blanco and R. S. Lokey, Curr. Opin. Chem. Biol., 2017, 38, 141–147 CrossRef CAS.
- S. R. Dennison, M. Whittaker, F. Harris and D. A. Phoenix, Curr. Protein Pept. Sci., 2006, 7, 487–499 CrossRef CAS.
- T. Rozek, J. H. Bowie, J. C. Wallace and M. J. Tyler, Rapid Commun. Mass Spectrom., 2000, 14, 2002–2011 CrossRef CAS.
- J. Doyle, C. S. Brinkworth, K. L. Wegener, J. A. Carver, L. E. Llewellyn, I. N. Olver, J. H. Bowie, P. A. Wabnitz and M. J. Tyler, Eur. J. Biochem., 2003, 270, 1141–1153 CrossRef CAS.
- X. Xu and R. Lai, Chem. Rev., 2015, 115, 1760–1846 CrossRef CAS.
- P. W. Soballe, W. L. Maloy, M. L. Myrga, L. S. Jacob and M. Herlyn, Int. J. Cancer, 1995, 60, 280–284 CrossRef CAS.
- E. Strandberg, D. Horn, S. Reisser, J. Zerweck, P. Wadhwani and A. S. Ulrich, Biophys. J., 2016, 111, 2149–2161 CrossRef CAS.
- S. Riedl, R. Leber, B. Rinner, H. Schaider, K. Lohner and D. Zweytick, Biochim. Biophys. Acta, 2015, 1848, 2918–2931 CrossRef CAS.
- A. Zachowski, Biochem. J., 1993, 294, 1–14 CrossRef CAS.
- K. A. Camilio, Ø. Rekdal and B. Sveinbjörnsson, Oncoimmunology, 2014, 3, e29181 CrossRef.
- S. C. Chan, L. Hui and H. M. Chen, Anticancer Res., 1998, 18, 4467–4474 CAS.
- Y. Wu, R. Huang, J. M. Jin, L. J. Zhang, H. Zhang, H. Z. Chen, L. L. Chen and X. Luan, Front. Chem., 2020, 8, 519 CrossRef CAS.
- Z. Q. Wang, H. W. An, D. Y. Hou, M. D. Wang, X. Z. Zeng, R. Zheng, L. Wang, K. L. Wang, H. Wang and W. H. Xu, Adv. Mater., 2019, 31, e1807175 CrossRef.
- I. Genovese, A. Ilari, Y. G. Assaraf, F. Fazi and G. Colotti, Drug Resist. Updates, 2017, 32, 23–46 CrossRef.
- A. A. Peters, S. Jamaludin, K. Yapa, S. Chalmers, A. P. Wiegmans, H. F. Lim, M. Milevskiy, I. Azimi, F. M. Davis, K. S. Northwood, E. Pera, D. L. Marcial, E. Dray, N. J. Waterhouse, P. J. Cabot, T. J. Gonda, P. A. Kenny, M. A. Brown, K. K. Khanna, S. J. Roberts-Thomson and G. R. Monteith, Oncogene, 2017, 36, 6490–6500 CrossRef CAS.
- S. Luo, D. Zhangsun, X. Zhu, Y. Wu, Y. Hu, S. Christensen, P. J. Harvey, M. Akcan, D. J. Craik and J. M. McIntosh, J. Med. Chem., 2013, 56, 9655–9663 CrossRef CAS.
- Y. Liu, J. Qian, Z. H. Sun, D. T. Zhangsun and S. L. Luo, Mar. Drugs, 2019, 17, 256 CrossRef CAS.
- J. Qian, Y. Q. Liu, Z. H. Sun, D. T. Zhangsun and S. L. Luo, Eur. J. Pharmacol., 2019, 865, 172674 CrossRef CAS.
- Y. F. Zhu, Q. X. Pan, H. Meng, Y. S. Jiang, A. Q. Mao, T. Wang, D. Hua, X. Q. Yao, J. Jin and X. Ma, Pharmacol. Res., 2015, 93, 36–42 CrossRef CAS.
- P. Anand, A. Grigoryan, M. H. Bhuiyan, B. Ueberheide, V. Russell, J. Quinonez, P. Moy, B. T. Chait, S. F. Poget and M. Holford, PLoS One, 2014, 9, e94122 CrossRef.
- P. Anand, P. Filipenko, J. Huaman, M. Lyudmer, M. Hossain, C. Santamaria, K. Huang, O. O. Ogunwobi and M. Holford, Mar. Drugs, 2019, 17, 587 CrossRef CAS.
- S. Kwon, F. Bosmans, Q. Kaas, O. Cheneval, A. C. Conibear, K. J. Rosengren, C. K. Wang, C. I. Schroeder and D. J. Craik, Biotechnol. Bioeng., 2016, 113, 2202–2212 CrossRef CAS.
- K. Dave and A. Lahiry, Curr. Top. Med. Chem., 2012, 12, 845–851 CrossRef CAS.
- C. Janke and M. M. Magiera, Nat. Rev. Mol. Cell Biol., 2020, 21, 307–326 CrossRef CAS.
- A. L. Risinger and L. Du, Nat. Prod. Rep., 2020, 37, 634–652 RSC.
- H. Luesch, R. E. Moore, V. J. Paul, S. L. Mooberry and T. H. Corbett, J. Nat. Prod., 2001, 64, 907–910 CrossRef CAS.
- K. Yang, B. Chen, D. A. Gianolio, J. E. Stefano, M. Busch, C. Manning, K. Alving, R. C. Gregory, W. H. Brondyk, R. J. Miller and P. K. Dhal, Org. Biomol. Chem., 2019, 17, 8115–8124 RSC.
- C. Brenner and G. Kroemer, Science, 2000, 289, 1150–1151 CrossRef CAS.
- I. Kerkis, M. A. Hayashi, D. S. A. Prieto, A. Pereira, J. P. De Sa, A. J. Zaharenko, G. Radis-Baptista, A. Kerkis and T. Yamane, BioMed Res. Int., 2014, 2014, 675985 Search PubMed.
- V. Fadel, P. Bettendorff, T. Herrmann, W. J. de Azevedo, E. B. Oliveira, T. Yamane and K. Wuthrich, Toxicon, 2005, 46, 759–767 CrossRef CAS.
- F. D. Nascimento, L. Sancey, A. Pereira, C. Rome, V. Oliveira, E. B. Oliveira, H. B. Nader, T. Yamane, I. Kerkis, I. L. Tersariol, J. L. Coll and M. A. Hayashi, Mol. Pharm., 2012, 9, 211–221 CrossRef CAS.
- F. L. Tansi, M. P. Filatova, D. O. Koroev, O. M. Volpina, S. Lange, C. Schumann, U. K. Teichgraber, S. Reissmann and I. Hilger, J. Cell. Biochem., 2019, 120, 6528–6541 CrossRef CAS.
- J. Gesell, M. Zasloff and S. J. Opella, J. Biomol. NMR, 1997, 9, 127–135 CrossRef CAS.
- I. Kerkis, M. A. Hayashi, D. S. A. Prieto, A. Pereira, J. P. De Sa, A. J. Zaharenko, G. Radis-Baptista, A. Kerkis and T. Yamane, BioMed Res. Int., 2014, 2014, 675985 Search PubMed.
- S. L. Crampton, E. G. Adams, S. L. Kuentzel, L. H. Li, G. Badiner and B. K. Bhuyan, Cancer Res., 1984, 44, 1796–1801 CAS.
- S. Alonso-Alvarez, E. Pardal, D. Sanchez-Nieto, M. Navarro, M. D. Caballero, M. V. Mateos and A. Martin, Drug Des., Dev. Ther., 2017, 11, 253–264 CrossRef CAS.
- T. Chavakis, D. B. Cines, J. S. Rhee, O. D. Liang, U. Schubert, H. P. Hammes, A. A. Higazi, P. P. Nawroth, K. T. Preissner and K. Bdeir, FASEB J., 2004, 18, 1306–1308 CrossRef CAS.
- I. Rady, I. A. Siddiqui, M. Rady and H. Mukhtar, Cancer Lett., 2017, 402, 16–31 CrossRef CAS.
- A. Laederach, A. H. Andreotti and D. B. Fulton, Biochemistry, 2002, 41, 12359–12368 CrossRef CAS.
- P. M. Hwang, N. Zhou, X. Shan, C. H. Arrowsmith and H. J. Vogel, Biochemistry, 1998, 37, 4288–4298 CrossRef CAS.
- L. E. Seber, B. W. Barnett, E. J. McConnell, S. D. Hume, J. Cai, K. Boles and K. R. Davis, PLoS One, 2012, 7, e35409 CrossRef CAS.
- R. J. Fahy and M. D. Wewers, Immunol. Res., 2005, 31, 75–89 CrossRef CAS.
- Y. Wang, X. Chen, Y. Y. Yan, X. C. Zhu, M. M. Liu and X. H. Liu, J. Med. Chem., 2020, 63, 3327–3347 CrossRef CAS.
- M. B. Hossain, D. van der Helm, J. Antel, G. M. Sheldrick, S. K. Sanduja and A. J. Weinheimer, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 4118–4122 CrossRef CAS.
- M. B. Potts, E. A. McMillan, T. I. Rosales, H. S. Kim, Y. H. Ou, J. E. Toombs, R. A. Brekken, M. D. Minden, J. B. MacMillan and M. A. White, Nat. Chem. Biol., 2015, 11, 401–408 CrossRef CAS.
- P. C. Jimenez, D. V. Wilke, P. C. Branco, A. Bauermeister, P. Rezende-Teixeira, S. P. Gaudencio and L. V. Costa-Lotufo, Br. J. Pharmacol., 2020, 177, 3–27 CrossRef CAS.
- A. Losada, J. J. Berlanga, J. M. Molina-Guijarro, A. Jimenez-Ruiz, F. Gago, P. Aviles, C. de Haro and J. F. Martinez-Leal, Biochem. Pharmacol., 2020, 172, 113744 CrossRef CAS.
- Z. R. Jiang, L. Wang, X. S. Liu, C. Chen, B. L. Wang, W. L. Wang, C. Hu, K. L. Yu, Z. P. Qi, Q. W. Liu, A. L. Wang, J. Liu, G. C. Hong, W. C. Wang and Q. S. Liu, Acta Pharm. Sin. B, 2020, 10, 488–497 CrossRef.
- B. Sitohy, J. A. Nagy and H. F. Dvorak, Cancer Res., 2012, 72, 1909–1914 CrossRef CAS.
- X. L. Zhang, A. M. Jiang, B. H. Qi, H. Yu, Y. Y. Xiong, G. L. Zhou, M. S. Qin, J. F. Dou and J. F. Wang, Appl. Microbiol. Biotechnol., 2018, 102, 4817–4827 CrossRef CAS.
- T. T. Dinh, D. H. Kim, H. X. Luong, B. J. Lee and Y. W. Kim, Bioorg. Med. Chem. Lett., 2015, 25, 4016–4019 CrossRef CAS.
- Y. Wu, M. F. Han, C. Liu, T. Y. Liu, Y. F. Feng, Y. Zou, B. Li and H. L. Liao, RSC Adv., 2017, 7, 17514–17518 RSC.
- S. L. Shi, Y. Y. Wang, Y. Liang and Q. F. Li, World J. Gastroenterol., 2006, 12, 1694–1698 CrossRef CAS.
- J. Chen, X. M. Xu, C. B. Underhill, S. Yang, L. Wang, Y. Chen, S. Hong, K. Creswell and L. Zhang, Cancer Res., 2005, 65, 4614–4622 CrossRef CAS.
- C. Y. Vargas, G. J. Rodriguez, P. Y. Umana, C. A. Leal, R. G. Almanzar, C. J. Garcia and M. Z. Rivera, Molecules, 2017, 22, 1641 CrossRef.
- M. J. Sheng, Y. J. Zhao, A. C. Zhang, L. Y. Wang and G. Z. Zhang, J. Pept. Sci., 2014, 20, 803–810 CrossRef CAS.
- J. Qi, W. S. Li, K. J. Lu, F. Y. Jin, D. Liu, X. L. Xu, X. J. Wang, X. Q. Kang, W. Wang, G. F. Shu, F. Han, X. Y. Ying, J. You, J. S. Ji and Y. Z. Du, Nano Lett., 2019, 19, 4949–4959 CrossRef CAS.
- S. Fernandez-Tome, F. Xu, Y. H. Han, B. Hernandez-Ledesma and H. Xiao, Int. J. Mol. Sci., 2020, 21, 537 CrossRef CAS.
- S. B. Vuyyuri, C. Shidal and K. R. Davis, Curr. Opin. Pharmacol., 2018, 41, 27–33 CrossRef CAS.
- E. Piktel, K. Niemirowicz, U. Wnorowska, M. Wątek, T. Wollny, K. Głuszek, S. Góźdź, I. Levental and R. Bucki, Arch. Immunol. Ther. Exp., 2016, 64, 33–46 CrossRef CAS.
- H. Kaplon, M. Muralidharan, Z. Schneider and J. M. Reichert, mAbs, 2020, 12, 1703531 CrossRef.
- Cancer Discovery, 2019, 9, OF2, DOI:10.1158/2159-8290.CD-NB2019-076.
- P. Kaloyannidis, M. Hertzberg, K. Webb, A. Zomas, R. Schrover, M. Hurst, I. Jacob, T. Nikoglou and J. M. Connors, Br. J. Haematol., 2020, 188, 540–549 CrossRef CAS.
- S. Pernas, M. Martin, P. A. Kaufman, M. Gil-Martin, P. P. Gomez, S. Lopez-Tarruella, L. Manso, E. Ciruelos, J. A. Perez-Fidalgo, C. Hernando, F. O. Ademuyiwa, K. Weilbaecher, I. Mayer, T. J. Pluard, G. M. Martinez, L. Vahdat, J. Perez-Garcia, A. Wach, D. Barker, S. Fung, B. Romagnoli and J. Cortes, Lancet Oncol., 2018, 19, 812–824 CrossRef.
- H. W. Liao, C. Garris, C. Pfirschke, S. Rickelt, S. Arlauckas, M. Siwicki, R. H. Kohler, R. Weissleder, V. Sundvold-Gjerstad, B. Sveinbjornsson, Ø. Rekdal and M. J. Pittet, Cell Stress, 2019, 3, 348–360 CrossRef CAS.
- K. W. Yang, B. Chen, D. A. Gianolio, J. E. Stefano, M. Busch, C. Manning, K. Alving, R. C. Gregory, W. H. Brondyk, R. J. Miller and P. K. Dhal, Org. Biomol. Chem., 2019, 17, 8115–8124 RSC.
- R. B. Pereira, N. M. Evdokimov, F. Lefranc, P. Valentao, A. Kornienko, D. M. Pereira, P. B. Andrade and N. Gomes, Mar. Drugs, 2019, 17, 329 CrossRef CAS.
- J. L. Iovanna and D. Closa, Oncoimmunology, 2017, 6, e1358840 CrossRef.
- S. Scala, Clin. Cancer Res., 2015, 21, 4278–4285 CrossRef CAS.
- T. Yamazaki, J. M. Pitt, M. Vetizou, A. Marabelle, C. Flores, Ø. Rekdal, G. Kroemer and L. Zitvogel, Cell Death Differ., 2016, 23, 1004–1015 CrossRef CAS.
- B. Sveinbjornsson, K. A. Camilio, B. E. Haug and Ø. Rekdal, Future Med. Chem., 2017, 9, 1339–1344 CrossRef CAS.
- M. R. Aronson, A. W. Simonson, L. M. Orchard, M. Llinas and S. H. Medina, Acta Biomater., 2018, 80, 269–277 CrossRef CAS.
- X. Yu, Y. F. Dai, Y. F. Zhao, S. H. Qi, L. Liu, L. S. Lu, Q. M. Luo and Z. H. Zhang, Nat. Commun., 2020, 11, 1110 CrossRef CAS.
- F. Araste, K. Abnous, M. Hashemi, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Controlled Release, 2018, 292, 141–162 CrossRef CAS.
- H. L. Jin, C. Wan, Z. W. Zou, G. F. Zhao, L. L. Zhang, Y. Y. Geng, T. Chen, A. Huang, F. G. Jiang, J. P. Feng, J. F. Lovell, J. Chen, G. Wu and K. Y. Yang, ACS Nano, 2018, 12, 3295–3310 CrossRef CAS.
- Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- L. A. Carvajal, D. B. Neriah, A. Senecal, L. Benard, V. Thiruthuvanathan, T. Yatsenko, S. R. Narayanagari, J. C. Wheat, T. I. Todorova, K. Mitchell, C. Kenworthy, V. Guerlavais, D. A. Annis, B. Bartholdy, B. Will, J. D. Anampa, I. Mantzaris, M. Aivado, R. H. Singer, R. A. Coleman, A. Verma and U. Steidl, Sci. Transl. Med., 2018, 10, eaao3003 CrossRef.
- Y. Wu, Y. H. Li, X. Li, Y. Zou, H. L. Liao, L. Liu, Y. G. Chen, D. Bierer and H. G. Hu, Chem. Sci., 2017, 8, 7368–7373 RSC.
- H. K. Cui, Y. Guo, Y. He, F. L. Wang, H. N. Chang, Y. Wang, F. M. Wu, C. L. Tian and L. Liu, Angew. Chem., Int. Ed., 2013, 52, 9558–9562 CrossRef CAS.
- A. Gori, C. I. Wang, P. J. Harvey, K. J. Rosengren, R. F. Bhola, M. L. Gelmi, R. Longhi, M. J. Christie, R. J. Lewis, P. F. Alewood and A. Brust, Angew. Chem., Int. Ed., 2015, 54, 1361–1364 CrossRef CAS.
- K. Holland-Nell and M. Meldal, Angew. Chem., Int. Ed., 2011, 50, 5204–5206 CrossRef CAS.
- Y. Gilad, M. Firer and G. Gellerman, Biomedicines, 2016, 4, 11 CrossRef.
- J. Habault and J. L. Poyet, Molecules, 2019, 24, 927 CrossRef.
- H. Zhao, X. Qin, D. Yang, Y. H. Jiang, W. H. Zheng, D. Y. Wang, Y. Tian, Q. S. Liu, N. H. Xu and Z. G. Li, Cell Death Discovery, 2017, 3, 17037 CrossRef CAS.
- Y. Chen, X. Xu, S. Hong, J. Chen, N. Liu, C. B. Underhill, K. Creswell and L. Zhang, Cancer Res., 2001, 61, 2434–2438 CAS.
- M. R. Felício, O. N. Silva, S. Gonçalves, N. C. Santos and O. L. Franco, Front. Chem., 2017, 5, 5 Search PubMed.
- B. Mishra, S. Reiling, D. Zarena and G. S. Wang, Curr. Opin. Chem. Biol., 2017, 38, 87–96 CrossRef CAS.
- D. Nejman, I. Livyatan, G. Fuks, N. Gavert, Y. Zwang, L. T. Geller, A. Rotter-Maskowitz, R. Weiser, G. Mallel, E. Gigi, A. Meltser, D. M. Douglas, I. Kamer, V. Gopalakrishnan, T. Dadosh, S. Levin-Zaidman, S. Avnet, T. Atlan, Z. A. Cooper, R. Arora, A. P. Cogdill, M. A. W. Khan, G. Ologun, Y. Bussi, A. Weinberger, M. Lotan-Pompan, O. Golani, G. Perry, M. Rokah, K. Bahar-Shany, E. A. Rozeman, C. U. Blank, A. Ronai, R. Shaoul, A. Amit, T. Dorfman, R. Kremer, Z. R. Cohen, S. Harnof, T. Siegal, E. Yehuda-Shnaidman, E. N. Gal-Yam, H. Shapira, N. Baldini, M. G. I. Langille, A. Ben-Nun, B. Kaufman, A. Nissan, T. Golan, M. Dadiani, K. Levanon, J. Bar, S. Yust-Katz, I. Barshack, D. S. Peeper, D. J. Raz, E. Segal, J. A. Wargo, J. Sandbank, N. Shental and R. Straussman, Science, 2020, 368, 973–980 CrossRef CAS.
- P. C. Barata and B. I. Rini, Ca-Cancer J. Clin., 2017, 67, 507–524 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |