
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,2-Bis-perfluoroalkylations of alkenes and alkynes with perfluorocarboxylic anhydrides via the formation of perfluoroalkylcopper intermediates†
Takuma
Tagami‡
 a,
Yuma
Aoki‡
a,
Shintaro
Kawamura
*ab and
Mikiko
Sodeoka
*ab
a,
Yuma
Aoki‡
a,
Shintaro
Kawamura
*ab and
Mikiko
Sodeoka
*ab
aCatalysis and Integrated Research Group, RIKEN Center for Sustainable Resource Science, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: sodeoka@riken.jp
bSynthetic Organic Chemistry Laboratory, RIKEN Cluster for Pioneering Research, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
First published on 15th September 2021
Abstract
A novel, Cu-mediated protocol toward the 1,2-bis-perfluoroalkyaltion of alkenes/alkynes was developed. The method proceeded with perfluorocarboxylic anhydrides as inexpensive and readily available perfluoroalkyl sources. Diacyl peroxide was generated in situ from the perfluorocarboxylic anhydrides and H2O2. The key step in this reaction is the formation of a stable perfluoroalkylcopper intermediate that is achieved with the aid of a bipyridyl ligand. Subsequent reaction of the intermediate with perfluoroalkyl-containing alkyl or vinyl radicals affords the desired products.
Introduction
Perfluoroalkyl-containing organic molecules are of interest as pharmaceuticals1 and organic functional materials2 because of their improved physical, chemical, and biological properties derived from their fluorine atoms. Among perfluoroalkyl molecules, alkanes and alkenes bearing two perfluoroalkyl groups on their vicinal carbons are expected to exhibit unique functions;3 thus methods toward the 1,2-bis-perfluoroalkyaltion of alkenes and alkynes have been actively studied for nearly 50 years.4–12 Recently, various 1,2-bis-trifluoromethylations have been reported due to the availability of sophisticated trifluoromethylating reagents such as Langlois,8 Umemoto,9 and Togni10 reagents, and trifluoromethylcopper complexes.11,12 However, such reagents for 1,2-bis-perfluoroalkylations are not as accessible. Thus, to the best of our knowledge, novel 1,2-bis-perfluoroalkylations that can introduce long perfluoroalkyl chains have not been reported in the last two decades, and the substrate scopes of these processes are still very limited.6a,d,fIn an attempt to remedy this disadvantage of perfluoroalkylation reactions, our group has been investigating the use of perfluorocarboxylic anhydrides as inexpensive and readily available perfluoroalkyl sources for these processes.13,14 We have realised the efficient radical perfluoroalkylation of alkenes by the in situ generation of diacyl peroxides from the reaction of the acid anhydrides and urea·H2O2 (Scheme 1a). The key step in successfully conducting these reactions was the taming of a highly reactive alkyl radical intermediate containing a perfluoroalkyl group, which was transformed into the corresponding carbocation by single-electron transfer (SET) with a Cu(II) species. As part of our interest in perfluoroalkylations driven by perfluorocarboxylic anhydrides, we envisaged a novel protocol for the 1,2-bis-perfluoroalkylation of alkenes and alkynes using perfluorocarboxylic anhydrides (Scheme 1b), which is described herein.
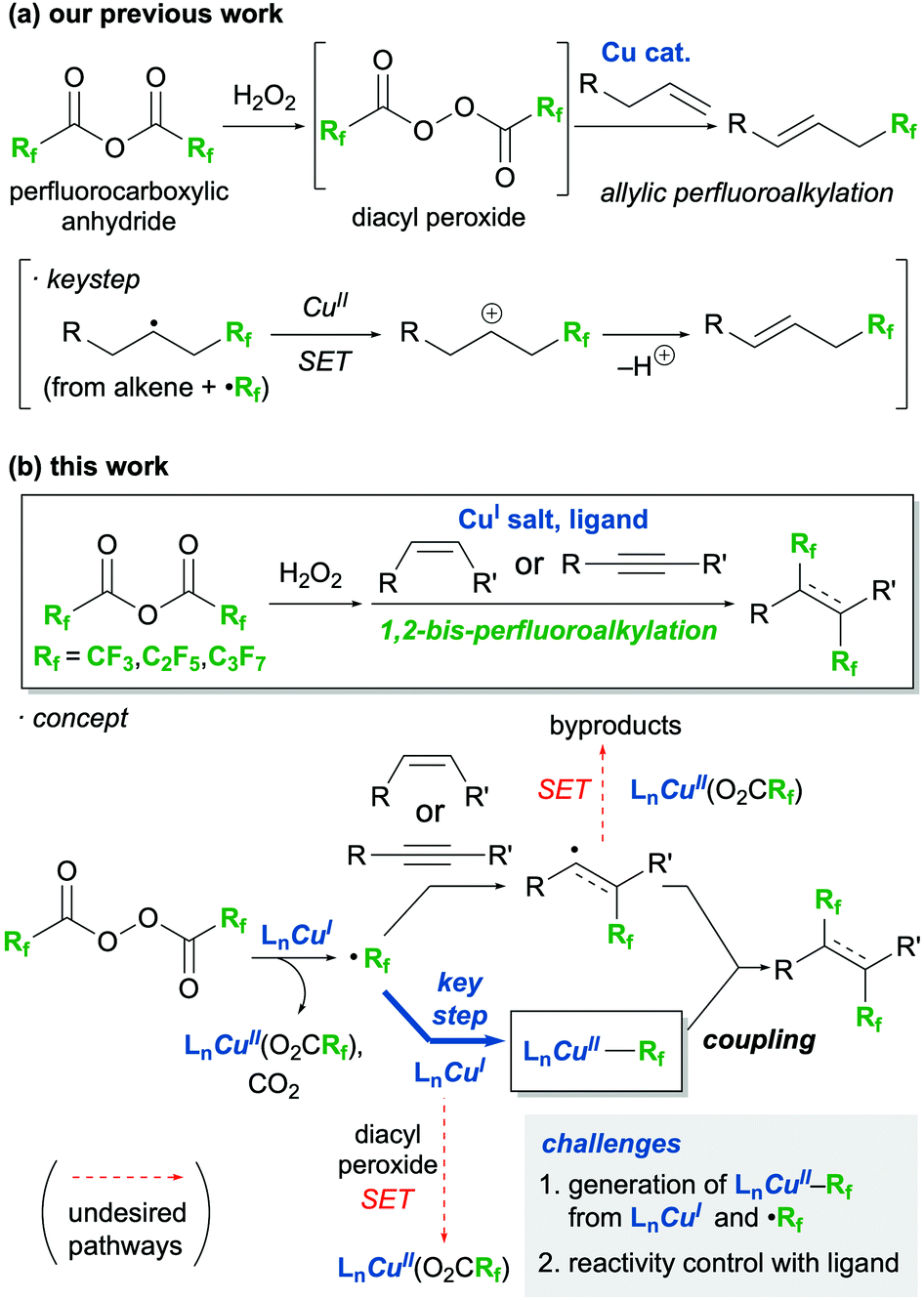 | ||
| Scheme 1 Perfluoroalkylation by using perfluorocarboxylic anhydrides (a) previous work (b) this work. | ||
According to recent literature,9–12 a similar process, 1,2-bis-trifluoromethylation, requires not only electrophilic CF3 reagents but also CF3 nucleophiles or CF3–Cu(II) coupling partners to introduce a second CF3 group. In 2019, Han and Lee achieved the 1,2-bis-trifluoromethylation of alkenes using the Umemoto reagent and CF3SiMe3 as the electrophilic and nucleophilic reagents, respectively, in the presence of a copper catalyst and a stoichiometric amount of silver fluoride.9 Zhu and Li performed the 1,2-bis-trifluoromethylation of alkynes using the Togni reagent and (bpy)Zn(CF3)2 (bpy = 2,2′-bipyridine) with a copper catalyst in 2020.10 Tsui developed a protocol for the 1,2-bis-trifluoromethylation of arynes with Cu(I)CF3 in the presence of an oxidant to afford unique 1,2-bis-trifluoromethylarenes.11 In this reaction, a CF3–Cu(II) intermediate was postulated to be formed and was thought to be responsible for the formation of CF3-containing aryl radicals; a second equivalent of CF3–Cu(II) then reacts with these radicals and affords the products. Cook developed an elegant method for the 1,2-bis-trifluoromethylation of terminal alkynes with (bpy)Cu(III)(CF3)3, where the photolysis of the latter simultaneously generated a CF3 radical and a CF3–Cu(II) intermediate.12 Inspired by these previous works, we designed a methodology for the Cu-mediated 1,2-bis-perfluoroalkylation of alkenes and alkynes with perfluorodiacyl peroxides. This unprecedented approach involves the generation of a perfluoroalkylcopper(II) intermediate (Rf–Cu(II)) that occurs through the capturing of perfluoroalkyl radicals with a Cu(I) salt (Scheme 1b, concept).15 However, there are a few, formidable challenges to this approach. First, the same Cu(I) species must contribute to two different processes simultaneously: the formation of perfluoroalkyl radicals with diacyl peroxide by SET and the formation of the Rf–Cu(II) intermediate through the capturing of the perfluoroalkyl radical. The second disadvantage of the proposed method is that the concentration balance between the Rf–Cu(II) and alkyl/vinyl radical intermediates, generated from perfluoroalkyl radicals reacting with the alkene/alkyne, must be precisely tuned to introduce a second perfluoroalkyl group. This balance is required because in the absence of the Rf–Cu(II) intermediate the alkyl/vinyl radicals, which are highly reactive, can participate in undesirable side reactions. To solve these issues, we envisioned the use of a ligand to control the reactivity and stability of the copper species.
Results and discussion
We preliminarily examined a model reaction of alkene 1a with trifluoroacetic anhydride (TFAA)/urea·H2O2 in the presence of 2.0 equiv. of [Cu(CH3CN)4]PF6 (Scheme 2). The desired product (2a) was obtained in 30% yield; however, it was accompanied by the formation of allylic trifluoromethylation product 3a,13a which was obtained in almost the same yield (28%). Next, the ligands of the Cu salt were screened to alter the reactivity of the complex (Table 1; see also Table S1 in ESI†). As a result, bipyridyl-type ligands were found to enhance the selectivity and yield of 2a (entries 2–4). Among them, bpy gave the best results (entry 2). In contrast, reactions employing pyridine or aliphatic, multidentate amine ligands preferentially provided 3a (entries 1, 5, and 6), whereas those conducted with phosphine ligands suppressed the reaction (entries 7 and 8). | ||
| Scheme 2 A preliminary result of the reaction using a simple Cu(I) salt. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Ligandb | Yieldc (%) |
2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a :3a |
|
| 2a | 3a (E/Z) | |||
| a Run on a 0.2 mmol scale. b 4.0 equiv. of ligand were used unless otherwise noted. c The yields were determined by NMR analysis. d Isolated yield was 58% yield. e PMDTA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine. | ||||
| 1 | Pyridine (8 equiv.) | 2 | 27 (90/10) | 7/93 |
| 2 | 2,2′-Bipyridine (bpy) | 75d | 4 (91/9) | 94/6 |
| 3 | 4,4′-Dimethyl-2,2′-bipyridine | 64 | 7 (91/9) | 90/10 |
| 4 | 1,10-Phenanthroline | 51 | 1 (n.d.) | 98/2 |
| 5 | TMEDA | 5 | 36 (88/12) | 12/88 |
| 6 | PMDTAe | 1 | 5 (88/12) | 17/83 |
| 7 | PPh3 (8.0 equiv.) | n.d. | n.d. | — |
| 8 | DPPE | n.d. | n.d. | — |
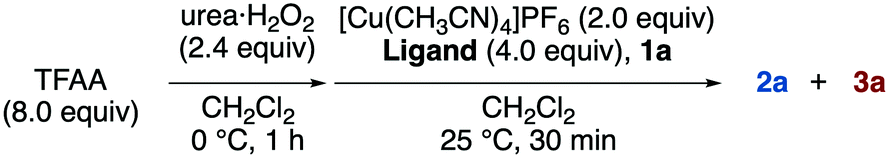
With the optimal conditions (Table 1, entry 2) in hand, the substrate scope was then investigated, as shown in Scheme 3.16,17 The functional group tolerance was confirmed by the reaction of several alkenes, which afforded the desired products bearing esters (2a,b), ketones (2c), phthalimides (2d), amides (2e), and sulphonamides (2f). A cyclic internal alkene could also be employed in the reaction, and the corresponding product (2g) was obtained. N-Tosyl allylamine, which was found to induce intramolecular amino- and carbo-trifluoromethylations in our previous work,13b successfully provided the 1,2-bis-trifluoromethylated product 2h in the present study. Perfluorocarboxylic anhydrides bearing longer alkyl chains were also employed in place of TFAA, providing the corresponding 1,2-bis-perfluoroalkylated molecules 2i and 2j. In addition, the reactions of terminal alkynes (4) smoothly proceeded to give 1,2-bis-perfluoroalkylated alkenes 5a–g with complete E-selectivity. The reaction also showed sufficient reactivity toward internal alkynes, affording the corresponding tetrasubstituted alkenes (5h–5k). To the best of our knowledge, the only other example of a 1,2-bis-perfluoroalkylation of an internal alkyne is that of arynes reported by Tsui's group.11 In all cases of the present study, E-selectivity was preferred; however, both the type of perfluoroalkyl group and the substituents on the triple bond of the alkyne were found to affect the stereoselectivity.
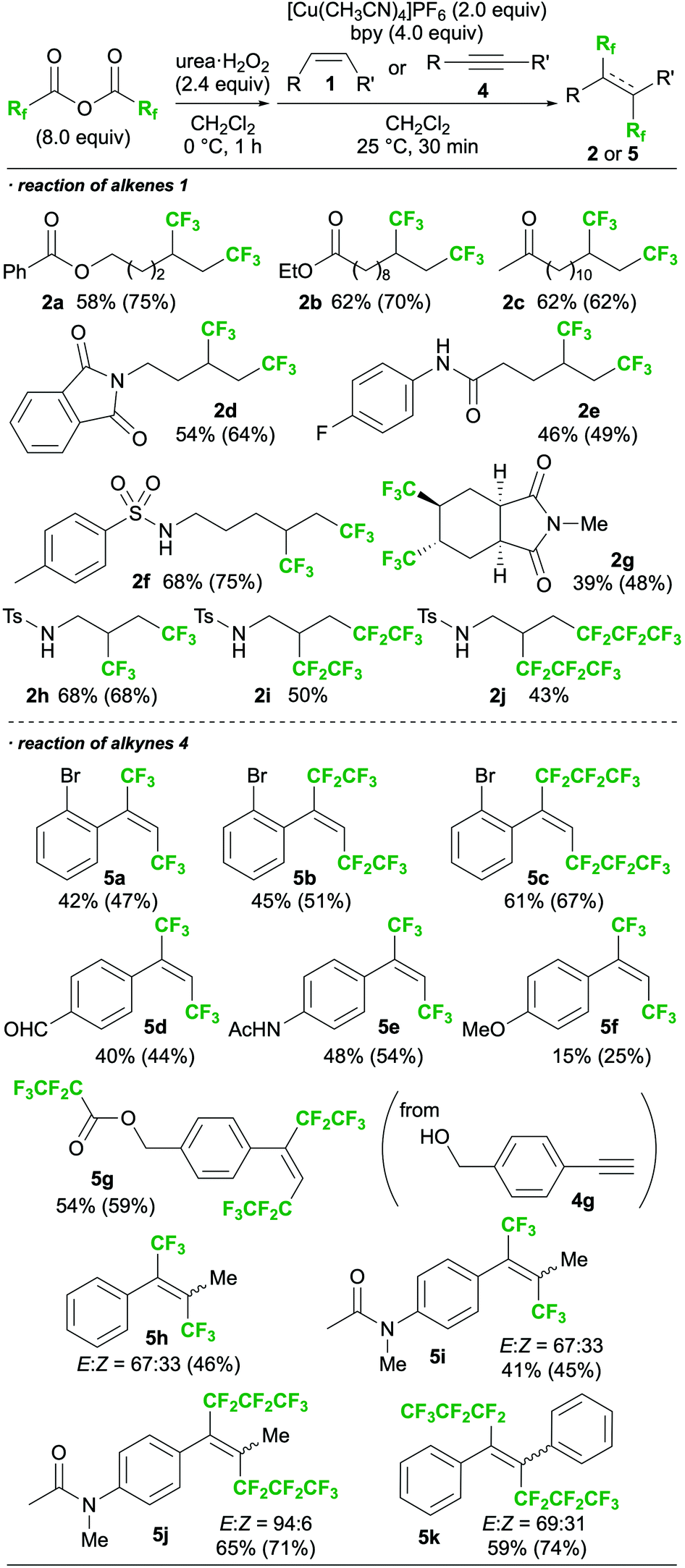 | ||
| Scheme 3 Substrate scope of the proposed method (NMR yields are shown in parentheses). | ||
Finally, the reaction mechanism was investigated using the 1,2-bis-trifluoromethylation of alkynes as a model reaction. We assumed that a stable, cationic copper(I) complex ligating two bpy ligands (A) is formed under the optimal conditions.18,19 At the beginning of the reaction, SET between bis(trifluoroacetyl) peroxide (BTFAP) and A generates a CF3 radical and [(bpy)2Cu(O2CCF3)]+ (B) (Scheme 4).13,20 The CF3 radicals react with the alkyne to afford a vinyl radical intermediate.21 Simultaneously, [(bpy)2Cu(II)CF3]+ (C) can be formed by the reaction of a CF3 radical with A. The free energies of activation obtained by density functional theory (DFT) calculations (M06(CPCM = DCM)/6-311+G(d,p), SDD for Cu) indicated that the capturing of CF3 radicals by A is kinetically preferable to vinyl radical formation (ΔG‡Cu(II) = +5.5 kcal mol−1vs. ΔG‡vinyl = +9.4 kcal mol−1). Furthermore, the bpy ligand was found to remarkably stabilize intermediate C; the free energy change for the formation of intermediate C (ΔGCu(II)) was −16.1 kcal mol−1, whereas the free energy changes for the formation of bpy-free Cu(II)–CF3 complexes [Cu(CH3CN)4(CF3)]+ and (CH3CN)2Cu(O2CCF3)(CF3) (ΔGCu(II)) were −2.8 and −8.8 kcal mol−1, respectively.19 The reversibility of this step would lead to an imbalance between the concentrations of the vinyl radical and the Cu(II)–CF3 species, which is due to the high thermodynamic stability of the former (ΔG‡vinyl = −31.9 kcal mol−1). Thus, bpy was considered to play a crucial role in controlling the selectivity of the reaction by stabilising intermediate C and suppressing this reverse reaction.
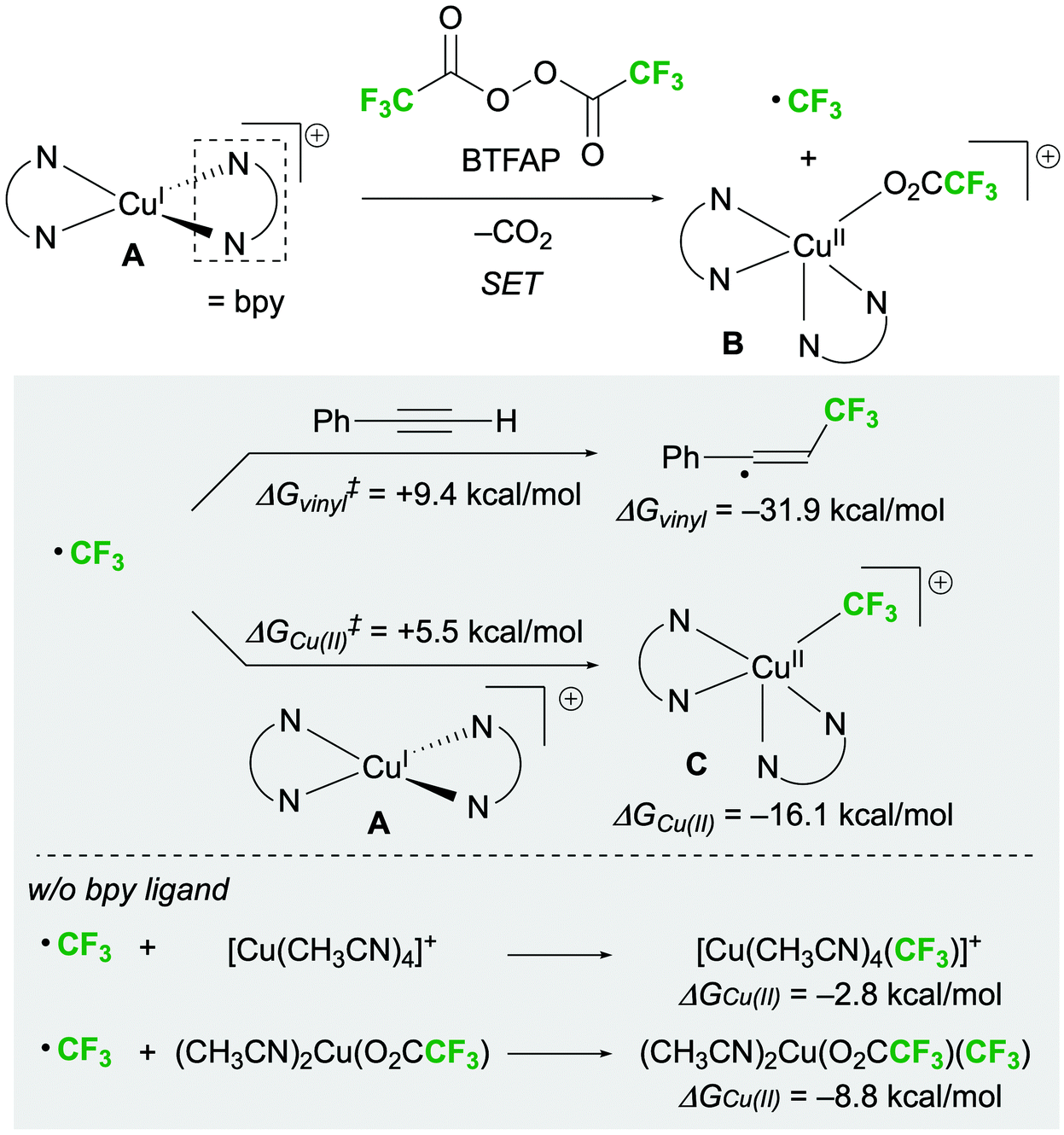 | ||
| Scheme 4 Proposed pathways of the formations of CF3–Cu(II) species with CF3 radicals. | ||
The trifluoromethylation of the vinyl radical was then evaluated and two possible pathways were proposed (Scheme 5a). If SET between Cu(I) intermediate A and the diacyl peroxide is the dominant process over the formation of CF3–Cu(II) intermediate C, the double addition of two CF3 radicals to the triple bond (path b) might proceed instead of a coupling process (path a).6,8,22 Further, when the reaction proceeds via path b, the stereoselectivity outcome would only depend on the substrate structure. We observed that bipyridyl-type ligands such as bpy, 6,6′-dimethyl-2,2′-bipyridine, and 1,10-phenanthroline increased the E-selectivity of the reaction of internal alkyne 4h (Scheme 5b). In particular, sterically demanding 6,6′-dimethyl-2,2′-bipyridine greatly increased the E-selectivity but decreased the product yield. This result confirms that path a involving CF3–Cu(II) intermediate C is the predominant pathway.
 | ||
| Scheme 5 Mechanistic investigation of the trifluoromethylation of vinyl radicals: (a) proposed mechanism and (b) ligand effect on the stereoselectivity outcome. | ||
Several attempts were made to detect the CF3–Cu(II) species via spectroscopic analyses of the reaction mixture, but all attempts failed. On the other hand, the 19F NMR analysis of the crude mixture after workup suggested the formation of (bpy)Cu(CF3)3, although its yield (as determined by 19F NMR analysis) was only 5%.23 This result strongly suggests the formation of CF3–Cu species during the reaction. To evaluate the reactivity of (bpy)Cu(CF3)3 under the optimal conditions (Table 1, entry 2), it was used as the reagent instead of a diacyl peroxide; however, no reactivity was observed (Scheme 6a). In addition, the reaction with heptafluorobutyric anhydride in the presence of (bpy)Cu(CF3)3 gave only the 1,2-bis-heptafluoropropylation product, proving that (bpy)Cu(CF3)3 did not participate in the reaction (Scheme 6b). Stable (bpy)Cu(CF3)3 was concluded to be a terminal by-product derived from the reactive CF3–Cu(II) intermediate (C).19
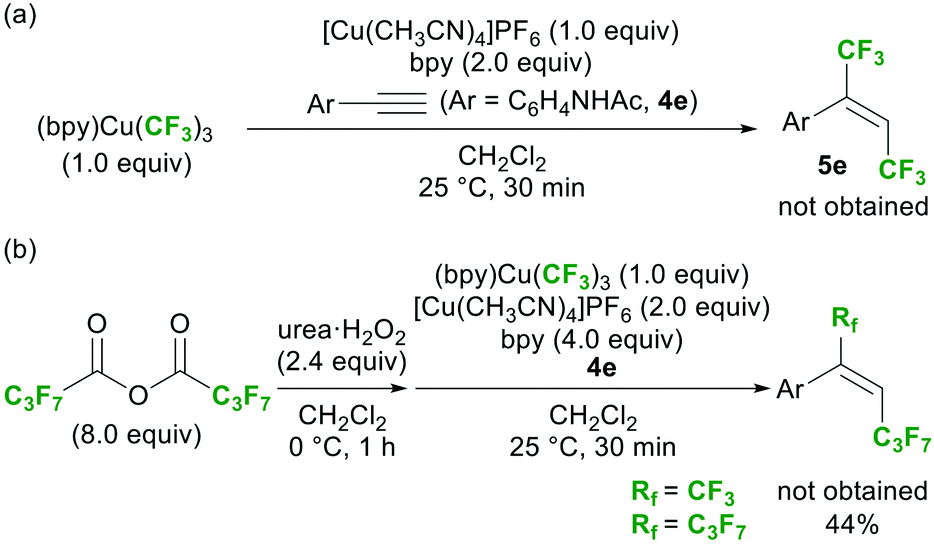 | ||
| Scheme 6 Examination of the reactivity of (bpy)Cu(CF3)3 under the optimal conditions (Table 1, entry 2). | ||
Next, the pathway for the reaction of vinyl radicals with C is discussed. Cook et al. proposed that their photo-induced 1,2-bis-trifluoromethylation of alkynes with (bpy)Cu(CF3)3 proceeds through the capturing of a vinyl radical with (bpy)(L)Cu(II)–CF3 (L = CF3 or OSO3H) and subsequent reductive elimination.12 Thus, as described above, we hypothesized that the coupling process in the present study proceeds in a similar manner: the capturing of the vinyl radical with intermediate C, followed by reductive elimination of Cu(III) intermediate D (Scheme 7, upper arrow).24 In addition to this, we also considered another possible mechanism involving SET between the vinyl radical and intermediate C, in which the resulting vinyl cation and CF3–Cu(I) intermediate E react via nucleophilic addition to give the desired product (Scheme 7, lower arrow).9 The sufficiently small free energy change obtained by DFT calculations suggested that Cu(III) intermediate D is formed by the reaction of C with the vinyl radical (ΔGCu(III) = −2.5 kcal mol−1), which is followed by reductive elimination (ΔG‡RE = +7.3 kcal mol−1). Notably, in the second trifluoromethylation step, no positive effects were induced by bpy on the stability of D and on the rate of the reductive elimination.19 On the other hand, the free energy change for vinyl cation formation by SET (ΔGox) was +28.3 kcal mol−1, suggesting that this process is either very slow or could not proceed.19
 | ||
| Scheme 7 Possible pathways of the reaction between the vinyl radical and [(bpy)2Cu(II)CF3]+ (C). | ||
Conclusions
We have achieved the Cu-mediated 1,2-bis-perfluoroalkylation of alkenes/alkynes using perfluorocarboxylic anhydrides as inexpensive and readily available perfluoroalkyl sources. The addition of the bpy ligand to the Cu salt dramatically improved the yields of the desired products, which allowed us to obtain various 1,2-bis-perfluoroalkylated molecules (2 and 5), including unique tetrasubstituted alkenes (5h–5k). In this study, we demonstrated an unprecedented approach for generating a key species in perfluoroalkylation, an Rf–Cu(II) intermediate (C for Rf = CF3), by the reaction of perfluoroalkyl radicals with a Cu(I) intermediate (A). This efficiently introduced a second Rf group onto the perfluoroalkyl-containing alkyl or vinyl radical via a Cu(III) intermediate (D for Rf = CF3). DFT studies proved that bpy ligands play a crucial role in stabilising Rf–Cu(II) intermediate (C for Rf = CF3), allowing for an efficient and selective transformation. We believe that this method is useful for the practical synthesis of 1,2-bis-perfluoroalkylated compounds and will contribute to the development of new bioactive molecules and functional materials. In addition, this methodology can control radical species with copper, which is a process that will contribute to future, new reaction designs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (No. 21K06489) and the project funding provided by RIKEN. TT is grateful to the SPDR Program in RIKEN. The computational study was conducted on HOKUSAI-GW (RIKEN).Notes and references
- For recent reviews, see: (a) H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem. – Eur. J., 2019, 25, 11797 CrossRef CAS PubMed; (b) Y. Pan, ACS Med. Chem. Lett., 2019, 10, 1016 CrossRef CAS PubMed; (c) K. Haranahalli, T. Honda and I. Ojima, J. Fluorine Chem., 2019, 217, 29 CrossRef CAS PubMed; (d) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633 CrossRef CAS PubMed; (e) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- For a recent book, see: New Fluorinated Carbons: Fundamentals and Applications, ed. O. V. Boltalina and T. Nakajima, Elsevier, Amsterdam, Netherlands, 2016 Search PubMed.
- For synthetic methods for 1,2-bis-perfluoroalkylated molecules, except for the 1,2-bis-perfluoroalkylation, see: (a) B. Gao, Y. Zhao, C. Ni and J. Hu, Org. Lett., 2014, 16, 102 CrossRef CAS; (b) B. Zhao, Y. Li, D.-H. Tu, W. Zhang, Z.-T. Liu and J. Lu, Tetrahedron Lett., 2016, 57, 4345 CrossRef CAS; (c) Y. Li, B. Zhao, K. Dai, D.-H. Tu, B. Wang, Y.-Y. Wang, Z.-T. Liu, Z.-W. Liu and J. Lu, Tetrahedron, 2016, 72, 5684 CrossRef CAS; (d) M. Yamamoto, D. C. Swenson and D. J. Burton, J. Fluorine Chem., 2016, 185, 213 CrossRef CAS; (e) W.-R. Zhu, Z.-W. Zhang, W.-H. Huang, N. Lin, Q. Chen, K.-B. Chen, B.-C. Wang, J. Weng and G. Lu, Synthesis, 2019, 51, 1969 CrossRef CAS.
- For a recent book, see: Organofluorine Chemistry: Synthesis, Modeling, and Applications, ed. K. J. Szabó and N. Selander, Wiley-VCH, Weinheim, Germany, 2021 Search PubMed.
- For recent reviews of perfluoroalkyaltions, see: (a) S. Kawamura and M. Sodeoka, Bull. Chem. Soc. Jpn., 2019, 92, 1245 CrossRef CAS; (b) Á. L. Mudarra, S. Martínez de Salinas and M. H. Pérez-Temprano, Synthesis, 2019, 51, 2809 CrossRef; (c) H. Mei, J. Han, S. White, G. Butler and V. A. Soloshonok, J. Fluorine Chem., 2019, 227, 109370 CrossRef CAS; (d) Q.-S. Gu, Z.-L. Li and X.-Y. Liu, Acc. Chem. Res., 2020, 53, 170 CrossRef CAS PubMed; (e) S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2020, 26, 11065 CrossRef CAS PubMed; (f) Y. Kuninobu and T. Torigoe, Bull. Chem. Soc. Jpn., 2021, 94, 532 CrossRef CAS.
- For Kolbe-electrolysis-type 1,2-bis-perfluoroalkylations of electron-deficient alkenes with perfluorocarboxylic acids, see: (a) C. J. Brookes, P. L. Coe, D. M. Owen, A. E. Pedler and J. C. Tatlow, J. Chem. Soc., Chem. Commun., 1974, 3, 323 RSC; (b) R. N. Renaud and P. J. Champagne, Can. J. Chem., 1975, 53, 529 CrossRef CAS; (c) R. N. Renaud, P. J. Champagne and M. Savard, Can. J. Chem., 1979, 57, 2617 CrossRef CAS; (d) P. L. Coe, D. M. Owen and A. E. Pedler, J. Chem. Soc., Perkin Trans. 1, 1983, 1995 RSC; (e) K. Uneyama, O. Morimoto and H. Nanbu, Tetrahedron Lett., 1989, 30, 109 CrossRef CAS; (f) K. Uneyama, S. Watanabe, Y. Tokunaga, K. Kitagawa and Y. Sato, Bull. Chem. Soc. Jpn., 1992, 65, 1976 CrossRef CAS; (g) W. Dmowski, A. Biernacki, T. Kozlowski, P. Gluziński and Z. Urbańczyk-Lipkowska, Tetrahedron, 1997, 53, 4437 CrossRef CAS; (h) K. Arai, K. Watts and T. Wirth, ChemistryOpen, 2014, 3, 23 CrossRef CAS PubMed; see also: (i) M. Elsherbini and T. Wirth, Acc. Chem. Res., 2019, 52, 3287 CrossRef CAS PubMed.
- For the 1,2-bis-trifluoromethylation of cyclohexene with Hg(CF3)2 and Te(CF3)2, see: D. Naumann, B. Wilkes and J. Kischkewitz, J. Fluorine Chem., 1985, 30, 73 CrossRef CAS.
- B. Yang, X.-H. Xu and F.-L. Qing, Org. Lett., 2015, 17, 1906 CrossRef CAS PubMed.
- H. Oh, A. Park, K.-S. Jeong, S. B. Han and H. Lee, Adv. Synth. Catal., 2019, 361, 2136 CrossRef CAS.
- H. Shen, H. Xiao, L. Zhu and C. Li, Synlett, 2020, 31, 41 CrossRef CAS.
- X. Yang and G. C. Tsui, Chem. Sci., 2018, 9, 8871 RSC.
- (a) S. Guo, D. I. AbuSalim and S. P. Cook, Angew. Chem., Int. Ed., 2019, 58, 11704 CrossRef CAS; see also: (b) J. He, T. N. Nguyen, S. Guo and S. P. Cook, Org. Lett., 2021, 23, 702 CrossRef CAS.
- (a) S. Kawamura and M. Sodeoka, Angew. Chem., Int. Ed., 2016, 55, 8740 CrossRef CAS; (b) S. Kawamura, K. Dosei, E. Valverde, K. Ushida and M. Sodeoka, J. Org. Chem., 2017, 82, 12539 CrossRef CAS PubMed; (c) E. Valverde, S. Kawamura, D. Sekine and M. Sodeoka, Chem. Sci., 2018, 9, 7115 RSC; (d) S. Kawamura, C. J. Henderson, Y. Aoki, D. Sekine, S. Kobayashi and M. Sodeoka, Chem. Commun., 2018, 54, 11276 RSC; see also: (e) S. Kawamura, S. Mukherjee and M. Sodeoka, Org. Biomol. Chem., 2021, 19, 2096 RSC.
- (a) J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6, 7919 CrossRef PubMed; (b) J. W. Beatty, J. J. Douglas, R. Miller, R. C. McAtee, K. P. Cole and C. R. J. Stephenson, Chem, 2016, 1, 456 CrossRef CAS; (c) R. C. McAtee, J. W. Beatty, C. C. McAtee and C. R. J. Stephenson, Org. Lett., 2018, 20, 3491 CrossRef CAS; (d) A. C. Sun, E. J. McClain, J. W. Beatty and C. R. J. Stephenson, Org. Lett., 2018, 20, 3487 CrossRef CAS; see also: (e) D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295 CrossRef CAS.
- (a) C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010 CrossRef CAS PubMed; (b) D. J. P. Kornfilt and D. W. C. MacMillan, J. Am. Chem. Soc., 2019, 141, 6853 CrossRef CAS; (c) Y. Liu, Z. Han, Y. Yang, R. Zhu, C. Liu and D. Zhang, Mol. Catal., 2021, 499, 111294 CrossRef CAS.
- The isolated yields were slightly lower than the NMR yields due to the difficulty in isolating the pure product from a mixture containing impurities with similar polarity .
- 1-Octyne and 1-(ethynylsulfonyl)-4-methylbenzene did not provide the desired products .
- (a) X. Lin, C. Hou, H. Li and Z. Weng, Chem. – Eur. J., 2016, 22, 2075 CrossRef CAS PubMed; (b) X. Lin, Z. Li, X. Han and Z. Weng, RSC Adv., 2016, 6, 75465 RSC.
- For more details, see the ESI.†.
- (a) H. Zhu, F. Teng, C. Pan, J. Cheng and J.-T. Yu, Tetrahedron Lett., 2016, 57, 2372 CrossRef CAS; (b) Y. Li, Y. Han, H. Xiong, N. Zhu, B. Qian, C. Ye, E. A. B. Kantchev and H. Bao, Org. Lett., 2016, 18, 392 CrossRef CAS PubMed; (c) C. Ye, Y. Li and H. Bao, Adv. Synth. Catal., 2017, 359, 3720 CrossRef CAS; (d) M. Israr, C. Ye, M. T. Muhammad, Y. Li and H. Bao, Beilstein J. Org. Chem., 2018, 14, 2916 CrossRef CAS PubMed; (e) C. Ye, Y. Li, X. Zhu, S. Hu, D. Yuan and H. Bao, Chem. Sci., 2019, 10, 3632 RSC; (f) X. Zhu, W. Deng, M.-F. Chiou, C. Ye, W. Jian, Y. Zeng, Y. Jiao, L. Ge, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2019, 141, 548 CrossRef CAS PubMed; (g) X. Zhu, M. Su, Q. Zhang, Y. Li and H. Bao, Org. Lett., 2020, 22, 620 CrossRef CAS PubMed; see also the references cited therein and (h) H. Liu, J.-T. Yu and C. Pan, Chem. Commun., 2021, 57, 6707 RSC.
- Radical trapping using TEMPO as the additive suggested the formation of vinyl radicals, where the TEMPO adduct of the vinyl radical was detected by ESI-MS analysis. See Scheme S1 in the ESI.†.
- The thermal fragmentation of the diacyl peroxide can generate a CF3 radical. However, heating the reaction mixture in the absence of the copper salt at a higher temperature (60 °C) did not give 2a. In addition, reducing the equivalents of [Cu(CH3CN)4]PF6 from 2.0 to 1.0 resulted in a significant decrease in yield and selectivity.
- The complex was identified by comparing the intensities and coupling patterns of the 19F NMR signals from the CF3 groups at the apical and equatorial positions with those in the literature (see Fig. S1 in ESI):† (a) A. M. Romine, N. Nebra, A. I. Konovalov, E. Martin, J. Benet-Buchholz and V. V. Grushin, Angew. Chem., Int. Ed., 2015, 54, 2745 CrossRef CAS PubMed; (b) N. Nebra and V. V. Grushin, J. Am. Chem. Soc., 2014, 136, 16998 CrossRef CAS PubMed.
- S. Liu, H. Liu, S. Liu, Z. Liu, C. Lu, X. Leng, Y. Lan and Q. Shen, J. Am. Chem. Soc., 2020, 142, 9785 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary data, experimental procedures, NMR spectra, computational details. See DOI: 10.1039/d1ob01529j |
| ‡ These authors equally contributed. |
| This journal is © The Royal Society of Chemistry 2021 |