
Two-dimensional materials for electrochemical CO2 reduction: materials, in situ/operando characterizations, and perspective
Zuolong
Chen†
,
Xiyang
Wang†
 ,
Joel P.
Mills
,
Cheng
Du
,
Jintae
Kim
,
John
Wen
and
Yimin A.
Wu
*
,
Joel P.
Mills
,
Cheng
Du
,
Jintae
Kim
,
John
Wen
and
Yimin A.
Wu
*
Department of Mechanical and Mechatronics Engineering, Waterloo Institute for Nanotechnology, Materials Interface Foundry, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada. E-mail: yimin.wu@uwaterloo.ca
First published on 11th November 2021
Abstract
Electrochemical CO2 reduction (CO2 ECR) is an efficient approach to achieving eco-friendly energy generation and environmental sustainability. This approach is capable of lowering the CO2 greenhouse gas concentration in the atmosphere while producing various valuable fuels and products. For catalytic CO2 ECR, two-dimensional (2D) materials stand as promising catalyst candidates due to their superior electrical conductivity, abundant dangling bonds, and tremendous amounts of surface active sites. On the other hand, the investigations on fundamental reaction mechanisms in CO2 ECR are highly demanded but usually require advanced in situ and operando multimodal characterizations. This review summarizes recent advances in the development, engineering, and structure–activity relationships of 2D materials for CO2 ECR. Furthermore, we overview state-of-the-art in situ and operando characterization techniques, which are used to investigate the catalytic reaction mechanisms with the spatial resolution from the micron-scale to the atomic scale, and with the temporal resolution from femtoseconds to seconds. Finally, we conclude this review by outlining challenges and opportunities for future development in this field.
 Zuolong Chen | Zuolong Chen graduated from the University of Toronto in 2020 with a MASc degree and joined Prof. Yimin Wu's group at the University of Waterloo to pursue his doctoral studies in 2021. His research focuses on the syntheses and applications of nanomaterials and thin films for the electrocatalytic and photocatalytic conversions of CO2 to valuable fuels. |
 Xiyang Wang | Xiyang Wang received his BS degree (2014) and Ph.D. degree (2019) in the Department of Chemistry at Jilin University. After that, he worked as a postdoctoral fellow at Jilin University in 2019. Currently, he is working as a postdoctoral fellow under the supervision of Professor Yimin Wu in the Department of Mechanical and Mechatronics Engineering at the University of Waterloo. His research interests focus on the structural design and structure–activity relationships in transition metal oxides (powders and thin films) applied in CO2 reduction and automotive exhaust purification. |
 Joel P. Mills | Joel P. Mills graduated from the University of Waterloo with a Bachelor's degree in Mechanical Engineering in 2020 and currently is a Master student working in catalyst and cell design for CO2 conversion under the supervision of Professor Yimin Wu at the University of Waterloo. |
 Cheng Du | Cheng Du received his Ph.D. in analytical chemistry from Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (2019). He has been a postdoc in Prof. Yimin Wu's group at the University of Waterloo since 2020. He was also co-supervised by Professor John Wen. His research interests include the controllable synthesis of nanomaterials and their application in electrocatalysis and photocatalysis. |
 Jintae Kim | Jintae Kim is a mechanical engineering undergraduate at the University of Waterloo. To pursue his interest in materials engineering, he has joined Prof. Yimin Wu's group at the University of Waterloo as co-operative program student. |
 John Wen | Prof John Wen is a Professor in the Department of Mechanical & Mechatronics Engineering and cross-appointed in the Department of Chemical Engineering at the University of Waterloo. He is the director of the Waterloo Laboratory for Emerging Energy Research (LEER) and has already worked in a multidisciplinary field for developing novel energy technologies focusing on nanotechnology and renewables. Prior to Waterloo he was a postdoctoral fellow at the Massachusetts Institute of Technology where he worked with the Green group at Chemical Engineering for modeling combustion and reaction kinetics. |
 Yimin A. Wu | Prof. Yimin A. Wu obtained his DPhil in Materials from the University of Oxford in 2013. From 2013–2014, he worked as a SinBeRise Postdoctoral Fellow at the University of California and Lawrence Berkeley National Laboratory. From 2014–2018, he worked at Argonne National Laboratory. In 2018, he joined the faculty of University of Illinois and held a joint appointment at Argonne National Laboratory. Since July 2019, he has been a professor at the University of Waterloo. Professor Wu's research focuses on the design of materials interfaces for energy and enivormental materials; novel electronic, photonic, responsive materials; and energy efficient neuromorphic computing. |
1 Introduction
The historically unprecedented atmospheric CO2 levels of current times have resulted in global warming and extinction risks to many animal and plant species.1,2 Therefore, the fixation and conversion of CO2 is critical in realizing energy and environmental sustainability. Over the past decades, many CO2 reduction reaction (CO2RR) strategies have been developed to produce high-value fuels or commodity chemicals to address this problem. Among these approaches, the electrochemical CO2 reduction (CO2 ECR) is a well-studied strategy that employs electrochemical electrolyzers to convert CO2 to value-added fuels using renewable energy and electrochemical catalysts. Compared to other CO2 conversion approaches, CO2 ECR has attracted great attention due to many of its advantages: (1) mild work environment at room temperature and atmospheric pressure, (2) the ability to yield various high-value-added chemicals, especially HCOOH, CO, CH4, CH3OH, CH3CH3OH, C2H4, etc. (3) tunable chemical reactions – parameters such as electrolytes, operating voltage, and reactants can be easily adjusted based on the desired reactions, (4) the potential to utilize renewable energy as the energy source and industrial waste as the reactants, and (5) variety of available options for reactor designs.3–5 However, in CO2RR, molecule CO2 is chemically inert that the activation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in CO2 requires high overpotentials and high energy at 750 kJ mol−1.6 Further complicating the reaction, the highly intricate electrochemical CO2 reduction processes occur in an aqueous environment and involve multiple electrons and protons transfer in various reaction paths, producing a wide variety of chemical products.6,7 To this day, the mass production of multicarbon CO2RR products, such as ethanol and ethylene, is still challenging and far from real-life applications, as only the catalysts with superior morphological and electronic structure are capable of achieving such highly demanded reactions.6,8,9 The adsorption, interaction, and desorption of CO2 and multiple reaction intermediates on catalyst surfaces are the key steps in CO2 ECR processes and are determined by such catalyst properties.10 Therefore, engineering low-cost, green electrocatalysts with excellent durability, high activity, and desired selectivity is crucial in developing the next-generation CO2 reduction reaction (CO2RR) applications.
O bond in CO2 requires high overpotentials and high energy at 750 kJ mol−1.6 Further complicating the reaction, the highly intricate electrochemical CO2 reduction processes occur in an aqueous environment and involve multiple electrons and protons transfer in various reaction paths, producing a wide variety of chemical products.6,7 To this day, the mass production of multicarbon CO2RR products, such as ethanol and ethylene, is still challenging and far from real-life applications, as only the catalysts with superior morphological and electronic structure are capable of achieving such highly demanded reactions.6,8,9 The adsorption, interaction, and desorption of CO2 and multiple reaction intermediates on catalyst surfaces are the key steps in CO2 ECR processes and are determined by such catalyst properties.10 Therefore, engineering low-cost, green electrocatalysts with excellent durability, high activity, and desired selectivity is crucial in developing the next-generation CO2 reduction reaction (CO2RR) applications.
The demand for high-efficiency catalysts for electrochemical CO2RR has stimulated tremendous research efforts in the field.9 Over the past decades, the research focus shifted from bulk materials to nanomaterials and then from nanomaterials to advanced two-dimensional materials. Early works on the subject focused on metal bulk materials such as Cu, Au, Pd, and Ag, which all demonstrated the catalytic activities of such catalysts to convert CO2 into hydrocarbons, CO, methanol, etc. However, the activity, durability, and selectivity of these catalysts are low for CO2 reduction to meet practical demand.11–15 To address these shortcomings, the rapid development of CO2RR nanocatalysts has improved the performance of CO2RR in the catalytic activity, selectivity, and stability due to a large amount of surface active sites of nanocatalysts.16–20 However, the current state-of-the-art systems are still far from practical applications.
Ultra-thin two-dimensional (2D) catalysts with atomic-scale thicknesses have demonstrated outstanding capabilities and performance as CO2 ECR catalysts. Compared with nanoparticles and their bulk materials, 2D materials are excellent catalyst candidates, owing to their intrinsic advantages such as ultrahigh specific surface area, abundant exposed surface atoms, atomically flat facet, and high electrical conductivity.3,21,22 Many structural, chemical, and electronic properties can be fine-tuned based on the desired reaction paths and products via various strategies, including defect engineering, doping, surface structural modification, and surface functionalization.23–27 On the other hand, the study of 2D catalysts for CO2RR can also help us understand the relationships between active sites and the catalysis mechanism and performance at the atomic scale.3 Hence 2D solids are usually considered as an ideal model catalyst to study CO2RR mechanisms. The emergence of 2D materials offers a promising opportunity for the further development and applications of electrochemical CO2 reduction.
Meanwhile, in situ and operando characterizations have become the essential tools to understand CO2 RR mechanisms and evaluate catalyst performance. The electrocatalytic reductions of CO2 are extremely complicated: multiple reaction pathways can lead to the generations of various reaction intermediates and products, requiring the transfer of multiple protons and electrons. During CO2 reduction reactions, advanced in situ and operando characterizations can monitor the generations of both reaction intermediates and products, identify catalytic active sites under the real time, probe the state and properties of catalysts, and reveal the reaction pathways in dynamic reaction environment.28,29 While many such advanced techniques have been developed and widely utilized for CO2 ECR studies, new concepts and techniques have attracted much attention for the discoveries of novel catalysts and studies of fundamental reaction mechanisms.
In this review, we summarize the research progress on four types of representative 2D materials employed in electrochemical CO2RRs: (1) transition metals (TMs), (2) transition metal oxides (TMOs), (3) transition metal dichalcogenides (TMDs), and (4) other types of 2D materials (MXenes, metal organic framework, covalent organic frameworks). Then, we describe current regulating strategies and structure–activity correlations of 2D materials for electrocatalytic CO2RR. In addition, several state-of-the-art in situ and operando characterization techniques are reviewed. Finally, this review will conclude with a perspective on upcoming challenges, opportunities, and trends of 2D materials of future 2D electrochemical CO2 reduction catalysts research.
2 Nanostructured two-dimensional materials for CO2 electroreduction
To date, numerous two-dimensional (2D) materials have been developed and utilized in CO2 ECR applications, leading to many exciting and promising findings and material candidates. In general, there are five categories of 2D materials that have demonstrated promising CO2 ECR performance: a. 2D transition metals, b. 2D transition metal oxides, c. 2D transition metal dichalcogenides (TMDs), d. 2D carbon-based materials, and e. metal–organic frameworks and covalent organic frameworks. Fig. 1 shows representative transmission electron microscopy (TEM) and atomic force microscopy (AFM) images of these five categories of 2D materials reviewed in this article.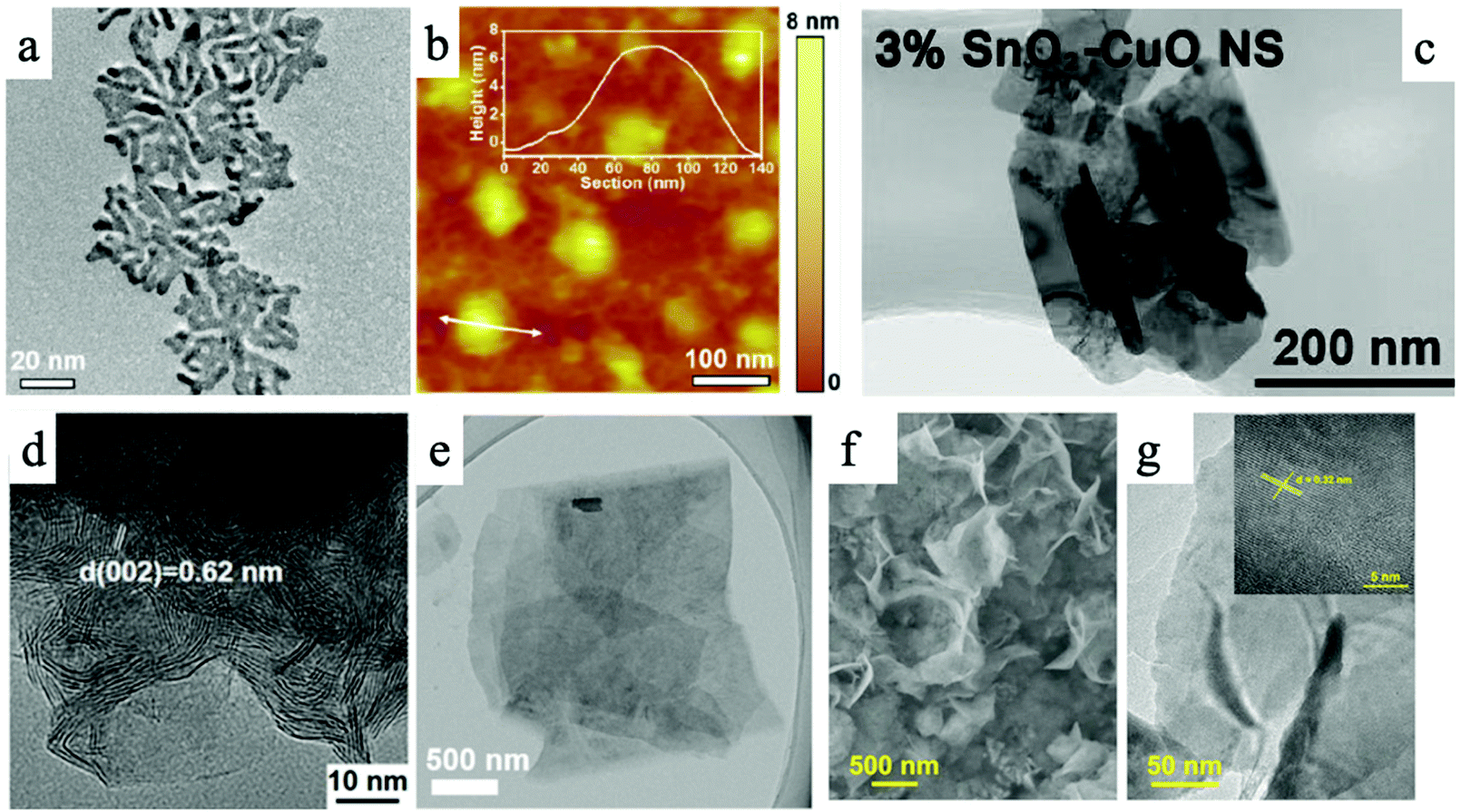 | ||
| Fig. 1 An overview of different types of 2D materials. (a) and (b) TEM and AFM images of 2D palladium–copper alloy nanodendrites; (c) SnO2 modified two-dimensional CuO nanosheet; (d) TEM image of MoS2 nanosheet; (e) TEM image of Ultrathin 2D Mxene. (f) and (g) TEM images of Bi-MOF ultrathin nanosheet.30–34 | ||
2.1 Two-dimensional 2D transition metals (TMs)
Transition metals have been extensively studied for CO2RR for decades.35 Notably, transition metal nanosheets are highly effective electrocatalysts for CO2 ECR due to their abundant active sites and high conductivity.36 Such advantages can benefit CO2 ECR via different mechanisms. By optimizing the electronic properties, Bi nanosheets have exhibited high selectivity towards formic acid in CO2RR and a high faradaic efficiency of 92% in 0.5 M KHCO3 solution at ∼1.5 V (versus SCE). Such 2D nanosheets also generated lower onset potential and about five times higher current density compared to commercial Bi nanoparticles.37 Density functional theory (DFT) calculations proved that 10 nm-thick Bi nanosheets have a higher density of states (DOS) at the Fermi level than bulk Bi, leading to improved CO2RR performance with high selectivity and excellent stability for electrocatalysis CO2 reduction.38 The morphological advantages of 2D TMs have also enhanced CO2 ECR performance. DFT calculations for Bi nanosheets synthesized via a molten-salt-assisted method suggested that the edge sites of Bi nanosheets demonstrate superior activity than terrace sites.39 In terms of surface properties, Bi nanosheets obtained by liquid-phase exfoliate method exhibited greater hydrophilicity than Bi bulk materials, thus promoting the electron transfer between solid and liquid. These Bi nanosheets produced a high current density of 16.5 mA cm−2 at −1.1 V (versus RHE), a faradaic efficiency of 86.0% for formate production, and well-preserved catalytic activity after 10 h of testing.36 In addition, advanced 2D structures have shown promising performance in CO2 ECR. 2D mesoporous Bi nanosheets prepared via the cathodic conversion method demonstrated a large current density of ∼18 mA cm−2 at −1.1 V, a high faradaic efficiency for formate conversion of almost 100%, and an excellent durability of 12 h.40 A recent report of hydrangea-like superstructure consisting of ultrathin Bi nanosheets demonstrated a faradaic selectivity of over 95% with a wide potential window ranging from −0.78 V to −1.18 V and a high current density of over 300 mA cm2.41In a recent report, 50 nm-thick Ag nanosheets with hierarchical structures demonstrated a large surface area for catalytic reactions and gas transport/diffusion, resulting in a high faradaic efficiency of 95% for CO2 conversion into CO and a current density that is 37 times higher than that of Ag nanoparticles at −0.6 V (versus RHE).42 Multilayered Zn nanosheets also showed high faradaic efficiency of ∼86% for CO product at 1.13 V (versus RHE) and good stability during a 7-hour test. The high density in edge sites in Zn nanosheets resulted in a density 9 times higher than that of bulk Zn foil.43 Ultrathin copper nanosheets prepared by galvanic replacement method also exhibited high selectivity and activity for CO2 reduction, where the optimized hierarchical Cu electrodes possessed a faradaic efficiency of 74.1% for CO product and a current density of 23.0 mA cm−2 at −1.0 V (versus RHE).44 Abundant vacancy defects in ultrathin Cu nanosheets can improve the initial kinetics of CO formation and accelerate the mass and electrons transport in the CO2 reduction process. Moreover, hybrid Cu/Ni(OH)2 nanosheets, which consist of atomically thick Cu nanosheets loaded on Ni(OH)2, also displayed excellent activity, selectivity, and durability (22 hours) in electrocatalytic CO2 reduction.45 The nanosheets provided a large current density of 4.3 mA cm−2 at −0.5 V (versus RHE) and a high faradaic efficiency of 92% for CO product.45 Ultrathin 2D hierarchical Pd/SnO2 nanosheets, namely Pd nanosheets partially capped by SnO2 nanoparticles, displayed a maximum faradaic efficiency (FE) of ∼54.8% at −0.24 V (versus RHE) and high stability for electroreduction of CO2 into CH3OH.46 The enhanced performance of Pd/SnO2 nanosheets could be attributed to the abundance of Pd–O–Sn interfaces. Notably, recent advances in 2D alloys have demonstrated enhanced CO2 ECR performance than their single-component counterparts. A palladium–copper alloy nanodendrite system exhibited a high Formate selectivity of over 90% at a low cathodic over potential of −0.4 VRHE.30Fig. 1a and b demonstrate the morphology of this nanodenrite system. DFT calculations revealed that Cu atoms can weaken the *CO adsorption to avoid CO surface poisoning and promote the *OCHO adsorption on neighboring Pd atoms.30 Such synergistic effects shall inspire more research endeavors in 2D alloy systems in the future.
Ultrathin metal nanosheets are prone to oxidation, which leads to a rapid decay in cyclability. However, many studies have also demonstrated through theory calculations and various characterizations that MOx layer on the surface of transition metals plays a vital role in electrocatalytic CO2.47–49 Recently, Xie et al. successfully prepared 4-atoms-thick Co nanosheets (∼0.84 nm) with different surface structures by a ligand-confined growth strategy.50 Partially oxidized Co nanosheets showed an onset potential of −0.68 V for formate production in 0.1 M Na2SO4 solution, a high current density of 10.59 mA cm−2, a faradaic efficiency of 90.1% towards formate, and durability of over 40 h.50 Furthermore, catalytic activity for these 4-atom-thick Co nanosheets showed a 260-fold improvement compared to bulk metal Co, stemming from a large number of surface active sites in ultrathin 2D nanosheets and the existence of surface Co oxides. Partially oxidized Co nanosheets with a smaller Tafel slope of ∼59 mV dec−1 show a fast pre-equilibrium for the formation of intermediate CO2˙−, suggesting that surface Co oxide in nanosheets can accelerated CO2 activation and facilitate the rate-determining step for H+ transfer in chemical reaction.50
2.2 Two-dimensional 2D transition metal oxides (TMOs)
Transition metal oxides have been extensively studied for usage in photoelectrochemical and electrochemical CO2 reduction reactions because of their controlled structure, tunable energy band gap, and low cost. Their unfilled orbitals and active d electrons can energetically facilitate the bonding between metal atoms and CO2 molecules.51 However, traditional bulk transition metal oxides’ low specific surface area and poor electrical conductivity are two key drawbacks that limit their activity and stability in CO2 reduction.52 Therefore, the emergence of two-dimensional (2D) transition metal oxides provides a promising opportunity to develop CO2 reduction reaction. It has been reported that 3D hierarchical SnO2 nanosheets, at a moderate overpotential of 0.88 V, demonstrated a high faradaic efficiency of 87% for the production of formate and a partial current density of 45 mA cm−2.53 This outstanding performance stemmed from the large surface area and fast electron transfer in 2D nanosheets. 1.72 nm-thick Co3O4 nanosheets prepared by solvothermal reaction also exhibited high activity for electrocatalytic CO2 reduction that the current density reached 0.68 mA cm−2 at −0.88 V (versus SCE).53 The formate faradaic efficiency was over 60% in 20 h.52 Based on this, engineered Vo-rich single-unit-cell layer Co3O4 possessed higher current densities of 2.7 mA cm−2 and 85% selectivity for formate product at −0.87 V (versus SCE).54 DFT calculations and experiments demonstrated that abundant oxygen vacancies could promote the adsorption of CO2 on Co3O4 and the desorption of HCOO−, while further lowering the rate-limiting activation energy, leading to improvement in activity and durability in the catalytic reaction. Some other researchers also found that ZnO nanosheets with rich oxygen vacancies display a high current density of −16.1 mA cm−2 at −1.1 V (versus RHE) and a high faradaic efficiency of 83% for CO production compared with ZnO nanosheets and ZnO nanosheets with poor oxygen vacancies.55 DFT calculations and various characterizations clarified that large surface specific area and rich oxygen vacancies could increase surface active sites and facility the activation of CO2 molecular.55 Up to now, many other ultrathin transition metal oxide nanosheets made of TiO2,56 WO3,57 Bi2WO6,58 ZnGa2O4,59 SnNb2O6,60 or BiVO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 61 have been successfully prepared via a variety of methods and these 2D materials all also exhibited excellent performance for photocatalytic CO2 reduction. Some optimization strategies, such as doping, have also demonstrated enhancing effect on CO2 ECR performance using 2D metal oxide catalysts. By modifying 2D CuO with SnO2, Lan et al. recently reported a high faradaic efficiency of 22% for C2H4 at a low over potential of −1.0 VRHE.31 The outstanding performance was attributed to the facilitated CO2-substrate bonding on Sn modified CuO. Fig. 1c shows the morphology of the catalyst studied in the research.31
61 have been successfully prepared via a variety of methods and these 2D materials all also exhibited excellent performance for photocatalytic CO2 reduction. Some optimization strategies, such as doping, have also demonstrated enhancing effect on CO2 ECR performance using 2D metal oxide catalysts. By modifying 2D CuO with SnO2, Lan et al. recently reported a high faradaic efficiency of 22% for C2H4 at a low over potential of −1.0 VRHE.31 The outstanding performance was attributed to the facilitated CO2-substrate bonding on Sn modified CuO. Fig. 1c shows the morphology of the catalyst studied in the research.31
2.3 Two-dimensional 2D transition metal dichalcogenides (TMDs)
Transition-metal dichalcogenides (TMDs) are a promising class of two-dimensional materials which are broadly applied in electrocatalytic reactions because of their unique metal–sulfur covalent bond, atomically flat facet, and high specific surface area.21 Notably, WS2 and MoS2 are two representative 2D TMDs electrocatalysts for CO2 reduction, and the main products in such systems are CO chemicals.62 A representative TEM image of MoS2 nanosheet is shown in Fig. 1d.32 To note, the use of 2D TMDs electrocatalysts, unlike other types of materials, requires an ionic liquid (1-ethyl-3-methylimidazolium tetrafluoroborate, EMIM-BF4) in the electrolyte. The ionic liquid plays a crucial role in forming the [EMIM-CO2]+ complex in acidic environments, which could effectively suppress the HER side reaction.62,63 Abbasi et al. demonstrated that metal-terminated edges in TMDs are mainly responsible for the remarkable dominated catalytic activity in CO2 reduction due to high density of d-electrons and metallic character via DFT calculations. A high spin-polarized partial DOS for Mo atoms at edge sites of MoS2 could improve the catalysis efficiency of CO2 reduction, and the rate-determining step in CO2 reduction is the desorption of CO* species adsorbed on edge Mo atom of MoS2. It is found that the electron-transfer properties in 2D transition metal dichalcogenides determined the catalytic activity of CO2 reduction.64 Doping 5% Nb element into MoS2 could facilitate the shift of the d-band center of edge Mo atom toward a less negative value, leading to a weakened binding of CO and 1 order improvement for CO turnover frequency. Asadi et al. evaluated the CO2 electrocatalytic activities of a series of 2D TMDs, including MoS2, WS2, MoSe2, and WSe2 nanoflakes and revealed that the electron transfer properties of TMDs are critical in enhancing their CO2 reduction performances.63 With the lowest work function among all nanoflakes prepared in this study, WSe2 nanoflakes terminated with W atoms exhibited the excellent performance in the conversion of CO2 into CO, showing a high current density of 18.95 mA cm−2, a faradaic efficiency of 24%, and a turnover frequency of 0.28 s−1 for CO formation at −0.164 V (versus RHE).63 Furthermore, ternary TMD alloys demonstrated enhanced CO2 reduction performance than their binary TMD counterparts. Notably, for CO production performed at −1.15 V (versus RHE), MoSeS alloy monolayers displayed a higher faradaic efficiency (45.2%) for CO production than MoS2 monolayers (16.6%) and MoSe2 monolayers (30.5%), and its current density (43 mA cm−2) was about 1.3 and 2.7 times higher than that of MoSe2 and MoS2 monolayers, respectively.65 This improvement is attributed to the optimized d-band electronic structure of Mo atoms in alloys. Such optimization increases density of states near conduction band edge and improves electron transfer.65 Theoretical calculations further illustrated that the off-center charge around Mo atoms not only better stabilized the COOH* intermediate, but also liberated the rate-limiting CO desorption step.65 In this aspect, Nørskov and coworkers found that breaking the linear scaling relations between the binding energies of intermediates could enhance the catalytic efficiency of CO2 reductions and promote the stabilization of intermediates, such as COOH*, CHO*, COH*, and CO*.66 By theoretical calculations, they demonstrated that doped MoS2 can create two binding sites, namely metal and sulfuring binding sites, and thus two different linear scaling relationships.66 The binding of CO and CHO* on the sulfur site can form different moieties: a close-shell “SCO” moiety for CO and an open-shell “SCHO” moiety for CHO*. “SCO” moiety weakens the neighboring S–Ni bond and increases the distortion, while “SCHO” moiety showed little impact on the neighboring S–Ni interactions and minimal distortion energy. This discovery should also fit other metal/p-block materials, where two binding sites are present.Notably, many other ultrathin two-dimensional transition metal dichalcogenides have been applied in photocatalytic CO2 reductions, which offers an important opportunity for photoelectrocatalytic CO2 reduction. One-unit-cell ZnIn2S4 layers with poor zinc vacancies and with rich zinc vacancies were prepared via hydrothermal method to study photocatalytic CO2 reduction.67 ZnIn2S4 with rich zinc vacancies exhibited 3.6 times higher than ZnIn2S4 with poor vacancies in formate yield, and its catalytic activity remain unchanged after 24 h photocatalysis.67 Whereafter, atomically thin CuIn5S8 and that with sulfur deficient were synthesized to measure photocatalytic CO2 reduction. 2D CuIn5S8 generally contained poor selectivity for CO and CH4 from CO2, while those with sulfur deficiencies showed higher activity and selectivity. Experimental results demonstrated that charge-enriched Cu–In dual sites in sulfur-deficient CuIn5S8 could promote the formation of stable Cu–C–O–In intermediate, which made it possess near 100% selectivity towards photocatalytic production of CH4 from CO2.68 This regulation not only changed the reaction pathway to form CH4 instead of CO in CO2 reduction but also decreased the overall activation energy barrier in catalytic reaction. Furthermore, 2D SnS2 nanosheets were used to study photocatalytic CO2 reduction. Partially oxidized SnS2 nanosheets showed excellent catalytic activity for CO production, which near 2.3 and 2.6 times higher than the poorly oxidized SnS2 nanosheets and the SnS2 nanosheets.69 These works confirmed the potential application of untapped two-dimensional transition metal dichalcogenides in photoelectrocatalytic CO2 reduction, which may also achieve high-efficiency conversion of important C2 production.
2.4 Two-dimensional 2D carbon-based materials
Two-dimensional carbon-based materials, such as graphene and g-C3N4, have attracted intense attention in CO2 reduction due to their outstanding electrical conductivity, low cost, and adjustable surface structure.70 Despite these qualities, two-dimensional pristine graphene is relatively chemically inert, and it has a weak absorption for molecular CO2 and poor catalytic activity for electrochemical CO2 reduction. The well delocalized π bonding network in pristine graphene possesses a high activation energy barrier for the endothermic absorption of key intermediates (e.g., COOH*, OCHO*).71 Specifically, the undoped zigzag edge of graphene displays a relatively high energy barrier of ∼1.3 eV for CO2 absorption in electrochemical CO2 reductions.72 Therefore, engineering the electronic structure of graphene is of vital importance to improve electrocatalytic activity. The enhancement approaches will be discussed in later sections.MXenes, a category of emerging two-dimensional solid materials made up of transition metal carbides, carbonitrides, and nitrides, received much attention in the field of electrocatalysis.73 The general formula of MXene is Mn+1XnTx (n = 1, 2, 3), where M is a transition metal, X represents carbon and/or nitrogen, and T denotes the surface terminations, such as O, OH, and F.74 MXenes layers are attached together via van der Waals forces. MXenes materials are generally prepared by selective etching of specific atomic layers in layered precursors (MAX phases), and their surface contains various functional group after treatment. Therefore, the surface termination in MXenes has significant impacts on the electronic and catalytic properties.25,75 Li et al. first predicted the outstanding CO2 capture capability of two-dimensional MXenes from group IV to VI series through density functional theory calculations. Furthermore, they found that the Cr3C2 and Mo3C2 MXenes possess high selectivity and activity for electrocatalytic CO2 reduction, and these surfaces are more inclined to chemisorbing CO2 than H2O.76 In the conversion process of CO2 → CH4, the formation of OCHO˙ and HOCO˙ radical species is a part of the spontaneous reactions in the early hydrogenation steps, and it is considered the rate-determining step for CO2 reduction. DFT results showed that MXenes (Cr3C2 and Mo3C2) with surface functional groups (e.g., –O or –OH) can dramatically decrease the overpotentials in electrocatalytic CO2 reduction compared to bare Cr3C2 and Mo3C2, reducing the energy input from 1.05 and 1.31 eV to 0.35 and 0.54 eV for Cr3C2 and Mo3C2, respectively.76 Chen et al. systematically studied electrocatalytic activity for CO2 reduction reaction on 17 different MXenes with –OH termination groups via the first-principles approach.25 Among these catalysts, Sc2C(OH)2 and Y2C(OH)2 showed the least negative limiting potential of −0.53 and −0.61 V in catalyzing CO2 reduction into CH4.25 Furthermore, they found that the adsorbed species can be better stabilized by the H atoms in –OH− terminated MXenes and that the strong –H binding can suppress the competing HER reaction. Such mechanisms can dramatically improve the activity, selectivity, and stability of electrocatalytic CO2 reduction. Bader charge analyses confirmed that less charge migration in MXene catalysts during the potential-limiting step (*(H)COOH → *CO elementary step) can well promote the catalytic performance for CO2RR.25
Handoko et al. also used DFT calculations to predict the property of two-dimensional MXenes in electrocatalytic CO2 reduction, where W2CO2 and Ti2CO2 were found to be the most promising MXene catalysts with low overpotential and good selectivity for CO2 reduction to CH4.77 DFT results showed that the accessible *HCOOH pathway is more favorable than the *CO route in electrocatalytic CO2 reduction process and that the binding energies of *COOH and/or *HCOOH are the important limiting step of MXene catalysts.77 Furthermore, their study also showed that O-termination functional groups on MXenes have vital effects on stabilizing the reaction intermediates and are the key reasons why MXenes possess good activity and high selectivity for CO2 reduction. Soon afterwards, Xiao et al. also systematically investigated the CO2 reduction reaction in 2D MXenes (M = Sc, V, Cr, Mn, Zr, Nb, Mo, Mo2Ti, Hf, Ta, and W) via DFT calculations.78 They found that the transition metal in catalysts has important effects on the active site of CO2 reduction and that the surface bicarbonate species on MXenes can suppress HER reaction for improving the selectivity of CO2RR. Furthermore, two possible catalytic mechanisms for CO2 reduction into CH4 were also proposed:78
Mechanism I:
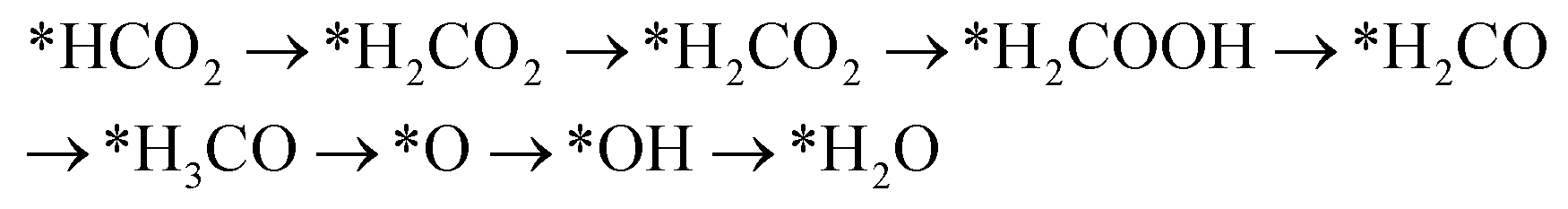
Mechanism II:
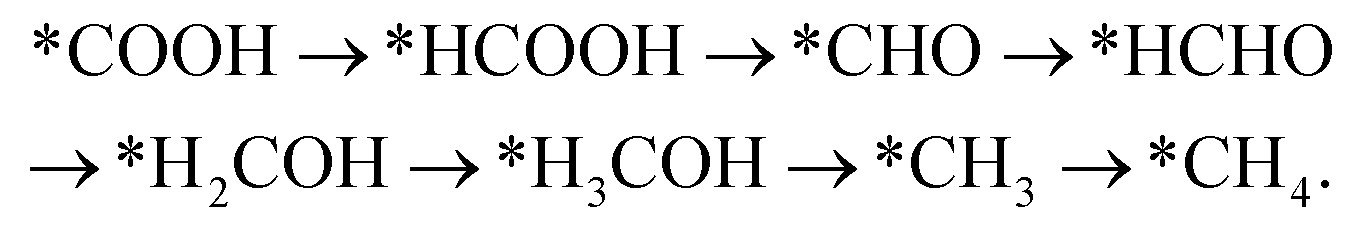
Among these MXenes, Sc3C2, Ta3C2, and Ti3C2 are considered efficient electrocatalysts for reducing CO2 to CH4. Other materials, such as Mo3C2, V3C2, Mo2TiC2, and W3C2, are predicted to possess lower overpotentials of 0.74 V, 0.45 V, 0.17 V, and 0.41 V in CO2RR, respectively.78 In addition, single atom catalyst systems have also been studied with MXene. Recently, single atom copper immobilized MXene Ti3C2Clx demonstrated a high faradaic efficiency of 59.1% for CH3OH.33 X-ray absorption spectroscopy and DFT calculations revealed that single atom copper can lower the energy barrier for the conversion of HCOOH* to absorbed CHO* intermediate due to its unsaturated electronic structure. Fig. 1e shows the TEM image of the 2D MXene with copper single atom.
2.5 Metal–organic frameworks and covalent organic frameworks
Metal–organic frameworks (MOF) composed of metal and organic ligands are a class of porous, crystalline materials and have been intensively studied in various fields due to their tunable structures, vast porosity, and large surface area.79 Notably, ultrathin 2D MOF nanosheets with larger surface area, adjustable chemical compositions and electronic structures, and more uniform pores can provide numerous surface active sites and greatly promote ion and electron transfers, making them highly suitable materials for CO2 conversion applications.80,81 Cui et al. systemically studied CO2 reduction activity for a family of emerging two-dimensional metal–organic frameworks (MOFs) via density functional theory (DFT) calculations.82 They found that Mo-based MOF exhibits good activity of CO2 activation, and the energy barrier in Mo-based MOF for CO2 reduced into methane is only 0.27 eV.82 These corresponding energy inputs in CO2 reduction are substantially lower than traditional pure metal catalysts and some single atom catalysts with noble metal. Recently, a layered, bimetallic 2D conjugated metal–organic framework (2D c-MOF) with copper-phthalocyanine as ligand (CuN4) and zinc-bis(dihydroxy) complex (ZnO4) as linkage (PcCu-O8-Zn) was created and exhibited a high CO selectivity of 88% with excellent durability.83 This performance is attributed to the synergistic effects between highly efficient ZnO4 catalysts and CuN4 centers. This study reveals the feasibility and advantages of utilizing bimetallic MOF catalysts. Similar to MOFs, 2D covalent organic frameworks have also exhibited outstanding CO2 reduction efficiency and selectivity. Lin et al. developed covalent organic frameworks (COFs) composed of cobalt porphyrin catalysts linked by organic struts and demonstrated the advantages of such materials in catalytic CO2 reduction.84 As a result of the π conjugation and π–π stacking in the COF, the charge-carrier mobility is significantly improved, resulting in a high FEco of 90% and a turnover number of up to 290000 at −0.67 V vs. RHE, demonstrating a 26-time increase in activity compared with molecular cobalt complex.84 In addition, other 2D electrocatalysts can also be derived from metal organic frameworks and used for CO2 ECR applications. For instance, 2D bismuthine has been derived from a Bi-based metal organic framework and demonstrated a high faradaic efficiency of more than 96% for formate production with large current densities and stability.34Fig. 1f and g show the morphologies of such MOF-derived structures. This example has provided novel approaches to utilize MOF in the field of CO2 ECR.
3 Regulating strategies for 2D electrocatalysts for CO2 reduction reaction
Many advanced strategies have been introduced to improve the CO2 electrocatalytic performance of two-dimensional materials by increasing the number of active sites and enhancing the activities of active sites. Generally, common approaches include structure and phases engineering, defect engineering, and nano composites.85,86 The desired outcomes of such optimizations are (1) higher current density at low overpotentials towards high-value products, (2) greater selectivity in single products and inhibiting the competitor HER reaction, and (3) enhancing long-term durability in catalytic processes.3 This section will review some advanced strategies to optimize 2D solid catalysts for CO2 electrocatalytic reductions.3.1 Structure engineering
Surface functionalization in solid catalysts is one of the most powerful methods to directly influence catalytic properties via modulating the electric and absorption properties of material surfaces. A recent study by Cheng et al. used a surface nitrogen-injection approach to reach high activity and selectivity for CO2 conversion into formate. The nitrogen abundant Sn nanosheets (N–Sn) showed a 5-times higher current density and a 2.45-times higher faradaic efficiency than that of pristine 2D Sn nanosheets, resulting in a high productivity of 1358 μmol h−1 cm−2.27 The nitrogen abundant Sn surface has a higher CO2 adsorption capacity than pristine Sn surface, because of the coordination between Sn and N atoms. Additionally, the undercoordinated Sn and defect sites created a higher active surface area of 90.79 mF cm−2 in N–Sn, much higher than the 64.18 mF cm−2 found in pristine S nanosheets.27 DFT calculations revealed that the valence electrons of S atoms were increased from 6 to 6.86, suggesting a more positively charged Snδ+ in N–Sn samples that resulted in higher reaction activity and selectivity.27Surface defect engineering has been a hot research topic in heterogeneous catalysis. Current research suggests that the abundant oxygen vacancies in 2D transition metal oxides, such as Co3O4 and Bi2O3, can promote the CO2 adsorption on catalyst surface and accelerate the desorption of products from surface, thus reducing the rate-limiting activation energy and improves the activity and durability in CO2 reduction reaction.87,88 A recent study on oxygen defective Bi2O3 nanosheets revealed that oxygen vacancies can result in a negative adsorption energy of −0.3 eV on the catalyst surface and promote the generation of CO2˙− radicals via a single-electron transfer process.88 The oxygen defective Bi2O3 demonstrated localized electrons around the oxygen vacancies, thus enhancing the interactions with CO2 on catalyst surface.88 The impacts of oxygen vacancies on CO2 ECR performance were also investigated with Co3O4 atomic layers. It was discovered that the oxygen(II) vacancy decreased the activation barrier by stabilizing the HCOO−* intermediates and promoting the proton transfer. Consequently, a high formate selectivity of 85% was achieved in vacancy rich 2D nanocatalyst.87
Furthermore, rich cationic vacancies and sulfur vacancies can promote the formation of the intermediate in CO2 reduction using 2D nanocatalysts, leading to improved selectivity, activity, and stability of CO2 reduction. Peng et al. reported the enhanced CO2 ECR to n-propanol using CuS 2D catalysts with double sulfur vacancies.89 It was discovered that the highly dense double sulfur vacancies in copper sulfide (100) planes can act as electrocatalytic centers to stabilize CO* and OCCO* dimers and promote the CO–OCCO coupling to yield C3 products. This phenomenon was not observed in CuS with single or no sulfur vacancies. Consequently, a high faradaic efficiency of 15.4% for n-propanol was observed.89 In addition, some other research showed that defect engineering in graphene is of vital importance for tuning the electronic structure and catalytic performance.90,91
The morphologies of 2D materials can play an important role in determining the CO2 ECR performance. Structure engineering can significantly enhance CO2 ECR performance by modulating the number of exposed active sites and affecting the chemisorption. Using a halogen-assisted calcination approach, Shu et al. synthesized 2D nitrogen-doped carbon from ZIF-8 precursor with a large electrochemical surface area of 788 to 1413 m2 and a high G–N ratio of 42.27 to 55.06%.92 When the surface area increased from 788 m2 g−1 in the N/C-1100 sample to 1413 m2 g−1 in the N/C–Cl-1100 sample, the resulted current density showed a pronounce improvement, and the CO selectivity increased from 91.2% at −0.7 V to 99.5% at −0.5V.92Fig. 2a shows the FE of catalysts with different surface areas. Additionally, the highest G–N ratio was also found in the N/C–Cl-1100 sample, suggesting a high active sites exposure. It was proven in this study that the increased G–N exposure can significantly promote the CO2 electrochemical conversion to CO. This significant enhancement demonstrated the effect of surface area and active site exposure on the catalytic performance in 2D materials.
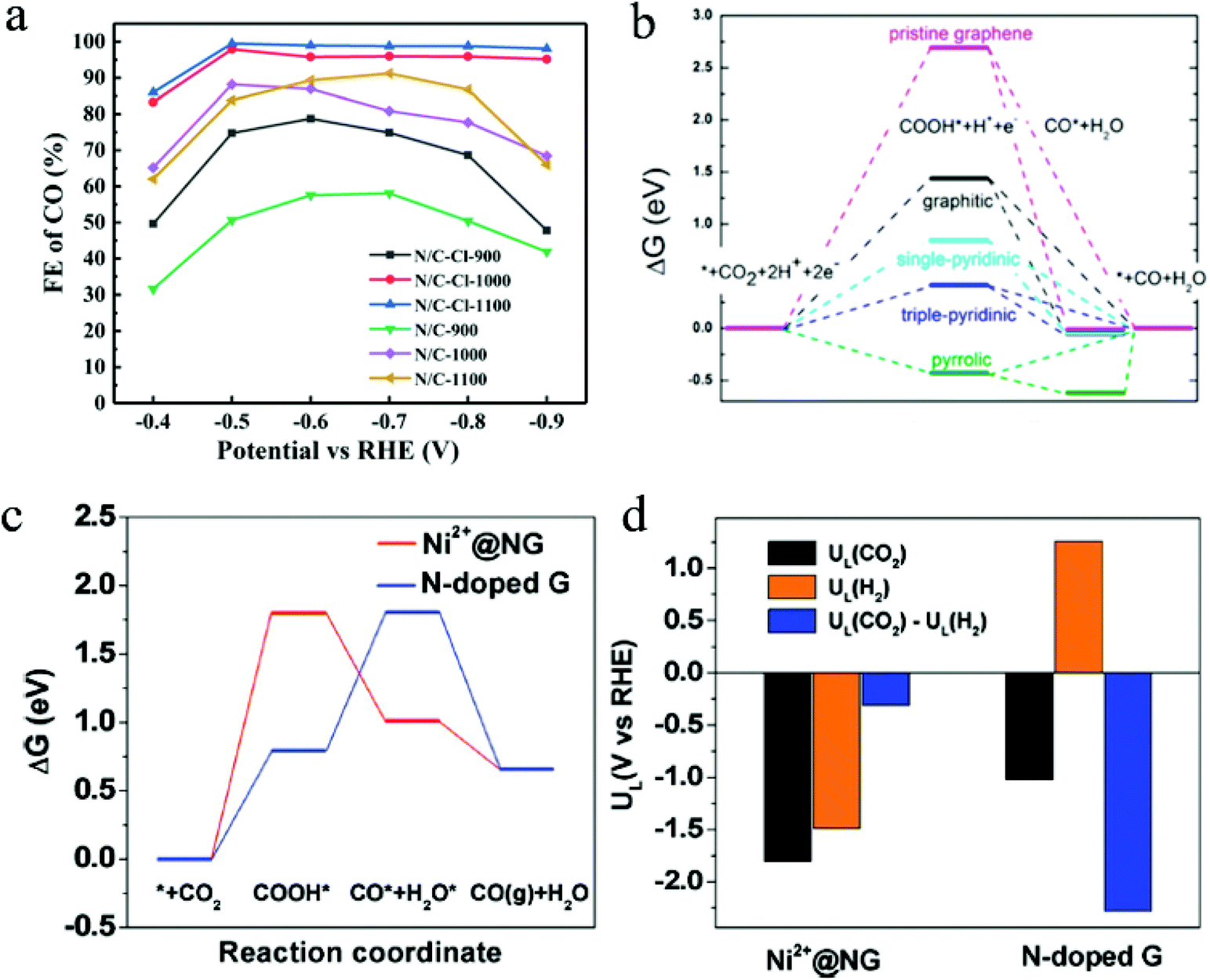 | ||
| Fig. 2 (a) FE of catalysts with different surface areas. Copyright 2021, Springer Nature; (b) free energy diagram of CO2 ECR to CO on different NG surfaces. Copyright 2016, American Chemical Society; (c) free energy diagrams and (d) the calculated limiting potentials for CO2RR and HER on composite and pristine graphene surfaces. Copyright 2018, John Wiley and Sons. All panels reproduced with permission.23,92,96 | ||
Modulating the exposure of active edges sites is another effective approach to enhance CO2 ECR performance. DFT calculations have suggested that edge sites are highly active catalytic sites for CO2 ECR using nanoparticles, calling for tunable 2D structures with high edges-to-corner site ratios.93 Layered 2D structures have demonstrated superior performance to their bulk counterparts in CO2 ECR, due to the exposure of more edges and defects for catalytic reactions.85,94 Recently, Li et al. synthesized layered Sb nanosheets by cathodically exfoliating bulk Sb crystals and demonstrated enhanced CO2 ECR to formate performance.94 With the same layer thickness, smaller Sb nanosheets with an average lateral size of 170 nm showed a 3.6-times higher peak current (mass loading normalized) than showed in larger Sb nanosheets with an average lateral size of 485 nm at a potential of −0.96 V, resulting in a FE of 84% for formate at −1.06 V.94 The morphology-dependent CO2 ECR performance of nanoplates was also observed with triangular silver nanoplates. Liu and coworkers discovered that Ag nanoplates demonstrated higher faradaic efficiency and energy efficiency than their Ag nanoparticle counterparts with similar sizes.93 This enhancement was attributed to the larger amount of edge sites and higher edge-to-corner ratios in nanosheets, as the adsorption of CO2 and the formation of COOH* are more promoted on edge sites.93
The exposure of different facets is also a critical parameter in CO2 ECR that determines the CO2 ECR efficiency and selectivity. Surface reconstruction has become an effective approach to program the desired exposure of facets, thus enhancing the CO2 ECR of 2D materials. A recent work demonstrated that the preferred exposure of Bi (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) facets causes an increased FE of 90% for formate at a high current density of 200 mA cm−2, due to the lower reaction energy barrier (0.03 eV) for CO2 ECR exhibited on the Bi (10) facet than the energy barrier (0.74 eV) on the Bi (121) facet.26 Moreover, the stepped Bi (10) surface also showed a Gibbs free binding energy of −0.01 eV for HCOO*.26 This value is very close to the thermodynamic minimum. The facet-dependent performance was also observed in other nanosheets. For Ag nanoplates, the Gibbs free energy (ΔG) for forming COOH* was found to be lower on Ag (100) than on Ag (111), making Ag (100) the preferred facet for CO2 ECR using nanoplates.95
0) facets causes an increased FE of 90% for formate at a high current density of 200 mA cm−2, due to the lower reaction energy barrier (0.03 eV) for CO2 ECR exhibited on the Bi (10) facet than the energy barrier (0.74 eV) on the Bi (121) facet.26 Moreover, the stepped Bi (10) surface also showed a Gibbs free binding energy of −0.01 eV for HCOO*.26 This value is very close to the thermodynamic minimum. The facet-dependent performance was also observed in other nanosheets. For Ag nanoplates, the Gibbs free energy (ΔG) for forming COOH* was found to be lower on Ag (100) than on Ag (111), making Ag (100) the preferred facet for CO2 ECR using nanoplates.95
In 2D materials, doping is an extremely effective approach to tune the catalytic performance. Unlike in bulk materials, where dopants are located within the materials, doped 2D materials have more dopants exposed on the surface that can affect the overall catalytic performance. Nitrogen doping is a successful approach to enhance the CO2 ECR performance using 2D carbon materials. In a recent study, three types of N dopants on graphene were prepared and investigated by Wu et al.: graphitic N, pyridinic N, and pyrrolic N, where the lowest uphill free energy barrier of COOH* adsorption was observed in samples with triple-pyridinic N dopants.96Fig. 2b shows the free energy diagram of CO2 ECR to CO on different graphenes. It was found that the positively charge C atoms adjacent to N dopants are the active sites for CO2 ECR. Additionally, DFT calculations suggested that N doping can significantly reduce the energy barrier for COOH* adsorption, and the direct protonation of COOH* requires significantly lower energy barrier (−0.5 eV) to form CO gas and H2O than that needed for the Heyrovski step to form H2 (−2.0 eV).96 Thus, this doping approach can greatly promote the CO formation and inhibit the competing HER. Besides, through computational approaches, Liu and coworkers discovered that pyrrolic-nitrogen doped graphene showed a lowest theoretical over potential of 0.24 V for CO2 ECR to HCOOH among all N-doped graphene.97 This result suggests that pyrrolic-nitrogen doping is a promising approach for CO2 ECR to formate. However, the role of active site in N-doped graphene materials is still controversial in CO2 electrochemical reduction reactions. Chai and Guo found that the graphic N is the effective active site in electrochemical CO2 reduction and has a lower activation barrier (0.58 eV).72 Furthermore, Sreekanth et al. also studied CO2 electrochemical reduction of the B-doped graphene in aqueous 0.1 M KHCO3 solutions.98 Their research results showed that boron dopants changed asymmetric charge and spin density distribution of pristine graphene, and B/C atoms with positive spin density in graphene are important active sites.98
3.2 Composite materials and alloys
Engineered composite materials with heterostructures can often achieved enhanced CO2 ECR performance by promoting charge transfer and the coupling effects between different phases. The general regulation parameters mainly involve compositions and alloys. Loading metals/metal oxides on two-dimensional graphene is a very common method to improve the electrical conductivity of metal oxides and increase the active sites of graphene. Bi et al. reported that the surface immobilization of Ni on nitrogen-dope graphene (Ni2+@NG) demonstrated significantly improved CO2 ECR performance with a high current density of 30 mA cm−2, compared with that of pristine N-doped graphene (<10 mA cm−2).23 The FE for CO also experienced a drastic increase from below 60% to 92% in Ni2+ @NG.23 By comparing the free energy barrier for the formation of *COOH, *CO, and *H, the authors found out that Ni2+@NG showed a lower free energy barrier for the formation of adsorbed *CO than N-doped graphene did. Besides, compared with its pristine counterpart, Ni2+@NG composite 2D catalysts exhibited a higher preference towards CO2RR over HER, as suggested by the limiting potential calculations. Fig. 2c and 1d show free energy diagrams on different graphene surfaces and the calculated limiting potentials for CO2RR and HER, respectively.Loading isolated metal atoms on graphene is an emerging regulation strategy in heterogeneous catalysis.99 These composite graphene materials have demonstrate superior CO2RR performance due to their high electrical conductivity, abundant active sites, and maximum atom efficiency. This strategy is regarded as one of the most effective strategies for improving the activity, stability, and selectivity in electrochemical CO2 reduction. Combined with the attributes of single-atom catalysts and 2D materials, some researchers also synthesized an atomically dispersed Fe on nitrogen-doped graphene (Fe/NG), where graphene oxide (GO) absorbed iron ions (a mixture of GO and FeCl3) is annealed in an atmosphere for Ar/NH3 at 700–800 °C.100 Fe/NG catalyst had a large number of nitrogen-confined atomic Fe moieties on the surface as active sites, resulting in a high faradaic efficiency of ∼80% for CO production at −0.60 V versus RHE.100 Theoretical calculations and experimental results suggested that the isolated Fe–N4 moiety in nitrogen-doped graphene as the activity center that enhanced CO2 adsorption and improved CO2 activation. Based on these works, a heterogeneous molecular Co electrocatalyst (NapCo@SNG) was prepared by immobilizing planar CoII-2,3-naphthalocyanine complexes (NapCo) on S/N/O heteroatoms doped graphene (SNG) through axial coordination.101 Sulfoxide dopants in NapCo@SNG can enhance the electron transfer between NapCo and doped graphene, causing an increase of ∼3 times in the turnover frequency of Co sites when compared with carboxyl-NapCo moiety and a high faradaic efficiency for CO production of ∼97% at −0.8 V versus RHE.101 In addition, Cu/N-doped graphene composite materials (p-NG/Cu) were introduced by combining monodisperse Cu nanoparticles and pyridinic-N rich graphene (p-NG).102 The p-NG catalyst demonstrated the reduction activity and selectivity to ethylene compared with Cu and p-NG, with a faradaic efficiency of ∼19% for C2H4 production and a 79% ethylene selectivity at ∼0.9 V versus RHE.102 The interaction between p-NG and ∼7 nm Cu NPs facilitated CO2 reduction, hydrogenation, and C–C coupling in the formation process of C2H4.
Monolayer ternary transition metal dichalcogenide (TMD) alloys have demonstrated enhanced CO2 ECR performance compared with binary TMDs. Xu et al. synthesized MoSeS alloy monolayers with shortened Mo–S and extended Mo–Se bonds that can tune the d-band electronic structure of Mo atom.65 By engineering the band structure in this way, the density of states around the conduction band edge showed a drastic increase, resulting in faster electron transfers due to the lowered work function (4.24 eV) and decreased charge-transfer resistance.65 Moreover, the overlap between the d-orbitals of Mo and the p-orbitals of Se atoms was reduced by the off-center charge density. DFT calculations revealed that the biased electron density around Mo atom in alloy monolayers can help stabilize COOH* intermediates and facilitate the CO desorption. Consequently, at an overpotential of −1.15 V vs. RHE, a high current density of 43 mA cm−2 and a high faradaic efficiency of 45.2% for CO were observed in ternary TMD alloy monolayers.65 Nevertheless, the low FE still needs to be improved, and the overpotential needs to be lowered to inhibit the competing HER.
4 Electrolyzers for electrocatalytic CO2 reductions
Electrolyzers also play a decisive role in determining the CO2 reduction performance of 2D materials. So far, various electrolyzer configurations have been developed and utilized in CO2 ECR applications. The H-type cell, a simple configuration that employs an ion-exchange membrane to separate anodic and cathodic reactions, has been a popular electrolyzer configuration for 2D catalyst development and screening.26,89,103,104 Catalysts can be deposited on carbon paper to form the cathode for CO2 ECR, and the reaction products are collected directly from the cells to evaluate CO2 ECR performance. Nevertheless, the use of H-cells is greatly limited by their low current densities (<100 mA cm−2) because of the low solubility of CO2 in liquid electrolytes.105 In order to solve this issue, liquid-phase flow cells have been developed to directly use CO2 gas as the CO2 source in CO2 ECR. Gas diffusion electrodes are applied in this configuration to allow for gas-phase CO2 diffusions through the porous electrode and reactions taking place on the catalyst–electrolyte interface.106 Ion exchange membranes, i.e., anion exchange membrane, cation exchange membrane, and bipolar membrane, are utilized to separate the anodic and cathodic chambers. Improved CO2 ECR stabilities, current densities, and faradaic efficiencies have been widely reported with the use of flow cells.105,107 In addition, modified H-cell and flow cell configurations are the most common reaction cells for in situ and operando investigations.27,28,108,109 On the other hand, the membrane electrode assembly (MEA) is a novel gas-phase electrolyzer that uses a solid polymer electrolyte to separate the anode and cathode. The catalyst on the gas diffusion electrode is directly pressed against the membrane to form the “zero-gap” configuration that reduces the ohmic losses and maximizes the energetic efficiency.110 Consequently, high current densities (>1 A cm−2) and faradaic efficiencies have been reported using MEA.111–113 However, the operation of gap-phase reactors requires humidity that can be provided either by liquid electrolytes in the anode and/or humidified gas.105 The separation of CO2 ECR products from liquid electrolytes can be eliminated using all-solid-state electrolyzers, which adopt solid electrolytes to facilitate the continuous production of pure liquid products.114 Remarkably, highly concentrated (∼98 wt%) formic acid and acetic acid have been produced with this solid-state electrolyzers.115,116 It can be projected that the developments and optimizations of novel electrolyzers can enhance the CO2 ECR performance of 2D materials and facilitate in situ and operando studies to better understand reaction mechanisms.5 Advanced in situ and operando characterization technology
During CO2 reduction reactions, catalysts are in metastable states and subjected to constant changes in their chemical and structural properties in a complex reaction environment. Such changes include surface reconstruction, catalysts deactivation, agglomeration, phase segregation, metal reconstruction, and vacancies formation.117–123 Many reaction intermediates are formed and consumed during the CO2RR process to yield various final products via different pathways.28 On the other hand, the changes in catalysts’ chemical and structural properties can affect their catalytic performance, determining their effectivity and selectivity.124 As illustrated in Fig. 3,29 the elementary transferring of electrons and protons, the adsorption and desorption of intermediates and products, the mass transfer of molecules in the reaction environment, and other processes in catalytic reactions, all take place in different time and length scales. It is critical to study all such processes in a time resolved manner to gain insightful information about CO2 catalysis. Nowadays, thanks to the developments of highly advanced in situ and operando characterization methods, fundamental studies of CO2RR at various time and length scales have become feasible. In situ characterizations refer to the characterizations of catalysts under conditions relevant to catalytic operation conditions. Such characterizations might not be simultaneous to reactions. However, operando characterizations are simultaneous characterizations of working catalysts during real catalytic reactions.125,126 In this section, state-of-the-art in situ and operando spectroscopy and microscopy methods are reviewed, and future research trends are proposed with the goal of achieving better understandings the mechanism and design guidelines of 2D materials in CO2RR. We summarized the spatial and temporal resolutions of these advanced characterization techniques in Table 1.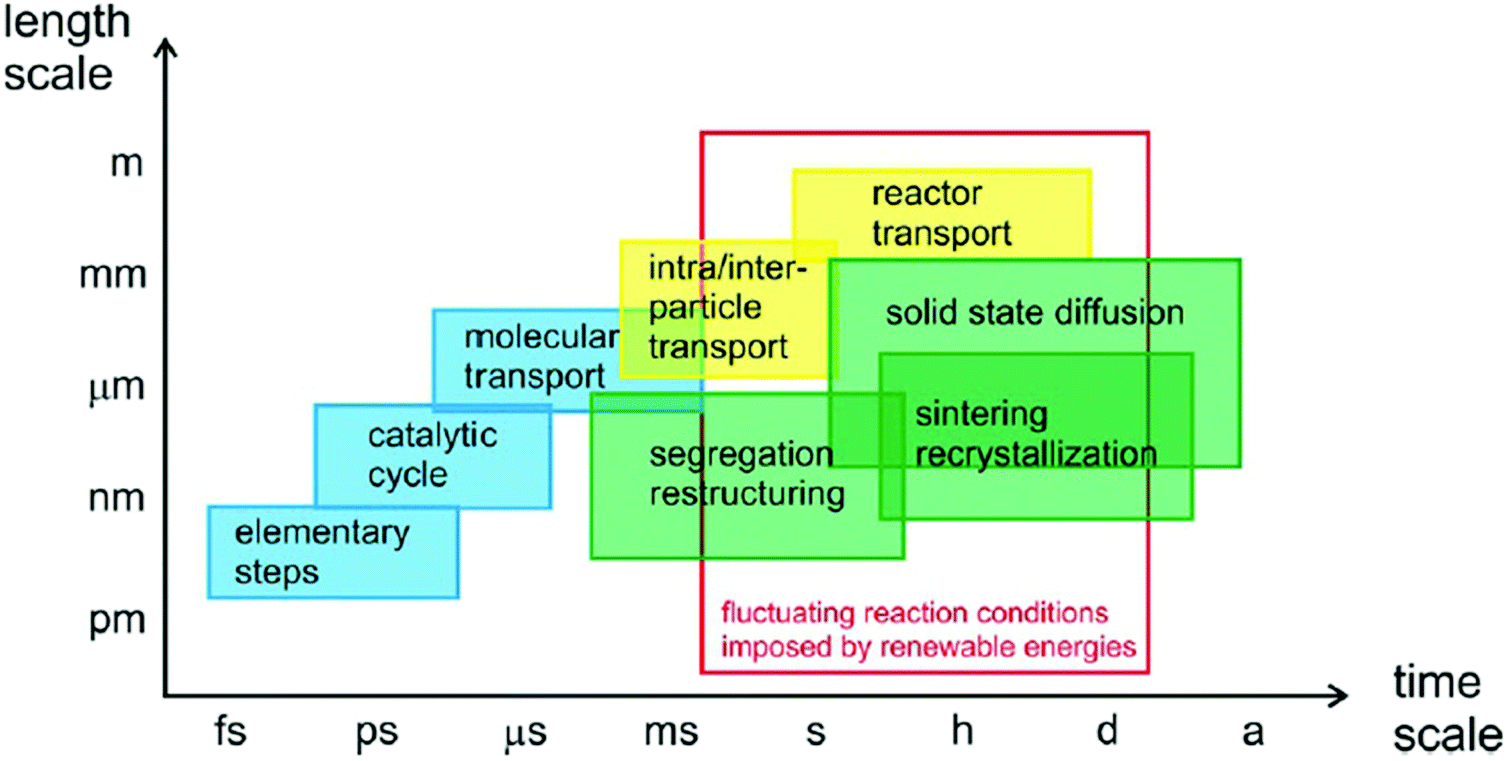 | ||
| Fig. 3 Time scale of various processes in catalytic reactions. All panels reproduced with permission.29 Copyright 2017, John Wiley and Sons. | ||
| Infrared | Raman | Synchrotron X-Ray | FEL X-Ray | Mass spectroscopy | |
|---|---|---|---|---|---|
| In situ and operando spectroscopy | |||||
| Temporal resolution | <1 ms | <1 ms | <1 ns | <10 fs | s |
| Spatial resolution | 1 μm | 1 μm | <15 nm | <15 nm | >20 μm |
| TEM | SECM | STM | AFM | STXM | CDI | |
|---|---|---|---|---|---|---|
| In situ and operando microscopy and imaging | ||||||
| Temporal resolution | 200 fs | 100 μs | 1 μs | 100 μs | <30 ps | 10 fs |
| Spatial resolution | <0.1 nm | 50 nm | 1 nm | 1 nm | 7 nm | 10 nm |
| 10−4 strain | ||||||
5.1 In situ scanning probe microscopy
 | ||
| Fig. 4 (a) SECM operation principle; (b) the SG/TC mode all panels reproduced with permission.128 Copyright 2020, Springer Nature. | ||
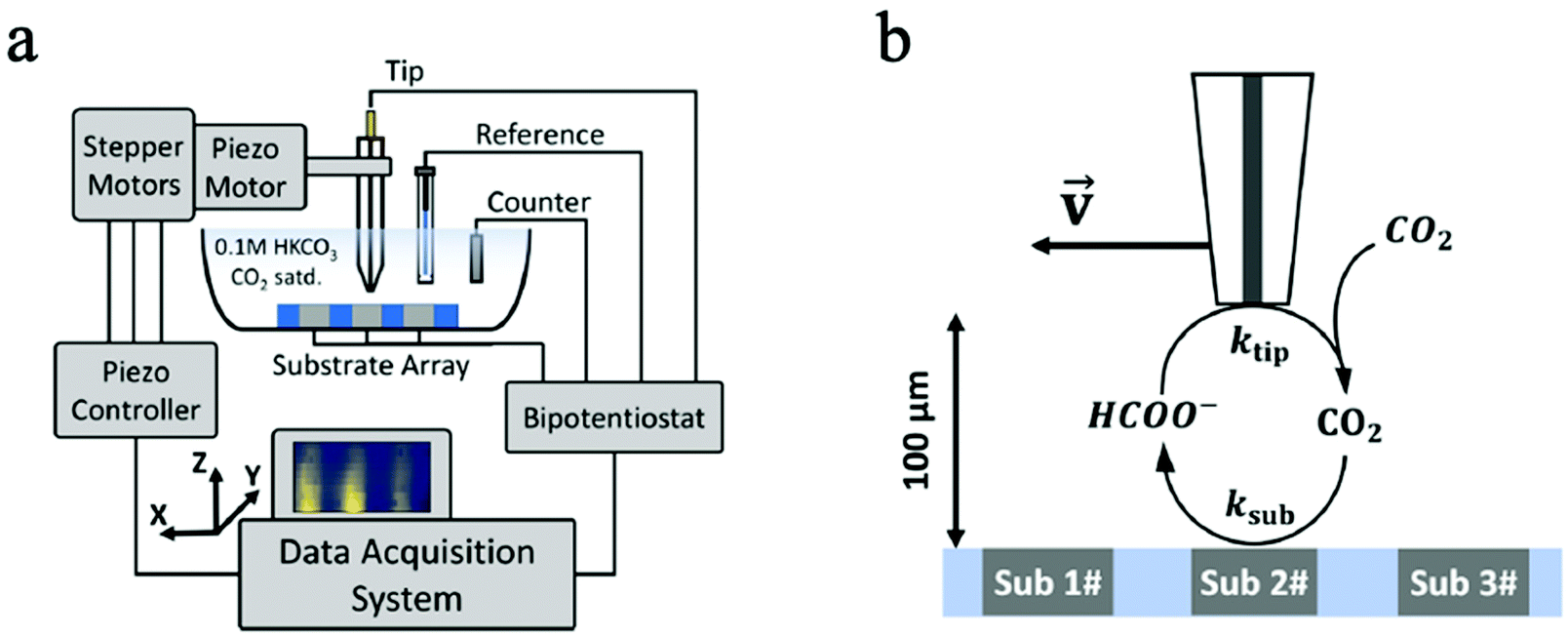
In another CO2 ECR study, Kim et al. employed SECM to monitor the CO2 ECR performance of Au nanoparticles and discovered a high selectivity of CO production without the interference of HER at low overpotentials.133 Likewise, the time-resolved quantitative probing of CO2RR products has also been demonstrated by Shaughnessy et al., who utilized SECM in the Substrate Generation-Tip Collection mode to investigate the potential dependent CO production of a In0−In2O3 composite catalyst.130 Also, Sreekanth et al. employed SECM to detect the CO selectivity of Au catalyst of different surface roughness in different pH environment.129,134 Due to its time-resolved detection ability, SECM was used in TG/SC mode to quantitatively probe the unstable, short-lived CO2˙− intermediate in CO2 ECR for the first time. Such intermediates have a high dimerization rate of 6.0 × 108 M−1 s−1, a short half-life of around 10 ns, and are critical intermediates in CO2RR. The successful monitoring of these intermediates can greatly benefit the studies of fundamental CO2RR mechanisms.131 However, the spatial resolution of this noncontact technique is restricted by the mass transport of products from the sample surface to the probe tip, and therefore the probe size and probe-sample distance are the main limiting factors of in situ ECM.135 Besides, the SG/TC model is also limited by low collection efficiency and interferences between the sample and probe tip.136
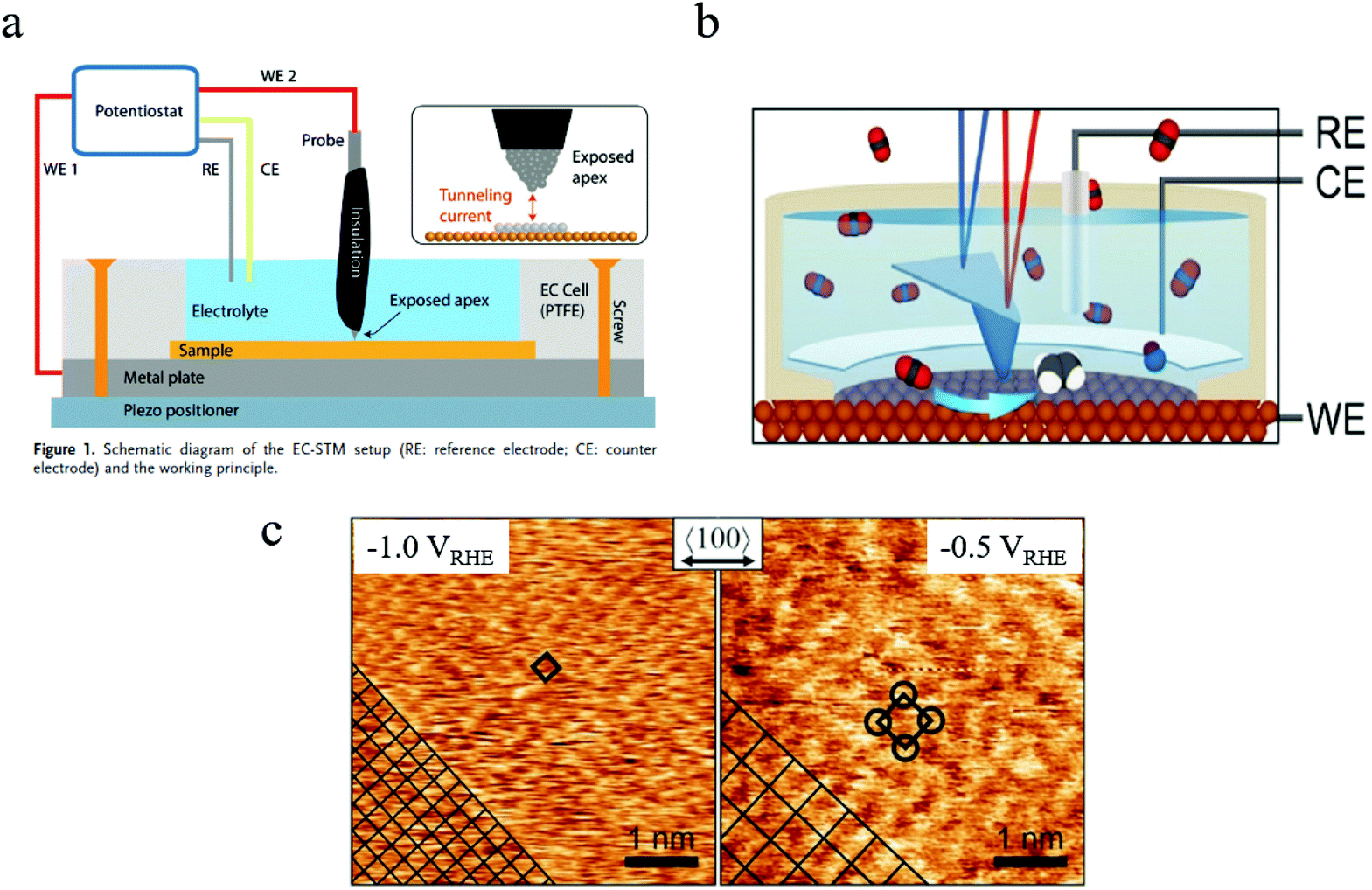 | ||
| Fig. 5 (a) Schematic of in situ EC-STM setup. Copyright 2018, John Wiley and Sons; (b) schematic of in situ EC-AFM; (c) morphology of Cu surface at different potentials characterized by in situ AFM. Copyright 2020, John Wiley and Sons. All panels reproduced with permission.139,143 | ||
5.2 In situ and operando microscopy and imaging
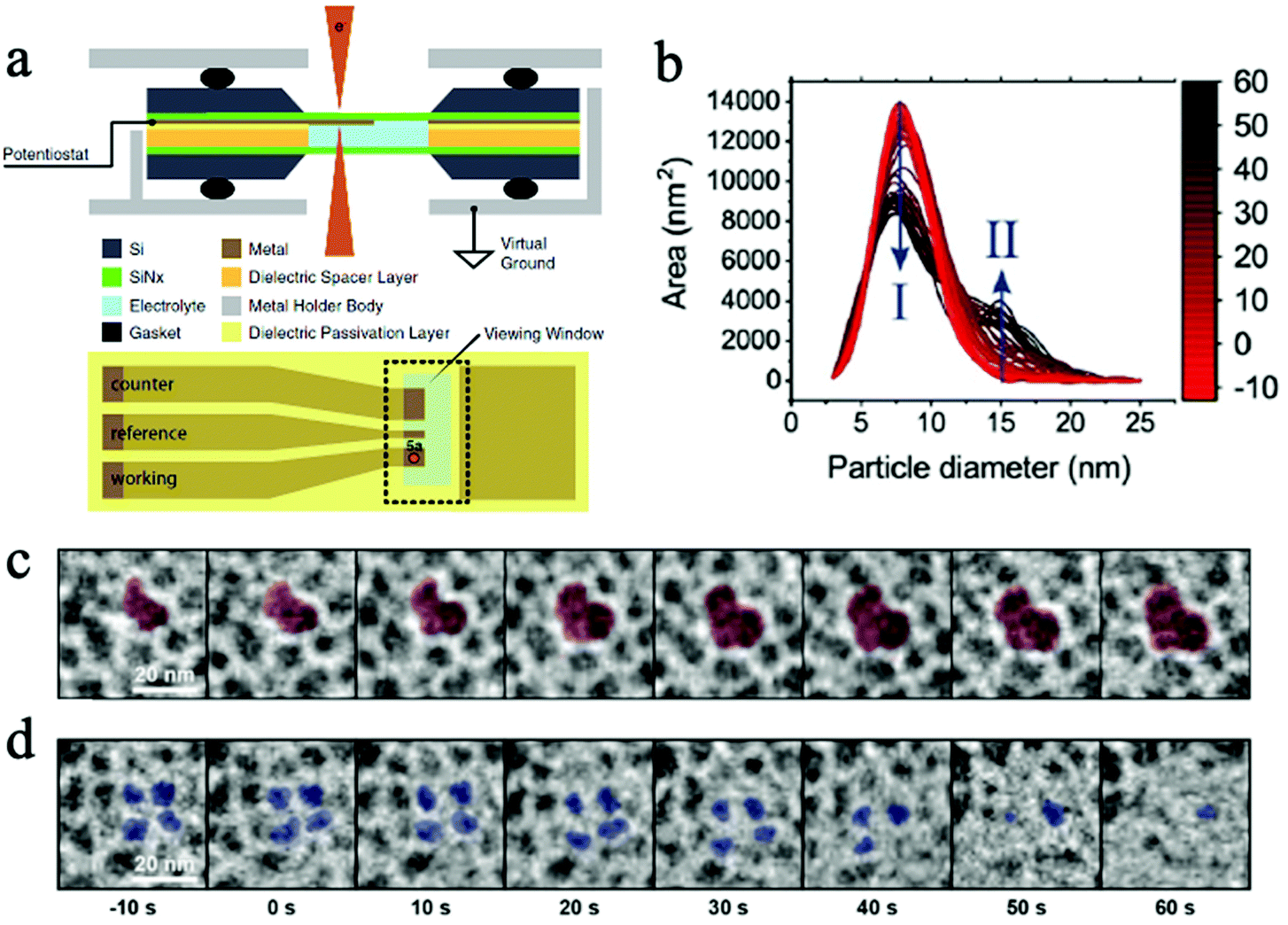 | ||
| Fig. 6 (a) The schematic of a micro electrochemical cell for in situ TEM. Copyright 2017, IOP Publishing, Inc.; (b) the change in primary (I) and secondary (II) particle sizes as experiment progresses; (c) the growth of secondary particles, and (d) the shrinkage of primary particles. Copyright 2020, John Wiley and Sons. All panels reproduced with permission.152,154 | ||
Benefited from operando TEM technology, our group studied the facet-dependent CO2 catalytic reduction behaviors of Cu2O nanoparticles and discovered that the (110) facet of Cu2O nanoparticles is catalytically active in CO2RR, while the (100) facet is not active.148 The operando TEM was used to identify nanoparticles and facets of interest and guided the use of focused electron beam and nano-focused synchrotron X-ray beams to desired facets for localized spectroscopic investigations using electron energy loss spectroscopy (EELS) and X ray absorption spectroscopy (XAS).148 In a recent publication by Wang et al., a micro electrochemical cell (“in situ TEM E-chip cell”) was employed to investigate the real-time morphological changes in 2D CuO nanosheets in a CO2 ECR environment.153 They discovered the potential- and time-dependent conversions of 2D CuO nanosheets into nanoscale fragments, which consecutively agglomerate and form Cu dendrites. The formation of undercoordinated Cu sites was found to promote the CO2 electrochemical conversions into C2+ products. This operando TEM successfully revealed the operation condition–morphology-performance relationship of CO2 ECR using CuO nanosheets as the catalyst. Additionally, Vavra and coworkers recently monitored the dissolution/redeposition of Cu nanocatalyst in CO2 ECR.154 They found that, at the open circuit voltage, the oxidation and the dissolution of Cu nanoparticles took place at the same time, yielding Cu2O and dissolved Cu ions in the initial electrolyte. Later, at higher cathodic potentials, the dissolved Cu+ and Cu2+ ions are reduced back to solid Cu phase, generating larger secondary Cu nanoparticles. It was also proposed that the Cu surface oxide, Cu2O, participated in the reconstruction process of Cu nanocatalyst as the CO2 ECR experiment progressed.154 With the help of time resolved monitoring at the nanoscale, the catalyst phase change mechanisms were investigated. Fig. 6b depicts the change in primary and secondary particles. Fig. 6c and d show the in situ observation of the morphological change in primary and secondary particles.
Nevertheless, there are still some limitations in such in situ and operando TEM characterizations. For example, it is still challenging to get high-quality 3D reconstructed images using in situ and operando TEM. One reason is due to the limited tilting angles inside TEM if using electrochemical cell. The gas bubbles generated in CO2RR might affect the local reaction environment, and this impact is much amplified in the micro reaction system where only a small amount of electrolyte is present.124 Additionally, the CO2 saturation in electrolyte results in deteriorated resolution and lowered contrast, due to inelastic electron scattering in liquids.154 It was also reported in the same publication that a high electron dose can damage the catalysts within a short amount of time. Hence, the electron dose needs to be fine-tuned to avoid damaging the catalysts while achieving an optimal resolution.
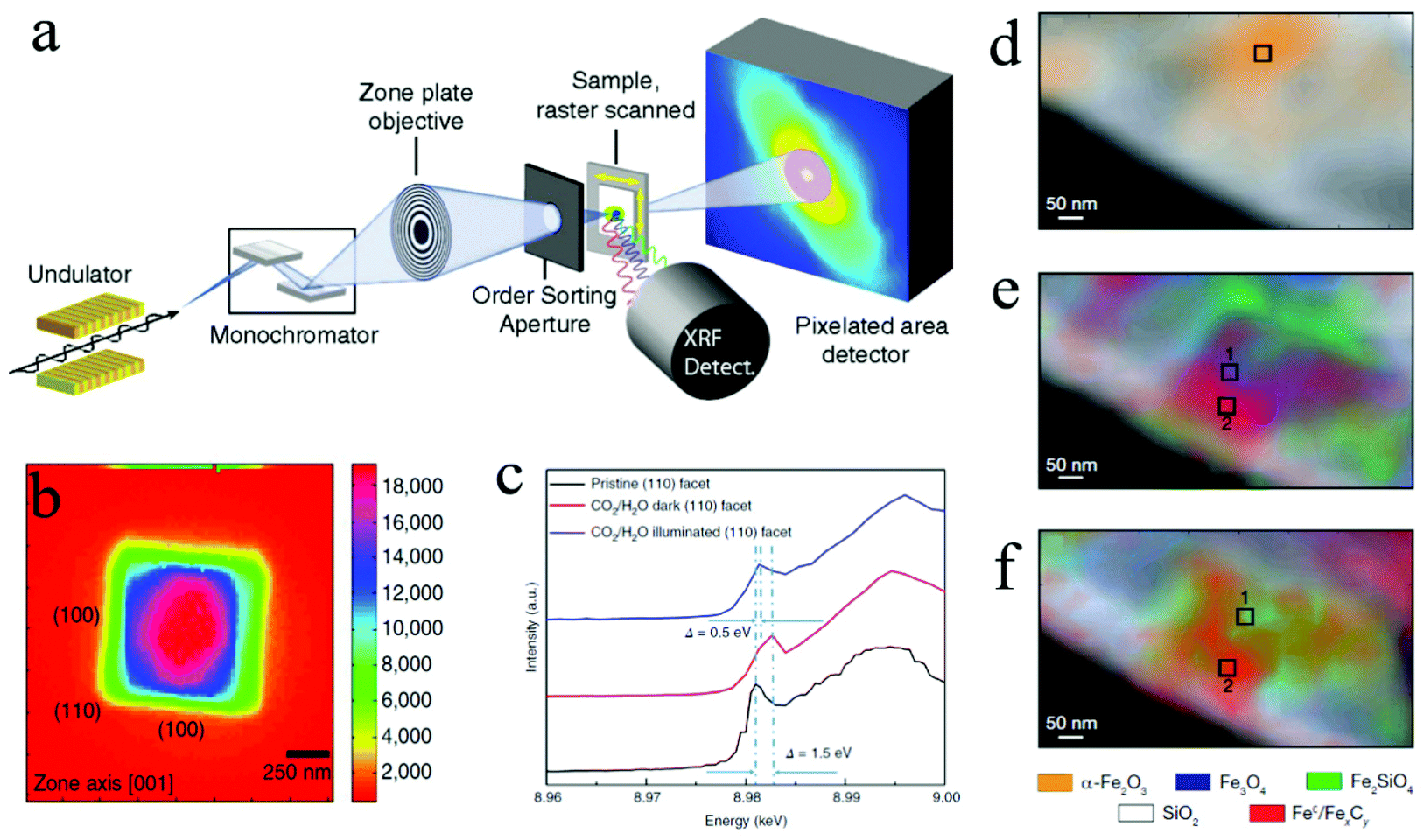 | ||
| Fig. 7 (a) STXM and SFXM setup; Copyright 2014, National Academy of Sciences; (b) SFXM image of a Cu2O nanoparticle; (c) scanning X-ray nanospectroscopy of the (110) facet; Copyright 2019, Springer Nature; (d) to (f) chemical contour images of the catalyst under different treatments; Copyright 2008, Springer Nature. All panels reproduced with permission.148,156,160 | ||
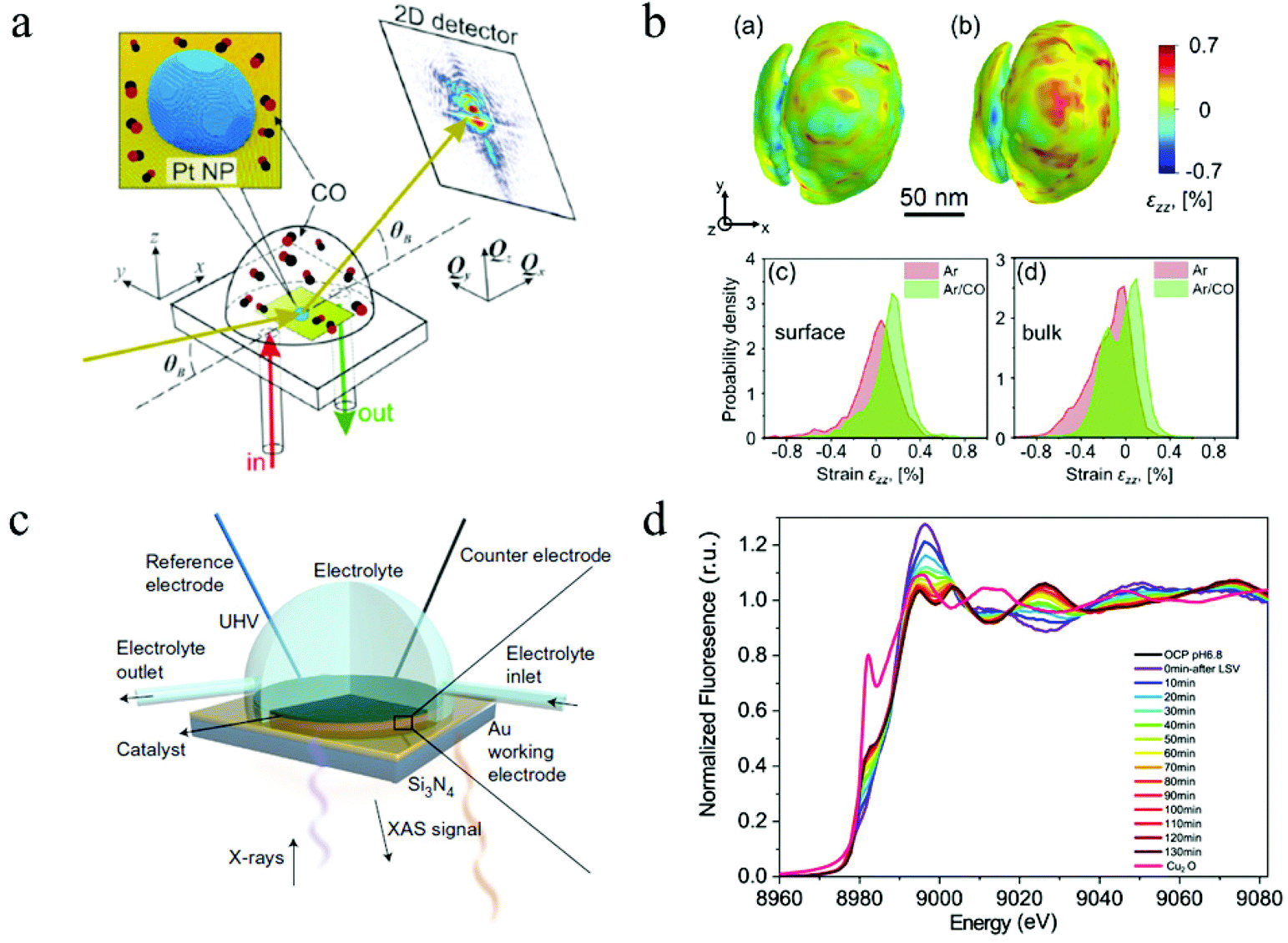 | ||
| Fig. 8 (a) The schematic of CXDI imaging. (b) 3D CXDI imaging of Pt NP under different conditions and the strain field in near surface region and in the bulk. Copyright 2019, American Chemical Society; (c) in situ XAS electrochemical cell. Copyright 2017, Springer Nature; (d) time-resolved XAS spectrum. Copyright 2021, Springer Nature. All panels reproduced with permission.153,164,171 | ||
5.3 In situ and operando X-ray spectroscopy
Hard X-rays with photon energies higher than 5 keV have strong penetration power. Consequently, hard X-ray absorption spectroscopy is highly desired in situ and operando studies that involve complex reaction environments with the presence of liquid, gas, and solid. In XAS, the absorption spectrum ranges immediately before and after the absorption edge is defined as the X-ray absorption near edge structure (XANES), which can characterize the density of states, thus revealing the valence states (at the absorption edge) and orbital hybridization due to coordination (pre-edge).173 The extended X-ray absorption fine structure (EXAFS) covers the energy region above the absorption edge and can characterize the local atomistic structure, i.e., the neighboring atoms to the bound atoms and the interatomic distances between them. For in situ and operando CO2 ECR studies, specially designed electrochemical cells with X-ray windows are required to provide a reaction environment that can be characterized by both electrochemical workstations and XAS. Such windows are usually made from Si3N4 to ensure no blockage for X-ray to pass through. Fig. 8c shows a design of this cell.171In situ XAS has been utilized in the studies of 2D materials for CO2 ECR as a powerful tool to probe the oxidation states and local coordination environment, thus determining the active sites during CO2 electrochemical reduction reactions. For instance, a recent study on a bimetallic 2D conjugated metalorganic framework (2D c-MOF) demonstrated the synergistic effects between its CuN4 ligand and ZnO4 complex (ZnO4) linkage with a high CO selectivity of 88%.108 In this study, EXAFS spectrum showed an intensity decrease in Zn–O bond, suggesting interactions between ZnO4 sites and reaction intermediates. With additional infrared absorption studies, the authors were able to conclude that ZnO4 sites are catalytic reactive sites. Another investigation on 2D bismuth metal–organic framework employed in situ XAS to probe the stability of Bi3+ during CO2 ECR. XANES analysis proved that the Bi3+ oxidation state stayed unchanged at a high potential of ∼−0.9 V for 90 min, revealing a significantly enhanced Bi3+ stability in the prepared MOF structure than in other 2D materials.104 Moreover, Wang and coworkers recently studied the highly selective CO2 ECR reaction mechanism of 2D CuO nanosheet.153 By using in situ XAS, they discovered that the CuO nanosheet gradually and directly converted into metallic Cu under a potential of −0.84 V (RHE) with no Cu2O intermediates detected.153Fig. 8d shows the time-resolved information on the catalyst under a CO2 ECR condition. EXAFS also showed that the coordination number of the produced Cu0 is smaller than that in bulk fcc Cu0, suggesting the existence of lattice defects associated with undercoordinated Cu atoms, which attributed to the enhanced efficiency and selectivity towards C2+ products. These in-depth mechanism studies demonstrated the abilities and advantages of in situ and operando XAS employed in CO2 ECR studies using 2D materials.
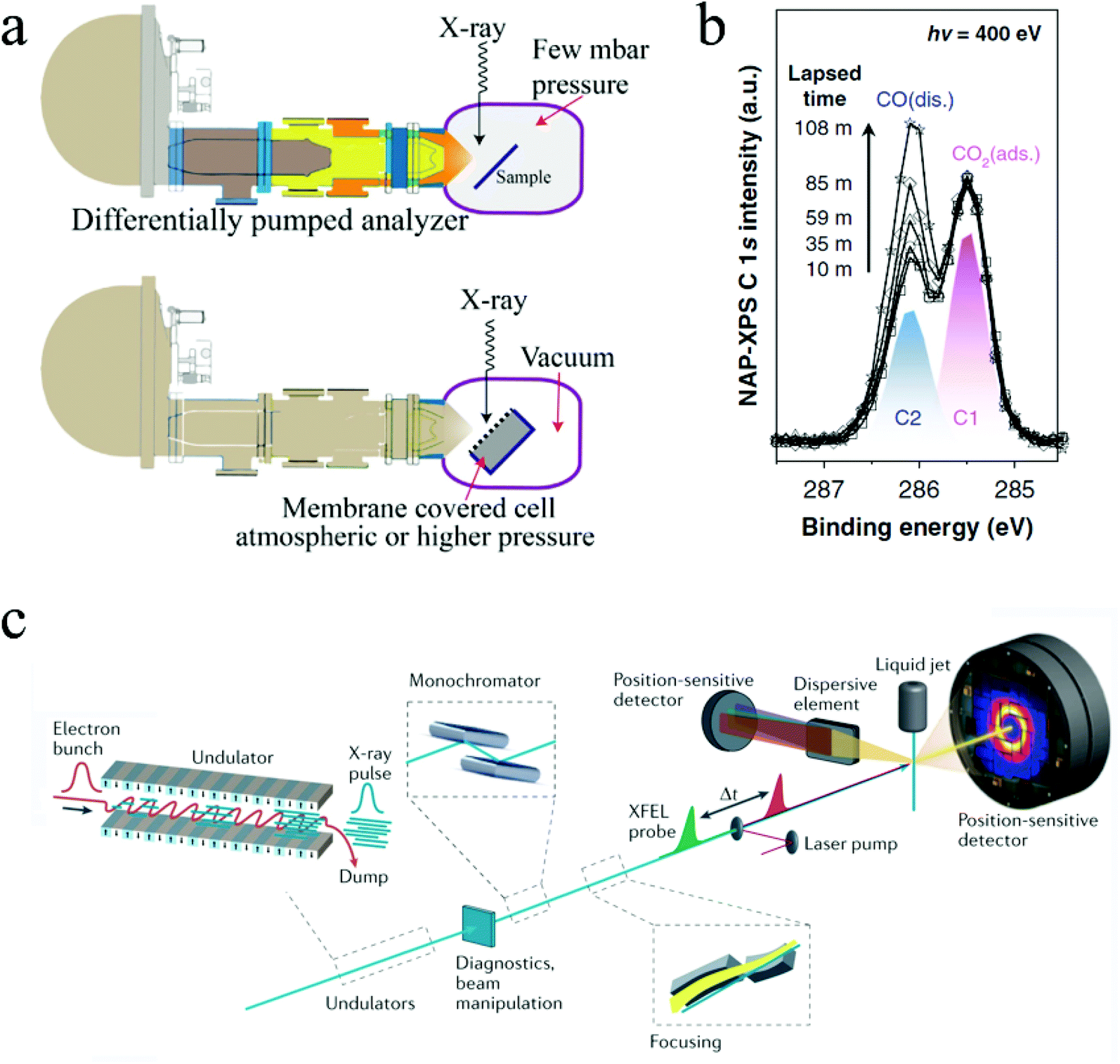 | ||
| Fig. 9 (a) Two NAP-XPS systems: differential pumping system and environmental cell system. Copyright 2018, John Wiley and Sons. (b) Time resolved NAP-XPS showing the formation of dissociated CO absorption. Copyright 2020, Springer Nature; (c) configuration of the Free Electron Laser (FEL) system. Copyright 2021, Springer Nature. All panels reproduced with permission.178,181,183 | ||
In situ XPS has been employed in some fundamental studies to scrutinize CO2 ECR mechanisms on different catalysts. For example, Favaro et al. used in situ ambient pressure X-ray photoelectron spectroscopy to investigate how subsurface oxide below Cu surface affects the CO2 activation process.180In situ XPS examined the binding energy shifts of C 1s and O 1s photoelectron peaks on different surfaces. It was found that the presence of an optimal amount of subsurface oxide can form a mixed Cu+ and Cu0 structure that stabilized the bent CO2, thus promoting the CO2 activation.180 Similarly, Kim and coworkers used in situ XPS to directly monitor the formation of dissociated CO* from chemisorbed CO2 on Rh surfaces in a time-resolved manner.181 Notably, this experiment was conducted under a 0.1 millibars environment. Fig. 9b shows the time-resolved monitoring of the formation of the dissociated CO adsorption on Rh surface.181 Note that all the above examples used synchrotron radiation sources for the in situ ambient pressure XPS characterization, especially with the presence of liquid in the system. Synchrotron light sources can modulate the photon energy and thus characterize the samples in different depths, enabling both depth-resolved and time-resolved characterizations.182
5.4 Free electron laser (FEL) and “pump/probe” technique
Free Electron Lasers (FEL) can provide extremely coherent laser beams with ultrashort pulse duration (femtoseconds), ultrahigh brilliance, and widely turntable wavelengths ranging from microwave frequencies to hard X-rays.184,185 They can generate much brighter electromagnetic radiation than other sources available nowadays.186 With such unique advantages, FELs can be used for both imaging and spectroscopy, offering ultrafast time-resolved investigations of material properties and molecular dynamics with a temporal resolution in the scale of femtoseconds. In X-ray Free Electron Lasers (XFEL), a high energy relativistic electron beam is first generated by an electron source and then passes through a periodically alternating magnetic field, which is provided by the undulator/wiggler and forces the electrons to move along a sinusoidal path. The undulating motions of electrons then emit highly coherent radiations with wavelengths that can be modulated by the electron energy and/or the magnetic field strength. Fig. 9c illustrates the configuration of the FEL system.183 There are only a few FEL facilities in the world nowadays, located in Asia, Europe, and the U.S.A. Notably, the European XFEL, located in Germany, has a high beam energy of 8.5–17.5 GeV and a short pulse duration of 3–150 fs. The photon energy is ranged from 240 eV to 25000 eV, making it a powerful tool for probing various information mentioned above in situ catalytic investigations.
These unique advantages of FEL enable it to be used in the ultrafast pump/probe technique. In this technique, first, an ultrashort light pulse that lasts for less than 100 fs interacts with the material and initiates photochemical reactions, thus exciting the system to a non-equilibrium state. This step is called the “pump” step. Then the non-equilibrium state is probed by a following ultrashort light pulse that monitors the changes that occurred due to the excitation. The temporal intervals of these two pulses can be modulated to investigate the time-dependent dynamics of the reactions. Besides using coherent X ray as pump and probe, the ultrafast electron diffraction (UED) use the laser pump and electron beam probe to study the molecular structural dynamics.187 Consequently, the field of “femtochemistry” was constructed to investigate the fundamental reaction mechanisms in chemical reactions.188 The pump and probe technique have been adopted in CO2RR studies to mainly investigate the carrier dynamics of photocatalysts, as such systems can be easily excited by pump pulses.189,190 However, many other reactants and catalysts in other catalytic systems cannot be excited by light pulses. Fortunately, hopes for probing electrocatalytic reactions have been brought by some pioneering studies. Ultrafast XAS, which can be realized by the pump/probe spectroscopy, has been utilized in the in situ monitoring of the sub-femtosecond motions of valence electrons. The monitoring of such motions can help reveal the fundamental reaction mechanism of CO2 ECR using 2D materials.
5.5 Other in situ and operando spectrometry
Fig. 10a illustrates the mechanism of this advanced technique. By combing ATR and the Surface Enhanced Infrared Absorption Spectroscopy (SEIRAS), ATR-SEIRAS has demonstrated enhanced time resolution and sensitivity for operando studies, less interference from the bulk electrolyte, and the reduced IR attenuation and transport limitations.196 The high IR absorption cross-section of C–O bonds makes IR very suitable for CO2 ECR studies that produce formate, CO, as well as other common intermediates and products in CO2RR.198 SEIRAS has been widely utilized in CO2 ECR studies for the investigations of fundamental reaction mechanisms and reaction species. For instance, SEIRAS demonstrated that the adsorbed hydrogen could partially displace adsorbed carbon monoxide on the Cu surface during CO2RR, while the displacement can't occur in the reversed direction. Hence, it was proposed that hydrogen has a higher absorption strength than that of CO.199
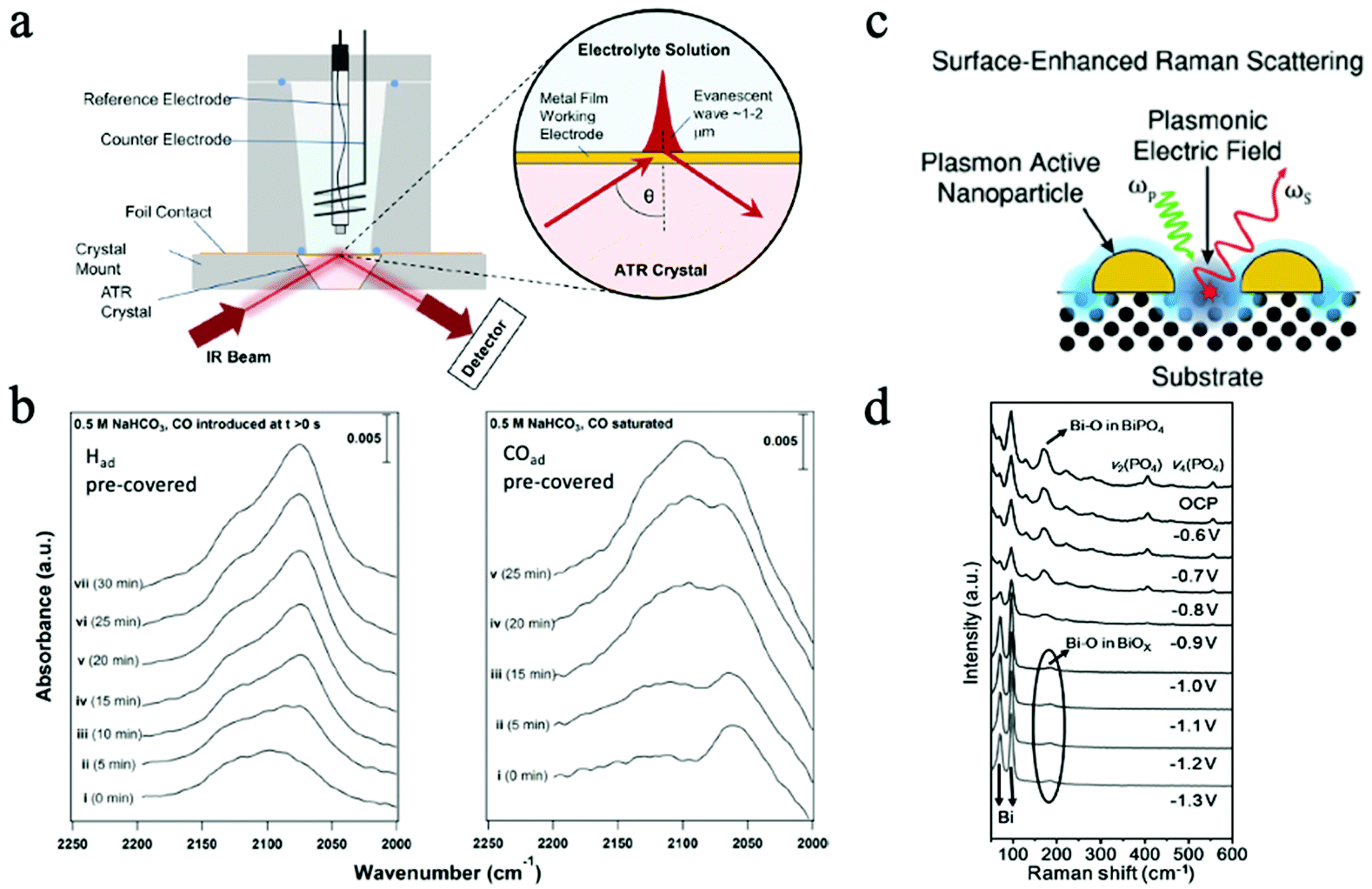 | ||
| Fig. 10 (a) Mechanism of in situ ATR. Copyright 2017, crown; (b) time-resolved SEIRA spectra of Had accumulation on CO-adsorbed Cu surface. Copyright 2016, American Chemical Society (c) mechanism of SERS. Copyright 2021, Royal Society of Chemistry; (d) operando Raman spectra under different applied potential. All panels reproduced with permission.198–201 Copyright 2021, 2021 Wiley and Sons. | ||
Fig. 10b shows the time-resolved SEIRA spectra of hydrogen and CO on Had pre-covered and CO pre-covered Cu surfaces, proving that hydrogen can displace adsorbed carbon monoxide.199 Additionally, SEIRS also revealed that the surface C-bound and O-bound reaction intermediates can affect the overall CO2RR reaction selectivity. Regardless of the powerful characterization ability, one drawback of in situ and operando IR in catalytic reactions is the limited access to low wavenumbers, which can help obtain information on the structural properties of surface oxides and hydroxides.196 Moreover, the focused IR beam area is on the micron scale, which is relatively large for precisely probing nanomaterials. Hence, the measured peak only shows the averaged data within the whole beam area.
Fig. 10c illustrates the schematic of this operation. As an enhanced technique, SERS operates based on the SERS enhancement effects, which are induced by the amplified local electric field as a result of the resonant excitations of surface plasmons and the non-resonant lightning rod effects due to the concentrated field at enhancement sources, i.e., surface features that maximize electromagnetic enhancement.198 A significant enhancement in the Raman signal can be achieved in this way, making this technique highly desired in catalytic reactions to monitor the surface absorbates and reaction species.200 Nevertheless, the dominant enhancement sources can also interfere with the characteristic signals from samples. Consequently, SHINERS was developed to isolate the enhancement sources (such as sharp tip/surfaces) to minimize the interference, making SERS applicable to all kinds of surfaces and morphologies.202 On the other hand, by combing SERS and scanning probe microscopy tips, tip-enhanced Raman spectroscopy (TERS) was introduced to provide highly sensitive and spatial-resolved characterizations by involving a scanning probe microscopy tip. With a spatial resolution as high as 1–10 nm, TERS is extremely helpful in in situ characterizations of nanocatalysts.203,204In situ Raman characterizations have been demonstrated with different 2D materials for CO2 ECR and provided information on the reaction mechanisms and reactive sites. For example, in a recent study, bismuth phosphate (BiPO4)-derived 2D Nanosheets were examined by operando Raman spectroscopy during CO2 ECR at different potentials. As shown in Fig. 10d, the disappeared Raman peaks associated with the PO4 group and the shifts of peaks assigned to Bi–O bond suggested the formation of Bi and BiOx during CO2 ECR.201 The abundance of Bi–O on the surface was attributed to the enhanced CO2 ECR performance. Many reaction intermediates, such as *CO and *HCOO, have been identified in in situ studies as key intermediates in CO2 ECR.201 Even though TERS is a highly advanced in situ characterization technique, many limitations, such as the short tip lifetime and the inability to characterize single-molecule level features, still need to be overcome in the future.203 Similar to IR, Raman spectroscopy is also limited by the large beam area with average data when characterizing nanomaterials, which make lack the heterogeneity information from samples.
6 Challenges and outlook
In recent years, 2D materials have been intensively investigated and utilized in CO2 electrochemical reductions to value-added products. With the emergence of many innovative 2D catalysts, exceptional progress has been achieved via various approaches to enhance the activity, selectivity, and overall performance of such 2D catalysts, providing promising paradigms for the next generation CO2 conversion systems. Nevertheless, the development of 2D catalysts for CO2 ECR still faces many challenges, bringing promising opportunities in this field of study. Most of the current studies are only focused on the catalyst material aspects, such as the types of materials, morphologies, defects, and compositions. However, catalytic systems are often very complex and include numerous components that need to be integrated into the studies of 2D catalysts. Many parameters, including the reactor design, electrolytes, and local reaction environment, can all determine the overall CO2 ECR performance.In CO2 ECR, the local reaction environment can be constructed by both solid/liquid and solid/gas/liquid interfaces. With the adsorption, reaction, and desorption all taking place at these interfaces, these reactions are all governed by the synergistic effects of all the components in the local environment. It has been reported that the interactions between solutions and reactants can critically affect CO2 performance. For instance, the types and sizes of both anions and cations in electrolytes have demonstrated governing effects on the selectivity of CO2 ECR via affecting the adsorptions of reaction intermediates, altering the charge distributions on catalysts surface, and shifting the reaction free-energy landscape of Cu catalysts.205–207 Moreover, the electrical fields created in the Helmholtz layer and the use of ionic liquids have demonstrated various effects.208,209 Organic surfactants like cetyltrimethylammonium bromide have also enhanced CO2 ECR performance by inhibiting the competing HER at the catalyst surface.210 The interactions between liquid electrolytes and solid catalysts and the generated liquid–solid interfaces can all determine the CO2 ECR performance. With their unique 2D morphologies and substantial dopant and vacancy exposures, 2D nanocatalysts might induce distinctive reaction behaviors in such a highly localized reaction environment. Such investigations on the local reaction environment are required to enhance the performance of 2D CO2 catalysts further.
Researchers might also investigate additional novel enhancing strategies, such as catalyst modifiers and strain engineering, in the study of 2D materials. Recently, polymers have shown exceptional optimizing capabilities by modulating the heterogeneous catalytic surfaces and stabilizing metal nanocatalysts.211–213 Hydrophilic polymeric modifiers can enhance the selectivity for formic acid, while cationic hydrophobic modifiers can promote the formation of CO on Cu catalysts.212 Moreover, polymeric N-heterocyclic carbenes (NHC), such as polydentate and monodentate, have exhibited stabilizing effects on Au and Pd nanocatalysts due to the formation of metal–carbene bonds that inhibit the nanoclustering of nanoparticles.211 Such findings especially pertain to the use of 2D catalysts. The mechanisms and approaches proposed in novel optimization paradigms may also be applied to 2D nanocatalysts to achieve organic modifications at the atomic scale. Lattice strains have demonstrated the potential to alter the band structures of 2D materials and break the linear scaling relationship for CO2 ECR, thus enhancing the selectivity and activity.214,215 However, it is still challenging to scrutinize the underlying mechanisms due to the difficulties of decoupling strain from other electronic effects in 2D materials with mismatched lattice.169 More simulation and experimental approaches need to be combined to investigate such optimization behaviors. Additionally, applying strain on 2D catalysts with desired magnitude and distribution remains difficult to realize.
With the introduction of gas diffusion layers and polymer ion-exchange membranes in membrane electrode assembly (MEA) electrolyzers, gas-phase electrolyzers have enhanced the current density and stability of CO2 ECR.105 Remarkably, some novel all-solid-state reactors that utilize polymer electrolytes can significantly facilitate the separation of liquid products in continuous productions.114 However, to date, most investigations on 2D catalysts have been conducted in liquid phase electrolyzers. The opportunities brought by such advanced reactors need to be seized in the study of 2D materials for CO2 ECR. It must be pointed out that, in these solid-state reactors, the interactions between solid catalysts and solid electrolytes, the adsorption, desorption, and the mass transport of reaction species, as well as the local reaction environment are all different at the solid–solid interfaces from those at traditional liquid–solid interfaces. The behaviors and performance of 2D nanocatalysts in these advanced reaction environments and reactors need to be characterized and optimized to achieve superior CO2 ECR efficiency and selectivity in future industrial-scale CO2 ECR systems.
Due to the complexity of the catalytic reactions, many scientific problems cross orders of magnitude in times and length scales. Thus, we urge to use in situ and operando multimodal characterizations to get comprehensive, insightful information across different characterization platforms. Advanced in situ and operando multimodal characterizations of 2D nanocatalysts can help achieve fundamental understandings of CO2 ECR mechanisms, especially when coupled with modern DFT calculations. Even though such characterizations often require advanced facilities such as synchrotron X-ray sources, electron microscopy, and free electron laser, the information corrected can reveal the underlying reaction active sites, pathways, intermediates, local environment, and morphological changes of 2D catalysts. Therefore, it is essential to actively employ operando multimodal characterizations in future studies to better design 2D nanocatalysts and beyond. More importantly, such in situ and operando characterizations conducted in flow cells and MEA electrolyzers remain extremely challenging. To develop 2D nanocatalysts used in industrial scale electrolyzers, specially designed apparatuses are highly demanded by in situ and operando studies.
Finally, to realize the utilization of 2D materials in real-life, industrial-scale CO2 conversion systems, the mass production of 2D materials with high efficiencies, high selectivity and high stability must be achieved in the coming future. Yet, the production of 2D nanomaterials is still very challenging due to the aggregation and restacking caused by electrostatic and van der Waals interactions. Moreover, the productivity is still too low to achieve cost-effective productions and utilizations of 2D materials for CO2 conversion. On the other hand, the feasibility of mass production should be kept in mind when designing next-generation 2D nanocatalysts.
Conflicts of interest
The authors do not have conflict of interests to declare.Acknowledgements
Yimin A. Wu thanks funding support from Natural Sciences and Engineering Research Council of Canada (NSERC) (RGPIN-2020-05903, GECR-2020-00476), and National Research Council of Canada Materials for Clean Fuels Challenge Program (CH-MCF-123). Cheng Du acknowledges the support from CUTRIC (Canadian Urban Transit Research & Innovation Consortium) through a MITACS accelerate postdoc program.References
- C. D. Thomas, A. Cameron, R. E. Green, M. Bakkenes, L. J. Beaumont, Y. C. Collingham, B. F. Erasmus, M. F. De Siqueira, A. Grainger, L. Hannah, L. Hughes, B. Huntley, A. S. Van Jaarsveld, G. F. Midgley, L. Miles, M. A. Ortega-Huerta, A. T. Peterson, O. L. Phillips and S. E. Williams, Nature, 2004, 427, 145–148 CrossRef CAS PubMed.
- P. M. Cox, R. A. Betts, C. D. Jones, S. A. Spall and I. J. Totterdell, Nature, 2000, 408, 184–187 CrossRef CAS PubMed.
- Z. Y. Sun, T. Ma, H. C. Tao, Q. Fan and B. X. Han, Chem, 2017, 3, 560–587 CAS.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- D. F. Gao, R. M. Aran-Ais, H. S. Jeon and B. Roldán Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- C. Du, X. Wang, W. Chen, S. Feng, J. Wen and Y. A. Wu, Mater. Today Adv., 2020, 6, 24 Search PubMed.
- F. P. Pan and Y. Yang, Energy Environ. Sci., 2020, 13, 2275–2309 RSC.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 1695–1698, DOI:10.1246/cl.1985.1695.
- R. Kortlever, I. Peters, C. Balemans, R. Kas, Y. Kwon, G. Mul and M. T. Koper, Chem. Commun., 2016, 52, 10229–10232 RSC.
- J. L. Qiao, Y. Y. Liu, F. Hong and J. J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328–330, 10.1039/c39840000328.
- D. Sun, X. Xu, Y. Qin, S. P. Jiang and Z. Shao, ChemSusChem, 2020, 13, 39–58 CrossRef CAS PubMed.
- A. Salehi-Khojin, H. R. M. Jhong, B. A. Rosen, W. Zhu, S. C. Ma, P. J. A. Kenis and R. I. Masel, J. Phys. Chem. C, 2013, 117, 1627–1632 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldán Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- H. Mistry, R. Reske, Z. Zeng, Z. J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- H. Mistry, R. Reske, P. Strasser and B. Roldán Cuenya, Catal. Today, 2017, 288, 30–36 CrossRef CAS.
- H. S. Jeon, I. Sinev, F. Scholten, N. J. Divins, I. Zegkinoglou, L. Pielsticker and B. R. Cuenya, J. Am. Chem. Soc., 2018, 140, 9383–9386 CrossRef CAS PubMed.
- Y. Sun, S. Gao, F. Lei and Y. Xie, Chem. Soc. Rev., 2015, 44, 623–636 RSC.
- H. Tao, Y. Gao, N. Talreja, F. Guo, J. Texter, C. Yan and Z. Sun, J. Mater. Chem. A, 2017, 5, 7257–7284 RSC.
- W. Bi, X. Li, R. You, M. Chen, R. Yuan, W. Huang, X. Wu, W. Chu, C. Wu and Y. Xie, Adv. Mater., 2018, 30, e1706617 CrossRef PubMed.
- Z. Lin, B. R. Carvalho, E. Kahn, R. Lv, R. Rao, H. Terrones, M. A. Pimenta and M. Terrones, 2D Mater., 2016, 3, 022002 CrossRef.
- H. Chen, A. D. Handoko, J. Xiao, X. Feng, Y. Fan, T. Wang, D. Legut, Z. W. Seh and Q. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 36571–36579 CrossRef CAS PubMed.
- F. P. García De Arquer, O. S. Bushuyev, P. De Luna, C.-T. Dinh, A. Seifitokaldani, M. I. Saidaminov, C.-S. Tan, L. N. Quan, A. Proppe, M. G. Kibria, S. O. Kelley, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1802858 CrossRef PubMed.
- H. Cheng, S. Liu, J. Zhang, T. Zhou, N. Zhang, X. S. Zheng, W. Chu, Z. Hu, C. Wu and Y. Xie, Nano Lett., 2020, 20, 6097–6103 CrossRef CAS PubMed.
- A. D. Handoko, F. X. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- K. F. Kalz, R. Kraehnert, M. Dvoyashkin, R. Dittmeyer, R. Glaser, U. Krewer, K. Reuter and J. D. Grunwaldt, ChemCatChem, 2017, 9, 17–29 CrossRef CAS PubMed.
- R. Zhou, X. Fan, X. Ke, J. Xu, X. Zhao, L. Jia, B. Pan, N. Han, L. Li, X. Liu, J. Luo, H. Lin and Y. Li, Nano Lett., 2021, 21, 4092–4098 CrossRef CAS PubMed.
- Y. Lan, G. Niu, F. Wang, D. Cui and Z. Hu, ACS Appl. Mater. Interfaces, 2020, 12, 36128–36136 CrossRef CAS PubMed.
- W. Huang, D. Zhou, H. Yang, X. Liu and J. Luo, ACS Appl. Energy Mater., 2021, 4, 7492–7496 CrossRef CAS.
- Q. Zhao, C. Zhang, R. Hu, Z. Du, J. Gu, Y. Cui, X. Chen, W. Xu, Z. Cheng, S. Li, B. Li, Y. Liu, W. Chen, C. Liu, J. Shang, L. Song and S. Yang, ACS Nano, 2021, 15, 4927–4936 CrossRef CAS PubMed.
- C. Cao, D. D. Ma, J. Jia, Q. Xu, X. T. Wu and Q. L. Zhu, Adv. Mater., 2021, 33, 2008631 CrossRef CAS PubMed.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- W. J. Zhang, Y. Hu, L. B. Ma, G. Y. Zhu, P. Y. Zhao, X. L. Xue, R. P. Chen, S. Y. Yang, J. Ma, J. Liu and Z. Jin, Nano Energy, 2018, 53, 808–816 CrossRef CAS.
- H. Zhang, Y. Ma, F. J. Quan, J. J. Huang, F. L. Jia and L. Z. Zhang, Electrochem. Commun., 2014, 46, 63–66 CrossRef CAS.
- P. Su, W. Xu, Y. Qiu, T. Zhang, X. Li and H. Zhang, ChemSusChem, 2018, 11, 848–853 CrossRef CAS PubMed.
- L. C. Yi, J. X. Chen, P. Shao, J. H. Huang, X. X. Peng, J. W. Li, G. X. Wang, C. Zhang and Z. H. Wen, Angew. Chem., Int. Ed., 2020, 59, 20112–20119 CrossRef CAS PubMed.
- H. Yang, N. Han, J. Deng, J. Wu, Y. Wang, Y. Hu, P. Ding, Y. Li, Y. Li and J. Lu, Adv. Energy Mater., 2018, 8, 1801536 CrossRef.
- C. J. Peng, G. Zeng, D. D. Ma, C. Cao, S. Zhou, X. T. Wu and Q. L. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 20589–20597 CrossRef CAS PubMed.
- C.-Y. Lee, Y. Zhao, C. Wang, D. R. G. Mitchell and G. G. Wallace, Sustainable Energy Fuels, 2017, 1, 1023–1027 RSC.
- T. T. Zhang, X. F. Li, Y. L. Qiu, P. P. Su, W. B. Xu, H. X. Zhong and H. M. Zhang, J. Catal., 2018, 357, 154–162 CrossRef.
- J. Pan, Y. M. Sun, P. L. Deng, F. Yang, S. H. Chen, Q. Zhou, H. S. Park, H. F. Liu and B. Y. Xia, Appl. Catal., B, 2019, 255, 117736 CrossRef CAS.
- L. Dai, Q. Qin, P. Wang, X. Zhao, C. Hu, P. Liu, R. Qin, M. Chen, D. Ou, C. Xu, S. Mo, B. Wu, G. Fu, P. Zhang and N. Zheng, Sci. Adv., 2017, 3, e1701069 CrossRef PubMed.
- W. Zhang, Q. Qin, L. Dai, R. Qin, X. Zhao, X. Chen, D. Ou, J. Chen, T. T. Chuong, B. Wu and N. Zheng, Angew. Chem., Int. Ed., 2018, 57, 9475–9479 CrossRef CAS PubMed.
- W. Deng, L. Zhang, L. Li, S. Chen, C. Hu, Z. J. Zhao, T. Wang and J. Gong, J. Am. Chem. Soc., 2019, 141, 2911–2915 CrossRef CAS PubMed.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, ACS Catal., 2015, 5, 3148–3156 CrossRef CAS.
- A. Dutta, A. Kuzume, V. Kaliginedi, M. Rahaman, I. Sinev, M. Ahmadi, B. Roldán Cuenya, S. Vesztergom and P. Broekmann, Nano Energy, 2018, 53, 828–840 CrossRef CAS.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed.
- J. Jia, C. Qian, Y. Dong, Y. F. Li, H. Wang, M. Ghoussoub, K. T. Butler, A. Walsh and G. A. Ozin, Chem. Soc. Rev., 2017, 46, 4631–4644 RSC.
- S. Gao, X. Jiao, Z. Sun, W. Zhang, Y. Sun, C. Wang, Q. Hu, X. Zu, F. Yang, S. Yang, L. Liang, J. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 698–702 CrossRef CAS PubMed.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- S. Gao, Z. Sun, W. Liu, X. Jiao, X. Zu, Q. Hu, Y. Sun, T. Yao, W. Zhang, S. Wei and Y. Xie, Nat. Commun., 2017, 8, 14503 CrossRef CAS PubMed.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed.
- N. Sakai, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2004, 126, 5851–5858 CrossRef CAS PubMed.
- X. Y. Chen, Y. Zhou, Q. Liu, Z. D. Li, J. G. Liu and Z. G. Zou, ACS Appl. Mater. Interfaces, 2012, 4, 3372–3377 CrossRef CAS PubMed.
- Y. Zhou, Z. P. Tian, Z. Y. Zhao, Q. Liu, J. H. Kou, X. Y. Chen, J. Gao, S. C. Yan and Z. G. Zou, ACS Appl. Mater. Interfaces, 2011, 3, 3594–3601 CrossRef CAS PubMed.
- Q. Liu, D. Wu, Y. Zhou, H. Su, R. Wang, C. Zhang, S. Yan, M. Xiao and Z. Zou, ACS Appl. Mater. Interfaces, 2014, 6, 2356–2361 CrossRef CAS PubMed.
- S. Y. Zhu, S. J. Liang, J. H. Bi, M. H. Liu, L. M. Zhou, L. Wu and X. X. Wang, Green Chem., 2016, 18, 1355–1363 RSC.
- S. Gao, B. Gu, X. Jiao, Y. Sun, X. Zu, F. Yang, W. Zhu, C. Wang, Z. Feng, B. Ye and Y. Xie, J. Am. Chem. Soc., 2017, 139, 3438–3445 CrossRef CAS PubMed.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Kral, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470 CrossRef CAS PubMed.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- P. Abbasi, M. Asadi, C. Liu, S. Sharifi-Asl, B. Sayahpour, A. Behranginia, P. Zapol, R. Shahbazian-Yassar, L. A. Curtiss and A. Salehi-Khojin, ACS Nano, 2017, 11, 453–460 CrossRef CAS PubMed.
- J. Xu, X. Li, W. Liu, Y. Sun, Z. Ju, T. Yao, C. Wang, H. Ju, J. Zhu, S. Wei and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 9121–9125 CrossRef CAS PubMed.
- X. Hong, K. R. Chan, C. Tsai and J. K. Nørskov, ACS Catal., 2016, 6, 4428–4437 CrossRef CAS.
- X. Jiao, Z. Chen, X. Li, Y. Sun, S. Gao, W. Yan, C. Wang, Q. Zhang, Y. Lin, Y. Luo and Y. Xie, J. Am. Chem. Soc., 2017, 139, 7586–7594 CrossRef CAS PubMed.
- X. D. Li, Y. F. Sun, J. Q. Xu, Y. J. Shao, J. Wu, X. L. Xu, Y. Pan, H. X. Ju, J. F. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- X. Jiao, X. Li, X. Jin, Y. Sun, J. Xu, L. Liang, H. Ju, J. Zhu, Y. Pan, W. Yan, Y. Lin and Y. Xie, J. Am. Chem. Soc., 2017, 139, 18044–18051 CrossRef CAS PubMed.
- L. D. Ramírez-Valencia, E. Bailón-García, F. Carrasco-Marín and A. F. Pérez-Cadenas, Catalysts, 2021, 11, 351 CrossRef.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- G. L. Chai and Z. X. Guo, Chem. Sci., 2016, 7, 1268–1275 RSC.
- H. Wang and J. M. Lee, J. Mater. Chem. A, 2020, 8, 10604–10624 RSC.
- Y. Gogotsi and B. Anasori, ACS Nano, 2019, 13, 8491–8494 CrossRef CAS PubMed.
- J. L. Hart, K. Hantanasirisakul, A. C. Lang, B. Anasori, D. Pinto, Y. Pivak, J. T. Van Omme, S. J. May, Y. Gogotsi and M. L. Taheri, Nat. Commun., 2019, 10, 522 CrossRef CAS PubMed.
- N. Li, X. Chen, W. J. Ong, D. R. MacFarlane, X. Zhao, A. K. Cheetham and C. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS PubMed.
- A. D. Handoko, K. H. Khoo, T. L. Tan, H. M. Jin and Z. W. Seh, J. Mater. Chem. A, 2018, 6, 21885–21890 RSC.
- Y. Xiao and W. Zhang, Nanoscale, 2020, 12, 7660–7673 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- X. X. Tang, L. Zhao, W. W. Sun and Y. Wang, J. Power Sources, 2020, 477, 228919 CrossRef CAS.
- M. K. Lee, M. Shokouhimehr, S. Y. Kim and H. W. Jang, Adv. Energy Mater., 2021, 2003990, DOI:10.1002/aenm.202003990.
- Q. Y. Cui, G. Q. Qin, W. H. Wang, K. R. Geethalakshmi, A. J. Du and Q. Sun, Appl. Surf. Sci., 2020, 500, 143993 CrossRef CAS.
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech, E. Brunner, I. M. Weidinger, J. Zhang, A. V. Krasheninnikov, S. Kaskel, R. Dong and X. Feng, Nat. Commun., 2020, 11, 1409 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- Y. Zhang, L. Li, S. X. Guo, X. Zhang, F. Li, A. M. Bond and J. Zhang, ChemSusChem, 2020, 13, 59–77 CrossRef CAS PubMed.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- S. Gao, Z. Sun, W. Liu, X. Jiao, X. Zu, Q. Hu, Y. Sun, T. Yao, W. Zhang, S. Wei and Y. Xie, Nat. Commun., 2017, 8, 14503 CrossRef CAS PubMed.
- S. Chen, H. Wang, Z. Kang, S. Jin, X. Zhang, X. Zheng, Z. Qi, J. Zhu, B. Pan and Y. Xie, Nat. Commun., 2019, 10, 788 CrossRef CAS PubMed.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T. K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS PubMed.
- S. Siahrostami, K. Jiang, M. Karamad, K. R. Chan, H. T. Wang and J. Norskov, ACS Sustainable Chem. Eng., 2017, 5, 11080–11085 CrossRef CAS.
- H. Terrones, R. Lv, M. Terrones and M. S. Dresselhaus, Rep. Prog. Phys., 2012, 75, 062501 CrossRef PubMed.
- Z. Y. Shu, G. Y. Ye, J. Wang, S. Q. Liu, Z. He, W. W. Zhu, B. Liu and M. Liu, Ionics, 2021, 27, 3089–3098 CrossRef CAS.
- S. Back, M. S. Yeom and Y. Jung, ACS Catal., 2015, 5, 5089–5096 CrossRef CAS.
- F. Li, M. Xue, J. Li, X. Ma, L. Chen, X. Zhang, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14718–14722 CrossRef CAS PubMed.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J. L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X. D. Zhou, R. Vajtai, B. I. Yakobson, J. Lou and P. M. Ajayan, Nano Lett., 2016, 16, 466–470 CrossRef CAS PubMed.
- Y. Liu, J. Zhao and Q. Cai, Phys. Chem. Chem. Phys., 2016, 18, 5491–5498 RSC.
- N. Sreekanth, M. A. Nazrulla, T. V. Vineesh, K. Sailaja and K. L. Phani, Chem. Commun., 2015, 51, 16061–16064 RSC.
- C. M. Zhao, Y. Wang, Z. J. Li, W. X. Chen, Q. Xu, D. S. He, D. S. Xi, Q. H. Zhang, T. W. Yuan, Y. T. Qu, J. Yang, F. Y. Zhou, Z. K. Yang, X. Q. Wang, J. Wang, J. Luo, Y. F. Li, H. H. Duan, Y. Wu and Y. D. Li, Joule, 2019, 3, 584–594 CrossRef CAS.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- J. Wang, X. Huang, S. Xi, J. M. Lee, C. Wang, Y. Du and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 13532–13539 CrossRef CAS PubMed.
- Q. Li, W. L. Zhu, J. J. Fu, H. Y. Zhang, G. Wu and S. H. Sun, Nano Energy, 2016, 24, 1–9 CrossRef CAS.
- K. Jiang, S. Siahrostami, T. T. Zheng, Y. F. Hu, S. Hwang, E. Stavitski, Y. D. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. T. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- F. Li, G. H. Gu, C. Choi, P. Kolla, S. Hong, T. S. Wu, Y. L. Soo, J. Masa, S. Mukerjee, Y. S. Jung, J. S. Qiu and Z. Y. Sun, Appl. Catal., B, 2020, 277, 119241 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, e1807166 CrossRef PubMed.
- K. Liu, W. A. Smith and T. Burdyny, ACS Energy Lett., 2019, 4, 639–643 CrossRef CAS PubMed.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech, E. Brunner, I. M. Weidinger, J. Zhang, A. V. Krasheninnikov, S. Kaskel, R. Dong and X. Feng, Nat. Commun., 2020, 11, 1409 CrossRef CAS PubMed.
- J. J. Velasco-Velez, R. V. Mom, L. E. Sandoval-Diaz, L. J. Falling, C. H. Chuang, D. Gao, T. E. Jones, Q. Zhu, R. Arrigo, B. Roldán Cuenya, A. Knop-Gericke, T. Lunkenbein and R. Schlogl, ACS Energy Lett., 2020, 5, 2106–2111 CrossRef CAS PubMed.
- J. B. Vennekoetter, R. Sengpiel and M. Wessling, Chem. Eng. J., 2019, 364, 89–101 CrossRef CAS.
- F. P. Garcia de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef CAS PubMed.
- L. Li, A. Ozden, S. Guo, F. P. GarcíA De Arquer, C. Wang, M. Zhang, J. Zhang, H. Jiang, W. Wang, H. Dong, D. Sinton, E. H. Sargent and M. Zhong, Nat. Commun., 2021, 12, 5223 CrossRef CAS PubMed.
- D. Kim, W. Choi, H. W. Lee, S. Y. Lee, Y. Choi, D. K. Lee, W. Kim, J. Na, U. Lee, Y. J. Hwang and D. H. Won, ACS Energy Lett., 2021, 6, 3488–3495 CrossRef CAS.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. T. Liang, E. Stavitsk, H. N. Alshareef and H. T. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- P. Zhu, C. Xia, C.-Y. Liu, K. Jiang, G. Gao, X. Zhang, Y. Xia, Y. Lei, H. N. Alshareef, T. P. Senftle and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2010868118 CrossRef CAS PubMed.
- L. Fan, C. Xia, P. Zhu, Y. Lu and H. Wang, Nat. Commun., 2020, 11 DOI:10.1038/s41467-020-17403-1.
- S. F. Tan, S. W. Chee, Z. Baraissov, H. M. Jin, T. L. Tan and U. Mirsaidov, Adv. Funct. Mater., 2019, 29, 1903242 CrossRef.
- J. Yin, S. Y. Shan, L. F. Yang, D. Mott, O. Malis, V. Petkov, F. Cai, M. S. Ng, J. Luo, B. H. Chen, M. Engelhard and C. J. Zhong, Chem. Mater., 2012, 24, 4662–4674 CrossRef CAS.
- T. Avanesian, S. Dai, M. J. Kale, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 4551–4558 CrossRef CAS PubMed.
- W. Zhan, J. Wang, H. Wang, J. Zhang, X. Liu, P. Zhang, M. Chi, Y. Guo, Y. Guo, G. Lu, S. Sun, S. Dai and H. Zhu, J. Am. Chem. Soc., 2017, 139, 8846–8854 CrossRef CAS PubMed.
- M. D. Argyle and C. H. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef CAS.
- S. Popovic, M. Smiljanic, P. Jovanovic, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef CAS PubMed.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- S. W. Chee, T. Lunkenbein, R. Schlogl and B. R. Cuenya, J. Phys.: Condens. Matter, 2021, 33, 153001 CrossRef CAS PubMed.
- Y. Zhu, J. Wang, H. Chu, Y.-C. Chu and H. M. Chen, ACS Energy Lett., 2020, 5, 1281–1291 CrossRef CAS.
- M. A. Bañares, Catal. Today, 2005, 100, 71–77 CrossRef.
- A. Preet and T. E. Lin, Catalysts, 2021, 11, 594 CrossRef CAS.
- F. D. Mayer, P. Hosseini-Benhangi, C. M. Sanchez-Sanchez, E. Asselin and E. L. Gyenge, Commun. Chem., 2020, 3, 155 CrossRef CAS.
- S. Narayanaru, J. Chinnaiah, K. L. Phani and F. Scholz, Electrochim. Acta, 2018, 264, 269–274 CrossRef CAS.
- C. I. Shaughnessy, D. T. Jantz and K. C. Leonard, J. Mater. Chem. A, 2017, 5, 22743–22749 RSC.
- T. Kai, M. Zhou, Z. Duan, G. A. Henkelman and A. J. Bard, J. Am. Chem. Soc., 2017, 139, 18552–18557 CrossRef CAS PubMed.
- D. T. Jantz and K. C. Leonard, Ind. Eng. Chem. Res., 2018, 57, 7431–7440 CrossRef CAS.
- Y. Kim, A. Jo, Y. Ha, Y. Lee, D. Lee, Y. Lee and C. Lee, Electroanalysis, 2018, 30, 2861–2868 CrossRef CAS.
- N. Sreekanth and K. L. Phani, Chem. Commun., 2014, 50, 11143–11146 RSC.
- L. I. Stephens, N. A. Payne and J. Mauzeroll, Anal. Chem., 2020, 92, 3958–3963 CrossRef CAS PubMed.
- D. Polcari, P. Dauphin-Ducharme and J. Mauzeroll, Chem. Rev., 2016, 116, 13234–13278 CrossRef CAS PubMed.
- D. Wang and L. J. Wan, J. Phys. Chem. C, 2007, 111, 16109–16130 CrossRef CAS.
- H. Feng, X. Xu, Y. Du and S. X. Dou, Electrochem. Energy Rev., 2021, 4, 249–268 CrossRef CAS.
- Y. Liang, J. H. K. Pfisterer, D. McLaughlin, C. Csoklich, L. Seidl, A. S. Bandarenka and O. Schneider, Small Methods, 2019, 3, 1800387 CrossRef.
- C. F. Tsang, A. C. Javier, Y.-G. Kim, J. H. Baricuatro, K. D. Cummins, J. Kim, G. Jerkiewicz, J. C. Hemminger and M. P. Soriaga, J. Electrochem. Soc., 2018, 165, J3350–J3354 CrossRef CAS.
- Y.-G. Kim and M. P. Soriaga, J. Electroanal. Chem., 2014, 734, 7–9 CrossRef CAS.
- Y.-G. Kim, J. H. Baricuatro, A. Javier, J. M. Gregoire and M. P. Soriaga, Langmuir, 2014, 30, 15053–15056 CrossRef CAS PubMed.
- G. H. Simon, C. S. Kley and B. Roldán Cuenya, Angew. Chem., Int. Ed., 2021, 60, 2561–2568 CrossRef CAS PubMed.
- M. R. Nellist, F. A. L. Laskowski, J. J. Qiu, H. Hajibabaei, K. Sivula, T. W. Hamann and S. W. Boettcher, Nat. Energy, 2018, 3, 46–52 CrossRef CAS.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldán Cuenya, Angew. Chem., Int. Ed., 2018, 57, 6192–6197 CrossRef CAS PubMed.
- Y. A. Wu, Z. Yin, M. Farmand, Y.-S. Yu, D. A. Shapiro, H.-G. Liao, W.-I. Liang, Y.-H. Chu and H. Zheng, Sci. Rep., 2017, 7, 42527 CrossRef CAS PubMed.
- Y. A. Wu, L. Li, Z. Li, A. Kinaci, M. K. Y. Chan, Y. Sun, J. R. Guest, I. McNulty, T. Rajh and Y. Liu, ACS Nano, 2016, 10, 3738–3746 CrossRef CAS PubMed.
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C. J. Sun, Z. H. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Z. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS.
- B. D. A. Levin, D. Haiber, Q. Liu and P. A. Crozier, Microsc. Microanal., 2020, 26, 134–138 CrossRef CAS PubMed.
- K. Jungjohann and C. B. Carter, in Transmission Electron Microscopy, Springer International Publishing, 2016, pp. 17–80, DOI:10.1007/978-3-319-26651-0_2.
- M. J. Williamson, R. M. Tromp, P. M. Vereecken, R. Hull and F. M. Ross, Nat. Mater., 2003, 2, 532–536 CrossRef CAS PubMed.
- E. Fahrenkrug, D. H. Alsem, N. Salmon and S. Maldonado, J. Electrochem. Soc., 2017, 164, H358–H364 CrossRef CAS.
- X. Wang, K. Klingan, M. Klingenhof, T. Möller, J. Ferreira de Araújo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl, H. Dau and P. Strasser, Nat. Commun., 2021, 12, 794 CrossRef CAS PubMed.
- J. Vavra, T. H. Shen, D. Stoian, V. Tileli and R. Buonsanti, Angew. Chem., Int. Ed., 2021, 60, 1347–1354 CrossRef CAS PubMed.
- D. A. Shapiro, Y.-S. Yu, T. Tyliszczak, J. Cabana, R. Celestre, W. Chao, K. Kaznatcheev, A. L. D. Kilcoyne, F. Maia, S. Marchesini, Y. S. Meng, T. Warwick, L. L. Yang and H. A. Padmore, Nat. Photonics, 2014, 8, 765–769 CrossRef CAS.
- J. Deng, D. J. Vine, S. Chen, Y. S. G. Nashed, Q. Jin, N. W. Phillips, T. Peterka, R. Ross, S. Vogt and C. J. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2314–2319 CrossRef CAS PubMed.
- T. H. Yoon, Appl. Spectrosc. Rev., 2009, 44, 91–122 CrossRef CAS.
- B. Rösner, S. Finizio, F. Koch, F. Döring, V. A. Guzenko, M. Langer, E. Kirk, B. Watts, M. Meyer, J. Loroña Ornelas, A. Späth, S. Stanescu, S. Swaraj, R. Belkhou, T. Ishikawa, T. F. Keller, B. Gross, M. Poggio, R. H. Fink, J. Raabe, A. Kleibert and C. David, Optica, 2020, 7, 1602–1608 CrossRef.
- S. Finizio, S. Mayr and J. Raabe, J. Synchrotron Radiat., 2020, 27, 1320–1325 CrossRef CAS PubMed.
- E. De Smit, I. Swart, J. F. Creemer, G. H. Hoveling, M. K. Gilles, T. Tyliszczak, P. J. Kooyman, H. W. Zandbergen, C. Morin, B. M. Weckhuysen and F. M. F. De Groot, Nature, 2008, 456, 222–225 CrossRef CAS PubMed.
- D. Guay, J. Stewart-Ornstein, X. Zhang and A. P. Hitchcock, Anal. Chem., 2005, 77, 3479–3487 CrossRef CAS PubMed.
- A. Ulvestad, A. Singer, J. N. Clark, H. M. Cho, J. W. Kim, R. Harder, J. Maser, Y. S. Meng and O. G. Shpyrko, Science, 2015, 348, 1344–1347 CrossRef CAS PubMed.
- I. Robinson and R. Harder, Nat. Mater., 2009, 8, 291–298 CrossRef CAS PubMed.
- M. Abuin, Y. Y. Kim, H. Runge, S. Kulkarni, S. Maier, D. Dzhigaev, S. Lazarev, L. Gelisio, C. Seitz, M. I. Richard, T. Zhou, V. Vonk, T. F. Keller, I. A. Vartanyants and A. Stierle, ACS Appl. Nano Mater., 2019, 2, 4818–4824 CrossRef CAS.
- R. Harder, M. Liang, Y. Sun, Y. Xia and I. K. Robinson, New J. Phys., 2010, 12, 035019 CrossRef.
- W. Cha, A. Ulvestad, M. Allain, V. Chamard, R. Harder, S. J. Leake, J. Maser, P. H. Fuoss and S. O. Hruszkewycz, Phys. Rev. Lett., 2016, 117, 225501 CrossRef CAS PubMed.
- I. Robinson, J. Clark and R. Harder, J. Opt., 2016, 18, 054007 CrossRef.
- A. Ulvestad, A. Tripathi, S. O. Hruszkewycz, W. Cha, S. M. Wild, G. B. Stephenson and P. H. Fuoss, Phys. Rev. B, 2016, 93, 184105 CrossRef.
- R. P. Jansonius, L. M. Reid, C. N. Virca and C. P. Berlinguette, ACS Energy Lett., 2019, 4, 980–986 CrossRef CAS.
- S. Fernández, L. Gao, J. P. Hofmann, J. Carnis, S. Labat, G. A. Chahine, A. J. F. Van Hoof, M. W. G. M. (Tiny) Verhoeven, T. U. Schülli, E. J. M. Hensen, O. Thomas and M.-I. Richard, Nanoscale, 2019, 11, 331–338 RSC.
- X. Zheng, B. Zhang, P. De Luna, Y. Liang, R. Comin, O. Voznyy, L. Han, F. P. García de Arquer, M. Liu, C. T. Dinh, T. Regier, J. J. Dynes, S. He, H. L. Xin, H. Peng, D. Prendergast, X. Du and E. H. Sargent, Nat. Chem., 2018, 10, 149–154 CrossRef CAS PubMed.
- J. Timoshenko and B. Roldán Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed.
- M. Benfatto and C. Meneghini, in Synchrotron Radiation, Springer Berlin Heidelberg, 2015, ch. 7, pp. 213–240, DOI:10.1007/978-3-642-55315-8_7.
- J. J. Velasco-Velez, C. H. Wu, T. A. Pascal, L. F. Wan, J. Guo, D. Prendergast and M. Salmeron, Science, 2014, 346, 831–834 CrossRef CAS PubMed.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- J. Wang, C. Cheng, B. Huang, J. Cao, L. Li, Q. Shao, L. Zhang and X. Huang, Nano Lett., 2021, 21, 980–987 CrossRef CAS PubMed.
- L. P. Zhong, D. K. Chen and S. Zafeiratos, Catal. Sci. Technol., 2019, 9, 3851–3867 RSC.
- K. Roy, L. Artiglia and J. A. van Bokhoven, ChemCatChem, 2018, 10, 666–682 CrossRef CAS.
- A. Kolmakov, D. A. Dikin, L. J. Cote, J. Huang, M. K. Abyaneh, M. Amati, L. Gregoratti, S. Gunther and M. Kiskinova, Nat. Nanotechnol., 2011, 6, 651–657 CrossRef CAS PubMed.
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard 3rd, J. Yano and E. J. Crumlin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CAS.
- J. Kim, H. Ha, W. H. Doh, K. Ueda, K. Mase, H. Kondoh, B. S. Mun, H. Y. Kim and J. Y. Park, Nat. Commun., 2020, 11, 5649 CrossRef CAS PubMed.
- R. Blume, D. Rosenthal, J. P. Tessonnier, H. N. Li, A. Knop-Gericke and R. Schlogl, ChemCatChem, 2015, 7, 2871–2881 CrossRef CAS.
- U. Bergmann, J. Kern, R. W. Schoenlein, P. Wernet, V. K. Yachandra and J. Yano, Nat. Rev. Phys., 2021, 3, 264–282 CrossRef CAS PubMed.
- I. Matsuda and Y. Kubota, Chem. Lett., 2021, 50, 1336–1344 CrossRef CAS.
- G. Geloni, Z. Huang and C. Pellegrini, in X-Ray Free Electron Lasers, The Royal Society of Chemistry, 2017, pp. 1–44, 10.1039/9781782624097-00001.
- C. Pellegrini, A. Marinelli and S. Reiche, Rev. Mod. Phys., 2016, 88, 015006 CrossRef.
- J. Yang, R. Dettori, J. P. F. Nunes, N. H. List, E. Biasin, M. Centurion, Z. Chen, A. A. Cordones, D. P. Deponte, T. F. Heinz, M. E. Kozina, K. Ledbetter, M.-F. Lin, A. M. Lindenberg, M. Mo, A. Nilsson, X. Shen, T. J. A. Wolf, D. Donadio, K. J. Gaffney, T. J. Martinez and X. Wang, Nature, 2021, 596, 531–535 CrossRef CAS PubMed.
- A. H. Zewail, Science, 1988, 242, 1645–1653 CrossRef CAS PubMed.
- L. B. Hoch, P. Szymanski, K. K. Ghuman, L. He, K. Liao, Q. Qiao, L. M. Reyes, Y. Zhu, M. A. El-Sayed, C. V. Singh and G. A. Ozin, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E8011–E8020 CrossRef CAS PubMed.
- J. Sá and J. Szlachetko, Catal. Lett., 2013, 144, 197–203 CrossRef.
- E. L. Clark, M. R. Singh, Y. Kwon and A. T. Bell, Anal. Chem., 2015, 87, 8013–8020 CrossRef CAS PubMed.
- E. L. Clark and A. T. Bell, J. Am. Chem. Soc., 2018, 140, 7012–7020 CrossRef CAS PubMed.
- T. Herl and F. M. Matysik, ChemElectroChem, 2020, 7, 2498–2512 CrossRef CAS.
- M. Plodinec, H. C. Nerl, R. Farra, M. G. Willinger, E. Stotz, R. Schlogl and T. Lunkenbein, Microsc. Microanal., 2020, 26, 220–228 CrossRef CAS PubMed.
- J. Lu, X. Hua and Y. T. Long, Analyst, 2017, 142, 691–699 RSC.
- R. Kas, O. Ayemoba, N. J. Firet, J. Middelkoop, W. A. Smith and A. Cuesta, ChemPhysChem, 2019, 20, 2904–2925 CrossRef CAS PubMed.
- F. Zaera, Chem. Soc. Rev., 2014, 43, 7624–7663 RSC.
- A. J. Wain and M. A. O'Connell, Adv. Phys.: X, 2017, 2, 188–209 CAS.
- J. Heyes, M. Dunwell and B. Xu, J. Phys. Chem. C, 2016, 120, 17334–17341 CrossRef CAS.
- C. Hess, Chem. Soc. Rev., 2021, 50, 3519–3564 RSC.
- Y. Wang, Y. Li, J. Liu, C. Dong, C. Xiao, L. Cheng, H. Jiang, H. Jiang and C. Li, Angew. Chem., 2021, 60, 7681–7685 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- Z. Zhang, S. Sheng, R. Wang and M. Sun, Anal. Chem., 2016, 88, 9328–9346 CrossRef CAS PubMed.
- N. Kumar, C. S. Wondergem, A. J. Wain and B. M. Weckhuysen, J. Phys. Chem. Lett., 2019, 10, 1669–1675 CrossRef CAS PubMed.
- E. Pérez-Gallent, G. Marcandalli, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 16412–16419 CrossRef PubMed.
- S. Lee, D. Kim and J. Lee, Angew. Chem., 2015, 127, 14914–14918 CrossRef.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 CrossRef CAS.
- D. Bohra, J. H. Chaudhry, T. Burdyny, E. A. Pidko and W. A. Smith, Energy Environ. Sci., 2019, 12, 3380–3389 RSC.
- Y. Wang, M. Hatakeyama, K. Ogata, M. Wakabayashi, F. Jin and S. Nakamura, Phys. Chem. Chem. Phys., 2015, 17, 23521–23531 RSC.
- S. Banerjee, X. Han and V. S. Thoi, ACS Catal., 2019, 9, 5631–5637 CrossRef CAS.
- L. Zhang, Z. C. Wei, S. Thanneeru, M. Meng, M. Kruzyk, G. Ung, B. Liu and J. He, Angew. Chem., Int. Ed., 2019, 58, 15834–15840 CrossRef CAS PubMed.
- A. K. Buckley, M. Lee, T. Cheng, R. V. Kazantsev, D. M. Larson, W. A. Goddard III, F. D. Toste and F. M. Toma, J. Am. Chem. Soc., 2019, 141, 7355–7364 CrossRef CAS PubMed.
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- A. Khorshidi, J. Violet, J. Hashemi and A. A. Peterson, Nat. Catal., 2018, 1, 263–268 CrossRef.
- Z. Dai, L. Liu and Z. Zhang, Adv. Mater., 2019, 31, 1805417 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |