Semisynthesis and biological evaluation of a focused library of unguinol derivatives as next-generation antibiotics†
Received
9th December 2020
, Accepted 8th January 2021
First published on 8th January 2021
Abstract
In this study, we report the semisynthesis and in vitro biological evaluation of thirty-four derivatives of the fungal depsidone antibiotic, unguinol. Initially, the semisynthetic modifications were focused on the two free hydroxy groups (3-OH and 8-OH), the three free aromatic positions (C-2, C-4 and C-7), the butenyl side chain and the depsidone ester linkage. Fifteen first-generation unguinol analogues were synthesised and screened against a panel of bacteria, fungi and mammalian cells to formulate a basic structure activity relationship (SAR) for the unguinol pharmacophore. Based on the SAR studies, we synthesised a further nineteen second-generation analogues, specifically aimed at improving the antibacterial potency of the pharmacophore. In vitro antibacterial activity testing of these compounds revealed that 3-O-(2-fluorobenzyl)unguinol and 3-O-(2,4-difluorobenzyl)unguinol showed potent activity against both methicillin-susceptible and methicillin-resistant Staphylococcus aureus (MIC 0.25–1 μg mL−1) and are promising candidates for further development in vivo.
Introduction
Infections caused by multidrug-resistant bacteria are a major threat to global health. In the clinical setting, methicillin-resistant Staphylococcus aureus (MRSA) continues to be a leading cause of infection-related mortality.1 While vancomycin has been the treatment of choice for serious MRSA infections for decades, there have been increasing reports of vancomycin treatment failures due to the emergence and dissemination of resistant strains.2,3 Concerningly, treatment failure with daptomycin, a drug of last resort for MRSA, can occur in more than 20% of cases.3,4 Therefore, there is a pressing need for the discovery and development of new antibiotics with novel modes of action to combat these deadly superbugs.5
There are many approaches currently being explored to identify new classes of antibiotics, including in situ cultivation of uncultured microbes,6,7 identification and prioritisation of novel organisms by chemotaxonomy8–14 and activation of silent biosynthetic gene clusters (BGCs) using synthetic biology and bioinformatics tools.15,16 An alternate strategy involves revisiting some of the old antibiotic scaffolds that were discovered many decades ago, during a time of plenty, that were abandoned in favour of more promising leads. Re-examining these neglected historic scaffolds through the lens of modern drug discovery platforms has proven to be an effective method of bringing new antibiotic classes to the market.17 Notable antibiotic revivals include linezolid (2000), daptomycin (2003) and lefamulin (2019), which belong to chemical classes first reported in 1978,18 198719 and 1952,20 respectively.
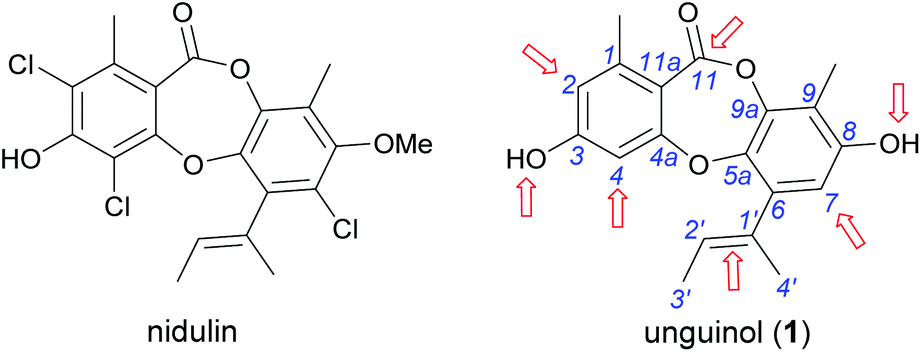
In our ongoing search for new antibiotic leads, we recently reported our work on expanding chemical space around the nidulin antibiotic pharmacophore.21 Nidulin is a trichlorinated depsidone antibiotic, first identified in 1945 from the fungus Aspergillus unguis.22 While nidulin has reported antibacterial activity against Mycobacterium tuberculosis23 and MRSA,24 the compound has received only modest attention since its initial discovery and the scaffold has not been systematically investigated as an antibiotic lead. In our recent study, manipulating the halide ion concentration in the cultivation medium of A. unguis led to the production of 12 previously unreported nidulin analogues, along with 11 known nidulin analogues. Biological testing of this small library revealed a number of interesting trends in potency and selectivity that warranted further investigation. In this study, we have employed a semisynthetic approach to expand the structure activity relationship (SAR) of the nidulin pharmacophore. Starting from the closely related metabolite unguinol, we have generated a library of 15 analogues by modifying 7 different locations around the unguinol core (Fig. 1). All semisynthetic analogues were screened for in vitro activity against a panel of bacteria, fungi and mammalian cell lines. In vitro antimicrobial testing revealed 3-O-benzylunguinol is fifteen times more potent than ampicillin against S. aureus. Further exploration of benzylation of unguinol with halogen-substituted benzyl bromide yielded more potent antibiotics, 3-O-(2-fluorobenzyl)unguinol and 3-O-(2,4-difluorobenzyl)unguinol.
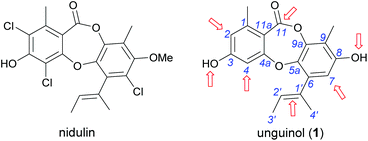 |
| | Fig. 1 Chemical structures of nidulin and unguinol (1) and the initial sites selected for semisynthetic modification of 1 (arrowed). | |
Results and discussion
First-generation semisynthetic unguinol analogues
We initiated our semisynthetic program starting with unguinol (1), which is the major metabolite of A. unguis and could be isolated in reasonable quantities by large-scale cultivation of the organism. Compound 1 is a nonchlorinated analogue of nidulin, containing two free hydroxy groups (3-OH and 8-OH), three unsubstituted aromatic positions (H-2, H-4 and H-7), one double bond (Δ1′,2′) in the butenyl side chain and one ester linkage. These 7 locations were selected as the initial sites for semisynthetic modification to assess the contributions of each group towards the antibiotic activity of the depsidone scaffold.
Oxidation and reduction.
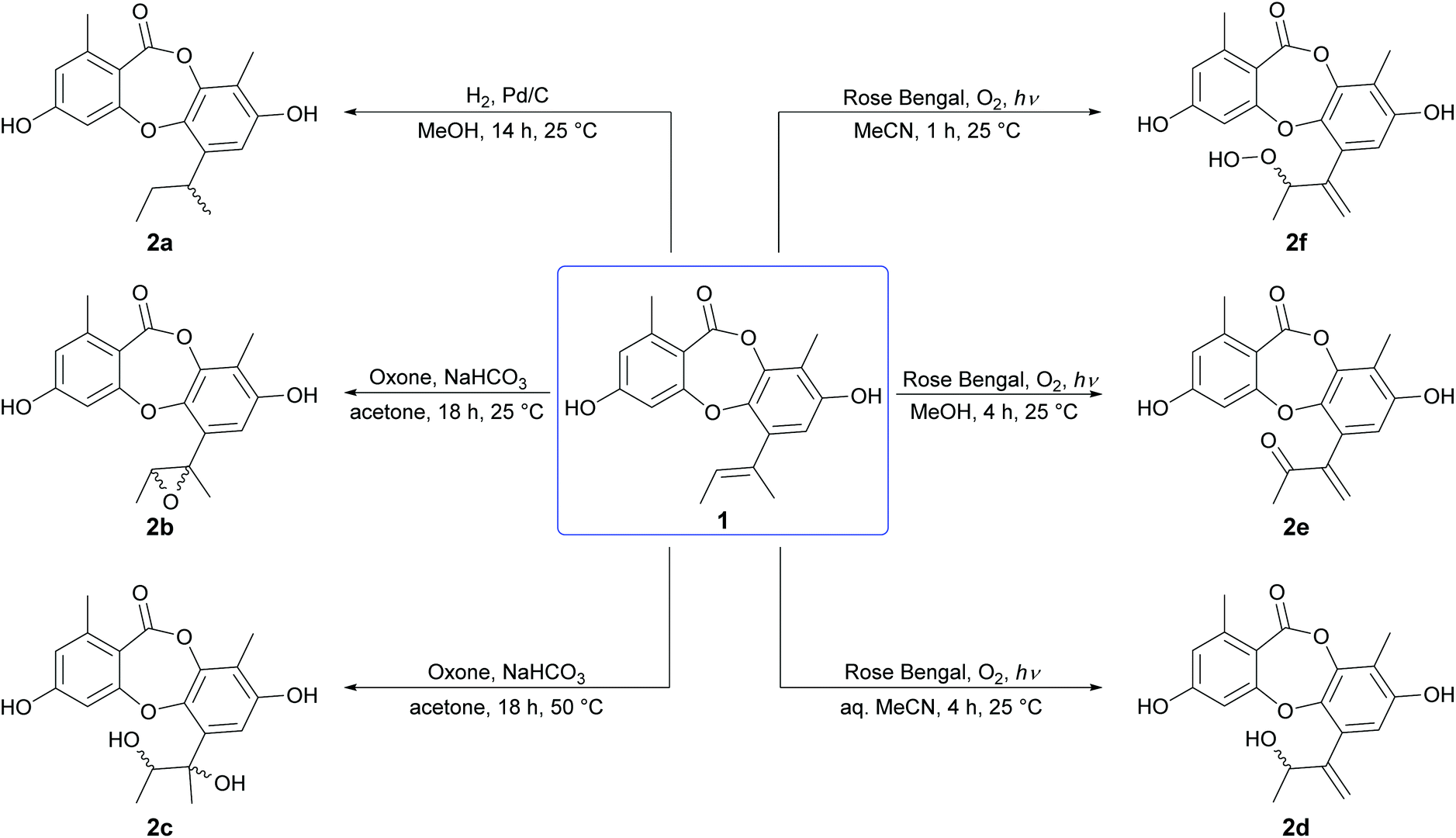
Initially, the Δ1′,2′ double bond in the butenyl side chain of 1 was reduced by catalytic hydrogenation (H2, Pd/C), to give a racemic mixture of 1′,2′-dihydrounguinol (2a) in quantitative yield (Scheme 1). Next, the Δ1′,2′ double bond of 1 was oxidised at 25 °C with dimethyldioxirane (DMDO), which was generated in situ from basic acetone and Oxone, to give cis-1′,2′-epoxyunguinol (2b) as a racemic mixture in 53% yield. Repeating the DMDO oxidation at 50 °C yielded a small quantity of 1′,2′-dihydroxyunguinol (2c), formed from 2b by nucleophilic ring-opening of the epoxide under the basic reaction conditions. LCMS analysis of crude reaction mixture revealed two diastereomers had formed in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, although only the major could be isolated in sufficient quantities for characterisation. Oxidation of the Δ1′,2′ double bond of 1 by molecular oxygen in the presence of UV light (254 nm) and Rose Bengal photosensitizer in aqueous MeCN, MeOH and anhydrous MeCN yielded three unguinol derivatives, 2′-hydroxy-Δ1′,4′-unguinol (2d), 2′-oxo-Δ1′,4′-unguinol (2e) and 2′-hydroperoxy-Δ1′,4′-unguinol (2f), respectively. The dye-sensitized photooxidation of alkenes has been studied extensively25,26 and the distribution of products observed can be rationalised by addition of singlet oxygen to the Δ1′,2′ double bond of 1 to yield allylic hydroperoxide 2f (Schenck ene reaction), followed by subsequent thermal decomposition to give alcohol 2d or dehydration to give ketone 2e.
:1 ratio, although only the major could be isolated in sufficient quantities for characterisation. Oxidation of the Δ1′,2′ double bond of 1 by molecular oxygen in the presence of UV light (254 nm) and Rose Bengal photosensitizer in aqueous MeCN, MeOH and anhydrous MeCN yielded three unguinol derivatives, 2′-hydroxy-Δ1′,4′-unguinol (2d), 2′-oxo-Δ1′,4′-unguinol (2e) and 2′-hydroperoxy-Δ1′,4′-unguinol (2f), respectively. The dye-sensitized photooxidation of alkenes has been studied extensively25,26 and the distribution of products observed can be rationalised by addition of singlet oxygen to the Δ1′,2′ double bond of 1 to yield allylic hydroperoxide 2f (Schenck ene reaction), followed by subsequent thermal decomposition to give alcohol 2d or dehydration to give ketone 2e.
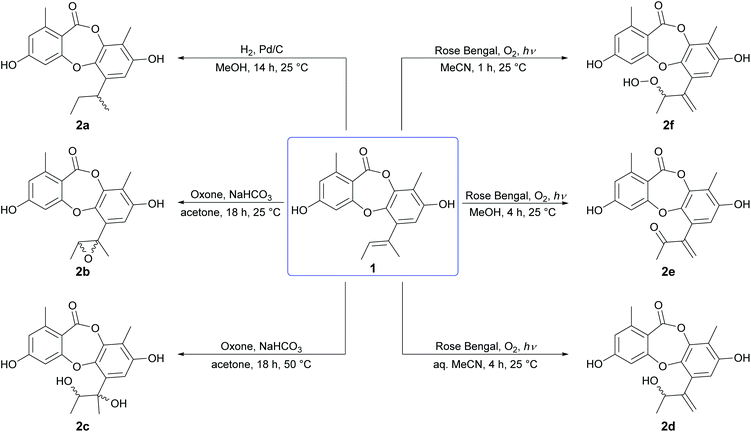 |
| | Scheme 1 Oxidation and reduction of the butenyl side chain of unguinol (1). | |
Halogenation.
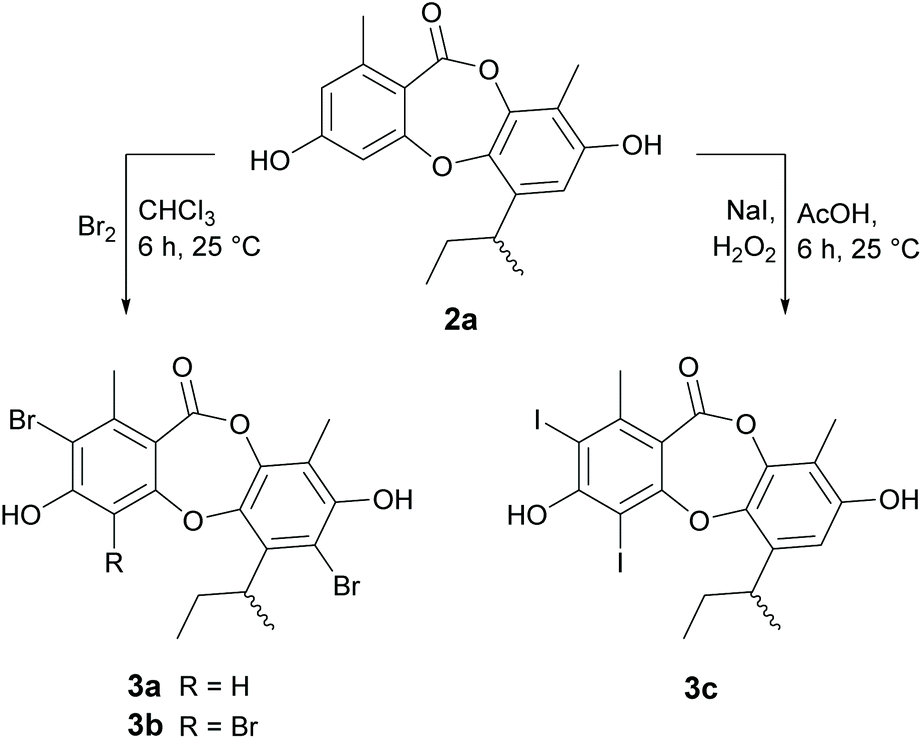
In our previous paper,21 we reported a series of novel brominated depsidones (mono- and dibromounguinol), which were obtained by supplementing the culture media with potassium bromide. Significantly, we found that these brominated depsidones showed improved antibacterial activities against Bacillus subtilis and S. aureus when compared with unguinol. To explore this effect more fully, we synthesised several brominated and iodinated unguinol analogues, as shown in (Scheme 2). As reduction of the butenyl side chain of 1 did not have any significant effects on antibacterial activity, we conducted the halogenation reactions on hydrogenated analogue 2a to avoid complicating side reactions. Treatment of 2a with bromine in chloroform at 25 °C yielded a mixture of 2,7-dibromo-1′,2′-dihydrounguinol (3a) and 2,4,7-tribromo-1′,2′-dihydrounguinol (3b) in 16% and 19% yield, respectively. While LCMS analysis of the crude reaction mixture suggested that trace amounts of monobrominated products had formed under these reaction conditions, varying the reaction temperature, solvent and equivalents of bromine did not yield sufficient quantities of these products for isolation and characterisation. Similarly, treatment of 2a with NaI and H2O2 in acetic acid at 25 °C yielded 2,4-diiodo-1′,2′-dihydrounguinol (3c) in 56% yield. LCMS analysis of the crude reaction mixture suggested trace amounts of monoiodinated products had formed, but there was no evidence for the formation of the triiodinated product, suggesting iodination at C-7 is not favoured under these conditions.
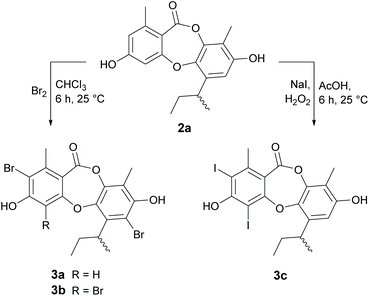 |
| | Scheme 2 Halogenation of 1′,2′-dihydrounguinol (2a). | |
Nucleophilic ring opening.
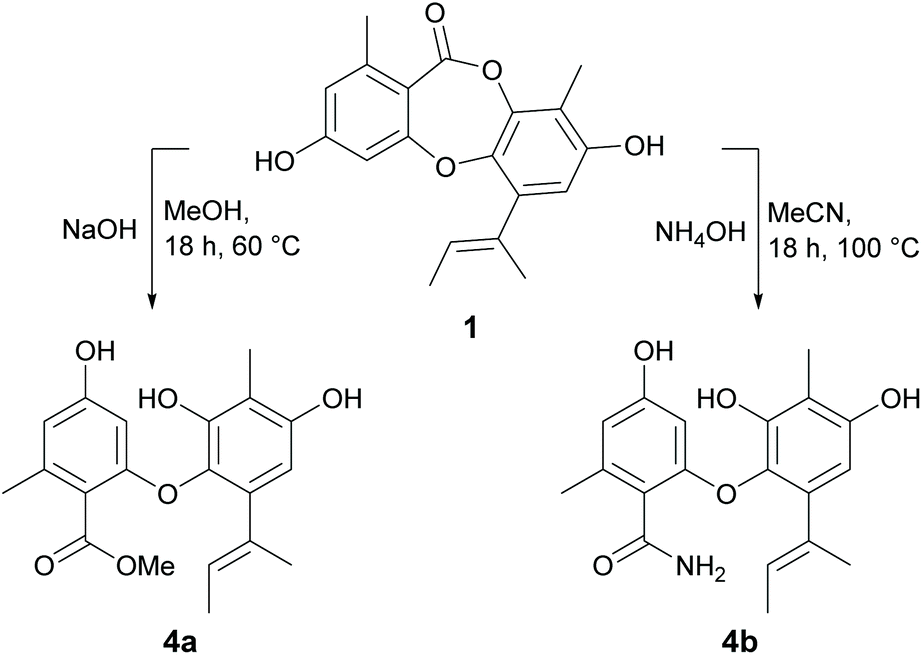
The depsidone scaffold contains a central seven-membered ring consisting of one ether and one ester linkage. Cleavage of the ether linkage yields depsides, while cleavage of the ester linkage yields diphenyl ethers. In our previous paper,21 we reported several naturally occurring depsides and diphenyl ethers with moderate to weak antibacterial activities. Therefore, to gain further insights into the SAR of diphenyl ethers, we next explored cleaving the ester linkage of 1 with sodium hydroxide in methanol and ammonium hydroxide in water to yield methyl unguinolate (4a) and unguinolamide (4b), respectively (Scheme 3). The presence of an additional exchangeable resonance in the 1H NMR spectra (δH 8.36 for 4a and 9.38 for 4b) attributable to 9a-OH confirmed that the depsidone ester linkages had been cleaved in both compounds. The presence of a methyl ester in 4a was evident from signals at δC 167.9 and 51.6 in 13C NMR spectrum, while the presence of a primary amide in 4b was evident from two additional exchangeable signals at δH 7.71 and 7.94. The free acid was isolated and characterised in our previous work21 as a natural product (unguinolic acid) and hence was not prepared synthetically in this work.
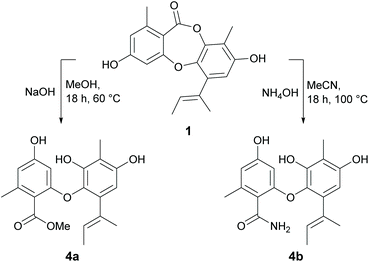 |
| | Scheme 3 Nucleophilic opening of the ester linkage of unguinol (1). | |
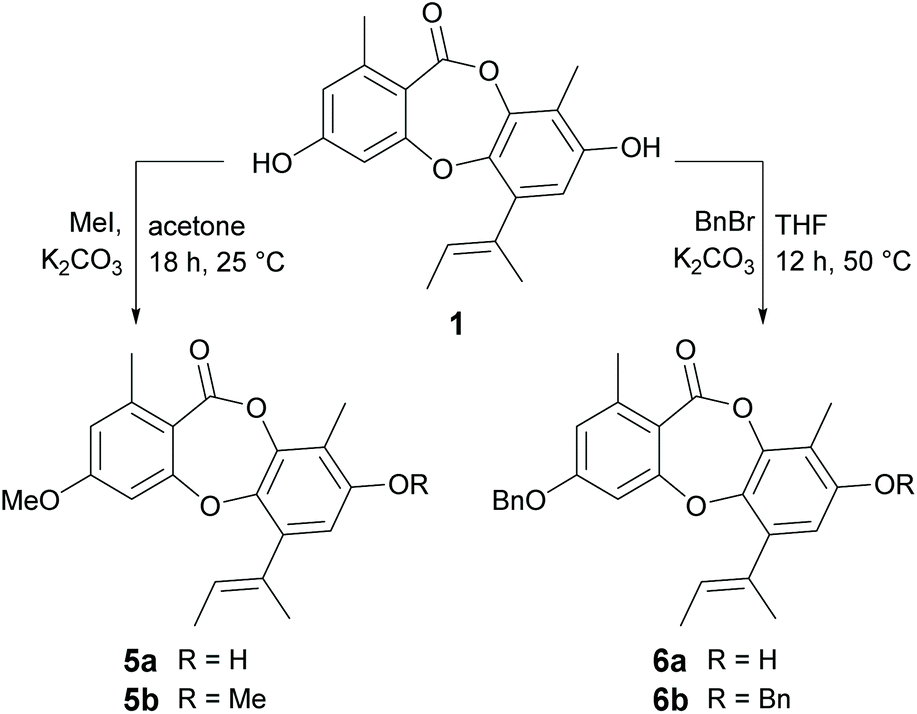
Methylation and benzylation.
We next turned our attention to derivatising the two free hydroxy groups (3-OH and 8-OH) of 1 (Scheme 4). Treatment of 1 with methyl iodide (2 eq.) in K2CO3 resulted in a mixture of 3-O-methylunguinol (5a) and 3,8-di-O-methylunguinol (5b), but not the 8-O-monomethylated product. The structure of 5a was confirmed by diagnostic ROESY NMR correlations between 3-OMe and aromatic protons H-2 and H-4. Attempts to access 8-O-methylunguinol by first protecting 3-OH as the tert-butyldimethylsilyl (TBDMS) ether were unsuccessful. This apparent regioselectivity is interesting given the preference for 8-O-methylation in Nature, with the only naturally occurring 3-O-methylated unguinol analogue reported to date being aspergillusidone B.27,28 Next, benzylation of 1 was carried out using benzyl bromide and K2CO3 at 50 °C in THF (Scheme 4). Under these reaction conditions, a mixture of mono- and dibenzylated products was formed, with 3-O-benzylunguinol (6a) isolated as the major product. The position of benzyl group on 3-OH was confirmed by diagnostic HMBC correlations from the benzyl methylene protons to C-3 (δC 161.7) and a ROESY correlation from the methylene protons to H-2. Interestingly, repeating the benzylation reaction using either acetone or acetonitrile as the solvent yielded 3,8-di-O-dibenzylunguinol (6b) as the major product.
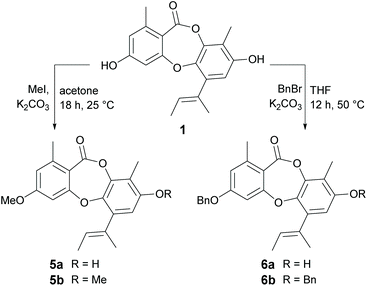 |
| | Scheme 4 Methylation and benzylation of unguinol (1). | |
Bioassay of first-generation semisynthetic unguinol analogues
The fifteen first-generation semisynthetic unguinol analogues were tested for in vitro activity against the Gram-positive bacteria B. subtilis (ATCC 6633) and S. aureus (ATCC 25923), the Gram-negative bacterium Escherichia coli (ATCC 25922), the fungi Candida albicans (ATCC 10231) and Saccharomyces cerevisiae (ATCC 9763), and mouse NS-1 myeloma (ATCC TIB-18) cells (Table 1). The compounds exhibited a wide range of antibacterial activities against the Gram-positive bacteria, but no activity was observed for any of the analogues against E. coli or C. albicans.
Table 1
In vitro biological activities of first-generation semisynthetic unguinol analogues
| Compounds |
MIC (μg mL−1) |
|
B. subtilis (ATCC 6633) |
S. aureus (ATCC 25923) |
S. cerevisiae (ATCC 9763) |
NS-1 (ATCC TIB-18) |
|
Not tested; – no activity up to 100 μg mL−1.
|
| Nidulin |
0.8 |
6.3 |
— |
27.2 |
| Unguinol (1) |
3.1 |
12.5 |
50 |
25 |
| 1′,2′-Dihydrounguinol (2a) |
3.1 |
25 |
50 |
25 |
|
cis-1′,2′-Epoxyunguinol (2b) |
25 |
100 |
— |
25 |
| 1′,2′-Dihydroxyunguinol (2c) |
— |
— |
— |
— |
| 2′-Hydroxy-Δ1′,4′-unguinol (2d) |
50 |
100 |
— |
50 |
| 2′-Oxo-Δ1′,4′-unguinol (2e) |
1.6 |
1.6 |
— |
<0.1 |
| 2′-Hydroperoxy-Δ1′,4′-unguinol (2f) |
3.1 |
3.1 |
— |
<0.1 |
| 2,7-Dibromo-1′,2′-dihydrounguinol (3a) |
2.1 |
2.1 |
4.2 |
4.2 |
| 2,4,7-Tribromo-1′,2′-dihydrounguinol (3b) |
1.2 |
4.6 |
— |
9.2 |
| 2,4-Diiodo-1′,2′-dihydrounguinol (3c) |
1.1 |
4.6 |
9.1 |
36.4 |
| Methyl unguinolate (4a) |
25 |
100 |
— |
50 |
| Unguinolamide (4b) |
25 |
— |
— |
<0.1 |
| 3-O-Methylunguinol (5a) |
6.3 |
12.5 |
12.5 |
25 |
| 3,8-Di-O-methylunguinol (5b) |
— |
— |
— |
16.1 |
| 3-O-Benzylunguinol (6a) |
1.6 |
0.2 |
— |
3.1 |
| 3,8-Di-O-benzylunguinol (6b) |
50 |
— |
— |
|
| Ampicillin |
0.2 |
3.1 |
|
|
| Clotrimazole |
|
|
0.4 |
|
| 5-Fluorouracil |
|
|
|
0.1 |
Reduction of the butenyl side chain of 1 by catalytic hydrogenation yielded 2a, which was equipotent against B. subtilis but two-fold less potent against S. aureus. Oxidation of the butenyl side chain of 1 yielded five oxygenated derivatives, 2b–2f. While epoxidation (2b) and dihydroxylation (2c) of the double bond significantly reduced antibacterial activity, the allylic ketone (2e) and hydroperoxide (2f) derivatives showed similar activity against B. subtilis and up to eight-fold increased activity against S. aureus. This was also accompanied by significantly increased cytotoxicity against mammalian tumour cells, with activities more potent than the positive control 5-fluorouracil. Dibromination (3a) and tribromination (3b) of 2a also significantly improved the antibacterial activity against both of the Gram-positive bacteria, but again was accompanied by increased cytotoxicity against NS-1 cells. Interestingly, 3a also showed twelve-fold improved activity against S. cerevisiae compared to 2a, while 3b showed no antifungal activity. Di-iodination (3c) of 2a yielded a similar improvement in antibacterial activity as dibromination, but with no increase in cytotoxicity. Nucleophilic opening of the ester linkage of 1 with NaOH/MeOH and NH4OH/MeCN to give methyl unguinolate (4a) and unguinolamide (4b), respectively, resulted in a significant decrease in antibacterial activity against both of the Gram-positive bacteria, highlighting the importance of the seven-membered depsidone ring system.
Methylation of 1 at 3-OH (5a) resulted in a two-fold decrease in activity against B. subtilis, with no change in activity against S. aureus. However, methylation of 1 at both 3-OH and 8-OH (5b) abolished activity against both of the Gram-positive bacteria, suggesting at least one free hydroxy group is essential for activity. Benzylation of 1 at 3-OH (6a) resulted in a modest increase in activity against B. subtilis, but a very significant increase in activity against S. aureus. Indeed, 6a (MIC 0.2 μg mL−1) was found to be over thirty-fold more active than nidulin (MIC 6.3 μg mL−1) and over sixty-fold more active than unguinol (MIC 12.5 μg mL−1) against S. aureus. Benzylation of 1 at both 3-OH and 8-OH (6b) also abolished all antibacterial activity, as was observed for dimethylation.
Second-generation semisynthetic unguinol analogues
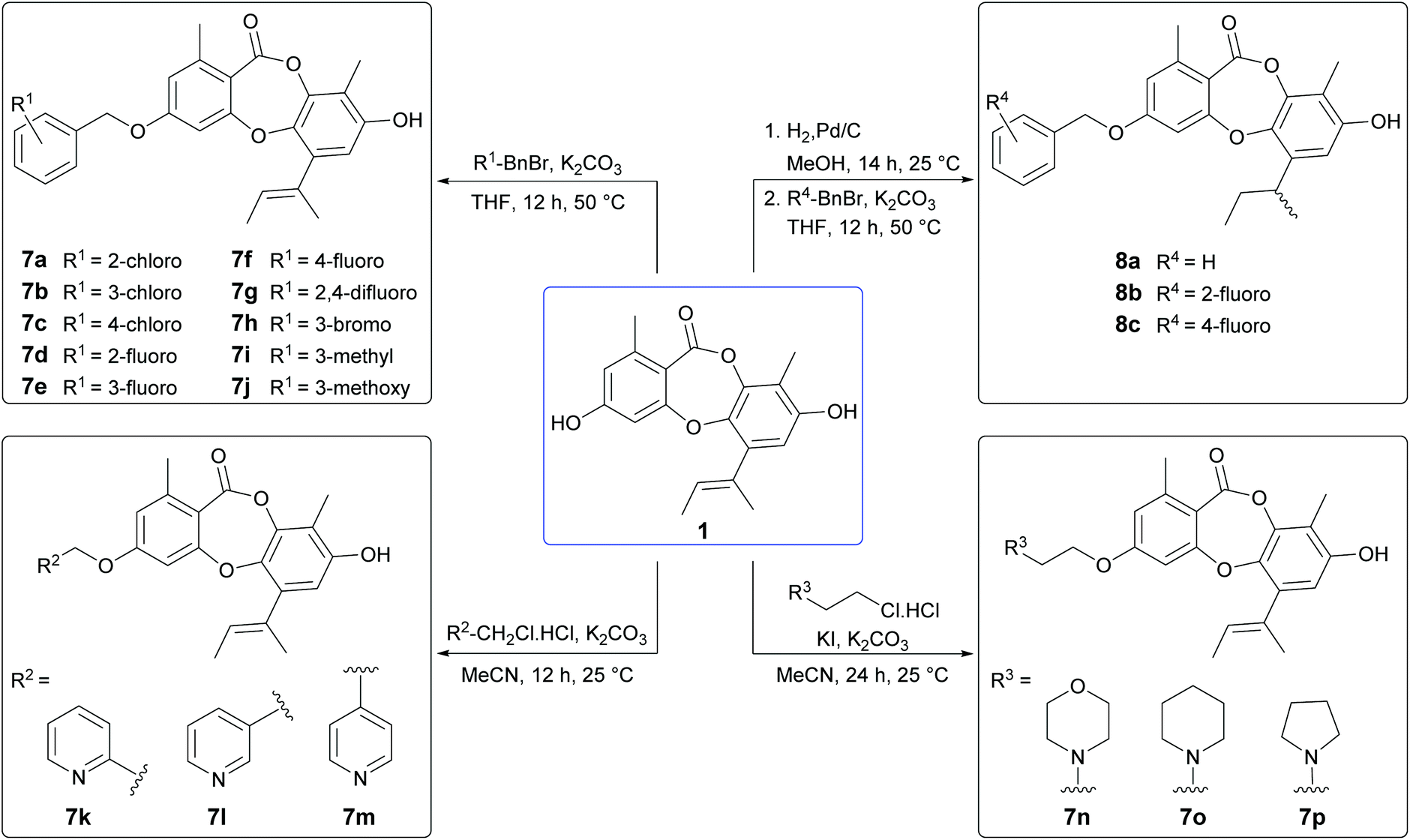
From our preliminary SAR studies, it was evident that benzylation of the 3-OH group of 1 significantly improved antibacterial activity, particularly against S. aureus. Inspired by these initial results, we next explored benzylation of 1 with a range of ortho-, meta- and para-substituted benzyl bromides (Scheme 5). Reaction of 1 with five equivalents of each benzyl bromide in THF at 50 °C yielded ten second-generation 3-O-benzylated derivatives, 7a–7j as the major products. Three picolyl derivatives (7k–7m) were also synthesised by reaction of 1 with 2-, 3-, and 4-picolyl chloride in MeCN at 25 °C. Alkylation of the 3-OH group of 1 by reaction with 4-(2-chloroethyl)morpholine, 1-(2-chloroethyl)piperidine and 1-(2-chloroethyl)pyrrolidine in MeCN at 25 °C yielded 7n–7p. Finally, benzylation of the 3-OH of 2a was performed using three different benzyl bromides, yielding 8a–8c.
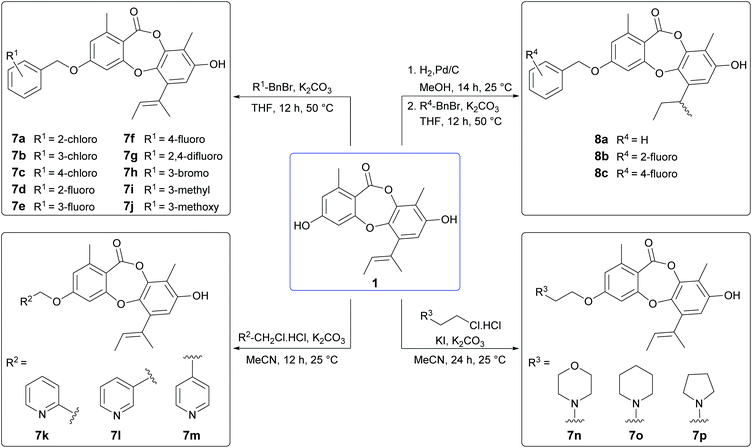 |
| | Scheme 5 Synthesis of second-generation unguinol analogues. | |
Bioassay of second-generation semisynthetic unguinol analogues
The nineteen second-generation unguinol congeners were initially tested for in vitro against the same panel of bacteria, fungi and mammalian cells as the first-generation analogues (Table 2). No activity was observed for any of the second-generation compounds against the tested Gram-negative bacterial or fungal species. The ten 3-O-benzylated unguinol analogues 7a–7j exhibited significant antibacterial activity against Gram-positive bacteria, with MICs ranging from <0.1–6.3 μg mL−1, and showed modest cytotoxicity against mouse NS-1 myeloma cells, with MICs ranging from 6.3–12.5 μg mL−1. Significantly, all ten benzylated analogues showed potent activity against S. aureus, with MICs ranging from <0.1–0.8 μg mL−1. The fluorobenzyl analogues 7d and 7f were the most promising leads, with activities superior to the antibiotic standards gentamicin (MIC 0.4 μg mL−1) and ampicillin (MIC 3.1 μg mL−1), and a selectivity index of 125. The 2-, 3- and 4-picolyl derivatives of 1 (7k–7m, respectively) showed 8- to 16-fold less activity against S. aureus compared to 6a, with only 7k retaining any activity against B. subtilis (MIC 6.3 μg mL−1). The 3-O-alkylamino derivatives 7n–7p also exhibited significantly reduced antibacterial activity compared to 6a. Intriguingly, this was accompanied by a dramatic increase in cytotoxicity against mouse myeloma NS-1 cells (MIC 0.2 μg mL−1), comparable in potency to the standard compound 5-fluorouracil (MIC 0.1 μg mL−1). The appearance of this potent and selective mammalian cytotoxicity is indicative of a mode of action distinct from the antibacterial activity of the unguinol scaffold. It has been recently reported that 8-O-alkylation of nornidulin results in moderate cytotoxicity against African green monkey kidney (Vero) cells.29 Similarly, the 1′,2′-dihydro analogues of 6a, 7d and 7f (8a–8c, respectively) showed slightly decreased antibacterial activities accompanied by markedly increased cytotoxicity against NS-1 cells (MIC 0.8 μg mL−1).
Table 2
In vitro biological activities of second-generation semisynthetic unguinol analogues
| Compound |
MIC (μg mL−1) |
|
B. subtilis (ATCC 6633) |
S. aureus (ATCC 25923) |
MRSA (ATCC 33592) |
NS-1 (ATCC TIB-18) |
|
Not tested; – no activity up to 100 μg mL−1.
|
| 3-O-Benzylunguinol (6a) |
1.6 |
0.2 |
0.4 |
3.1 |
| 3-O-(2-Chlorobenzyl)unguinol (7a) |
3.1 |
0.2 |
0.2 |
12.5 |
| 3-O-(3-Chlorobenzyl)unguinol (7b) |
3.1 |
0.8 |
0.4 |
6.3 |
| 3-O-(4-Chlorobenzyl)unguinol (7c) |
1.6 |
0.2 |
0.2 |
12.5 |
| 3-O-(2-Fluorobenzyl)unguinol (7d) |
0.8 |
0.1 |
0.1 |
12.5 |
| 3-O-(3-Fluorobenzyl)unguinol (7e) |
1.6 |
0.4 |
0.4 |
6.3 |
| 3-O-(4-Fluorobenzyl)unguinol (7f) |
1.6 |
<0.1 |
0.1 |
12.5 |
| 3-O-(2,4-Difluorobenzyl)unguinol (7g) |
0.8 |
0.2 |
0.2 |
12.5 |
| 3-O-(3-Bromobenzyl)unguinol (7h) |
6.3 |
1.6 |
1.6 |
6.3 |
| 3-O-(3-Methylbenzyl)unguinol (7i) |
3.1 |
0.8 |
0.8 |
6.3 |
| 3-O-(3-Methoxybenzyl)unguinol (7j) |
6.3 |
1.6 |
1.6 |
6.3 |
| 3-O-(2-Picolyl)unguinol (7k) |
6.3 |
1.6 |
1.6 |
12.5 |
| 3-O-(3-Picolyl)unguinol (7l) |
— |
3.1 |
6.3 |
25 |
| 3-O-(4-Picolyl)unguinol (7m) |
— |
1.6 |
3.1 |
12.5 |
| 3-O-(4-Morpholinoethyl)unguinol (7n) |
6.3 |
12.5 |
6.3 |
0.2 |
| 3-O-(1-Piperidinylethyl)unguinol (7o) |
6.3 |
25 |
6.3 |
0.2 |
| 3-O-(1-Pyrrolidinylethyl)unguinol (7p) |
12.5 |
100 |
50 |
0.2 |
| 3-O-Benzyl-1′,2′-dihydrounguinol (8a) |
3.1 |
3.1 |
1.6 |
0.8 |
| 3-O-(2-Fluorobenzyl)-1′,2′-dihydrounguinol (8b) |
3.1 |
3.1 |
1.6 |
0.8 |
| 3-O-(4-Fluorobenzyl)-1′,2′-dihydrounguinol (8c) |
3.1 |
3.1 |
1.6 |
0.8 |
| Ampicillin |
0.2 |
3.1 |
— |
|
| Gentamicin |
|
0.4 |
25 |
|
| 5-Fluorouracil |
|
|
|
0.1 |
We next explored the activity of the nineteen second-generation unguinol analogues against MRSA (ATCC 33592). Encouragingly, all ten 3-O-benzylated unguinol analogues 7a–7j retained equal potency against MRSA (MICs 0.1–0.8 μg mL−1) and were significantly more active than the standard gentamicin (MIC 25 μg mL−1). The picolyl derivatives 7k–7m also showed similar potencies against MRSA, while the alkylamino derivatives 7n–7p and the hydrogenated benzyl derivatives 8a–8c showed slightly improved potencies against MRSA. A subset of the 3-O-benzylated unguinol analogues (6a, 7a, 7c, 7d and 7g) was screened against one additional strain of MRSA (USA300), one additional strain of methicillin-sensitive S. aureus (MSSA; ATCC 49775) and two strains of Enterococcus faecium (ATCC 19434, E734) (Table 3), as well as two strains of Pseudomonas aeruginosa (PA01, ATCC 27853) and two additional strains of E. coli (ATCC 25322, ATCC 35218). No Gram-negative activity, or activity against either E. faecium, was detected for any of the compounds up to 16 μg mL−1. The analogues all showed good activities against both MRSA and MSSA (MICs 0.5–2 μg mL−1), comparable to the control compound daptomycin (MIC 0.5 μg mL−1). It is noteworthy that the MICs for the 3-O-benzylated unguinol analogues increased 32-fold in the presence of 10% foetal calf serum.
Table 3
In vitro antibacterial activity of benzylunguinol derivatives against MSSA, MRSA and E. faecium
| Compound |
MIC (μg mL−1) |
| MRSAa |
MSSAb |
E. faecium
|
E. faecium
|
|
USA300.
ATCC 49775.
ATCC 19434.
E734.
|
|
1
|
8 |
8 |
>16 |
>16 |
|
6a
|
1 |
1 |
>16 |
>16 |
|
7a
|
1 |
2 |
>16 |
>16 |
|
7c
|
0.5 |
1 |
>16 |
>16 |
|
7d
|
0.5 |
1 |
>16 |
>16 |
|
7g
|
0.5 |
0.5 |
>16 |
>16 |
| Daptomycin |
0.5 |
0.5 |
>2 |
>2 |
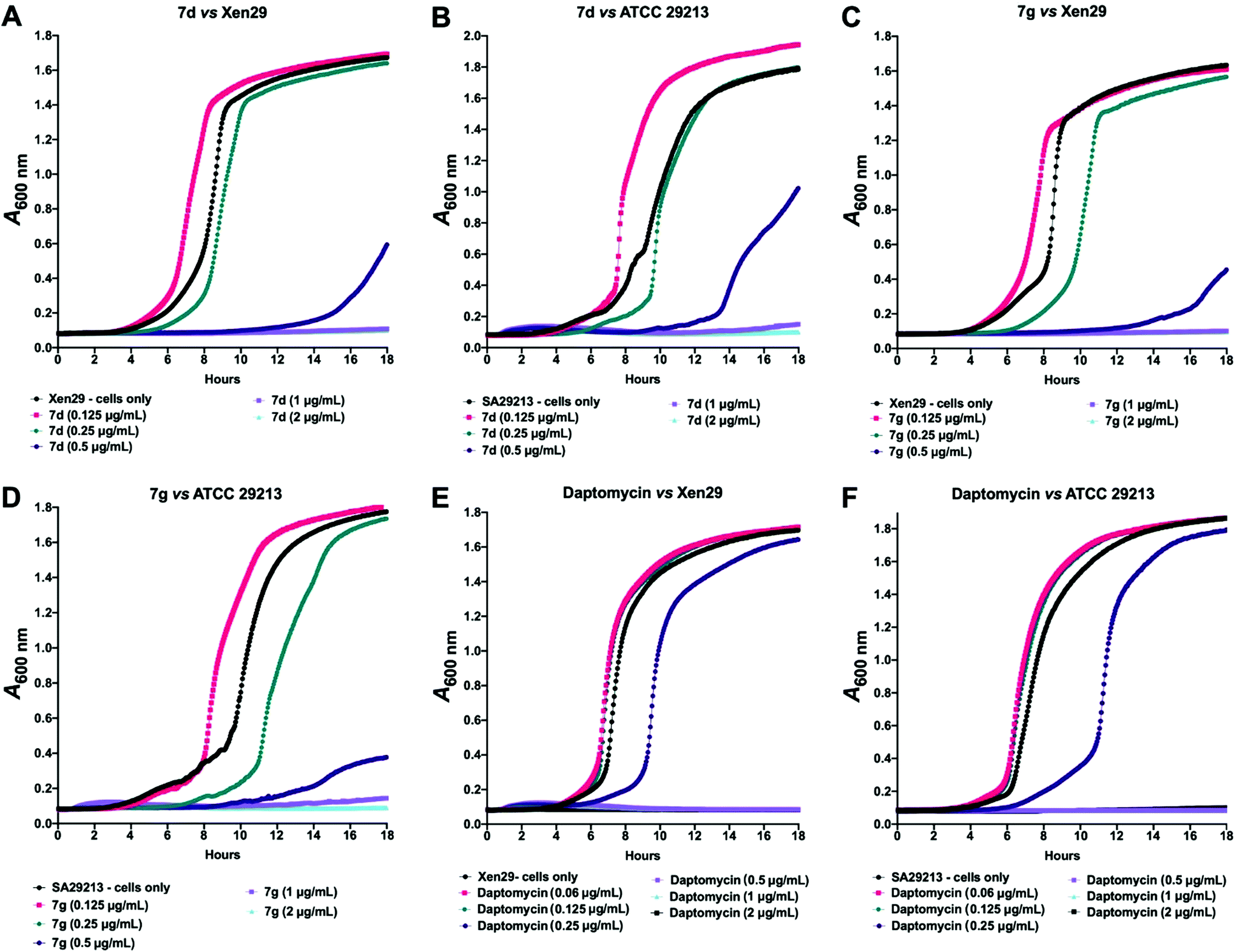
Given these findings, further in-depth evaluation of the antibacterial activities of 7d and 7g was carried out with an expanded list of S. aureus isolates and strains to obtain a clearer picture of the potency and selectivity of these analogues. The results show potent activity for both compounds, returning MIC range, MIC50 and MIC90 values comparable to the daptomycin standard (Table 4). The potency of 7d and 7g was further investigated in a kinetic assay to measure the time- and concentration-dependent activity of the two compounds against two S. aureus ATCC strains using daptomycin as a comparator. The results show a time- and concentration-dependent inhibition of growth for 7d and 7g, consistent with features of bacteriostatic drugs. As expected, daptomycin displayed patterns of a bactericidal drug (Fig. 2).
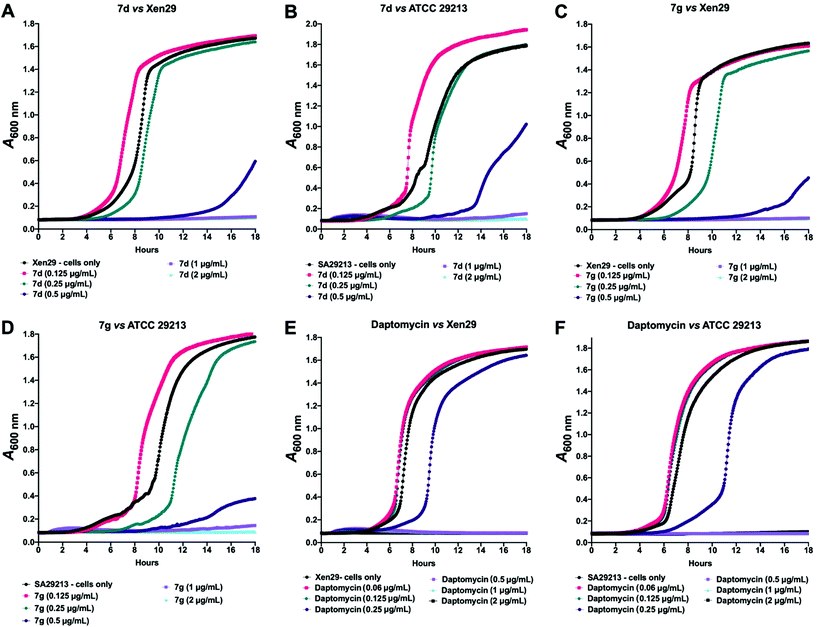 |
| | Fig. 2 Kinetic assay showing time- and concentration-dependent inhibition of S. aureus ATCC 12600 (Xen29; A, C and E) and ATCC 29213 (B, D and F) by 7d and 7g, with daptomycin as a comparator. Results show 7d and 7g are bacteriostatic, while daptomycin is bactericidal. | |
Table 4 MIC range, MIC50 and MIC90 against S. aureus (20 MRSA and 3 MSSA)
| Compound |
MIC (μg mL−1) |
| MIC range |
MIC50 |
MIC90 |
|
7d
|
0.25–1 |
0.5 |
0.5 |
|
7g
|
0.25–1 |
0.5 |
0.5 |
| Daptomycin |
0.25–1 |
0.5 |
0.5 |
| Ampicillin |
0.125–>16 |
>16 |
>16 |
A preliminary investigation into the suitability of 7d and/or 7g for administration as a drug was conducted by assessing their cytotoxicity to mammalian cells in fresh human red blood cells (RBCs), human embryonic kidney (HEK293) cell line and human epithelial liver (Hep G2) cell line. At the highest concentration (128 μg mL−1), 7d and 7g did not result in haemolysis of RBCs and both returned IC50 values of 32 μg mL−1 against the HEK293 and Hep G2 cell lines. These desirable cytotoxicity profiles promote exploration of 7d and 7g for in vivo safety and subsequent efficacy testing in relevant animal models.
Conclusions
In this study, we have completed the semisynthesis and in vitro biological evaluation of thirty-four derivatives of the fungal depsidone antibiotic, unguinol. Our SAR studies against a panel of microorganisms and mammalian cells revealed that at least one free hydroxy group is essential for antibacterial activity. We have demonstrated that by modification of the butenyl side chain of unguinol, we can target more potent and selective antitumor activity, while the introduction of bulkier halogens abolishes selectivity. Most notably, we have demonstrated that introduction of a 3-O-benzyl group exploits a putative additional hydrophobic pocket present in Gram-positive bacteria, affording a more potent and selective antibiotic class. The binding pocket appears to be optimal for fluorine-substituted benzyl analogues. Two of these analogues, 3-O-(2-fluorobenzyl)unguinol and 3-O-(2,4-difluorobenzyl)unguinol, demonstrated potent activity against MSSA and MRSA at concentrations comparable to those of a leading clinically effective Gram-positive antibiotic, daptomycin. Our assays also show these two compounds appear to be bacteriostatic and exhibit desirable mammalian cytotoxicity profiles, supporting their consideration for in vivo safety and drug efficacy testing in animal models of disease. While the mode of action of unguinol and its analogues remains unknown, our SAR results suggest this family of depsidones may act by binding to a target shared by prokaryotes, lower eukaryotes and higher eukaryotes. It is noteworthy that many lichen symbionts use variants of the depsidone scaffold to similar effect.30–32 This broad chemotherapeutic specificity represents an efficient use of resources, enabling the fungus to ward off a wide taxonomic framework of competitors. In Nature, potency and selectivity are implicit to each metabolite, which is an inverse template of its site of action. Employing a cohort of bioassays to help illuminate previously unrecognised aspects of potency and selectivity is an effective strategy for reviving existing chemical classes as potential new drugs.
Experimental
General experimental details
UV–vis spectra were acquired in MeOH on a Varian Cary 4000 spectrophotometer or a Jasco V-760 spectrophotometer in a 10 × 10 mm quartz cuvette. IR spectra were recorded on a Jasco FT/IR-6000 FTIR (ATR) spectrometer. Photooxidation reactions were performed using a Philips TUV PL-S 11 W/2P UVC light. 1H NMR and 13C NMR spectra were recorded in 5 mm Pyrex tubes (Wilmad, USA) on either a Bruker Avance II DRX-600K 600 MHz or Bruker Avance III HD 500 MHz spectrometer. All NMR spectra were obtained at 25 °C, processed using Bruker Topspin 3.5 software and referenced to residual solvent signals (DMSO-d6δH 2.49/δC 39.5 ppm). High resolution electrospray ionisation mass spectra (HRESIMS) were obtained on a Q Exactive Plus hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) by direct infusion. Electrospray ionisation mass spectra (ESIMS) were acquired on an Agilent 1260 UHPLC coupled to an Agilent 6130B single quadrupole mass detector. Analytical HPLC was performed on a gradient Agilent 1260 Infinity quaternary HPLC system equipped with a G4212B diode array detector. The column was an Agilent Poroshell 120 EC-C18 (4.6 × 50 mm, 2.7 μm) eluted with a 1 mL min−1 gradient of 10–100% MeCN/water (0.01% TFA) over 8.33 min. Semipreparative HPLC was performed on a gradient Agilent 1260 Infinity quaternary HPLC system coupled to a G4212B diode array detector. The column used in the purification of the compounds was an Agilent Zorbax SB-C18 (9.4 × 250 mm, 5 μm) eluted isocratically at 4.18 mL min−1. Preparative HPLC was performed on a gradient Shimadzu HPLC system comprising two LC-8A preparative liquid pumps with static mixer, SPD-M10AVP diode array detector and SCL-10AVP system controller with standard Rheodyne injection port. The columns used were a Grace Discovery Hypersil C18 spring column (150 × 50 nm, 5 μm) eluted isocratically at 60 mL min−1, an Agilent Zorbax SB-C18 column (150 × 50 nm, 5 μm) eluted isocratically at 60 mL min−1 and an Agilent Zorbax SB-C18 column (2 × 250 mm, 5 μm) eluted isocratically at 20 mL min−1. Silica flash chromatography was performed on a Biotage Isolera Four system coupled with a variable UV (200–400 nm) detector.
Isolation and purification of unguinol
A. unguis MST-FP511 was grown on pearl barley, which had been boiled in distilled water for 12 min and sterilised (120 °C for 40 min), in 60 × 250 mL Erlenmeyer flasks, with each flask containing 50 g of barley. Agar squares from a 7-day-old Petri plate of A. unguis were used as the inoculum for the flasks. The cultures were incubated for 21 days at 24 °C, then the grains were pooled and extracted with acetone (2 × 4 L) and the combined extracts were evaporated under vacuum to produce an aqueous slurry (2 L). The slurry was partitioned against ethyl acetate (2 × 2 L) and the ethyl acetate was reduced in vacuo to give the crude extract (55.6 g). The crude extract was redissolved in 90% MeOH/H2O (500 mL) and partitioned against hexane (2 × 500 mL) to remove lipids, yielding an enriched extract (35.7 g). The enriched extract was adsorbed onto silica gel (40 g), which was then loaded onto a silica gel column (100 g, 300 × 50 mm). The column was washed once with hexane (500 mL), then eluted with 50% hexane/CHCl3 (500 mL), 25% hexane/CHCl3 and CHCl3 (500 mL), followed by a stepwise gradient of 1, 2, 4, 8, 16, 32 and 100% MeOH/CHCl3 (500 mL each step), to yield 11 fractions (Fr. 1–11). Fraction 6 (2.1 g) was purified by isocratic preparative HPLC (Hypersil C18, isocratic 60% MeCN/H2O containing 0.01% TFA, 60 mL min−1) to yield 1 (tR 14.62 min; 384 mg).
Semisynthesis of unguinol analogues
1′,2′-Dihydrounguinol (2a).
Unguinol (1; 50 mg, 0.15 mmol) was dissolved in methanol (5 mL) and 10% palladium on carbon catalyst (3.2 mg) was added. The reaction mixture was stirred overnight under an atmosphere of H2 (balloon) at 25 °C. The reaction mixture was filtered through Celite and the solvent was removed in vacuo to yield 1′,2′-dihydrounguinol (2a; 50 mg, quant.) as a colourless solid, which was used without further purification. UV (MeOH) λmax (logε) 203 (5.08), 223 (4.78), 265 (4.43) nm; IR (ATR) νmax 3310, 2963, 1698, 1608, 1576, 1427, 1336, 1256, 1210, 1151, 1108, 1086 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 10.61 (s, 1H), 9.49 (s, 1H), 6.57 (dd, J = 2.4, 0.8 Hz, 1H), 6.54 (d, J = 2.4 Hz, 1H), 6.49, (s, 1H), 3.25 (m, 1H), 2.33 (s, 3H), 2.03 (s, 3H), 1.51 (m, 2H), 1.11 (d, J = 7.0 Hz, 3H), 0.79 (t, J = 7.3 Hz, 3H), 13C NMR (150 MHz, DMSO-d6): δ 163.1, 162.5, 161.8, 152.9, 144.7, 142.8, 141.0, 136.1, 115.6, 113.6, 111.6, 108.0, 104.4, 32.4, 29.8, 21.4, 20.7, 12.1, 9.1. HRESI(+)MS m/z 329.1381 [M + H]+ (calculated for C19H21O5+ 329.1384).
cis-1′,2′-Epoxyunguinol (2b).
To a stirred mixture of unguinol (1; 15 mg, 46 μmol), NaHCO3 (23 mg, 0.27 mmol) and acetone (5 mL) at 0 °C, a solution of Oxone (84 mg, 0.27 mmol) in water (3 mL) was added dropwise. The resulting mixture was stirred at 25 °C for 18 h and then extracted with ethyl acetate (3 × 3 mL). The organic layer was dried over MgSO4 and the solvent was evaporated under nitrogen. The residue (13.8 mg) was purified by semipreparative C18 HPLC with isocratic 80% MeCN/H2O (4.18 mL min−1), yielding a racemic mixture of cis-1′,2′-epoxyunguinol (2b; tR = 17.5 min; 8.0 mg, 53%) as white amorphous solid. UV (MeOH) λmax (logε) 202 (4.92), 221 (4.57), 267 (4.23) nm; IR (ATR) νmax 3461, 2989, 1699, 1619, 1579, 1424, 1384, 1381, 1254, 1217, 1188, 1160, 1104 cm−1; 1H NMR (500 MHz, DMSO-d6): δ 10.76 (br s, 1H), 9.72 (br s, 1H), 6.58 (s, 1H), 6.57 (d, J = 2.4 Hz, 1H), 6.46 (d, J = 2.4 Hz, 1H), 2.85 (q, J = 5.4 Hz, 1H), 2.33 (s, 3H), 2.03 (s, 3H), 1.56 (s, 3H), 1.47 (d, J = 5.4 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 162.6, 162.2, 162.1, 152.8, 144.9, 142.9, 140.2, 132.7, 115.9, 115.4, 111.1, 108.3, 104.4, 59.4, 59.0, 20.7, 18.8, 13.8, 9.2. HRESI(−)MS m/z 341.1032 [M − H]− (calculated for C19H17O6−, 341.1031).
1′,2′-Dihydroxyunguinol (2c).
To a stirred solution of unguinol (1; 15 mg, 46 μmol) in acetone (5 mL) was added NaHCO3 (38 mg) and a solution of Oxone (141 mg, 0.46 mmol) in water (2 mL). The reaction mixture was stirred at 50 °C for 18 h and then extracted with ethyl acetate (3 × 3 mL). The organic layer was dried over anhydrous MgSO4, and reduced to dryness under nitrogen. The residue (12.5 mg) was purified by semipreparative HPLC with isocratic 80% MeCN/H2O plus 0.01% TFA (4.18 mL min−1), yielding 1′,2′-dihydroxyunguinol (2c; tR = 15.8 min; 2.2 mg, 14.7%) as a colourless solid. UV (MeOH) λmax (logε) 203 (4.91), 222 (4.59), 268 (4.28) nm; IR (ATR) νmax 3674, 2985, 2901, 1393, 1251, 1066 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 10.56 (s, 1H), 9.44 (s, 1H), 6.88 (s, 1H), 6.73 (d, J = 2.4 Hz, 1H), 6.56 (d, J = 2.4 Hz, 1H), 4.23 (p, J = 6.4 Hz, 1H), 2.36 (s, 3H), 2.05 (s, 3H), 1.43 (s, 3H), 1.02 (d, J = 6.4 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 163.3, 162.5, 161.5, 151.7, 145.0, 143.4, 141.1, 136.7, 115.5, 114.6, 111.3, 110.6, 105.7, 75.8, 70.9, 24.5, 21.1, 17.7, 9.3. HRESI(−)MS m/z 359.1135 [M − H]− (calculated for C19H19O7−, 359.1136).
2′-Hydroxy-Δ1′,4′-unguinol (2d).
Unguinol (1; 40 mg, 123 μmol) was dissolved in 95% aqueous MeCN (10 mL) and Rose Bengal (4 mg, 4 μmol) was added. The reaction mixture was irradiated with UV light (254 nm) for 4 h then reduced to dryness under nitrogen. The residue was redissolved in MeOH (1 mL) and purified by preparative HPLC running with isocratic 60% MeCN/H2O, 20 mL min−1 yielding 2′-hydroxy-Δ1′,4′-unguinol (2d; tR = 5.75 min, 3.5 mg, 8.75%) as a colourless solid. UV (MeOH) λmax (logε) 206 (5.11), 243 (4.62) nm; IR (ATR) νmax 2976, 1724, 1607, 1576, 1422, 1332, 1247, 1213, 1153, 1104 cm−1; 1H NMR (500 MHz, DMSO-d6): δ 10.55 (s, 1H), 9.63 (s, 1H), 6.54 (d, J = 2.4 Hz, 1H), 6.49 (s, 1H), 6.44 (d, J = 2.4 Hz, 1H), 5.52 (s, 1H), 5.12 (d, J = 4.7 Hz, 1H), 4.95 (s, 1H), 4.59 (p, J = 6.5 Hz, 1H), 2.32 (s, 3H), 2.05 (s, 3H), 1.05 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 163.0, 162.3, 161.7, 152.4, 150.3, 144.4, 143.2, 140.2, 131.1, 115.6, 115.3, 113.2, 111.4, 111.3, 104.9, 67.9, 22.7, 20.6, 9.23. HRESI(−)MS m/z 341.1032 [M − H]− (calculated for C19H17O6−, 341.1031).
2′-Oxo-Δ1′,4′-unguinol (2e).
Unguinol (1; 60 mg, 184 μmol) was dissolved in MeOH (10 mL) and Rose Bengal (20 mg, 20 μmol) was added. The reaction mixture was transferred to glass reaction chamber and exposed to UV light (254 nm) for 4 h. The solution was then reduced to dryness under nitrogen and purified via preparative HPLC running with an isocratic gradient 60% MeCN/H2O (60 mL min−1) yielding 2′-oxo-Δ1′,4′-unguinol (2e; tR = 6.35 min, 1.8 mg, 3.0%) as a colourless solid. UV (MeOH) λmax (logε) 204 (4.71), 222 (4.39), 263 (4.08) nm; IR (ATR) νmax 3398, 2920, 2851, 1698, 1673, 1618, 1575, 1427, 1352, 1328, 1256, 1197, 1157, 1105 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 6.51 (d, J = 2.1 Hz, 1H) 6.47 (s, 1H), 6.32 (s, 1H), 6.17 (d, J = 2.1 Hz, 1H), 5.85 (s, 1H), 2.40 (s, 3H), 2.30 (s, 3H), 2.08 (s, 3H); 13C NMR (150 MHz, DMSO-d6): δ 198.3, 162.5, 162.1, 152.7, 145.5, 144.6, 142.9, 140.1, 128.0, 127.5, 116.4, 115.9, 111.7, 110.6, 104.3, 26.9, 20.7, 9.3. HRESI(−)MS m/z 339.0871 [M − H]− (calculated for C19H15O6−, 339.0874).
2′-Hydroperoxy-Δ1′,4′-unguinol (2f).
Unguinol (1; 65 mg, 199 μmol) was dissolved in MeCN (10 mL) and Rose Bengal (0.5 mg, 0.5 μmol) was added. The reaction mixture was transferred to glass UV reaction chamber and exposed to UV light (254 nm) for 1 h. The solution was reduced to dryness under nitrogen, redissolved in MeCN (1 mL) and purified by preparative HPLC running with an isocratic gradient running 50% MeCN/H2O (20 mL min−1), yielding 2′-hydroperoxy-Δ1′,4′-unguinol (2f; tR = 8.02 min, 9.4 mg, 14.5%) as a colourless solid. UV (MeOH) λmax (logε) 205 (4.95), 226 (4.66), 264 (4.32) nm; IR (ATR) νmax 3166, 2984, 1726, 1608, 1576, 1424, 1331, 1284, 1214, 1153, 1105 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 11.66 (br s, 1H), 9.97 (br s, 1H), 6.54 (s, 1H), 6.51 (d, J = 2.3 Hz, 1H), 6.42 (d, J = 2.3 Hz, 1H), 5.53 (dq, J = 1.6, 1.2 Hz, 1H), 5.13 (d, J = 1.6 Hz, 1H), 4.81 (q, J = 6.5 Hz, 1H), 2.31 (s, 3H), 2.06 (s, 3H), 1.16 (d, J = 6.5 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 163.1, 162.6, 162.3, 152.4, 145.2, 144.4, 143.2, 140.3, 130.2, 116.5, 115.8, 115.6, 111.6, 110.7, 104.9, 81.9, 20.6, 17.9, 9.24. ESI; HRESI(−)MS m/z 357.0980 [M − H]− (calculated for C19H17O7−, 357.0980).
2,7-Dibromo-1′,2′-dihydrounguinol (3a) and 2,4,7-tribromo-1′,2′-dihydrounguinol (3b).
1′,2′-Dihydrounguinol (2a; 10 mg, 30 μmol) was dissolved in chloroform (3 mL) and a solution of bromine in chloroform (2.1 M; 33 μL, 70 μmol) was added. The reaction mixture was stirred at 25 °C for 6 h. The crude reaction mixture was washed with brine (3 × 3 mL) and the organic layer was dried over MgSO4, filtered and reduced to dryness under nitrogen. The residue (9.1 mg) was purified by semipreparative HPLC with isocratic 60% MeCN/H2O plus 0.01% TFA (4.18 mL min−1). After separation, 2,7-dibromo-1′,2′-dihydrounguinol (3a; tR = 22.8 min; 1.64 mg, 16.4%) and 2,4,7-tribromo-1′,2′-dihydrounguinol (3b; tR = 23.5 min; 1.9 mg, 19%) were isolated. Compound 3a was isolated as white solid; UV (MeOH) λmax (logε) 203 (4.74), 221 (4.58), 283 (4.05), 322 (4.10) nm; IR (ATR) νmax 2965, 1732, 1596, 1567, 1418, 1338, 1226, 1179 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 11.63 (s, 1H), 9.23 (s, 1H), 6.86 (s, 1H), 3.74 (br s, 1H), 2.44 (s, 3H), 2.16 (s, 3H), 1.99 (m, 1H), 1.81 (m, 1H), 1.34 (d, J = 7.2 Hz, 3H), 0.82 (br s, 3H); 13C NMR (150 MHz, DMSO-d6): δ 161.7, 161.3, 158.6, 149.9, 143.5, 142.3, 134.0, 116.7, 113.2, 111.3, 104.6, 34.1, 26.2, 21.6, 18.0, 12.7, 10.7. HRESI(−)MS m/z 482.9443 [M − H]− (calculated for C19H1779Br2O5−, 482.9448). Compound 3b was isolated as white solid; UV (MeOH) λmax (logε) 205 (4.54), 223 (4.36), 251 (4.07), 322 (4.14) nm; IR (ATR) νmax 2963, 1732, 1560, 1419, 1363, 1338, 1289, 1227 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 11.15 (s, 1H), 9.30 (s, 1H), 4.44 (br s, 1H), 2.41 (s, 3H), 2.19 (s, 3H), 2.00 (m, 1H), 1.76 (m, 1H), 1.30 (d, J = 7.2 Hz, 3H), 0.70 (t, J = 7.3 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 161.3, 158.7, 156.0, 150.4, 143.4, 142.2, 141.8, 134.2, 116.7, 114.1, 113.0, 108.4, 100.4, 33.6, 25.7, 22.3, 18.0, 12.3, 10.7. HRESI(−)MS m/z 560.8553 [M − H]− (calculated for C19H1679Br3O5−, 560.8553).
2,4-Diiodo-1′,2′-dihydrounguinol (3c).
1′,2′-Dihydrounguinol (2a; 10 mg, 30 μmol) was dissolved in acetic acid (3 mL) and NaI (130 mg, 88 μmol) was added. An aqueous solution of H2O2 (30%; 0.5 mL, 0.15 mmol) was added dropwise to the well-stirred solution, and the reaction mixture was stirred at 25 °C for 6 h. The mixture was treated with an aqueous sodium thiosulfate solution (2 × 3 mL) and extracted with ethyl acetate (3 × 3 mL). The organic layer was dried over anhydrous MgSO4, and reduced to dryness under nitrogen. The residue was purified by silica flash chromatography using a SNAP-10 g cartridge. The whole reaction product was dissolved in 1 mL n-hexane/ethyl acetate (50:50) and separated with n-hexane and ethyl acetate gradient system (7% to 60%), 12 mL min−1, yielding 2,4-diiodo-1′,2′-dihydrounguinol (3c; tR = 12.5 min, 5.6 mg, 56%) as a colourless solid. UV (MeOH) λmax (logε) 205 (4.85), 231 (4.41), 281 (3.88) nm; IR (ATR) νmax 2961, 1727, 1590, 1560, 1420, 1334, 1226, 1177 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 10.47, (br s, 1H), 9.61 (s, 1H), 6.52 (s, 1H), 3.98 (m, 1H), 2.45 (s, 3H), 2.06 (s, 3H), 1.49 (m, 2H), 1.05 (d, J = 6.9 Hz, 3H), 0.79 (t, J = 7.3 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 163.0, 162.1, 160.4, 153.4, 146.3, 142.5, 142.1, 136.5, 113.9, 108.3, 92.3, 76.9, 32.9, 29.8, 28.2, 21.6, 11.7, 9.1. HRESI(−)MS m/z 578.9172 [M − H]− (calculated for C19H17I2O5−, 578.9171).
Methyl unguinolate (4a).
Unguinol (1; 50 mg, 153.0 μmol) was dissolved in 2 M KOH in MeOH (10 mL) and transferred to clear glass scintillation vial and the reaction mixture was heated to 60 °C for 18 h. The reaction was quenched by diluting with H2O (50 mL) and recovered with ethyl acetate (50 mL). The organic layer was dried over Na2SO4 and reduced to dryness in vacuo followed by purification with preparative HPLC (Alltima, 20 mL min−1, 0.01% TFA) with an isocratic gradient running 50% MeCN/H2O, yielding methyl unguinolate (4a; tR = 23.89 min, 8.7 mg, 11.9%) as white residue. UV (MeOH) λmax (logε) 215 (4.48), 248 (4.00), 285 (3.37) nm; IR (ATR) νmax 3231, 2918, 1695, 1605, 1429, 1327, 1265, 1208, 1152 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.56 (s, 1H), 9.09 (s, 1H), 8.36 (s, 1H), 6.19 (s, 1H), 6.19 (d, J = 2.2 Hz, 1H), 5.73 (d, J = 2.2 Hz, 1H), 5.41 (qq, J = 6.8, 1.4 Hz, 1H), 3.77 (s, 3H), 2.17 (s, 3H), 1.96 (s, 3H), 1.75 (dq, J = 1.4, 1.1 Hz, 3H), 1.55 (dq, J = 6.8, 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 167.9, 159.0, 157.7, 152.6, 148.0, 137.9, 136.0, 133.2, 131.8, 123.5, 113.4, 110.7, 110.0, 105.8, 98.8, 51.6, 19.7, 16.6, 13.7, 9.0. HRESI(−)MS m/z 357.1340 [M − H]− (calculated for C20H21O6−, 357.1343).
Unguinolamide (4b).
Unguinol (1; 60 mg, μmol) was dissolved in MeCN (10 mL) and transferred to clear glass scintillation vial. Concentrated aqueous ammonia (25%; 8 mL) was added and reaction mixture was heated to 100 °C for 18 h. The reaction mixture was neutralised with HCl, diluted with H2O (50 mL) and extracted with ethyl acetate (50 mL). The organic layer was dried over Na2SO4 and reduced to dryness in vacuo. The residue purified via preparative HPLC (Alltima, 20 mL min−1, 0.01% TFA) with an isocratic gradient running 60% MeCN/H2O, yielding unguinolamide (4b; tR = 11.99 min, 7.1 mg, 12.1%) as white residue. UV (MeOH) λmax (logε) 208 (4.62), 244 (4.09), 283 (3.49) nm; IR (ATR) νmax 3172, 1648, 1588, 1425, 1378, 1320, 1267, 1209, 1157, 1104 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.43 (s, 1H), 9.38 (s, 1H), 9.03 (s, 1H), 7.94 (s, 1H), 7.71 (s, 1H), 6.22 (d, J = 2.2 Hz, 1H) 6.17 (s, 1H), 5.82 (d, J = 2.2 Hz, 1H), 5.54 (qq, J = 6.8, 1.5 Hz, 1H), 2.22 (s, 3H), 1.91 (s, 3H), 1.79 (dq, J = 1.5, 1.1 Hz, 3H), 1.59, (dq, J = 6.8, 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 170.2, 158.0, 156.5, 152.6, 148.4, 136.7, 135.8, 133.5, 132.5, 123.7, 118.5, 110.6, 110.2, 105.0, 100.0, 19.6, 16.6, 13.8, 8.8. HRESI(−)MS m/z 342.1341 [M − H]− (calculated for C19H20NO5−, 342.1347).
3-O-Methylunguinol (5a) and 3,8-di-O-methylunguinol (5b).
Unguinol (1; 15 mg, 46 μmol) was dissolved in acetone (5 mL) and iodomethane (6 μL, 0.09 mmol) and excess K2CO3 (5 mg) were added. The reaction mixture was stirred at 25 °C for 12 hours then filtered. The filtrate was reduced to dryness under nitrogen. The residue was purified by silica chromatography using a SNAP-10 g cartridge. The whole reaction product was dissolved in 1 mL n-hexane/ethyl acetate (50:50). The mixture was separated with n-hexane and ethyl acetate gradient system (2% to 20%), 12 mL min−1, yielding 3-O-methylunguinol (5a; 3.6 mg, tR = 23.5 min, 24%) and 3,8-di-O-methylunguinol (5b; 3.0 mg, tR = 17.8 min, 20%) as solid white powder. 3-O-Methylunguinol (5a): UV (MeOH) λmax (logε) 205 (4.81), 225 (4.55), 263 (4.20) nm; IR (ATR) νmax 3385, 2927, 2353, 1710, 1602, 1424, 1333, 1253, 1205, 1144, 1101, 102.3 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (s, 1H), 6.77 (d, J = 2.5 Hz, 1H), 6.42 (s, 1H), 6.39 (d, J = 2.5 Hz, 1H), 5.45 (qq, J = 6.8, 1.5 Hz, 1H) 3.76 (s, 3H), 2.38 (s, 3H), 2.04 (s, 3H), 2.00 (dq, J = 1.5, 1.2 Hz, 3H), 1.78 (dq, J = 6.8, 1.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.6, 162.2, 152.6, 144.4, 143.0, 140.1, 135.3, 132.4, 125.0, 114.6, 114.2, 113.2, 110.7, 103.0, 55.6, 20.5, 17.6, 13.6, 9.1. HRESI(−)MS m/z 339.1237 [M − H]− (calculated for C20H19O5−, 339.1238). 3,8-Di-O-methylunguinol (5b): UV (MeOH) λmax (logε) 205 (4.81), 225 (4.55), 263 (4.20) nm; IR (ATR) νmax 2934, 1731, 1607, 1573, 1484, 1445, 1415, 1378, 1324, 1296, 1235, 1216, 1198, 1147, 1126 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 6.78 (d, J = 2.5 Hz, 1H), 6.57 (s, 1H), 6.42 (d, J = 2.5 Hz, 1H), 5.53 (qq, J = 6.8, 1.5 Hz, 1H) 3.77 (s, 3H), 3.76 (s, 3H), 2.38 (s, 3H), 2.07 (s, 3H), 2.04 (dq, J = 1.5, 1.2 Hz, 3H), 1.80 (dq, J = 6.8, 1.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.7, 162.6, 162.0, 154.2, 144.6, 142.8, 141.2, 135.5, 132.4, 125.6, 116.3, 114.3, 113.0, 107.2, 103.0, 56.0, 55.7, 20.5, 17.7, 13.7, 9.0. HRESI(+)MS m/z 355.1535 [M + H]+ (calculated for C21H23O5+, 355.1540).
General procedure for benzylation of unguinol.
A mixture of unguinol (1; 15 mg, 46 μmol), benzyl bromide (0.43 mmol) and excess K2CO3 (5 mg) in tetrahydrofuran (5 mL) was stirred at 50 °C for 12 h. The crude reaction mixture was filtered and the filtrate was reduced to dryness under nitrogen. The crude reaction mixture was purified by silica chromatography using a SNAP-10 g cartridge with a n-hexane/ethyl acetate gradient system (1% to 20%), 12 mL min−1, yielding a mixture of 3-O-benzylated unguinol and 3,8-di-O-benzylated unguinol.
3-O-Benzylunguinol (6a).
Isolated as white powder, tR = 22.3 min, 6.0 mg, 40%. UV (MeOH) λmax (logε) 206 (5.08), 262 (4.50) nm; IR (ATR) νmax 2923, 2360, 1728, 1605, 1569, 1420, 1378, 1323, 1291, 1252, 1215, 1176, 1145, 1103 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (br s, 1H), 7.39, (m, 2H), 7.38 (m, 2H), 7.32 (m, 1H), 6.87 (d, J = 2.6 Hz, 1H), 6.46 (d, J = 2.6 Hz, 1H) 6.41 (s, 1H), 5.45 (qq, J = 6.7, 1.5 Hz, 1H), 5.14, (s, 2H), 2.32 (s, 3H), 2.04 (s, 3H), 1.93 (dq, J = 1.5, 1.1 Hz, 3H), 1.75, (dq, J = 6.7, 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.7, 152.6, 144.4, 143.0, 140.1, 136.0, 135.2, 132.2, 128.5, 128.0, 127.5, 125.1, 115.2, 114.6, 113.4, 110.7, 103.7, 69.6, 20.5, 17.5, 13.7, 9.1. HRESI(+)MS m/z 417.1689 [M + H]+ (calculated for C26H25O5+, 417.1696).
3,8-Di-O-benzylunguinol (6b).
Isolated as white powder, tR = 12.8 min, 4.0 mg, 27%. UV (MeOH) λmax (logε) 206 (4.94), 260 (4.34) nm; IR (ATR) νmax 3673, 2924, 2360, 2339, 1733, 1650, 1607, 1573, 1484, 1452, 1417, 1377, 1324, 1252, 1218, 1146, 1120 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 7.39 (m, 2H), 7.38 (m, 4H), 7.33 (m, 1H), 7.41 (m, 2H), 7.31 (m, 1H), 6.88, (d, J = 2.4 Hz, 1H), 6.68 (s, 1H), 6.48, (d, J = 2.4 Hz, 1H), 5.51 (qq, J = 6.8, 1.4 Hz, 1H), 5.15 (s, 2H), 5.08, (s, 2H), 2.38 (s, 3H), 2.13 (s, 3H), 1.96 (dq, J = 1.4, 1.1 Hz, 3H), 1.77, (dq, J = 6.8, 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.6, 162.0, 161.8, 153.3, 144.6, 142.9, 141.4, 137.0, 136.1, 135.3, 132.1, 128.5, 128.4, 128.1, 127.8, 127.5, 127.3, 125.7, 116.9, 115.4, 113.3, 108.8, 103.9, 69.9, 69.6, 20.6, 17.6, 13.8, 9.2. HRESI(+)MS m/z 507.2162 [M + H]+ (calculated for C33H31O5+, 507.2166).
3-O-(2-Chlorobenzyl)unguinol (7a).
Isolated as white powder, tR = 21.5 min, 7.8 mg, 52%. UV (MeOH) λmax (logε) 204 (4.82), 258 (4.25) nm; IR (ATR) νmax 3673, 3364, 2972, 2360, 2339, 1701, 1606, 1570, 1420, 1380, 1326, 1253, 1216, 1147, 1101 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (br s, 1H), 7.50, (m, 2H), 7.39 (m, 1H), 7.36 (m, 1H), 6.90 (d, J = 2.6 Hz, 1H), 6.45 (d, J = 2.6 Hz, 1H) 6.41 (s, 1H), 5.45 (qq, J = 6.8, 1.4 Hz, 1H), 5.19, (s, 2H), 2.39 (s, 3H), 2.04 (s, 3H), 1.93 (dq, J = 1.4, 1.2 Hz, 3H), 1.73, (dq, J = 6.8, 1.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.4, 152.6, 144.5, 143.0, 140.1, 135.2, 133.3, 132.4, 132.1, 130.0, 129.8, 129.4, 127.4, 125.1, 115.2, 114.7, 113.7, 110.7, 103.4, 69.1, 20.5, 17.5, 13.6, 9.1. HRESI(+)MS m/z 451.1306 [M + H]+ (calculated for C26H2435ClO5+, 451.1306).
3-O-(3-Chlorobenzyl)unguinol (7b).
Isolated as white powder, tR = 21.7 min, 7.7 mg, 51.3%. UV (MeOH) λmax (logε) 203 (4.62), 263 (4.01) nm; IR (ATR) νmax 3356, 2926, 2360, 1696, 1610, 1567, 1478, 1417, 1378, 1366, 1323, 1293, 1265, 1215, 1197, 1146 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.63 (br s, 1H), 7.45 (m, 1H), 7.41 (m, 1H), 7.40 (m, 1H), 7.34 (m, 1H), 6.89 (d, J = 2.6 Hz, 1H), 6.45 (d, J = 2.5 Hz, 1H), 6.41 (s, 1H), 5.45 (qq, J = 6.7, 1.2 Hz, 1H), 5.17 (s, 2H), 2.38 (s, 3H), 2.04 (s, 3H), 1.91 (dq, J = 1.2, 1.0 Hz, 3H), 1.75 (dq, J = 6.7, 1.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.4, 152.6, 144.5, 143.0, 140.1, 138.7, 135.2, 133.2, 132.1, 130.4, 127.9, 127.0, 125.9, 125.1, 115.2, 114.7, 113.6, 110.7, 103.7, 68.6, 20.5, 17.5, 13.7, 9.1. HRESI(−)MS m/z 449.1162 [M − H]− (calculated for C26H2235ClO5−, 449.1161).
3-O-(4-Chlorobenzyl)unguinol (7c).
Isolated as white powder, tR = 21.4 min; 5.3 mg, 35.3%. UV (MeOH) λmax (logε) 202 (4.91), 258 (4.37) nm; IR (ATR) νmax 3672, 2971, 2901, 2360, 1730, 1607, 1570, 1491, 1421, 1379, 1324, 1255, 1218, 1148, 1102 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.61 (br s, 1H), 7.44, (m, 2H), 7.40 (m, 2H), 6.87 (d, J = 2.6 Hz, 1H), 6.43 (d, J = 2.6 Hz, 1H) 6.41 (s, 1H), 5.45 (qq, J = 6.8, 1.4 Hz, 1H), 5.15 (s, 2H), 2.38 (s, 3H), 2.04 (s, 3H), 1.92 (dq, J = 1.4, 1.0 Hz, 3H), 1.74, (dq, J = 6.8, 1.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.5, 152.6, 144.5, 143.0, 140.1, 135.2, 135.1, 132.6, 132.1, 129.2, 128.5, 125.1, 115.2, 114.7, 113.5, 110.7, 103.8, 68.7, 20.5, 17.5, 13.7, 9.1. HRESI(−)MS m/z 449.1164 [M − H]− (calculated for C26H2235ClO5−, 449.1161).
3-O-(2-Fluorobenzyl)unguinol (7d).
Isolated as white powder, tR = 22.1 min; 6.5 mg, 43.3%. UV (MeOH) λmax (logε) 205 (4.87), 263 (4.31) nm; IR (ATR) νmax 3672, 2971, 2360, 1729, 1606, 1570, 1493, 1421, 1380, 1325, 1253, 1216, 1181, 1146, 1104, cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (br s, 1H), 7.48 (ddd, J = 7.5, 7.5, 1.7 Hz, 1H), 7.42 (m, 1H), 7.25 (ddd, J = 10.9, 8.2, 1.1 Hz, 1H), 7.22 (ddd, J = 7.5, 7.5, 1.1 Hz, 1H), 6.89 (d, J = 2.5 Hz, 1H) 6.48 (d, J = 2.5 Hz, 1H), 6.42 (s, 1H), 5.46 (qq, J = 6.8, 1.4 Hz, 1H), 5.18 (s, 2H), 2.38 (s, 3H), 2.04 (s, 3H), 1.96 (dq, J = 1.4, 1.0 Hz, 3H), 1.75 (dq, J = 6.8, 1.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.4, 160.2, 152.6, 144.5, 143.0, 140.1, 135.2, 132.2, 130.6, 130.4, 125.1, 124.6, 122.9, 115.4, 115.1, 114.7, 113.6, 110.7, 103.6, 63.9, 20.5, 17.5, 13.6, 9.1. HRESI(+)MS m/z 435.1603 [M + H]+ (calculated for C26H24FO5+, 435.1602).
3-O-(3-Fluorobenzyl)unguinol (7e).
Isolated as white powder, tR = 22.8 min; 6.3 mg, 42.0%. UV (MeOH) λmax (logε) 207 (4.75), 262 (4.23) nm; IR (ATR) νmax 2919, 2360, 2340, 1719, 1608, 1568, 1488, 1475, 1416, 1376, 1347, 1324, 1290, 1254, 1238, 1218, 1147, 1128 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (br s, 1H), 7.42 (m, 1H), 7.22 (m, 2H), 7.15 (dddd, J = 9.2, 7.5, 2.5, 1.0 Hz, 1H), 6.88 (d, J = 2.4 Hz, 1H) 6.45 (d, J = 2.4 Hz, 1H), 6.41 (s, 1H), 5.44 (qq, J = 6.7, 1.3 Hz, 1H), 5.18 (s, 2H), 2.38 (s, 3H), 2.04 (s, 3H), 1.92 (dq, J = 1.3, 1.0 Hz, 3H), 1.74 (dq, J = 6.7, 1.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 162.1, 161.2, 152.6, 144.5, 143.0, 140.1, 139.0, 135.2, 132.1, 130.6, 125.1, 123.3, 115.2, 114.8, 114.7, 114.0, 113.6, 110.7, 103.8, 68.7, 20.5, 17.5, 13.7, 9.1. HRESI(−)MS m/z 433.1454 [M − H]− (calculated for C26H22FO5−, 433.1456).
3-O-(4-Fluorobenzyl)unguinol (7f).
Isolated as white powder, tR = 23.4 min; 7.1 mg, 47.3%. UV (MeOH) λmax (logε) 205 (4.82), 262 (4.29) nm; IR (ATR) νmax 3673, 2971, 2901, 2360, 1730, 1607, 1571, 1511, 1420, 1379, 1325, 1254, 1222, 1147, 1103 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.61 (br s, 1H), 7.44, (m, 2H), 7.21 (m, 2H), 6.87 (d, J = 2.5 Hz, 1H) 6.45 (d, J = 2.5 Hz, 1H), 6.41, (s, 1H), 5.46 (qq, J = 6.7, 1.4 Hz, 1H), 5.12 (s, 2H), 2.38 (s, 3H), 2.04 (s, 3H), 1.94 (dq, J = 1.4, 1.0 Hz, 3H), 1.75, (dq, J = 6.7, 1.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.8, 161.6, 152.5, 144.5, 143.0, 140.1, 135.2, 132.3, 132.2, 129.8, 125.1, 115.3, 115.1, 114.7, 113.4, 110.7, 103.8, 68.9, 20.5, 17.5, 13.7, 9.1. HRESI(−)MS m/z 433.1456 [M − H]− (calculated for C26H22FO5−, 433.1457).
3-O-(2,4-Difluorobenzyl)unguinol (7g).
Isolated as white powder, tR = 23.1 min; 8.4 mg, 56%. UV (MeOH) λmax (logε) 205 (5.00), 262 (4.45) nm; IR (ATR) νmax 3673, 3377, 2971, 2901, 2360, 2339, 1702, 1606, 1571, 1506, 1421, 1381, 1327, 1255, 1217, 1147, 1100 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (br s, 1H), 7.56 (m, 1H), 7.31 (ddd, J = 10.6, 9.3, 2.5 Hz, 1H), 7.12 (m, 1H), 6.89 (d, J = 2.5 Hz, 1H) 6.47 (d, J = 2.5 Hz, 1H), 6.42 (s, 1H), 5.46 (qq, J = 6.8, 1.4 Hz, 1H), 5.14 (s, 2H), 2.38 (s, 3H), 2.04 (s, 3H), 1.97 (dq, J = 1.4, 1.1 Hz, 3H), 1.76 (dq, J = 6.8, 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.4, 162.2, 161.3, 160.5, 152.6, 144.5, 143.0, 140.1, 135.2, 132.2, 131.9, 125.1, 119.4, 115.4, 115.0, 114.7, 113.7, 111.7, 110.7, 104.1, 103.6, 63.5, 20.5, 17.5, 13.6, 9.1. HRESI(−)MS m/z 451.1363 [M − H]− (calculated for C26H21F2O5−, 451.1363).
3-O-(3-Bromobenzyl)unguinol (7h).
Isolated as white powder, tR = 22.5 min; 6.8 mg, 45.3%. UV (MeOH) λmax (logε) 203 (4.76), 260 (4.17) nm; IR (ATR) νmax 2922, 2360, 2340, 1727, 1606, 1575, 1510, 1416, 1383, 1346, 1317, 1295, 1263, 1217, 1142, 1107 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (br s, 1H), 7.59 (dd, J = 1.6, 1.6 Hz, 1H), 7.52 (ddd, J = 7.7, 1.6, 1.3 Hz, 1H), 7.39 (ddd, J = 7.7, 1.6, 1.3 Hz, 1H), 7.34 (t, J = 7.7 Hz, 1H), 6.88 (d, J = 2.5 Hz, 1H), 6.44 (d, J = 2.5 Hz, 1H) 6.41 (s, 1H), 5.45 (qq, J = 6.8, 1.2 Hz, 1H), 5.16 (s, 2H), 2.38 (s, 3H), 2.04 (s, 3H), 1.91 (dq, J = 1.2, 1.1 Hz, 3H), 1.75 (dq, J = 6.8, 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.4, 152.6, 144.5, 143.0, 140.1, 138.9, 135.2, 132.1, 130.8, 130.7, 129.9, 126.3, 125.1, 121.7, 115.2, 114.7, 113.6, 110.7, 103.7, 68.5, 20.5, 17.5, 13.7, 9.1. HRESI(−)MS m/z 493.0656 [M − H]− (calculated for C26H2279BrO5−, 493.0656).
3-O-(3-Methylbenzyl)unguinol (7i).
Isolated as white powder, tR = 22.1 min; 6.5 mg, 43.3%. UV (MeOH) λmax (logε) 205 (4.88), 262 (4.30) nm; IR (ATR) νmax 2920, 2360, 2340, 1723, 1608, 1586, 1568, 1493, 1422, 1383, 1348, 1327, 1288, 1257, 1238, 1225, 1217, 1188, 1149 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (br s, 1H), 7.26 (t, J = 7.4 Hz, 1H), 7.19, (m, 1H), 7.16 (m, 1H), 7.14 (m, 1H), 6.86 (d, J = 2.5 Hz, 1H), 6.44 (d, J = 2.5 Hz, 1H), 6.41 (s, 1H), 5.45 (qq, J = 6.8, 1.4 Hz, 1H), 5.09 (s, 2H), 2.37 (s, 3H), 2.29 (s, 3H), 2.04 (s, 3H), 1.93 (dq, J = 1.4, 1.0 Hz, 3H), 1.75 (dq, J = 6.8, 1.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.7, 152.6, 144.4, 143.0, 140.1, 137.7, 136.0, 135.2, 132.2, 128.7, 128.4, 128.0, 125.1, 124.5, 115.2, 114.7, 113.4, 110.7, 103.7, 69.6, 20.9, 20.5, 17.5, 13.7, 9.1. HRESI(−)MS m/z 429.1706 [M − H]− (calculated for C27H25O5−, 429.1707).
3-O-(3-Methoxybenzyl)unguinol (7j).
Isolated as white powder, tR = 20.8 min; 5.7 mg, 38.0%. UV (MeOH) λmax (logε) 203 (4.80), 263 (4.23), 281 (4.08) nm; IR (ATR) νmax 3399, 2926, 2360, 2340, 1708, 1608, 1570, 1503, 1477, 1419, 1395, 1347, 1324, 1290, 1274, 1242, 1221, 1184, 1145, 1130 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (br s, 1H), 7.29 (t, J = 8.1 Hz, 1H), 6.94 (m, 2H), 6.89 (m, 1H), 6.88 (d, J = 2.5, 1H), 6.44 (d, J = 2.5, 1H), 6.41 (s, 1H), 5.45 (qq, J = 6.7, 1.1, 1H), 5.16 (s, 2H), 3.73 (s, 3H), 2.38 (s, 3H), 2.04 (s, 3H), 1.91 (dq, J = 1.2, 1.1 Hz, 3H), 1.75 (dq, J = 6.7, 1.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.7, 159.3, 152.6, 144.4, 143.0, 140.1, 137.6, 135.2, 132.1, 129.6, 125.1, 119.4, 115.2, 114.7, 113.4, 113.4, 112.9, 110.7 103.8, 69.4, 55.0, 20.5, 17.5, 13.7, 9.1. HRESI(−)MS m/z 445.1656 [M − H]− (calculated for C27H25O6−, 445.1656).
3-O-(2-Picolyl)unguinol (7k).
A mixture of unguinol (15 mg, 46 μmol), 2-(bromomethyl)pyridine (23 mg, 2 eq.) and excess K2CO3 (5 mg) in MeCN (5 mL) was stirred for 12 h at 25 °C. The crude reaction mixture was filtered and the filtrate was reduced to dryness under nitrogen. The residue was purified by semipreparative HPLC with isocratic 40% MeCN/H2O plus 0.01% TFA (4.18 mL min−1), yielding 3-O-(2-picolyl)unguinol (7k; tR = 17.8 min; 4.1 mg, 27.3%). UV (MeOH) λmax (logε) 205 (4.92), 263 (4.44) nm; IR (ATR) νmax 3673, 2985, 2901, 2360, 2339, 1730, 1608, 1572, 1420, 1326, 1252, 1215, 1150, 1105 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.61 (s, 1H), 8.57 (d, J = 4.4 Hz, 1H), 7.81 (td, J = 7.7, 1.7 Hz, 1H), 7.42 (d, J = 7.7 Hz, 1H), 7.34 (ddd, J = 7.7, 4.4, 1.0 Hz, 1H), 6.89 (d, J = 2.5 Hz, 1H) 6.46 (d, J = 2.5 Hz, 1H), 6.41 (s, 1H), 5.43 (qq, J = 6.7, 1.4 Hz, 1H), 5.21 (s, 2H), 2.38 (s, 3H), 2.04 (s, 3H), 1.91 (dq, J = 1.4, 1.0 Hz, 3H), 1.73 (dq, J = 6.7, 1.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.5, 155.6, 152.5, 149.2, 144.4, 143.0, 140.1, 137.0, 135.2, 132.1, 125.1, 123.1, 121.5, 115.3, 114.7, 113.6, 110.7, 103.6, 70.5, 20.5, 17.5, 13.6, 9.1. HRESI(+)MS m/z 418.1644 [M + H]+ (calculated for C25H24NO5+, 418.1649).
3-O-(3-Picolyl)unguinol (7l).
A mixture of unguinol (15 mg, 46 μmol), 3-(bromomethyl)pyridine (46 mg, 4 eq.) and excess K2CO3 (5 mg) in MeCN (5 mL) was stirred at 25 °C for 12 h. The crude reaction mixture was filtered and the filtrate was reduced to dryness under nitrogen and purified by semipreparative HPLC with isocratic 30% MeCN/H2O plus 0.01% TFA (4.18 mL min−1), yielding 3-O-(3-picolyl)unguinol (7l; tR = 19.4 min; 1.2 mg, 8.0%). UV (MeOH) λmax (logε) 204 (4.48), 260 (4.00) nm; IR (ATR) νmax 3673, 2985, 2901, 2360, 2339, 1732, 1606, 1407, 1251, 1148 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.61 (br s, 1H), 8.69 (br s, 2H), 7.83 (d, J = 7.7 Hz, 1H), 7.47 (br s, 1H), 6.90 (d, J = 2.5 Hz, 1H), 6.48 (d, J = 2.5 Hz, 1H), 6.41 (s, 1H), 5.45 (qq, J = 6.7, 1.4 Hz, 1H), 5.21 (s, 2H), 2.38 (s, 3H), 2.04 (s, 3H), 1.94 (dq, J = 1.4, 1.1 Hz, 3H), 1.76, (dq, J = 6.7, 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.4, 152.6, 149.1, 148.7, 144.5, 143.0, 140.1, 135.6, 135.2, 132.2, 132.2, 125.1, 124.1, 115.1, 114.7, 113.6, 110.7, 103.8, 67.3, 20.5, 17.5, 13.7, 9.1. HRESI(+)MS m/z 418.1643 [M + H]+ (calculated for C25H24NO5+, 418.1649).
3-O-(4-Picolyl)unguinol (7m).
A mixture of unguinol (1; 15 mg, 46 μmol), 4-(bromomethyl)pyridine (23 mg, 4 eq.) and excess K2CO3 (5 mg) in MeCN (5 mL) was stirred at 25 °C for 12 h. The crude reaction mixture was filtered and the filtrate was reduced to dryness under nitrogen and purified by semipreparative HPLC with isocratic 35% MeCN/H2O plus 0.01% TFA (4.18 mL min−1), yielding 3-O-(4-picolyl)unguinol (7m; tR = 22.1 min; 4.6 mg, 30.7%). UV (MeOH) λmax (logε) 204 (4.84), 256 (4.33) nm; IR (ATR) νmax 3671, 3203, 2986, 2901, 2359, 2090, 1611, 1517, 1410, 1253, 1150 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (s, 1H), 8.68 (br s, 2H), 7.41 (br s, 2H), 6.89 (d, J = 2.5 Hz, 1H) 6.44 (d, J = 2.5 Hz, 1H), 6.40 (s, 1H), 5.42 (qq, J = 6.7, 1.4 Hz, 1H), 5.24 (s, 2H), 2.38, (s, 3H), 2.04 (s, 3H), 1.89 (dq, J = 1.4, 1.1 Hz, 3H), 1.71 (dq, J = 6.7, 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.2, 152.6, 149.6, 145.4, 144.6, 143.0, 140.1, 135.2, 132.0, 125.1, 121.8, 115.2, 114.7, 113.8, 110.7, 103.7, 67.8, 20.5, 17.5, 13.7, 9.1. HRESI(+)MS m/z 418.1643 [M + H]+ (calculated for C25H24NO5+, 418.1649).
3-O-(4-Morpholinoethyl)unguinol (7n).
A mixture of unguinol (1; 20 mg, 61 μmol), 4-(2-chloroethyl)morpholine (11.4 mg, 1.0 eq.), K2CO3 (8.5 mg, 1.0 eq.) and KI (7.64 mg, 1.0 eq.) in MeCN was stirred for 72 h at 25 °C. The crude reaction mixture was filtered and the filtrate was reduced to dryness under nitrogen and purified by semipreparative HPLC with isocratic 15% MeCN/H2O plus 0.01% TFA (4.18 mL min−1), yielding 3-O-(4-morpholinoethyl)unguinol (7n; tR = 17.1 min; 5.3 mg, 34.6%). UV (MeOH) λmax (logε) 205 (5.05), 263 (4.51) nm; IR (ATR) νmax 2927, 1728, 1605, 1570, 1420, 1323, 1251, 1214, 1151, 1106 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.61 (s, 1H), 6.79 (d, J = 2.5 Hz, 1H) 6.42 (s, 1H), 6.40 (d, J = 2.5 Hz, 1H), 5.45 (qq, J = 6.7, 1.5 Hz, 1H), 4.11 (br s, 1H), 3.55 (br s, 4H), 2.66 (br s, 1H), 2.43 (br s, 2H), 2.37, (s, 3H), 2.04 (s, 3H), 2.00 (dq, J = 1.5, 1.1 Hz, 3H), 1.78 (dq, J = 6.7, 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 161.8, 152.6, 144.4, 143.0, 140.2, 135.3, 132.5, 125.0, 114.8, 114.7, 113.2, 110.7, 103.4, 66.0, 65.8, 56.4, 53.4, 20.5, 17.6, 13.7, 9.1. HRESI(+)MS m/z 440.2059 [M + H]+ (calculated for C25H30NO6+, 440.2068).
3-O-(1-Piperidinylethyl)unguinol (7o).
A mixture of unguinol (1; 15 mg, 46 μmol), 1-(2-chloroethyl)piperidine (8.5 mg, 1.0 eq.) K2CO3 (9.53 mg, 1.5 eq.) and KI (7.64 mg, 1.0 eq.) in MeCN was stirred at 25 °C for 24 h. The crude reaction mixture was filtered and the filtrate was reduced to dryness under nitrogen and purified by semipreparative HPLC with isocratic 15% MeCN/H2O plus 0.01% TFA (4.18 mL min−1), yielding 3-O-(1-piperidinylethyl)unguinol (7o; tR = 18.8 min; 3.4 mg, 22.7%). UV (MeOH) λmax (logε) 205 (4.97), 261 (4.45) nm; IR (ATR) νmax 2969, 2360, 2339, 1728, 1673, 1606, 1573, 1420, 1324, 1253, 1199, 1153, 1106 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.64 (s, 1H), 6.82 (s, 1H), 6.42 (s, 2H), 4.30 (br s, 2H), 5.46 (qq, J = 6.7, 1.3 Hz, 1H), 2.38 (s, 3H), 2.05 (s, 3H), 2.00 (dq, J = 1.3, 1.1 Hz, 3H), 1.78 (dq, J = 6.7, 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 152.6, 144.5, 143.0, 140.1, 135.3, 132.4, 125.0, 114.7, 114.7, 110.8, 103.7, 20.5, 17.6, 13.7, 9.1. HRESI(+)MS m/z 438.2272 [M + H]+ (calculated for C26H32NO5+, 438.2275).
3-O-(1-Pyrrolidinylethyl)unguinol (7p).
A mixture of unguinol (1; 15 mg, 46 μmol), 1-(2-chloroethyl)pyrrolidine (7.8 mg, 1.0 eq.) K2CO3 (9.53 mg, 1.5 eq.) and KI (7.64 mg, 1.0 eq.) was stirred at 25 °C for 24 h. The crude reaction mixture was filtered and the filtrate was reduced to dryness under nitrogen and purified by semipreparative HPLC with isocratic 15% MeCN/H2O plus 0.01% TFA (4.18 mL min−1), yielding 3-O-(1-pyrrolidinylethyl)unguinol (7p; tR = 19.5 min; 1.3 mg, 8.7%). UV (MeOH) λmax (logε) 205 (4.84), 226 (4.54), 266 (4.21) nm; IR (ATR) νmax 3673, 2971, 2901, 2360, 2339, 1730, 1675, 1607, 1418, 1253, 1201, 1156, 1105 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.66 (s, 1H), 6.85 (d, J = 2.4 Hz, 1H), 6.44 (d, J = 2.4 Hz, 1H), 6.43 (s, 1H), 5.46 (qq, J = 6.8, 1.5 Hz, 1H), 4.32 (br s, 2H), 3.57 (br s, 4H), 3.07 (br s, 2H), 2.40 (s, 3H), 2.05 (s, 3H), 2.00 (dq, J = 1.5, 1.0 Hz, 3H), 1.79 (dq, J = 6.8, 1.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.8, 162.2, 152.6, 144.6, 143.0, 140.1, 135.3, 132.4, 125.0, 114.7, 110.8, 103.8, 63.6, 53.8, 52.5, 22.4, 20.5, 17.6, 13.7, 9.1. HRESI(+)MS m/z 424.2111 [M + H]+ (calculated for C25H30NO5+, 424.2118).
3-O-Benzyl-1′,2′-dihydrounguinol (8a).
Isolated as white powder, tR = 20.5 min; 5.1 mg, 34%. UV (MeOH) λmax (logε) 203 (5.09), 265 (4.45) nm; IR (ATR) νmax 3356, 2962, 2929, 1728, 1607, 1570, 1428, 1378, 1336, 1256, 1214, 1149, 1112 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.50 (br s, 1H), 7.41 (m, 2H), 7.37 (m, 2H), 7.32 (m, 1H), 6.88 (d, J = 2.5 Hz, 1H) 6.83 (d, J = 2.5 Hz, 1H), 6.48 (s, 1H), 5.19 (d, J = 12.0 Hz, 2H) 3.32 (m, 1H), 2.39 (s, 3H), 2.03 (s, 3H), 1.48 (m, 2H), 1.05 (d, J = 6.8 Hz, 3H), 0.75 (t, J = 7.3 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.9, 162.3, 161.8, 152.9, 144.6, 142.7, 140.9, 136.2, 136.1, 128.4, 128.0, 127.5, 115.3, 113.6, 113.5, 108.1, 103.9, 69.7, 32.2, 29.8, 21.5, 20.6, 12.0, 9.1. HRESI(+)MS m/z 419.1848 [M + H]+ (calculated for C26H27O5+, 419.1853).
3-O-(2-Fluorobenzyl)-1′,2′-dihydrounguinol (8b).
Isolated as white powder, tR = 19.8 min; 5.7 mg, 38%. UV (MeOH) λmax (logε) 203 (5.08), 263 (4.44) nm; IR (ATR) νmax 3341, 2962, 2360, 2339, 1727, 1606, 1570, 1493, 1427, 1379, 1335, 1256, 1214, 1148, 1111 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.51 (br s, 1H), 7.52 (ddd, J = 7.6, 7.6, 1.7 Hz, 1H), 7.42 (m, 1H), 7.25 (ddd, J = 10.9, 8.2, 1.1 Hz, 1H), 7.21 (ddd, J = 7.6, 7.6, 1.0 Hz, 1H), 6.90 (d, J = 2.5 Hz, 1H) 6.88 (d, J = 2.5 Hz, 1H), 6.49, (s, 1H), 5.22 (d, J = 12.0 Hz, 2H) 3.32 (m, 1H), 2.39 (s, 3H), 2.04 (s, 3H), 1.49 (m, 2H), 1.07 (d, J = 6.8 Hz, 3H), 0.76 (t, J = 7.3 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.9, 162.3, 161.6, 160.3, 152.9, 144.7, 142.7, 140.9, 136.2, 130.6, 124.5, 122.9, 115.4, 115.2, 113.7, 113.6, 108.1, 103.8, 64.1, 32.2, 29.8, 21.5, 20.6, 11.9, 9.1. HRESI(+)MS m/z 437.1756 [M + H]+ (calculated for C26H26FO5+, 437.1758).
3-O-(4-Fluorobenzyl)-1′,2′-dihydrounguinol (8c).
Isolated as white powder, tR = 22.9 min; 6.4 mg, 42.6%. UV (MeOH) λmax (logε) 203 (5.10), 265 (4.43) nm; IR (ATR) νmax 3673, 3378, 2966, 2360, 2339, 1728, 1606, 1570, 1511, 1427, 1378, 1336, 1256, 1219 cm−1; 1H NMR (600 MHz, DMSO-d6): δ 9.50 (br s, 1H), 7.47 (m, 2H), 7.20 (m, 2H), 6.87 (d, J = 2.5 Hz, 1H) 6.82 (d, J = 2.5 Hz, 1H), 6.48, (s, 1H), 5.17 (d, J = 12.2 Hz, 2H) 3.28, (m, 1H), 2.39 (s, 3H), 2.03 (s, 3H), 1.48 (m, 2H), 1.05, (d, J = 6.8 Hz, 3H), 0.74 (t, J = 7.3 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 162.9, 162.3, 161.8, 161.7, 152.9, 144.6, 142.7, 140.9, 136.2, 132.4, 129.8, 115.3, 115.3, 113.6, 113.5, 108.1, 103.8, 68.9, 32.2, 29.8, 21.5, 20.6, 11.9, 9.1. HRESI(+)MS m/z 437.1756 [M + H]+ (calculated for C26H26FO5+, 437.1758).
Biological screening
Purified metabolites were dissolved in DMSO to provide stock solutions (10000 μg mL−1 or 1000 μg mL−1 depending on the amount of material available). An aliquot of each stock solution was transferred to the first lane of Rows B to G in a 96-well microtitre plate and two-fold serially diluted with DMSO across the 12 lanes of the plate to provide a 2048-fold concentration gradient. Bioassay medium was added to an aliquot of each test solution to provide a 100-fold dilution into the final bioassay, thus yielding a test range of 100 to 0.05 μg mL−1 in 1% DMSO. Row A contained no test compound (as a reference for no inhibition) and Row H was uninoculated (as a reference for complete inhibition).
MIC determination and time-dependent inhibition assays.
B. subtilis (ATCC 6633), five S. aureus (ATCC12600, ATCC 25923, ATCC 33592, ATCC 49775 and USA300), two E. faecium (ATCC 19434 and E734), two P. aeruginosa (PAO1 and ATCC 27853) and three E. coli (ATCC 25322, ATCC 25922 and ATCC 35218) strains were used as indicative species for antibacterial activity. A bacterial suspension (50 mL in 250 mL flask) was prepared in nutrient media by cultivation at 28 °C for 24 h with shaking at 250 rpm. The suspension was diluted to an absorbance of 0.01 absorbance units per mL, and 10 μL aliquots were added to the wells of a 96-well microtitre plate containing the test compounds dispersed in nutrient broth (Amyl) with resazurin (12.5 μg mL−1). The plates were incubated at 28 °C for 48 h, during which time the control wells with no test compound changed colour from a blue to light pink colour. MIC end points were determined visually. The absorbance was measured using Spectromax plate reader (Molecular Devices) at 605 nm and the IC50 values determined graphically. Our preliminary testing clearly showed 7d and 7g exhibited potent activity against the selected S. aureus strains. Therefore, these two compounds were subjected to MIC testing against an expanded panel of 20 clinical MRSA isolates plus 3 ATCC S. aureus strains in a standard plate microdilution MIC assay, using the Clinical Laboratory Standards Institute guidelines (CLSI 2017),33 essentially as described previously.6,34 The time- and concentration-dependent activities of 7d and 7g against two S. aureus ATCC strains were also determined in a kinetics assay by optical density (A600 nm) measurements on a Cytation 5 Multimode reader (BioTek).
Antifungal assay.
The yeasts C. albicans (ATCC 10231) and S. cerevisiae (ATCC 9763) were used as indicative species for antifungal activity. A yeast suspension (50 mL in 250 mL flask) was prepared in 1% malt extract broth by cultivation for 24 h at 250 rpm, 24 °C. The suspension was diluted to an absorbance of 0.005 and 0.03 absorbance units per mL for C. albicans and S. cerevisiae, respectively. Aliquots (20 μL and 30 μL) of C. albicans and S. cerevisiae, respectively were applied to the wells of a 96-well microtitre plate, which contained the test compounds dispersed in malt extract agar containing bromocresol green (50 μg mL−1). The plates were incubated at 24 °C for 48 h during which time the control wells containing no test compound change colour from a blue to yellow colour. MIC end points were determined visually. The absorbance was measured using Spectromax plate reader (Molecular Devices) at 620 nm and the IC50 determined graphically.
Cytotoxicity assay.
NS-1 (ATCC TIB-18) mouse myeloma cells were inoculated in 96-well microtitre plates (190 μL) at 50000 cells per mL in DMEM (Dulbecco's Modified Eagle Medium + 10% fetal bovine serum (FBS) + 1% penicillin/streptomycin (10000 U mL−1/10000 μg mL−1, Life Technologies Cat. no. 15140122), together with resazurin (250 μg mL−1; 10 μL) and incubated in 37 °C (5% CO2) incubator. The plates were incubated for 72 h during which time the control wells containing no test compound changed colour from a blue to pink colour. The absorbance of each well was measured at 605 nm using a Spectromax plate reader (Molecular Devices).
Haemolysis and real-time in vitro cytotoxicity assays.
Haemolysis assays for 7d and 7g were performed using fresh donor human red blood cells (RBCs), essentially as described previously.6,34 The toxicity profiles of the 2 compounds were also evaluated by measuring viability of HEK293 kidney and Hep G2 liver cell lines over a 20 h period in the presence of two-fold increasing concentrations of the compounds (2 μg mL−1–64 μg mL−1) on a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek) using the RealTime-Glo™ MT Cell Viability Assay reagent (Promega), as described previously.34
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
HRMS data were acquired by the Australian Proteome Analysis Facility, supported under the Australian Government's National Collaborative Research Infrastructure Strategy (NCRIS). This research was funded, in part, by the Australian Research Council (FT130100142 to AMP), the Cooperative Research Centres Projects scheme (CRCPFIVE000119) and Macquarie University (MQRTP scholarship to MTM).
Notes and references
- M. Bassetti, A. Carnelutti, N. Castaldo and M. Peghin, Expert Opin. Pharmacother., 2019, 20, 2317–2334 CrossRef CAS.
- S. Purrello, J. Garau, E. Giamarellos, T. Mazzei, F. Pea, A. Soriano and S. Stefani, J. Glob. Antimicrob. Resist., 2016, 7, 178–186 CrossRef CAS.
- S. Stefani, F. Campanile, M. Santagati, M. L. Mezzatesta, V. Cafiso and G. Pacini, Int. J. Antimicrob. Agents, 2015, 46, 278–289 CrossRef CAS.
- R. A. Seaton, F. Menichetti, G. Dalekos, A. Beiras-Fernandez, F. Nacinovich, R. Pathan and K. Hamed, Adv. Ther., 2015, 32, 1192–1205 CrossRef CAS.
- U. Theuretzbacher, K. Bush, S. Harbarth, M. Paul, J. H. Rex, E. Tacconelli and G. E. Thwaites, Nat. Rev. Microbiol., 2020, 18, 286–298 CrossRef CAS.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes and S. Epstein, Nature, 2015, 517, 455–459 CrossRef CAS.
- D. Nichols, N. Cahoon, E. Trakhtenberg, L. Pham, A. Mehta, A. Belanger, T. Kanigan, K. Lewis and S. Epstein, Appl. Environ. Microbiol., 2010, 76, 2445–2450 CrossRef CAS.
- C. L. Gilchrist, H. J. Lacey, D. Vuong, J. I. Pitt, L. Lange, E. Lacey, B. Pilgaard, Y. Chooi and A. M. Piggott, Fungal Genet. Biol., 2020, 143, 103435 CrossRef CAS.
- H. Li, C. L. Gilchrist, C. Phan, H. J. Lacey, D. Vuong, S. A. Moggach, E. Lacey, A. M. Piggott and Y. Chooi, J. Am. Chem. Soc., 2020, 142, 7145–7152 CrossRef CAS.
- H. J. Lacey, C. L. Gilchrist, A. Crombie, J. A. Kalaitzis, D. Vuong, P. J. Rutledge, P. Turner, J. I. Pitt, E. Lacey and Y. Chooi, Beilstein J. Org. Chem., 2019, 15, 2631–2643 CrossRef CAS.
- H. Li, C. L. Gilchrist, H. J. Lacey, A. Crombie, D. Vuong, J. I. Pitt, E. Lacey, Y. Chooi and A. M. Piggott, Org. Lett., 2019, 21, 1287–1291 CrossRef CAS.
- N. K. Chaudhary, J. I. Pitt, E. Lacey, A. Crombie, D. Vuong, A. M. Piggott and P. Karuso, J. Nat. Prod., 2018, 81, 1517–1526 CrossRef CAS.
- J. I. Pitt, L. Lange, A. E. Lacey, D. Vuong, D. J. Midgley, P. Greenfield, M. I. Bradbury, E. Lacey, P. K. Busk, B. Pilgaard, Y. Chooi and A. M. Piggott, PLoS One, 2017, 12, e0170254 CrossRef.
- H. J. Lacey, D. Vuong, J. I. Pitt, E. Lacey and A. M. Piggott, Aust. J. Chem., 2016, 69, 152–160 CrossRef CAS.
- K. Ochi and T. Hosaka, Appl. Microbiol. Biotechnol., 2013, 97, 87–98 CrossRef CAS.
- L. Foulston, Curr. Opin. Microbiol., 2019, 51, 1–8 CrossRef CAS.
- T. A. Wencewicz, Bioorg. Med. Chem., 2016, 24, 6227–6252 CrossRef CAS.
-
R. B. Fugitt and R. W. Luckenbaugh, U.S. Patent, 4128654, 1978 Search PubMed.
- M. Debono, M. Barnhart, C. Carrell, J. Hoffmann, J. Occolowitz, B. Abbott, D. Fukuda, R. Hamill, K. Biemann and W. Herlihy, J. Antibiot., 1987, 40, 761–777 CrossRef CAS.
- F. Kavanagh, A. Hervey and W. J. Robbins, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 570 CrossRef CAS.
- M. T. Morshed, D. Vuong, A. Crombie, A. E. Lacey, P. Karuso, E. Lacey and A. M. Piggott, Org. Biomol. Chem., 2018, 16, 3038–3051 RSC.
- J. M. Kurung, Science, 1945, 102, 11–12 CrossRef CAS.
- F. Dean, A. Robertson, J. C. Roberts and K. Raper, Nature, 1953, 172, 344–344 CrossRef CAS.
- Y. Zhang, J. Mu, Y. Feng, L. Wen and J. Han, Nat. Prod. Res., 2014, 28, 503–506 CrossRef CAS.
- M. Prein and W. Adam, Angew. Chem., Int. Ed. Engl., 1996, 35, 477–494 CrossRef CAS.
- E. L. Clennan, Tetrahedron, 2000, 47, 9151–9179 CrossRef.
- S. Sureram, S. Wiyakrutta, N. Ngamrojanavanich, C. Mahidol, S. Ruchirawat and P. Kittakoop, Planta Med., 2012, 78, 582–588 CrossRef CAS.
- P. Phainuphong, V. Rukachaisirikul, S. Phongpaichit, J. Sakayaroj, P. Kanjanasirirat, S. Borwornpinyo, N. Akrimajirachoote, C. Yimnual and C. Muanprasat, Tetrahedron, 2018, 74, 5691–5699 CrossRef CAS.
- M. Isaka, A. Yangchum, S. Supothina, S. Veeranondha, S. Komwijit and S. Phongpaichit, J. Antibiot., 2019, 72, 181–184 CrossRef CAS.
- B. Emsen, A. Aslan, B. Togar and H. Turkez, Pharm. Biol., 2016, 54, 1748–1762 CrossRef CAS.
- I. L. Lauinger, L. Vivas, R. Perozzo, C. Stairiker, A. Tarun, M. Zloh, X. Zhang, H. Xu, P. J. Tonge and S. G. Franzblau, J. Nat. Prod., 2013, 76, 1064–1070 CrossRef CAS.
- A. Cimmino, P. L. Nimis, M. Masi, L. De Gara, W. A. van Otterlo, R. Kiss, A. Evidente and F. Lefranc, Phytochem. Rev., 2019, 18, 1–36 CrossRef CAS.
-
Clinical and Laboratory Standards Institute, Performance standards for antimicrobial susceptibility testing, Clinical and Laboratory Standards Institute, Wayne, PA, 2017 Search PubMed.
- A. D. Ogunniyi, M. Khazandi, A. J. Stevens, S. K. Sims, S. W. Page, S. Garg, H. Venter, A. Powell, K. White and K. R. Petrovski, PLoS One, 2017, 12, e0183457 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, UV-vis spectra and HRMS spectra of all compounds. See DOI: 10.1039/d0ob02460k |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Hang T.
Nguyen
a,
Hang T.
Nguyen