
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a universal conductive platform for anchoring photo- and electroactive proteins using organometallic terpyridine molecular wires†
Margot
Jacquet
 a,
Miriam
Izzo
a,
Silvio
Osella
b,
Sylwia
Kozdra
c,
Paweł P.
Michałowski
c,
Dariusz
Gołowicz
d,
Krzysztof
Kazimierczuk
d,
Maciej T.
Gorzkowski
e,
Adam
Lewera
e,
Marian
Teodorczyk
c,
Bartosz
Trzaskowski
b,
Rafał
Jurczakowski
f,
Daniel T.
Gryko
*g and
Joanna
Kargul
*a
a,
Miriam
Izzo
a,
Silvio
Osella
b,
Sylwia
Kozdra
c,
Paweł P.
Michałowski
c,
Dariusz
Gołowicz
d,
Krzysztof
Kazimierczuk
d,
Maciej T.
Gorzkowski
e,
Adam
Lewera
e,
Marian
Teodorczyk
c,
Bartosz
Trzaskowski
b,
Rafał
Jurczakowski
f,
Daniel T.
Gryko
*g and
Joanna
Kargul
*a
aSolar Fuels Laboratory, Centre of New Technologies, University of Warsaw, Banacha 2C, 02-097 Warsaw, Poland. E-mail: j.kargul@cent.uw.edu.pl
bChemical and Biological Systems Simulation Laboratory, Centre of New Technologies, University of Warsaw, Banacha 2C, 02-097 Warsaw, Poland
cŁukasiewicz Research Network – Institute of Microelectronics and Photonics, Aleja Lotników 32/46, 02-668 Warsaw, Poland
dLaboratory of NMR Spectroscopy, Centre of New Technologies, University of Warsaw, Banacha 2C, 02-097 Warsaw, Poland
eCatalysis and Surface Chemistry Laboratory, Centre of Biological and Chemical Sciences, Faculty of Chemistry, University of Warsaw, ul. Żwirki i Wigury 101, 02-089 Warsaw, Poland
fElectrochemistry of New Materials, Centre of Biological and Chemical Sciences, Faculty of Chemistry, University of Warsaw, ul. Żwirki i Wigury 101, 02-089 Warsaw, Poland
gInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl
First published on 7th May 2021
Abstract
The construction of an efficient conductive interface between electrodes and electroactive proteins is a major challenge in the biosensor and bioelectrochemistry fields to achieve the desired nanodevice performance. Concomitantly, metallo-organic terpyridine wires have been extensively studied for their great ability to mediate electron transfer over a long-range distance. In this study, we report a novel stepwise bottom-up approach for assembling bioelectrodes based on a genetically modified model electroactive protein, cytochrome c553 (cyt c553) and an organometallic terpyridine (TPY) molecular wire self-assembled monolayer (SAM). Efficient anchoring of the TPY derivative (TPY-PO(OH)2) onto the ITO surface was achieved by optimising solvent composition. Uniform surface coverage with the electroactive protein was achieved by binding the cyt c553 molecules via the C-terminal His6-tag to the modified TPY macromolecules containing Earth abundant metallic redox centres. Photoelectrochemical characterisation demonstrates the crucial importance of the metal redox centre for the determination of the desired electron transfer properties between cyt and the ITO electrode. Even without the cyt protein, the ITO-TPY nanosystem reported here generates photocurrents whose densities are 2-fold higher that those reported earlier for ITO electrodes functionalised with the photoactive proteins such as photosystem I in the presence of an external mediator, and 30-fold higher than that of the pristine ITO. The universal chemical platform for anchoring and nanostructuring of (photo)electroactive proteins reported in this study provides a major advancement for the construction of efficient (bio)molecular systems requiring a high degree of precise supramolecular organisation as well as efficient charge transfer between (photo)redox-active molecular components and various types of electrode materials.
Introduction
Biohybrid technologies provide a promising approach to develop cost-effective, sustainable nanomaterials for a wide range of applications1–5 combining evolutionary optimised natural functionalities with advanced nanoengineering.6 In this context, metalloproteins play an important role due to their unique redox properties and fast (on ns-μs timescale) electron transfer (ET). One such protein, cytochrome c (cyt c) has been used as an electroactive component of various types of devices including biosensors and biosolar cells.7,8 This small protein containing a redox-active haem group is a crucial electron mediator in the mitochondrial respiratory chain and in photosynthesis. It has been intensively studied as a model system to understand the ET processes occurring in biological systems9 as well as in hybrid nanomaterials.10 Indeed, its ability to relay electrons has found many applications in various fields ranging from biosensors,11 biocatalysis,12 molecular memory13 and biophotovoltaics.14 In natural oxygenic photosynthesis, cyt c delivers water-derived electrons to the photo-oxidised primary electron donor of photosystem I (PSI), one of the key light-harvesting macromolecular components of the photosynthetic machinery. This feature has been utilised in artificial photosynthetic systems, whereby cyt c forms a bio-organic conductive interface between PSI and the electrode surface,15,16 at the same time orienting PSI so as to achieve a significant improvement of photocurrent output.17,18 A key factor for the optimal performance of such biohybrid devices is to ensure the efficient electronic communication between cyt c and the electrode surface, which can be achieved either via direct electron transfer (DET) or in a mediated fashion (MET). Depending on the nature of the ET many factors can influence the efficiency of this process such as the energetic compatibility with the electrode material, the surrounding solvent, as well as the mediator itself and its concentration in the case of the mediated ET.19 A common important factor is the nature of the interface between cyt c and the electrode surface that affects ET kinetic parameters and modifies the distance and orientation of the electroactive protein.11,20In the molecular wiring approach, the design of the molecular wire's structure together with the appropriate orientation of the photo-electroactive protein towards the electrode surface is important to achieve the highest possible charge transport efficiency and minimise wasteful back reactions. The rational approach (concluded both from the experimental data and theoretical calculations) for designing the molecular wire with improved conductivity is based on increasing the conjugation level of the electronic molecular structures.21–28 A promising strategy relies on the use of organometallic terpyridine (TPY) wires covalently attached to the conductive transparent material.29–31 The geometry of TPY complexes allows for the construction of a well-organised modular architecture that is amenable for the fine tuning of the conductive properties through the incorporation of various metallic redox centres.
It was reported that some self-assembled molecular wires, based on TPY complexes, can be highly conductive even up to ∼40 nm in length. This high conductivity was attributed to the presence of coordination redox-active centres in the highly rigid organic core.32 Remarkably, it was shown that there is no strong dependence between electron work function and the molecular length of the TPY wire.32 Another important factor to take into account is the development of the appropriate structure of the highly conductive TPY wire for the domain-specific anchoring of an electroactive protein such as cyt c. In this context, the bottom-up strategy based on step-by-step construction of the wire offers an advantage over other approaches as it permits to fine-tune the ET parameters by tuning the molecular structure of the wire.33,34 These structural and electronic features altogether make the coordinative TPY ligand-based conductive organic interface an excellent material of choice for efficient ET between redox-active proteins and the compatible electrode, together with its ability of strong and selective binding to protein molecules.
In this work, we report for the first time the rational design and cost-effective production of bio-hybrid nanodevices based on cyt c553 and well-organised metallo-organic terpyridine wires. Highly π-conjugated TPY ditopic ligands and phosphonate derivatives were synthesised and used as the conductive building blocks in the hybrid nanoarchitecture. The optimised step-by-step construction of the nano-assemblies on transparent oxide-based electrodes via the bottom up approach and using non-toxic Earth abundant metal redox centres is described. We demonstrate that these molecules form a well-defined surface-coverage SAM that can be used as a universal, highly conductive platform for the domain-specific anchoring of electroactive proteins. The efficient ET amenable for fine-tuning through the incorporation of metallic redox centres within the TPY structure paves the way for the application of this promising novel interface in various types of bionanodevices ranging from solar cells and solar-to-fuel devices to nanosensors.
Experimental
General materials
All chemical reagents and solvents used for synthesis were purchased from commercial sources (Aldrich, Acros and VWR) and used without further purification. ITO glass substrates were purchased from Ossila (15 mm × 20 mm, 1.1 mm thickness). Prior to use, ITO electrodes were cut in 15 × 10 mm for all following modifications, and in 7.5 mm × 10 mm for SIMS analysis. TPY-Br was synthesised by a combination of previously described procedures.30,35–37 Cyt c553 19AA protein was prepared and purified in our laboratory as previously described.38 The 1H and 13C NMR spectra were recorded on a 700 MHz and 176 MHz Agilent DirectDrive2 spectrometer equipped with a room-temperature HCN probe, temperature-controlled at 25 °C. 1H and 13C chemical shifts were calibrated to the residual solvent peak. Coupling constant values (J) are given in Hz and chemical shifts (δ) in ppm. The high-resolution electrospray ionisation mass spectroscopy (ESI-HRMS) analyses were performed on a Mariner mass spectrometer (PerSeptive Biosystems).Synthesis
ITO cleaning and activation
Commercial ITO surfaces were first cleaned by a detergent/solution (DSC) process; surfaces were scrubbed with a drop of Triton X-100, followed by successive sonication in diluted Triton X-100 solution in water, Mili-Q water and ethanol for 15 min each. Dried surfaces were then activated in an oxygen-fed plasma cleaner (Plasma processing reactor centre Dionex 2000 coupled to a solid-state power generator OEM-12A) operating at 100 W for 15 min, followed by direct immersion in the anchor solution.Anchoring optimisation
Cleaned and activated ITO surfaces were immersed in 1 mM solution of the different surface anchors overnight (TPY-PO(OEt)2 in CHCl3, TPY-PO(OH)2 in DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1) and TPY-PO(OH)2 in DMSO), then rinsed thoroughly with their respective solvent followed by ethanol and annealed at 140 °C for 30 min to complete the chemical bonding. Surfaces were then sonicated with 5% Et3N solution in ethanol for 15 min and rinsed generously with ethanol. The anchored ITO surfaces were immersed in 0.1 M solution of FeSO4·7H2O in water for 15 min followed by a copious rinsing with water and ethanol. The dried surfaces were then immersed in 1 mM solution of 2,2′:6′,2′′-terpyridine in CHCl3 for 15 min followed by a generous rinsing with CHCl3.
:H2O (1:1) and TPY-PO(OH)2 in DMSO), then rinsed thoroughly with their respective solvent followed by ethanol and annealed at 140 °C for 30 min to complete the chemical bonding. Surfaces were then sonicated with 5% Et3N solution in ethanol for 15 min and rinsed generously with ethanol. The anchored ITO surfaces were immersed in 0.1 M solution of FeSO4·7H2O in water for 15 min followed by a copious rinsing with water and ethanol. The dried surfaces were then immersed in 1 mM solution of 2,2′:6′,2′′-terpyridine in CHCl3 for 15 min followed by a generous rinsing with CHCl3.
Stepwise formation of the modified ITO electrode
Cleaned and activated ITO surfaces were immersed in 1 mM solution of TPY-PO(OH)2 in DMSO overnight, then rinsed thoroughly with DMSO followed by ethanol and annealed at 140 °C for 30 min. Surfaces were then sonicated with 5% Et3N solution in ethanol for 15 min and rinsed generously with ethanol. The anchored ITO surfaces were immersed in 0.1 M solution of the corresponding metal (FeSO4·7H2O or Co(NO3)2·6H2O) in water for 15 min followed by a copious rinsing with water and ethanol. The dried surfaces were then immersed in 0.1 mM solution of the ditopic ligand Lφ in CHCl3 overnight followed by a generous rinsing with CHCl3. The metalation step was reproduced as before with the corresponding metal salt. Biofunctionalisation was performed by incubating the modified ITO electrodes with 30 μM 19AA cyt c553 solution (25% glycerol in 5 mM phosphate buffer) for 2 h at room temperature followed by rinsing with 5 mM phosphate buffer (pH 7).Secondary ion mass spectrometry (SIMS)
SIMS measurements were performed employing a CAMECA SC Ultra instrument under ultra-high vacuum (UHV), usually of 4 × 10−10 mbar. The Cs + primary beam was rastered over 250 × 250 μm2 (the analysis area was limited to 200 × 200 μm2) and positive ion detection mode was used in the experiments and thus all species were measured as CsX + cluster ions. The intensity of the primary beam was 2 pA, and the impact energy was 5 keV. To perform lateral imaging measurements a highly uniform beam was required – the beam on the sample in the SC Ultra tool has a square shape and owing to the “variable rectangular shape concept” forms a homogeneous spot. The primary beam at the working point in the SC Ultra is formed by two stencils – well-shaped apertures. While the first one is used to choose the most intense and homogeneous part of the Gaussian-shaped ion beam, the second one changes the size of the spot.X-ray photoelectron spectroscopy (XPS)
XPS experiments were carried out using a SPECS Surface Nano Analysis GmbH (Berlin, Germany) instrument equipped with an XR 50 MF X-ray source and a μ-Focus 600 monochromator (600 mm Rowland Circle), using a monochromatised X-ray Al Kα emission line, photon energy 1486.6 eV, operating at 100 W, and a Phoibos 150 hemispherical analyser with a 150 mm radius, NAP version, equipped with a 2D-DLD detector. The system base pressure was in the 10−10 mbar range. Spectra were fitted using Gaussian–Lorentzian lineshapes using CasaXPS software, version 2.3.18PR1.0 and the quantitative analysis was performed using CasaXPS built-in Scofield relative sensitivity factors.Electrochemistry
Electrochemical experiments were performed with a Metrohm Autolab B.V. potentiostat/galvanostat in a custom-made Teflon three-electrode cell. Cyclic voltammetry was conducted under an argon atmosphere, using a glassy carbon rod as a counter electrode (CE) and an ITO surface as a working electrode (WE) connected with a conductive adhesive copper tape (6.4 mm width, 1181, 3 M) to provide electrical contact. For non-aqueous experiments, 0.1 M HFPTBA in CH3CN was used as the electrolyte support with an Ag/AgNO3 (0.01 M AgNO3) reference electrode (REF1). The reference electrode was calibrated with ferrocene prior to use with a Fc/Fc+ redox potential at 81 mV. For aqueous measurements, 0.1 M phosphate buffer (pH 7) was used as the electrolyte support with an Ag/AgCl (3 M KCl) reference electrode (REF2). The surface coverage Γ (mol cm−2) was calculated using the following eqn (1), where Ip is the current peak intensity, n is the number of electrons involved in the process (n = 1), F is the Faraday constant, R is the gas constant, T is the temperature (298 K), ν is the scan rate and A is the surface.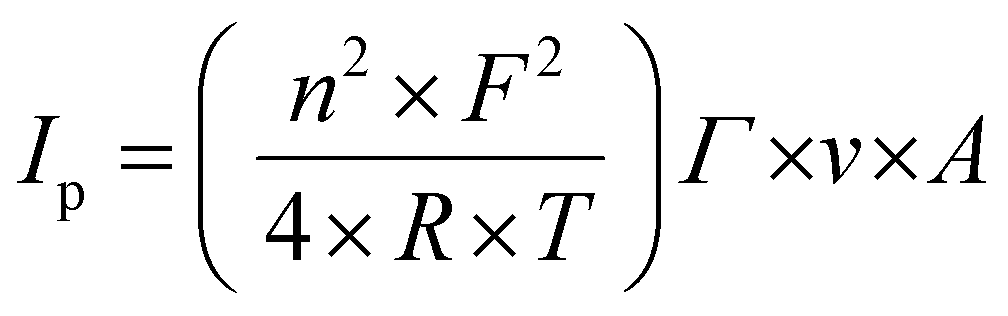 | (1) |
Electrochemical impedance measurements were conducted in 0.1 M phosphate buffer (pH 7) with 1 mM 1,1′-ferrocenedimethanol in a frequency range from 0.01 Hz to 0.1 MHz. Z-View 2 software was used to fit the impedance data with the equivalent circuit. Photoelectrochemical experiments were performed using a KL 2500 LCD halogen white light source (Schott) with a light intensity of 100 mW cm−2. The geometric surface area of the analysed samples was calculated as 0.4185 cm2. Photochronoamperometric experiments were performed under aerobic conditions at room temperature with a 5 mM phosphate buffer (pH 7) as the electrolyte support. Before each measurement, the open circuit potential (OCP) was recorded under dark conditions until a stable potential was achieved. During photochronoamperometric measurements, samples were illuminated at different potentials (vs. Ag/AgCl) with 30 s ‘light ON/OFF’ periods. The influence of the presence of O2 was studied firstly in deaerated solution (20 min bubbling with argon), then with a freshly aerated electrolyte solution.
Computational methods
A 15 ns-long NPT molecular dynamics was run with a 2 fs Time step using Desmond software (Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2020. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2020) with the OPLS2005 forcefield.43 The Nose–Hoover thermostat was used, keeping the temperature at 300 K, while the pressure was constrained using the Martyna–Tobias–Klein barostat at 1 bar. Long range non-bonding interactions (Coulomb and van der Waals) have been considered within a 1.5 nm cutoff. Water was modelled using the TIP3P model. TPY molecules forming the SAM were restrained with a force constant of 10 Hartree. To speed up the computations, the ITO surface was not considered explicitly, and hydrogen atoms were added to TPY to avoid radicals.
Results and discussion
Synthesis of the TPY ligands
The synthesis pathway used to obtain the anchor TPY-PO(OH)240 and the ditopic ligand Lφ used in the present study is shown in Scheme 1. The tTPY-Br intermediate was formed by a Hantzsch reaction in the presence of potassium hydroxide and ammonium hydroxide in ethanol.39 The phosphonate TPY-PO(OEt)2 was obtained by a palladium-catalysed cross-coupling between tTPY-Br and diethylphosphite in toluene with triethylamine. As the next step, deprotection was performed using bromotrimethylsilane in dichloromethane. The boronate ester TPY-BO2-C6H12 was obtained from a palladium-catalysed cross-coupling between tTPY-Br and bis(pinacolato)diboron in dimethylsulfoxide in the presence of potassium acetate.41 Finally, the ditopic ligand Lφ (Fig. S1–S4†) was synthesised by a Suzuki–Miyaura coupling between TPY-BO2-C6H12 and TPY-Br in a water-tetrahydrofuran mixture in the presence of potassium carbonate.42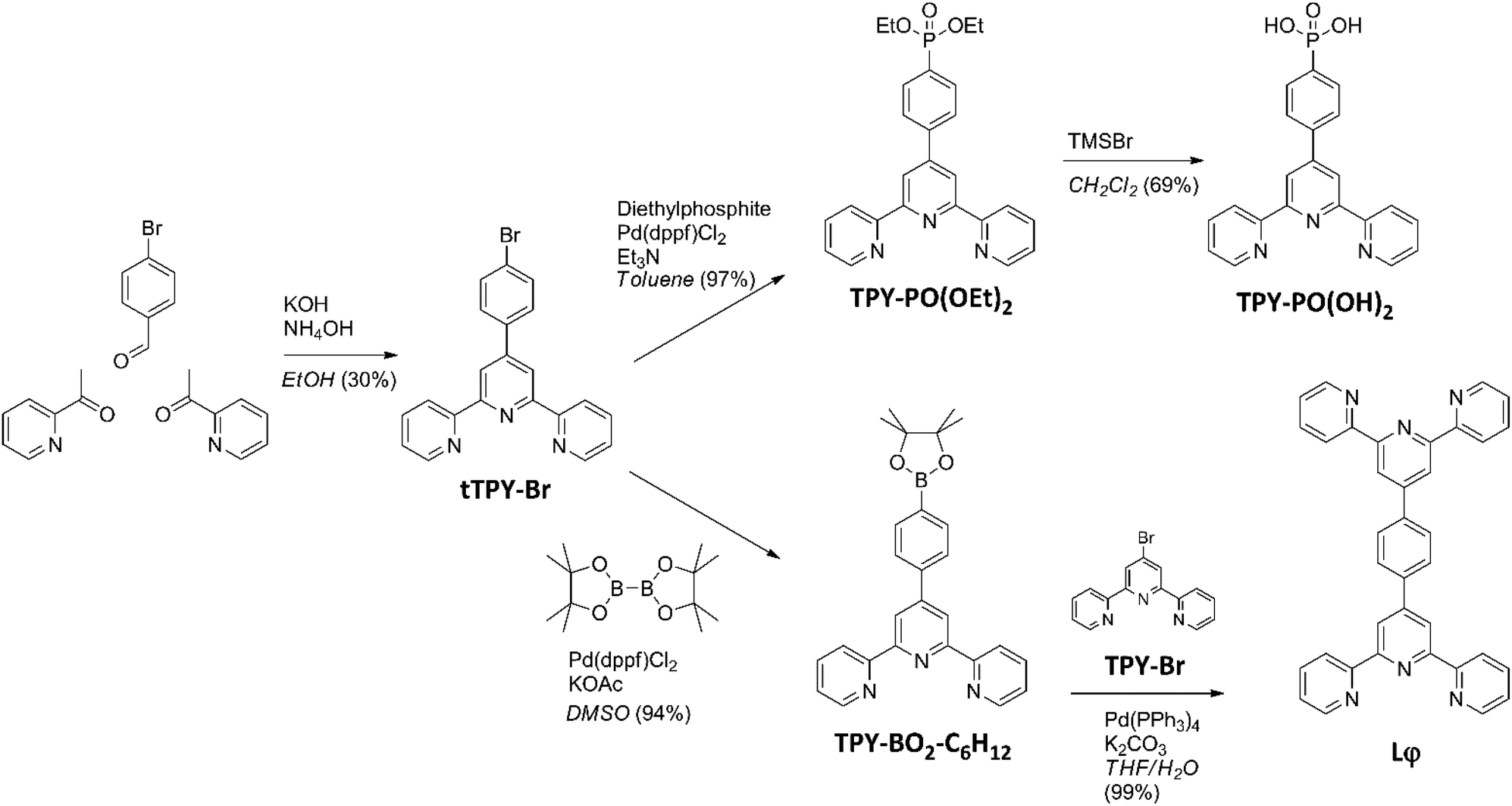 | ||
| Scheme 1 Synthesis pathway for the anchor TPY-PO(OH)2 and the ditopic ligand Lφ. | ||
Optimisation of the ITO surface anchoring process
Although the phosphonic-type attachment group showed efficient properties concerning stability and reactivity towards different types of electrode surfaces,45–47 the compound TPY-PO(OH)2 has been used in this study for the first time for direct anchoring onto the ITO surface. In 2010, Spampinato et al. reported a methodology to graft this anchor on an SiO2 surface using a priming step based on a zirconium phosphate monolayer. The anchor can then be attached either by the TPY moiety or by the phosphonic acid.40 In 2007, Wolpher et al., described properties of ruthenium(II) complexes grafted on nanostructured TiO2.48 However, the authors of the latter study directly attached the entire complex onto the surface in contrast to the step-by-step, bottom-up approach of the present study.In order to find the best conditions for ITO grafting, both TPY-PO(OEt)2 and TPY-PO(OH)2 compounds were investigated since both functions were reported for efficient attachment to various types of electrode surfaces via a phosphonate group.49–53 Commercially available ITO surfaces were first subjected to a detergent/solution cleaning (DSC) followed by an activation with oxygen plasma treatment to improve electroactivity and surface work function.54 The plasma-activated surfaces were functionalised using different conditions depending of the compound solubility: TPY-PO(OEt)2 in CHCl3, TPY-PO(OH)2 in DMF:H2O (1:1)40 or TPY-PO(OH)2 in DMSO, followed by an annealing treatment to increase chemical bond stability.55,56
Incorporation of the redox metal centre was performed in an aqueous solution of FeSO4·7H2O and the coordination sphere was finally completed by immersing the surfaces in a solution of 2,2′:6′,2′′-terpyridine.
To assess the efficiency of surface coverage using the abovementioned step-by-step approach, the freshly modified electrodes were analysed by secondary ion mass spectrometry (SIMS) to simultaneously map the presence of phosphorus and iron atoms. The SIMS analyses confirmed that efficient TPY anchoring and complex formation was accomplished on the ITO surface especially when DMSO was used as the solvent for the TPY-PO(OH)2 modifier (Fig. 1C). The mapping result obtained for TPY-PO(OEt)2 shows a strong heterogeneity between phosphorus and iron distribution with the non-functionalised area corresponding to the black spots in Fig. 1A. The higher concentration of phosphorus compared to iron could be rationalised by a partial protonation of the terpyridine or a misalignment of the terpyridine motif due to the acidic tendency of chloroform57 preventing the chelation of the iron centre. In contrast, both samples prepared from TPY-PO(OH)2 display better surface coverage (Fig. 1B and C) characterised by a higher concentration of phosphorus atoms. A particularly higher density and clear coverage homogeneity of phosphorus and iron atoms are demonstrated for the sample produced from DMSO solution (when compared to other solvents) displaying the presence of the same subtle shadow pattern in both phosphorus and iron maps (Fig. 1C). The observed mismatch between phosphorus and iron atoms in the case of the DMF:H2O solution could be explained by the poor solubility of the anchor, and the presence of residual compounds, which could hinder the access of iron.
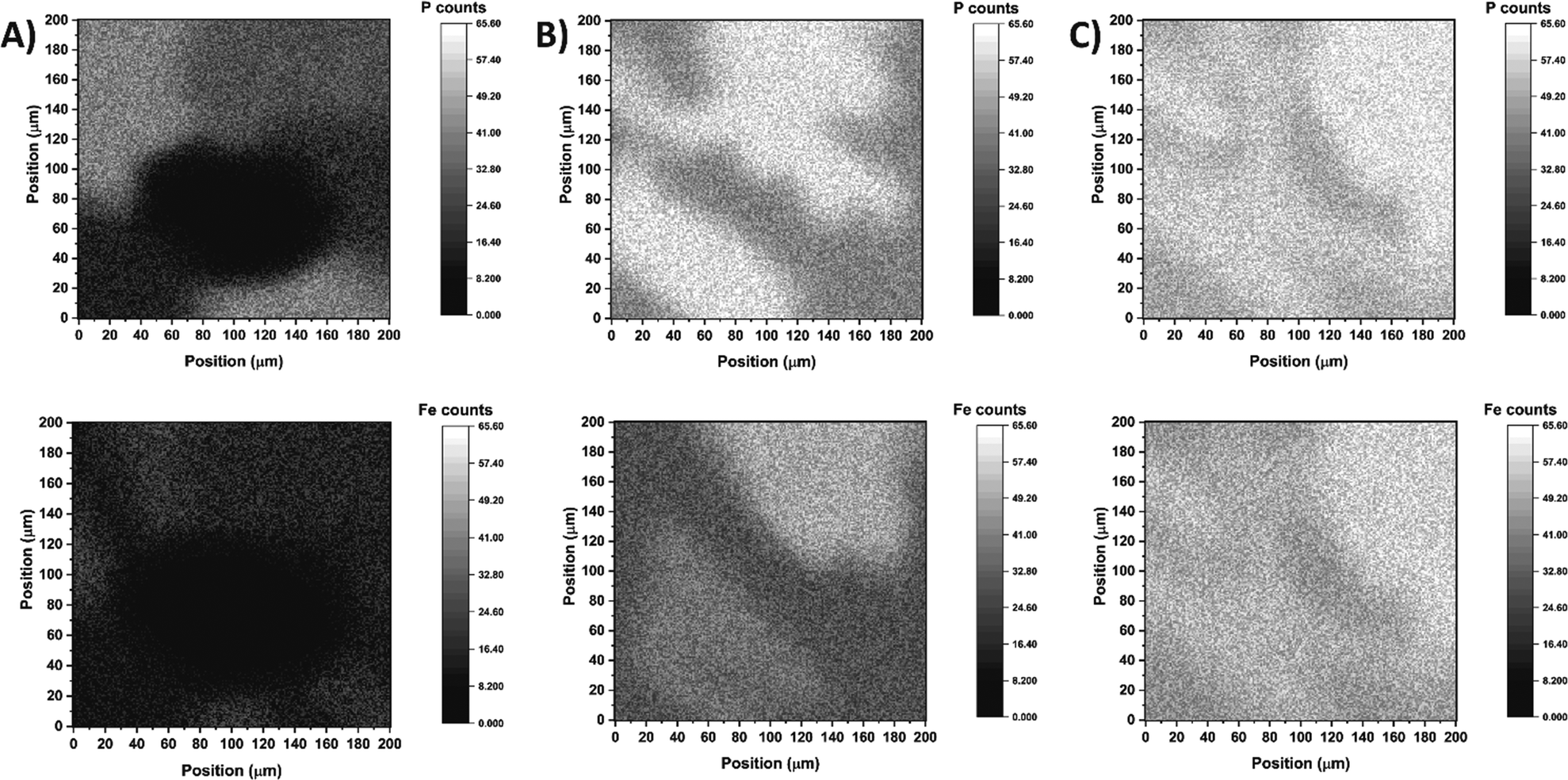 | ||
| Fig. 1 Secondary ion mass spectrometry analyses of ITO-TPY-Fe-TPY electrodes with phosphorus (top) and iron (bottom) atoms. (A) Surface obtained with TPY-PO(OEt)2 in CHCl3. (B) Surface obtained with TPY-PO(OH)2 in DMF:H2O (1:1). (C) Surface obtained with TPY-PO(OH)2 in DMSO. | ||
A similar sample of TPY-PO(OH)2 in DMSO with cobalt was also analysed (Fig. S6†), confirming the good surface coverage and efficient formation of cobalt complexes. Consequently, all following experiments on the functionalisation of the ITO electrodes were performed using TPY-PO(OH)2 solution in DMSO.
Construction and characterisation of cobalt and iron-based ITO electrodes
The methodology employed for the construction of cyt c553 biohybrid electrodes is depicted in Fig. 2. It was important to rationally design the final TPY molecular structure so as to achieve fine-tuning of the ET process within the SAM through incorporation of the metallic redox centres into the wire. To this end, two different metals were chosen for insertion into the structure of the TPY molecules, iron and cobalt, for their ability to easily form TPY complexes under mild conditions, which is optimal for the development of a viable, step-by-step, bottom-up strategy. Typically, cleaned and activated ITO surfaces were anchored with TPY-PO(OH)2 in DMSO, as previously described. Metalation was performed with either FeSO4·7H2O or Co(NO3)2·6H2O aqueous solution followed by the complexation with the ditopic ligand Lφ in CHCl3. Homo-dinuclear TPY wires were formed by a second metalation step. Finally, the cyt c553 protein molecules (Fig. S6†) were immobilised through binding of the C-terminal His6-tag moiety (genetically incorporated into the cyt structure, see the ESI†) to the metal centre in order to form a thin layer of this electroactive protein.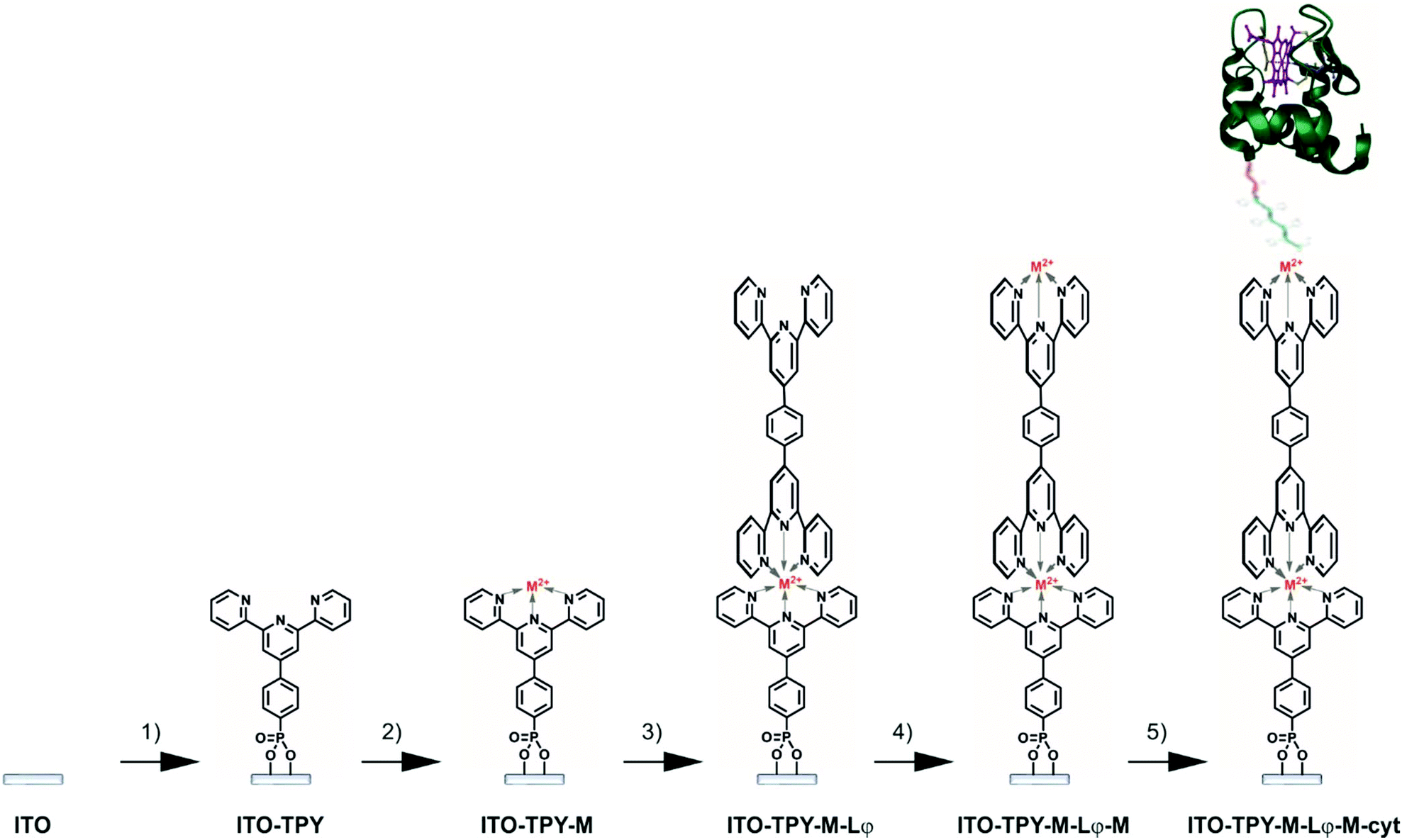 | ||
| Fig. 2 Stepwise preparation of functionalised ITO electrodes. (1) 1 mM TPY-PO(OH)2 in DMSO. (2) 0.1 M aqueous solution of FeSO4·7H2O or Co(NO3)2·6H2O. (3) 0.1 mM Lφ in CHCl3. (4) 0.1 M aqueous solution of FeSO4·7H2O or Co(NO3)2·6H2O. (5) 30 μM 19AA cyt c553 in phosphate buffer. | ||
X-ray photoelectron spectroscopy (XPS) analysis was performed on the bare ITO surface and on the full organometallic wires (ITO-TPY-Co-Lφ-Co and ITO-TPY-Fe-Lφ-Fe) in order to analyse the elemental composition of the surfaces (Fig. S7 and S8†). Due to the extremely low concentration of Co-TPY and Fe-TPY on the ITO surface, the registered spectra for those samples were dominated by signals of ITO components. Analysis of the Fe 2p region for the pure ITO and ITO-TPY-Fe-Lφ-Fe samples revealed overlapping of potential Fe 2p signals with strong In 2p1/2 and Sn 3p3/2 signals, making the detection of Fe impossible (Fig. S7†). The small Fe 2p signals are completely masked by the In 3p1/2 and Sn 3p3/2 signals.
The analysis of the Co 2p region (Fig. S8A†) in the ITO-TPY-Co-Lφ-Co sample revealed the Co 2p signals typical of Co(II) components, namely 2p3/2 and 2p1/2 doublets at 781.5 and 796.6 eV, respectively (versus 778.3 for Co 2p3/2 for metallic Co58), and two doublets of satellite peaks (present only for the oxidised Co) at 787.0 and 802.1 eV (first doublet) and 792.1 and 806.6 eV (second doublet), together with the Co LMM signal at 776.0 eV. Those signals were not present in the case of pure ITO (see Fig. S8A†). XP spectra registered for N 1s and P 2p regions (Fig. S8B†) allowed for a rough approximation of sample elemental composition. As both samples are dominated by In and Sn (components of ITO), O (component of ITO and part of the adventitious organic contamination), and C (mostly adventitious carbon), the general atomic composition did not represent the Co-TPY composition. Thus, we focused on Co, N and P ratios, as those elements are not common contaminants and were not present in significant amounts in the control ITO sample (see Fig. S8†). Using integrated Co 2p, N 1s and P 2p signals (Fig. S8†) and Casa build-in Scofield Relative Sensitivity Factors, equal to 1.8 for N 1s, 1.192 for P 2p and 19.16 for Co 2p, the Co:N:P atomic ratio was determined as close to 1.0:2.3:1.0, where the small amount of N and P originating from the reference ITO sample was subtracted from the N and P amounts determined from the N 1s and P 2p signals in the Co-TPY-ITO sample. The XPS quantitative analysis proved to be challenging due to the fact that a monolayer of TPY ligand wires yielded extremely low signals, and thus, determination of the peak area for such low intensity peaks may result in errors as high as ±25%.
The step-by-step construction of TPY-functionalised electrodes was followed by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) analyses which were used to explore the evolution of interfacial electronic processes (Fig. 3). Experiments were performed in the presence of a redox probe 1,1′-ferrocenedimethanol in phosphate buffer (pH 7) in a frequency range from 0.01 Hz to 0.1 MHz. The corresponding Nyquist plot and CV scans obtained for Fe-TPY-based electrodes are presented in Fig. 3 (see Fig. S9† for Co-TPY ligand-functionalised electrodes). The charge transfer resistance R2 and the double layer capacitance Cdl of the bare and modified ITO electrodes were calculated by fitting the impedance data with the equivalent circuit model (Fig. 3C) where R1 stands for the electrolyte resistance, CPE1 is the constant phase element corresponding to double layer capacitance59 existing between the solid/liquid phases, R2 corresponds to the charge transfer resistance between the electrolyte and the studied ITO electrode, and W1 represents Warburg impedance caused by the diffusion process. The obtained resistances (R1/R2) and capacitance Cdl (from CPE1 parameter Q and Φ59) values are summarised in Table 1. The evolution of the resistance values shown in Fig. 3A and in Table 1 is consistent with the step-by-step construction of the organometallic TPY ligand molecular wires (terminated with the anchored His6-tagged cyt c553). The bare ITO electrode possesses a small charge transfer resistance of 178 Ω cm−2 in accordance with the good conductivity of this material. After the attachment of TPY-phosphonic acid, the resistance rises to 323 Ω cm−2 and continues to increase after each step of functionalisation, reaching the final value of 1.09 kΩ cm−2 and 1.02 kΩ cm−2 respectively for Fe- and Co-containing TPY wires. Even though the construction of the organometallic interface hinders somewhat the charge transfer process, it is interesting to notice that the whole conductivity, which is inversely proportional to the resistivity, is still promising prior to the cyt c553 attachment. After the immobilisation of this protein, a significant increase of resistance is observed with the values of 3.73 kΩ cm−2 for iron and 2.39 kΩ cm−2 for cobalt nanoassemblies, which is correlated with the insulating properties of the dense protein backbone.60–62 The hindrance of ET due to the presence of cyt c553 is also visible from the CV analysis shown in Fig. 3B. For the Fe-based TPY wires, a well-defined quasi-reversible redox process is observed. After the biofunctionalisation step, a clear attenuation of the electrochemical processes is recorded with an increase of the peak-to-peak potential separation of 100 mV and a decrease of current intensity.
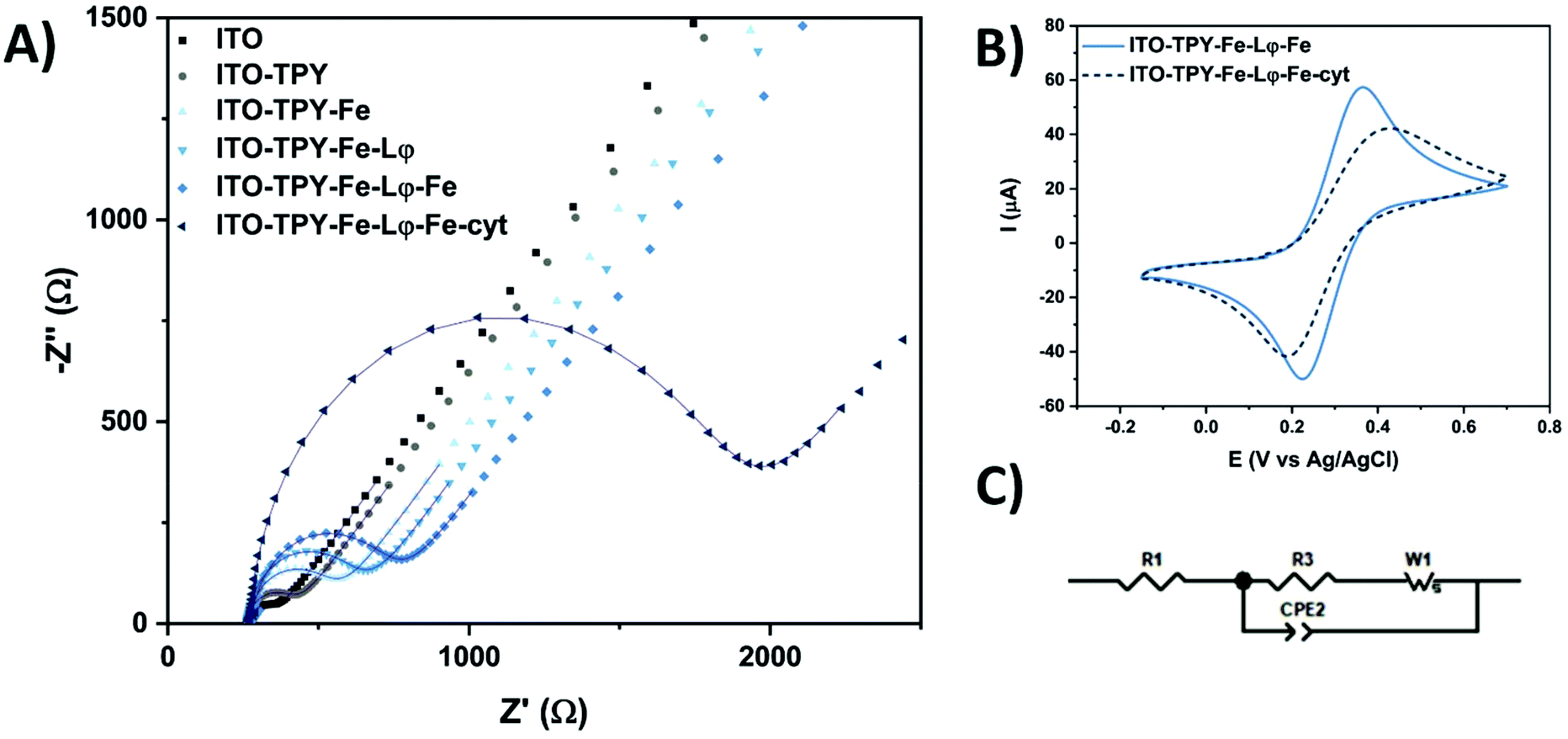 | ||
| Fig. 3 (A) Nyquist plot with fitted curves of electrochemical impedance spectra obtained for Fe-based ITO electrodes at different steps of functionalisation. (B) Cyclic voltammetry of ITO-TPY-Fe-Lφ-Fe and ITO-TPY-Fe-Lφ-Fe-cyt at 100 mV s−1. (C) Equivalent circuit model. All measurements were performed with 1 mM 1,1′-ferrocenedimethanol in 0.1 M phosphate buffer (pH 7) in the dark. | ||
| Samples | Dark | Light | ||||
|---|---|---|---|---|---|---|
| R1 (Ω cm−2) | R2 (kΩ cm−2) | C dl (μF cm−2) | R1 (Ω cm−2) | R2 (kΩ cm−2) | C dl (μF cm−2) | |
| ITO | 672 | 0.18 | 12.5 | 663 | 0.25 | 13.2 |
| ITO-TPY | 632 | 0.32 | 13.3 | 618 | 0.42 | 13.8 |
| ITO-TPY-Co | 627 | 0.61 | 14.0 | 642 | 0.71 | 14.2 |
| ITO-TPY-Fe | 639 | 0.65 | 13.8 | 639 | 0.77 | 14.0 |
| ITO-TPY-Co-Lφ | 628 | 0.84 | 14.9 | 626 | 0.90 | 14.8 |
| ITO-TPY-Fe-Lφ | 628 | 0.87 | 14.1 | 611 | 0.89 | 14.3 |
| ITO-TPY-Co-Lφ-Co | 644 | 1.02 | 14.0 | 638 | 1.02 | 14.1 |
| ITO-TPY-Fe-Lφ-Fe | 664 | 1.09 | 14.1 | 663 | 1.10 | 14.4 |
| ITO-TPY-Co-Lφ-Co-cyt | 632 | 2.39 | 14.7 | 628 | 2.26 | 14.7 |
| ITO-TPY-Fe-Lφ-Fe-cyt | 790 | 3.73 | 15.3 | 774 | 3.26 | 15.4 |
Interestingly, the double layer capacity of the interface does not significantly change upon the stepwise electrode functionalisation suggesting that the nanostructures with inbound cyt c553 molecules do not collapse at the electrode surface. Moreover, the Φ parameter of the CPE element is close to unity (0.92 < Φ < 0.96) suggesting the formation of a highly ordered architecture at the molecular level.63 The homogeneous electrode coverage with the bio-interface was confirmed by the calculation of the apparent fractional electrode coverage parameter ϴ,64 with the respective values of 0.93 for Co-TPY wires and 0.95 for the Fe-TPY counterparts.
The interface resistivity was also measured under light conditions for all the different configurations (Table 1). Similar to the bare ITO, all the electrodes devoid of cyt c553 possess a higher or similar resistance upon exposure to light compared to their respective resistance under dark conditions. In contrast, for both cyt-based electrodes noticeably lower resistivity values were observed, especially for the iron-based TPY interface, with a decrease from 3.73 kΩ cm−2 to 3.26 kΩ cm−2 (see Table 1). The observed lower resistivity in the presence of cyt c553 highlights the beneficial combination of TPY SAM and the redox protein under light irradiation, which is promising for further development of high-performance bio-photoelectrodes, such as those based on photosynthetic macromolecular machines.18,65
Additional electrochemical analyses were performed in acetonitrile with 0.1 M hexafluorophosphate tetrabutylammonium (HFPTBA) for Fe- and Co-TPY organometallic wires in order to study their redox behaviour upon immobilisation on ITO (Fig. 4 and Table 2). In solution, these TPY complexes possess a MIII/MII redox signature at E1/2 = 0.76 V and E1/2 = −0.06 V,66 respectively for iron and cobalt with a peak-to-peak potential separation of 60–70 mV, typical of a reversible system (n = 1). Upon their assembly on the ITO surface, the Fe-TPY complexes show a redox signature at E1/2 = 0.81 V (Fig. 4A) and the Co counterparts at E1/2 = −0.1 V (Fig. S10†). Their peak-to-peak potential separation values are very small with respective values of 5 mV and 12 mV for Fe and Co complexes, which indicates a surface process with fast ET kinetics and also points out a facile charge transport between the ITO electrode and the TPY SAM.31 The high reversibility of the ET process demonstrates the formation of the highly conductive TPY SAM on ITO, which is further confirmed by the observed linear dependency of current intensity with the scan rate67,68 (Fig. 4B and Fig. S10B†). From these analyses, the surface coverage of both Fe-and Co-TPY SAMs was determined as 1.65 × 10−11 mol cm−2 and 1.27 × 10−11 mol cm−2, respectively, corresponding to the values reported for similar architectures on ITO.69,70 Using Laviron's equation,71 the interfacial electron transfer kinetic constants kET of both complexes were determined with a similar value of 29 s−1. This electron rate is lower compared to the one published on gold72 or silicon73 probably due to the loss of conjugation induced by the tetrahedral phosphorus atom in the TPY anchor. Nevertheless, the kinetic constant values determined for both systems are still promising for the attachment of cytochrome c which was shown to yield smaller electron transfer rates when adsorbed74 (kET = 18 s−1) or wired75 (kET = 5.9 s−1) onto ITO.
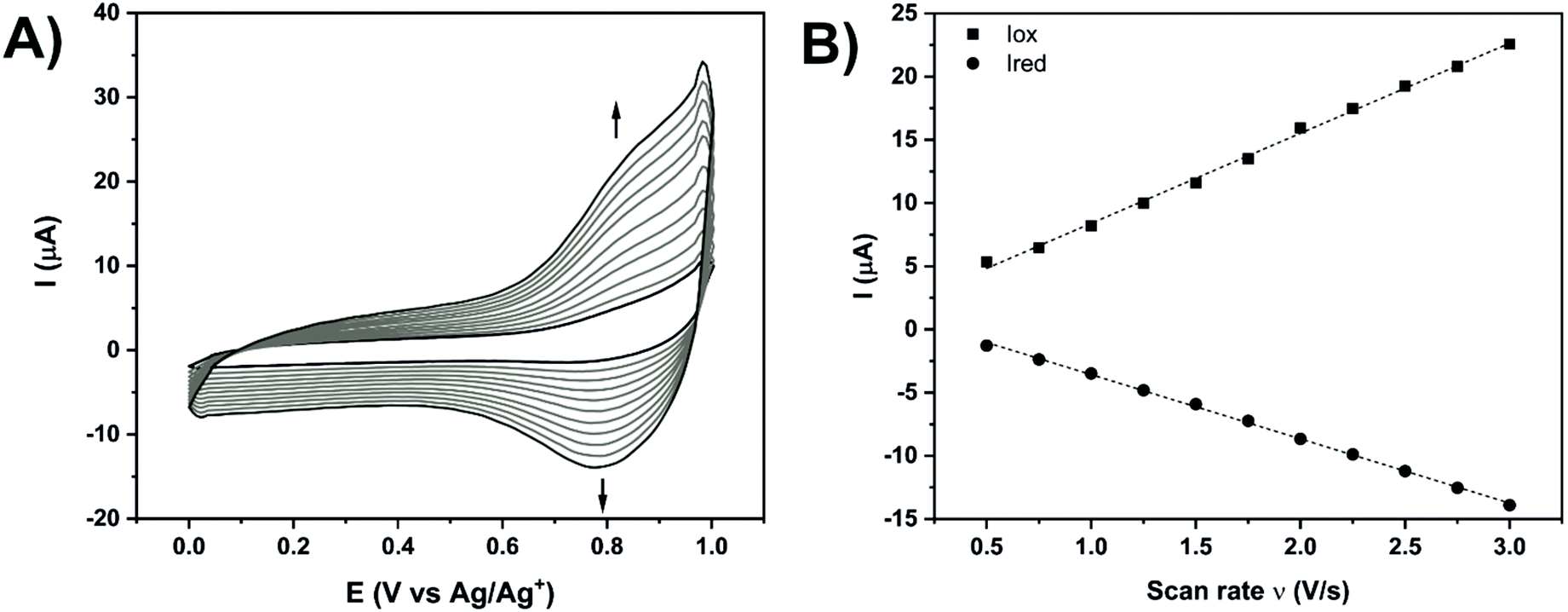 | ||
| Fig. 4 (A) Cyclic voltammetry of ITO-TPY-Fe-Lφ at different scan rates from 500 mV s−1 to 3 V s−1 in CH3CN + 0.1 M HFPTBA. (B) Linear relation between the current peak and scan rate obtained for ITO-TPY-Fe-Lφ. | ||
It was important to relate all the above data to the electrochemical properties of the immobilised TPY SAM under biocompatible conditions, as ultimately the developed TPY interface is destined for the domain-specific capturing of (photo)electroactive proteins including photosystems and other redox active enzymes of biotechnological and fundamental applications. To this end, a preliminary electrochemical analysis of the His6-tagged cyt c553 in solution was performed using an aqueous buffer (phosphate buffer, pH 7) as an electrolyte and a bare ITO as a WE (see Fig. 5A). As expected, in the potential range from −0.2 V to 0.6 V the bare ITO is redox inactive. After the addition of cyt c553, a well-defined redox system appears with an oxidative peak at 0.33 V and a reductive peak at 0.06 V. This redox signature, attributed to the FeIII/FeII couple in the haem group, is positively shifted compared to previously reported data on ITO found between −0.1 V and 0.1 V for horse heart and yeast cyt c.74,76,77 Although different, the redox peak detection confirms the ability of the modified cyt c553 to conduct ET with pristine ITO. Finally, both metal-based architectures with immobilised cyt were analysed under the same conditions with regards to the 5 mV s−1 scan rate.
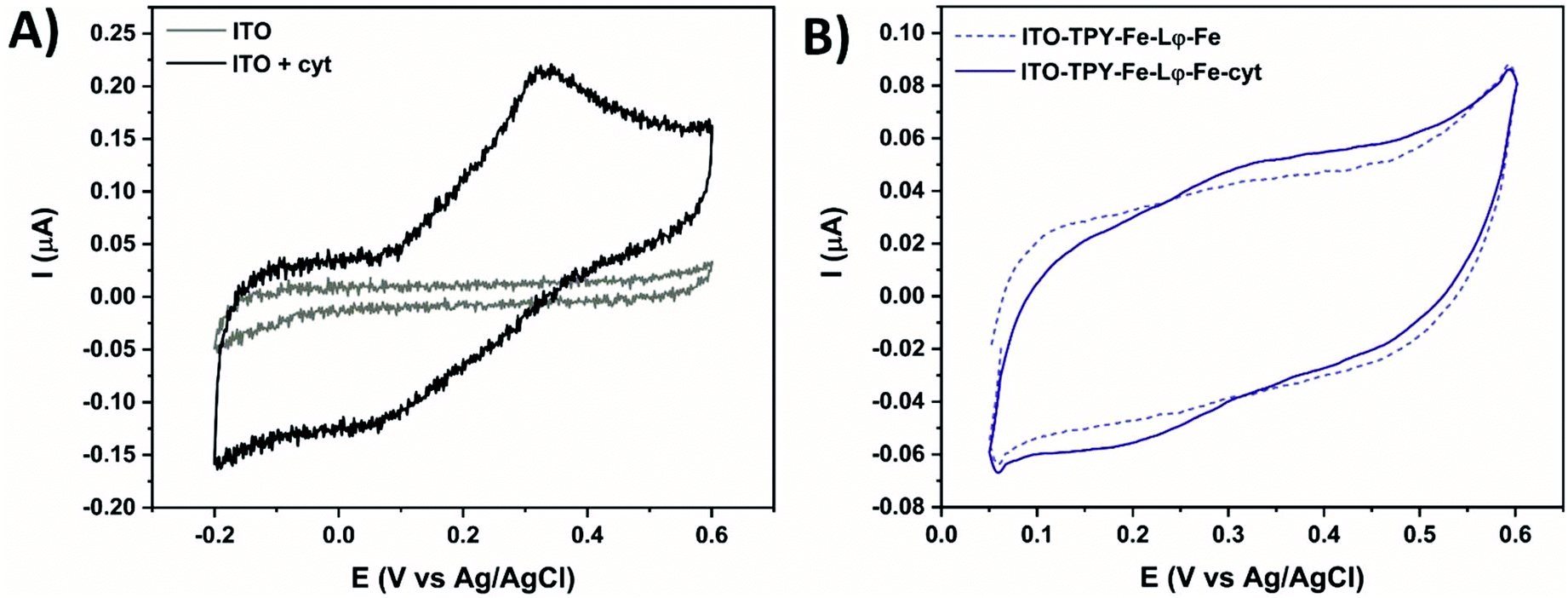 | ||
| Fig. 5 Cyclic voltammetry at 5 mV s−1 in 0.1 M phosphate buffer (pH 7) of the ITO electrode without and in the presence of 7 μM cyt c553 (A). Fe-based electrodes with and without immobilised cyt (B). | ||
Fig. 5B shows the CV of the Fe-based TPY-modified electrodes in the absence or presence of the anchored cyt c553. A clear signature of the immobilised redox active cyt protein is visible, with an oxidative peak at 0.33 V and a reductive peak at 0.2 V. The peak-to-peak potential separation of 130 mV is smaller compared to the one found for cyt c553 in solution (270 mV), which proves the immobilisation of the protein with an improvement of surface processes. Moreover, the smaller peak-to-peak potential separation proves the structural integrity of the immobilised cytochrome, whereas the formation of aggregates or unfolded cytochrome was shown to result in fully irreversible CV signatures.78,79 Concerning the Co-based nanoarchitecture, the electrochemical analyses revealed quenching of the ET process as confirmed by the absence of the cyt c553 redox signature (Fig. S11†).
The difference in the ET properties depending on the metal-centre used could be explained by a variable contribution of the ET depending on the metal centre used in the TPY SAM. In the case of the iron, this metal centre facilitates ET between the haem group of cyt and the ITO surface, whereas the incorporation of a cobalt redox centre into the TPY SAM inhibits this process. These electrochemical data clearly point to the importance of the SAM molecular structure for efficient ET to occur in the biohybrid electronic devices, whereby the presence of the specific metallic centre in the conductive SAM can greatly affect the kinetics and directionality of ET.80
Photocurrent measurements
In order to verify the suitability of the organometallic TPY-based SAM developed in this study for future applications, such as those involving the oriented light-harvester/charge separator, photosystem I,18 photochronoamperometry measurements were conducted for all the electrode configurations. All the analyses were conducted in an oxygenated aqueous buffer without any external electron mediator. The obtained photocurrent density values (J) are presented in Fig. 6.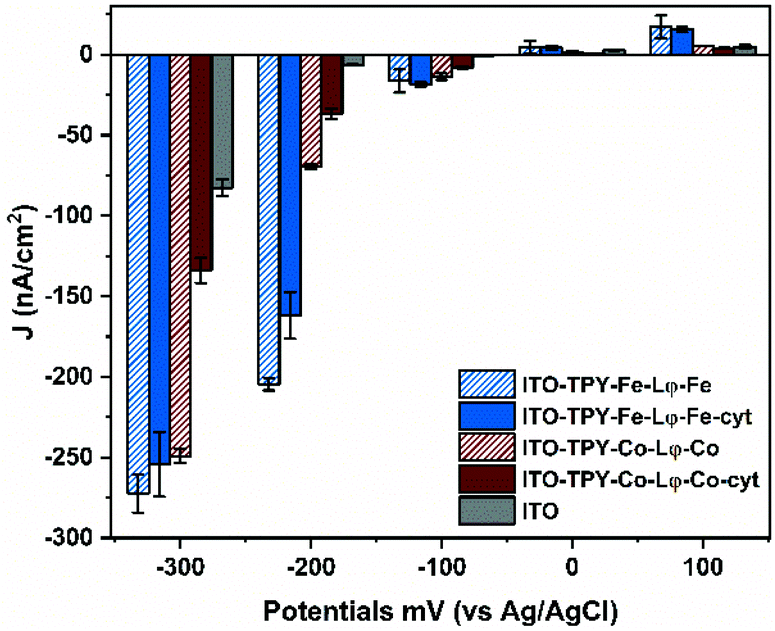 | ||
| Fig. 6 Photocurrent density generation of ITO electrodes at different applied potentials vs. Ag/AgCl. The current density values correspond to the average values from two independent measurements with error bars representing standard deviation (n = 2). | ||
Overall, all the TPY-functionalised electrodes are characterised by a preferential generation of the cathodic photocurrent. This feature is particularly prominent in the case of Fe-TPY SAM. Indeed at +100 mV, the highest anodic current was recorded for Fe-based TPY wires with a value of 17.2 nA cm−2, slightly above that of the corresponding electrode with captured cyt c553 (15.6 nA cm−2). At 0 mV, all electrodes behave similar to the bare ITO electrode with values oscillating between 0.7 to 4.4 nA cm−2. In the negative potential range, the presence of the Fe-TPY SAM yields a significant improvement of cathodic photocurrent generation, exhibiting over 20-fold higher photocurrent output compared to a bare ITO control (≈17 nA cm−2vs. 0.7 nA cm−2 at −100 mV). Interestingly, at −200 mV a photocurrent of ∼200 nA cm−2 was recorded for Fe-TPY-based electrodes, with a respective 30-fold and 24-fold higher production without and with cyt c553, respectively.
In comparison, the photocurrent production of Co-based wires is less pronounced at this potential, with an obvious difference between the electrodes without and with cyt c553 (69.8 nA cm−2vs. 36.8 nA cm−2). This trend is even more visible at −300 mV. While both Fe- and Co-based TPY-functionalised electrodes generate a similar photocurrent of ∼260 nA cm−2, the addition of cyt c553 onto the Co-based electrode results in a 2-fold decrease of the cathodic photocurrent. These observations are in accordance with the previous electrochemical analyses and confirm the role of the metal in the electron transfer process through the TPY wire in these nano-assemblies. At positive and negative potentials (Fig. S11† and Fig. 6), the presence of cobalt quenches the ET process between the cyt and the ITO electrode, whereas in the case of iron the metal centre facilitates the ET process (Fig. 5B and 6).
The observation of the ET process between ITO and cyt c that is promoted by Fe(II) embedded in the structure of TPY ligand molecular wire is closely related to the electron transfer step exerted by the easily oxidizable non-haem Fe present on the electron acceptor side of Type II photosynthetic reaction centres (photosystem II and the bacterial reaction centre81). In these macromolecular structures the non-haem Fe mediates ET occurring on a μs timescale82,83 between the two quinones (QA and QB) that are symmetrically located on either side of this metal centre. The removal of Fe or its replacement with other metals (e.g. Zn, Co) has been shown to alter the electron transfer rates from the intermediate acceptor to the primary acceptor QA,84,85 highlighting the importance of the non-haem iron for the native ET processes on the electron acceptor side of the photosynthetic reaction centres.
Previous electrochemical studies on other types of SAMs supported by DFT calculations explained that the electron flow can be tuned by the presence of different metal redox centres.80,86 In this case, the observed decrease in the photocurrent generation in the presence of cyt c553 can be explained by the interplay between the cathodic and anodic currents occurring simultaneously. In fact, for all the configurations, ET occurs from the electrode to the organometallic SAM (cathodic current). With the addition of cytochrome, the cathodic electron flow is preserved for Fe-based TPY SAM, whereas for a Co-containing nanoconstruct a competitive reverse electron flow (from cyt c553 to ITO) occurs (anodic current). Nevertheless, the effectiveness of covalently attached TPY wires for cathodic photocurrent generation is well demonstrated in the present work exceeding the photocurrent output obtained for other cyt-modified electrodes,80 even after plasmonic enhancement (92.3 nA cm−2 at −200 mV)65 or for PSI-based ITO electrodes in the presence of an external mediator (160 nA cm−2 at −300 mV).16
To support the hypothesis concerning the influence of the cyt c553 on the cathodic photocurrent generation, the energy diagrams of the Co- and Fe-based configurations were determined87 (Scheme 2).
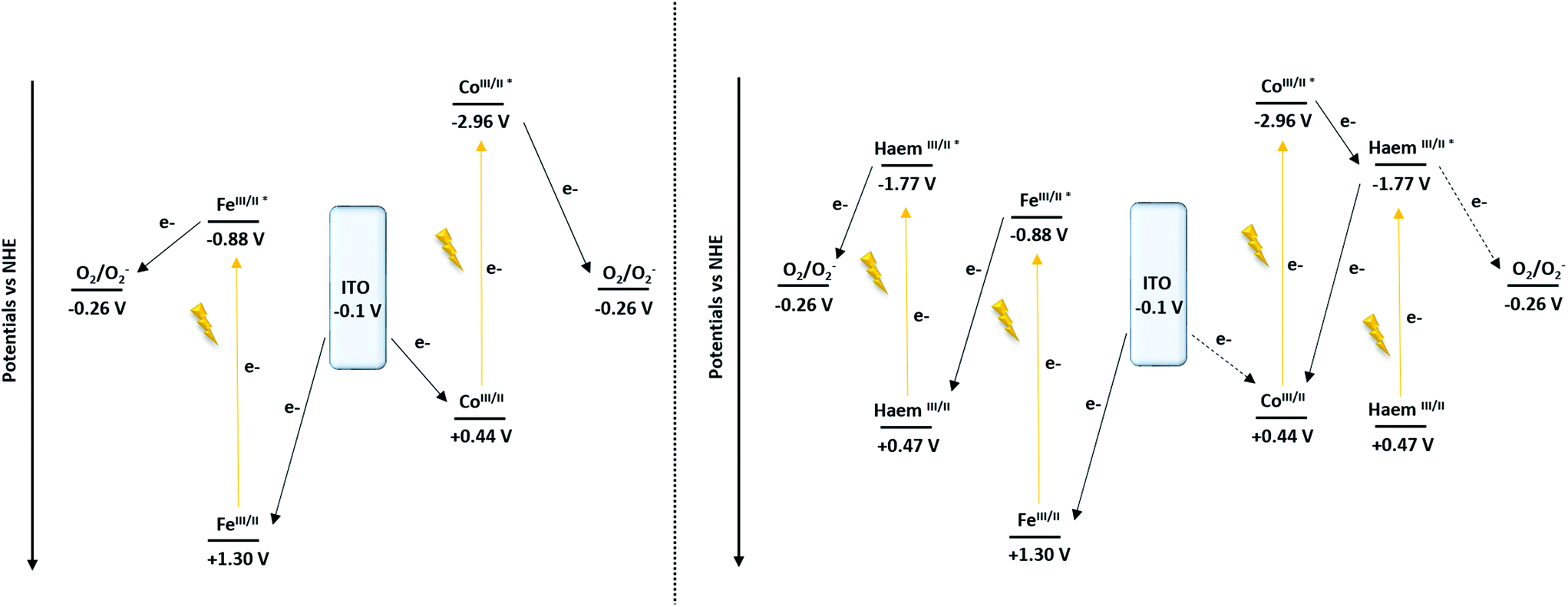 | ||
| Scheme 2 Proposed mechanism of the cathodic photocurrent recorded at −0.1 V vs. NHE (−0.3 V vs. Ag/AgCl) of the ITO-TPY devices without (left) and with the presence of immobilised cyt c553 (right). | ||
From the converted redox potentials of the Co- and Fe-TPY complexes immobilised on ITO vs. the SCE reference electrode (by adding 298 mV (ref. 88) to the values vs. Ag/Ag+), the energy of the highest occupied molecular orbital (HOMO) can be calculated. The respective values of −4.94 eV (+0.44 V vs. NHE) and −5.80 eV (+1.30 V vs. NHE) related to the vacuum energy level were determined using eqn (2) with an energy of −4.74 eV for SCE with respect to the zero-vacuum level. Considering the 0–0 transition energy ΔE00 of the metal-terpyridine chromophores (λ00Co-TPY = 365 nm (ref. 87) and λ00Fe-TPY = 568 nm (ref. 66)), the energy of the lowest occupied molecular level (LUMO) can be estimated at −1.54 eV (−2.96 V vs. NHE) and −3.62 eV (−0.88 V vs. NHE), respectively, related to the vacuum energy level using the eqn (2) and (3).
| EHOMO (eV) = −4.74 − E1/2 | (2) |
| ΔE00 = ELUMO − EHOMO = 1240/λ00 | (3) |
Similarly for cyt c553, after the conversion of the redox potential vs. SCE (by removing 39 mV to the value vs. Ag/AgCl) and with the 0–0 transition energy (λ00 = 553 nm, Fig. S6†), the energy levels of the HOMO and the LUMO were found at −4.97 eV (+0.47 V vs. NHE) and −2.72 eV (−1.77 V vs. NHE), respectively.
Concerning the nano-architectures devoid of cyt c553 (Scheme 2, left), under illumination (370 nm < λ < 790 nm) both metal-TPY complexes absorb light and are promoted to their respective excited states. In the next step, the electron transfer process occurs, ultimately yielding the reduction of oxygen molecules present in the electrolyte solution, as oxygen is a well-known electron acceptor.89,90 Finally, the initial redox state of the devices is regenerated by electron transfer from ITO, which results in a photo-induced cathodic current generation enhanced by the presence of O2 (Fig. S12†).
In the case of the Fe-based biohybrid devices (Scheme 2, right), the photo-induced electron mechanism depicts a “Z-scheme” similarly to the natural photosynthesis. Upon illumination the haem and Fe-TPY complex absorb light and are promoted to the excited states. While the excited state of the haem is able to reduce the oxygen (Fig. S12†), a favourable electron transfer occurs from the excited state of the Fe-TPY complex to regenerate the ground state of the haem. Finally, the ground state of the device is restored by the electron injection from the ITO substrate. On the other hand, for the Co-based nanosystem the presence of the haem induces a competitive ET process. After the excitation of the cobalt TPY complex, an electron transfer occurs to the excited state of the haem group from which point two mechanisms are possible: (1) either the electron is transferred to oxygen (Fig. S12†) or (2) a back-electron transfer (BET) reaction takes place to regenerate the initial redox state of the cobalt; thus, reducing the overall cathodic photocurrent output.
Computational analysis of the electron transfer mechanism
The QM/MM calculations reveal that for the TPY-Fe/haem pair frontier orbitals are localised over two different fragments, with the HOMO localised over the TPY molecule and the LUMO localised on the haem (Fig. 7). The energy of the HOMO was found at −4.70 eV and for the LUMO at −4.28 eV, leading to an energy gap of 0.42 eV. For the TPY-Co/haem interface, the situation is the opposite. The presence of cobalt ion leads to the presence of a diradical state of the TPY molecule, resulting in a different localisation of the frontier molecular orbitals, with the two semi-occupied SOMOs localised over the haem group and the LUMO localised over the TPY molecule. The energy for the SOMO are −4.55 and −4.39 eV for spin up and down, respectively, while the LUMO is found at −4.00 eV, leading to an energy gap of 0.39 eV. The computational results may be used to explain the difference in photogenerated current observed experimentally. When applying a cathodic bias (from ITO to cyt c553) the current is enhanced for the TPY-Fe/haem interface, as the internal electron flow and the external bias are parallel. For the TPY-Co/haem complex, the situation is different as due to the triplet states recombination91 a diminished ET is observed. This results in a lower photogenerated current for the TPY-Co/haem interface with respect to the TPY-Fe/haem counterpart. In addition, the energy gap found for both interfaces is relatively low, opening the possibility of an external bias, even small, to overcome the internal flux of electrons, as seen for the anodic bias.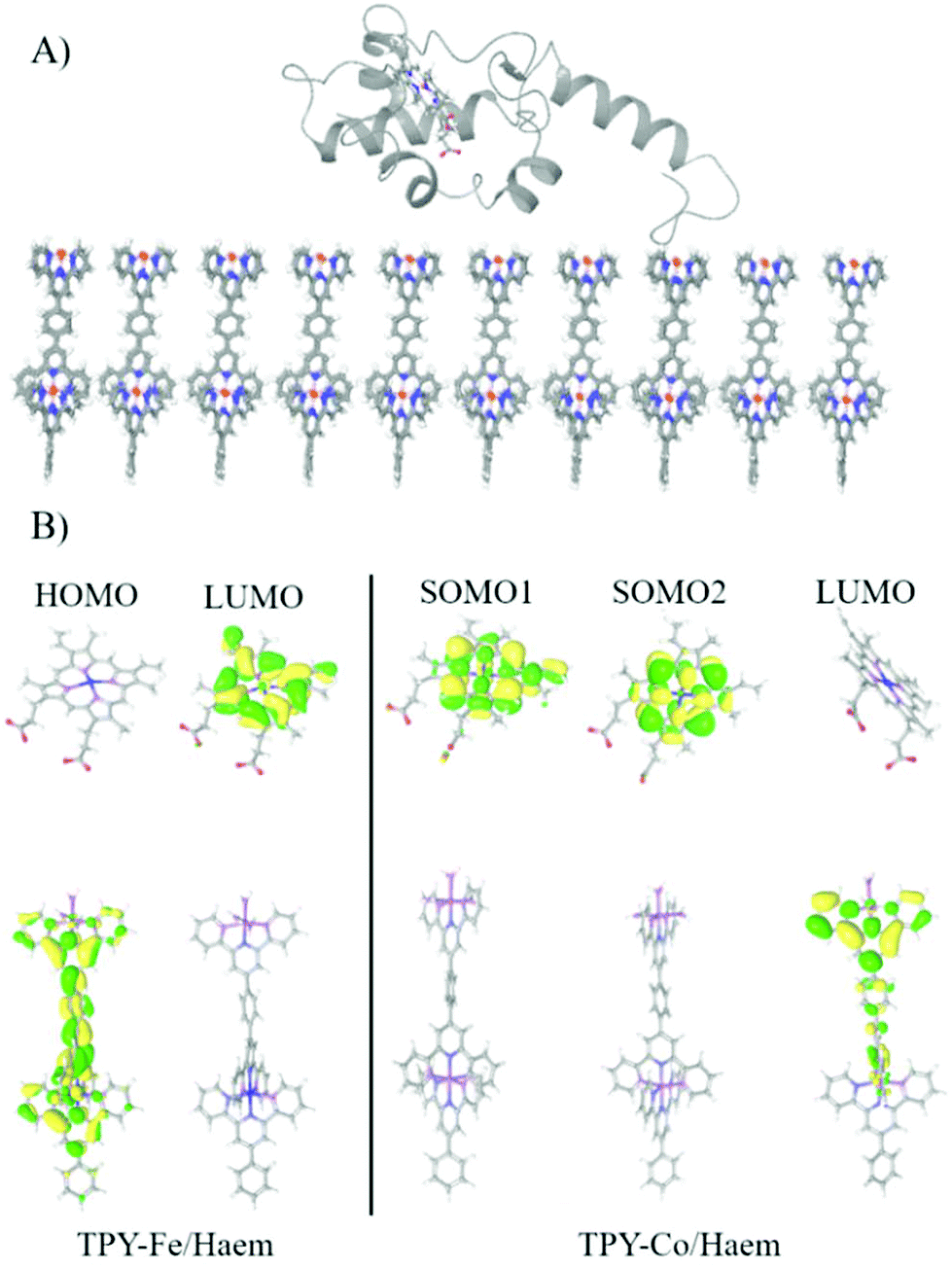 | ||
| Fig. 7 (A) The last snapshot of MD simulation showing the equilibrated interface. (B) Frontier molecular orbitals for TPY-Fe/haem and TPY-Co/haem interfaces. | ||
Several possible electronic-coupling pathways can be predicted in the heterogeneous cyt/TPY biomolecular system of this study (Fig. 8). The shortest distance found between haem and the TPY molecule (0.99 nm) is without the mediation of any amino acid present within the structure of cyt c553. The charged lysine residues close to haem, which were suggested to be important for the successful promotion of electrochemical activity,92 are further away from the haem-TPY interface. The closest to haem charged residue (E45) is found at 1.19 nm, again further than the closest haem-TPY distance, suggesting that the ET occurs via a ‘through-space’ rather than ‘through-bond’ mechanism.
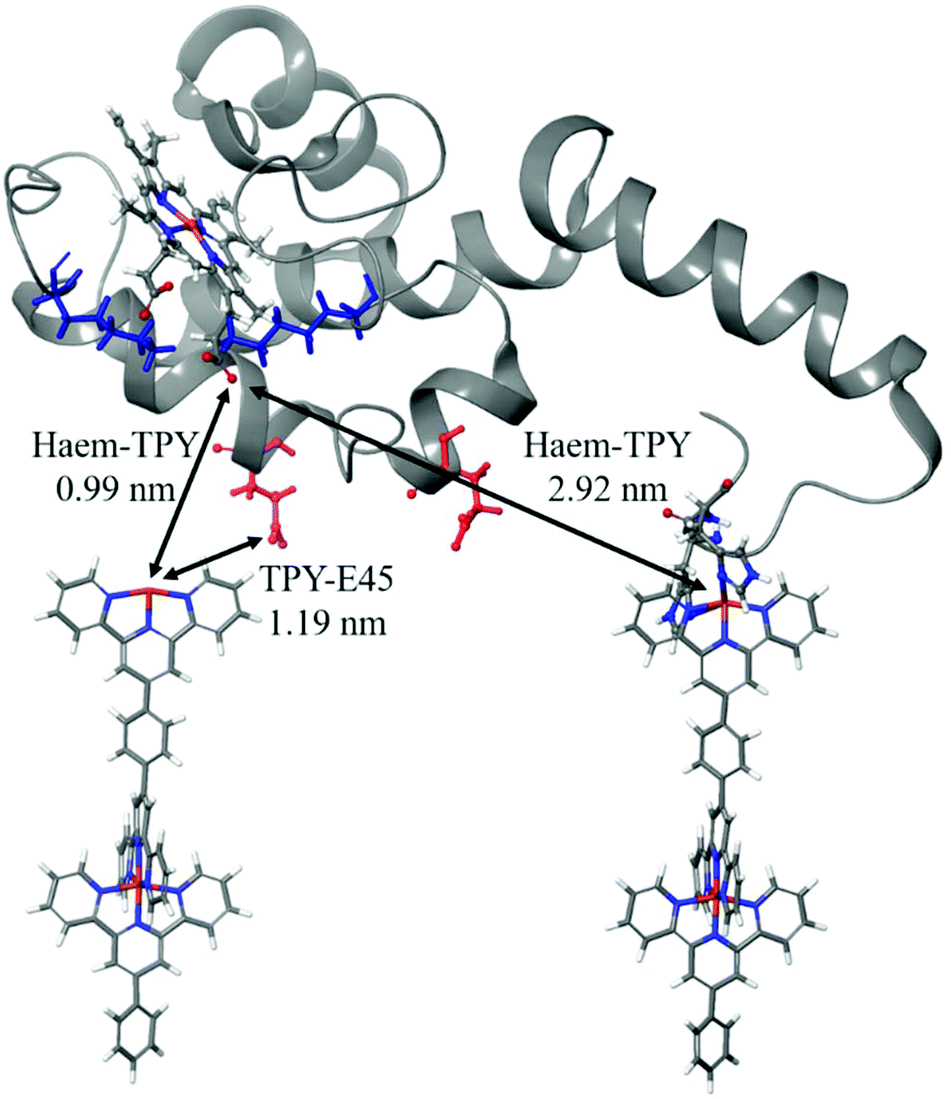 | ||
| Fig. 8 Electronic-coupling pathways within the cyt c/TPY biomolecular system. Charged amino acids with the shortest distance to haem and TPY are depicted in blue (for lysine residues bearing the formal +1 charge) and red (for glutamic acid residues bearing the formal −1 charge), respectively. The distances were measured from the carboxylic oxygen of the haem to the iron atom of the TPY molecule. | ||
This hypothesis is strengthened by a relatively large distance present between the haem group and the TPY molecule connected to the cyt holoprotein via the 19AA peptide linker,38 which is slightly below 3 nm and is 3-fold larger than the shortest haem-TPY distance. Following the protein backbone of the cyt c-peptide linker system, i.e., from the haem to the connected TPY molecular wire, a much longer distance than 3 nm is observed. The ET pathway through the protein backbone, although cannot be excluded, is likely to yield very low ET rates, far beyond the electrochemical detection range.
Conclusions
The construction of an efficient conductive interface between electrodes and electroactive proteins is a major challenge in the biosensor and biophotoelectrochemistry fields to achieve the desired performance of various types of nanodevices. As a major step towards this ambitious goal we report here the rational bottom-up approach to build a novel highly conductive interface based on metallo-organic TPY wires forming a uniform and well-structured SAM on the transparent ITO electrode. We show for the first time that the TPY-ligand-SAM can be successfully used as a universal platform for specific anchoring of the electroactive protein, cyt c553 on ITO. The ITO-TPY nanosystem, even without the cyt c553 protein, generates photocurrents whose densities are 30-fold higher that those reported for the pristine ITO electrodes and 2-fold higher than ITO electrodes functionalised with the well-known photoactive protein PSI in the presence of an external mediator.16 Interestingly, the nature of the metal centre in the TPY wires plays a crucial role in the photocurrent generation and the overall photocurrent output. While in the case of iron the combination of Fe-TPY and cyt c553 is beneficial for cathodic photocurrent generation, in the case of Co-TPY, due to BET between the haem group and the Co-TPY molecular wire, the cathodic photocurrent is diminished.In summary, the development of a universal chemical platform for anchoring and nanostructuring of electroactive proteins reported in this study provides a major advancement for the construction of efficient (bio)molecular systems requiring a high degree of precise supramolecular organisation as well as efficient charge transfer between (photo)redox-active molecular components and various types of electrode materials to ensure maximised power output.18 Importantly, the highly conductive photoactive Fe-TPY nanosystem described here is fully based on non-toxic and Earth-abundant elements and operates in a water-based electrolyte without external mediators, which makes it attractive for future applications such as sustainable solar-to-fuel devices.
Author contributions
Margot Jacquet conceptualised and performed experiments, analysed data and wrote the original draft. Miriam Izzo expressed, purified, and biochemically/spectroscopically analysed the cyt protein and performed biofunctionalization experiments. Silvio Osella performed MD and QM/MM analyses. Sylwia Kozdra, Paweł P. Michałowski and Marian Teodorczyk provided resources and performed SIMS analysis. Dariusz Gołowicz and Krzysztof Kazimierczuk performed NMR analyses. Adam Lewera and Maciej T. Gorzkowski performed XPS analyses. Rafał Jurczakowski, Bartosz Trzaskowski and Daniel T. Gryko analysed the data and reviewed the manuscript. Joanna Kargul conceived, conceptualised and supervised all aspects of the study, co-wrote, reviewed and edited the manuscript as well as obtained funding for this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the National Science Centre, Poland (OPUS14 grant no. UMO-2017/27/B/ST5/00472 to J.K., HARMONIA 2016/22/M/ST5/00431 to D.T.G. and SONATA UMO-2018/31/D/ST4/01475 to S.O.). We thank Gleb Andryianau for the initial suggestions on the organic synthesis pathways.Notes and references
- L. T. Wey, P. Bombelli, X. Chen, J. M. Lawrence, C. M. Rabideau, S. J. L. Rowden, J. Z. Zhang and C. J. Howe, ChemElectroChem, 2019, 6, 5375–5386 CrossRef CAS PubMed
.
- A. H. Teodor, B. D. Sherman, Z. Y. Ison, E. J. Ooi, J. J. Bergkamp and B. D. Bruce, Catalysts, 2020, 10, 1–30 CrossRef
- L. Ricotti, B. Trimmer, A. W. Feinberg, R. Raman, K. K. Parker, R. Bashir, M. Sitti, S. Martel, P. Dario and A. Menciassi, Sci. Robot., 2017, 2, 1–18 CrossRef PubMed
- S. Arshi, M. Nozari-Asbemarz and E. Magner, Catalysts, 2020, 10, 1232 CrossRef CAS
- J. Tschörtner, B. Lai and J. O. Krömer, Front. Microbiol., 2019, 10, 866 CrossRef PubMed
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed
- P. Manickam, A. Kaushik, C. Karunakaran and S. Bhansali, Biosens. Bioelectron., 2017, 87, 654–668 CrossRef CAS PubMed
- C. E. Lubner, A. M. Applegate, P. Knörzer, A. Ganago, D. A. Bryant, T. Happe and J. H. Golbeck, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20988–20991 CrossRef CAS PubMed
- J. C. Lee, H. B. Gray and J. R. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7760–7764 CrossRef CAS PubMed
- E. F. Bowden, F. M. Hawkridge and H. N. Blount, J. Electroanal. Chem., 1984, 161, 355–376 CrossRef CAS
- Z. S. Aghamiri, M. Mohsennia and H. A. Rafiee-Pour, Talanta, 2018, 176, 195–207 CrossRef CAS PubMed
- L. Tarpani, F. Bellezza, P. Sassi, M. Gambucci, A. Cipiciani and L. Latterini, J. Phys. Chem. B, 2019, 123, 2567–2575 CrossRef CAS PubMed
- T. Lee, S. U. Kim, J. Min and J. W. Choi, Adv. Mater., 2010, 22, 510–514 CrossRef CAS PubMed
- V. M. Friebe, D. Millo, D. J. K. Swainsbury, M. R. Jones and R. N. Frese, ACS Appl. Mater. Interfaces, 2017, 9, 23379–23388 CrossRef CAS PubMed
- R. A. Grimme, C. E. Lubner, D. A. Bryant and J. H. Golbeck, J. Am. Chem. Soc., 2008, 130, 6308–6309 CrossRef CAS PubMed
- A. Efrati, R. Tel-Vered, D. Michaeli, R. Nechushtai and I. Willner, Energy Environ. Sci., 2013, 6, 2950–2956 RSC
- K. R. Stieger, S. C. Feifel, H. Lokstein and F. Lisdat, Phys. Chem. Chem. Phys., 2014, 16, 15667–15674 RSC
- M. Kiliszek, E. Harputlu, M. Szalkowski, D. Kowalska, C. G. Unlu, P. Haniewicz, M. Abram, K. Wiwatowski, J. Niedziółka-Jönsson, S. MaćKowski, K. Ocakoglu and J. Kargul, J. Mater. Chem. A, 2018, 6, 18615–18626 RSC
- M. Fedurco, Coord. Chem. Rev., 2000, 209, 263–331 CrossRef CAS
- K. Nguyen and B. D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1553–1566 CrossRef CAS PubMed
- H. D. Sikes, J.
F. Smalley, S. P. Dudek, A. R. Cook, M. D. Newton, C. E. D. Chidsey and S. W. Feldberg, Science, 2001, 291, 1519–1524 CrossRef CAS PubMed
- K. Slowinski, H. K. Y. Fong and M. Majda, J. Am. Chem. Soc., 1999, 121, 7257–7261 CrossRef CAS
- S. Sek, A. Misicka and R. Bilewicz, J. Phys. Chem. B, 2000, 104, 5399–5402 CrossRef CAS
- J. J. Sumner, K. S. Weber, L. A. Hockett and S. E. Creager, J. Phys. Chem. B, 2000, 104, 7449–7454 CrossRef CAS
- A. Salomon, D. Cahen, S. Lindsay, J. Tomfohr, V. B. Engelkes and C. D. Frisbie, Adv. Mater., 2003, 15, 1881–1890 CrossRef CAS
- J. He, F. Chen, J. Li, O. F. Sankey, Y. Terazono, C. Herrero, D. Gust, T. A. Moore, A. L. Moore and S. M. Lindsay, J. Am. Chem. Soc., 2005, 127, 1384–1385 CrossRef CAS PubMed
- S. H. Choi, B. Kim and C. D. Frisbie, Science, 2008, 320, 1482–1486 CrossRef CAS PubMed
- G. Sedghi, K. Sawada, L. J. Esdaile, M. Hoffmann, H. L. Anderson, D. Bethell, W. Haiss, S. J. Higgins and R. J. Nichols, J. Am. Chem. Soc., 2008, 130, 8582–8583 CrossRef CAS PubMed
- R. Sakamoto, K. H. Wu, R. Matsuoka, H. Maeda and H. Nishihara, Chem. Soc. Rev., 2015, 44, 7698–7714 RSC
- S. Katagiri, R. Sakamoto, H. Maeda, Y. Nishimori, T. Kurita and H. Nishihara, Chem. – Eur. J., 2013, 19, 5088–5096 CrossRef CAS PubMed
- H. Maeda, R. Sakamoto and H. Nishihara, J. Electroanal. Chem., 2016, 779, 112–116 CrossRef CAS
- N. Tuccitto, V. Ferri, M. Cavazzini, S. Quici, G. Zhavnerko, A. Licciardello and M. A. Rampi, Nat. Mater., 2009, 8, 41–46 CrossRef CAS PubMed
- Y. Nishimori, K. Kanaizuka, T. Kurita, T. Nagatsu, Y. Segawa, F. Toshimitsu, S. Muratsugu, M. Utsuno, S. Kume, M. Murata and H. Nishihara, Chem. – Asian J., 2009, 4, 1361–1367 CrossRef CAS PubMed
- H. Maeda, R. Sakamoto and H. Nishihara, Coord. Chem. Rev., 2017, 346, 139–149 CrossRef CAS
- T. Wieprecht, J. Xia, U. Heinz, J. Dannacher and G. Schlingloff, J. Mol. Catal. A: Chem., 2003, 203, 113–128 CrossRef CAS
- E. C. Constable and M. D. Ward, J. Chem. Soc., Dalton Trans., 1990, 1405–1409 RSC
- K. T. Potts and D. Konwar, J. Org. Chem., 1991, 56, 4815–4816 CrossRef CAS
- J. D. J. Olmos, P. Becquet, D. Gront, J. Sar, A. Dąbrowski, G. Gawlik, M. Teodorczyk, D. Pawlak and J. Kargul, RSC Adv., 2017, 7, 47854–47866 RSC
- J. Wang and G. S. Hanan, Synlett, 2005, 8, 1251–1254 Search PubMed
- V. Spampinato, N. Tuccitto, S. Quici, V. Calabrese, G. Marletta, A. Torrisi and A. Licciardello, Langmuir, 2010, 26, 8400–8406 CrossRef CAS PubMed
- C. J. Aspley and J. a. G. Williams, New J. Chem., 2001, 25, 1136–1147 RSC
- W. Goodall, K. Wild, K. J. Arm and J. A. G. Williams, J. Chem. Soc., Perkin Trans., 2002, 2, 1669–1681 RSC
- J. L. Banks, H. S. Beard, Y. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef CAS PubMed
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS
- A. Forget, B. Limoges and V. Balland, Langmuir, 2015, 31, 1931–1940 CrossRef CAS PubMed
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322–6356 CrossRef CAS PubMed
- E. Hampson, J. M. Cameron, S. Amin, J. Kyo, J. A. Watts, H. Oshio and G. N. Newton, Angew. Chem., Int. Ed., 2019, 58, 18281–18285 CrossRef CAS PubMed
- H. Wolpher, S. Sinha, J. Pan, A. Johansson, M. J. Lundqvist, P. Persson, R. Lomoth, J. Bergquist, L. Sun, V. Sundström, B. Åkermark and T. Polívka, Inorg. Chem., 2007, 46, 638–651 CrossRef CAS PubMed
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13, 4367–4373 CrossRef CAS
- A. Lanzilotto, L. A. Büldt, H. C. Schmidt, A. Prescimone, O. S. Wenger, E. C. Constable and C. E. Housecroft, RSC Adv., 2016, 6, 15370–15381 RSC
- M. Waser, C. Siebenhaar, J. Zampese, G. Grundler, E. Constable, M. Height and U. Pieles, Chimia, 2010, 64, 328–329 CrossRef CAS PubMed
- R. Frantz, J. O. Durand, M. Granier and G. F. Lanneau, Tetrahedron Lett., 2004, 45, 2935–2937 CrossRef CAS
- G. Guerrero, P. H. Mutin, E. Framery and A. Vioux, New J. Chem., 2008, 32, 1519–1525 RSC
- S. A. Paniagua, P. J. Hotchkiss, S. C. Jones, S. R. Marder, A. Mudalige, F. S. Marrikar, J. E. Pemberton and N. R. Armstrong, J. Phys. Chem., 2008, 112, 7809–7817 CAS
- G. Guerrero, J. G. Alauzun, M. Granier, D. Laurencin and P. H. Mutin, Dalton Trans., 2013, 42, 12569–12585 RSC
- J. Lee, J. Bong, Y. G. Ha, S. Park and S. Ju, Appl. Surf. Sci., 2015, 330, 445–448 CrossRef CAS
- M. Mennicken, S. K. Peter, C. Kaulen, U. Simon and S. Karthäuser, J. Phys. Chem. C, 2019, 123, 21367–21375 CrossRef CAS
-
J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data. Eden Prairie, MN, USA: Physical Electronics Division, Perkin-Elmer Corp, 1992 Search PubMed
- B. Hirschorn, M. E. Orazem, B. Tribollet, V. Vivier, I. Frateur and M. Musiani, Electrochim. Acta, 2010, 55, 6218–6227 CrossRef CAS
- C. Xiang, Y. Zou, L. X. Sun and F. Xu, Electrochem. Commun., 2008, 10, 38–41 CrossRef CAS
- M. Murphy, K. Theyagarajan, P. Ganesan, S. Senthilkumar and K. Thenmozhi, Appl. Surf. Sci., 2019, 492, 718–725 CrossRef CAS
- Y. Zhao, Y. Hu, J. Hou, Z. Jia, D. Zhong, S. Zhou, D. Huo, M. Yang and C. Hou, J. Electroanal. Chem., 2019, 842, 16–23 CrossRef CAS
- T. Pajkossy and R. Jurczakowski, Curr. Opin. Electrochem., 2017, 1, 53–58 CrossRef CAS
- R. P. Janek, W. R. Fawcett and A. Ulman, Langmuir, 1998, 14, 3011–3018 CrossRef CAS
- M. Szalkowski, E. Harputlu, M. Kiliszek, C. G. Unlu, S. Mackowski, K. Ocakoglu, J. Kargul and D. Kowalska, J. Mater. Chem. C, 2020, 8, 5807–5814 RSC
- R. Farran, L. Le Quang, D. Jouvenot, F. Loiseau, R. Pansu, A. Deronzier and J. Chauvin, Inorganica Chim. Acta, 2017, 454, 197–207 CrossRef CAS
- K. Kanaizuka, M. Murata, Y. Nishimori, I. Mori, K. Nishio, H. Masuda and H. Nishihara, Chem. Lett., 2005, 34, 534–535 CrossRef CAS
- Y. Yamanoi, J. Sendo, T. Kobayashi, H. Maeda, Y. Yabusaki, M. Miyachi, R. Sakamoto and H. Nishihara, J. Am. Chem. Soc., 2012, 134, 20433–20439 CrossRef CAS PubMed
- M. Miyachi, M. Ohta, M. Nakai, Y. Kubota, Y. Yamanoi, T. Yonezawa and H. Nishihara, Chem. Lett., 2008, 37, 404–405 CrossRef CAS
- R. Farran, D. Jouvenot, F. Loiseau, J. Chauvin and A. Deronzier, Dalton Trans., 2014, 43, 12156–12159 RSC
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS
- T. Kurita, Y. Nishimori, F. Toshimitsu, S. Muratsugu, S. Kume and H. Nishihara, J. Am. Chem. Soc., 2010, 132, 4524–4525 CrossRef CAS PubMed
- H. Maeda, R. Sakamoto and H. Nishihara, Chem. – Eur. J., 2014, 20, 2761–2764 CrossRef CAS PubMed
- A. El Kasmi, M. C. Leopold, R. Galligan, R. T. Robertson, S. S. Saavedra, K. El Kacemi and E. F. Bowden, Electrochem. Commun., 2002, 4, 177–181 CrossRef CAS
- Y. Wang, X. Bian, L. Liao and J. Zhu, Microchim. Acta, 2012, 178, 277–283 CrossRef CAS
- X. Jiang, L. Zhang and S. Dong, Electrochem. Commun., 2006, 8, 1137–1141 CrossRef CAS
- N. Matsuda, H. Okabe, A. Omura, M. Nakano and K. Miyake, Anal. Sci., 2017, 33, 469–472 CrossRef CAS
- Á. Szucs and M. Novák, J. Electroanal. Chem., 1995, 384, 47–55 CrossRef
- S. Monari, A. Ranieri, C. A. Bortolotti, S. Peressini, C. Tavagnacco and M. Borsari, Electrochim. Acta, 2011, 56, 6925–6931 CrossRef CAS
- S. Osella, M. Kiliszek, E. Harputlu, C. G. Unlu, K. Ocakoglu, B. Trzaskowski and J. Kargul, J. Phys. Chem. C, 2019, 123, 8623–8632 CrossRef CAS
- T. Cardona, A. Sedoud, N. Cox and A. W. Rutherford, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 26–43 CrossRef CAS PubMed
- M. Y. Okamura, M. L. Paddock, M. S. Graige and G. Feher, Biochim. Biophys. Acta, Bioenerg., 2000, 1458, 148–163 CrossRef CAS
- R. De Wijn and H. J. Van Gorkom, Biochemistry, 2001, 40, 11912–11922 CrossRef CAS PubMed
- L. M. Utschig, S. R. Greenfield, J. Tang, P. D. Laible and M. C. Thurnauer, Biochemistry, 1997, 36, 8548–8558 CrossRef CAS PubMed
- R. J. Debus, G. Feher and M. Y. Okamura, Biochemistry, 1986, 25, 2276–2287 CrossRef CAS PubMed
- S. Osella, M. Kiliszek, E. Harputlu, C. G. Unlu, K. Ocakoglu, J. Kargul and B. Trzaskowski, J. Mater. Chem. C, 2018, 6, 5046–5054 RSC
- X. Chen, Y. C. Dai, Z. B. Zheng and K. Z. Wang, J. Colloid Interface Sci., 2013, 402, 107–113 CrossRef CAS PubMed
- V. V. Pavlishchuk and A. W. Addison, Inorganica Chim. Acta, 2000, 298, 97–102 CrossRef CAS
- Y. Kim, K. Liang, K. Law and D. G. Whitten, J. Phys. Chem., 1994, 98, 984–988 CrossRef CAS
- H. Imahori, H. Norieda, Y. Nishimura, I. Yamazaki, K. Higuchi, N. Kato, T. Motohiro, H. Yamada, K. Tamaki, M. Arimura and Y. Sakata, J. Phys. Chem. B, 2000, 104, 1253–1260 CrossRef CAS
- P. Salvatori, G. Marotta, A. Cinti, E. Mosconi, M. Panigrahi, L. Giribabu, M. K. Nazeeruddin and F. De Angelis, Inorganica Chim. Acta, 2013, 406, 106–112 CrossRef CAS
- P. M. Allen, H. Allen, O. Hill and N. J. Walton, J. Electroanal. Chem. Interfacial Electrochem., 1984, 178, 69–86 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr08870f |
| This journal is © The Royal Society of Chemistry 2021 |