
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PMDTA-catalyzed multicomponent synthesis and biological activity of 2-amino-4H-chromenes containing a phosphonate or phosphine oxide moiety†
Ádám
Tajti
 a,
Kármen Emőke
Szabó
a,
Nóra
Popovics-Tóth
a,
Javad
Iskanderov
a,
Franc
Perdih
b,
László
Hackler
Jr.
c,
Beáta
Kari
c,
László G.
Puskás
c and
Erika
Bálint
*a
a,
Kármen Emőke
Szabó
a,
Nóra
Popovics-Tóth
a,
Javad
Iskanderov
a,
Franc
Perdih
b,
László
Hackler
Jr.
c,
Beáta
Kari
c,
László G.
Puskás
c and
Erika
Bálint
*a
aDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, Budafoki út 8, 1111 Budapest, Hungary. E-mail: balint.erika@vbk.bme.hu
bFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, 1000 Ljubljana, Slovenia
cAvidin Ltd, Alsó kikötő sor 11/D, 6726 Szeged, Hungary
First published on 22nd July 2021
Abstract
A new approach for the preparation of (2-amino-3-cyano-4H-chromen-4-yl)phosphonate derivatives is described. The multicomponent reaction of salicylaldehydes, malononitrile and dialkyl phosphites catalyzed by pentamethyldiethylenetriamine (PMDTA) provided the bicyclic derivatives in high yields. The method developed did not require chromatographic separation, since the products could be recovered from the reaction mixture by simple filtration. Our approach made also possible condensation with secondary phosphine oxides, and this reaction has not been previously reported in the literature. The crystal structures of five derivatives were studied by single-crystal XRD analysis. The in vitro cytotoxicity on different cell lines and the antibacterial activity of the (2-amino-4H-chromen-4-yl)phosphonates synthesized were also explored. According to the IC50 values determined, several derivatives showed moderate or promising activity against mouse fibroblast (NIH/3T3) and human promyelocytic leukemia (HL-60) cells. Furthermore, three (2-amino-3-cyano-4H-chromen-4-yl)phosphine oxides were active against selected Gram-positive bacteria.
Introduction
Phosphorus-substituted heterocycles represent an important group within organophosphorus compounds. They have gained an increasingly growing interest due to their importance in synthetic, medicinal and polymer chemistry.1–3In the last few decades, (2-amino-4H-chromen-4-yl)phosphonates have received significant attention. These compounds are analogues of 2-amino-4H-chromenes, which have valuable applications as pharmaceutical agents,4 and are widely employed as cosmetics, pigments and potential biodegradable agrochemicals.5 A few (2-amino-4H-chromen-4-yl)phosphonates showed a moderate antioxidant effect6 and a modest anticancer activity against human lung adenocarcinoma (A549) and human epidermoid cancer (KB) cell lines.7 Therefore, the introduction of a phosphonate moiety on the 2-amino-4H-chromene ring may also have a synergistic effect on the various biological properties of this scaffold. Consequently, the development of green and efficient methods for the synthesis of (2-amino-4H-chromen-4-yl)phosphonates is of great importance.
Multicomponent reactions may represent an efficient method for the one-pot synthesis of complex ring systems.8 Due to their simplicity, high atom efficiency and the ability to provide easy access to large compound libraries, these transformations have attracted significant attention in the field of organic chemistry.9 (2-Amino-4H-chromen-4-yl)phosphonates may be prepared by the three-component reaction of salicylaldehyde derivatives, CH-acidic nitriles (e.g. malononitrile or ethyl cyanoacetate) and dialkyl- or trialkyl phosphites.10 Most procedures utilizing the above synthesis route were carried out in the presence of both a catalyst and a solvent. Diethylamine,11 dibutylamine,7 triethylamine,12 dimethylaminopyridine,13 imidazole,14 ethylenediamine diacetate,15 lithium hydroxide,16 potassium phosphate,17 magnesium oxide18 or indium chloride19 were tried out in ethanol, and iron oxide,20 iodine21 or β-cyclodextrin22 in water. Water may be considered as a green solvent; however, it requires an additional extraction step during the reaction workup. In a few cases, special solvents, such as PEG,23 ionic liquids24,25 or a mixture of urea and choline chloride,26 were used, which also served as a catalyst. Only four solvent-free variations can be found in the literature;27–30 however, in these procedures, special catalysts were used, or a simple catalyst was required in a large excess. The condensation of salicylaldehydes, malononitrile and trialkyl phosphites was carried out in the presence of a silica-bonded 2-HEAA-3 catalyst at room temperature27 or applying iodine at 50 °C.28 In other cases, dialkyl phosphites were used as phosphorus reagents, and the reactions were carried out applying ZnO nanorods at 100 °C,29 or in the presence of a large excess (3.5 equiv.) of tetramethylguanidine catalyst at 25 °C.30
Although many variations have been reported on the multicomponent synthesis of (2-amino-4H-chromen-4-yl)phosphonates, a suitable method using a simple basic catalyst under solvent-free conditions is still required. Furthermore, an extensive literature survey has revealed that there are no reports at all on the preparation of (2-amino-4H-chromen-4-yl)phosphine oxides.
Based on the above considerations, in this paper, we report on a fast, cheap and efficient PMDTA-catalyzed solvent-free process for the synthesis of novel (2-amino-3-cyano-4H-chromen-4-yl)phosphonates by the domino Knoevenagel–phospha-Michael reaction of salicylaldehydes, malononitrile and dialkyl phosphites. Our approach made also possible condensation with secondary phosphine oxides, and this reaction has not been previously reported in the literature. The biological activity of the synthesized compounds was investigated in terms of antibacterial activity and in vitro cytotoxicity assays.
Results and discussion
Synthesis of (2-amino-3-cyano-4H-chromen-4-yl)phosphonates
To optimize the three-component reaction of salicylaldehyde, malononitrile and diethyl phosphite, at first, the effect of various basic catalysts was studied (Table 1). The reactions were monitored by HPLC, and the reaction mixtures were analyzed by HPLC-MS. The condensations were carried out in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in the presence of 5 mol% of an organic base at 60 °C for 30 min in an oil bath. At first, organic bases containing one nitrogen atom were tested as catalysts (Table 1, entries 1–3). Using dipropylamine (DPA) in the absence of any solvent, the conversion was complete, and the reaction mixture contained 65% of 2-(2-hydroxybenzylidene)malononitrile (1) ([M + H]+ = 171.1) as an intermediate formed by the Knoevenagel condensation of salicylaldehyde with malononitrile and 35% of diethyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonate (2a) (Table 1, entry 1). Performing the three-component reaction in the presence of triethylamine (TEA) or dipropylethylamine (DIPEA), the ratio of intermediate 1 and product 2a was 62:38 or 58:42, respectively (Table 1, entries 2 and 3). After that, the condensation was investigated using organic bases containing two nitrogen atoms, such as dimethylaminopridine (DMAP), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), diazobicyclooctane (DABCO) or tetramethylethylenediamine (TMEDA) (Table 1, entries 4–7). In these reactions, the desired diethyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonate (2a) was formed in 27%, 41%, 53% and 58% yield, respectively. Finally, the domino Knoevenagel–phospha-Michael reaction was carried out by the addition of pentamethyldiethylenetriamine (PMDTA) as a catalyst containing three nitrogen atoms, and the proportion of product 2a was increased to 86% (Table 1, entry 8). As expected, the condensation was significantly affected by the number of basic nitrogen atoms in the catalyst, which provided more active sites, and a few steric effects were also observed. Among the catalysts tested, PMDTA was found to be the most effective; therefore, further experiments were performed in the presence of this catalyst. To study the effect of solvent, a reaction was also performed in ethanol, which is a common solvent used in the synthesis of similar derivatives, and in acetonitrile (Table 1, entries 9 and 10). In both reactions, the proportion of the expected chromenylphosphonate (2a) decreased dramatically, and only 35% or 30% of product 2a was present in the mixtures.
:1:1 in the presence of 5 mol% of an organic base at 60 °C for 30 min in an oil bath. At first, organic bases containing one nitrogen atom were tested as catalysts (Table 1, entries 1–3). Using dipropylamine (DPA) in the absence of any solvent, the conversion was complete, and the reaction mixture contained 65% of 2-(2-hydroxybenzylidene)malononitrile (1) ([M + H]+ = 171.1) as an intermediate formed by the Knoevenagel condensation of salicylaldehyde with malononitrile and 35% of diethyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonate (2a) (Table 1, entry 1). Performing the three-component reaction in the presence of triethylamine (TEA) or dipropylethylamine (DIPEA), the ratio of intermediate 1 and product 2a was 62:38 or 58:42, respectively (Table 1, entries 2 and 3). After that, the condensation was investigated using organic bases containing two nitrogen atoms, such as dimethylaminopridine (DMAP), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), diazobicyclooctane (DABCO) or tetramethylethylenediamine (TMEDA) (Table 1, entries 4–7). In these reactions, the desired diethyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonate (2a) was formed in 27%, 41%, 53% and 58% yield, respectively. Finally, the domino Knoevenagel–phospha-Michael reaction was carried out by the addition of pentamethyldiethylenetriamine (PMDTA) as a catalyst containing three nitrogen atoms, and the proportion of product 2a was increased to 86% (Table 1, entry 8). As expected, the condensation was significantly affected by the number of basic nitrogen atoms in the catalyst, which provided more active sites, and a few steric effects were also observed. Among the catalysts tested, PMDTA was found to be the most effective; therefore, further experiments were performed in the presence of this catalyst. To study the effect of solvent, a reaction was also performed in ethanol, which is a common solvent used in the synthesis of similar derivatives, and in acetonitrile (Table 1, entries 9 and 10). In both reactions, the proportion of the expected chromenylphosphonate (2a) decreased dramatically, and only 35% or 30% of product 2a was present in the mixtures.
|
|
|||||
|---|---|---|---|---|---|
| Entry | P-reagent | Catalyst [5 mol%] | Solvent | Product compositionb [%] | |
| 1 | 2a | ||||
| a The reaction was performed in the presence of salicylaldehyde (1.0 mmol), malononitrile (1.0 mmol), diethyl phosphite (1.0 mmol) and a basic catalyst (0.05 mmol). b Determined by HPLC analysis (256 nm). | |||||
| 1 | DEP | DPA | — | 65 | 35 |
| 2 | DEP | TEA | — | 62 | 38 |
| 3 | DEP | DIPEA | — | 58 | 42 |
| 4 | DEP | DMAP | — | 73 | 27 |
| 5 | DEP | DBU | — | 59 | 41 |
| 6 | DEP | DABCO | — | 47 | 53 |
| 7 | DEP | TMEDA | — | 42 | 58 |
| 8 | DEP | PMDTA | — | 14 | 86 |
| 9 | DEP | PMDTA | EtOH | 65 | 35 |
| 10 | DEP | PMDTA | MeCN | 70 | 30 |
| 11 | TEP | PMDTA | — | 100 | 0 |
| 12 | TEP | PMDTA | H2O | 27 | 73 |
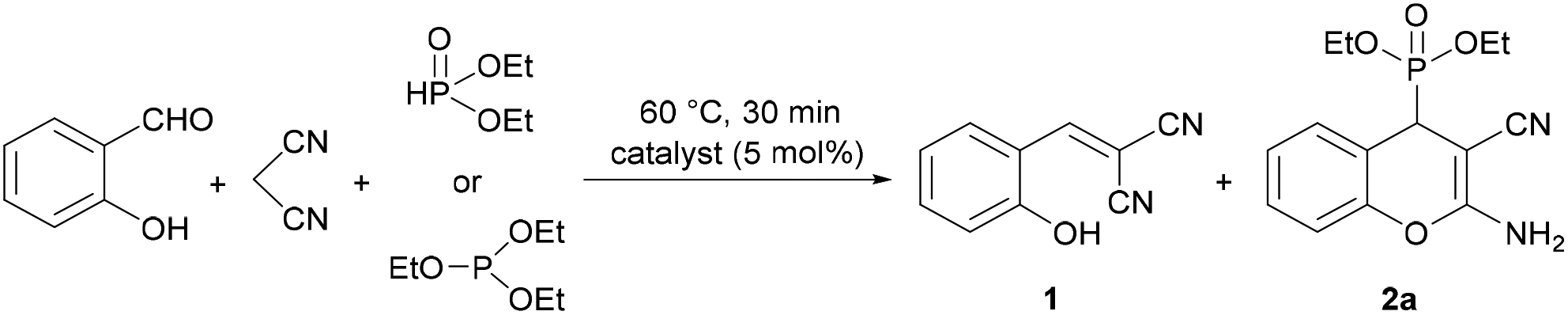
The presence of the solvent in the reaction carried out at 60 °C for 30 min reduced the reaction rate, thus, the solvent-free reaction allowed a higher proportion of product 2a under the same conditions (Table 1, entries 8 vs. 9 and 10). Although in a few literature examples, salicylaldehyde and malononitrile were reacted with triethyl phosphite (TEP) instead of DEP as the P-reagent, according to our experience only 2-(2-hydroxybenzylidene)malononitrile (1) was formed, and no product 2a in the reaction mixture was obtained at 60 °C for 30 min in the absence of any solvent (Table 1, entry 11). Therefore, under the conditions applied, the reactivities of DEP and TEP were significantly different. When the condensation was performed in water, 73% of chromenylphosphonate (2a) was present in the reaction mixture (Table 1, entry 12). This may be explained by the base-catalyzed hydrolysis of TEP to DEP in the presence of water, which then reacts in the condensation – a similar observation was previously described by our group in the case of Kabachnik–Fields reactions.31
The model reaction was further optimized with respect to the reaction time and temperature, as well as the catalyst amount; furthermore, the condensation was extended to various dialkyl phosphites and ethyl phenyl-H-phosphinate (Table 2).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Y1 | Y2 | T [°C] | t [min] | PMDTA [mol%] | Product compositionb [%] | Yieldc [%] | |
| 1 | 2 | |||||||
|
a The reaction was performed in the presence of salicylaldehyde (1.0 mmol), malononitrile (1.0 mmol), dialkyl phosphite (1.0 mmol) and PMDTA (0.05–0.1 mmol) at 60–80 °C for 15–60 min.
b Determined by HPLC analysis (256 nm).
c Isolated yield.
d The product was formed as a 42:58 mixture of two diastereomers.
|
||||||||
| 1 | EtO | EtO | 60 | 30 | 5 | 14 | 86 | — |
| 2 | EtO | EtO | 60 | 45 | 5 | 11 | 89 | — |
| 3 | EtO | EtO | 60 | 60 | 5 | 8 | 92 | — |
| 4 | EtO | EtO | 80 | 30 | 5 | 8 | 92 | — |
| 5 | EtO | EtO | 60 | 15 | 10 | 9 | 91 | — |
| 6 | EtO | EtO | 60 | 30 | 10 | 0 | 100 | 92 (2a) |
| 7 | MeO | MeO | 60 | 15 | 10 | 0 | 100 | 90 (2b) |
| 8 | BuO | BuO | 60 | 30 | 10 | 21 | 79 | — |
| 9 | BuO | BuO | 80 | 15 | 10 | 16 | 84 | — |
| 10 | BuO | BuO | 80 | 30 | 10 | 1 | 99 | 92 (2c) |
| 11 | iPrO | iPrO | 80 | 30 | 10 | 3 | 97 | 84 (2d) |
| 12 | BnO | BnO | 60 | 30 | 10 | 3 | 97 | 85 (2e) |
| 13 | PhO | PhO | 60 | 30 | 10 | 2 | 98 | 90 (2f) |
| 14 | EtO | Ph | 60 | 15 | 10 | 3 | 97 | 86d (2g) |
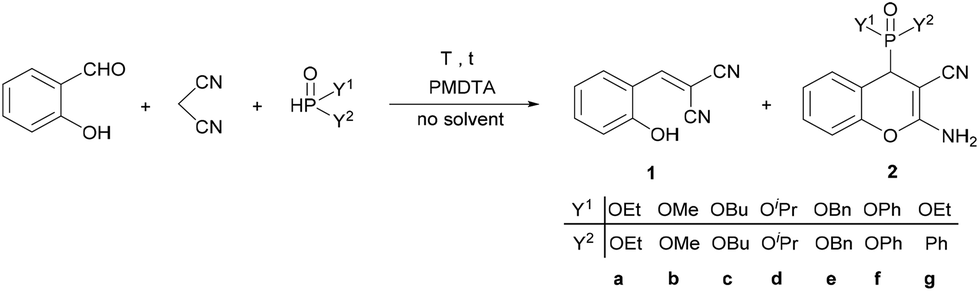
First, the reaction time was gradually increased from 30 min to 60 min, and then, a higher temperature of 80 °C was also tried out; however, the ratio of product 2a did not change significantly (Table 2, entries 1–4). By increasing the amount of catalyst to 10 mol% at 60 °C, a product ratio of 91% was reached after 15 min, which increased to 100% after 30 min (Table 2, entries 4 and 5). Diethyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonate (2a) was isolated in a yield of 92% (Table 2, entry 6). When carrying out the domino Knoevenagel–phospha-Michael reaction with dimethyl phosphite, a reaction time of 15 min was enough for a complete conversion, and the corresponding chromenylphosphonate (2b) was obtained in a yield of 90% (Table 2, entry 7). Applying dibutyl- or diisopropyl phosphite as the P-reagent, a reaction temperature of 80 °C and a reaction time of 30 min were necessary to efficiently obtain products 2c and 2d, respectively (Table 2, entries 8–11). The condensation with dibenzyl- and diphenyl phosphite was similar to the reaction of diethyl phosphite, and at 60 °C for 30 min the proportions of the corresponding (2-amino-3-cyano-4H-chromen-4-yl)phosphonates (2e and 2f) were 97% and 98%, respectively. Finally, the three-component reaction was performed using ethyl phenyl H-phosphinate at 60 °C for 15 min, and the desired chromenylphosphinate (2g) was obtained in a ratio of 97%, and it was isolated in a yield of 86% (Table 2, entry 14). Due to the second chiral center on the phosphorus atom, compound 2g was obtained as a mixture of two diastereomers in a ratio of 42:58 based on the 31P NMR spectrum.
In the next part of our work, the three-component reaction of substituted salicylaldehydes, malononitrile and dialkyl phosphites was studied under the optimal conditions determined for each phosphite (Scheme 1). Using diethyl phosphite, the condensations were complete at 60 °C for 30 min, similar to the reaction of salicylaldehyde. Starting from 5-fluoro- or 2-chlorosalicylaldehyde, the desired products (3a or 4a) were isolated in yields of 91% and 90%, respectively. 3-Bromosalicylaldehyde was found to be slightly less reactive at 60 °C for 30 min than salicylaldehydes bearing 5-F or 2-Cl substituents. Using 3-ethoxysalicylaldehyde, an excellent yield (96%) was obtained. According to our experiences, 3-hydroxy-substituted salicylaldehyde was less reactive as compared to other substituted salicylaldehydes, and product 7a was isolated in a yield of 82%. After that, 5-fluoro- and 2-chloro-salicylaldehyde was reacted with malononitrile and dibutyl phosphite at 80 °C for 30 min, and the corresponding dibutyl chromenylphosphonates (3c and 4c) were obtained in high yields (88% and 90%, respectively). Finally, the condensations were performed with dibenzyl phosphite at 60 °C for 30 min. As in the previous examples, starting from salicylaldehyde or 5-fluoro-, 2-chloro- or 3-ethoxysalicylaldehyde, better results were obtained (81–85% yield), while the derivative containing a bromine substituent at position eight (6e) was obtained in a yield of 70%. The method developed could be effectively applied for various substituted salicylaldehydes.
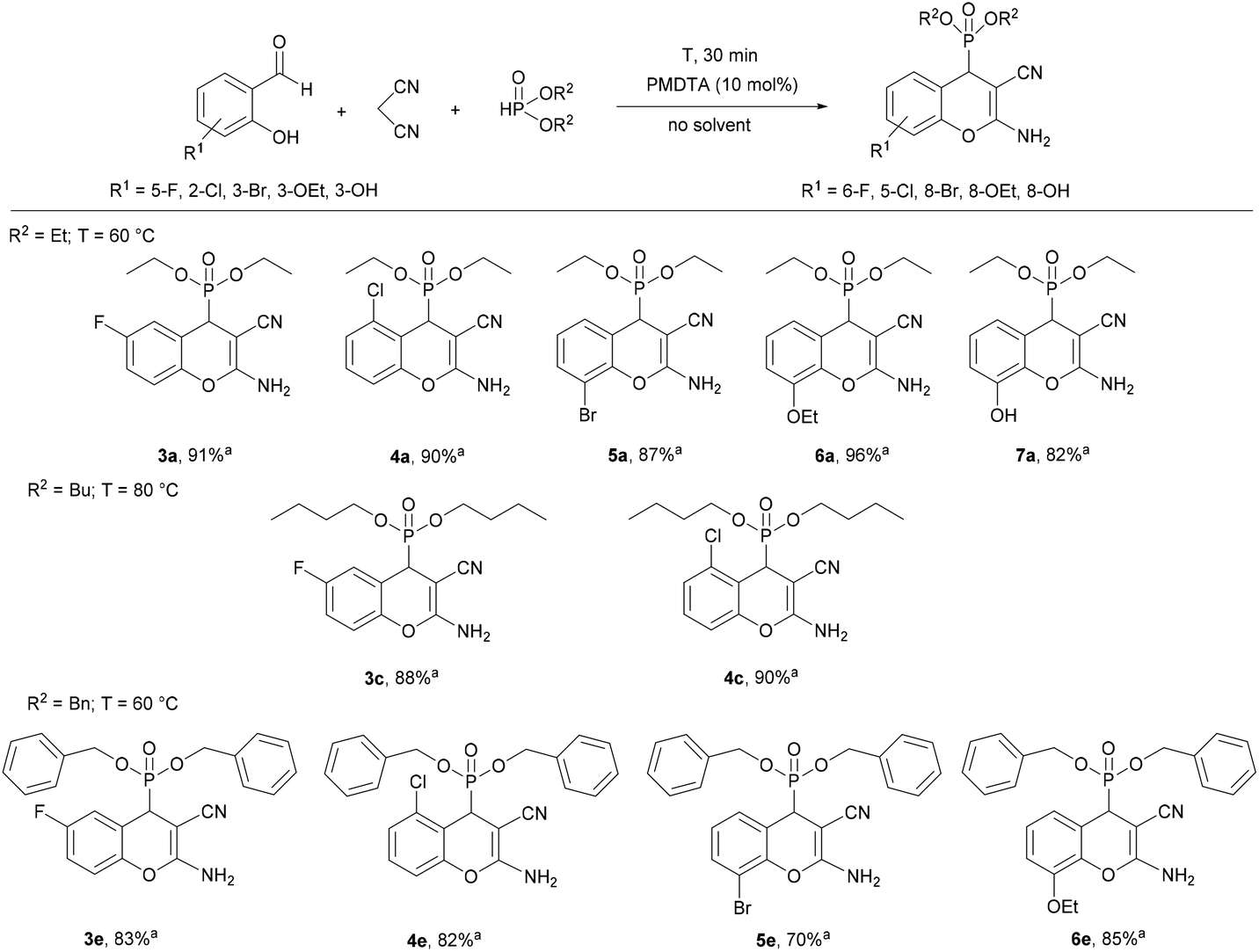 | ||
| Scheme 1 Condensation of salicylaldehydes, malononitrile and dialkyl phosphites. aIsolated yield. | ||
In contrast to the previous reports dealing with a similar topic, the multicomponent approach developed is a fast, cheap and simple solvent-free methodology for the synthesis of (2-amino-3-cyano-4H-chromen-4-yl)phosphonates. Besides a comprehensive optimization, we have provided exact product compositions, including the amount of the “Knoevenagel product” (1).
The formation of (2-amino-3-cyano-4H-chromen-4-yl)phosphonates can be explained by the proposed mechanism shown in Scheme 2. First, by the Knoevenagel condensation of salicylaldehyde and malononitrile in the presence of PMDTA, 2-(2-hydroxybenzylidene)malononitrile (1) is formed, and then, an intramolecular Pinner-like reaction (cyclization of the hydroxyl group of 1 on the cyano group) leads to iminocoumarine (III). Finally, the phospha-Michael addition of dialkyl phosphite, as the nucleophile, affords the phosphonium salt intermediate IV, which further transforms into the corresponding (2-amino-3-cyano-4H-chromen-4-yl)phosphonate.
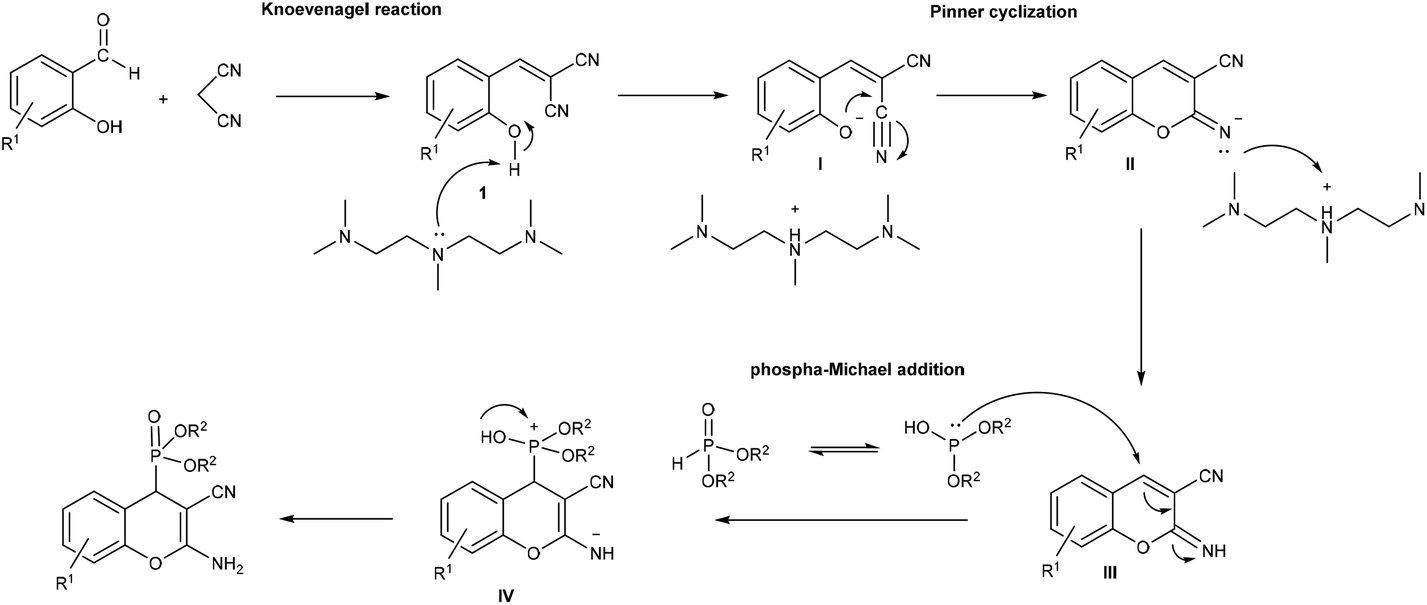 | ||
| Scheme 2 Proposed mechanism for the PMDTA-catalyzed synthesis of (2-amino-3-cyano-4H-chromen-4-yl)phosphonates. | ||
Synthesis of (2-amino-3-cyano-4H-chromen-4-yl)phosphine oxides
As a novel extension, the PMDTA-catalyzed domino Knoevenagel–phospha-Michael reaction was investigated with secondary phosphine oxides, such as diphenyl-, di(p-tolyl)-, bis(3,5-dimethylphenyl)- and di(naphthalen-2-yl)phosphine oxides. In these experiments, a small amount of acetonitrile as a solvent was used to overcome the heterogeneity. First, the condensation of salicylaldehyde, malononitrile and diphenylphosphine oxide was carried out in the presence of 5 mol% of PMDTA (Table 3). Stirring the reaction mixture at room temperature for 60 min, the conversion was 80%, and the mixture comprised 17% of the Knoevenagel intermediate (1) and 83% of the desired chromenylphosphine oxide (8a) (Table 3, entry 1). On increasing the reaction time to 180 min, the condensation was almost complete; however, the ratio of 1 and 8a was 11:89, respectively (Table 3, entry 1). Performing the reaction at 60 °C for 10 min, a complete conversion was achieved, and the proportion of the diphenyl (2-amino-3-cyano-4H-chromen-4-yl)phosphine oxide (8a) was 92%. By increasing the reaction time to 15 min, only product 8a was formed.
|
|
|||||
|---|---|---|---|---|---|
| Entry | T [°C] | t [min] | Conversionb [%] | Product compositionb [%] | |
| 1 | 8a | ||||
| a The reaction was performed in the presence of salicylaldehyde (1.0 mmol), malononitrile (1.0 mmol), diphenylphosphine oxide (1.0 mmol) and PMDTA (0.05 mmol) at 25–60 °C for 10–180 min. b Determined by HPLC analysis (256 nm). | |||||
| 1 | 25 | 60 | 80 | 17 | 83 |
| 2 | 25 | 180 | 96 | 11 | 89 |
| 3 | 60 | 10 | 100 | 8 | 92 |
| 4 | 60 | 15 | 100 | 0 | 100 |
Next, the three-component reaction of various salicylaldehydes, malononitrile and secondary phosphine oxides was studied under the optimized conditions (5 mol% PMTDA, 60 °C, 15 min) (Scheme 3). It was found that all the target derivatives (8a–12d) could be efficiently prepared using the developed procedure. It should be noted that the purification did not require chromatographic separation, and the products could be recovered from the reaction mixture by simple filtration. Altogether 20 new (2-amino-3-cyano-4H-chromen-4-yl)phosphine oxides (8a–12d) were synthesized in high yields (82–97%). These derivatives can be considered as a new family among O-heterocyclic organophosphorus compounds.
 | ||
| Scheme 3 Synthesis of (2-amino-3-cyano-4H-chromen-4-yl)phosphine oxides. aIsolated yield. | ||
The PMDTA-catalyzed multicomponent method developed is a fast, efficient and convenient methodology for the synthesis of (2-amino-3-cyano-4H-chromen-4-yl)phosphonates and a novel method for the preparation of (2-amino-3-cyano-4H-chromen-4-yl)phosphine oxides, which utilizes a cheap base catalyst, and in most cases, solvent-free conditions and short reaction time. The scope of the reaction was extended to 38 derivatives, of which 33 are new compounds.
X-ray diffraction study
In addition to the spectroscopic analysis, we have determined the crystal structures of compounds 2c, 6e and 8a–c. Single-crystal XRD analysis confirmed the molecular structures (Fig. 1 and Fig. S1†) and revealed the formation of intermolecular N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P and N–H⋯N hydrogen bonding leading to hydrogen-bonded chains or layers (Table S2†). Although the studied compounds possessed a similar chromenylphosphonate scaffold, their crystal structures showed a range of H-bonding interactions. The intermolecular N–H⋯OP hydrogen bonding between the amino group and the phosphonate oxygen atom was found to be a robust structural motif, since it was found in all structures under study. It was also observed that in structures 2c and 14a–c, the amino group as a hydrogen bond donor is involved in the formation of two interactions, while in 6e, the amino group is involved only in one interaction.
P and N–H⋯N hydrogen bonding leading to hydrogen-bonded chains or layers (Table S2†). Although the studied compounds possessed a similar chromenylphosphonate scaffold, their crystal structures showed a range of H-bonding interactions. The intermolecular N–H⋯OP hydrogen bonding between the amino group and the phosphonate oxygen atom was found to be a robust structural motif, since it was found in all structures under study. It was also observed that in structures 2c and 14a–c, the amino group as a hydrogen bond donor is involved in the formation of two interactions, while in 6e, the amino group is involved only in one interaction.
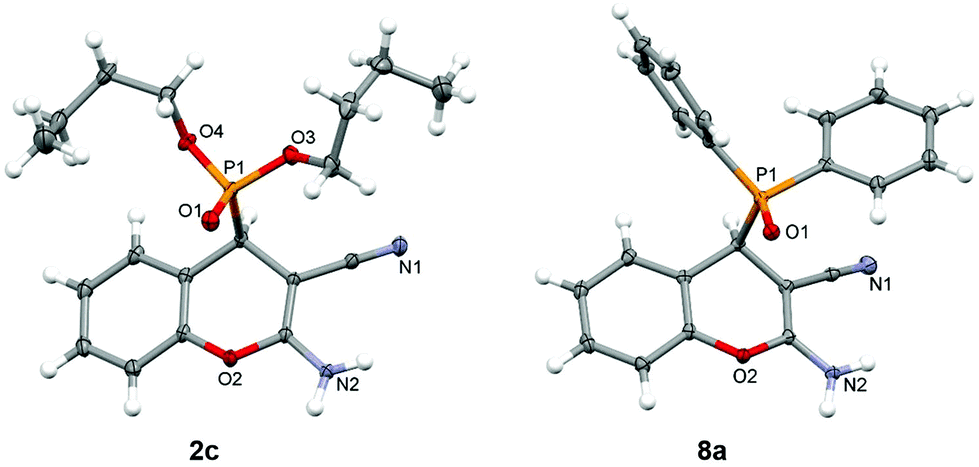 | ||
| Fig. 1 Molecular structures of compounds 2c and 8a. | ||
In compound 2c, the amino group is involved in the formation of the centrosymmetric N–H⋯N hydrogen bond with the cyano group and in the formation of the N–H⋯OP interaction with two adjacent molecules, thus enabling the formation of hydrogen-bonded layers (Fig. 2). On the other hand, in compound 6e, the amino group is involved only in the formation of centrosymmetric N–H⋯OP interactions, and the hydrogen-bonded chain is formed through the centrosymmetric C–H⋯N interaction connecting the chromenyl scaffold with the cyano group of the adjacent molecule (Fig. 2).
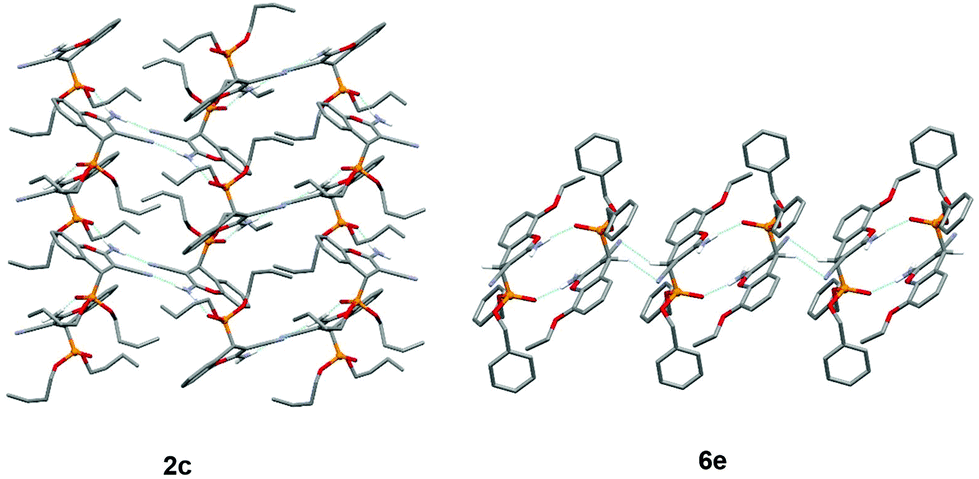 | ||
| Fig. 2 Supramolecular structures of compounds 2c and 6e. Blue dashed lines indicate hydrogen bonds. | ||
In the structures of 8a·CH3CN, 8b and 8c, three different packing motifs were observed. In 8a·CH3CN and 8c, the amino group is involved in a centrosymmetric N–H⋯N interaction with the cyano group of the adjacent molecule, and the hydrogen-bonded layer is formed through the N–H⋯OP interaction between the amino group and the phosphonate oxygen atom of the adjacent molecule (Fig. 3). Although in 8a and 8c, similar hydrogen bonds are present, and layer structures are formed, the supramolecular structure of 8a possesses a honeycomb-like structure, which is not the case in 8c. In 8b, a hydrogen-bonded chain is formed through two centrosymmetric hydrogen bonds both formed by the amino group, namely, a centrosymmetric N–H⋯N interaction with the cyano group and a centrosymmetric N–H⋯OP interaction with the phosphonate oxygen atom of two adjacent molecules (Fig. 3).
 | ||
| Fig. 3 Supramolecular structures of compounds 8a, 8b and 8c. Blue dashed lines indicate hydrogen bonds. | ||
Biological activity studies
The in vitro cytotoxicity and the antibacterial activity of the synthesized (2-amino-4H-chromen-4-yl)phosphonates and (2-amino-4H-chromen-4-yl)phosphine oxides were also investigated. The cytotoxicity evaluations were performed on three different cell lines, such as human lung adenocarcinoma (A549), mouse fibroblasts (NIH/3T3) as a healthy cell line and human promyelocytic leukemia (HL-60), using the fluorescent resazurin assay as described previously.32 Positive controls were doxorubicin for A549 and NIH/3T3 (IC50 = 0.31 ± 0.24 μM and 5.65 ± 0.81 μM, respectively) and bortezomib for HL60 (IC50 = 7.42 ± 2.60 nM). The antibacterial activity of the compounds was tested on green fluorescent protein (GFP) producing Bacillus subtilis (Gram-positive) and Escherichia coli (Gram-negative) bacterial cells. The GFP producing bacteria are effective tools for screening the antibacterial activity, since the GFP signal measured by fluorimetry is proportional to the number of bacterial cells. Active compounds kill bacterial cells, which results in a decrease in the GFP fluorescence signal; therefore it is suitable for evaluating the antimicrobial effects of different agents. Positive controls were doxycycline and gentamicin for Bacillus subtilis (IC50 = 0.04 ± 0.01 μM and 0.49 ± 0.14 μM) and Escherichia coli (IC50 = 0.10 ± 0.02 μM and 4.23 ± 0.99 μM) bacterial cells. The IC50 values (50% inhibiting concentration) determined are shown in Table 4.| Compound | R | In vitro cytotoxicity [IC50, μM] | Antibacterial activity (IC50, μM) | |||
|---|---|---|---|---|---|---|
| A549 | NIH/3T3 | HL-60 | B. subtilis | E. coli | ||
| a Data are expressed as mean ± standard deviation. | ||||||
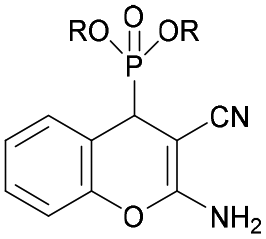
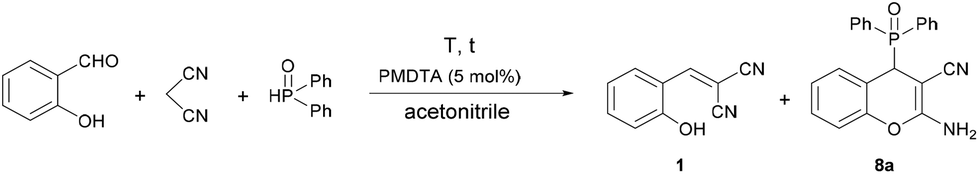
|
Me (2b) | >30 | >30 | >30 | >10 | >10 |
| Et (2a) | >30 | >30 | >30 | >10 | >10 | |
| Bu (2c) | >30 | >30 | >30 | >10 | >10 | |
| Bn (2e) | 26.46 ± 1.02 | 8.73 ± 1.17 | 6.25 ± 1.06 | >10 | >10 | |
| Ph (2f) | >30 | >30 | 28.18 ± 1.17 | >10 | >10 | |
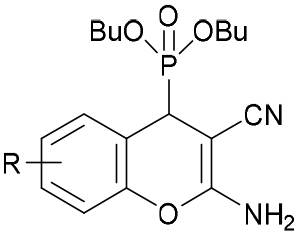
|
6-F (3c) | >30 | >30 | >30 | >10 | >10 |
| 5-Cl (4c) | >30 | >30 | 17.55 ± 1.70 | >10 | >10 | |
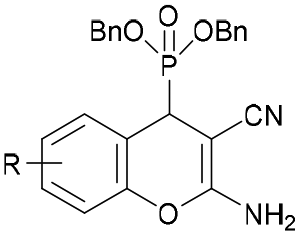
|
6-F (3e) | >30 | 21.2 ± 1.71 | 3.62 ± 1.38 | >10 | >10 |
| 5-Cl (4e) | >30 | 23.49 ± 1.09 | 7.51 ± 1.02 | >10 | >10 | |
| 8-Br (5e) | 28.65 ± 1.22 | 9.33 ± 1.18 | 4.79 ± 1.08 | >10 | >10 | |
| 8-OEt (6e) | >30 | 27.99 ± 1.06 | 14.37 ± 1.24 | >10 | >10 | |
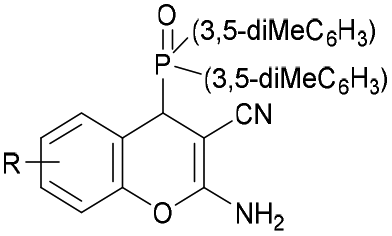
|
H (8c) | >30 | >30 | >30 | 8.92 ± 1.21 | >10 |
| 6-F (9c) | >30 | >30 | 10.06 ± 1.25 | 5.03 ± 1.28 | >10 | |
| 5-Cl (10c) | >30 | >30 | >30 | 5.29 ± 1.38 | >10 | |
| 8-Br (11c) | >30 | >30 | 9.8 ± 1.33 | >10 | >10 | |
| 8-OEt (12c) | >30 | >30 | >30 | >10 | >10 | |
| Doxorubicin | 0.31 ± 0.24 | 5.65 ± 0.81 | — | — | — | |
| Bortezomib | — | — | 7.42 × 10−3 ± 2.60 × 10−3 | — | — | |
| Doxycycline | — | — | — | 0.126 ± 0.029 | 0.10 ± 0.02 | |
| Gentamicin | — | — | — | 0.115 ± 0.001 | 4.23 ± 0.99 | |
Among the chromenylphosphonates containing an unsubstituted backbone (2a–c, 2e and 2f), the dibenzyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonate (2e) showed the highest activity. Although the anti-cancer effect of 2e against A549 (IC50 = 26.46 ± 1.02 μM) and HL-60 (IC50 = 6.25 ± 1.06 μM) cell lines was lower than those of the reference drugs (IC50 = 0.31 ± 0.24 μM and 7.42 × 10−3 ± 2.60 × 10−3 μM, respectively), the activity against NIH/3T3 cells (IC50 = 8.73 ± 1.17 μM) was close to the value of doxorubicin (IC50 = 5.65 ± 0.81 μM). In addition, the diphenyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonate (2f) showed a modest activity (IC50 = 28.18 ± 1.17 μM) against HL-60 cells. Next, the cytotoxic activities of 6-fluoro- or 5-chloro-substituted dibutyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonates (3c and 4c), as well as 6-fluoro-, 5-chloro-, 8-bromo- or 8-ethoxy-substituted dibenzyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonates (3e, 4e, 5e and 6e) were determined to investigate the effect of substituents on the 2-amino-4H-chromene backbone. The dibutyl (2-amino-3-cyano-6-fluoro-4H-chromen-4-yl)phosphonate (3c) was not active against the cell lines investigated, similar to the unsubstituted derivative (2c); however, the 5-chloro-substituted chromenylphosphonate butyl ester (4c) showed a moderate cytotoxicity (IC50 = 17.55 ± 1.70 μM) against HL-60 cells. The substituted dibenzyl chromenylphosphonates (3e, 4e, 5e and 6e) were more active than the butyl esters (3c and 4c). All substituted dibenzyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonates (3e, 4e, 5e and 6e) showed good or moderate activities against NIH/3T3 and HL-60 cells. Compounds containing a 6-fluoro (3e) or an 8-bromo (5e) substituent on the chromene ring were found to be the most active derivatives against the HL-60 cell line (IC50 = 3.62 ± 1.38 μM and 4.79 ± 1.08 μM, respectively).
Among the chromenylphosphine oxides, the 6-fluoro- or 5-chloro-substituted [bis(3,5-dimethylphenyl)](2-amino-3-cyano-4H-chromen-4-yl)phosphine oxides (9c and 10c) showed a moderate activity against the HL-60 cell line (IC50 = 10.06 ± 1.12 μM and 9.8 ± 1.33 μM, respectively).
According to the results of the antibacterial activity studies, none of the synthesized chromenylphosphonates and chromenylphosphine oxides were active against selected Escherichia coli bacteria; however, the growth of Bacillus subtilis bacteria was reduced by the [bis(3,5-dimethylphenyl)](2-amino-3-cyano-4H-chromen-4-yl)phosphine oxides (8c, 9c and 10c). The IC50 values obtained were in the range of 5–9 μM.
The most active compounds were the dibenzyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonates (2e, 3e, 4e, 5e and 6e), since they showed activity in the 8–9 μM range against NIH/3T3 cells and in the 3–7 μM range against HL-60 cells.
Conclusions
In summary, a facile and efficient PMDTA-catalyzed synthetic method was developed for the preparation of novel (2-amino-3-cyano-4H-chromen-4-yl)phosphonates by the domino Knoevenagel–phospha-Michael reaction. Our approach made also possible condensation with secondary phosphine oxides, and this reaction has not been reported previously in the literature. The model reaction of salicylaldehydes, malononitrile and dialkyl phosphites was optimized in detail, and then, the condensation was extended to various substituted salicylaldehydes and secondary phosphine oxides. The approach developed did not require chromatographic separation, since the products could be recovered from the reaction mixture by simple filtration. Altogether 18 (2-amino-3-cyano-4H-chromen-4-yl)phosphonate derivatives and 20 (2-amino-3-cyano-4H-chromen-4-yl)phosphine oxides were prepared in good to high yields, and fully characterized. Except five chromenylphosphonates (2a–d and 6a), all of them are new compounds. The crystal structure of compounds 2c, 6e and 8a–c was studied by single-crystal XRD analysis. The in vitro cytotoxicity and the antibacterial activity of the chromenylphosphonates and chromenylphosphine oxides synthesized were also investigated. Several chromenylphosphonates showed moderate and promising activities against the tested cell lines, especially the dibenzyl (2-amino-3-cyano-4H-chromen-4-yl)phosphonates, which had their IC50 values in the 8–9 micromolar range against NIH/3T3 cells, and in the 3–7 micromolar range against HL-60 cells. None of the prepared derivatives reduced the growth of Gram-negative bacteria; however, chromenylphosphine oxides containing 3,5-dimethylphenyl groups on the phosphorus atom were active against selected Gram-positive bacteria. Two of the latter derivatives also showed activity in the 10 micromolar range against HL-60 cells.Author contributions
E. B. and Á. T. planned the experiments, Á. T., K. E. Sz., N. P.-T. and J. I. carried out the experiments, F. P. performed the crystal structure analysis, B. K. and L. H. J. performed the biological evaluation (screening), E. B. and L. G. P. contributed reagents/materials/analysis tools, E. B. and Á. T. wrote the paper, and L. H. J. and L. G. P. reviewed the paper. All the authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was supported by the Hungarian Research Development and Innovation Office (FK123961), the bilateral Hungarian-Slovenian Science and Technology Cooperation project (2018-2.1.11-TÉT-SI-2018-00008) and the Slovenian Research Agency (P1-0230-0175). N. P.-T. was supported by the Servier-Beregi PhD Research Fellowship. E. B. was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00278/17/7) and the ÚNKP-20-5-BME-288 New National Excellence Program of the Ministry of Human Capacities. F. P. thanks the EN-FIST Centre of Excellence, Slovenia, for the use of the Supernova diffractometer.Notes and references
- Phosphorus Heterocycles II, in Topics in Heterocyclic Chemistry, ed. R. K. Bansal, Springer, Berlin, Germany, 2010, vol. 21 Search PubMed.
- T. E. Ali and S. M. Abdel-Kariem, ARKIVOC, 2015, 2015, 246 Search PubMed.
- L. Chen, X.-Y. Liu and Y.-X. Zou, Adv. Synth. Catal., 2020, 362, 1724 CrossRef CAS.
- N. Thomas and S. M. Zachariah, Asian J. Pharm. Clin. Res., 2013, 6, 11 CAS.
- A. Pragi, A. Varun, H. S. Lamba and W. Deepak, Int. J. Pharma Sci. Res., 2012, 3, 2947 Search PubMed.
- M. Rajasekhar, K. U. M. Rao, C. S. Sundar, N. M. B. Reddy, S. K. Nayak and C. S. Reddy, Chem. Pharm. Bull., 2012, 60, 845 CrossRef.
- R. M. N. Kalla, J.-S. Choi, J.-W. Yoo, S. J. Byeon, M. S. Heo and I. Kim, Eur. J. Med. Chem., 2014, 76, 61 CrossRef CAS PubMed.
- M. Haji, Beilstein J. Org. Chem., 2016, 12, 1269 CrossRef CAS PubMed.
- Multicomponent Reactions 1, in Science of Synthesis, ed. T. J. J. Müller, Thieme, Stuttgart, Germany, 2014 Search PubMed.
- S. U. Deshmukh, J. N. Sangshetti, S. V. Bhosale and R. P. Pawar, ChemistrySelect, 2021, 6, 617 CrossRef CAS.
- M. Kulkarni, V. Pandurangi, U. Desai and P. Wadgaonkar, C. R. Chim., 2012, 15, 745 CrossRef CAS.
- P. Kour, A. Kumar, R. Sharma, R. Chib, I. Ali Khan and V. Rai, Res. Chem. Intermed., 2017, 43, 1 CrossRef.
- P. Kour, A. Kumar and V. Rai, C. R. Chim., 2017, 20, 140 CrossRef CAS.
- F. Darvish, A. Abdollahzade and D. Saravani, Res. Chem. Intermed., 2017, 43, 1487 CrossRef CAS.
- S. Kolla and Y. Lee, Tetrahedron, 2012, 68, 226 CrossRef CAS.
- P. Dai, G. Zha, X. Lai, W. Liu, Q. Gan and Y. Shen, RSC Adv., 2014, 4, 63420 RSC.
- D. Gaikwad, K. Undale, T. Shaikh and D. Pore, C. R. Chim., 2011, 14, 865 CrossRef CAS.
- G. Brahmachari and S. Laskar, Phosphorus, Sulfur Silicon Relat. Elem., 2014, 189, 873 CrossRef CAS.
- P. Jayashree, G. Shanthi and P. Perumal, Synlett, 2009, 917–920 CAS.
- R. Mohammadi and M. Kassaee, J. Mol. Catal. A: Chem., 2013, 380, 152 CrossRef CAS.
- M. Rajasekhar, R. K. Maheswara, C. Sundar, N. Reddy, S. Nayak and C. Reddy, Chem. Pharm. Bull., 2015, 60, 854 CrossRef.
- S. Murthy, B. Madhav, V. Reddy and Y. Nageswar, Tetrahedron Lett., 2010, 51, 3649 CrossRef CAS.
- B. Das, P. Balasubramanyam, C. G. Reddy and N. Salvanna, Helv. Chim. Acta, 2011, 94, 1347 CrossRef CAS.
- S. Sobhani and M. Honarmand, J. Iran. Chem. Soc., 2012, 9, 661 CrossRef CAS.
- Y. Wang, Y. Wu, Y. Wang and L. Dai, Chin. J. Chem., 2012, 30, 1709 CrossRef CAS.
- S. Krishnammagari, B. Cho and Y. Jeong, Phosphorus, Sulfur Silicon Relat. Elem., 2018, 193, 306 CrossRef CAS.
- S. Sobhani and M. Honarmand, Catal. Lett., 2013, 143, 476 CrossRef CAS.
- S. Sobhani and R. Jahanshahi, Synth. Commun., 2013, 43, 3247 CrossRef CAS.
- M. Hosseini-Sarvari and A. Roosta, Comb. Chem. High Throughput Screening, 2014, 17, 47 CrossRef CAS PubMed.
- R. Kalla, S. Byeon, M. Heo and I. Kim, Tetrahedron, 2013, 69, 10544 CrossRef CAS.
- G. Keglevich, E. Bálint, R. Kangyal, M. Bálint and M. Milen, Heteroat. Chem., 2014, 25, 282 CrossRef CAS.
- G. J. Szebeni, A. Balázs, I. Madarász, G. Pocz, F. Ayaydin, I. Kanizsai, R. Fajka-Boja, R. Alföldi, L. Hackler and L. G. Puskás, Int. J. Mol. Sci., 2017, 18, 2105 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, characterization data and 31P, 1H and 13C NMR spectra. CCDC 2083632, 2083633, 2083634, 2083635 and 2083636. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob01204e |
| This journal is © The Royal Society of Chemistry 2021 |