
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Control of crystal size tailors the electrochemical performance of α-V2O5 as a Mg2+ intercalation host†
Ian D.
Johnson
 abc,
Natalie
Stapleton
a,
Gene
Nolis
cd,
Dustin
Bauer
a,
Prakash
Parajuli
ce,
Hyun Deog
Yoo
f,
Liang
Yin
cg,
Brian J.
Ingram
bc,
Robert F.
Klie
ce,
Saul
Lapidus
cg,
Jawwad A.
Darr
*a and
Jordi
Cabana
*cd
abc,
Natalie
Stapleton
a,
Gene
Nolis
cd,
Dustin
Bauer
a,
Prakash
Parajuli
ce,
Hyun Deog
Yoo
f,
Liang
Yin
cg,
Brian J.
Ingram
bc,
Robert F.
Klie
ce,
Saul
Lapidus
cg,
Jawwad A.
Darr
*a and
Jordi
Cabana
*cd
aDepartment of Chemistry, University College London, London, UK. E-mail: j.a.darr@ucl.ac.uk
bChemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, IL 60439, USA
cJoint Center for Energy Storage Research, Argonne National Laboratory, Argonne, IL 60439, USA. E-mail: jcabana@uic.edu
dDepartment of Chemistry, University of Illinois at Chicago, Chicago, IL 60607, USA
eDepartment of Physics, University of Illinois at Chicago, Chicago, IL 60607, USA
fDepartment of Chemistry and Chemical Institute for Functional Materials, Pusan National University, Busan 46241, Republic of Korea
gX-ray Science Division, Argonne National Laboratory, Argonne, IL 60439, USA
First published on 28th May 2021
Abstract
α-V2O5 has been extensively explored as a Mg2+ intercalation host with potential as a battery cathode, offering high theoretical capacities and potentials vs. Mg2+/Mg. However, large voltage hysteresis is observed with Mg insertion and extraction, introducing significant and unacceptable round-trip energy losses with cycling. Conventional interpretations suggest that bulk ion transport of Mg2+ within the cathode particles is the major source of this hysteresis. Herein, we demonstrate that nanosizing α-V2O5 gives a measurable reduction to voltage hysteresis on the first cycle that substantially raises energy efficiency, indicating that mechanical formatting of the α-V2O5 particles contributes to hysteresis. However, no measurable improvement in hysteresis is found in the nanosized α-V2O5 in latter cycles despite the much shorter diffusion lengths, suggesting that other factors aside from Mg transport, such as Mg transfer between the electrolyte and electrode, contribute to this hysteresis. This observation is in sharp contrast to the conventional interpretation of Mg electrochemistry. Therefore, this study uncovers critical fundamental underpinning limiting factors in Mg battery electrochemistry, and constitutes a pivotal step towards a high-voltage, high-capacity electrode material suitable for Mg batteries with high energy density.
1. Introduction
Rechargeable batteries, particularly those based on Li-ion chemistry, have spurred the development and commercialization of devices requiring portable power sources, such as electric vehicles (EVs).1 Excitingly, EVs powered by Li-ion batteries are predicted to become cost-competitive with conventional internal combustion engine (ICE) vehicles in the near future, and more recent EV models are approaching driving ranges achievable by ICE vehicles.2 Despite these significant advances, there is still a clear desire to move beyond Li-ion batteries; the decarbonization of transport would be further accelerated by more energy-dense, lower-cost, and more sustainable rechargeable technologies.3,4 The Mg battery is one potential technology able to surpass Li-ion batteries in these respects; several studies report that Mg metal deposits and strips with a lower tendency to form dendrites, the presence of which preclude the use of the energy-dense Li metal anode in Li-ion batteries.5–7 Hence, the combination of the energy-dense Mg metal anode with a suitably high energy-density cathode material, such as a metal oxide, yields higher theoretical energy densities than Li-ion batteries.3 Moreover, the significantly greater earth-abundance and availability of Mg (compared to Li) would mitigate concerns regarding the relative scarcity of Li supply and lower the raw materials cost of battery production.Stable Mg2+ intercalation hosts have largely consisted of sulfide materials;8–10 however, their operating voltages vs. Mg2+/Mg are too low to give a higher energy density than Li-ion batteries. Therefore, oxide intercalation hosts (which allow higher voltages vs. Mg2+/Mg) are of great interest as potential high energy-density cathode materials because of the potential for high capacity with high reversibility and efficiency.11–14 However, the literature has contained significant discrepancies in reported capacities and reactivities of metal oxides in Mg battery systems. Detailed analysis has revealed that many literature reports of reversible Mg intercalation were more likely to result from parasitic electrolyte decomposition and proton intercalation.15 Indeed, a recent perspective has highlighted the disparity between reported capacities of Mg battery cathodes and the capacity verified by elemental, redox, and structural changes in the oxide host.16 For this reason, reported capacities should not be directly equated with Mg intercalation activity without robust supporting evidence. When these requirements are taken into consideration, very few oxide cathode materials have demonstrated large, reversible Mg2+ intercalation, in part due to the susceptibility of oxides to undergo competing reactions with cycling, such as conversion and MgO formation,13 both of which introduce irreversibility and severe energy inefficiency.17
While many different structures and compositions of vanadium oxides and phosphates have been investigated for Mg batteries, most have only demonstrated reversible Mg intercalation in the presence of water (either in the electrolyte or contained in the structure), such as VOx nanotubes,18,19 β-NaV6O15,20 and VOPO4·xH2O.21 It should be stressed that a functional Mg battery cannot contain water due to the irreversible formation of a passivation layer on the Mg metal anode, essentially preventing reversible Mg plating and stripping. Moreover, separate studies by Verrelli et al. and Lopez et al. revealed that H2O impurities in the electrolyte did not increase Mg intercalation in the α-V2O5 and ζ-V2O5 cathode materials, respectively, and that any additional reactivity could be attributed to parasitic reactions and proton intercalation.15,22 Therefore, the postulated enhancement of Mg intercalation by water inclusion has been cast in serious doubt, and rigorous studies of electrode materials should be conducted in anhydrous environments.
Considering electrochemical cycling in dry conditions, Mg intercalation has been confirmed conclusively within the vanadium oxides α-V2O5,23 layered V4Nb18O55,24 tunnel ζ-V2O5,11,25 and spinel MgV2O4,26 using redox, elemental, and structural probes, but only at elevated temperatures (50 °C or 110 °C).11,23 However, we note that other spinel compounds (AM2O4, where A = Mg or Zn, M = Cr, Co, Fe, Mn, V) have also consistently demonstrated Mg removal on charge (∼200 mA h g−1).14,26–31 Comparing between these materials, the report of α-V2O5 is arguably the most striking with a high capacity of 300 mA h g−1 at 110 °C, where this degree of Mg intercalation was confirmed and quantified by a combination of EDS, FTIR, XRD and XAS analysis.23 The voltage hysteresis of the first cycle (1.35 V) was significantly higher than latter cycles (0.73 V), and inspection of the α-V2O5 particles revealed significant delamination of the α-V2O5 layers following the first discharge: it can therefore be hypothesized that the delamination of α-V2O5 reduced the observed voltage hysteresis. However, this delamination was suspected to have a detrimental impact on cycle stability, as the capacity degraded rapidly to 230 mA h g−1 after 20 cycles.
Given the above observations, it could also be hypothesized that the kinetics of Mg insertion contribute to voltage hysteresis, and that comparison of the electrochemistry of nanometric and micrometric α-V2O5 would clarify the relative contribution of these kinetic effects.32–36 Nanosizing the active material particles effectively reduces the diffusion distance required for Mg to penetrate the core of the particle, which should minimize the diffusion overpotential within an Mg-ion cell and reduce the degree of particle fracture with ion intercalation. Indeed, a recent report revealed nanosizing ζ-V2O5 results in reduced voltage hysteresis and increased capacity when cycled in Mg-containing electrolytes.25 Synthesized α-V2O5 has typically formed micron-long nanorods, micron-wide nanosheets and hollow microspheres when prepared from solution without a physical scaffold.33,36–38 In contrast, α-V2O5 prepared as part of a composite material has allowed for the formation of smaller, semi-spherical nanometric α-V2O5 crystallites, where the composite material provided heterogeneous nucleation sites for V2O5 crystallites and prevented their agglomeration and fusion.34,39–43 These templating agents have typically consisted of carbonaceous materials (such as graphene),34,39–41 and nanometric oxides, such as ZrO2 and TiO2.42,43 Therefore, it is clear that generating nanometric α-V2O5 crystallites can be achieved by use of templating agents such as TiO2, which can otherwise be challenging using conventional synthesis methodologies.
Herein, we compare the Mg-ion electrochemistry of micrometric α-V2O5 and an α-V2O5–TiO2 nanocomposite to evaluate sources of voltage hysteresis as a function of cycling, and reveal that the larger first cycle hysteresis can be avoided by sufficiently nanosizing the crystallites of α-V2O5. Moreover, the nanocomposite displayed an enhanced capacity retention, negating the detrimental impact of delamination on cycle life. Crucially, the voltage hysteresis of subsequent cycles of the nanocomposite was similar to that of micrometric samples, suggesting that additional kinetic or thermodynamic factors contributed to the observed hysteresis in later cycles. Given the lack of studies investigating diffusive contributions to hysteresis in Mg battery oxide cathode materials in dry conditions, where Mg intercalation is demonstrated with elemental, redox, and structural probes, this study is a critical step in determining the true limiting factors in cathode performance.
2. Experimental details
2.1. Synthesis
Synthesis: Two routes (labelled route 1 and 2, respectively) were employed to synthesize nano-V2O5, which are discussed in detail below. Route 1 involved the manufacture of a nanosized VO2 precursor via Continuous Hydrothermal Flow Synthesis (CHFS), which was oxidized to α-V2O5 in air in a subsequent step. Route 2 involved the direct synthesis of a hydrated V2O5 precursor in CHFS, which was heat-treated to remove intercalated water and produce a more crystalline α-V2O5.Synthesis Route 1: VO2 was synthesized via CHFS with an identical method described in previous publications.1,2 The dried VO2 powder was heat-treated at 600 °C for 5 hours (ramp rate 1 °C min−1) in air to produce V2O5 with a 140 nm average crystallite size.
Synthesis Route 2: A partially hydrated form of V2O5 with a TiO2 seed was synthesized directly using CHFS incorporating a Confined Jet Mixer (CJM) (diagrams of the CHFS mixing apparatus are included in Fig. S1a and S1b†).3 Firstly, a TiO2 sol was formed by CHFS. Two aqueous, room temperature precursor solutions of 0.6 M TiBALD (Aldrich, Dorset, U.K.) and 0.18 M KOH (Merck, Darmstadt, Germany) were pumped from pumps P2 and P3, respectively, at 40 mL min−1 each to combine in a tee-piece mixer. The combined precursors (at flow rate 80 mL min−1) flowed into the side arms of the CJM, where it combined with an 80 mL min−1 flow of deionized (D.I.) water heated to 310 °C. Nanoparticles formed rapidly in flow under transitional mixing conditions (Reynolds number of ∼3390) with a mixing temperature (Tmix) of 171 °C. The product sol from the CJM then passed through a 1 m long outlet pipe, giving a residence time of ca. 10.0 s at 171 °C, thereafter a pipe-in-pipe countercurrent heat exchanger cooled the product slurry to ca. 40 °C. Finally, the sol exited the apparatus via a back-pressure regulator (24 MPa pressure, Tescom), which maintained pressure throughout the CHFS apparatus. The precursors, concentrations and temperatures used in this reaction are indicated by reaction conditions (“1)” in Fig. S1a.† The product sol (0.15 M TiO2 in suspension) was used directly as a seed for the production of the nanocomposite.
To make the partially hydrated V2O5–TiO2 composite, the same reactor apparatus was used, with identical flow rates from each pump. However, the hot water feed was at 450 °C, giving turbulent mixing dynamics (Reynolds number >6900) and a reaction temperature of 335 °C. P1 was used to pump 0.5 M H2O2 (Fisher, Loughborough, U.K.) to generate an oxidizing environment. The P2 precursor was 0.08 M VOSO4 (Alfa Aesar, Loughborough, U.K.) and preformed 0.02 M TiO2 sol, and P3 pumped DI water only. The precursors, concentrations and temperatures used in this reaction are indicated by reaction conditions “2” in Fig. S1a.†
Post-synthesis, the slurries of VO2 and V2O5–TiO2 were allowed to settle, and placed in dialysis bags (Visking Dialysis Tubing, Medicell Membranes Ltd, London, UK) suspended in D.I. water. The D.I. water was regularly replaced to leach the aqueous waste products from the slurries until the conductivity of the slurry supernatant was below 100 μS m−1. The cleaned, wet pastes were freeze-dried by heating from −60 °C to 25 °C over 24 h under vacuum (<13 Pa, VirTis Genesis 35 XL, SP Scientific, New York, U.S.).
The freeze-dried, hydrated V2O5–TiO2 composite was heat-treated at 450 °C for 5 hours (ramp rate 1 °C min−1) in air to produce the V2O5–TiO2 composite with an average V2O5 crystallite size 25 nm.
2.2. Physical characterization
XRD analysis of the pristine powders was collected using a Stoe StadiP diffractometer in transmission mode (coupled θ–2θ geometry), using Mo-Kα radiation, with the sample sandwiched between two plastic foil disks held together with a thin layer of silicon grease. A pre-sample Ge (111) monochromator selected the Mo Kα1 radiation only (λ = 0.709 Å) and included a 0.5 mm collimator restricted to 3 mm height. The sample was rotated in the beam, and the diffraction intensity recorded using a Dectris Mython 1k silicon strip detector covering 18° in 2θ. Patterns were collected in the range 2–40°, with a step size of 0.5° in 2θ and a collection time of 20 s per step. Rietveld refinement (to extract lattice parameters and determine site occupancies) was performed with Material Analysis Using Diffraction (MAUD) software.4XRD patterns of the pristine, discharged and charged powders/electrodes were collected at the Advanced Photon Source (APS) at beamline 11-BM. The powder or electrode samples were loaded into Kapton capillaries for data collection, where the calibrated wavelength of X-ray radiation was 0.412799 or 0.457854 nm.
V L2,3-edge and O K-edge XAS was performed at the beamline 4-ID-C, Advanced Photon Source, Argonne National Laboratory, USA. At 4-ID-C, spectroscopy was completed simultaneously in both the Total-Electron-Yield (TEY) and Total-Fluorescence Yield (TFY) mode utilizing photocurrent for the TEY and a silicon drift diode detector for the TFY, in order to make direct surface to bulk comparisons. Data were obtained at a spectral resolution of ∼0.2 eV, with a 2 s dwell time. 3 scans were performed on each sample, at each absorption edge, and scans were averaged in order to maximize the signal to noise ratio. The V L2,3- and O K-edges were scanned in the range 500 to 560 eV. The V and O energy scales were normalized using a SrTiO3 standard.
Scanning transmission electron microscopy (STEM) imaging, energy dispersive X-ray (EDX) and electron energy loss (EEL) spectroscopy were performed on an aberration-corrected JEOL JEM-ARM200CF, equipped with a cold field emission gun operated at 200 kV, which allows a 73 pm spatial resolution and a 0.35 eV energy resolution. The microscope is equipped with high and low angle annular dark field detectors (HAADF/LAADF), bright field detector (BF), post-column Gatan Continuum spectrometer and an Oxford XMAX100TLE silicon drift detector (SDD). Imaging of the V2O5 samples is done along specific zone axes, which show unmixed atomic columns. STEM images were acquired simultaneously in HAADF, LAADF and ABF modes to identify both heavy elements, as well as light elements. The collection angles for HAADF, LAADF and ABF detectors were set at 90–370, 40–160 and 14–28 mrad, respectively. The EELS spectra were collected using a Gatan Quantum imaging filter with a convergence angle of 30 mrad and a collection angle of 35 mrad. The TEM samples were prepared in a glovebox under an argon environment to prevent any changes to the sample structure as the result of exposure to oxygen.
2.3. Electrochemical characterization
Electrodes were prepared by hand-grinding the samples, carbon black (Denka), and polyvinylidene difluoride (PVDF) (Kynar) in N-methylpyrrolidone (NMP) (Sigma-Aldrich) in a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:20 wt% ratio, which were then cast on 1/2′′ stainless steel 316 mesh electrodes and allowed to dry overnight in air. These were pressed (8 tons) and dried at 100 °C overnight under vacuum in the glovebox antechamber, and possessed active material loadings in the range 2.50–2.75 mg cm−2.
:20:20 wt% ratio, which were then cast on 1/2′′ stainless steel 316 mesh electrodes and allowed to dry overnight in air. These were pressed (8 tons) and dried at 100 °C overnight under vacuum in the glovebox antechamber, and possessed active material loadings in the range 2.50–2.75 mg cm−2.
Mg-ion coin cells contained an Activated Carbon Cloth (ACC) as a counter electrode (ACC-5092-20, Kynol, New York, US), and a glass fiber separator (VWR, grade 691). Discharge/charge of the cells was carried out at 110 °C in 0.5 M Mg(N(SO2)2(CF3)2)2-(C9H20N)(N(SO2)2(CF3)2) (Mg(TFSI)2-PY14TFSI) electrolyte with low H2O content (<50 ppm), with lower and upper voltage limits of −1.7 V and 1.2 V vs. carbon, respectively. The charge/discharge rate was galvanostatically controlled by a Biologic VMP3 potentiostat.
After oxidation or reduction of the electrodes, they were recovered and rinsed in acetonitrile five times, and dried at room temperature before XAS, XRD and EDX characterization.
3. Results and discussion
A two-step process utilizing Continuous Hydrothermal Flow Synthesis (CHFS) successfully produced a micrometric α-V2O5 sample and a nanometric α-V2O5–TiO2 composite as described in the Experimental section. PXRD analysis revealed the materials crystallized in the orthorhombic, α-V2O5 polymorph with the Pmmn space group (Fig. 1a, reference pattern PDF 01-072-0433). Very weak additional diffraction peaks were observed in the V2O5 sample, which were tentatively assigned to trace V6O11. This impurity may have arisen from the tendency for metal oxides to be reduced close to their melting points (the melting point of α-V2O5 is 690 °C). The inclusion of the TiO2 sol was evident in the PXRD pattern of the V2O5–TiO2 composite, which displayed additional broad peaks consistent with anatase TiO2 (reference pattern PDF 01-070-6826). Rietveld refinement of the three samples revealed lattice parameters consistent with literature values (Table S1 and Fig. S2†) for α-V2O5, and were able to provide average crystallite diameters, d, calculated from peak fitting within the refinements (which also accounted for instrumental broadening). These refinements gave d values of 140 nm and 25 nm for the V2O5 sample and the V2O5–TiO2 composite, respectively. A slight expansion of the a and c lattice parameters was evident in the V2O5–TiO2 composite compared to the other samples, which could either result from doping of Ti in the V2O5 structure, or from lattice-matching effects at the V2O5–TiO2 interface. It has been reported by Kryukova et al. that the V2O5–anatase interface forms along the V2O5 (301) and TiO2 (110) lattice planes; as the d-spacing for the latter (2.68 Å) exceeds the former (2.61 Å), it would be expected that the a and c lattice constants would expand in the V2O5–TiO2 composite due to lattice mismatch and resulting strain at the interface, although the possibility of Ti doping within the V2O5 lattice cannot be ruled out.44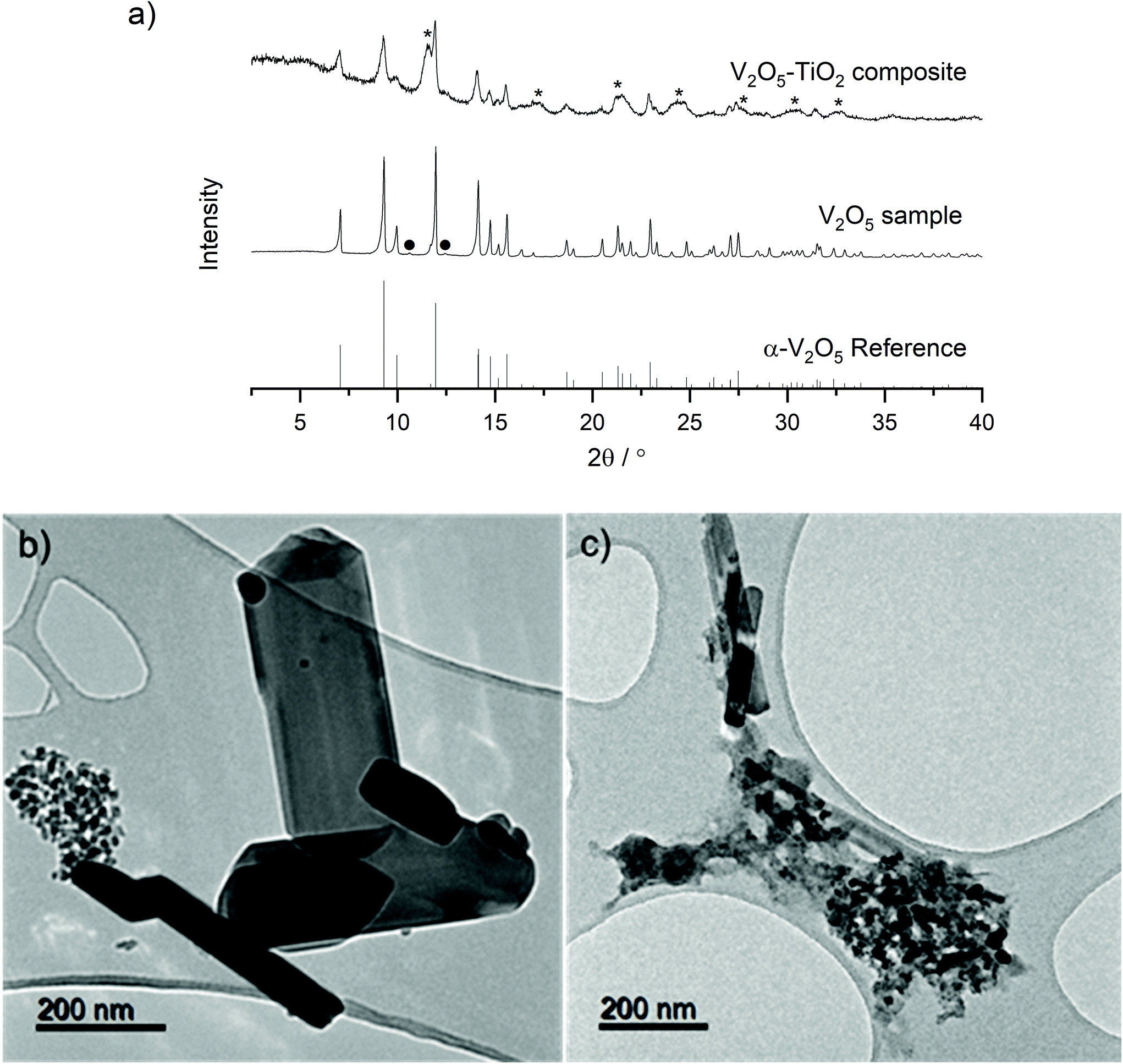 | ||
| Fig. 1 (a) XRD patterns (Mo-Kα radiation) of the V2O5 and V2O5–TiO2 materials, with an α-V2O5 reference pattern (PDF 01-072-0433). TiO2 diffraction peaks are indicated with asterisks (*), and the minor impurity peaks assigned to V6O11 indicated with ●; (b) TEM images of V2O5; (c) TEM images of V2O5–TiO2. | ||
TEM analysis revealed the V2O5 sample generally formed large rods (ca. 500 nm long with widths from 50 nm to 200 nm), although a small proportion of smaller particle clusters (ca. 20 nm diameter) were also present (Fig. 1b). In contrast, the V2O5–TiO2 composite formed a mixture of thin rods (with lengths in the range 200 nm to 400 nm and ca. 50 nm wide) and clusters of smaller particles ca. 20 nm in diameter (Fig. 1c). Given Rietveld analysis found an average particle size of 25 nm for the V2O5–TiO2 composite, it is suggested that the smaller particles were the majority phase in the sample. EDS analysis of a 250 nm cluster of the V2O5–TiO2 composite confirmed a molar ratio of 0.9:1 V2O5:TiO2 (Fig. S3†), which was vanadium-deficient compared to the precursor solution (2:1 V2O5:TiO2 ratio). Rietveld phase analysis corroborated the observations from EDS, revealing an approximate molar ratio of 0.75:1 of crystalline V2O5:TiO2 in the composite, and suggested the hydrothermal conversion of the vanadium precursor was approximately 50% of that of Ti. EDS mapping of this cluster revealed a relatively homogenous distribution of V and Ti, which implied intimate mixing of the V2O5 and TiO2 phases on the nm-scale, and that the TiO2 nanoparticulate phase successfully acted as a nucleation seed for V2O5.
Raman spectroscopy was used to further elucidate local structural differences between the V2O5 and V2O5–TiO2 samples. All peaks in the Raman spectrum of V2O5 could be indexed with good agreement to crystalline V2O5 (Fig. 2).45,46 In contrast, the Raman peaks were both broadened and slightly red-shifted in the V2O5–TiO2 composite compared to the V2O5 sample, and contained contributions from the anatase phase (Table 1).47 This observation suggests that the V2O5–TiO2 composite not only possessed smaller crystallite sizes than the V2O5 sample, but a greater degree of crystalline disorder.28
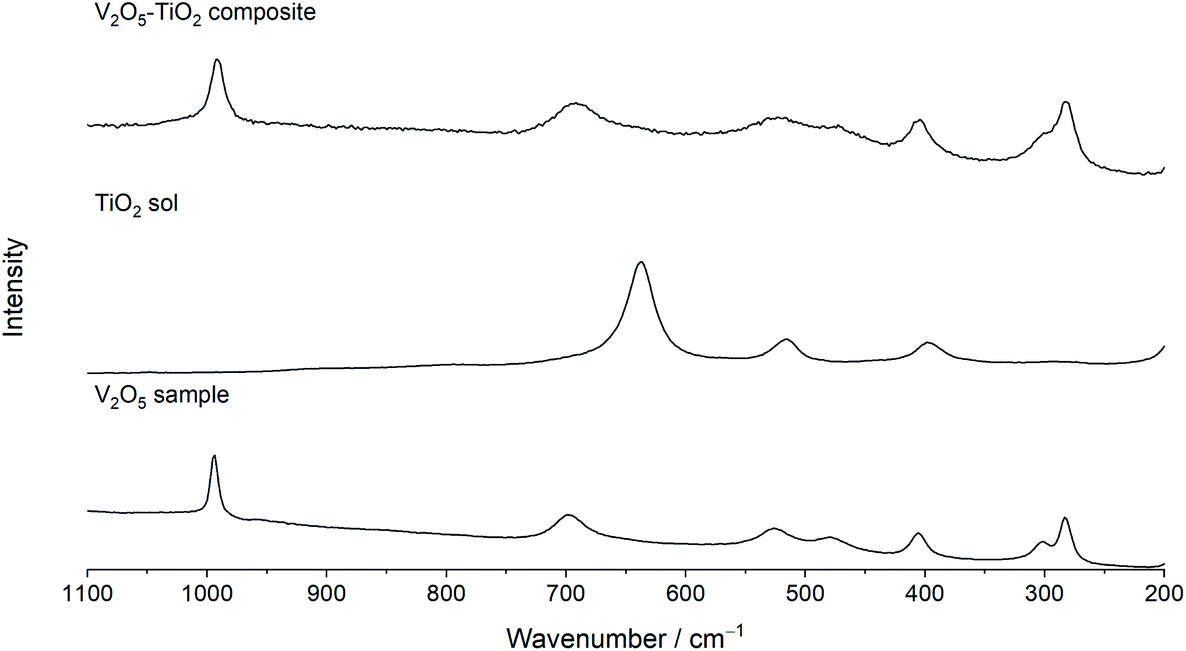 | ||
| Fig. 2 Raman spectra of the V2O5 sample, a TiO2 sol reference, and the V2O5–TiO2 composite, showing the Raman spectra analyzed in the range 200–1100 cm−1. | ||
| Symmetry | Vibrational mode | V(600/140)/cm−1 | TiO2 sol/cm−1 | V–Ti(450/25)/cm−1 |
|---|---|---|---|---|
| A1g, B2g | V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching O stretching |
994 | — | 993 |
| B1g, B3g | V–O–V asymmetric stretching | 698 | — | 694 |
| Eg | Ti–O stretching | — | 637 | 642(br) |
| A1g | V–O stretching | 526 | — | 521 |
| A1g, B1g | Ti–O stretching | — | 516 | — |
| A1g | V–O–V symmetric stretching | 479 | — | 471 |
| A1g | V–O–V angle bending | 405 | — | 405 |
| B1g | O–Ti–O angle bending | — | 396 | — |
| B2g, A1g | Ladder puckering and VO swinging |
301 | — | 299 |
| B1g, B3g | Ladder distortions | 283 | — | 281 |
Electrochemical analysis of the two materials under study was performed in conditions previously found to give high degrees of Mg insertion (110 °C in 0.5 Mg(TFSI)2 in PY14TFSI ionic liquid electrolyte).23,24 For the first discharge/charge cycle of the V2O5 sample, the behavior resembled that observed in bulk α-V2O5 previously by Yoo et al.,23i.e. a capacity of >200 mA h g−1 with a very large calculated voltage hysteresis of 1.68 V (Fig. 3a). The hysteresis was quantified with eqn (1), where ΔV is the voltage hysteresis, CChar is the charge capacity, and CDis is the discharge capacity.
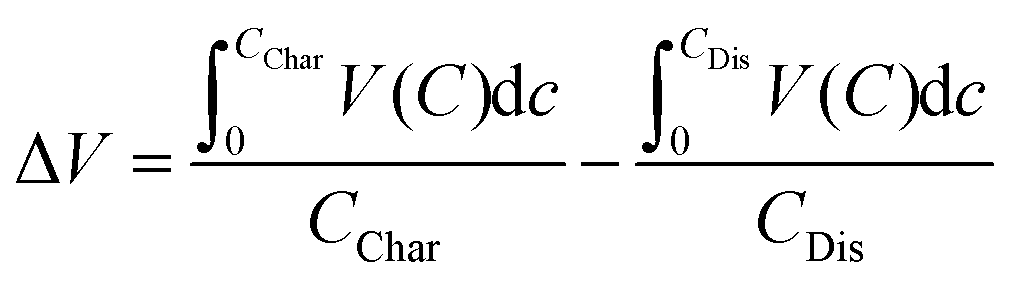 | (1) |
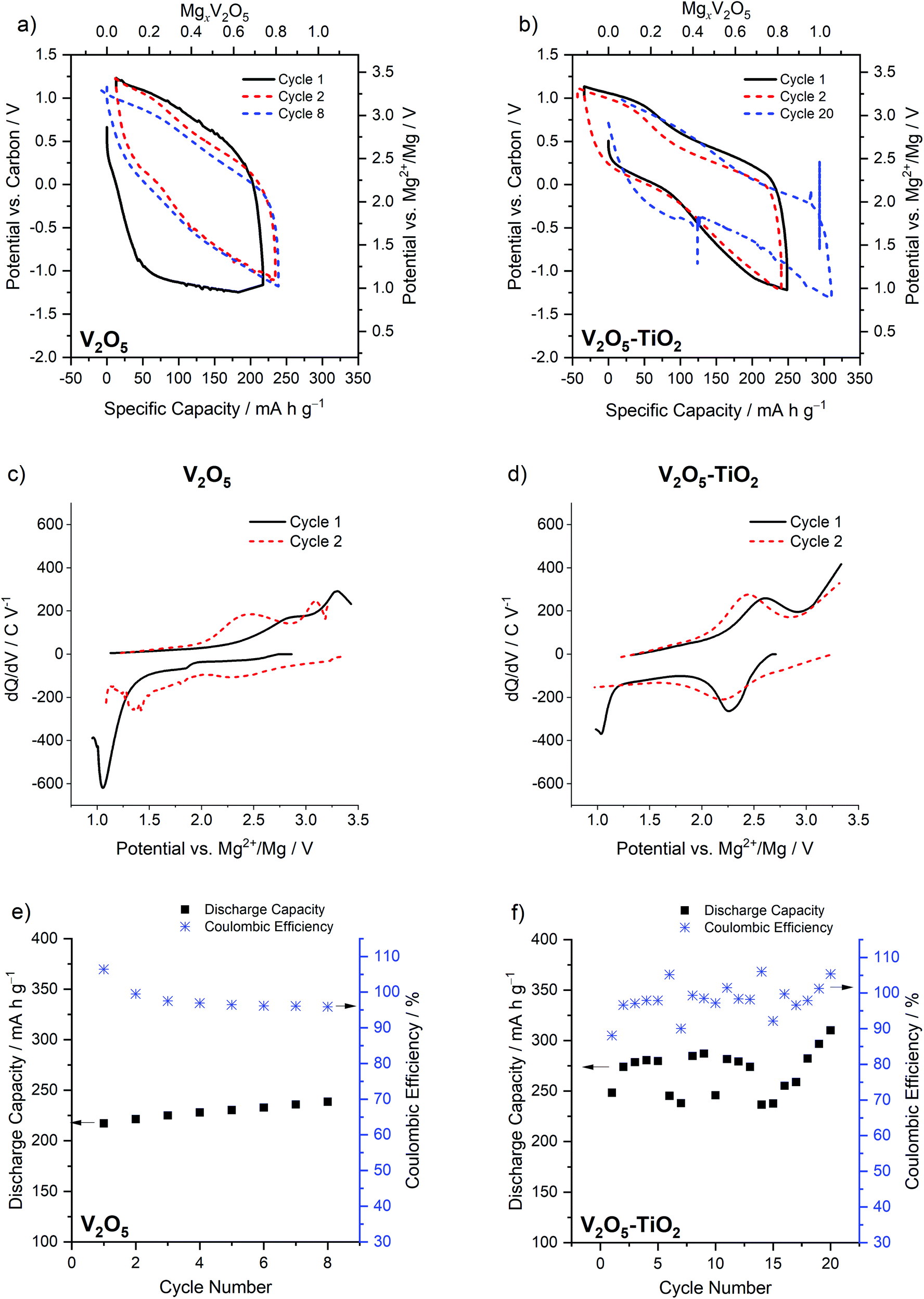 | ||
| Fig. 3 Voltage profiles of (a) the V2O5 sample and (b) the V2O5–TiO2 composite; dQ/dV plots of (c) the V2O5 sample and (d) the V2O5–TiO2 composite; capacity as a function of cycle number for (e) the V2O5 sample and (f) the V2O5–TiO2 composite. All electrochemical measurements were performed at 110 °C. | ||
This hysteresis manifested as a single prominent maximum at a low potential of 1.03 V vs. Mg2+/Mg in the dQ/dV plot, followed by maxima at 2.4 V and 3.1 V vs. Mg2+/Mg on the subsequent charge cycle (Fig. 3c). On the second cycle of the V2O5 sample, the calculated hysteresis was much reduced (0.98 V, Fig. 3a), as indicated by changes in the dQ/dV plot: a new maximum appeared at 2.15 V vs. Mg2+/Mg on discharge, and 2.45 and 3.1 V vs. Mg2+/Mg on charge (Fig. 3c). In contrast, the V2O5–TiO2 composite possessed significantly reduced hysteresis in comparison to the V2O5 sample on the first cycle (1.05 V, Fig. 3b), but only reduced by a small amount on the second cycle (0.81 V). This was reflected in the dQ/dV profiles of the V2O5–TiO2 composite, where the first cycle was almost identical to the second (Fig. 3d). It should be noted, however, that the hysteresis of the V2O5–TiO2 composite on the second cycle (0.81 V) was similar to the previous report of micrometric particles on the second cycle (0.73 V). The capacity of the V2O5–TiO2 composite (normalized to the mass of V2O5) was significantly enhanced compared to the V2O5 sample (Fig. 3e and f), although there was greater cycle-to-cycle variation in capacity for the V2O5–TiO2 composite, with values appearing to oscillate between values ca. 275 and 245 mA h g−1. This implied there was potentially a variance in the resistance of electrical contacting of the V2O5–TiO2 electrode between cycles. This is evidenced by the voltage spike, followed by a subtly different discharge voltage value, visible on the discharge step of cycle 20 (Fig. 3). Therefore, this variable contacting contributed to the voltage hysteresis, meaning specific cycles achieved a lower capacity before reaching the voltage limit. Overall, the V2O5–TiO2 composite achieved an average capacity of 270 mA h g−1 over 20 cycles at C/20, i.e. higher than that observed in the V2O5 sample (230 mA h g−1 over 8 cycles).12,23
To investigate structural, compositional, and redox changes in the V2O5–TiO2 composite, electrodes of the material were harvested following discharge and a discharge–charge cycle for ex situ analysis. The electrochemical response of the electrodes generated for this purpose is shown in Fig. S4.† Comparing PXRD patterns of the pristine V2O5–TiO2 composite with synchrotron PXRD of the discharged and charged electrodes revealed significant loss of crystallinity with Mg insertion and removal (Fig. 4a), with the majority of the remaining features corresponding to the anatase TiO2 phase. This is consistent with the previous observations made by Yoo et al.; in their study, the diffraction peaks of macroscopic V2O5 were significantly broadened with cycling. Careful examination of the discharged electrode PXRD pattern revealed a peak at Q = 1.92 Å−1 (highlighted by the red dot in Fig. 4b), which corresponds exactly to the most intense diffraction peak of the discharged MgV2O5 phase.23 This peak disappeared with charging, and the most intense diffraction peak of α-V2O5 reappeared at Q = 1.84 Å−1 (highlighted by the blue dot in Fig. 4b). Therefore, PXRD provides some evidence for similar structural changes occurring with Mg insertion and removal in the V2O5–TiO2 composite compared to the microscale V2O5 reported previously.
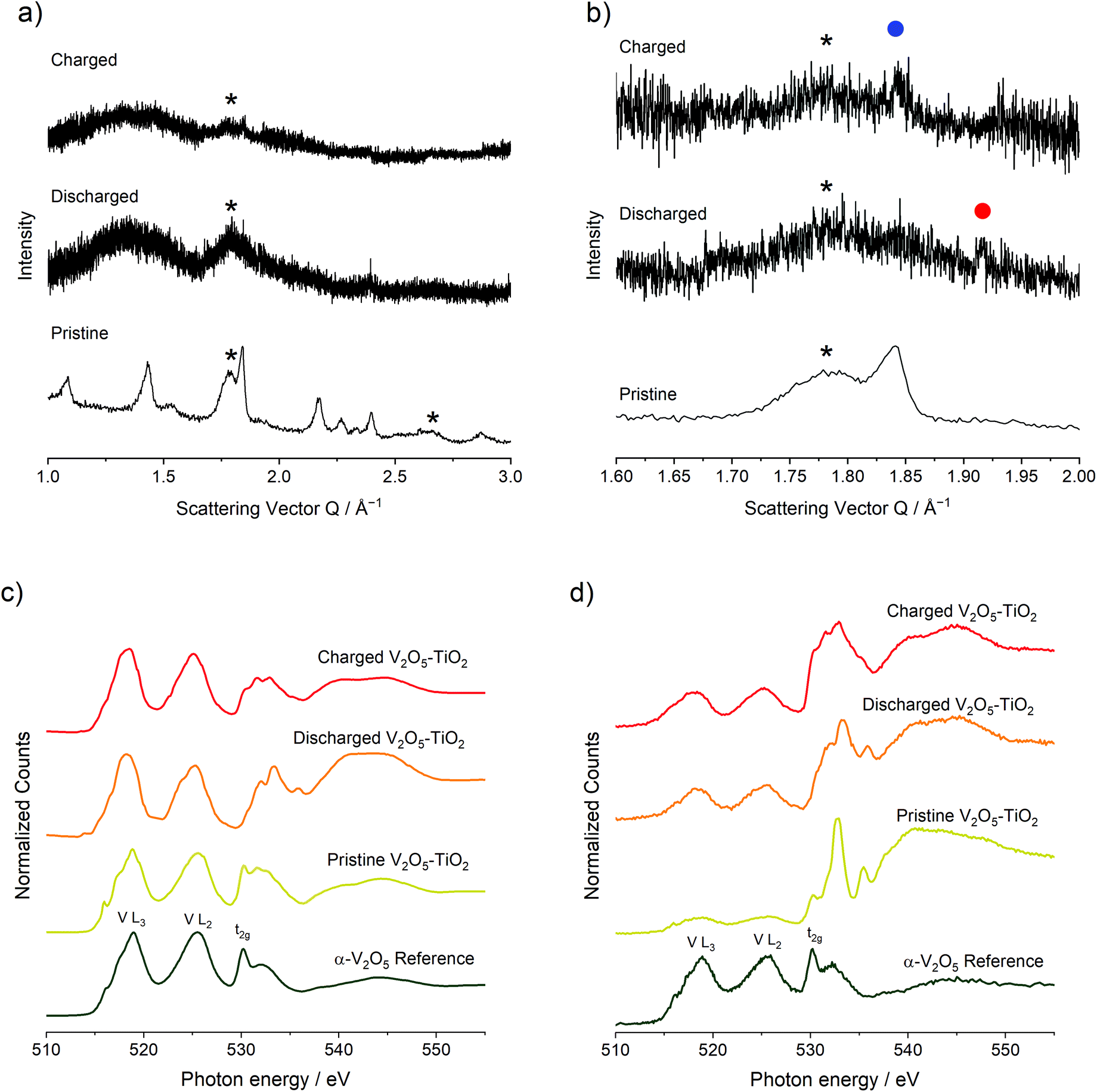 | ||
| Fig. 4 (a) XRD patterns of the pristine V2O5–TiO2 composite and the magnesiated V2O5–TiO2 electrode (b) Voltage vs. capacity plot for the V2O5–TiO2 discharged electrode. The red dot highlights a peak in the discharged PXRD pattern at Q = 1.92 Å−1, and the blue dot highlights a peak in the charged PXRD pattern at Q = 1.84 Å−1. (c) TEY XAS for the pristine, discharged and charged electrodes, corresponding to surface states for the V L2,3 and O K edges of V2O5–TiO2 samples, compared to an α-V2O5 standard. (d) TFY XAS for the pristine, discharged and charged electrodes, corresponding to bulk states for the V L2,3 and O K edges of V2O5–TiO2 samples, compared to an α-V2O5 standard. | ||
XAS analysis was conducted on the pristine, discharged and charged electrodes of the V2O5–TiO2 composite on the V and Ti L2,3- and O K-edges (Fig. 4c and d). XAS data were collected using both total electron and fluorescence yield detectors. Signals from total electron yields (TEY) correspond to the chemical state of the first 5–10 nm into the electrode, whereas total fluorescence yields (TFY) probes approximately 100 nm deep, thus having a notable contribution from the bulk crystal structure, especially considering the particle size of the materials. The Ti L2,3-edge of all V2O5–TiO2 samples and electrodes (Fig. S5†) was consistent with a Ti4+ state, indicating TiO2 did not contribute to redox activity, and is not discussed further. The V L-edge is characterized by V L3 and L2 spectral features, which correspond to transitions from V core 2p3/2 (L3, ∼519 eV) and 2p1/2 (L2, ∼525 eV) levels to unoccupied V 3d states (electron yield, Fig. 4c), which are split by a spin–orbit coupling of the V 2p atomic orbitals of about 7 eV. These parameters were similar to the V2O5 standard shown and to other experimental and theoretical reports of oxides with V5+.11,23,48–55 The signal of V L2,3 features in fluorescence yield (Fig. 4d) was less intense due to self-absorption effects, but had similar positions and shapes as the electron yield. This similarity of peak position and shape indicates that V5+ existed at the surface and the bulk.
For the discharged V2O5–TiO2 composite electrode, the V L3 and L2 features generally red-shifted in both fluorescence and electron yields. In TEY mode (Fig. 4c) the L3 feature broadened and red-shifted by about 1 eV; meanwhile the center of gravity of the L2 feature red-shifted by 0.5 eV and a shoulder near 523 eV appeared. Overall, the V L-edge spectra for discharged V2O5–TiO2 are similar to compounds containing V3+.14,50,54–56 Since the expected state is V4+ following discharge, these results suggests that the surface of the electrodes was at least partially reduced beyond V4+. Over-reduction of the V2O5 surface was also observed for macroscopic V2O5 particles previously.23 In TFY mode (Fig. 4d), the changes in V L3 and L2 features were smaller than observed in TEY, again supporting that the surface was reduced more than the bulk. Overall, these results suggested that significant redox reactions occurred in both the surface and bulk of the particles. For the charged electrode, the L3 feature was blue-shifted to 518.5 eV and the center of gravity of the L2 feature blue-shifted by 0.4 eV in TEY mode (Fig. 4c). Blue-shifts were also observed in TFY mode (Fig. 4d), although the degree of oxidation was lower than was observed in TEY mode. Overall, the original oxidation state of V was not recovered on charge, implying that V only partially re-oxidized back to the V5+ oxidation state following charge, and that the reaction was somewhat diffusion limited due to the relatively high reactivity of the surface compared to the bulk. This minor irreversibility, and inability to obtain the original transition metal oxidation state on charge, was further evidenced by changes in the O K-edge on discharge and charge (ESI†). This irreversibility has been commonly observed in metal oxide electrodes with Mg electrolytes.23,24
Fig. 4c and d also depicts integrated O K-edge X-ray absorption spectra acquired for pristine and electrochemically cycled V2O5–TiO2 samples, compared with standard α-V2O5.7 O K-edge features result from excited electron transitions from the core O 1s levels to unoccupied O 2p states which start to form the conduction band (528–555 eV).8,9 O K-edge spectra are divided into two parts. The first part, called O 2p–M 3d region goes from absorption edge to 5–7 eV above the threshold. Since this region is attributed to O 2p states, which are hybridized with V and Ti 3d states, the many spectral lines present resulted from the crystal field splitting of d orbitals of both metals. Despite that fact, the spectral feature ascribed to the t2g band (530.2 eV) is present for both α-V2O5 and pristine V2O5–TiO2 in both electron and fluorescence yields. This supports V L-edge analysis, where V5+ existed at both the surface and bulk levels. The second region appears at higher energies and is attributed to O 2p states mixed with metal 4sp states.8 For the discharged electrode, the t2g band (530.2 eV) reduces in intensity in electron yield as the lines between 532–535 eV move to slightly higher energy. This trend was also observed in the electrochemical reduction of V2O5 to form MgV2O5.10 In fluorescence yield, the spectral line at 531.5 eV gains significant intensity, and the peak at 532.8 eV remains quite prominent. Correspondingly, the O 2p–M sp red-shifted by ca. 1 eV upon discharge, consistent with a decrease of the binding energy of the core electrons when the compound was reduced. For the charged electrode, the t2g band (530.2 eV) spectral event increased in intensity relative to the other spectral features for both electron and fluorescence yields. Similar phenomena were observed when MgV2O5 was charged, consistent with Mg2+ deintercalation and V oxidation.10 The fact that the spectrum did not return to the pristine state indicated chemical irreversibility, consistent with V L2,3-edge observations.
EDS analysis was performed on the pristine, discharged and charged V2O5–TiO2 composite electrodes to investigate changes in stoichiometry with electrochemical cycling. The electron image revealed the structure of the composite was retained after discharge (Fig. S6a†), with no evidence of the delamination that affected previous reports of micrometric α-V2O5.23 In keeping with the electrochemistry, the Mg content of the V2O5–TiO2 composite increased on discharge and decreased on charge, and EDS mapping revealed a uniform dispersion of Mg within individual particles upon discharge, consistent with Mg intercalation (Fig. S6b–e†). Quantification of the Mg content relative to V revealed significant particle-to-particle variation in the discharged electrode, whereas the charged electrode revealed a much smaller variance (Tables S2 and 3†). This observation was reflected by the error bars in the average compositions, Mg0.28(14)V2O5 and Mg0.075(11)V2O5 for the discharged and charged states, respectively. Given the significant variation in Mg content observed in the discharged electrode, it is unclear whether the average Mg content found by EDS accurately described the element quantity and distribution in the sample. However, if this Mg content is representative of the discharged electrode, this would suggest a significant proportion of the observed electrochemical capacity arose from parasitic side-reactions. We suggest that specific α-V2O5 particles in the electrode underwent significant Mg intercalation, and that further studies should be conducted to quantify the degree of Mg intercalation within cycled Mg-ion electrode particles.
Overall, the electrochemical analyses revealed the positive impact of TiO2 inclusion on the α-V2O5 cathode material first cycle hysteresis. As TiO2 was proven to be electrochemically inactive by XAS analysis, and the changes to the V2O5 unit cell and crystallinity were relatively modest with TiO2 inclusion, we suggest the principle benefit of TiO2 to the electrochemistry was nanosizing the α-V2O5 phase. This implies that the energy required to delaminate the V2O5 layers contributed significantly to the first cycle hysteresis observed in micrometric V2O5 samples.23 However, there was no clear benefit to hysteresis of nanosizing beyond the first cycle. The greater effect of nanosizing on the first-cycle hysteresis (as opposed to later cycles) is supported by a previous investigation of ζ-V2O5: while reducing particle size from 150 nm-wide, micron-long rods to 100 nm diameter crystallites significantly reduced the first-cycle voltage hysteresis from 1.65 V to 1.15 V, the effect on the second cycle was much smaller (reduced from 1.29 V to 0.94 V).25 Therefore, the presence of voltage hystereses >0.7 V, even at elevated temperatures (110 °C) and with significantly nanosized particles, indicates that other energetic barriers contribute to Mg intercalation in V2O5. These barriers could originate from either inherent kinetics (such as Mg transfer from electrolyte to electrode) or thermodynamic factors associated with the electrochemical reactions that are not affected by particle size.
While these additional sources of hysteresis can be probed using techniques such as electrochemical impedance spectroscopy (EIS) or potentiostatic intermittent titration (PITT) within Li-ion systems, the lack of fully stable electrolytes at the high voltages provided by metal oxides preclude their exploitation to investigate these phenomena in Mg batteries. It is suggested that future works should seek to probe transfer of Mg between the electrode and the electrolyte in carefully controlled conditions to enable the identification and quantification of these contributions. For example, use of EIS/PITT with thin-films of metal oxides with well-defined interfaces with the electrolyte may enable the subtraction of any electrolyte decomposition effects, and allow for extraction of the kinetics of Mg transfer and a greater understanding of the sources of hysteresis during cycling of metal oxides in Mg batteries.
4. Conclusions
The effect of crystallite size on the electrochemistry of α-V2O5 in Mg electrolytes was investigated using Continuous Hydrothermal Flow Synthesis (CHFS). Seeding of the V2O5 using a TiO2 sol in the CHFS process successfully reduced the crystallite size to 25 nm, which reduced the first cycle voltage hysteresis from 1.66 V to 1.08 V, and increased the achievable stable cycle capacity up to 310 mA h g−1 compared to 230 mA h g−1 observed for micrometric V2O5. However, the observed hysteresis on later cycles remained high (0.81 V on the second cycle), showing no improvement in hysteresis compared to micrometric α-V2O5. Therefore, this study reveals that nanosizing α-V2O5 was only partially able to mitigate the energy barriers to Mg insertion and removal, and that these additional sources of hysteresis are likely to contribute to the observed hysteresis. Indeed, mechanical formatting of the α-V2O5 particles appears to significantly contribute to the first cycle hysteresis. Therefore, the conventional assertion that bulk transport of Mg within oxide cathodes limits electrochemical Mg insertion appears to be an oversimplification. As a result, other sources of hysteresis (such as Mg transfer between electrode and electrolyte) should be investigated to advance α-V2O5 towards a practical Mg battery cathode material.Author contributions
IDJ wrote the original draft of the manuscript. IDJ, NS and DB synthesized the materials within this study. IDJ, NS, GN and HDY performed the electrochemistry on the materials within this study, and HDY designed the electrochemical protocols for this work. IDJ, NS, LY and SL performed the XRD analysis of the pristine and cycled electrode materials. GN and JC performed the XAS analysis and interpretation of the pristine and cycled electrode materials. PP and RFK performed the TEM/EDS analysis and interpretation. JAD and JC supervised, administered, and acquired funding support for this project. JC leads the Mg battery research team at the University of Illinois at Chicago. JAD is academic lead of the synthesis team at University College London, developed CHFS process, and is a co-inventor of the CJM mixer that was used in this work. All authors reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
IDJ, NS, DB and JAD would like to thank the EPSRC for funding the JUICED project (EP/R023662/1). IDJ, GN, PP, HDY, LY, BJI, RK, SL and JC were supported as part of the Joint Center for Energy Storage Research (JCESR, a U.S. Department of Energy, Energy Innovation Hub). IDJ would also like to thank the Materials Modelling and Molecular Doctoral Training Centre (EP/G036675/1) and the STFC for providing funding support for travel within the collaboration (STFC/MDC Futures Early Career Award, ST/N002385/1). HDY acknowledges support from the National Research Foundation (NRF-2021R1C1C1005446 and NRF-2018R1A5A1025594) of the Korean Ministry of Science and ICT. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, under Contract No. DE-AC02-06CH11357. Argonne National Laboratory is a U.S. Department of Energy, Office of Science Laboratory operated under Contract No. DE-AC02-06CH11357.References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- G. Berckmans, M. Messagie, J. Smekens, N. Omar, L. Vanhaverbeke and J. Van Mierlo, Energies, 2017, 10, 1314 CrossRef.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS PubMed.
- M. S. Whittingham, Chem. Rev., 2014, 114, 11414–11443 CrossRef CAS PubMed.
- C. Ling, D. Banerjee and M. Matsui, Electrochim. Acta, 2012, 76, 270–274 CrossRef CAS.
- M. Matsui, J. Power Sources, 2011, 196, 7048–7055 CrossRef CAS.
- Z. Takehara, J. Power Sources, 1997, 68, 82–86 CrossRef CAS.
- E. Levi, Y. Gofer and D. Aurbach, Chem. Mater., 2010, 22, 860–868 CrossRef CAS.
- D. Aurbach, I. Weissman, Y. Gofer and E. Levi, Chem. Rec., 2003, 3, 61–73 CrossRef CAS PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- J. L. Andrews, A. Mukherjee, H. D. Yoo, A. Parija, P. M. Marley, S. Fakra, D. Prendergast, J. Cabana, R. F. Klie and S. Banerjee, Chem, 2018, 4, 564–585 CAS.
- N. Sa, H. Wang, D. L. Proffit, A. L. Lipson, B. Key, M. Liu, Z. Feng, T. T. Fister, Y. Ren, C. J. Sun, J. T. Vaughey, P. A. Fenter, K. A. Persson and A. K. Burrell, J. Power Sources, 2016, 323, 44–50 CrossRef CAS.
- M. E. Spahr, P. Novák, O. Haas and R. Nesper, J. Power Sources, 1995, 54, 346–351 CrossRef CAS.
- L. Hu, I. D. Johnson, S. Kim, G. M. Nolis, J. Freeland, H. D. Yoo, T. T. Fister, L. McCafferty, T. E. Ashton, J. A. Darr and J. Cabana, Nanoscale, 2019, 11, 639–646 RSC.
- R. Verrelli, A. P. Black, C. Pattanathummasid, D. S. Tchitchekova, A. Ponrouch, J. Oró-Solé, C. Frontera, F. Bardé, P. Rozier and M. R. Palacín, J. Power Sources, 2018, 407, 162–172 CrossRef CAS.
- I. D. Johnson, B. J. Ingram and J. Cabana, ACS Energy Lett., 2021, 6, 1892–1900 CrossRef CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, 170–193 CrossRef PubMed.
- L. Jiao, H. Yuan, Y. Wang, J. Cao and Y. Wang, Electrochem. Commun., 2005, 7, 431–436 CrossRef CAS.
- L. Jiao, H. Yuan, Y. Si, Y. Wang, J. Cao, X. Gao, M. Zhao, X. Zhou and Y. Wang, J. Power Sources, 2006, 156, 673–676 CrossRef CAS.
- A. Medina, M. Cabello, R. Alcántara, C. Pérez-Vicente and J. L. Tirado, J. Electrochem. Soc., 2020, 167, 070512 CrossRef CAS.
- L. Zhou, Q. Liu, Z. Zhang, K. Zhang, F. Xiong, S. Tan, Q. An, Y. M. Kang, Z. Zhou and L. Mai, Adv. Mater., 2018, 30, e1801984 CrossRef PubMed.
- M. Lopez, H. D. Yoo, L. Hu, J. L. Andrews, S. Banerjee and J. Cabana, ACS Energy Lett., 2020, 5, 3357–3361 CrossRef CAS.
- H. D. Yoo, J. R. Jokisaari, Y.-S. S. Yu, B. J. Kwon, L. Hu, S. Kim, S.-D. D. Han, M. Lopez, S. H. Lapidus, G. M. Nolis, B. J. Ingram, I. L. Bolotin, S. Ahmed, R. F. Klie, J. T. Vaughey, T. T. Fister and J. Cabana, ACS Energy Lett., 2019, 4, 1528–1534 CrossRef CAS.
- I. D. Johnson, G. Nolis, K. McColl, Y. A. Wu, D. Thornton, L. Hu, H. D. Yoo, J. W. Freeland, F. Corà, J. K. Cockcroft, I. P. Parkin, R. F. Klie, J. Cabana and J. A. Darr, Inorg. Chem., 2020, 59, 9783–9797 CrossRef CAS PubMed.
- I. D. Johnson, G. Nolis, L. Yin, H. D. Yoo, P. Parajuli, A. Mukherjee, J. L. Andrews, M. Lopez, R. F. Klie, S. Banerjee, B. J. Ingram, S. Lapidus, J. Cabana and J. A. Darr, Nanoscale, 2020, 12, 22150–22160 RSC.
- L. Hu, J. R. Jokisaari, B. J. Kwon, L. Yin, S. Kim, H. Park, S. H. Lapidus, R. F. Klie, B. Key, P. Zapol, B. J. Ingram, J. T. Vaughey and J. Cabana, ACS Energy Lett., 2020, 5, 2721–2727 CrossRef CAS.
- R. D. Bayliss, B. Key, G. Sai Gautam, P. Canepa, B. J. Kwon, S. H. Lapidus, F. Dogan, A. A. Adil, A. S. Lipton, P. J. Baker, G. Ceder, J. T. Vaughey and J. Cabana, Chem. Mater., 2020, 32, 663–670 CrossRef CAS.
- B. J. Kwon, K. C. Lau, H. Park, Y. A. Wu, K. L. Hawthorne, H. Li, S. Kim, I. L. Bolotin, T. T. Fister, P. Zapol, R. F. Klie, J. Cabana, C. Liao, S. H. Lapidus, B. Key and J. T. Vaughey, Chem. Mater., 2020, 32, 1162–1171 CrossRef CAS.
- S. Okamoto, T. Ichitsubo, T. Kawaguchi, Y. Kumagai, F. Oba, S. Yagi, K. Shimokawa, N. Goto, T. Doi and E. Matsubara, Adv. Sci., 2015, 2, 1500072 CrossRef PubMed.
- T. Hatakeyama, N. L. Okamoto, K. Shimokawa, H. Li, A. Nakao, Y. Uchimoto, H. Tanimura, T. Kawaguchi and T. Ichitsubo, Phys. Chem. Chem. Phys., 2019, 21, 23749–23757 RSC.
- Z. Feng, X. Chen, L. Qiao, A. L. Lipson, T. T. Fister, L. Zeng, C. Kim, T. Yi, N. Sa, D. L. Proffit, A. K. Burrell, J. Cabana, B. J. Ingram, M. D. Biegalski, M. J. Bedzyk and P. Fenter, ACS Appl. Mater. Interfaces, 2015, 7, 28438–28443 CrossRef CAS PubMed.
- J. Yao, Y. Li, R. C. Massé, E. Uchaker and G. Cao, Energy Storage Mater., 2018, 11, 205–259 CrossRef.
- J. Pan, M. Li, Y. Luo, H. Wu, L. Zhong, Q. Wang and G. Li, Mater. Res. Bull., 2016, 74, 90–95 CrossRef CAS.
- D. Su, Y. Zhao, D. Yan, C. Ding, M. Ning, J. Zhang, J. Li and H. Jin, J. Alloys Compd., 2017, 695, 2974–2980 CrossRef CAS.
- M. Lee, S. K. Balasingam, H. Y. Jeong, W. G. Hong, H.-B.-R. Lee, B. H. Kim and Y. Jun, Sci. Rep., 2015, 5, 8151 CrossRef CAS PubMed.
- S. Liang, Y. Hu, Z. Nie, H. Huang, T. Chen, A. Pan and G. Cao, Nano Energy, 2015, 13, 58–66 CrossRef CAS.
- W. G. Menezes, D. M. Reis, T. M. Benedetti, M. M. Oliveira, J. F. Soares, R. M. Torresi and A. J. G. Zarbin, J. Colloid Interface Sci., 2009, 337, 586–593 CrossRef CAS PubMed.
- X. Zhang, J.-G. Wang, H. Liu, H. Liu and B. Wei, Materials, 2017, 10, 77 CrossRef PubMed.
- W. Bi, G. Gao, Y. Wu, H. Yang, J. Wang, Y. Zhang, X. Liang, Y. Liu and G. Wu, RSC Adv., 2017, 7, 7179–7187 RSC.
- Y. Cheng, Y. Shao, V. Raju, X. Ji, B. L. Mehdi, K. S. Han, M. H. Engelhard, G. Li, N. D. Browning, K. T. Mueller and J. Liu, Adv. Funct. Mater., 2016, 26, 3446–3453 CrossRef CAS.
- X. Du, G. Huang, Y. Qin and L. Wang, RSC Adv., 2015, 5, 76352–76355 RSC.
- K. Lee and G. Cao, J. Phys. Chem. B, 2005, 109, 11880–11885 CrossRef CAS PubMed.
- Y. N. Ko, S. H. Choi, Y. C. Kang and S. Bin Park, ACS Appl. Mater. Interfaces, 2013, 5, 3234–3240 CrossRef CAS PubMed.
- G. N. Kryukova, D. O. Klenov and G. A. Zenkovets, React. Kinet. Catal. Lett., 1997, 60, 179–187 CrossRef CAS.
- G. N. Kryukova, G. A. Zenkovets, G. Mestl and R. Schlögl, React. Kinet. Catal. Lett., 2003, 80, 161–169 CrossRef CAS.
- P. Shvets, O. Dikaya, K. Maksimova and A. Goikhman, J. Raman Spectrosc., 2019, 50, 1226–1244 CrossRef CAS.
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321–324 CrossRef.
- M. Abbate, H. Pen, M. T. Czyzyk, F. M. F. de Groot, J. C. Fuggle, Y. J. Ma, C. T. Chen, F. Sette, A. Fujimori, Y. Ueda and K. Kosuge, J. Electron Spectrosc. Relat. Phenom., 1993, 62, 185–195 CrossRef CAS.
- Y. Ueda, F. M. F. de Groot, J. C. Fuggle, K. Kosuge, Y. J. Ma, C. T. Chen, F. Sette, M. T. Czyżyk, M. Abbate, A. Fujimori and H. Pen, J. Electron Spectrosc. Relat. Phenom., 2002, 62, 185–195 Search PubMed.
- X. Sun, L. Blanc, G. M. Nolis, P. Bonnick, J. Cabana and L. F. Nazar, Chem. Mater., 2018, 30, 121–128 CrossRef CAS.
- L. R. De Jesus, G. A. Horrocks, Y. Liang, A. Parija, C. Jaye, L. Wangoh, J. Wang, D. A. Fischer, L. F. J. Piper, D. Prendergast and S. BanerjeeNature Publishing Group, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- L. R. De Jesus, J. L. Andrews, A. Parija and S. Banerjee, ACS Energy Lett., 2018, 3, 915–931 CrossRef CAS.
- J. M. Velazquez, C. Jaye, D. A. Fischer and S. Banerjee, J. Phys. Chem. C, 2009, 113, 7639–7645 CrossRef CAS.
- M. G. Brik, K. Ogasawara, T. Ishii, H. Ikeno and I. Tanaka, Radiat. Phys. Chem., 2006, 75, 1564–1570 CrossRef CAS.
- D. Maganas, M. Roemelt, M. Hävecker, A. Trunschke, A. Knop-Gericke, R. Schlögl and F. Neese, Phys. Chem. Chem. Phys., 2013, 15, 7260–7276 RSC.
- M. Abbate, Braz. J. Phys., 1994, 24, 785–795 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr03080a |
| This journal is © The Royal Society of Chemistry 2021 |