
Electrochemical gelation of quantum dots using non-noble metal electrodes at high oxidation potentials†
Chathuranga C.
Hewa-Rahinduwage
,
Karunamuni L.
Silva
,
Xin
Geng
,
Stephanie L.
Brock
 * and
Long
Luo
*
* and
Long
Luo
*
Department of Chemistry, Wayne State University, Detroit, Michigan 48202, USA. E-mail: sbrock@chem.wayne.edu; long.luo@wayne.edu
First published on 30th November 2021
Abstract
Relative to conventional chemical approaches, electrochemical assembly of metal chalcogenide nanoparticles enables the use of two additional levers for tuning the assembly process: electrode material and potential. In our prior work, oxidative and metal-mediated pathways for electrochemical assembly of metal chalcogenide quantum dots (QDs) into three-dimensional gel architectures were investigated independently by employing a noble-metal (Pt) electrode at relatively high potentials and a non-noble metal electrode at relatively low potentials, respectively. In the present work, we reveal competition between the two electrogelation pathways under the condition of high oxidation potentials and non-noble metal electrodes (including Ni, Co, Zn, and Ag), where both pathways are active. We found that the electrogel structure formed under this condition is electrode material-dependent. For Ni, the major phase is oxidative electrogel, not a potential-dependent mixture of oxidative and metal-mediated electrogel that one would expect. A mechanistic study reveals that the metal-mediated electrogelation is suppressed by dithiolates, a side product from the oxidative electrogelation, which block the Ni electrode surface and terminate metal ion release. In contrast, for Co, Ag, and Zn, the electrode surface blockage by dithiolates is less effective than for Ni, such that metal-mediated electrogelation is the primary gelation pathway.
 Long Luo | Dr. Long Luo is currently an assistant professor in the Department of Chemistry at Wayne State University. He received his B.S. (2009) in applied chemistry from Beijing University of Aeronautics and Astronautics and his Ph.D. (2011–2014) in chemistry with Prof. Henry S. White from the University of Utah. Before joining Wayne State University in 2017, he worked as a postdoctoral fellow in the laboratory of Prof. Richard M. Crooks at the University of Texas at Austin. He received the National Science Foundation CAREER Award, NIH Maximizing Investigators’ Research Award (MIRA), and Wayne State University Academy of Scholars Junior Faculty Award and was selected as the Langmuir inaugural Early Career Advisory Board Member. His current research interests include developing electrochemical sensors, electrochemical synthesis of functional nanomaterials and organic drug molecules. |
Introduction
Nanoparticle (NP)-based gels are macroscopic porous structures consisting of interconnected matter (NP building blocks) and pore networks in which the pores may be filled with solvent (i.e., wet gels) or air (i.e., aerogels). Among various 3-dimensionally connected NP assemblies, NP-based gels are unique because they can achieve macroscopic sizes while fully retaining the size-specific properties of the initial nanobuilding blocks.1 The extensive porosity also ensures that each NP in the gel network is accessible to the ambient, leading to high performance in applications that require high surface areas, including electrocatalysis,2–10 (photo)catalysis,11,12 surface-enhanced Raman scattering sensing,13 photo-electrochemical sensing,14 and chemiresistive gas sensing.15,16 Furthermore, the structural features of NP-based gels on different length scales, including the shape and size of NP building blocks, the fractal dimension of the NP network, and the pore size and shape, can be controlled synthetically, offering unprecedented tunability for functional materials.17Synthesis of NP-based gels by assembly requires fine control over the repulsive and attractive forces between colloidal NPs in solution to progressively reduce the repulsive forces (e.g., steric or electrostatic) until they are overcome by the attractive ones (e.g., van der Waals forces, hydrophobic/hydrophilic interactions, and permanent covalent bonding).18 Such control has been previously achieved by slowly stripping ligands off the NP surface through chemical or photooxidation;19,20 adding destabilizers such as alcohol, H2O2, NaBH4, and dopamine;3 or crosslinkers such as metal ions,21 polymers,22,23 and dihydrazide/aldehyde pairs;18 or simply freeze-drying a colloidal NP solution.11,24
Recently, we demonstrated that the controlled assembly of NPs into an NP-based gel was also feasible using electrochemistry.15,25Scheme 1 illustrates two electrochemical gelation or electrogelation mechanisms for metal chalcogenide quantum dots (QDs). In the first mechanism, a positive potential is applied at an inert electrode such as Pt to drive the electrochemical removal (oxidation) of surface-bound thiolate ligand “protecting groups” as dithiolates, exposing the metal ions on the surface of the metal chalcogenide QDs (e.g., Cd on CdS QDs). Following the solvation of these metal ions, the “core” chalcogenides are further electrochemically oxidized to form interparticle dichalcogenide bonds (e.g., 2S2− − 2e → S22− for CdS QDs in Scheme 1(i)). To initiate this oxidative electrogelation or OE-gelation, the electrode potential (E) should be more positive than the oxidation potentials of thiolate ligands and the core chalcogenide (EOE). The QDs in the formed electrogel (OE-gel) are electrically wired by the dichalcogenide bonds.
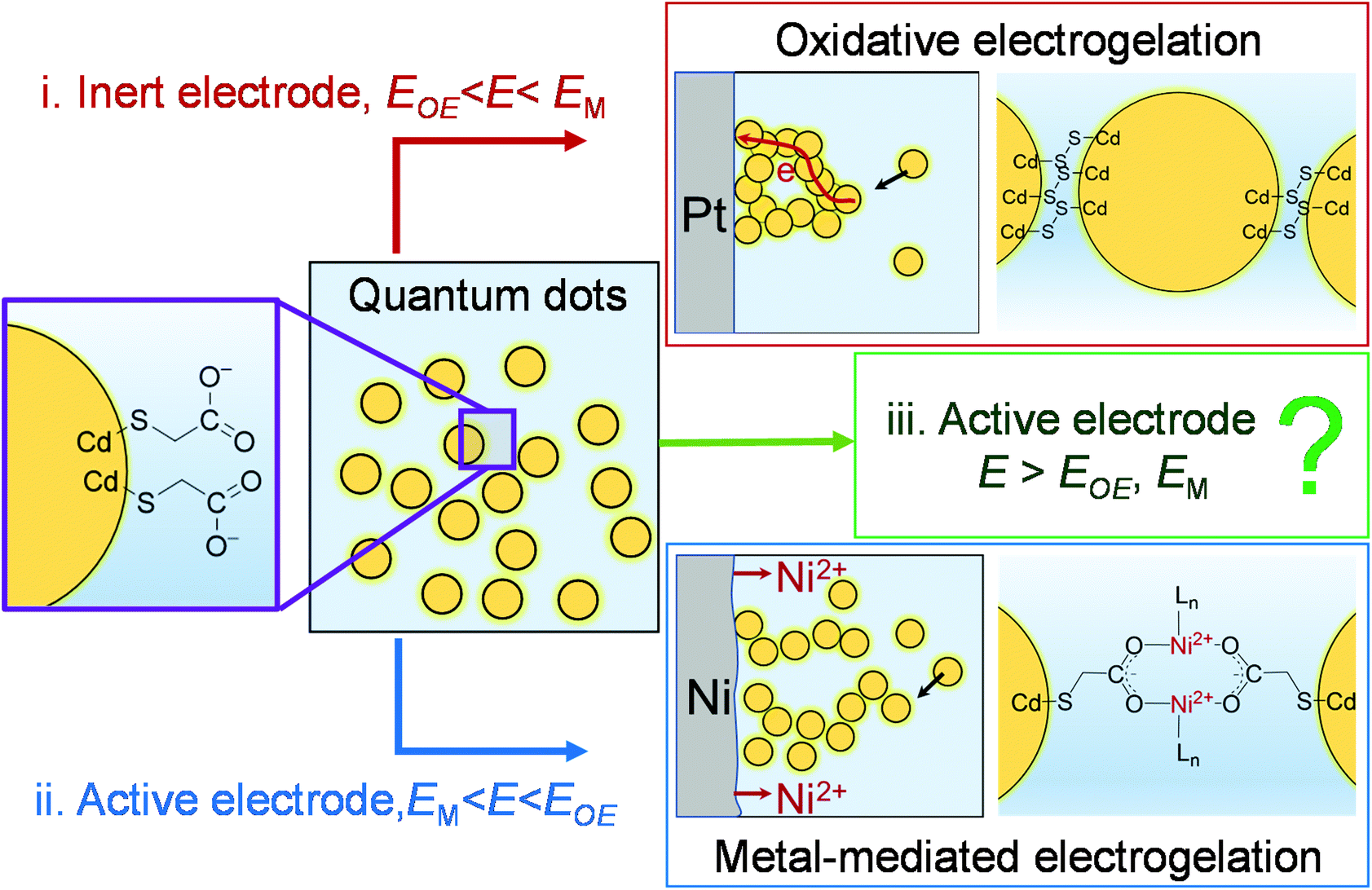 | ||
| Scheme 1 Electrochemical gelation of metal chalcogenide QDs by different mechanisms. (i) Oxidative electrogelation (OE-gelation): the potential of an inert electrode (e.g., Pt) is sufficiently positive to oxidatively remove the thiolate ligands and form dichalcogenides bonds between QDs. (ii) Metal-mediated electrogelation (ME-gelation): an active electrode (e.g., Ni) undergoes electrodissolution due to its low oxidation potential, releasing metal ions to crosslink ligand-capped QDs bearing pendant carboxylates by forming coordination bonds between metal ions and QD-bound carboxylates. (iii) The focus of this study: the potential of an active electrode is positive enough to drive both OE- and ME-gelation. E: electrode potential; EOE: the potential required for OE-gelation; and EM: the electrode oxidation potential. | ||
In the second mechanism, a non-noble metal electrode with a relatively low oxidation potential (EM), such as Ni, Co, Ag, and Zn, is used to in situ electrogenerate metal ion crosslinkers (Ni2+, Co2+, Ag+ or Zn2+) within a colloidal solution of QDs capped with ligands featuring pendant carboxylate groups, forming a metal-mediated electrogel (ME-gel). Unlike the OE-gel, the QDs in a ME-gel are connected by redox-inactive coordinate bonds between Lewis acidic metal cationic linkers and Lewis basic carboxylate terminals of surface-capping ligands (Scheme 1(ii)). To form ME-gels, E should be set between EM and EOE (i.e., EM < E < EOE) so that only the active electrode dissolves to provide metal ions, and QDs cannot be oxidized to trigger the OE-gelation mechanism.
In this work, we studied a special scenario where E is positive enough to electrodissolve the electrode itself, remove the thiolate ligands on QDs, and form dichalcogenide bonds between QDs (i.e., E > EOE, EM in Scheme 1(iii)). We found, surprisingly, that the structure of electrogel formed under this scenario depends on the specific non-noble metal electrode material. Thus, for Ni, the electrogel is dominated by OE-gel rather than the mixture of OE-gel and ME-gel that one would expect. A mechanistic study reveals that the ME-gelation is significantly suppressed by dithiolates, which are produced as a side product from the OE-gelation. The dithiolates block the Ni electrode surface, thereby preventing continued electrodissolution of the electrode. However, for other non-noble metals studied: Co, Ag, and Zn, the electrode surface blockage by dithiolates is less effective than Ni, making ME-gelation the primary gelation pathway.
Results and discussion
Synthesis and characterization of CdS QD electrogels
According to our previous findings, ME-gelation of CdS QDs capped with 11-mercaptoundecanoic acid (MUA) was observed at E > ∼0.5 V vs. Ag/AgCl using a Ni electrode25 and the OE-gelation of these QDs required E > 1.6 V using a Pt electrode.15 Therefore, to conduct a comparative study of QD gelation in regimes governed by ME-gelation only vs. ME- + OE-gelation (i.e., corresponding to the scenarios defined Scheme 1(ii) and (iii), respectively), we used a Ni electrode at two different potentials: 1.0 V and 2.0 V.Fig. 1a shows the QD ME-gel formation on a Ni wire electrode at E = 1 V in a nearly monodisperse CdS QD solution (QD diameter = 2.9 ± 0.4 nm, Fig. S1 and S2a†). A thin layer of gel started appearing on the electrode surface after applying the electrode potential for 10 min and grew thicker with time. The presence of an induction time for gelation arises from the need for a critical stoichiometry of 0.5 metal ions: 1 QD in solution to initiate ME-gelation.25 Under high-magnification scanning transmission electron microscopy (STEM), ∼1–2 nm gaps between CdS QDs are noticeable in the three-dimensional mesoporous gel network, which is characteristic of ME-gels that form by ligand-Ni2+-ligand linkages between QDs (Fig. 1b). The coordination bonds between the MUA ligand and Ni2+ were confirmed by dispersion of the 1 V gel in an aqueous solution of ethylenediaminetetraacetic acid (EDTA), which competes with the carboxylate-terminated thiolate ligands for binding with Ni2+ and disrupts the gel network (Fig. S3b and e). No dissolution was observed for 1 V gels in pure water (control experiment, Fig. S4b and e). To confirm that only Ni oxidation is active at 1 V, we carried out linear sweep voltammetry of MUA-capped QDs and Ni wire (Fig. S5a†). The voltammogram of CdS QDs shows two anodic peaks at 1.5 V and 1.9 V, which correspond to the thiolate ligand oxidation and oxidative crosslinking of QDs, respectively.15 In comparison, the oxidation of Ni wire started at as low as ∼0.4 V (Fig. S5b†), confirming our premise.
 | ||
| Fig. 1 Electrogelation of MUA-capped CdS QDs using a Ni electrode at E = 1 V and 2 V Ag/AgCl/sat. KCl reference electrode. (a and b) photographs and STEM images of a QD gel grown at 1 V. (c and d) photographs and STEM images of a QD gel grown at 2 V. The yellow dashed lines in the high-magnification STEM images highlight the boundaries of individual QDs in the gel. | ||
When E was increased to 2 V, the QD gel appeared on the Ni electrode within a couple of min. The gel rapidly grew in the first 10 min, but its growth slowed down afterward (Fig. 1c). As shown in the high-resolution STEM images (Fig. 1d), the QDs in the 2 V gel are directly connected without the obvious gaps found in the 1 V gel. We also tested the stability of the 2 V gel in an EDTA solution and found that it did not disperse (Fig. S3c, f and S4c, f†). Both results suggest the 2 V gel structurally resembles an OE-gel where QDs are crosslinked via short interparticle covalent dichalcogenide bonds. This conclusion is supported by inductively coupled plasma mass spectrometry (ICP-MS) analysis revealing a significantly lower Ni2+/QD ratio in the 2 V gel (ca. 8) vs. the 1 V gel (ca. 150), as shown in Fig. 2a. Elemental mapping of the 1 V and 2 V gels using STEM energy-dispersive X-ray spectroscopy are consistent with the localization of Ni at the particle interfaces for the 1 V gel, whereas the Ni signal of the 2 V gel is both much weaker (consistent with the low Ni-content obtained from ICP-MS) and more uniform (Fig. S6†). This finding further confirms that QDs in the 1 V gel are mainly connected via ligand-Ni2+-ligand linkages, and those in the 2 V gel are not.
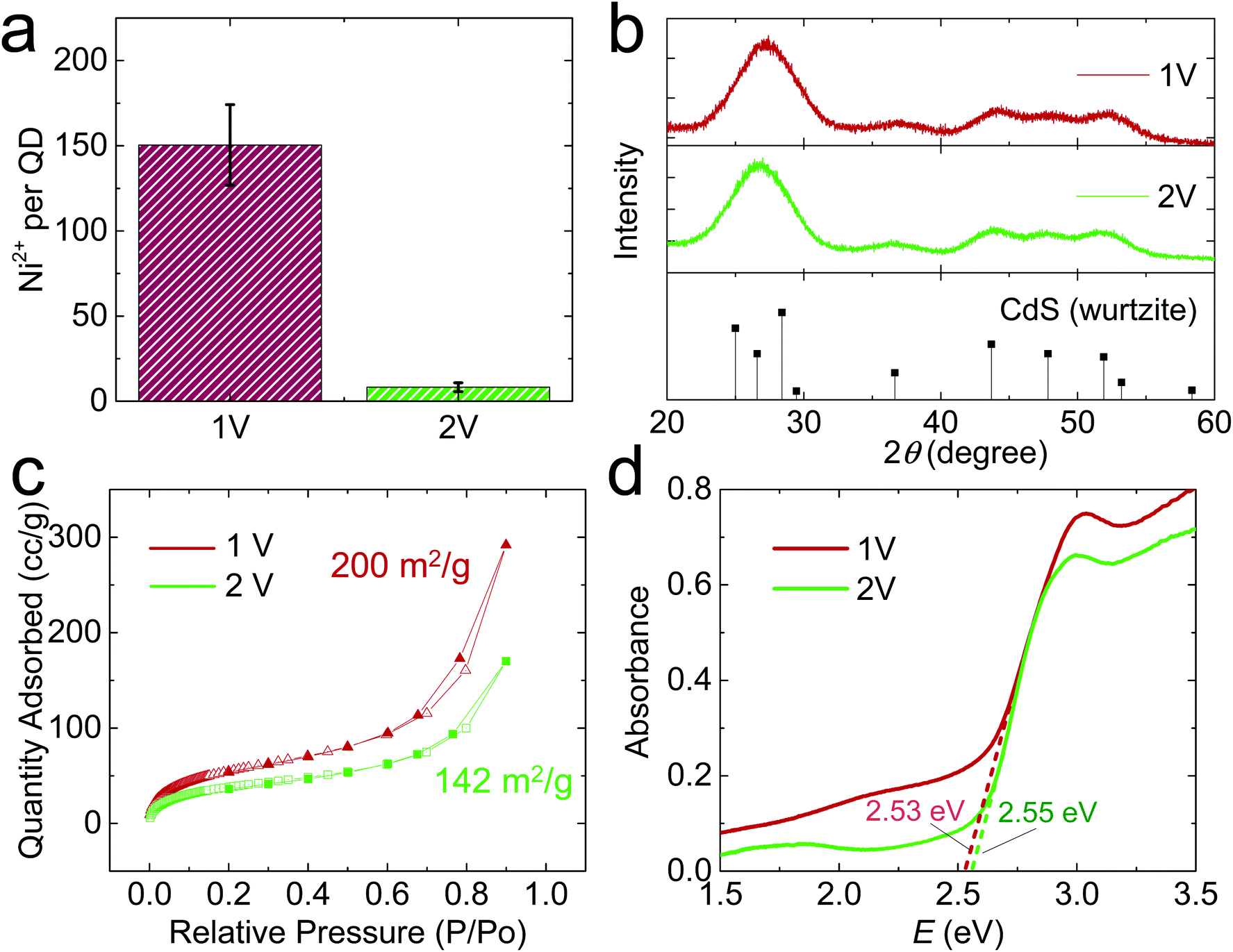 | ||
| Fig. 2 Compositional and structural characterization of QD electrogels synthesized at 1 V and 2 V. (a) Ni2+/QD ratio in the gels. (b) Powder X-ray diffraction (PXRD) patterns. The stick diagram shows the PXRD pattern of hexagonal CdS (wurtzite) as a reference. (c) Nitrogen adsorption–desorption isotherms. (d) Solid-state diffuse reflectance data (converted to absorption). | ||
The infrared (IR) spectra were collected to further identify the ligand–metal interaction in the gels (Fig. S7†). For identical mass loading, we observed much weaker C–H and carboxylate stretching peaks for the 2 V gel relative to the 1 V gel, consistent with oxidative removal of a portion of the MUA ligands during 2 V gelation. In addition, we found a wavenumber difference of 145 cm−1 between asymmetric and symmetric stretching vibrations of the carboxylate groups, indicating the presence of bridging bidentate coordination between Ni2+ and ligands in both gels.25
We also characterized the crystallinity of electrochemically prepared CdS aerogels using powder X-ray diffraction (PXRD). Both 1 V and 2 V gels exhibited the characteristic peaks of hexagonal CdS (PDF 00-001-0780, Fig. 2b). In addition, their PXRD peak widths were similar, suggesting the average crystallite size does not significantly differ under different electrode potentials, consistent with the electron microscopic result that the QD building blocks in both gels are ∼3 nm in diameter (Fig. S2b†). The surface area of the gels was analyzed by nitrogen physisorption, which exhibited a type-IV isotherm, characteristic of a mesoporous material (Fig. 2c). The 1 V and 2 V gels had surface areas of 200 and 142 m2 g−1, respectively, based on the Brunauer–Emmett–Teller model.26 The bandgap values of the electrogel and chemgel were measured by diffuse reflectance spectroscopy (Fig. 2d). The absorption onsets for 1 V and 2 V gels are nearly identical: 2.53 and 2.55 eV, despite the chemically distinct interfacial connectivity between QD building blocks in the gels produced at 1 V and 2 V. The UV/Vis spectra of the two gels also show a peak at 413 nm (Fig. S8a†), but their photoluminescence (PL) spectra (Fig. S8b†) are significantly different. We observed two PL peaks for both samples: a narrow one at ∼470 nm for the characteristic band edge emission and a broad one at ∼545 nm for the trap state emission. However, the 1 V gel shows overall much weaker PL intensity than the 2 V one because thiolate ligands on the 1 V gel effectively scavenge the photogenerated holes in QD gel, quenching its luminescence.27 The 2 V gel is partially denuded of thiolate ligands and Cd ions, with interparticle bonds achieved largely by disulfide linkages. The transformation of the surface results in a large change in the emission intensity of the trap-state. Augmentation of trap-state emission relative to band-edge emission is common for oxidative gelation; recovery of band-edge emission intensity can be achieved by annealing the gels28 or washing with pyridine.29
Electrochemical impedance spectroscopy (EIS) was further used to evaluate the electrochemical processes in a QD solution at 1 V and 2 V. The EIS results shown in Nyquist plots (Fig. S9†) have two semicircles for each potential, characteristic of the behavior of redox-active electrodes.30 The redox-active material on the glassy carbon electrode should be a QD gel thin film formed during the EIS measurement. The first semicircle at high frequency is independent of the bias potential (i.e., 1 V or 2 V) and thus assigned to the electrolyte resistance, and the second one to the electron transfer resistance of QDs, which shrunk significantly at 2 V relative to 1 V due to the facile oxidation of QDs at 2 V, further confirming the different electrogelation mechanisms at 1 V and 2 V.
Electrogelation mechanism
Based on the facile electrooxidation of Ni to Ni2+ at 2 V, we expected gelation conducted at 2 V to proceed by a combination of OE- and ME-mechanisms. However, the ultralow Ni content (Ni2+/QD ratio = 8) in the 2 V gel suggests little-to-no ME-gelation is occurring.We initially considered whether oxidative removal of the MUA/Ni2+/MUA linkers from the 2 V gel could be responsible for the low Ni content. To test this premise, we prepared a metal-mediated gel with a similar Ni2+/QD ratio as the 1 V gel by mixing Ni2+ and QDs at a molar ratio of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (green bar, Fig. 3a), then added 150 equivalents of tetranitromethane (TNM), a commonly used oxidant for chemically induced oxidative QD gelation. We found that the Ni2+/QD ratio in the gel decreased from 164 to 128 (orange bar, Fig. 3a), suggesting that the oxidative removal of the MUA/Ni2+/MUA linkers occurs but is a minor contributor to any Ni2+ losses. Indeed, to reproduce the ultralow Ni2+/QD ratio in the 2 V gel by chemical means, we had to significantly decrease the equivalents of Ni2+ relative to the oxidant; for example, a mixture of 15 Ni2+: 150 TNM:1 QD produced a Ni2+/QD ratio of 7 (red bar, Fig. 3a). This finding suggests the low Ni content in the 2 V gel is due to the limited Ni2+ supply at 2 V. However, control experiments in Fig. 3b indicate that the average current values (iaverage) for Ni electrooxidation in the absence of QDs (green bar) is comparable to that for QD electrogelation at 2 V with a Pt or Ni wire electrode (red and orange bars). That is, in the absence of QDs, the ratio of Ni2+ ions to oxidizing equivalents produced at 2V should be roughly equivalent.
:1 (green bar, Fig. 3a), then added 150 equivalents of tetranitromethane (TNM), a commonly used oxidant for chemically induced oxidative QD gelation. We found that the Ni2+/QD ratio in the gel decreased from 164 to 128 (orange bar, Fig. 3a), suggesting that the oxidative removal of the MUA/Ni2+/MUA linkers occurs but is a minor contributor to any Ni2+ losses. Indeed, to reproduce the ultralow Ni2+/QD ratio in the 2 V gel by chemical means, we had to significantly decrease the equivalents of Ni2+ relative to the oxidant; for example, a mixture of 15 Ni2+: 150 TNM:1 QD produced a Ni2+/QD ratio of 7 (red bar, Fig. 3a). This finding suggests the low Ni content in the 2 V gel is due to the limited Ni2+ supply at 2 V. However, control experiments in Fig. 3b indicate that the average current values (iaverage) for Ni electrooxidation in the absence of QDs (green bar) is comparable to that for QD electrogelation at 2 V with a Pt or Ni wire electrode (red and orange bars). That is, in the absence of QDs, the ratio of Ni2+ ions to oxidizing equivalents produced at 2V should be roughly equivalent.
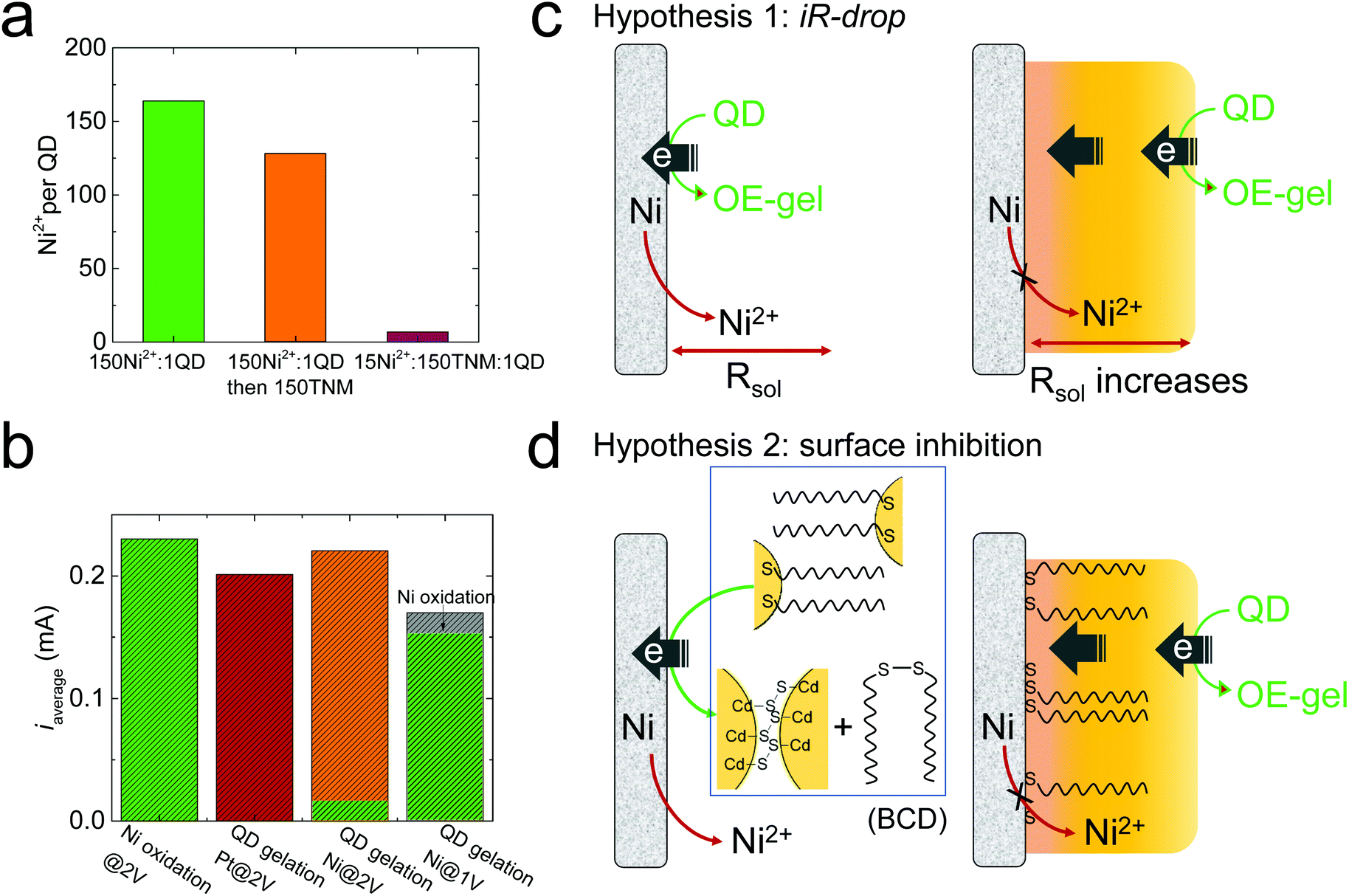 | ||
| Fig. 3 Electrogelation mechanism study. (a) The Ni2+/QD ratio in the QD gels prepared by mixing Ni2+ and QDs at a molar ratio of 150:1 (green bar), and with an additional 150 equivalents of chemical oxidant, tetranitromethane (TNM, orange bar), and with reduced equivalents of Ni2+ (15Ni2+:150TNM:1 QD, red bar). (b) The average current (iaverage) for Ni oxidation in the absence of QDs at 2 V (green bar), QD electrogelation using a Pt electrode at 2 V (red bar), and QD electrogelation using a Ni electrode at 2 V (orange/green two-color bar) and 1 V (gray/green two-color bar). The green portion of the two-color bars represents the specific current for Ni oxidation obtained by analyzing the total amount of Ni2+ released during electrogelation. (c and d) Two hypotheses that explain the suppressed Ni2+ release during QD electrogelation at 2 V. | ||
To verify that the QDs are involved in suppressing Ni oxidation at 2 V, we analyzed the total released Ni2+ during QD gelation. We found that only ∼6% of the total current (i.e., 0.01 mA) was used to generate Ni2+ at 2 V, whereas nearly all the current (0.15 mA out of 0.17 mA) was used for Ni oxidation at 1 V (the green portion in the two-color bars in Fig. 3a represents the specific current for Ni oxidation). Note that the specific current for Ni oxidation at 2 V is only ∼6% of that at 1 V, confirming Ni electrooxidation to Ni2+ was drastically suppressed at 2 V relative to 1 V.
To elucidate the mechanism behind the suppressed Ni2+ release during QD electrogelation at 2 V, we tested the following two hypotheses, as illustrated in Fig. 3c and d.
To test Hypothesis 1, we monitored the solution resistance (R) during electrogelation at 2 V to see if R gradually increased as the gel grew thicker over time, as predicted. However, the value of R stayed relatively constant at ∼400 Ω during electrogelation (Fig. 4a). In addition, we measured the percentage of the total current used to generate Ni2+during electrogelation (QNi%), which should decrease with time as well, according to Hypothesis 1. However, QNi% did not show the predicted decrease and stayed below ∼10% (Fig. 4b). These data confirm that the suppressed Ni2+ release at 2 V is not arising from a QD-gel-induced iR drop.
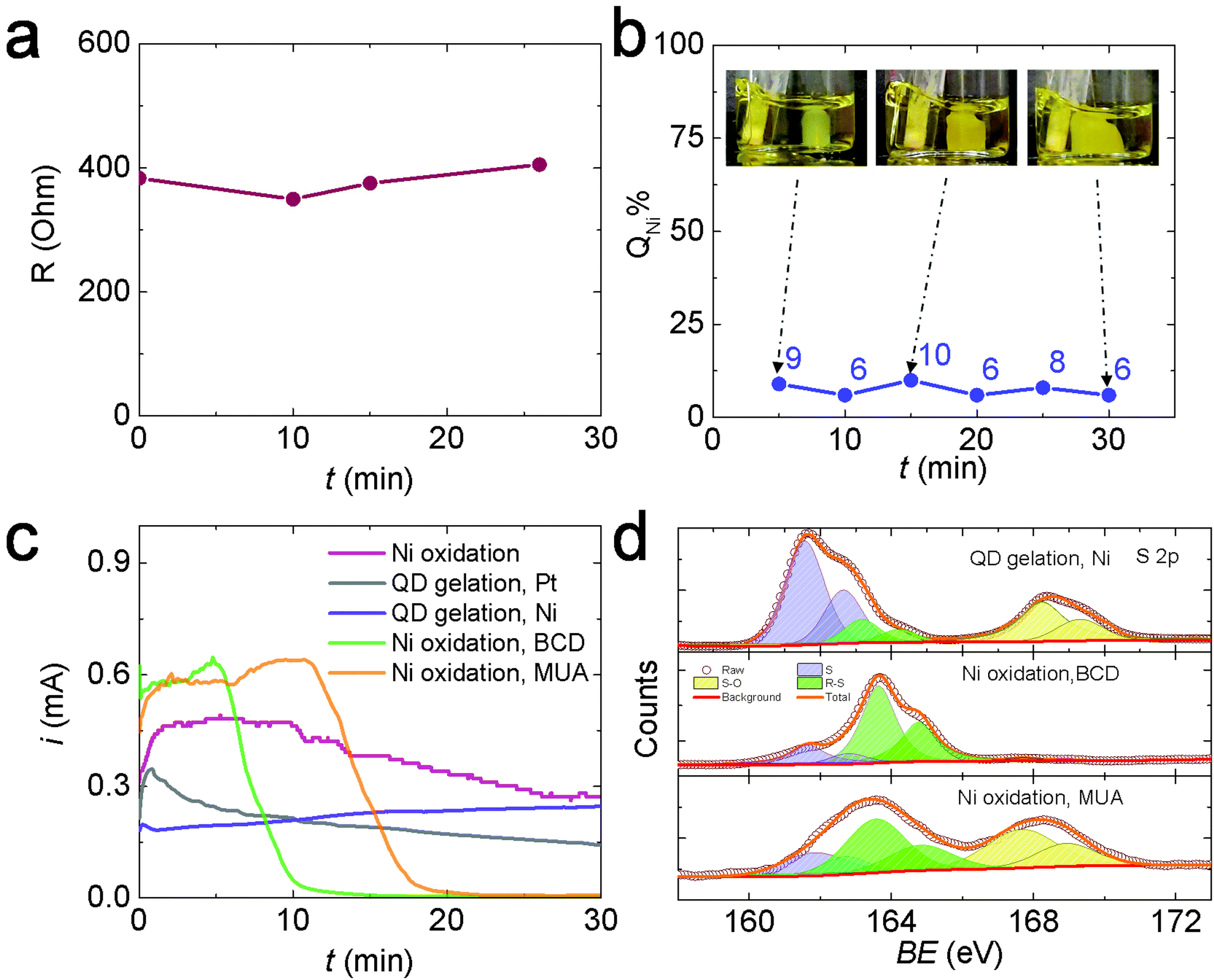 | ||
| Fig. 4 (a) Solution resistance (R) as a function of the electrogelation time (t) at 2 V. (b) Percentage charge used for Ni oxidation to Ni2+ during electrogelation (QNi%) at 2 V after 5 to 30 min. (c) i–t traces for OE-gelation of CdS QDs at 2 V using a Pt electrode (gray line), Ni oxidation in methanol (containing 0.01 M TBAPF-6 but no QDs) at 2 V (red line), QD electrogelation using Ni at 2 V (purple line), and Ni oxidation at 2 V in a 5 mM BCD solution (green line) and 10 mM MUA solution (orange line). (d) XPS spectra of Ni electrodes used for QD electrogelation vs. oxidation in BCD or MUA solutions at 2 V. | ||
To test hypothesis 2, we first evaluated the inhibition effect of BCD on Ni electrooxidation. Specifically, we performed Ni electrooxidation in a QD-free solution containing 5 mM BCD. The concentration of 5 mM was estimated from the experimentally measured CdS QD concentration of 36 μM and the theoretical maximum MUA coverage on a 3.5 nm-diameter CdS QD of ∼270 MUA assuming the MUA/surface Cd ratio = 1, yielding an equivalent MUA concentration of ∼10 mM, or BCD concentration of 5 mM. Fig. 4c shows the current during Ni electrooxidation in the presence of BCD (green line) decreases rapidly after ∼5 min and is nearly completely suppressed by 10 min. A higher BCD concentration further accelerated the inhibition process (Fig. S10†). Similar inhibition was also observed when 10 mM MUA was present, which is not surprising because MUA is oxidized in situ to BCD. The initial BCD formation step extended the time needed to shut down the current to ∼20 min (orange line, Fig. 4c). In comparison, the current during Ni electrooxidation in the absence of BCD or MUA (pink line) and the current during OE-gelation (grey line) only a gradual decrease in current over time. Collectively, these data suggest that the presence of BCD is responsible for the suppressed Ni oxidation at 2 V, lending support to hypothesis 2.
Hypothesis 2 further supposes that the BCD species formed in solution, or a byproduct thereof, are depositing on exposed surfaces of the Ni electrode (i.e., those areas that are not interfacially bound to the colloidal gel network) and suppressing nickel oxidation. Accordingly, we used X-ray photoelectron spectroscopy (XPS) to analyze the surface species on the Ni electrodes after performing electrooxidation at 2 V in a QD solution, a BCD solution, and an MUA solution, each for 30 min. Fig. 4d and Table S1† show the XPS spectra and the results of peak fitting. Three S species were identified in all samples: atomic S at ∼162 eV, thiolate (R–S) at ∼163 eV, and oxidized S (S–O) at 168 eV.31–33 The relative ratios of these S species varied by the electrooxidation condition. Prior studies on the adsorption of thiols and disulfides on a Ni surface from the gas phase have shown that the S–S bond in disulfides and the sulfhydryl (S–H) bond in thiols break below room temperature, forming a self-assembled thiolate (R–S–) monolayer.31,34–37 Similar self-assembly of thiols and disulfides also occurs on Ni surfaces in solution.38 The C–S bonds in the surface bonded thiolates can undergo spontaneous scission to form atomic S and release hydrocarbon at room temperature. The Ni surface promotes the C–S bond scission because its d-band is populated near the Fermi level, resulting in a significant electron density transfer from the Ni d-band to the antibonding molecular orbitals of thiolate molecules, thus weakening, elongating, and ultimately cleaving the S–C bond.39 After S formation, the center of the d-band of Ni is lowered in energy with respect to the clean surface, i.e., the surface is “passivated”, allowing adsorption of intact thiolates.39 Meanwhile, under the electrooxidation conditions, thiolates can also be transformed into sulfinates (SO2−, XPS binding energy = 165.5 eV) and sulfonates (SO3−, XPS binding energy = 168 eV) possibly via a chemical reaction between electrogenerated nickel oxides and thiolates.38 All the chemical and electrochemical processes discussed above lead to the formation of an electrically insulating layer of atomic S, R–S, and S–O species on the Ni electrode surface, inhibiting the Ni electrodissolution and suppressing the ME-gelation pathway. However, OE-gelation is not significantly affected by these processes because the OE-gel can itself serve as the electrode to support the continued growth of OE-gel from its attachment points on the Ni surface. As a result, the 2 V gel formed at a Ni electrode is a de facto OE-gel.
Electrogelation using other non-noble metal electrodes
Thiolates are also known to self-assemble on the surface of other non-noble metals such as Co, Ag, and Zn.40–42 Like Ni, Co, Ag, and Zn can be conveniently converted to their ionic forms under mild oxidation potentials (<1 V vs. Ag/AgCl), so we carried out QD electrogelation using these metals as electrodes for comparison. QD gel formation was observed for Co, Ag, and Zn at both 1 V and 2 V (Fig. 5a–f). The darkened hue of the gel prepared using an Ag electrode is attributed to some degree of cation exchange of the CdS QDs with Ag+, which is known to be facile, to produce Ag2S.43,44 In contrast to the Ni case, the distances between QDs in these 1 V and 2 V gels did not show any apparent differences according to their TEM images. Indeed, noticeable gaps exist between QDs in all samples, suggesting the presence of ligand–metal ion-ligand linkage. The analysis of metal content in these gels is consistent with this observation. As shown in Fig. 5g, the metal ion/QD ratios (Mn+/QD, purple bars) are comparable for gels prepared at 1 V and 2 V using a Co, Ag, or Zn electrode (Co: 64 vs. 81; Zn: 155 vs. 200; and Ag: 191 vs. 263, respectively). In addition, the QM% values for all three electrodes at 2 V are close to 90% or above. These results indicate that the ME-gelation remains the primary gelation pathway even at 2 V for Co, Ag, and Zn electrodes, in direct contradiction to the above Ni results.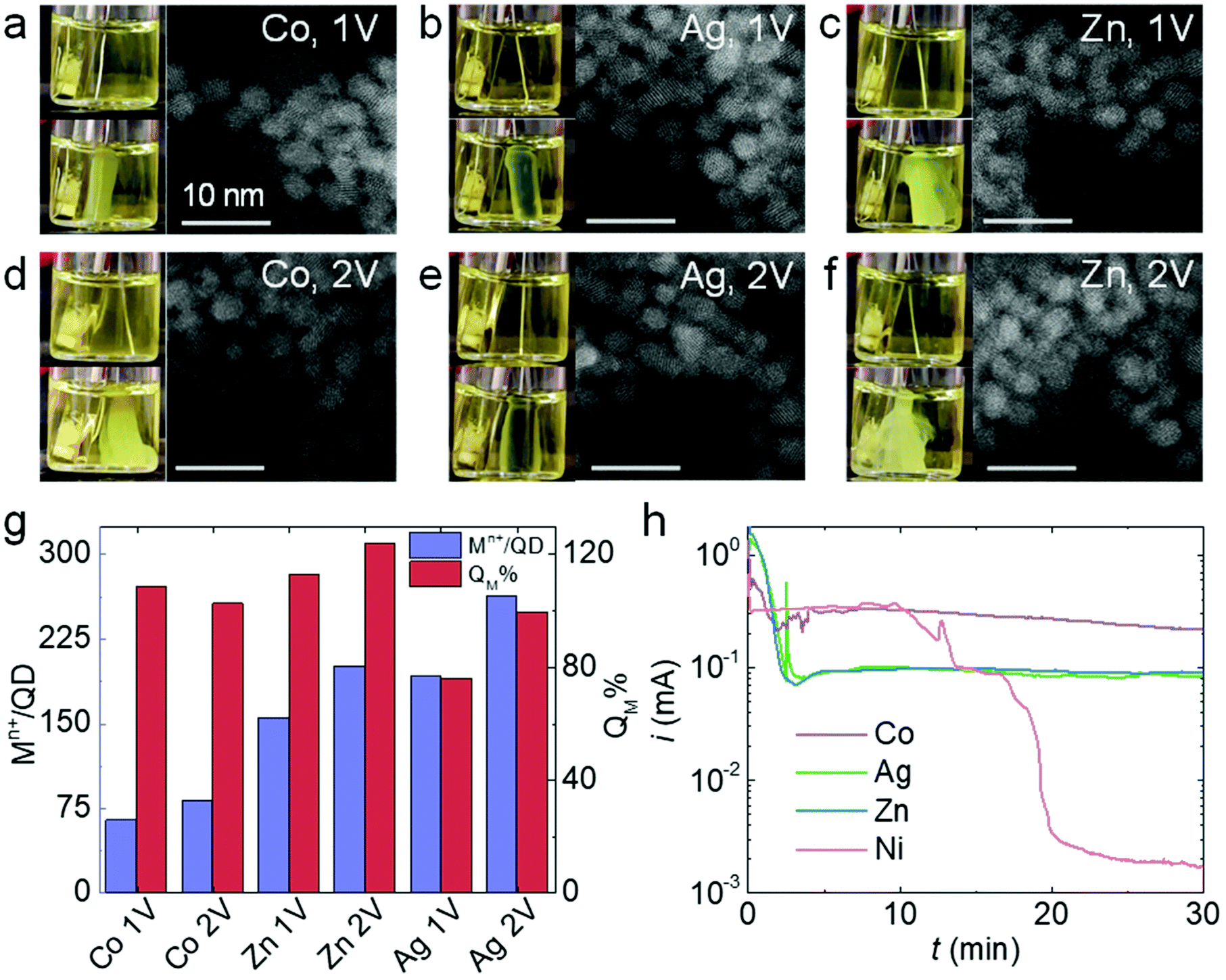 | ||
| Fig. 5 Electrogelation of CdS QDs using Co, Ag, and Zn electrodes. (a–f) Photographs and STEM micrographs of the gels produced using Co, Ag, and Zn electrodes at 1 V and 2 V after 6 min gelation time. (g) Metal ions to QD ratios in the gels (Mn+/QD, purple) and the percentage of charge used for generating metal ions during electrogelation (QM%, red) using Co, Ag, and Zn electrodes at 1 V or 2 V. (h) i–t traces for electrooxidation of Co (purple), Ag (green), Zn (blue), and Ni (red) electrodes in a 10 mM MUA solution at 2 V. | ||
As previously discussed, the adsorption and reaction of BCD electrogenerated by OE-gelation on the Ni electrode inhibits Ni2+ release, shutting down the ME-gelation pathway. To assess the inhibition effect of BCD on the Co, Ag, and Zn electrodes, we conducted the same electrooxidation experiment in an MUA solution at 2 V for 30 min as in Fig. 4c. Although Co, Ag, and Zn electrodes all exhibited a current drop, their current levels stayed around ∼0.1–0.3 mA, rather than diminishing to ∼1 μA as for Ni (Fig. 5h), suggesting the sulfur species only partially block the Co, Ag, and Zn surfaces. The origin of the unique behavior of Ni is likely attributable to its higher activity relative to Co, Ag, and Zn in promoting C–S bond scission of the adsorbed thiolates at room temperature,35,39,42,45 producing a dense insulating layer that is not formed with the other metals.
Conclusion
We have studied the electrochemical gelation of QDs using non-noble metal electrodes including Ni, Co, Ag, and Zn at high oxidation potentials. We found that the electrogelation mechanism shifted from ME-gelation at 1 V to OE-gelation at 2 V when a Ni electrode was used. The mechanism switch is caused by the inhibition effect of BCD, a product from OE-gelation, on the electrodissolution of Ni to Ni2+ at 2 V, which suppresses the ME-gelation pathway due to the limited availability of the Ni2+ crosslinkers. However, for Co, Ag, and Zn electrodes, ME-gelation remains the dominant gelation pathway at 2 V because the electrogenerated BCD cannot inhibit the electrooxidation of these electrodes as effectively as Ni, possibly due to their low activity in promoting C–S bond breakage of the adsorbed thiolates at room temperature.Experimental section
Chemicals and materials
Tetrabutylammonium hexafluorophosphate (TBAPF-6), OmniTrace nitric acid [HNO3, 67–71%], tetranitromethane (TNM), bis(trimethylsilyl)sulfide (TMS), 1-tetradecylphosphonic acid (TDPA), trioctylphosphine oxide [TOPO, 99%], 11-mercaptoundecanoic acid (MUA), and tetramethylammonium hydroxide (TMAH) were purchased from Millipore Sigma; Ag wire [0.010′′ diameter, 99.99%] was purchased from A–M Systems; Co wire [0.25 mm diameter, 99.995%], Ni wire [0.25 mm diameter, 99.98%], and Pt wire [0.25 mm diameter, 99.99%] were purchased from Alfa-Aesar; Ni(II) chloride hexahydrate [NiCl2·6H2O, 99%] was purchased from Avantor chemicals; ethylenediamine tetra-acetic acid, disodium salt dihydrate [Na2EDTA, >99%] was purchased from Fisher Scientific chemicals; cadmium oxide [CdO, 99.99%], and trioctylphosphine [TOP >85%] were purchased from Strem chemicals; and the deionized [DI, 18.2 MΩ cm, TOC <3 ppb] water was used for washing electrodes and aqueous solution preparation.Synthesis of QDs
A modified hot injection method was used for CdS QD synthesis.46,47 The synthesis procedure and the purification steps are described in the ESI.†QD ligand exchange
After the synthesis, the trioctylphosphine oxide (TOPO) ligand shell on QDs was exchanged with MUA by following the method explained in ESI.† MUA-capped QDs were dispersed in methanol, and the concentration was determined using literature-reported absorptivity values.48 Before gelation, the QD concentration was adjusted to 40 μM by serial dilution.QD OE-gelation using a Pt electrode
A CHI 650E potentiostat was used to apply the potentials (bulk electrolysis technique) for the gelation. The iR drop was manually compensated before every run.The gelation mixture was prepared by mixing 900 μL of 40 μM QD solution with 100 μL of 0.1 M TBAPF-6 in methanol. TBAPF-6 was added as the electrolyte to reduce the solution resistance during electrochemical reactions. TBAPF-6 was selected because it was found to be the optimal electrolyte that provides a good ionic conductivity without sacrificing the stability of the colloidal QDs. Other electrolytes such as LiClO4, KCl, tetrabutylammonium chloride, and tetrabutylammonium bromide easily destabilize the QD solution, resulting in precipitation. An Ag/AgCl/sat. KCl electrode was used as the reference electrode. Two Pt wires (d = 0.25 mm) were used as the working and counter electrodes. Pt electrodes were cleaned before the gelation by running 50 cyclic voltammograms between 1.1 and −0.23 V in a 0.1 M H2SO4 aqueous solution at a scan rate of 0.1 V s−1. Then the Pt electrodes were washed with plenty of DI water and methanol, dried, and used for electrogelation. The counter electrode was made into a shape of a loop to enhance its surface area. OE-gelation was carried out by applying a potential of 2 V.
QD electrogelation using Ni, Co, Zn, and Ag electrodes
Like OE-gelation using a Pt electrode, QD electrogelation using Ni, Co, Zn, and Ag electrodes was carried out by first cleaning the electrode. These metal wires (d = 0.25 mm) were polished with a 400-grit 3 M sandpaper to remove the surface oxide layer. The wires were then dipped in DI water and sonicated for 1 minute. Next, the wire electrodes were washed with plenty of DI water and methanol, dried, and used for the electrogelation by applying an electrode potential of1 V or 2 V. The gelation mixture was also prepared by mixing 900 μL of 40 μM QD solution with 100 μL of 0.1 M TBAPF6 in methanol. An Ag/AgCl/sat. KCl electrode was used as the reference electrode and the Pt wire electrode as the counter electrode. Current–time traces of these wires electrodes were collected by dipping the clean metal wires in corresponding electrolyte solutions. The pH of all MUA- and BCD-containing electrolyte solutions was adjusted to 10 using TMAH.Chemical gelation of QDs
The preparation of metal-ion crosslinked gels was performed by direct addition of Ni2+ ions (from NiCl2) to a colloidal MUA-capped CdS QD dispersion to form a metal-mediated chemical gel (MC-gel). First, the Ni2+ ion solution (0.05 M) was prepared by dissolving 0.30 g of NiCl2·6H2O in 25.0 mL of absolute ethanol. 54 μL of freshly prepared 0.05 M Ni2+ ion solution was added to 0.5 mL of MUA-capped CdS QD solution (36 μM) dispersed in methanol (CdS QDs:Ni2+ = 1:150) in a glass vial. The mixture was shaken vigorously for 2 seconds, and gel formation occurred within 60 seconds. The wet gel was kept in the dark in a desiccator to prevent photooxidative gelation for 24 h before solvent exchange.
The preparation of oxidatively assembled chemical gel (OC-gel) was performed by adding 10.8 μL of freshly prepared 3% TNM (in acetone) to 0.5 mL of MUA-capped CdS QD solution (36 μM) dispersed in methanol (CdS QDs:TNM = 1:150) in a glass vial. The mixture was shaken vigorously for 2 seconds, and gel formation occurred within 30 minutes. The wet gel was kept in the dark in a desiccator to prevent photooxidative gelation for 24 h before solvent exchange.
To assess the possible oxidative removal of ligand–metal ion-ligands from a MC-gel, we first added 54 μL of 0.05 M Ni2+ solution to a 0.5 mL of MUA-capped CdS QD solution (36 μM) dispersed in methanol (CdS QDs:Ni2+ = 1:150) in a glass vial to form the MC-gel. After 10 min, 10.8 μL of 3% TNM was added to the gel solution and the mixture was shaken vigorously for 2 seconds. The low Ni content gel with a Ni2+/QD ratio of 7 was prepared by adding 10.8 μL of 3% TNM and 2.5 μL of Ni2+ solutions simultaneously to a 0.5 mL of 36 μM MUA-capped CdS QD solution.
EDTA test
A 0.01 M EDTA solution was prepared by dissolving 0.093 g of Na2EDTA in 25.0 mL of DI water, and the pH was adjusted to 12 using 1 M NaOH solution. 0.9 mL of EDTA solution was added to a vial containing gel and mixed gently (Ni2+:EDTA = ∼1:1). As a control, H2O was added to a separate set of vials containing the gels.
Aerogel preparation
The supernatant QD dispersion in the gelation vial was removed carefully without completely drying the gel. The vial was then filled with acetone without breaking the gel into pieces. Next, to wash any excess QDs and remaining methanol, the supernatant was replaced with another new portion of acetone. The vial was stored in the dark, and the acetone washing was continued twice a day for four days. Next, the wet gel was placed in a Tousimis Autosamdri-931 critical point dryer to replace acetone in the sample with liquid CO2 completely. Finally, the sample was dried by bringing the CO2 to a supercritical state.Inductively coupled plasma mass spectrometry (ICP-MS) analysis
Samples for ICP-MS analysis were prepared by digesting the QD gels in conc. HNO3 for 6 hours and diluting with a 2% HNO3 solution to get the concentration into the calibration range of 1 ppb to 200 ppb. The Mn+/QD ratio in the gel (M = Ni, Co, Ag, and Zn, and n is the charge of the corresponding ion) was calculated as below. First, the Cd2+ and Mn+ concentrations obtained in ppb were converted into the number of atoms using their molecular weight and the Avogadro number. Then, the volume of a QD was calculated using the particle radius determined from the UV-vis peak absorbance.48 Next, the number of Cd atoms present in a QD was calculated using the volume of a QD, CdS density (4.8 g cm−3), and molecular weight. The number of QDs present in the sample was found after dividing Cd2+ by Cd atoms per QD. Finally, the number of Mn+ ions was divided by the number of QDs.The percentage of the total current used to generate Mn+ during electrogelation (QM%) was calculated using the following equation.
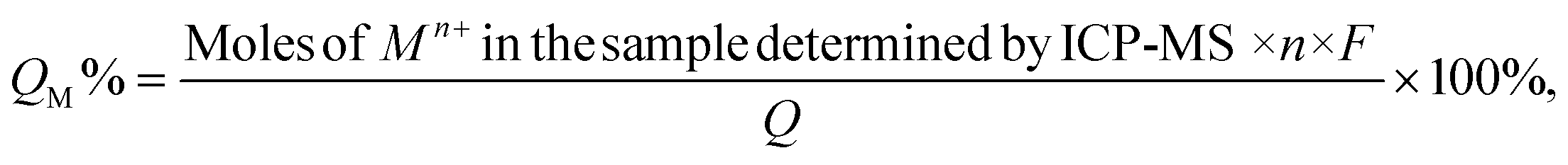
Transmission electron microscopy
QD dispersion and gels were diluted and drop cast onto the Formvar/Carbon-coated 200 mesh Cu grids (Ted Pella, USA) to collect TEM and STEM images using JEOL 3100R05 electron microscope. ImageJ 1.51j8 software was used to analyze the particle size distributions in QD dispersion and the gel.Powder X-ray diffraction
Aerogel samples were mounted on a Bruker D2 Phaser X-ray diffractometer to collect the PXRD data. The powder diffraction file (PDF) database of the International Center for Diffraction Data was used to confirm the collected data by comparison.Nitrogen physisorption isotherms
The aerogel surface areas were measured by fitting nitrogen physisorption isotherm data to a Brunauer–Emmett–Teller (BET) model. Data was collected using a Micrometrics 3Flex Version 5.02 analyzer at 77 K. Samples were degassed for 14 hours at 150 °C before collecting the data.Diffuse reflectance and UV/Vis
A JASCO V-570 UV/VIS/NIR spectrometer with an integrating sphere was used to collect the reflectance and UV/Vis data. In the diffuse reflectance measurements, aerogel samples were diluted with BaSO4. In the UV/Vis measurements, aerogel samples were prepared by sonication in methanol to obtain a uniform dispersion.X-ray photoelectron spectroscopy
The electrode samples were washed with methanol and dried in ambient for 24 hours before collecting the spectra using an Al K-alpha X-ray Photoelectron Spectrometer (Thermo Scientific, K-alpha). Data were analyzed using Thermo Avantage v5.9921 software, and all the spectra were calibrated according to the C 1s peak at 284.8 eV.Photoluminescence (PL)
PL data were collected using a JASCO FP-6500 spectrofluorometer. MUA-capped CdS aerogel samples were dispersed in methanol by sonication in the dark inside a vial filled with Ar. The UV/Vis spectra were obtained for both CdS QDs and gel samples before the PL measurements. Both samples were diluted to get a maximum absorbance around 0.5 using methanol. Samples were excited at the wavelength of maximum absorbance (413 nm).Fourier-transform infrared (FTIR) Spectroscopy
FTIR data of on the aerogels (1 V and 2 V) were collected in transmission mode using a KBr pellet prepared with a mass ratio of 1 mg of sample to 500 mg of KBr using a Bruker Tensor 27 FTIR spectrophotometer.Linear sweep voltammetry (LSV)
LSV data of QDs was collected using a CHI 650E potentiostat, with a 3 mm-diameter glassy carbon electrode as the working electrode, a Pt wire as the counter electrode, and an Ag/AgCl/sat. KCl as the reference electrode. The glassy carbon electrode was first polished with a series of alumina powders (1, 0.3, and 0.01 μm) and then sonicated and washed with a large amount of DI water and methanol. A blank run was performed using the cleaned glassy carbon electrode dipped in the electrolyte. The CdS QD-coated carbon electrode was prepared by drop-casting 12 μL of ∼400 μM CdS QD solution onto a clean carbon electrode, followed by drying in air. The electrolyte solution was 10 mM TBAPF-6 in methanol solution. The scan rate was 0.1 V s−1.Electrochemical impedance spectroscopy (EIS)
Electrochemical impedance spectroscopy (EIS). EIS data were collected at a bias potential of 1 V and 2 V in MUA-capped CdS QD dispersion (40 μM) at room temperature using a CHI 650E potentiostat. A Pt wire and an Ag/AgCl electrode were used as the counter and the reference electrodes. The frequency range was set from 100 kHz to 1 Hz with an excitation amplitude of ±5 mV. Data were collected at 12 points per decade.Conflicts of interest
The authors declare there are no conflicts of interest.Acknowledgements
This work was financially supported by the start-up funds of L. L., Wayne State University (WSU), National Science Foundation (NSF) grants to S. L. B. (#1709776, #1904775), and WSU Rumble Fellowships to C. C. H. and K. L. S. The authors thank E. Nikolla at WSU for performing physisorption measurements, and T. Ma at the Michigan Center for Materials Characterization, University of Michigan, for TEM imaging using a JEOL3100R05 funded by NSF grant # 723032. This work also made use of an XPS partially funded by NSF grant #1849578, a JEOL-2010 TEM supported by NSF grant #0216084, a JEOL JSM-7600F SEM supported by NSF grant #0922912, and the PXRD Facility supported by NSF grant #1427926.References
- F. Matter, A. L. Luna and M. Niederberger, Nano Today, 2020, 30, 100827 CrossRef CAS.
- D. Müller, D. Zámbó, D. Dorfs and N. C. Bigall, Small, 2021, 17, 2007908 CrossRef PubMed.
- X. Jiang, R. Du, R. Hübner, Y. Hu and A. Eychmüller, Matter, 2021, 4, 54–94 CrossRef CAS.
- R. Du, W. Jin, H. Wu, R. Hübner, L. Zhou, G. Xue, Y. Hu and A. Eychmüller, J. Mater. Chem. A, 2021, 9, 17189–17197 RSC.
- R. Du, J.-O. Joswig, X. Fan, R. Hübner, D. Spittel, Y. Hu and A. Eychmüller, Matter, 2020, 2, 908–920 CrossRef PubMed.
- R. Du, J. Wang, Y. Wang, R. Hübner, X. Fan, I. Senkovska, Y. Hu, S. Kaskel and A. Eychmüller, Nat. Commun., 2020, 11, 1590 CrossRef CAS PubMed.
- R. Du, W. Jin, R. Hübner, L. Zhou, Y. Hu and A. Eychmüller, Adv. Energy Mater., 2020, 10, 1903857 CrossRef CAS.
- X. Fan, S. Zerebecki, R. Du, R. Hübner, G. Marzum, G. Jiang, Y. Hu, S. Barcikowki, S. Reichenberger and A. Eychmüller, Angew. Chem., 2020, 132, 5755–5760 CrossRef.
- B. Cai and A. Eychmüller, Adv. Mater., 2019, 31, 1804881 CrossRef PubMed.
- R. Du, X. Fan, X. Jin, R. Hübner, Y. Hu and A. Eychmüller, Matter, 2019, 1, 39–56 CrossRef.
- R. Du, J. O. Joswig, R. Hübner, L. Zhou, W. Wei, Y. Hu and A. Eychmüller, Angew. Chem., 2020, 132, 8370–8377 CrossRef.
- X. Fan, B. Cai, R. Du, R. Hübner, M. Georgi, G. Jiang, L. Li, M. Samadi Khoshkhoo, H. Sun and A. Eychmüller, Chem. Mater., 2019, 31, 10094–10099 CrossRef CAS.
- L. Zhou, Y. Peng, N. Zhang, R. Du, R. Hübner, X. Wen, D. Li, Y. Hu and A. Eychmüller, Adv. Opt. Mater., 2021, 9, 2100352 CrossRef CAS.
- A. Schlosser, L. C. Meyer, F. Lübkemann, J. F. Miethe and N. C. Bigall, Phys. Chem. Chem. Phys., 2019, 21, 9002–9012 RSC.
- C. C. Hewa-Rahinduwage, X. Geng, K. L. Silva, X. Niu, L. Zhang, S. L. Brock and L. Luo, J. Am. Chem. Soc., 2020, 142, 12207–12215 CrossRef CAS PubMed.
- X. Geng, S. Li, L. Mawella-Vithanage, T. Ma, M. Kilani, B. Wang, L. Ma, C. C. Hewa-Rahinduwage, A. Shafikova, E. Nikolla, G. Mao, S. L. Brock, L. Zhang and L. Luo, Nat. Commun., 2021, 12, 4895 CrossRef CAS PubMed.
- P. Rusch, D. Zambo and N. C. Bigall, Acc. Chem. Res., 2020, 53, 2414–2424 CrossRef CAS PubMed.
- Z. M. Sherman, A. M. Green, M. P. Howard, E. V. Anslyn, T. M. Truskett and D. J. Milliron, Acc. Chem. Res., 2021, 54, 798–807 CrossRef CAS PubMed.
- I. U. Arachchige and S. L. Brock, Acc. Chem. Res., 2007, 40, 801–809 CrossRef CAS PubMed.
- I. U. Arachchige and S. L. Brock, J. Am. Chem. Soc., 2006, 128, 7964–7971 CrossRef CAS PubMed.
- D. Zambo, A. Schlosser, P. Rusch, F. Lubkemann, J. Koch, H. Pfnur and N. C. Bigall, Small, 2020, 16, e1906934 CrossRef PubMed.
- C. A. Saez Cabezas, G. K. Ong, R. B. Jadrich, B. A. Lindquist, A. Agrawal, T. M. Truskett and D. J. Milliron, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8925 CrossRef CAS PubMed.
- C. A. Saez Cabezas, Z. M. Sherman, M. P. Howard, M. N. Dominguez, S. H. Cho, G. K. Ong, A. M. Green, T. M. Truskett and D. J. Milliron, Nano Lett., 2020, 20, 4007–4013 CrossRef CAS PubMed.
- A. Freytag, S. Sánchez-Paradinas, S. Naskar, N. Wendt, M. Colombo, G. Pugliese, J. Poppe, C. Demirci, I. Kretschmer, D. W. Bahnemann, P. Behrens and N. C. Bigall, Angew. Chem., Int. Ed., 2016, 55, 1200–1203 CrossRef CAS PubMed.
- C. C. Hewa-Rahinduwage, K. L. Silva, S. L. Brock and L. Luo, Chem. Mater., 2021, 33, 4522–4528 CrossRef CAS.
- L. D. Gelb and K. Gubbins, Langmuir, 1998, 14, 2097–2111 CrossRef CAS.
- I.-S. Liu, H.-H. Lo, C.-T. Chien, Y.-Y. Lin, C.-W. Chen, Y.-F. Chen, W.-F. Su and S.-C. Liou, J. Mater. Chem., 2008, 18, 675–682 RSC.
- J. L. Mohanan, I. U. Arachchige and S. L. Brock, Science, 2005, 307, 397–400 CrossRef CAS PubMed.
- I. U. Arachchige and S. L. Brock, J. Am. Chem. Soc., 2007, 129, 1840–1841 CrossRef CAS PubMed.
- B.-A. Mei, J. Lau, T. Lin, S. H. Tolbert, B. S. Dunn and L. Pilon, J. Phys. Chem. C, 2018, 122, 24499–24511 CrossRef CAS.
- T. S. Rufael, D. R. Huntley, D. R. Mullins and J. L. Gland, J. Phys. Chem. B, 1998, 102, 3431–3440 CrossRef CAS.
- X. Chu, P. Guo, Y. Pei, S. Yan, H. Hu, M. Qiao, K. Fan, B. Zong and X. Zhang, J. Phys. Chem. C, 2007, 111, 17535–17540 CrossRef CAS.
- S. Kane and J. Gland, Surf. Sci., 2000, 468, 101–108 CrossRef CAS.
- F. Blobner, P. N. Abufager, R. Han, J. Bauer, D. A. Duncan, R. J. Maurer, K. Reuter, P. Feulner and F. Allegretti, J. Phys. Chem. C, 2015, 119, 15455–15468 CrossRef CAS.
- T. S. Rufael, D. R. Huntley, D. R. Mullins and J. L. Gland, J. Phys. Chem., 1995, 99, 11472–11480 CrossRef CAS.
- A. Fernandez, J. P. Espinos, A. R. Gonzalez-Elipe, M. Kerkar, P. B. J. Thompson, J. Ludecke, G. Scragg, A. V. d. Carvalho, D. P. Woodruff, M. Fernandez-Garcia and J. C. Conesa, J. Condens. Matter Phys., 1995, 7, 7781–7796 CrossRef CAS.
- C. J. Fisher, D. P. Woodruff, R. G. Jones, B. C. C. Cowie and V. Formoso, Surf. Sci., 2002, 496, 73–86 CrossRef CAS.
- Z. Mekhalif, J. Riga, J. J. Pireaux and J. Delhalle, Langmuir, 1997, 13, 2285–2290 CrossRef CAS.
- C. Vericat, M. E. Vela, G. Corthey, E. Pensa, E. Cortés, M. H. Fonticelli, F. Ibañez, G. E. Benitez, P. Carro and R. C. Salvarezza, RSC Adv., 2014, 4, 27730–27754 RSC.
- A. N. Caruso, L. G. Wang, S. S. Jaswal, E. Y. Tsymbal and P. A. Dowben, J. Mater. Sci., 2006, 41, 6198–6206 CrossRef CAS.
- M. M. Walczak, C. Chung, S. M. Stole, C. A. Widrig and M. D. Porter, J. Am. Chem. Soc., 1991, 113, 2370–2378 CrossRef CAS.
- P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y. T. Tao, A. N. Parikh and R. G. Nuzzo, J. Am. Chem. Soc., 1991, 113, 7152–7167 CrossRef CAS.
- F. Lubkemann, P. Rusch, S. Getschmann, B. Schremmer, M. Schafer, M. Schulz, B. Hoppe, P. Behrens, N. C. Bigall and D. Dorfs, Nanoscale, 2020, 12, 5038–5047 RSC.
- N. Nair and B. R. Sankapal, New J. Chem., 2016, 40, 10144–10152 RSC.
- S. Devillers, A. Hennart, J. Delhalle and Z. Mekhalif, Langmuir, 2011, 27, 14849–14860 CrossRef CAS PubMed.
- J. Joo, H. B. Na, T. Yu, J. H. Yu, Y. W. Kim, F. Wu, J. Z. Zhang and T. Hyeon, J. Am. Chem. Soc., 2003, 125, 11100–11105 CrossRef CAS PubMed.
- L. Korala and S. L. Brock, J. Phys. Chem. C, 2012, 116, 17110–17117 CrossRef CAS PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06615c |
| This journal is © The Royal Society of Chemistry 2021 |