
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFusarochromene, a novel tryptophan-derived metabolite from Fusarium sacchari†
James W.
Marshall
 a,
Kate M. J.
de Mattos-Shipley
*b,
Iman A. Y.
Ghannam
ac,
Asifa
Munawar
b,
Jonathan C.
Killen
a,
Colin M.
Lazarus
b,
Russell J.
Cox
ad,
Christine L.
Willis
a and
Thomas J.
Simpson
*a
a,
Kate M. J.
de Mattos-Shipley
*b,
Iman A. Y.
Ghannam
ac,
Asifa
Munawar
b,
Jonathan C.
Killen
a,
Colin M.
Lazarus
b,
Russell J.
Cox
ad,
Christine L.
Willis
a and
Thomas J.
Simpson
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: tom.simpson@bristol.ac.uk
bSchool of Biological Sciences, University of Bristol, 24 Tyndall Avenue, Bristol, BS8 1TQ, UK. E-mail: kd4495@bristol.ac.uk
cChemistry of Natural and Microbial Products Department, Pharmaceutical and Drug Industries Research Division, National Research Centre, El-Bohouth Street, Dokki, Cairo, 12622, Egypt
dInstitute for Organic Chemistry and BMWZ, Leibniz University of Hannover, Schneiderberg 38, 30167, Hannover, Germany
First published on 23rd October 2020
Abstract
Fusarochromene isolated from the plant pathogenic fungus, Fusarium sacchari is closely related to a group of mycotoxins including fusarochromanone previously isolated from various Fusaria spp. Despite their assumed polyketide biogenesis, incorporation studies with 13C-labelled acetate, glycerol and tryptophans show that fusarochromene is unexpectedly derived via oxidative cleavage of the aromatic amino acid tryptophan. A putative biosynthetic gene cluster has been identified.
Introduction
Fusaria comprise a large group of filamentous fungi which produce a diverse range of natural products, including many known to be mycotoxins, e.g. trichothecins, fumonosins, zearalenone and enniatins.1,2 In addition, many are phytotoxic. As part of our ongoing series of projects to study biologically active and biosynthetically unusual metabolites in fungi, e.g. fusarins, squalestatins, tropolones, maleidrides, strobilurins and cryptosporioptides,3 we decided to investigate the production of toxins from these economically damaging organisms. Fusarium sacchari is widely reported as a devastating pathogen of sugar cane, typically reducing the sugar content by up to 65% and causing plant death in some cases.4 Infection of sugar cane by Fusarium spp., known as Pokkah boeng disease, is considered a major problem to commercial sugar cane cultivation in many countries such as the Philippines, Mexico, India and Brazil.5With the aim of better understanding the toxin-producing potential of this important fungal pathogen, cultures of Fusarium sacchari were subjected to chemical extraction and HPLC analysis. This led to the identification of a novel metabolite named fusarochromene and the subsequent elucidation of its unexpected biosynthetic origins.
Results and discussion
A strain which was initially believed to be Colletotrichum falcatum, but reclassified as Fusarium sacchari,6 obtained from the first Fungal Culture Bank, University of The Punjab, Lahore, Pakistan, was grown on Czapek Dox (CD), Complete (CM), and mannitol yeast extract (MY) media. Extracts from all three media showed the presence of two major compounds. The first (typically 10 mg L−1) showed NMR signals characteristic of an n-butyl group and a 1,3,5-substituted aromatic ring. Its molecular weight, 179 Da was consistent with one nitrogen, and it was readily identified as the common metabolite fusaric acid (5-butylpyridine-2-carboxylic acid).7,8The second compound (typically 30 mg L−1) has a molecular formula C17H22N2O4 (HRMS [M + Na]+ 341.1484). The 1H NMR spectrum (Fig. S1†) showed the presence of three singlet methyl groups, two pairs of diastereotopic methylenes, both adjacent to the same stereogenic methine centre, two cis-coupled olefinic protons, two ortho-coupled aromatic hydrogens and four exchangeable hydrogens. The 13C NMR spectrum showed sixteen chemical environments, including one ketone (199.6 ppm), one other ester/amide carbonyl (170.6 ppm), six aromatic, two olefinic, and three aliphatic carbons (1°, 2° and 3°) all attached to heteroatoms, a further methylene and three methyl carbons.
HSQC and HMBC spectra gave correlations (Fig. 1) which were consistent with structure 1. An alternative structure in which the nitrogen and oxygen functionalities on the aromatic ring are reversed was also consistent with these correlations (Fig. S2†). The structures were distinguished by rerunning the spectra in d6-DMSO which gave a much sharper signal (7.45 ppm) for the NH2 protons than in CDCl3. This allowed nOe enhancements to be observed (Fig. S3†) only when the olefinic hydrogen (H-4, 6.80 ppm) was irradiated. Irradiation of the aromatic hydrogens (6.05 and 7.40 ppm) gave only mutual nOes. This confirmed structure 1, which we have called fusarochromene. Fusarochromene 1 could now be seen to be similar to the known fusarachromanone (3)9 and several related compounds (4–10)10–12 isolated from various Fusaria, but mainly Fusarium equiseti (Fig. 2).
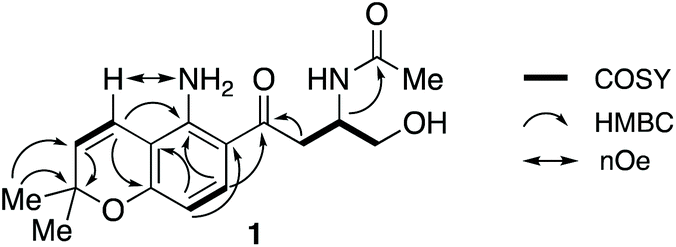 | ||
| Fig. 1 Key COSY, HMBC and nOe correlations in fusarochromene 1. | ||
 | ||
| Fig. 2 Fusraochromene and related structures. | ||
Fusarochromanone was originally described as a mycotoxin causing tibial dyschondroplasia (TD) in chickens,13 but has since been shown to have a wide range of other potent biological activities including inhibition of angiogenesis, prevention of cell reproduction and apoptosis in many cancer cells including glioblastomas and melanomas.14–16 It has been proposed to be an important new lead compound for the treatment of cancer.15 Interestingly, fusarochromene shares many structural features with the amide containing chromene 11 isolated from the higher plant Amyris plumieri,17 described as a chemo-preventive anticancer agent via inhibition of cytochrome P450 CYP1, which catalyzes critical conversions to the ultimate carcinogen.
Fusaraochromanone 3, based on an unpublished X-ray crystallographic study,14 was reported to have the R stereochemistry. Fusarochromene 1 is optically active, [α]D −22 (c 0.0014, CHCl3). To investigate the absolute configuration, we studied a number of variations of the Mosher's method using fusarochromene 1 itself and derivatives based on serine and aspartate as suitable models for the secondary amine side chain, but these gave contradictory results. Fusarochromene 1 failed all attempts at crystallization, but the p-bromobenzoate derivative 2 was crystalline, and after very slow evaporation, suitable crystals were obtained and the X-ray structure shown in Fig. 3 was determined. This confirmed the R stereochemistry. The Flack parameter was measured as −0.005 giving a 99.9% certainty in the stereochemical assignment. As no optical rotations are reported for fusarochromanone, no facile stereochemical correlation with fusarochromene can be made.
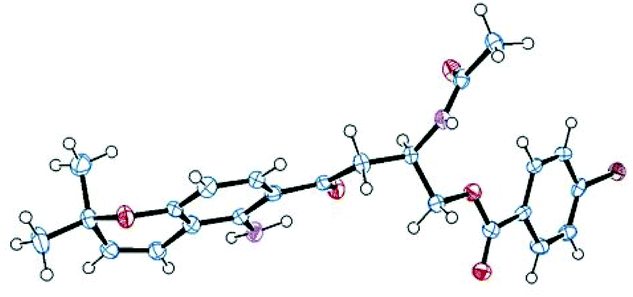 | ||
| Fig. 3 X-ray crystal structure of (R)-p-bromobenzoyl-fusarochromene 2. | ||
Initial retro-biosynthetic analysis, in particular the alternating oxidation pattern of the side chain and aromatic ring, and the prenyl group suggested a meroterpenoid (mixed polyketide–terpenoid) origin.18 However, incorporation of [1,2-13C2]acetate resulted in labelling of only the prenyl and N-acetyl groups (Scheme 1a, Fig. S4†) with no label at all in either the aromatic ring or side chain carbons. While labelling of the chromene ring is consistent with a terpenoid origin, a polyketide synthase cannot be involved. Further retro-biosynthetic analysis, after removing the prenyl, phenol and N-acetyl moieties gives the simplified skeleton (15) shown in Scheme 1, and bond dissection adjacent to the ketone gives anthranilic acid 12 and an amino acid, either serine or aspartic acid 13, possibly as its reduced equivalent, aspartinol. Incorporation of [U-13C6]glucose and analysis of fragments showing contiguous labelling has proved a useful method of detecting cryptic biosynthetic precursors.19 Following the known pathways5 of synthesis of anthranilate and aspartate from glucose, the labelling pattern in fusarochromene should be as shown in Scheme 1b(i). In particular, the aromatic ring would contain three contiguous labelled carbons, a single isolated labelled carbon with the remaining two carbons forming a triad with the side chain ketone. The remaining three carbons in the side chain would also be mutually coupled.
 | ||
| Scheme 1 Labelling patterns in fusarochromene from (a) [1,2-13C2]acetate, (b) [U-13C]glycerol, and (c) [1-13C]- and [2-13C]tryptophans. | ||
Accordingly, cultures of F. sacchari were supplemented with [U-13C6]glucose, but this resulted in very poor titres and isolation of insufficient fusarochromene 1 for informative NMR spectra to be obtained. [U-13C3]glycerol has been used as an alternative to uniformly labelled glucose,6 and control experiments showed that addition of unlabeled glycerol had no detrimental effect on metabolite production. The actual labelling pattern obtained on feeding [U-13C3]glycerol is shown in Scheme 1b(ii) and Fig. S5,† and agreed with that predicted apart from two crucial positions. No incorporation of 13C label was observed at either of the side chain C11 or aromatic ring C5 positions. While the lack of label at C5 could be accounted for by dilution of the label by the large quantity of sugars with natural abundance 13C levels during the pentose phosphate pathway,20 the lack of a coupled carbon at C11 was not consistent with the proposed pathway. An alternative would be that, coupled with decarboxylation of anthranilate, aspartate provided all four side chain carbons. An intact C3 unit would still be expected at C12–C14 but C11 would show a single isotopic enrichment. However, on feeding [4-13C]aspartate to F. sacchari, no label was incorporated at C11 of the isolated fusrarochromene.
Thus, further revision of the possible pathway was required. In 1969, Yoshida and Katagiri demonstrated that 14C-labelled tryptophan was efficiently incorporated into the quinoxaline (QXC) chromophore of triostin,21 a cyclodepsipetide antibiotic produced by Streptomyces s-2-210L. It was subsequently shown22 that QXC biosynthesis involves degradation of tryptophan 14 to intermediates such as β-hydroxy-kynurenine (Scheme 2a), which are similar to the core structure of fusarochromene 1. A key step is the cleavage of the C2–C3 bond of tryptophan by tryptophan 2,3-dioxygenase (TDO).23 A labelling pattern, based on the expected labelling of tryptophan from [U-13C3]glycerol,24,25 is summarized in Scheme 1c. This is now consistent with that observed. To confirm the involvement of tryptophan, both [1-13C]- and [2-13C]tryptophans were synthesised from [1-13C]- and [2-13C]glycine respectively via modifications of literature methods (ESI and Scheme S1†) and fed to cultures of F. sacchari. Both were specifically incorporated in very high enrichment levels into C14 (25 fold) and C13 (100 fold) respectively of fusarochromene (Fig. S6 and S7†). Interestingly, the same approach of using [U-13C]glucose was used to identify tryptophan as a precursor to the quinolactacins, quinolone metabolites (see Fig. 4 below) of Penicillium sp. EPF-6.26
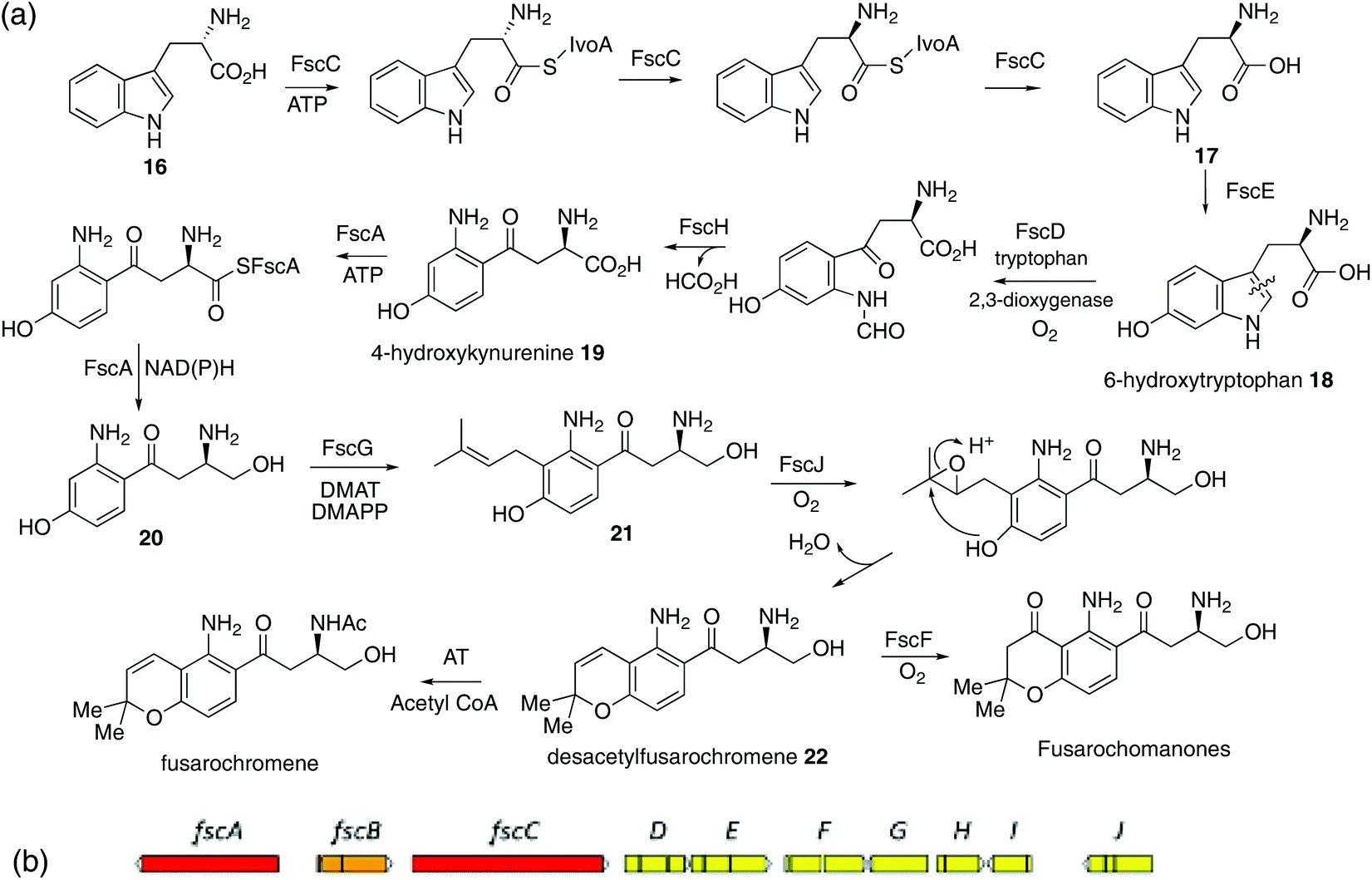 | ||
| Scheme 2 (a) Proposed pathway to fusarochromene via 6-hydroxytryptophan 18 and 4-hydroxykynurenine 19. (b) A putative fusarochromanone biosynthetic gene cluster (BGC) identified in the genome of Fusarium equiseti. Predicted gene functions are shown in Table 1. Red: NRPS-like enzyme. Orange: Putative regulator. Yellow: Catalytic enzyme. | ||
 | ||
| Fig. 4 Fungal metabolites biosynthesised via tryptophan cleavage; quinolactacins A 23 and nanagelenin 24. | ||
With this result to hand, we searched the publicly available F. equiseti genome for a potential biosynthetic gene cluster (BGC) encoding homologues of known tryptophan dioxygenase (TDO) and dimethylallyl diphosphate (prenyl) transferase (DMAT) enzymes. This immediately revealed a gene cluster (Scheme 2b, Tables 1 and S1†) encoding 10 predicted proteins of significance. One of these, FscD, has significant homology with indoleamine 2,3-dioxygenases and so is almost certainly a TDO. FscG similarly shows high homology to DMATs. Associated with these are FscE with homology to IvoC, a N-acetyltryptophan 6-hydroxylase,27 and FscH homologous to kynurenine formidase which deformylates N-formyl-kynurenine to kynurenine.28 The biosynthesis of fusarochromene would require the epimerization of L-tryptophan 16 to D-tryptophan 17 and consistent with that is the presence of FscC. FscC, has significant homology to IvoA, an NRPS-like enzyme recently described as a tryptophan N-acetyltransferase,29 but subsequently shown to be an L-tryptophan epimerase30 with no transferase activity. It contains 4 domains: A, PCP, E and C*. Thus adenylation (A) and transfer to peptidyl carrier protein (PCP) for epimerization (E) and release (C*) converts L- to D-tryptophan. FscA is another NRPS-like enzyme which contains A, PCP and C-terminal thioester reductase (R) domains, an architecture characteristic of other fungal carboxylate reductases.31,32
| Gene | Putative function | Homologue (accession number)a |
|---|---|---|
| a See Table S1† for further details. | ||
| fscA | NRPS-like oxidoreductase | ApmA36 (A0A1W6BT53) |
| fscB | Putative transcription factor | AurF37 (A0A0M4LAF6) |
| fscC | NRPS-like Tryptophan epimerase | IvoA29,30 (C8V7P4.1) |
| fscD | Tryptophan 2,3-dioxygenase | IDO38 (P47125) |
| fscE | P450 (tryptophan 6-hydroxylase) | IvoC29 (C8V7P3) |
| fscF | P450 | Apf739 (S0DPM1) |
| fscG | DMATS-type prenyltransferase | XptA40 (Q5AY46) |
| fscH | Kynurenine formamidase-like hydrolase | KFA28 (Q04066) |
| fscI | Oxidoreductase | TIC3241 (A2RVM0) |
| fscJ | Aromatic peroxidase/chloroperoxidase | DotB42 (M2XZY2.1) |
Taken with the other activities encoded by the fsc cluster, the pathway shown in Scheme 2a can thus be proposed. Epimerization of L-trytophan 16 to D-tryptophan 17 which is then hydroxylated to give the known 6-hydroxytryptophan 18.33 This undergoes cleavage of the pyrrole ring, deformylation to 4-hydroxykyrunenine 19 and reduction of the carboxyl to primary alcohol 20. That hydroxylation occurs as a first step, is suggested by the failure of either L- or D-kynurenine labelled with 2H or 13C to be incorporated.34 Prenylation to 21 and chromene ring formation, the latter catalysed by the oxidoreductase FscI, would give desacetyl-fusarochromene 22. The biosynthesis of the fusarochromanone group can be rationalised via the same pathway. Epoxidation (FscF) of the chromene double bond and rearrangement would convert the chromene to the chroman-4-one system. The presence of a tryptophan epimerase would suggest that fusarochromanone 3 also has the R stereochemistry. Elimination of the side chain secondary amine function followed by reduction of the resulting double bond explains the formation of, e.g. compounds 7 and 9. No specific acetyltransferases have been found near the fsc BGC, but multiple predicted proteins containing the N-acetyltransferase super family domain were located in the genome. One such, located on scaffold 7, has some homology (29.3% protein identity) with Ypa3 which is an N-acetyltransferase from yeast which specifically acetylates D-amino acids, but has very relaxed substrate specificity (Fig. S9†).35
A number of actinomycetes are known to produce metabolites by tryptophan cleavage, e.g. triostin21 (vide supra), antimycin,43 sibiromycin44,45 and quinomycin.46 The quinolactacins (e.g. quinolactacin A 23), whose biosynthetic gene cluster has been identified recently,47 were the only fungal metabolites known to be produced via the cleavage of tryptophan until the recently reported nanagelenin 24 from Aspergillus nanagensis.48 The biosynthetic pathways of the quinolactacins, nanagelenins and now fusarochromene, share the requirement for IDO-catalysed oxidative cleavage but are otherwise quite diverse, as demonstrated by the general lack of homology in their respective gene clusters (see ESI; Fig. S10–S15†). Features that are unique to the fusarochromene pathway include epimerisation to give D-tryptophan and the presence of a pathway specific kynurenine formamidase in the putative fusarochromene BGC, which is absent from both the nan and qul clusters.
Conclusions
These results indicate a new biosynthetic pathway which rationalises the formation of an important group of fungal toxins via cleavage of the pyrrole ring of tryptophan, a relatively rare occurrence among fungal secondary metabolites. Indeed, this is the first case of such a pathway involving a D-amino acid. Knowledge of fusarochromene biosynthesis allowed identification of a putative biosynthetic gene cluster (BGC), involving two modified NRPS components and a series of coordinated redox and allylation steps. Identification of the likely BGC responsible for these toxins will facilitate efforts to analyse or disrupt toxin production in pathogenic strains.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank MRC (MR/N029909/1) (KMJdeM-S), EPSRC (EP/E000177/1) (JCK), the Ministry of Higher Education (MOHE) – Egypt (IAYG), and the Higher Education Commission of Pakistan (AM) for financial support. JWM thanks BBSRC and Mr Mark Evans for PhD scholarships. X-ray crystallography was carried out by Dr Mairi Haddow, School of Chemistry, University of Bristol.Notes and references
- A. Bertero, A. Moretti, L. J. Spicer and F. Caloni, Toxins, 2018, 10, 244 CrossRef.
- M. Li, R. Yu, X. Bai, H. Wang and H. Zhang, Nat. Prod. Rep., 2020 10.1039/D0NP00038H.
- (a) Z. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef CAS; (b) R. J. Cox, F. Glod, D. Hurley, C. M. Lazarus, T. P. Nicholson, B. A. M. Rudd, T. J. Simpson, B. Wilkinson and Y. Zhang, Chem. Commun., 2004, 2260–2261 RSC; (c) J. Davison, A. al-Fahad, M. Cai, Z. Song, S. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7642–7467 CrossRef CAS; (d) K. Williams, A. J. Szwalbe, N. P. Mulholland, J. L. Vincent, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Angew. Chem., Int. Ed., 2016, 55, 6784–6788 CrossRef CAS; (e) R. Nofiani, K. de Mattos-Shipley, K. E. Lebe, L.-C. Han, Z. Iqbal, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Nat. Commun., 2018, 9, 3940–3951 CrossRef; (f) C. Greco, K. de Mattos-Shipley, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Sci., 2019, 10, 2930–2039 RSC.
- R. C. Ploetz, Phytopathology, 2006, 96, 648–652 CrossRef.
- S. Vishwakarma, P. Kumar, A. Nigam, A. Singh and A. Kumar, J. Plant Pathol. Microbiol., 2013, 4, 2 Search PubMed.
- J. F. Leslie, B. A. Summerell, S. Bullock and F. J. Doe, Mycologia, 2005, 97, 718–724 CrossRef.
- C. Bacon, J. Porter, W. Norred and J. Leslie, Appl. Environ. Microbiol., 1996, 62, 4039–4043 CrossRef CAS.
- I. A. Y. Ghannam, H. F. Roaiah, M. M. Hanna, S. S. El-Nakkady and R. J. Cox, Int. J. Pharm. Technol., 2014, 6, 6528–6535 CAS.
- S. V. Pathre, W. B. Gleason, Y.-W. Lee and C. J. Mirocha, Can. J. Chem., 1986, 64, 1258–1261 CrossRef.
- W. Xie, C. J. Mirocha and Y. Wen, J. Nat. Prod., 1995, 58, 124–127 CrossRef CAS.
- W. Wu, P. Nelson, M. Cook and E. Smalley, Appl. Environ. Microbiol., 1990, 56, 2989–2993 CrossRef CAS.
- P. Krogh, D. Christensen, B. Hald, B. Harlou, C. Larsen, E. Pedersen and U. Thrane, Appl. Environ. Microbiol., 1989, 55, 3184–3188 CrossRef CAS.
- W. Wu, M. E. Cook, Q. Chu and E. B. Smalley, Avian Dis., 1993, 302–309 CrossRef CAS.
- E. Mahdavian, P. Palyok, S. Adelmund, T. Williams-Hart, B. D. Furmanski, Y.-J. Kim, Y. Gu, M. Barzegar, Y. Wu and K. N. Bhinge, BMC Res. Notes, 2014, 7, 1–11 CrossRef.
- E. Mahdavian, M. Marshall, P. M. Martin, P. Cagle, B. A. Salvatore and Q. A. Quick, Int. J. Mol. Med., 2014, 34, 880–885 CrossRef CAS.
- Y. Gu, M. Barzegar, X. Chen, Y. Wu, C. Shang, E. Mahdavian, B. A. Salvatore, S. Jiang and S. Huang, Oncotarget, 2015, 6, 42322 CrossRef.
- S. Badal, S. Williams, G. Huang, S. Francis, P. Vendantam, O. Dunbar, H. Jacobs, T. Tzeng, J. Gangemi and R. Delgoda, Fitoterapia, 2011, 82, 230–236 CrossRef CAS.
- R. Geris and T. J. Simpson, Nat. Prod. Rep., 2009, 26, 1063–1094 RSC.
- H. G. Floss, P. J. Keller and J. M. Beale, J. Nat. Prod., 1986, 49, 957–970 CrossRef CAS.
- L. Stryer, Biochemistry, W.H. Freeman, New York, 3rd edn, 1998 Search PubMed.
- T. Yoshida and K. Katagiri, Biochemistry, 1969, 8, 2645–2651 CrossRef CAS.
- K. Watanabe, K. Hotta, A. P. Praesuth, K. Koketsu, A. Migita, C. N. Boddy, C. C. Wang, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2006, 2, 423–428 CrossRef CAS.
- J. Basran, I. Efimov, N. Chauhan, S. J. Thackray, J. L. Krupa, G. Eaton, G. A. Griffith, C. G. Mowat, S. Handa and E. L. Raven, J. Am. Chem. Soc., 2011, 133, 16251–16257 CrossRef CAS.
- E. R. Radwanski and R. L. Last, Plant Cell, 1995, 7, 921 CAS.
- V. A. Higman, J. Flinders, M. Hiller, S. Jehle, S. Markovic, S. Fiedler, B.-J. van Rossum and H. Oschkinat, J. Biomol. NMR, 2009, 44, 245–260 CrossRef CAS.
- T. Sasaki, S. Takahashi, K. Uchida, S. Funayama, M. Kainosho and A. Nakagawa, J. Antibiot., 2006, 59, 418–427 CrossRef CAS.
- N. J. McCorkindale, D. Hayes, G. A. Johnston and A. J. Clutterbuck, Phytochemistry, 1983, 22, 1026–1028 CrossRef CAS.
- M. Wogulis, E. R. Chew, P. D. Donohoue and D. K. Wilson, Biochemistry, 2008, 47, 1608–1621 CrossRef CAS.
- C. T. Sung, S.-L. Chang, R. Entwistle, G. Ahn, T.-S. Lin, V. Petrova, H.-H. Yeh, M. B. Praseuth, Y.-M. Chiang and B. R. Oakley, Fungal Genet. Biol., 2017, 101, 1–6 CrossRef CAS.
- Y. Hai, M. Jenner and Y. Tang, J. Am. Chem. Soc., 2019, 141, 16222–16226 CrossRef CAS.
- J. Casqueiro, S. Gutiérrez, O. Bañuelos, F. Fierro, J. Velasco and J. F. Martín, Mol. Gen. Genet., 1998, 259, 549–556 CrossRef CAS.
- Z. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef CAS.
- A. Ishihara, N. Sugai, T. Bito, N. Ube, K. Ueno, Y. Okuda and E. Fukushima-Sakuno, Biosci. Biotechnol. Biochem., 2019, 83, 1800–1806 CrossRef CAS.
- I. A. Y. Ghannam, PhD thesis, Cairo University, 2014.
- G. Y. Yow, T. Uo, T. Yoshimura and N. Esaki, Arch. Microbiol., 2004, 182, 396–403 CrossRef CAS.
- W. Li, A. Fan, L. Wang, P. Zhang, Z. Liu, Z. An and W. B. Yin, Chem. Sci., 2018, 9, 2589–2594 RSC.
- X. M. Mao, Z. J. Zhan, M. N. Grayson, M. C. Tang, W. Xu, Y. Q. Li, W. B. Yin, H. C. Lin, Y. H. Chooi, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2015, 137, 11904–11907 CrossRef CAS.
- H. J. Yuasa and H. J. Ball, J. Mol. Evol., 2011, 72, 160–168 CrossRef CAS.
- E. M. Niehaus, S. Janevska, K. W. von Bargen, C. M. Sieber, H. Harrer, H. U. Humpf and B. Tudzynski, PLoS One, 2014, 9, e103336 CrossRef.
- J. F. Sanchez, R. Entwistle, J.-H. Hung, J. Yaegashi, S. Jain, Y.-M. Chiang, C. C. Wang and B. R. Oakley, J. Am. Chem. Soc., 2011, 133, 4010–4017 CrossRef CAS.
- F. Hörmann, M. Küchler, D. Sveshnikov, U. Oppermann, Y. Li and J. Soll, J. Biol. Chem., 2004, 279, 34756–34762 CrossRef.
- P. Chettri, K. C. Ehrlich, J. W. Cary, J. Collemare, M. P. Cox, S. A. Griffiths, M. A. Olson, P. J. de Wit and R. E. Bradshaw, Fungal Genet. Biol., 2013, 51, 12–20 CrossRef CAS.
- R. F. Seipke and M. I. Hutchings, Beilstein J. Org. Chem., 2013, 9, 2556–2563 CrossRef.
- A. Mesentsev, V. Kuljaeva and L. Rubasheva, J. Antibiot., 1974, 27, 866–873 CrossRef CAS.
- W. Li, A. Khullar, S. Chou, A. Sacramo and B. Gerratana, Appl. Environ. Microbiol., 2009, 75, 2869–2878 CrossRef CAS.
- D. Martin, S. Mizsak, C. Biles, J. Stewart, L. Baczynskyj and P. Meulman, J. Antibiot., 1975, 28, 332–336 CrossRef CAS.
- F. Zhao, Z. Liu, S. Yang, N. Ding and X. Gao, Angew. Chem., Int. Ed., 2020, 59, 19108–19114 CrossRef CAS.
- H. Li, C. L. Gilchrist, C.-S. Phan, H. J. Lacey, D. Vuong, S. A. Moggach, E. Lacey, A. M. Piggott and Y.-H. Chooi, J. Am. Chem. Soc., 2020, 142, 7145–7152 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2022413. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob02031a |
| This journal is © The Royal Society of Chemistry 2021 |