
Tandem molecular self-assembly for selective lung cancer therapy with an increase in efficiency by two orders of magnitude†
Debin
Zheng
a,
Jingfei
Liu
a,
Yinghao
Ding
b,
Limin
Xie
a,
Yingying
Zhang
a,
Yaoxia
Chen
a,
Rong
Peng
a,
Miao
Cai
a,
Ling
Wang
b,
Huaimin
Wang
 *c,
Jie
Gao
*a and
Zhimou
Yang
*a
*c,
Jie
Gao
*a and
Zhimou
Yang
*a
aState Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Bioactive Materials, Ministry of Education, College of Life Sciences, Collaborative Innovation Center of Chemical Science and Engineering, and National Institute of Functional Materials, Nankai University, Tianjin 300071, P. R. China. E-mail: chemgaojie@nankai.edu.cn; yangzm@nankai.edu.cn
bCollege of Pharmacy, Nankai University, Tianjin 300071, P. R. China
cKey Laboratory of Precise Synthesis of Functional Molecules of Zhejiang Province, School of Science, Westlake University; Institute of Natural Sciences, Westlake Institute for Advanced Study, No. 18 Shilongshan Road, Hangzhou 310024, Zhejiang Province, P. R. China. E-mail: wanghuaimin@westlake.edu.cn
First published on 24th May 2021
Abstract
In situ self-assembly of prodrug molecules into nanomedicine can elevate the therapeutic efficacy of anticancer medications by enhancing the targeting and enrichment of anticancer drugs at tumor sites. However, the disassembly and biodegradation of nanomedicine after enrichment prevents the further improvement of the efficiency, and avoiding such disassembly and biodegradation remains a challenge. Herein, we rationally designed a tandem molecular self-assembling prodrug that could selectively improve the therapeutic efficacy of HCPT against lung cancer by two orders of magnitude. The tandem molecular self-assembly utilized an elevated level of alkaline phosphatase and reductase in lung cancer cells. The prodrug first self-assembled into nanofibers by alkaline phosphatase catalysis and was internalized more efficiently by lung cancer cells than free HCPT. The resulting nanofiber was next catalyzed by intracellular reductase to form a more hydrophobic nanofiber that prevented the disassembly and biodegradation, which further significantly improved the efficacy of HCPT against lung cancer both in vitro and in vivo.
Introduction
Nanomedicine-based therapies have been studied extensively to address delivery problems with chemotherapeutics by improving the selectivity and increasing the drug concentration at desired locations.1–5 For the time being, however, successfully translating nanomedicine in the clinic is still challenging.6 One of the most significant factors delaying the clinical translation is the poor penetration of nanomedicines into tumors after intravenous injection.7–9 In recent years, inspired by the approach of prodrugs that focus on enhancing the tumor permeability and penetrability of drugs,10–13in situ self-assembly (SA) of drug–peptide conjugates has been developed and emerged as a promising method in targeted drug delivery for cancer treatment by integrating the advantages of prodrugs and nanomedicines.14–18 The drug–peptide conjugates serve as both prodrugs and nanomedicines by tumor site selective induction of SA, including environmental factors such as differences in pH,19,20 temperature or ionic strength,21–23 elevated levels of redox molecules such as GSH, H2O2 and other ROS species,24,25 and overexpressed enzymes such as phosphatases,26–29 proteases30 and esterases.31 These approaches work very well in enhancing the penetration of nanomedicines and enriching the concentration of drugs at desired sites.10,32–34 However, the complex biological environment often causes the disassembly or biodegradation of the SA nanomedicine after enrichment in disease sites, which would lead to fast clearance of the drug and hinder the further improvement of the therapeutic efficacy. Avoiding such undesirable disassembly or biodegradation of the SA nanomedicine remains a challenge.35–38Recently, in situ SA has evolved into tandem SA by connecting two steps of in situ SA. In several pioneering works, the first in situ SA triggered by enzymes overexpressed by disease sites promoted the targeting and cellular internalization of the SA nanomaterials, and the second tandem SA caused changes in the molecular chemical structure, morphology, or surface charge of the nanomaterial, which would ultimately lead to better selectivity or retention of the material.39–41 Here, we described a novel tandem SA based approach to specifically deliver HCPT to lung tumors by in situ self-assembling HCPT at the tumor site and then preventing its disassembly and biodegradation inside the cancer cells by tandem SA (Scheme 1). The specific delivery and prolonged retention at lung tumor sites simultaneously improved the therapeutic efficacy of HCPT by two orders of magnitude.
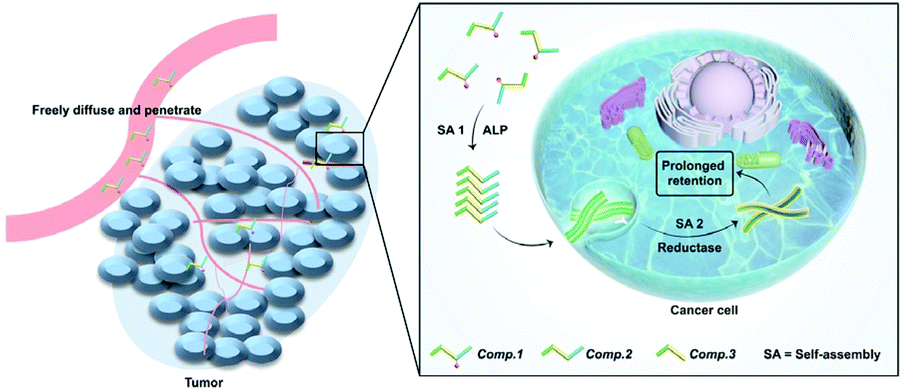 | ||
| Scheme 1 Schematic of the tandem molecular self-assembly of the prodrug Comp. 1. | ||
Experimental
Peptide synthesis
The drug–peptide conjugates were prepared through SPPS. The F-moc group was removed by piperidine. HBTU served as the coupling reagents. The Fmoc-protected azobenzene group as amino acid can be used in SPPS. Finally, the liquid reagent composed of 95% of trifluoroacetic acid, 2.5% of PhSCH3 and 2.5% of water was reacted with resin of crude products in the dark for 60 minutes. After purification and freeze-drying, pure products were obtained.Tandem molecular self-assembly
The Comp. 1 solution was firstly incubated at 37 °C for 5 min (pH = 7.4, 100 μM in PBS). The first step of self-assembly was induced by adding alkali phosphatase (ALP, 10 U mL−1) into the work solution for 10 hours at 37 °C. After that, sodium dithionite (SDT, 20 mM) was added at 37 °C to initiate the second step of SA for 2 hours.Bio-TEM
2 × 105 of lung cancer cells (A549) were seeded in a 6 well Petri dish. The cells were incubated with Comp. 1 or Comp. 2, 4, and 5 (100 μM) for 6 h. After that, the cells were collected using a cell scraper and then fixed with glutaraldehyde (2.5%), and finally observed by transmission electron microscopy (TEM).Cell uptake
1 × 105 of A549 cells were seeded in a CLSM dish. After the cells adhered, the cells were treated with Comp. 1, 2, 4 or 5 (100 μM) for 1, 2, 4 and 6 h, respectively. After that, the cells were incubated with Red dot 1 (1×, diluted with DMEM) to stain the nuclei for 5–10 min. The cells were washed with DMEM three times, and then CLSM was employed to detect fluorescence intensity. We utilized Image J to analyze relative fluorescence intensity.Cell proliferation
Different cells were seeded in a 96-well plate and incubated with the compound for 48 hours after cell attachment. The medium containing the compound was removed. We used 100 μL of cold trichloroacetic acid solution (10%) to fix the cells at 4 °C overnight. We poured the fixative out and washed with water. We put it into an oven at 37 °C to dry it. Next, we used 0.4% SRB solution (100 μL) to stain the fixed cells for 25 min to form an SRB-bound protein, and then washed excessive SRB with 300 μL of AcOH solution (1%). After drying it, 10 mM Tris base solution was added into each well to solubilize the SRB-bound protein for 30 minutes. We measured the OD 570 nm via a microplate reader.Tumor penetration study in vivo
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Nankai University and approved by the Centre of Tianjin Animal Experiment Ethics Committee, and the accreditation number of the laboratory is SYXK(Jin) 2019-0003 promulgated by Tianjin Science and Technology Commission. BALB/c nude mice bearing about 200 mm3 of A549 tumor were injected with Comp. 6, 7, 8 and 9 (dose shown in Table S1,†i.v.). After 24 h, the mice were sacrificed to obtain the tumor tissue for frozen sections. The tumor samples were sectioned into 8 μm thickness on the equatorial plane. Then, the samples were fixed with acetone for 15 min and washed twice with PBS, 5% BSA blocking solution prevented nonspecific binding at room temperature for 1 h and the blood vessel was marked with the FITC-CD31 antibody for 1 h. After washing with PBS 3 times, the nucleus of the sample was stained with DAPI for 10 min.Anti-tumor therapy
A549 tumor-bearing nude mice (tumor volume ∼100 mm3) were randomly divided into six groups (n = 5), then they were intravenously injected with Comp. 1, 2, 4, 5, free HCPT and PBS at 1, 4, 7 and 10 days. Tumor volume and weight were monitored every 2 days for 19 days. After the whole observation period, the mice were sacrificed, and solid tumors were separated, and then fixed with 4% formaldehyde for hematoxylin and eosin (H&E) staining and immunofluorescence analysis.Results and discussion
Prodrug design and synthesis
In our prior work,41 we designed a peptide (GFFpYG–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–ERGD) that could self-assemble in a stepwise manner controlled by the catalysis of ALP and intracellular reductase. The ALP is reported to be highly overexpressed by a variety of cancer cells and mainly located on or outside the surface of the cell membrane. Meanwhile, lung cancer cells exhibit elevated expression of intracellular reductase. The peptide therefore could tandemly respond to those enzymes and selectively formed nanomaterials inside lung cancer cells. In this study, we conjugated 10-hydroxycamptothecin (HCPT) with the above-mentioned peptide. The obtained molecule HCPT–GFFpYG–NN–ERGD (Comp. 1 in Fig. 1A) was a prodrug with tandem self-assembling ability.
N–ERGD) that could self-assemble in a stepwise manner controlled by the catalysis of ALP and intracellular reductase. The ALP is reported to be highly overexpressed by a variety of cancer cells and mainly located on or outside the surface of the cell membrane. Meanwhile, lung cancer cells exhibit elevated expression of intracellular reductase. The peptide therefore could tandemly respond to those enzymes and selectively formed nanomaterials inside lung cancer cells. In this study, we conjugated 10-hydroxycamptothecin (HCPT) with the above-mentioned peptide. The obtained molecule HCPT–GFFpYG–NN–ERGD (Comp. 1 in Fig. 1A) was a prodrug with tandem self-assembling ability.
 | ||
| Fig. 1 (A) Enzyme instructed chemical structure conversions from Comp. 1 to 2 by ALP and then Comp. 2 to 3 by SDT. TEM and optical images of (B) Comp. 2 acquired by ALP catalysis and (C) Comp. 3 acquired by SDT catalysis, scale bar = 100 μm. | ||
We first synthesized the Fmoc-azo-benzene (Comp. S2) as the reductase responsive group and glutaric anhydride modified 10-hydroxycamptothecin as the drug capping group. Both of them can be directly used in solid phase peptide synthesis (SPPS). Therefore, after standard SPPS and purification by reversed-phase high-performance liquid chromatography (HPLC), we obtained the pure compound of Comp. 1. Other control compounds were synthesized via a comparable process, including HCPT–GFFYG–NN–ERGD (only reductase-response, namely Comp. 2), HCPT–GFFpYGERGD (only ALP-response, namely Comp. 4), and HCPT–GFFYGERGD (non-ALP- and non-reductase-response, namely Comp. 5).
Enzyme instructed tandem self-assembly
A yellow clear solution (100 μM, 0.018 wt%) was formed by Comp. 1 in phosphate buffered saline (PBS, pH = 7.4), which was lower than its critical aggregation concentration (CAC = 198.01 μM, Fig. S11A†). The transmission electron microscopy (TEM) images showed non-assembled structures in the PBS solution of Comp. 1 (Fig. S12A†). It remained a yellow clear solution (Fig. 1B), after adding ALP (10 U mL−1) to the solution of Comp. 1 for 10 hours at 37 °C. The LC-MS trace indicated that Comp. 1 could be converted into Comp. 2 by ALP (Fig. S13†). The value of CAC decreased to 53.3 μM (Fig. S10B†), due to changes in hydrophobicity. We observed nanofibers with diameters of 8–14 nm in the resulting solution of Comp. 2 through TEM (Fig. 1B). After the addition of sodium dithionite (Na2S2O4, 20 mM), which was used to mimic reductase,42 the yellow solution of Comp. 2 became light yellow and more transparent within 2 hours, probably due to the conversion of azo benzene to aniline (Fig. 1C). The LC-MS trace shown in Fig. S14† revealed that most of the Comp. 2 had been converted into Comp. 3. The TEM images in Fig. 1C showed nanofibers with diameters of 6–10 nm. These observations demonstrated the tandem molecular self-assembling behavior of Comp. 1 catalyzed by ALP and reductase.Intracellular tandem self-assembly in lung cancer cells
To study whether tandem molecular SA could enrich HCPT in lung cancer cells, we utilized confocal laser scanning microscopy (CLSM) and bio-TEM to reveal the distribution of different compounds in live A549 cells. As shown in Fig. 2A, we could observe obvious green fluorescence dots in the cytoplasm of A549 cells after incubating the cells with Comp. 1 (100 μM) for 6 hours, while the cells treated with the same amount of control compounds (Comp. 2, 4 or 5) or free HCPT showed much weaker fluorescence. The quantified fluorescence intensity from CLSM images could indicate the relative cellular concentration of different compounds. As shown in Fig. 2B, the Comp. 1 group exhibited the highest fluorescence intensity, which indicated the highest cellular drug concentration. Meanwhile, the free HCPT group showed the least fluorescence intensity, mainly due to the extremely water-insoluble and poor cellular internalization efficiency of the molecule. Interestingly, the Comp. 4 group showed higher fluorescence intensity than the groups of Comp. 2 and 5. According to many studies, the ALP catalyzed self-assembling molecules were internalized much more than the unassembled ones by the ALP over-expressing cancer cells,43 because they entered the cells through endocytosis. Since both Comp. 1 and 4 are ALP responsive self-assembling molecules, it is reasonable that these two molecules are internalized by the A549 cells more than non-ALP responsive molecules. However, the fluorescence intensity of Comp. 4 was still much lower than that of Comp. 1. The reason for the differences in fluorescence intensity could be attributed to two factors: more cellular internalization and less biodegradation inside the cells. | ||
| Fig. 2 (A) CLSM images of A549 cells treated with 100 μM of Comp. 1, 2, 4, 5 and HCPT for 6 h (λexc. = 405 nm for green channel, and λexc. = 633 nm for red channel, white scale bar = 25 μm). (B) Corrected total cell fluorescence (CTCF, quantified from the gray scale of (A), one-way ANOVA, compared to the Comp. 1 group, mean ± SD, ***P < 0.001, **P < 0.01, N = 5). (C). Bio-TEM image of A549 cells after administration of Comp. 1 for 6 hours, the red arrow points to nanofibers. | ||
In order to elucidate the reason, we monitored the fluorescence intensity of cells treated with different compounds in a time dependent manner. As shown in Fig. S16 and S17,† after treating the cells with different compounds for a short time period (2 hours), the fluorescence intensities of the Comp. 1 and 4 group were already larger than those of other groups, indicating that the cells could uptake Comp. 1 and 4 more than other compounds. However, since the CAC value for Comp. 4 after adding ALP was larger than that for Comp. 1 (102.6 μM, Fig. S11C and D†), which indicated that its self-assembling ability was weaker than that of Comp. 1, the cellular uptake of Comp. 4 was less than that of Comp. 1. After a relatively long time of treatment (4 hours), the differences were further increased between the Comp. 1 group and other groups. We then removed the compounds from the cell culture medium to evaluate the biodegradation of the compounds inside the cell. As shown in Fig. S18,† after removing the compounds for 4 hours, the fluorescence intensity of Comp 2, 4 and 5 decreased a lot, while Comp. 1 and HCPT remained unchanged, suggesting that Comp. 1 and HCPT could resist the biodegradation inside the cells. The chemical structure of the self-assembled molecules changes under the catalysis of reductase inside the cell. The shedding of hydrophilic ERGD will cause the formation of more hydrophobic nanofibers, which may be one of the reasons for the resistance to biodegradation. Furthermore, as shown in Fig. 2C, the bio-TEM images of A549 cells post administration of Comp. 1 for 6 hours exhibited short nanofibers with diameters of 7–10 nm in the cytoplasm. In contrast, no such nanostructures were found in the other control groups (Fig. S15†). These observations demonstrated the formation of the second nanofiber of Comp. 1 and its strong anti-biodegradation ability after the tandem SA process, indicating that tandem SA could significantly enrich HCPT in A549 cells for a longer time because of more internalization and stronger biodegradation resistance of Comp. 1.
We then studied the selectivity of the tandem self-assembled nanomedicines. We chose three cancer cell lines with overexpression of ALP on the cell membrane and lower expression of reductase inside the cell than the A549 cell (HeLa, MCF-7 and PC-3) and treated them with Comp. 1 for 6 hours. The confocal images in Fig. S19† show that weak green fluorescence was observed in the cytoplasm of HeLa, MCF-7 and PC-3 cells. The quantified fluorescence intensities (Fig. S20†) of these cell lines were significantly lower than those of A549 cells. These observations indicated that Comp. 1 had excellent selectivity to A549 cells because of the overexpression of both ALP and reductase in lung cancer cells.
The above explorations showed that the tandem self-assembling strategy could effectively retain SA nanomedicines for a longer time inside the cancer cells, which might improve the efficiency of drugs to inhibit cancer cells. We therefore examined the cytotoxicity of Comp. 1 and other compounds against A549 cells. Compared to Comp. 2, 4, 5 and free HCPT with IC50 values of 4.9 μM, 3.5 μM, 4.7 μM, and 5.7 μM, respectively, Comp. 1 exhibited the most remarkable suppression to A549 cells with an IC50 value as low as 63 nM (Fig. 3A). The inhibition efficacy of A549 cells treated with Comp. 1 was about 91 fold higher than that of free HCPT and 45–75 fold higher than that of other compounds, demonstrating the strong potency of Comp. 1 for killing A549 cells. Next, we examined the ratio of the IC50 value for Comp. 1 and free HCPT. As shown in Fig. 3B, the IC50 values of Comp. 1 against PC-3, MCF-7 and HeLa cells were only 2.4, 0.8 and 22 fold higher than that of free HCPT, respectively. Furthermore, we tested the compatibility and cell uptake of Comp. 1 towards the LO2 normal cells, as shown in Fig. S21.† The toxicity and cell uptake of Comp. 1 was not higher than that of free HCPT towards LO2 cells at the same concentration. These results clearly showed the excellent selectivity of Comp. 1 to lung cancer cells and demonstrated that the improved potency of Comp. 1 was due to the tandem self-assembling process triggered by the elevated level of ALP and reductase in lung cancer cells.
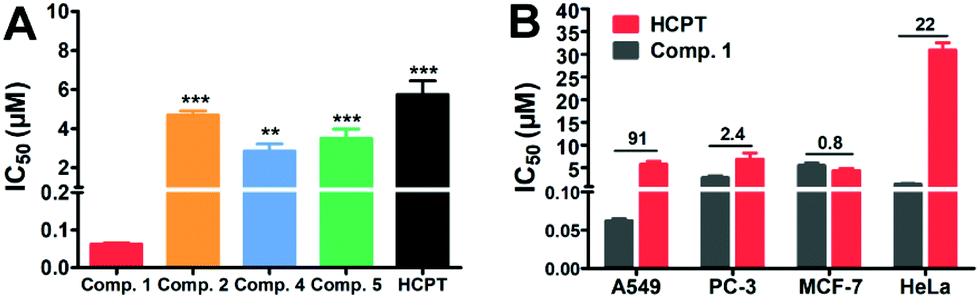 | ||
| Fig. 3 Cytotoxicity of the tandem self-assembling prodrug. (A) IC50 value of A549 cells treated with Comp. 1, 2, 4, 5 and free HCPT. (B) IC50 value of Comp. 1 and HCPT to different cell lines. (One-way ANOVA, compared to the Comp. 1 group, mean ± SD, ***P < 0.001, **P < 0.01. N = 3.) | ||
Evaluation of penetration, accumulation and retention of tandem molecules at tumor sites
According to many studies, the efficacy of nanomedicines depends on their ability of penetration, accumulation and retention at the tumor site.10 Tandem in situ self-assembly of prodrugs enables the drug to penetrate and further retain. Inspired by this, we evaluated the penetration and retention of Comp. 1 on multicellular spheroids (MCSs) of A549 cells that mimic solid tumors. The MCSs were incubated with different compounds (100 μM) for 6 hours. As shown in Fig. S22,† green fluorescence signals of Comp. 1 were observed at different depths and reached the interior of MCSs (80 μm). Since Comp. 1 was a small hydrophilic molecule, it could diffuse freely inside the MCSs and then tandemly self-assemble to form stable nanofibers instructed by ALP and reductase, which further improved the accumulation and retention in the MCSs. As a comparison, the fluorescence intensities of the A549 MCSs incubated with Comp. 2, 4 and 5 were much lower than that in the Comp. 1 group at the corresponding depths.We then investigated the body distribution of the tandem self-assembling molecules on A549 tumor-bearing BALB/c nude mice (Table S1†). The cyanine-5.5 (Cy-5.5) labeled tandem self-assembling molecule (Comp. 6, Fig. S6†) and three control compounds without tandem SA were synthesized and utilized for IVIS Lumina II imaging (Comp. 7, 8 and 9, Fig. S8–S10†). As shown in Fig. 4A, Comp. 6, 8 and 9 achieved fast whole-body distribution within 4 hours with high concentration due to excellent hydrophilicity. 24 hours after the injection, we observed the strongest fluorescence intensity from Comp. 6 at the tumor site and the fluorescence signals could last up to 36 hours. The fluorescence intensities of Comp. 8 and 9 were weaker than that of Comp. 6 after at 8 hours and later disappeared within 36 hours. Meanwhile, only weak fluorescence could be observed in the Comp. 7 group due to the poor solubility of the molecule. These results demonstrated the excellent tumor accumulation and retention characteristic of the tandem self-assembling molecule Comp. 6. Furthermore, the excised normal organs and tumor tissues were harvested 36 hours post injection, in order to assess the distribution of different compounds ex vivo. As shown in Fig. S23,†Comp. 6 possessed the highest retention and accumulation capacity among all groups, which is consistent with the results of IVIS images in vivo. We also observed that Comp. 6, 8 and 9 were mainly metabolized by the kidneys, while the poorly soluble Comp. 7 was also metabolized by the liver in addition to the kidneys. We also obtained frozen sections of the excised solid tumors after intravenously injecting Comp. 6–9 for 24 hours. As shown in Fig. 4B and Fig. S24,†Comp. 6 (red fluorescence) could efficiently retain in larger tumor areas compared to other control compounds without tandem SA. As a result, it was concluded that the tandem molecular SA strategy can improve the ability of accumulation, retention and penetration of the prodrug.
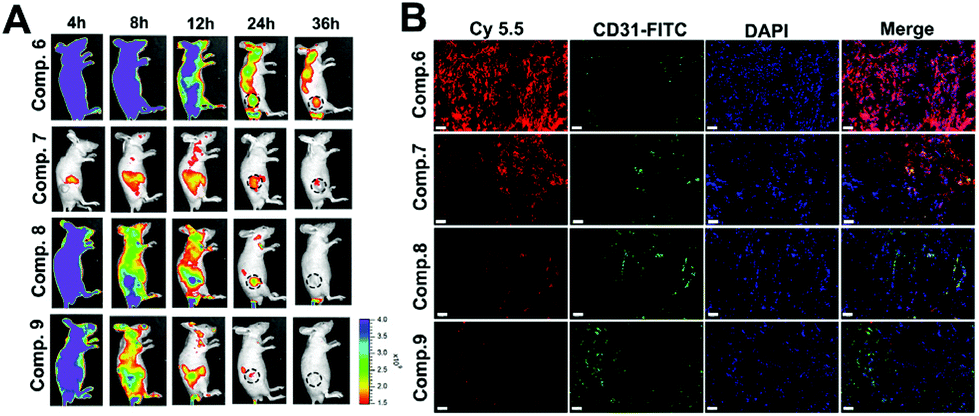 | ||
| Fig. 4 (A) Representative pictures of NIR-fluorescence imaging of A549 tumor bearing nude mice with the treatment of Comp. 6, 7, 8 and 9 at different time points after the intravenous administration. (B) CLSM images of cryo-sections of A549 tumor tissue after intravenous administration of Comp. 6, 7, 8 and 9 for 24 h, the DAPI-stained nucleus (blue channel), FITC-CD31-stained blood vessel (green channel), Cy5.5 (red channel), and overlay image, scale bar = 50 μm. | ||
Evaluation of anti-tumor efficacy and biosafety in vivo
Encouraged by the efficient cancer cell inhibition and excellent tumor penetration, accumulation and retention capacity, we evaluated the therapeutic potential of the tandem self-assembling prodrug to the solid tumor in a BALB/c nude mouse model bearing A549 cells. Six groups of mice with an average tumor size of 100 mm3 were treated with Comp. 1, 2, 4, 5, free HCPT and PBS via the tail vein (once every three days, 3 mg kg−1 HCPT-equivalent dose, Table S2,†N = 5). As shown in Fig. 5B and S25,† the tumor volume began to decrease after the first administration of Comp. 1. Tumor volume slightly increased to 189 mm3 at the 19th day. The tumor volume in the groups of Comp. 2, 4, 5 and free HCPT were about 423 mm3, 451 mm3, 451 mm3 and 499 mm3, respectively, which were more than 2.5 fold larger than that of the Comp. 1 group. The tumor volume of PBS groups reached about 951 mm3 at the 19th day, which was 5-fold larger compared to the Comp. 1 group. Meanwhile, as shown in Fig. 5C and Fig. S26B,† the average tumor weight of the Comp. 1 group was 141.5 mg, which was significantly lower than that of the other groups. We then verified whether Comp.1 could improve the survival rate relative to free HCPT. The BLAB/c nude mice bearing A549 tumors were randomly divided into three groups (N = 6), including Comp. 1, free HCPT and PBS groups. After the administration of therapeutic agents and monitoring for 34 days (Fig. S27†), the first death in the PBS group occurred on the 23rd day, and all the mice died on the 27th day. Meanwhile, the first death in the free HCPT group occurred on the 27th day, and all of the mice in this group died on the 31st day. However, only one mouse in the Comp. 1 group died on the 29th day during the entire monitoring period. These results clearly demonstrated that the tandem self-assembling prodrug inhibited the growth of lung tumor efficiently and improved the survival rate of animals greatly.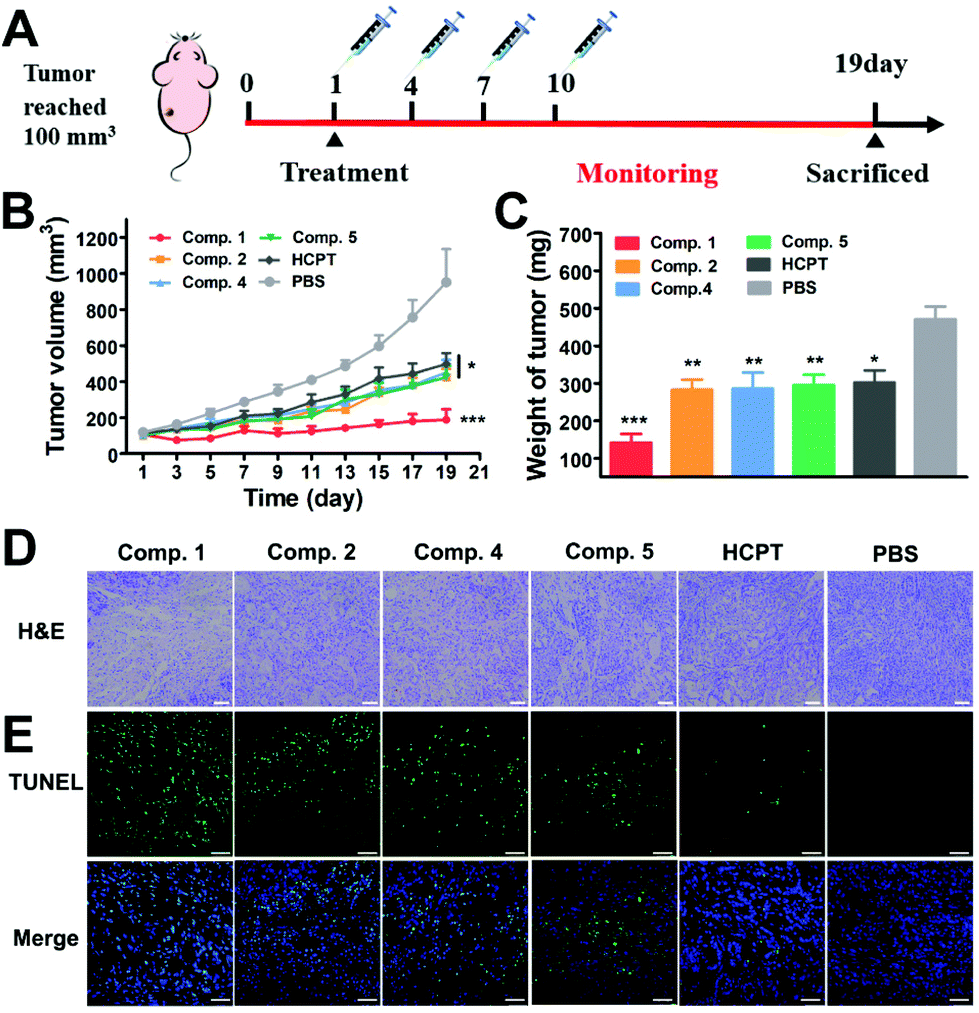 | ||
| Fig. 5 (A) Treatment schedule for inhibition of A549 tumors in BALB/c nude mice. The treatment of A549 tumor bearing nude mice by the intravenous injection of Comp. 1, 2, 4, 5, HCPT and PBS at a dose equaled 3 mg kg−1 equivalent of HCPT. (B) Tumor volume. (C) Tumor weight. (D) H&E staining of tumor tissues, scale bar = 100 μM. (E) TUNEL analyses of tumor tissues, scale bar = 50 μM. | ||
We further used Hematoxylin and Eosin (H&E) staining and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling assay (TUNEL) to assess the anti-tumor efficacy of different compounds. All groups of mice were sacrificed on the 19th day after administration, and tumor tissues were harvested. H&E staining of tumor tissue (Fig. 5D) in the PBS group presented intact tumor tissue morphology, where compact tumor cells speckled with large amounts of extracellular matrix maintaining cancer cell normal growth. Among the groups treated with different prodrugs and free HCPT, the Comp. 1 group displayed the largest tissue necrotic area and the least tumor cellularity, exhibiting the most prominent anti-tumor efficacy of tandem self-assembling prodrugs. Meanwhile, as shown in Fig. 5E, the results of TUNEL also showed that Comp. 1 induced a large number of tumor cell apoptosis, while other compounds or the free HCPT could not. These results together demonstrated that tandem molecular SA could significantly improve the tumor inhibition efficiency of anticancer drugs.
In order to test the biosafety of various therapeutic agents used in this study, body weights of mice were continuously recorded for 19 days (once every two days). As shown in Fig. S26A,† all groups showed no obvious body weight change during the whole treatment period. In addition, the main organs (heart, liver, spleen, lungs, and kidneys) of the mice were collected after completing the treatment monitoring. H&E staining images (Fig. S28†) show that there were no obvious cell damage and histopathological changes compared to the saline group. To further assess the biosafety of various formulations of HCPT in vivo, normal BALB/c nude mice were injected in all therapeutic agents three times (3 mg kg−1 HCPT-equivalent dose, once every two days), and the blood samples were collected and analyzed to obtain the clinical blood parameters and blood biochemical values. As shown in Fig. S29,† the clinical hematology markers of Comp. 1, 2, 4, 5 and free HCPT groups, including red blood cell (RBC) count, white blood cell (WBC) count, hemoglobin (HGB), and platelets (PLT) count showed a negligible difference compared to the PBS group, and the sample detection values are all within the index reference range. For the blood biochemical index, the levels of aspartate transaminase (AST), alanine transaminase (ALT), alkaline phosphatase (ALP), total bilirubin (TBIL) and direct bilirubin (DBIL) represented the liver parenchymal injury. The levels of urea (UA), blood urea nitrogen (BUN), and creatinine (CRE) were commonly used to evaluate the kidney condition. As shown in Fig. S30,† although there is a certain difference in the detection values for each group, all the values of the above blood biochemistry indicators are within the normal reference range of the index. These observations demonstrated trustworthy biosafety of the peptide-based prodrug with tandem molecular SA.
Conclusions
In summary, we developed a tandem molecular SA strategy to significantly improve the anti-tumor efficiency of HCPT by two orders of magnitude. The designed peptide–HCPT prodrug could self-assemble into fibrous nanomedicine by ALP catalysis and thus be up-taken by lung cancer cells much more than free HCPT. Then, the internalized nanofibers could transform into another nanomedicine by reductase instructed self-assembling, which prevented the degradation or disassembly of the nanomedicine. At the cellular level, the tandem self-assembled prodrug had good selectivity and greatly increased the IC50 of HCPT due to the increased internalization and retention of the drug. As a result, it could improve the tumor accumulation and retention of the drug in vivo, which effectively inhibited the lung tumor growth without side effects. We envision that the tandem molecular SA strategy can facilitate the development of selective and effective nanomedicines for precise chemotherapeutics.Author contributions
D. B. Zheng, J. F. Liu, Y. H. Ding, Y. Y. Zhang and R. Peng synthesized and characterized the compounds. D. B. Zheng, J. F. Liu and L. M. Xie performed the cell imaging and experiments. D. B. Zheng, Y. X. Chen and M. Cai analysed the results and performed the scheme illustration. L. Wang, H. M. Wang, J. Gao and Z. M. Yang guided the research and wrote the manuscripts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Fund for Distinguished Young Scholars (31825012), the NSFC (51773097, 82022038, and 31961143004), and the Young Elite Scientists Sponsorship Program by Tianjin (TJSQNTJ-2017-16).Notes and references
- J. J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- E. K. Lim, T. Kim, S. Paik, S. Haam, Y. M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS PubMed.
- K. Sato, M. P. Hendricks, L. C. Palmer and S. I. Stupp, Chem. Soc. Rev., 2018, 47, 7539–7551 RSC.
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS PubMed.
- V. J. Venditto and F. C. Szoka, Adv. Drug Delivery Rev., 2013, 65, 80–88 CrossRef CAS PubMed.
- M. F. Flessner, J. Choi, K. Credit, R. Deverkadra and K. Henderson, Clin. Cancer Res., 2005, 11, 3117–3125 CrossRef CAS PubMed.
- C. H. Heldin, K. Rubin, K. Pietras and A. Ostman, Nat. Rev. Cancer, 2004, 4, 806–813 CrossRef CAS PubMed.
- Y. Matsumoto, J. W. Nichols, K. Toh, T. Nomoto, H. Cabral, Y. Miura, R. J. Christie, N. Yamada, T. Ogura, M. R. Kano, Y. Matsumura, N. Nishiyama, T. Yamasoba, Y. H. Bae and K. Kataoka, Nat. Nanotechnol., 2016, 11, 533–538 CrossRef CAS PubMed.
- Q. Zhou, S. Shao, J. Wang, C. Xu, J. Xiang, Y. Piao, Z. Zhou, Q. Yu, J. Tang, X. Liu, Z. Gan, R. Mo, Z. Gu and Y. Shen, Nat. Nanotechnol., 2019, 14, 799–809 CrossRef CAS PubMed.
- R. W. Chakroun, F. Wang, R. Lin, Y. Wang, H. Su, D. Pompa and H. Cui, ACS Nano, 2019, 13, 7780–7790 CrossRef CAS PubMed.
- F. H. Wang, H. Su, R. Lin, R. W. Chakroun, M. K. Monroe, Z. Y. Wang, M. Porter and H. G. Cui, ACS Nano, 2020, 14, 10083–10094 CrossRef CAS PubMed.
- S. Li, Y. Zhang, S. H. Ho, B. Li, M. Wang, X. Deng, N. Yang, G. Liu, Z. Lu, J. Xu, Q. Shi, J. Y. Han, L. Zhang, Y. Wu, Y. Zhao and G. Nie, Nat. Biomed. Eng., 2020, 4, 732–742 CrossRef CAS PubMed.
- H. Wang, Z. Feng and B. Xu, Adv. Drug Delivery Rev., 2017, 110–111, 102–111 CrossRef CAS PubMed.
- C. Liang, X. Yan, R. Zhang, T. Xu, D. Zheng, Z. Tan, Y. Chen, Z. Gao, L. Wang, X. Li and Z. Yang, J. Controlled Release, 2020, 317, 109–117 CrossRef CAS PubMed.
- P. Schiapparelli, P. Zhang, M. Lara-Velazquez, H. Guerrero-Cazares, R. Lin, H. Su, R. W. Chakroun, M. Tusa, A. Quinones-Hinojosa and H. Cui, J. Controlled Release, 2020, 319, 311–321 CrossRef CAS PubMed.
- T. Wang, Y. Li, J. Wang, Y. Xu, Y. Chen, Z. Lu, W. Wang, B. Xue, Y. Li and Y. Cao, ACS Biomater. Sci. Eng., 2020, 6, 6800–6807 CrossRef CAS PubMed.
- Q. Guo, Y. Liu, G. Mu, L. Yang, W. Wang, J. Liu and J. Liu, Biomater. Sci., 2020, 8, 5638–5646 RSC.
- Y. Cong, L. Ji, Y. J. Gao, F. H. Liu, D. B. Cheng, Z. Y. Hu, Z. Y. Qiao and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 4632–4637 CrossRef CAS PubMed.
- R. Kubota, S. Torigoe, S. Liu and I. Hamachi, Chem. Lett., 2020, 49, 1319–1323 CrossRef CAS.
- P. Nasrollahi, K. Khajeh, E. Tamjid, M. Taleb, M. Soleimani and G. J. Nie, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 1170–1177 CrossRef CAS PubMed.
- W. Ji, C. Yuan, S. Zilberzwige-Tal, R. Xing, P. Chakraborty, K. Tao, S. Gilead, X. Yan and E. Gazit, ACS Nano, 2019, 13, 7300–7309 CrossRef CAS PubMed.
- Y. Li, Q. Zou, C. Yuan, S. Li, R. Xing and X. Yan, Angew. Chem., Int. Ed., 2018, 57, 17084–17088 CrossRef CAS PubMed.
- Q. Wu, Z. G. He, X. Wang, Q. Zhang, Q. C. Wei, S. Q. Ma, C. Ma, J. Li and Q. G. Wang, Nat. Commun., 2019, 10, 240 CrossRef PubMed.
- X. Xia, X. Yang, P. Huang and D. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 18301–18308 CrossRef CAS PubMed.
- H. M. Wang, Z. Q. Q. Feng and B. Xu, Angew. Chem., Int. Ed., 2019, 58, 5567–5571 CrossRef CAS PubMed.
- J. F. Shi, G. Fichman and J. P. Schneider, Angew. Chem., Int. Ed., 2018, 57, 11188–11192 CrossRef CAS PubMed.
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 5032 CrossRef PubMed.
- J. Zhang, S. Liu, H. Li, X. Tian and X. Li, Langmuir, 2020, 36, 11316–11323 CrossRef CAS PubMed.
- H. W. An, L. L. Li, Y. Wang, Z. Wang, D. Hou, Y. X. Lin, S. L. Qiao, M. D. Wang, C. Yang, Y. Cong, Y. Ma, X. X. Zhao, Q. Cai, W. T. Chen, C. Q. Lu, W. Xu, H. Wang and Y. Zhao, Nat. Commun., 2019, 10, 4861 CrossRef PubMed.
- X. Han, K. Cheng, Y. Xu, Y. Wang, H. Min, Y. Zhang, X. Zhao, R. Zhao, G. J. Anderson, L. Ren, G. Nie and Y. Li, J. Am. Chem. Soc., 2020, 142, 2490–2496 CrossRef CAS PubMed.
- Y. B. Cai, H. S. Shen, J. Zhan, M. L. Lin, L. H. Dai, C. H. Ren, Y. Shi, J. F. Liu, J. Gao and Z. M. Yang, J. Am. Chem. Soc., 2017, 139, 2876–2879 CrossRef CAS PubMed.
- D. B. Cheng, D. Wang, Y. J. Gao, L. Wang, Z. Y. Qiao and H. Wang, J. Am. Chem. Soc., 2019, 141, 4406–4411 CrossRef CAS PubMed.
- Z. Feng, X. Han, H. Wang, T. Tang and B. Xu, Chem, 2019, 5, 2442–2449 CAS.
- T. Suma, J. Cui, M. Mullner, S. Fu, J. Tran, K. F. Noi, Y. Ju and F. Caruso, J. Am. Chem. Soc., 2017, 139, 4009–4018 CrossRef CAS PubMed.
- G. Rong, C. Wang, L. Chen, Y. Yan and Y. Cheng, Sci. Adv., 2020, 6, eaaz1774 CrossRef CAS PubMed.
- B. C. Evans, K. M. Hocking, K. V. Kilchrist, E. S. Wise, C. M. Brophy and C. L. Duvall, ACS Nano, 2015, 9, 5893–5907 CrossRef CAS PubMed.
- C. Palm, M. Jayamanne, M. Kjellander and M. Hallbrink, Biochim. Biophys. Acta, 2007, 1768, 1769–1776 CrossRef CAS PubMed.
- J. Zhan, Y. Cai, S. He, L. Wang and Z. Yang, Angew. Chem., Int. Ed., 2018, 57, 1813–1816 CrossRef CAS PubMed.
- W. Tang, Z. Zhao, Y. Chong, C. Wu, Q. Liu, J. Yang, R. Zhou, Z. X. Lian and G. Liang, ACS Nano, 2018, 12, 9966–9973 CrossRef CAS PubMed.
- D. Zheng, Y. Chen, S. Ai, R. Zhang, Z. Gao, C. Liang, L. Cao, Y. Chen, Z. Hong, Y. Shi, L. Wang, X. Li and Z. Yang, Research, 2019, 2019, 4803624 CAS.
- F. Zhou, T. Fu, Q. Huang, H. Kuai, L. Mo, H. Liu, Q. Wang, Y. Peng, D. Han, Z. Zhao, X. Fang and W. Tan, J. Am. Chem. Soc., 2019, 141, 18421–18427 CrossRef CAS PubMed.
- J. Gao, J. Zhan and Z. Yang, Adv. Mater., 2020, 32, e1805798 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of the compounds, NMR, HR-MS spectrum and CAC of the compounds, LC-MS spectrum and bio-TEM image; photographs of tumor tissue. See DOI: 10.1039/d1nr01174j |
| This journal is © The Royal Society of Chemistry 2021 |