
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of apratoxin A and B using Matteson's homologation approach†
Oliver
Andler
and
Uli
Kazmaier
 *
*
Organic Chemistry, Saarland University, P.O. Box 151150, 66041 Saarbrücken, Germany. E-mail: u.kazmaier@mx.uni-saarland.de
First published on 17th May 2021
Abstract
Apratoxin A and B, two members of an interesting class of marine cyclodepsipeptides are synthesized in a straightforward manner via Matteson homologation. Starting from a chiral boronic ester, the polyketide fragment of the apratoxins was obtained via five successive homologation steps in an overall yield of 27% and very good diastereoselectivity. This approach is highly flexible and should allow modification also of this part of the natural products, while previous modifications have been carried out mainly in the peptide fragment.
Introduction
The apratoxins are a class of cyclodepsipeptides produced by cyanobacteria.1 The first described member of this group, apratoxin A, was isolated by Moore and Paul et al. from the marine cyanobacterium Lyngbya majuscula in 2001.2 It showed potent cytotoxicity towards a range of tumor cell lines in the sub nanomolar range,3 acting as a broad-spectrum Sec61 inhibitor4 targeting HER/ErbB family proteins.5 Over the following years, a wide range of further apratoxins have been isolated.6 Some of these structures are shown in Fig. 1.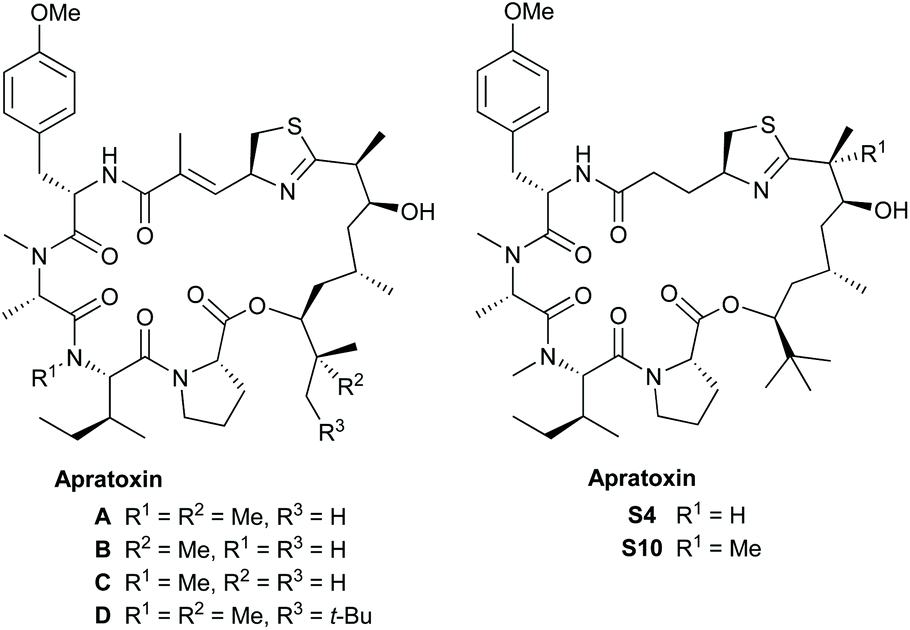 | ||
| Fig. 1 Selected apratoxins. | ||
The apratoxins form a 25-membered ring consisting of a pentapeptide and a rather unusual polyketide fragment, containing, in most cases, a terminal t-butyl group. The apratoxins differ mainly in their methylation pattern (apratoxins A–D) or in the composition of the peptide fragment. While most apratoxins embody an unusual unsaturated prolonged cysteine unit, in some of the members of the apratoxin S family, artificial derivatives obtained by a medicinal chemistry campaign, this unit is (structurally) reduced.7 The unusual polyketide fragment is common to most family members. In case of apratoxin D, its carbon chain is even one carbon longer. Detailed studies indicate that an iron-dependent methyltransferase directs the tert-butyl group formation by initiating the assembly of the apratoxin A polyketide starter unit.8
Not surprisingly, the excellent cytotoxicities in the low nanomolar range, and the interesting mode of action initiated synthetic investigations for the synthesis of the polyketide fragment of the apratoxins,9 the apratoxins themselves10 as well as derivatives for SAR studies.11 Common to all syntheses of apratoxin A is the late-stage assembly of the thiazoline moiety, which is oxidatively sensitive and potentially prone to unwanted side reactions, e.g. epimerization of the adjacent stereogenic centre. To synthesise the polyketide fragment, asymmetric cuprate additions or allyl isomerisations are used to introduce the internal methyl group, while the two stereogenic centres adjacent to the thiazoline ring are generated via different versions of aldol reactions.
Results and discussion
Since a couple of years our group is involved in the synthesis of natural products,12 especially cyclic peptides with interesting biological activities.13 Recently, we have become interested in the synthesis of peptide/polyketide hybrids14 such as the apratoxins, with a focus on aldol-free polyketide synthesis. The aldol reaction,15 or alternatively an allylation/ozonolysis sequence,16 is perfectly suited to generating the 3, 5, 7, …-polyhydroxylated carboxylic acids found in many natural products, with methyl groups between the hydroxy functionalities, but these reactions are more or less restricted to this substitution pattern. With the synthesis of lagunamide A,17 we could show that also other approaches, such as Matteson homologations,18 which are more flexible, are well suited to the synthesis of complex polyketides. This elegant stereoselective prolongation of chiral boronic esters was introduced by Donald Matteson 40 years ago (Scheme 1).19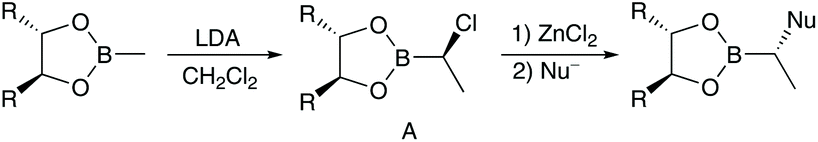 | ||
| Scheme 1 Matteson homologation. | ||
A key step of this protocol is the highly stereoselective formation of an α-chloro boronic ester A, which can be subjected to nucleophilic substitution under SN2-conditions with a wide range of nucleophiles such as Grignard reagents, alkoxides or certain enolates.20 Continual application of this procedure allows the stepwise stereoselective incorporation of a wide range of substituents and functionalities into a growing carbon chain.
We attempted to prepare the polyketide fragment of apratoxin A and B using the above-mentioned Matteson homologation approach. Starting from diisopropyl tert-butylboronate 121 transesterification with (S,S)-DICHED (dicyclohexylethane-1,2-diol)22 gave the chiral boronic ester 2 (Scheme 2). While homologation of sterically demanding 2 to the α-chloroboronic ester proceeded smoothly under the usual conditions, the subsequent substitution to the alkoxyboronic ester 3 was sluggish and incomplete when we used only a slight excess (1.3 eq.) of NaOPMB. In contrast, with a larger excess (3.0 eq.) of alkoxide, we observed full conversion and could isolate 3 in excellent yield.
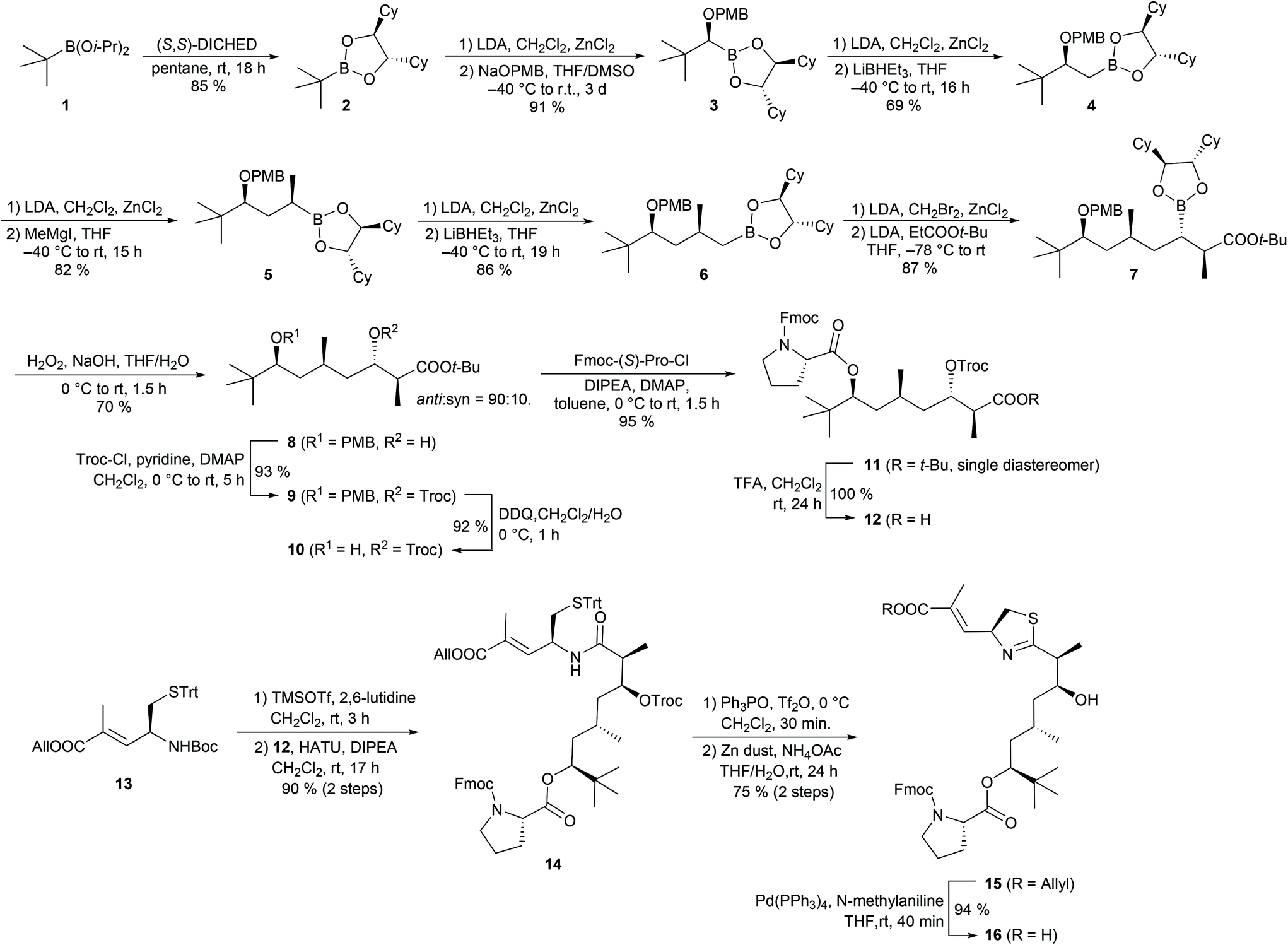 | ||
| Scheme 2 Synthesis of the apratoxin polyketide fragment. | ||
In the following step, we introduced a CH2 group by treating the intermediate α-chloroboronic ester with LiBHEt3. Next, we used a methyl Grignard reagent as a nucleophile, which cleanly yielded 5. Introduction of a second CH2 unit gave the prolonged boronic ester 6 in good yield. In the following homologation step, we applied the lithium enolate of tert-butyl propionate as a nucleophile. Similar reactions of simple α-haloboronic esters with enolates have already been described by Matteson et al.20a,b but, surprisingly, this method has not been applied to the synthesis of more complex natural products so far. A reason for this may be that this reaction requires the use of α-bromoboronic esters which are more reactive, but also more prone to epimerization than their chloro-analogues.23 Fortunately, we could completely suppress epimerization by quenching the formation of the α-bromoboronic ester at low temperature (−55 °C) and immediately treated the crude product with the enolate. While the homologations from 2 to 6 provided the products as single diastereomers, we obtained 8 as a mixture of syn and anti isomers, but with good anti selectivity (d.r. = 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 after oxidation of the boronic ester to alcohol 8). O-Troc protection of 8 followed by oxidative cleavage of the PMB ether provided alcohol 10, which we reacted with Fmoc-(S)-Pro-Cl24 to ester 11 in almost quantitative yield and without epimerization of the α-stereogenic centre of proline. Acidic cleavage of the tert-butyl ester gave access to crude carboxylic acid 12. N-Boc deprotection of the modified cysteine building block 13, followed by coupling with acid 12, yielded amide 14, which is a known intermediate of the apratoxin A synthesis reported by Doi et al.10c,11c To convert 14 into a thiazoline, we applied Kelly's method using Tf2O and Ph3PO.25 For the subsequent cleavage of the Troc carbamate, we examined several conditions, e.g. with a Zn/Cu couple or Me3SnOH26 as a deprotection reagent, but Doi's protocol using zinc dust and aqueous NH4OAc10c,11c gave 15 in the best yield. Pd-catalysed cleavage of the allyl ester provided carboxylic acid 16.
:1 after oxidation of the boronic ester to alcohol 8). O-Troc protection of 8 followed by oxidative cleavage of the PMB ether provided alcohol 10, which we reacted with Fmoc-(S)-Pro-Cl24 to ester 11 in almost quantitative yield and without epimerization of the α-stereogenic centre of proline. Acidic cleavage of the tert-butyl ester gave access to crude carboxylic acid 12. N-Boc deprotection of the modified cysteine building block 13, followed by coupling with acid 12, yielded amide 14, which is a known intermediate of the apratoxin A synthesis reported by Doi et al.10c,11c To convert 14 into a thiazoline, we applied Kelly's method using Tf2O and Ph3PO.25 For the subsequent cleavage of the Troc carbamate, we examined several conditions, e.g. with a Zn/Cu couple or Me3SnOH26 as a deprotection reagent, but Doi's protocol using zinc dust and aqueous NH4OAc10c,11c gave 15 in the best yield. Pd-catalysed cleavage of the allyl ester provided carboxylic acid 16.
We were also interested in preparing apratoxin B27 for which, to our knowledge, there has not yet been a total synthesis reported. Starting from Boc-(S)-Ile-OAllyl (17b),28 we synthesised the tripeptide building block 19b in two HATU-mediated peptide couplings, achieving excellent overall yield (Scheme 3). The corresponding apratoxin A tripeptide 19a, in which (S)-Ile is replaced by N-Me-(S)-Ile, was prepared according to known procedures.10c,11c
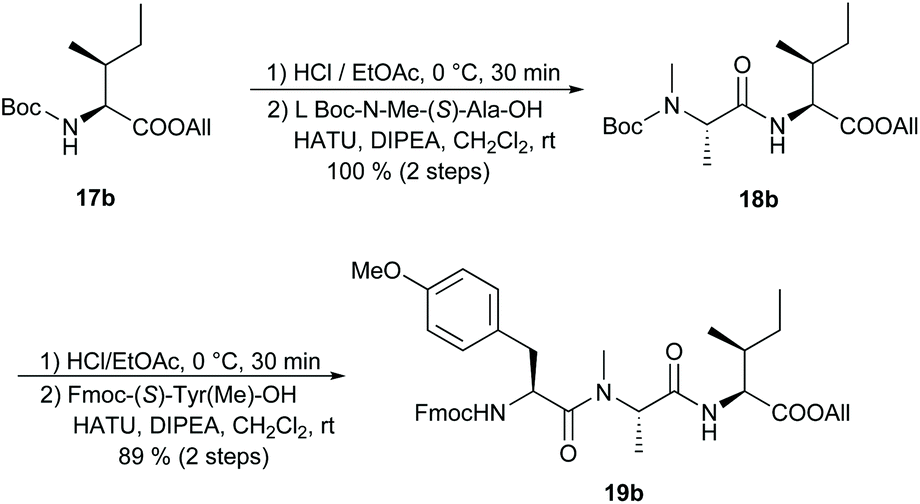 | ||
| Scheme 3 Synthesis of the apratoxin B tripeptide. | ||
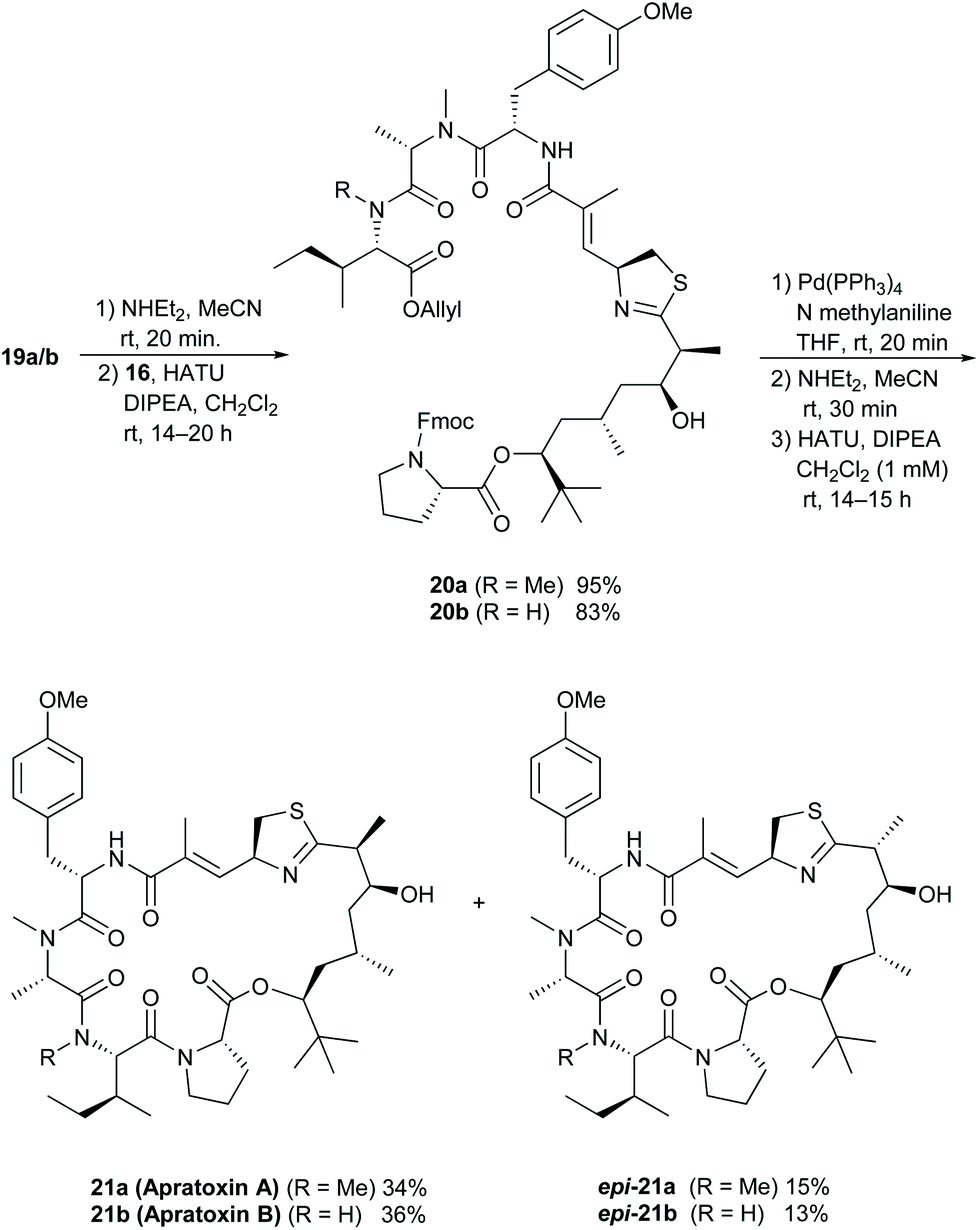
Coupling of the polyketide fragment 16 with Fmoc-deprotected 19a or 19b yielded the linear precursors 20a and 20b, which we cyclised to the natural products after cleavage of the allyl and Fmoc protecting groups (Scheme 4). As reported in previous syntheses,10c,d,11c,d we observed partial epimerization of the stereogenic center adjacent to the thiazoline moiety during the steps from 15 to 21, and we obtained the cyclisation products as mixtures of two diastereomers 21a/b and epi-21a/b. Interestingly, no water elimination to the α,β-unsaturated thiazoline occurred under these conditions. In both cases, we were able to separate the isomers via preparative HPLC to isolate the diastereomerically pure natural products.
 | ||
| Scheme 4 Synthesis of apratoxin A and B. | ||
Conclusions
In conclusion: we have shown that the Matteson homologation is perfectly suited to generating the unusual polyketide fragment of the apratoxins. Starting from a chiral boronic ester (2), we were able to prepare the desired building block 8 of apratoxin A and B via five successive Matteson homologations and subsequent oxidation, achieving an overall yield of 27% and very good diastereoselectivity. Obviously, even a sterically demanding tert-butyl group is well accepted in the boronic ester and, therefore, this protocol should also be perfectly suited for the synthesis of libraries of derivatives for SAR studies, simply by changing the nucleophilic coupling partners in the homologation steps. Further applications are currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Saarland University and the DFG (Grant Ka 880/13-1) is gratefully acknowledged.Notes and references
- (a) M. Gutiérrez, T. L. Suyama, N. Engene, J. S. Wingerd, T. Matainaho and W. H. Gerwick, J. Nat. Prod., 2008, 71, 1099–1103 CrossRef PubMed; (b) S. Matthew, P. J. Schupp and H. Luesch, J. Nat. Prod., 2008, 71, 1113–1116 CrossRef CAS PubMed (correction: J. Nat. Prod. 2018, 81, 217–217); (c) C. C. Thornburg, E. S. Cowley, J. Sikorska, L. A. Shaala, J. E. Ishmael, D. T. A. Youssef and K. L. McPhail, J. Nat. Prod., 2013, 76, 1781–1788 CrossRef CAS PubMed; (d) E. M. Tarsis, E. J. Rastelli, S. E. Wengryniuk and D. M. Coltart, Tetrahedron, 2015, 71, 5029–5044 CrossRef CAS.
- H. Luesch, W. Y. Yoshida, R. E. Moore, V. J. Paul and T. H. Corbett, J. Am. Chem. Soc., 2001, 123, 5418–5423 CrossRef CAS PubMed.
- (a) Y. Liu, B. K. Law and H. Luesch, Mol. Pharmacol., 2009, 76, 91–104 CrossRef CAS PubMed; (b) H. Luesch, S. K. Chanda, R. M. Raya, P. D. DeJesus, A. P. Orth, J. R. Walker, J. C. Izpisúa Belmonte and P. G. Schultz, Nat. Chem. Biol., 2006, 2, 158–167 CrossRef CAS PubMed; (c) X. Wan, J. D. Serrill, I. R. Humphreys, M. Tan, K. L. McPhail, I. G. Ganley and J. E. Ishmael, Mar. Drugs, 2018, 16, 77 CrossRef PubMed.
- (a) K.-C. Huang, Z. Chen, Y. Jiang, S. Akare, D. Kolber-Simonds, K. Condon, S. Agoulnik, K. Tendyke, Y. Shen, K.-M. Wu, S. Mathieu, H.-W. Choi, X. Zhu, H. Shimizu, Y. Kotake, W. H. Gerwick, T. Uenaka, M. Woodall-Jappe and K. Nomoto, Mol. Cancer Ther., 2016, 15, 1208–1216 CrossRef CAS PubMed; (b) A. O. Paatero, J. Kellosalo, B. M. Dunyak, J. Almaliti, J. E. Gestwicki, W. H. Gerwick, J. Taunton and V. O. Paavilainen, Cell Chem. Biol., 2016, 23, 561–566 CrossRef CAS PubMed.
- S. Kazemi, S. Kawaguchi, C. E. Badr, D. R. Mattos, A. Ruiz-Saenz, J. D. Serrill, M. M. Moasser, B. P. Dolan, V. O. Paavilainen, S. Oishi, K. L. McPhail and J. E. Ishmael, Biochem. Pharmacol., 2021, 183, 114317 CrossRef CAS PubMed.
- (a) Q. Chen, Y. Liu and H. Luesch, ACS Med. Chem. Lett., 2011, 2, 861–865 CrossRef CAS PubMed; (b) C. C. Thornburg, E. S. Cowley, J. Sikorska, L. A. Shaala, J. E. Ishmael, D. T. A. Youssef and K. L. McPhail, J. Nat. Prod., 2013, 76, 1781–1788 CrossRef CAS PubMed.
- (a) B. Qiu, A. Tan, A. B. Veluchamy, Y. Li, H. Murray, W. Cheng, C. Liu, J. M. Busoy, Q.-Y. Chen, S. Sistla, W. Hunziker, C. M. G. Cheung, T. Y. Wong, W. Hong, H. Luesch and X. Wang, Invest. Ophthalmol. Visual Sci., 2019, 60, 3254–3263 CrossRef CAS PubMed; (b) W. Cai, Q.-Y. Chen, L. H. Dang and H. Luesch, ACS Med. Chem. Lett., 2017, 8, 1007–1012 CrossRef CAS PubMed (correction: ACS Med. Chem. Lett. 2017, 8, 1342).
- (a) M. A. Skiba, A. P. Sikkema, N. A. Moss, C. L. Tran, R. M. Sturgis, L. Gerwick, W. H. Gerwick, D. H. Sherman and J. L. Smith, ACS Chem. Biol., 2017, 12, 3039–3048 CrossRef CAS PubMed; (b) M. A. Skiba, A. P. Sikkema, N. A. Moss, A. N. Lowell, M. Su, R. M. Sturgis, L. Gerwick, W. H. Gerwick, D. H. Sherman and J. L. Smith, ACS Chem. Biol., 2018, 13, 1640–1650 CrossRef CAS PubMed.
- (a) J. Chen and C. J. Forsyth, Org. Lett., 2003, 5, 1281–1283 CrossRef CAS PubMed; (b) Z. Xu, Z. Chen and T. Ye, Tetrahedron: Asymmetry, 2004, 15, 355–363 CrossRef CAS; (c) A. Gilles, J. Martinez and F. Cavelier, C. R. Chim., 2011, 14, 437–440 CrossRef CAS; (d) S. Dey, S. E. Wengryniuk, E. M. Tarsis, B. D. Robertson, G. Zhou and D. M. Coltart, Tetrahedron Lett., 2015, 56, 2927–2929 CrossRef CAS.
- (a) J. Chen and C. J. Forsyth, J. Am. Chem. Soc., 2003, 125, 8734–8735 CrossRef CAS PubMed; (b) J. Chen and C. J. Forsyth, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12067–12072 CrossRef CAS PubMed; (c) T. Doi, Y. Numajiri, A. Munakata and T. Takahashi, Org. Lett., 2006, 8, 531–534 CrossRef CAS PubMed; (d) T. Doi, Yo. Numajiri, T. Takahashi, M. Takagi and K. Shin-ya, Chem. – Asian J., 2011, 6, 180–188 CrossRef CAS PubMed; (e) B. D. Robertson, S. E. Wengryniuk and D. M. Coltart, Org. Lett., 2012, 14, 5192–5195 CrossRef CAS PubMed; (f) Y. Masuda, J. Suzuki, Y. Onda, Y. Fujino, M. Yoshida and T. Doi, J. Org. Chem., 2014, 79, 8000–8009 CrossRef CAS PubMed; (g) P. Wu, W. Cai, Q.-Y. Chen, S. Xu, R. Yin, Y. Li, W. Zhang and H. Luesch, Org. Lett., 2016, 18, 5400–5403 CrossRef CAS PubMed; (h) T. Doi, Y. Masuda and M. Yoshida, J. Synth. Org. Chem., Jpn., 2018, 76, 1170–1175 CrossRef CAS.
- (a) B. Zou, J. Wei, G. Cai and D. Ma, Org. Lett., 2003, 5, 3503–3506 CrossRef CAS PubMed; (b) D. Ma, B. Zou, G. Cai, X. Hu and J. O. Liu, Chem. – Eur. J., 2006, 12, 7615–7626 CrossRef CAS PubMed; (c) Y. Numajiri, T. Takahashi and T. Doi, Chem. – Asian J., 2009, 4, 111–125 CrossRef CAS PubMed; (d) T. Doi, Chem. Pharm. Bull., 2014, 62, 735–743 CrossRef CAS PubMed; (e) M. Yoshida, Y. Onda, Y. Masuda and T. Doi, Biopolymers, 2016, 104, 404–414 CrossRef PubMed; (f) R. Yin, W. Zhang, G. Liu, P. Wu, C. Lau and Y. Li, Tetrahedron, 2016, 72, 3823–3831 CrossRef CAS; (g) P. Wu, H. Xu, Z. Li, Y. Zhou, Y. Li and W. Zhang, Tetrahedron Lett., 2017, 58, 3333–3336 CrossRef CAS; (h) Y. Onda, Y. Masuda, M. Yoshida and T. Doi, J. Med. Chem., 2017, 60, 6751–6765 CrossRef CAS PubMed; (i) Z.-Y. Mao, C.-M. Si, Y.-W. Liu, H.-Q. Dong, B.-G. Wie and G.-Q. Lin, J. Org. Chem., 2017, 82, 10830–10845 CrossRef CAS PubMed; (j) E. J. Rastelli and D. M. Coltart, Tetrahedron, 2018, 74, 2269–2290 CrossRef CAS; (k) W. Cai, R. Ratnayake, M. H. Gerber, Q.-Y. Chen, Y. Yu, H. Derendorf, J. G. Trevino and H. Luesch, Invest. New Drugs, 2019, 37, 364–374 CrossRef CAS PubMed.
- (a) A. Ullrich, Y. Chai, D. Pistorius, Y. A. Elnakady, J. E. Herrmann, K. J. Weissman, U. Kazmaier and R. Müller, Angew. Chem., Int. Ed., 2009, 48, 4422–4425 ( Angew. Chem. , 2009 , 121 , 4486–4489 ) CrossRef CAS PubMed; (b) A. Ullrich, J. Herrmann, R. Müller and U. Kazmaier, Eur. J. Org. Chem., 2009, 6367–6378 CrossRef CAS; (c) P. Servatius, T. Stach and U. Kazmaier, Eur. J. Org. Chem., 2019, 3163–3168 CrossRef CAS; (d) P. Servatius, L. Junk and U. Kazmaier, Synlett, 2019, 1289–1302 CAS.
- (a) P. Barbie and U. Kazmaier, Org. Lett., 2016, 18, 204–207 CrossRef CAS PubMed; (b) L. Junk and U. Kazmaier, Angew. Chem., Int. Ed., 2018, 57, 11432–11435 ( Angew. Chem. , 2018 , 130 , 11602–11606 ) CrossRef CAS PubMed; (c) L. Junk and U. Kazmaier, J. Org. Chem., 2019, 84, 2489–2500 CrossRef CAS PubMed; (d) A. Kiefer, C. D. Bader, J. Held, A. Esser, J. Rybniker, M. Empting, R. Müller and U. Kazmaier, Chem. – Eur. J., 2019, 25, 8894–8902 CAS.
- (a) L. Karmann, K. Schultz, J. Herrmann, R. Müller and U. Kazmaier, Angew. Chem., Int. Ed., 2015, 54, 4502–4507 ( Angew. Chem. , 2015 , 127 , 4585–4590 ) CrossRef CAS PubMed; (b) D. Becker and U. Kazmaier, Eur. J. Org. Chem., 2015, 2591–2602 CrossRef CAS; (c) S. Kappler, L. Karmann, C. Prudel, J. Herrmann, G. Caddeu, R. Müller, A. Vollmar, S. Zahler and U. Kazmaier, Eur. J. Org. Chem., 2018, 6952–6965 CrossRef CAS.
- (a) G. Ehrlich, J. Hassfeld, U. Eggert and M. Kalesse, Chem. – Eur. J., 2008, 14, 2232–2247 CrossRef CAS PubMed; (b) H. Steinmetz, K. Gerth, R. Jansen, N. Schläger, R. Dehn, S. Reinecke, A. Kirschning and R. Müller, Angew. Chem., Int. Ed., 2011, 50, 532–536 ( Angew. Chem. , 2011 , 123 , 553–557 ) CrossRef CAS PubMed; (c) I. Paterson and N. Y. S. Lam, J. Antibiot., 2018, 71, 215–233 CrossRef CAS PubMed; (d) S. Scheeff and D. Menche, Org. Lett., 2019, 21, 271–274 CrossRef CAS PubMed; (e) N. Y. S. Lam and I. Paterson, Eur. J. Org. Chem., 2020, 2310–2320 CrossRef CAS.
- (a) W. R. Roush, A. D. Palkowitz and K. Ando, J. Am. Chem. Soc., 1990, 112, 6348–6359 CrossRef CAS; (b) A. Arefolov and J. S. Panek, J. Am. Chem. Soc., 2005, 127, 5596–5603 CrossRef CAS PubMed; (c) A.-M. R. Dechert-Schmitt, D. C. Schmitt, X. Gao, T. Itoh and M. J. Krische, Nat. Prod. Rep., 2014, 31, 504–513 RSC; (d) M. Pantin, J. G. Hubert, T. Söhnel, M. A. Brimble and D. P. Furkert, J. Org. Chem., 2017, 82, 11225–11229 CrossRef CAS PubMed; (e) K. Spielmann, G. Niel, R. M. de Figueiredo and J.-M. Campagne, Chem. Soc. Rev., 2018, 47, 1159–1173 RSC.
- (a) J. Gorges and U. Kazmaier, Org. Lett., 2018, 20, 2033–2036 CrossRef CAS PubMed; (b) O. Andler and U. Kazmaier, Chem. – Eur. J., 2021, 27, 949–953 CrossRef CAS PubMed.
- Reviews: (a) D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed; (b) S. Kirupakaran, H.-G. Korth and C. Hirschhäuser, Synthesis, 2018, 50, 2307–2322 CrossRef CAS.
- (a) R. Ray and D. S. Matteson, Tetrahedron Lett., 1980, 21, 449–450 CrossRef CAS; (b) D. S. Matteson and R. Ray, J. Am. Chem. Soc., 1980, 102, 7590–7591 CrossRef CAS.
- (a) D. S. Matteson, K. M. Sadhu and M. L. Peterson, J. Am. Chem. Soc., 1986, 108, 810–819 CrossRef CAS; (b) D. S. Matteson and T. J. Michnick, Organometallics, 1990, 9, 3171–3177 CrossRef CAS; (c) D. S. Matteson and J.-J. Yang, Tetrahedron: Asymmetry, 1997, 8, 3855–3861 CrossRef CAS; (d) H. W. Man, W. C. Hiscox and D. S. Matteson, Org. Lett., 1999, 1, 379–382 CrossRef CAS PubMed.
- H. C. Brown, M. Srebnik and T. E. Cole, Organometallics, 1986, 5, 2300–2303 CrossRef CAS.
- W. C. Hiscox and D. S. Matteson, J. Org. Chem., 1996, 61, 8315–8316 CrossRef CAS PubMed.
- (a) D. S. Matteson and H.-W. Man, J. Org. Chem., 1994, 59, 5734–5741 CrossRef CAS; (b) D. S. Matteson, J. Organomet. Chem., 1999, 581, 51–65 CrossRef CAS.
- L. A. Carpino, B. J. Cohen, K. E. Stephens, S. Y. Sadat-Aalaee, J. H. Tien and D. C. Langridge, J. Org. Chem., 1986, 51, 3732–3734 CrossRef CAS.
- S. You, H. Razavi and J. W. Kelly, Angew. Chem., Int. Ed., 2003, 42, 83–85 ( Angew. Chem. , 2003 , 115 , 87–89 ) CrossRef CAS PubMed.
- B. M. Trost, C. A. Kalnmals, J. S. Tracy and W. Bai, Org. Lett., 2018, 20, 8043–8046 CrossRef CAS PubMed.
- H. Luesch, W. Y. Yoshida, R. E. Moore and V. J. Paul, Bioorg. Med. Chem., 2002, 10, 1973–1978 CrossRef CAS PubMed.
- J. Hur, J. Jang, J. Sim, W. S. Son, H. Ahn, T. S. Kim, Y. Shin, C. Lim, S. Lee, H. An, S. Kim, D.-C. Oh, E. Jo, J. Jang, J. Lee and Y. Suh, Angew. Chem., Int. Ed., 2018, 57, 3069–3073 ( Angew. Chem. , 2018 , 130 , 3123–3127 ) CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization, copies of 1H and 13C NMR spectra and HPLC chromatograms. See DOI: 10.1039/d1ob00713k |
| This journal is © The Royal Society of Chemistry 2021 |