
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantiospecific deoxyfluorination of cyclic α-OH-β-ketoesters†
Christopher
Mairhofer
a,
Victoria
Haider
a,
Thomas
Bögl
b and
Mario
Waser
 *a
*a
aJohannes Kepler University Linz, Institute of Organic Chemistry, Altenbergerstraße 69, 4040 Linz, Austria. E-mail: mario.waser@jku.at
bJohannes Kepler University Linz, Institute of Analytical Chemistry, Altenbergerstraße 69, 4040 Linz, Austria
First published on 18th November 2020
Abstract
We herein report the deoxyfluorination of cyclic α-hydroxy-β-ketoesters using diethylaminosulfur trifluoride (DAST). The reaction proceeds with excellent levels of stereospecificity, giving the configurationally inverted α-fluoro-β-ketoesters in high yields under operationally simple conditions.
The deoxyfluorination of alcohol derivatives is one of the most commonly applied methods to access organofluorine compounds.1,2 A broad variety of complementary strategies for the deoxyfluorination of Csp2–OH and Csp3–OH functionalities by making use of different, nowadays often commercially available, reagents have been reported over the last few decades.1–6 The most prominently used agent for this purpose is diethylaminosulfur trifluoride (DAST, 1), which acts by simultaneously activating the hydroxyl group while delivering a nucleophilic fluoride anion.3–5 Besides, the last years have seen significant progress in the development of alternative (sometimes less sensitive) and maybe cheaper reagents,6 as illustrated in e.g. a very recent report using CuF2 in combination with Lewis bases.6g
Over the last years our group has focused on the stereoselective synthesis of α-hydroxylated and α-fluorinated β-ketoesters 2 and 3 (starting from β-ketoesters 4) by using bifunctional chiral quaternary ammonium salt catalysts.7,8 Within these studies, we generally achieved higher enantioselectivities for the α-hydroxylated products 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 than for the α-fluorinated 3.7 We thus wondered, if it may be possible to convert the alcohols 2 into organofluorine compounds 3 by means of a stereospecific deoxyfluorination process (Scheme 1). However, and surprising to us, despite of all the achievements in the field of (stereospecific) deoxyfluorinations of alcohols, α-hydroxy-carbonyl substrates have been sparingly used only.9,10 When looking at these previous reports it becomes obvious that the main obstacle, when targeting the conversion of 2 into 3, will be the suppression of the deoxygenation and difluorination of the carbonyl-group. However, by looking at some of the earlier reports, it may sound feasible that carefully balanced reaction conditions (temperature, order of addition of the reagents, …) may be fruitful to carry out the deoxyfluorination of compounds 2 without touching the carbonyl group, thus filling this long-standing application gap in the toolbox of enantiospecific deoxyfluorination reactions.
8 than for the α-fluorinated 3.7 We thus wondered, if it may be possible to convert the alcohols 2 into organofluorine compounds 3 by means of a stereospecific deoxyfluorination process (Scheme 1). However, and surprising to us, despite of all the achievements in the field of (stereospecific) deoxyfluorinations of alcohols, α-hydroxy-carbonyl substrates have been sparingly used only.9,10 When looking at these previous reports it becomes obvious that the main obstacle, when targeting the conversion of 2 into 3, will be the suppression of the deoxygenation and difluorination of the carbonyl-group. However, by looking at some of the earlier reports, it may sound feasible that carefully balanced reaction conditions (temperature, order of addition of the reagents, …) may be fruitful to carry out the deoxyfluorination of compounds 2 without touching the carbonyl group, thus filling this long-standing application gap in the toolbox of enantiospecific deoxyfluorination reactions.
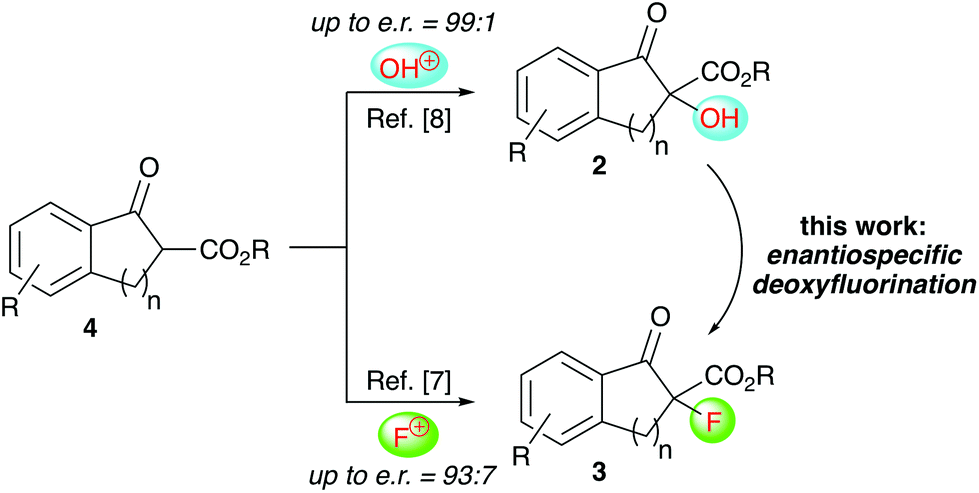 | ||
| Scheme 1 Our previously developed α-hydroxylation and α-fluorination of β-ketoesters 4 and the herein investigated deoxyfluorination of alcohols 2 to access α-F-β-ketoesters 3. | ||
We started our investigations by optimizing the deoxyfluorination of enantioenriched (S)-2a (obtained as reported previously8) with DAST (1), one of the cheapest commercially available established deoxyfluorination agents.11 As outlined in Table 1, all reactions were carried out at room temperature using dry CH2Cl2 as the solvent. Other solvents (e.g. THF or toluene) were tested too, but did not allow for any reasonable conversion and non-reproducible results were obtained thereby. First experiments where DAST (1) was added to a solution of 2a showed that conversion is mainly depending on the amount of 1 (compare entries 1–3) as longer reaction times were not beneficial in case of the slowly converting experiments. These results suggest that DAST partially decomposes under the conditions. Noteworthy however, all the reactions turned out to be very clean and “spot-to-spot”, with no other fluorinated products being formed. In each case the reaction proceeded with high levels of stereospecificity, giving the configurationally inverted (R)-3a. Absolute configuration of starting material (S)-2a and product (R)-3a were assigned by comparison of optical rotation and HPLC retention time orders with previous reports.7,8,12,13 The stereochemical course (inversion or retention of configuration) of DAST-mediated deoxyfluorinations has been a matter of discussion4,5 and mostly depends on the nature of the substrate. For our target transformation the observed inversion suggests a clean SN2-type mechanism14 and as can be seen from all the results summarized in Table 1, the observed levels of enantiospecificity were always satisfying (see Scheme 2 for the proposed mechanistic scenario). In order to improve the conversion, we next changed the order of addition (entries 4–6). Adding substrate 2a to DAST (add. order B) had a beneficial effect, allowing for high conversion by using just two equivalents of reagent 1 (NMR yields determined by addition of an internal standard were in the same range as the conversion of 2a and the isolated yields after column chromatography were higher than 70% in all these cases).
 | ||
| Scheme 2 Application scope employing the conditions shown in Table 1, entry 6 (all reactions were run using 0.05–0.1 mmol 2) and the proposed stereospecific inversion mechanism. | ||
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 (eq.) | Addition orderb | Conv.c | ee (2a)d | ee (3a)d | ese [%] |
| a All reactions were run using 0.05 mmol 2a in a total volume of 2.5 mL CH2Cl2 under Ar. b Addition order A: Dropwise addition of 1 in CH2Cl2 (1.25 mL) to 2a in CH2Cl2 (1.25 mL) over 15 min; B: Dropwise addition of 2a in CH2Cl2 (1.25 mL) to 1 in CH2Cl2 (1.25 mL) over 15 min; C: Dropwise addition of 2a in CH2Cl2 (1.25 mL) to 2 eq. 1 in CH2Cl2 (1.25 mL) over 15 min followed by stirring for 20 h and addition of another 2 eq. of 1 and stirring for further 20 h (40 h total reaction time). c Determined by 1H NMR of the crude reaction mixture. d Determined by HPLC using a chiral stationary phase. e 100 × ee (3a)/ee (2a); absolute configuration was assigned as described previously.7,8,12,13 | ||||||
| 1 | 1.1 | A | 0 | — | — | — |
| 2 | 4 | A | 85 | 90.2 | 88.9 | 98.5 |
| 3 | 8 | A | 95 | 90.2 | 89.5 | 99.2 |
| 4 | 1.1 | B | 84 | 88.5 | 88.1 | 99.6 |
| 5 | 2 | B | 91 | 90.2 | 89.0 | 98.6 |
| 6 | 4 | C | >95 | 95.3 | 93.1 | 98.0 |
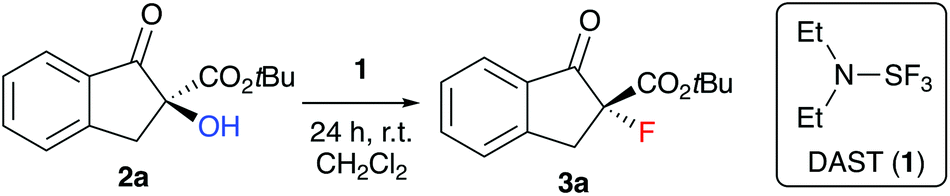
Interestingly, when carrying out the reaction with other deoxyfluorination methods, like the above mentioned CuF2 protocol6g or by using PyFluor,6c absolutely no formation of product 3a could be achieved under otherwise identical conditions. Unfortunately, the reactions with two equivalents of 1 often stalled at around 90% conversion (and more restricted conversions were later observed during the investigation of the application scope as well). Thus, we finally used a slightly larger excess of 1 (entry 6, add. order C), which allowed for robust and reproducible reaction conditions with very high levels of enantiospecifity (repeating the reaction several times always resulted in >98.0% es).
With reliable and highly stereospecific conditions at hand, we next investigated the application scope of this methodology (Scheme 2). A variety of differently substituted indanone-based α-hydroxy-β-ketoesters 2 were well tolerated, giving the corresponding configurationally inverted products 3 in satisfying yields and with high levels of enantiospecificity in most cases. Interestingly, the fluorine containing 3d and 3h were obtained with slightly lower es values, but still in an acceptable range. In sharp contrast to the other substrates, the 5-methoxy-substituted 2i performed very slow only, and even adding additional amounts of DAST did not allow for a higher conversion and yield of product 3i. Most likely the strong electron donating effect of the methoxy group para to the carbonyl group leads to an increased contribution of the enolate resonance structure, which as a result leads to a lower nucleophilicity of the α-OH group and thus slows down the deoxyfluorination process. Unfortunately, when using tetralone-based starting material 2o, even large excesses of DAST did not allow for any product formation and resulted in more or less quantitative recovery of alcohol 2o. A possible explanation maybe that the nucleophilic fluoride attack to the in situ activated alcohol has to proceed via a pseudo-axial trajectory on the 6-ring system, where 1,2- and 1,3-(pseudo)-diaxial interactions are much stronger than on the indanone-based 5-ring systems.
In conclusion, we have developed an operationally simple method for the stereospecific deoxyfluorination of enantioenriched α-hydroxy-β-ketoesters 2, by reaction with DAST (1). This protocol works well for a variety of indanone-based ketoesters 2, without any occurrence of carbonyl-group deoxofluorination or any other side reactions.
General reaction procedure
A solution of α-hydroxy-β-ketoester 2 (0.1 mmol) in dry CH2Cl2 (2.5 mL) is added dropwise over 15 min to a stirred solution of DAST (1; 200 μL, 1 M in CH2Cl2) in 2.5 mL dry CH2Cl2 at room temperature (Ar-atmosphere). After stirring for 20 h, another portion of DAST (1; 200 μL, 1 M in CH2Cl2) is added and the mixture is stirred for additional 20 h (40 h total reaction time). The reaction is quenched with 5 mL saturated aq. NaHCO3, and the aqueous phase is extracted with dichloromethane (3 × 5 mL). The combined organic phases are dried over anhydrous Na2SO4, filtered and the solvent is removed in vacuo. The residue is purified by silica gel column chromatography (heptanes/EtOAc) to afford the targeted fluorinated products 3 in the reported yields and enantiospecificities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the Austrian Science Funds (FWF): Project No. P30237 (financed by the Austrian National Foundation for Research, Technology and Development and Research Department of the State of Upper Austria) and Project No. P31784. The used NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”).Notes and references
- For selected reviews on nucleophilic fluorination methods covering deoxyfluorinations as well:
(a) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC
; (b) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS
- For reviews on deoxyfluorinations:
(a) N. Al-Maharik and D. O'Hagan, Aldrichimica Acta, 2011, 44, 65–75 CAS
- W. J. Middleton, J. Org. Chem., 1975, 40, 574–578 CrossRef CAS
- R. P. Singh and J. M. Shreeve, Synthesis, 2002, 2561–2578 CAS
- L. Baptista, G. F. Bauerfeldt, G. Arbilla and E. C. Silva, J. Mol. Struct., 2006, 761, 73–81 CrossRef CAS
- Selected more recent examples on the development of new reagents and methods:
(a) T. Umemoto, R. P. Singh, Y. Xu and N. Saito, J. Am. Chem. Soc., 2010, 132, 18199–18205 CrossRef CAS
- J. Novacek and M. Waser, Eur. J. Org. Chem., 2014, 802–809 CrossRef CAS
-
(a) J. Novacek, J. A. Izzo, M. J. Vetticatt and M. Waser, Chem. – Eur. J., 2016, 22, 17339–17344 CrossRef CAS
- With SF4:
(a) W. R. Hasek, W. C. Smith and V. A. Engelhardt, J. Am. Chem. Soc., 1960, 82, 543–551 CrossRef CAS
- With DAST:
(a) F. A. Davis, P. Zhou and C. K. Murphy, Tetrahedron Lett., 1993, 34, 3971–3974 CrossRef CAS
- Depending on supplier, quantities, and conditions, DAST can be obtained for less than € 2 per gram from standard chemical suppliers within the European Union.
- Pioneering report for the assignment of compounds 2: M. R. Acocella, O. G. Mancheno, M. Bella and K. A. Jørgensen, J. Org. Chem., 2004, 69, 8165–8167 CrossRef CAS
- Pioneering report for the assignment of compounds 3: Y. Hamashima, K. Yagi, H. Takano, L. Tamas and M. Sodeoka, J. Am. Chem. Soc., 2002, 124, 14530–14531 CrossRef CAS
- For a recent review on stereospecific nucleophilic displacement reactions on tertiary stereocenters: V. Lanke and I. Marek, Chem. Sci., 2020, 11, 9378–9385 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and analytical data. See DOI: 10.1039/d0ob02152k |
| This journal is © The Royal Society of Chemistry 2021 |