DOI:
10.1039/D0PY01519A
(Paper)
Polym. Chem., 2021,
12, 1086-1096
Make or break: Mg(II)- and Zn(II)-catalen complexes for PLA production and recycling of commodity polyesters†
Received
30th October 2020
, Accepted 23rd December 2020
First published on 12th January 2021
Abstract
Recently we reported a series of highly active Al(III)-complexes bearing a catalen ligand support for lactide polymerisation, observing unprecedented activity in the melt. Herein we report diversification of the metal to furnish a series of well-defined dimeric Zn(II)- and Mg(II)-complexes, which were fully characterised by X-ray crystallography and NMR spectroscopy. The production of biocompatible atactic PLA from rac-LA in solution and under industrially preferred solvent-free conditions was demonstrated, typically observing good activity and Mn control with a broad range of dispersities (Đ = 1.08–2.04). Mg(II)-Complexes were shown to facilitate the relatively mild methanolysis of PLA, achieving up to 64% conversion to Me-LA within 8 h at 80 °C in THF. Further kinetic analysis found [Mg(1,3)]2 to have kapp values of 0.628 ± 0.0536 {4 wt% cat. loading} and 0.265 ± 0.0193 h−1 {8 wt% cat. loading} respectively for the rate of consumption of PLA. Preliminary work extended polymer scope to PET from various sources, demonstrating catalyst versatility.
Introduction
Inexpensive, lightweight and robust, plastics remain a tremendous source of social and economic value, providing 1.6 million jobs and turning over € 360 billion in 2018 in Europe alone.1,2 However, the current plastics economy remains fundamentally limited, dominated by petroleum-based products operating within a linear model, which underpins mounting environmental concerns.3–8 Ocean plastics typify the severity of current plastic pollution with the Great Pacific Garbage Patch (GPGB), an accumulation zone of ocean plastics, encompassing ca. 1.6 million km2, equivalent to three times the area of France.9 This has stimulated considerable research into renewable and environmentally friendly alternatives, for example poly(lactic acid) (PLA). Traditionally produced from the metal-mediated ring-opening polymerisation (ROP) of lactide (LA), a bio-based monomer feedstock, PLA has subsequently found commercial use in both the packaging and biomedical sector.3,10–14 Recent research has primarily sought to address toxicity concerns associated with the industry standard, Sn(Oct)2, through the development of biocompatible and environmentally benign alternatives.15–17 Consequently, a diverse range of metals have been explored, including Al(III)18–35 and group(IV),36–49 with a particular focus on retaining activity whilst maintaining stereocontrol. Metals most pertinent to this report include Zn(II) and Mg(II), which are cheap and non-toxic. Zinc complexes have previously been shown to be highly effective initiators for the ROP of LA, generally observing high activity in solution.50–64 Pioneering work by Coates and co-workers reported a highly heteroselective Zn(II)-complex bearing a β-diiminate ligand, achieving up to Pr = 0.94 in the ROP of rac-LA at 0 °C.65 Dizinc catalysts supported by macrocyclic ligands developed by Williams and co-workers remain the fastest reported to date under solution conditions, exhibiting high TOFs (up to 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1) at room temperature in THF.66 The most isoselective Zn(II)-complex known is an aminophenolate initiator reported by Ma and co-workers, which achieved Pr = 0.07 at −40 °C.67 McKeown et al.68 have reported a series of simple Zn(II)-Schiff Base complexes capable of achieving TOFs in excess of 100000 h−1 under immortal conditions in the melt. Recently, Hermann et al.69 reported a highly active and robust zinc-guanidine complex capable of producing colourless, high molecular weight (Mn ∼150000 g mol−1) PLA within minutes under solvent-free conditions (kp = 1.43 ± 0.09 L mol−1 s−1), culminating in the fastest reported system to date, significantly outperforming Sn(Oct)2. Magnesium complexes have also received significant interest in the ROP of LA.70–82 Chisholm and co-workers have reported numerous Mg(II)-complexes for the production of heterotactic PLA (Pr = 0.90–0.96).51,83–85 Both Coates et al.50 (Zn(II), Pr = 0.94; Mg(II), atactic PLA) and Ma et al.58 (Zn(II), Pr = 0.20; Mg(II), Pr = 0.81) have previously shown metal exchange to dramatically impact stereocontrol. Despite such promising advancements, the widespread use of PLA remains limited by a high production cost.3,11 Additionally, if not disposed of appropriately, PLA is a potential contributor to the plastic waste crisis.3,86 Plastic pollution mitigation requires the industry adopts a circular model, one concerned with material recapture and reuse, with recycling a potential solution.1,3,7 Whilst mechanical recycling is traditionally employed, its long-term suitability is limited by eventual material downcycling.2,87 A possible alternative is chemical recycling, which enables value-added products such as lactate esters, lactic acid and acrylic acid to be accessed.3,88,89 Lactic acid has been identified as a platform chemical, whilst lactate esters have been cited as potential green solvent replacements owing to their low toxicity and biodegradability.90–93 It is anticipated the potential for enhanced socio-economic performance will drive market penetration and reduce PLA production costs.94 Indeed, the ethyl lactate market is projected to reach $ 92 million by 2024 and currently trades at £ 2.54–3.49 per kg relative to £ 1.69 per kg for virgin PLA.91,95 PLA recycling processes include hydrolysis96–103 and alcoholysis.104–115 Hydrogenation116–118 and hydrosilylation119 processes exploiting ruthenium and iridium have also been reported. Simple, commercially available metal salts and precursors (e.g. FeCl3) have been shown to facilitate the transesterification of PLA, typically in the presence of methanol to afford methyl lactate (Me-LA).120–122 Sobota and co-workers demonstrated the transesterification of PLA using a wide range of alcohols in the presence of Mg(II) and Ca(II) pre-catalysts, typically operating under high temperature and pressure regimes.111 Organocatalysts have also been exploited in PLA degradation, for example triazabicyclodecene (TBD), 4-(dimethylamino)pyridine (DMAP) and tetramethylammonium methyl carbonate.112–114 McKeown et al.123 recently demonstrated a homoleptic Zn(II)-complex bearing a propylenediamine backbone to be highly active for PLA methanolysis, achieving 84% conversion to Me-LA (YMe-LA) within 1 h at 50 °C. The corresponding ethylenediamine analogue exhibited significantly reduced activity (YMe-LA = 12% in 6 h) under comparable conditions (40 °C), highlighting the importance of structure-activity relationships.109,123 However, the use of metal-based catalysts for this purpose remains rare despite the plethora reported for LA polymerisation. There is also a clear appetite to diversify metal scope to address potential long-term availability concerns associated with zinc.124
000 h−1) at room temperature in THF.66 The most isoselective Zn(II)-complex known is an aminophenolate initiator reported by Ma and co-workers, which achieved Pr = 0.07 at −40 °C.67 McKeown et al.68 have reported a series of simple Zn(II)-Schiff Base complexes capable of achieving TOFs in excess of 100000 h−1 under immortal conditions in the melt. Recently, Hermann et al.69 reported a highly active and robust zinc-guanidine complex capable of producing colourless, high molecular weight (Mn ∼150000 g mol−1) PLA within minutes under solvent-free conditions (kp = 1.43 ± 0.09 L mol−1 s−1), culminating in the fastest reported system to date, significantly outperforming Sn(Oct)2. Magnesium complexes have also received significant interest in the ROP of LA.70–82 Chisholm and co-workers have reported numerous Mg(II)-complexes for the production of heterotactic PLA (Pr = 0.90–0.96).51,83–85 Both Coates et al.50 (Zn(II), Pr = 0.94; Mg(II), atactic PLA) and Ma et al.58 (Zn(II), Pr = 0.20; Mg(II), Pr = 0.81) have previously shown metal exchange to dramatically impact stereocontrol. Despite such promising advancements, the widespread use of PLA remains limited by a high production cost.3,11 Additionally, if not disposed of appropriately, PLA is a potential contributor to the plastic waste crisis.3,86 Plastic pollution mitigation requires the industry adopts a circular model, one concerned with material recapture and reuse, with recycling a potential solution.1,3,7 Whilst mechanical recycling is traditionally employed, its long-term suitability is limited by eventual material downcycling.2,87 A possible alternative is chemical recycling, which enables value-added products such as lactate esters, lactic acid and acrylic acid to be accessed.3,88,89 Lactic acid has been identified as a platform chemical, whilst lactate esters have been cited as potential green solvent replacements owing to their low toxicity and biodegradability.90–93 It is anticipated the potential for enhanced socio-economic performance will drive market penetration and reduce PLA production costs.94 Indeed, the ethyl lactate market is projected to reach $ 92 million by 2024 and currently trades at £ 2.54–3.49 per kg relative to £ 1.69 per kg for virgin PLA.91,95 PLA recycling processes include hydrolysis96–103 and alcoholysis.104–115 Hydrogenation116–118 and hydrosilylation119 processes exploiting ruthenium and iridium have also been reported. Simple, commercially available metal salts and precursors (e.g. FeCl3) have been shown to facilitate the transesterification of PLA, typically in the presence of methanol to afford methyl lactate (Me-LA).120–122 Sobota and co-workers demonstrated the transesterification of PLA using a wide range of alcohols in the presence of Mg(II) and Ca(II) pre-catalysts, typically operating under high temperature and pressure regimes.111 Organocatalysts have also been exploited in PLA degradation, for example triazabicyclodecene (TBD), 4-(dimethylamino)pyridine (DMAP) and tetramethylammonium methyl carbonate.112–114 McKeown et al.123 recently demonstrated a homoleptic Zn(II)-complex bearing a propylenediamine backbone to be highly active for PLA methanolysis, achieving 84% conversion to Me-LA (YMe-LA) within 1 h at 50 °C. The corresponding ethylenediamine analogue exhibited significantly reduced activity (YMe-LA = 12% in 6 h) under comparable conditions (40 °C), highlighting the importance of structure-activity relationships.109,123 However, the use of metal-based catalysts for this purpose remains rare despite the plethora reported for LA polymerisation. There is also a clear appetite to diversify metal scope to address potential long-term availability concerns associated with zinc.124
We recently reported a series of highly active Al(III)-complexes supported by a catalen framework for lactide polymerisation, observing unprecedented activity in the melt.35 However, these complexes are inactive for PLA methanolysis. Herein, we report diversification of the metal to Zn(II) and Mg(II) in pursuit of catalysts active for PLA degradation, for which there is existing literature precedent.109,111,123 Consequently, a range of well-defined dimeric Zn(II)- and Mg(II)-catalen complexes were prepared, employing a new and emerging class of ligands in the area. Their application to the ROP of rac-LA in solution, and under industrially preferred melt conditions, is discussed. The relatively mild metal-mediated methanolysis of PLA into Me-LA is reported. Preliminary work diversifying polyester scope is also demonstrated.
Results and discussion
Synthesis
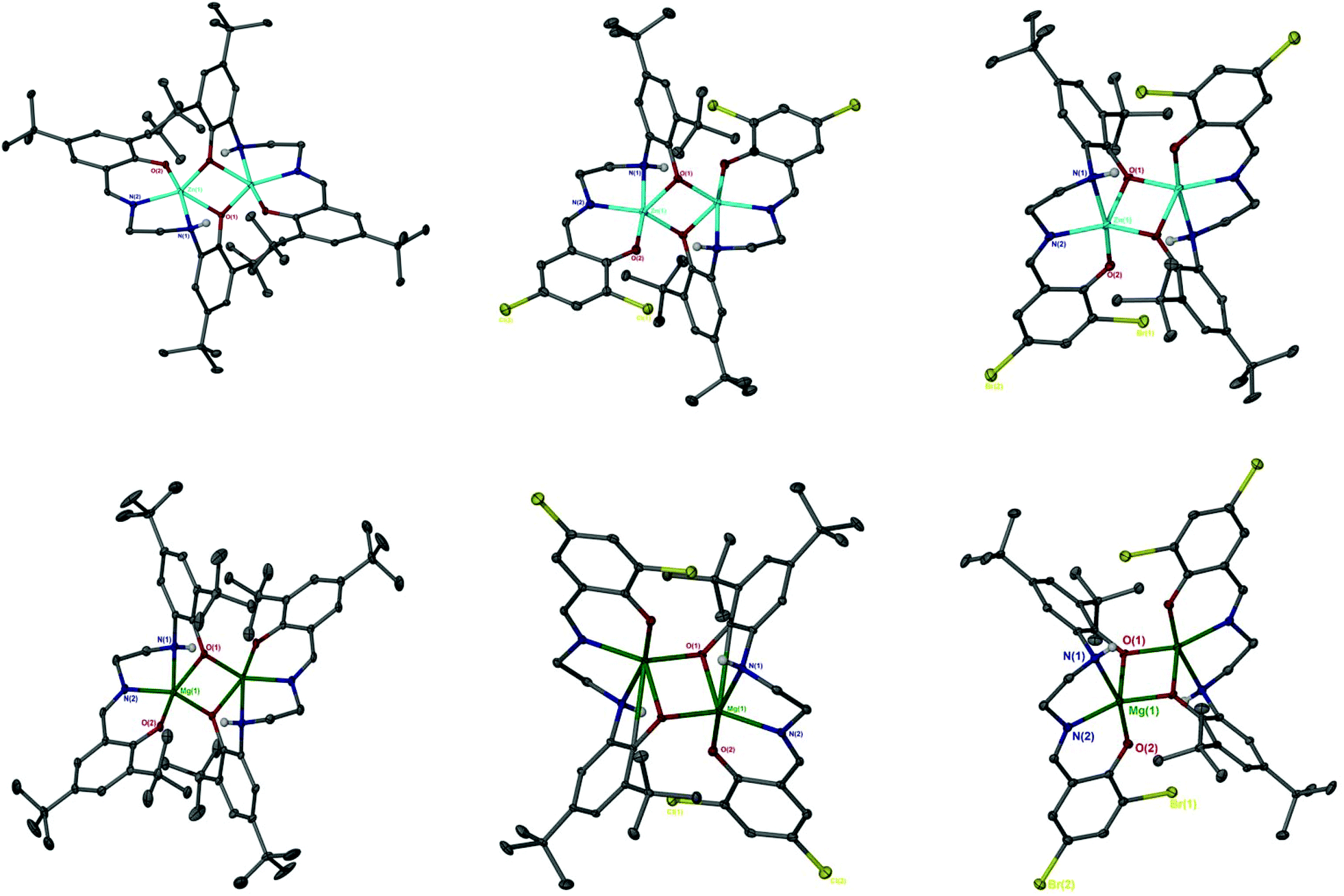
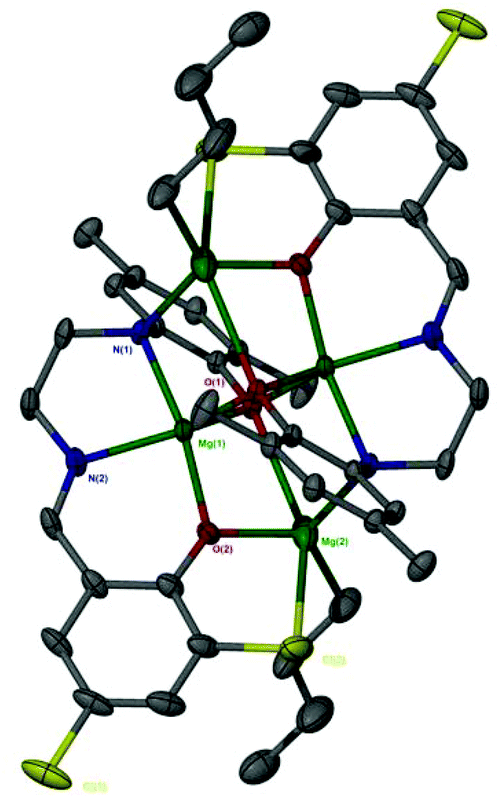
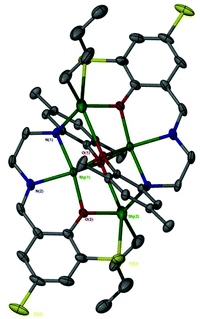
The catalen ligands were prepared via a simple two-step synthesis (Scheme 1), exploiting successive condensation reactions, and characterised by 1H NMR spectroscopy and mass spectrometry (MS). 1H NMR spectroscopic analysis revealed a characteristic singlet at ca. δ = 8.40 ppm, corresponding to a HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N resonance, confirming formation of the imine. The –CH2 resonances were observed as two distinct triplets between ca. δ = 3.40 and 3.80 ppm. Dimeric complexes of Zn(II) and Mg(II) were then prepared in anhydrous toluene and recrystallised from the reaction solvent (Scheme 1). All complexes were characterised by single crystal X-ray diffraction (XRD) as shown in Fig. 1. Selected bond lengths and angles for [Zn(1–3)]2 and [Mg(1–3)]2 are provided in the ESI.† A Zn(1)–N(2) and Mg(1)–N(2) bond length of ca. 2 Å confirmed retention of the imine functionality present in 1–3H2 upon coordination to the metal centre. In all instances, τ5 values tended towards 1, indicative of a distorted trigonal bipyramidal geometry (see ESI†). Interestingly, a highly unusual tetrameric Mg(II)-complex based on an amine-deprotonated derivative of 2H2 was isolated and characterised by XRD (Fig. 2). Direct concentration of the same solution led to the isolation of [Mg(2)]2 in good purity. The tetramer exhibited an Mg(1)–N(1) bond length of ca. 0.20 Å shorter relative to [Mg(2)]2 as expected {Mg(1)–N(1): tetramer, 2.078(4); [Mg(2)]2, 2.2181(19)}. Tetrameric analogues based on 1,3H2 and Zn(II) were not observed, whilst synthesis reattempts were unsuccessful. 1H NMR spectroscopic analysis of [Zn(1–3)]2 and [Mg(1–3)]2 revealed characteristic singlets at ca. δ = 8.00 and 4.50 ppm corresponding to ArCHN and –NH resonances respectively. More interestingly, diasterotopic –CH2 resonances between ca. δ = 3.50 and 4.00 ppm were observed, indicating the ligand is locked in position once coordinated (see ESI†). 13C{1H} NMR spectroscopic analysis was consistent with 1H NMR and XRD analysis. It is proposed the dimeric structure observed in the solid-state is retained in solution. Zn(II)- and Mg(II)-complexes were in generally good agreement with elemental analysis (EA) data obtained, demonstrating their purity. However, C% values were consistently low on [Mg(2–3)]2, potentially due to air and moisture sensitivity confounded by high hygroscopicity, consistent with the Al(III)-catalens.35
N resonance, confirming formation of the imine. The –CH2 resonances were observed as two distinct triplets between ca. δ = 3.40 and 3.80 ppm. Dimeric complexes of Zn(II) and Mg(II) were then prepared in anhydrous toluene and recrystallised from the reaction solvent (Scheme 1). All complexes were characterised by single crystal X-ray diffraction (XRD) as shown in Fig. 1. Selected bond lengths and angles for [Zn(1–3)]2 and [Mg(1–3)]2 are provided in the ESI.† A Zn(1)–N(2) and Mg(1)–N(2) bond length of ca. 2 Å confirmed retention of the imine functionality present in 1–3H2 upon coordination to the metal centre. In all instances, τ5 values tended towards 1, indicative of a distorted trigonal bipyramidal geometry (see ESI†). Interestingly, a highly unusual tetrameric Mg(II)-complex based on an amine-deprotonated derivative of 2H2 was isolated and characterised by XRD (Fig. 2). Direct concentration of the same solution led to the isolation of [Mg(2)]2 in good purity. The tetramer exhibited an Mg(1)–N(1) bond length of ca. 0.20 Å shorter relative to [Mg(2)]2 as expected {Mg(1)–N(1): tetramer, 2.078(4); [Mg(2)]2, 2.2181(19)}. Tetrameric analogues based on 1,3H2 and Zn(II) were not observed, whilst synthesis reattempts were unsuccessful. 1H NMR spectroscopic analysis of [Zn(1–3)]2 and [Mg(1–3)]2 revealed characteristic singlets at ca. δ = 8.00 and 4.50 ppm corresponding to ArCHN and –NH resonances respectively. More interestingly, diasterotopic –CH2 resonances between ca. δ = 3.50 and 4.00 ppm were observed, indicating the ligand is locked in position once coordinated (see ESI†). 13C{1H} NMR spectroscopic analysis was consistent with 1H NMR and XRD analysis. It is proposed the dimeric structure observed in the solid-state is retained in solution. Zn(II)- and Mg(II)-complexes were in generally good agreement with elemental analysis (EA) data obtained, demonstrating their purity. However, C% values were consistently low on [Mg(2–3)]2, potentially due to air and moisture sensitivity confounded by high hygroscopicity, consistent with the Al(III)-catalens.35
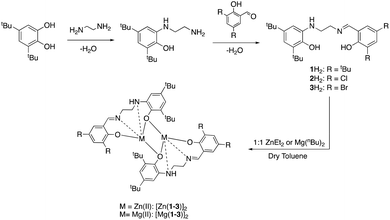 |
| | Scheme 1 Catalen ligand preparation and subsequently derived Zn(II)- and Mg(II)-complexes. | |
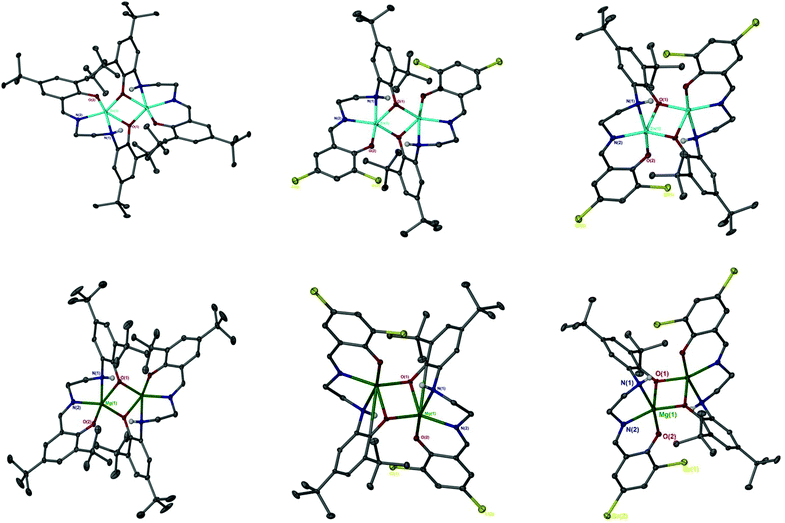 |
| | Fig. 1 Solid-state structures of [Zn(1–3)]2 (top left to right) and [Mg(1–3)]2 (bottom left to right). Ellipsoids shown at 30% probability with the exception of [Mg(2)]2, which is shown at 50% probability. All hydrogen atoms except those bound to nitrogen or involved in hydrogen bonding, as noted for [Mg(2–3)]2, have been omitted for clarity. | |
 |
| | Fig. 2 Solid-state structure of a tetrameric Mg(II)-complex based on an amine-deprotonated derivative of 2H2. Ellipsoids shown at 30% probability. All hydrogen atoms and methyl groups of the tBu groups have been omitted for clarity. | |
Polymerisation of rac-LA
All Zn(II)- and Mg(II)-complexes were trialled in the ROP of rac-LA under solvent-free conditions (130 and 180 °C) in alignment with industrial practices (Table 1). Industrially, solvents are a significant source of waste and thus catalysts that operate under melt conditions are highly desirable.3,15,68,69 All initiators were also tested out under solution conditions (80 °C) (Table 2). The lactide monomer, rac-LA, was recrystallised from anhydrous toluene once prior to use and benzyl alcohol (BnOH) was employed as a co-initiator. Conversion was determined via analysis of the methine region (ca. δ = 4.9–5.2 ppm) using 1H NMR spectroscopy. To ensure one –OBn moiety was associated per metal centre, both Zn(II)- and Mg(II)-complexes were treated as monomeric. It is proposed [Zn(1–3)]2 and [Mg(1–3)]2 operate via an activated-monomer mechanism. To investigate this the stability of [Zn(1)]2 with excess BnOH was studied using 1H NMR (CDCl3), which confirmed [Zn(1)]2 to be stable at both room temperature and 80 °C (see ESI†). Due to a poor isolation yield (0.065 g, 14%), [Mg(2)]2 was not pursued in polymerisation studies but was expected to exhibit comparable activity to [Mg(3)]2 based on previous work.35
Table 1 Melt polymerisation of rac-LA using [Zn(1–3)]2 and [Mg(1,3)]2
| Init. |
Time/min |
[rac-LA]:[M]:[BnOH] |
Conv.a/% |
M
n,theo
/g mol−1 |
M
n
/g mol−1 |
Đ
|
P
r
|
| Reaction conditions: rac-LA (1.0 g), solvent-free (130 °C). Determined via1H NMR spectroscopy. Theoretical average number molecular weight (Mn) dependent on conversion and co-initiator added {(Mr,LA × 3 × %conv) + Mn,BnOH}. Determined via GPC analysis (in THF). Determined via homonuclear decoupled NMR spectroscopy. Insufficient polymeric material isolated for material characterisation. N.B. {[rac-LA]:[M]:[BnOH] = 3000:1:10} were performed at 180 °C. [M]:[BnOH] = 1:1 corresponds to 1 equivalent of BnOH per metal centre. Monomer conversion plateau between 60–70% can be attributed to reduced catalyst activity coupled with mass transfer limitations. |
| [Zn(1)]2 |
20 |
300:1:1 |
69 |
29900 |
28600 |
1.37 |
0.57 |
| 90 |
3000:1:10 |
68 |
29500 |
23500 |
2.04 |
0.53 |
| [Zn(2)]2 |
17 |
300:1:1 |
63 |
27300 |
21600 |
1.24 |
0.54 |
| 60 |
3000:1:10 |
69 |
29900 |
25150 |
1.58 |
0.52 |
| [Zn(3)]2 |
16 |
300:1:1 |
69 |
29900 |
23050 |
1.37 |
0.57 |
| 50 |
3000:1:10 |
60 |
26000 |
22750 |
1.91 |
0.52 |
| [Mg(1)]2 |
20e |
300:1:1 |
69 |
29900 |
— |
— |
— |
| 120 |
3000:1:10 |
64 |
27750 |
8750 |
1.58 |
0.49 |
| [Mg(3)]2 |
8 |
300:1:1 |
71 |
30800 |
17900 |
1.50 |
0.54 |
| 70 |
3000:1:10 |
69 |
29900 |
16800 |
2.00 |
0.50 |
Table 2 Solution polymerisation of rac-LA using [Zn(1–3)]2 and [Mg(1,3)]2
| Init. |
Time/h |
[rac-LA]:[M]:[BnOH] |
Conv.a/% |
M
n,theo
/g mol−1 |
M
n
/g mol−1 |
Đ
|
P
r
|
| Reaction conditions: rac-LA (0.5 g), solvent (toluene, 80 °C). Determined via1H NMR spectroscopy. Theoretical average number molecular weight (Mn) dependent on conversion and co-initiator added {(Mr,LA × %conv) + Mn,BnOH}. Determined via GPC analysis (in THF). Determined via homonuclear decoupled NMR spectroscopy. Bimodal GPC observed, distributions treated together for reported Mn and Đ values. Insufficient polymeric material isolated for material characterisation. N.B. [M]:[BnOH] = 1:1 corresponds to 1 equivalent of BnOH per metal centre. |
| [Zn(1)]2 |
8 |
100:1:1 |
60 |
8750 |
12400e |
1.08e |
0.61 |
| [Zn(2)]2 |
8f |
100:1:1 |
31 |
4550 |
— |
— |
— |
| [Zn(3)]2 |
8f |
100:1:1 |
21 |
3150 |
— |
— |
— |
| [Mg(1)]2 |
1.5 |
100:1:1 |
94 |
13650 |
13900 |
1.17 |
0.46 |
| [Mg(3)]2 |
8 |
100:1:1 |
77 |
12000 |
9400 |
1.63 |
0.48 |
All Zn(II)-complexes exhibited good activity at 130 °C, achieving reasonably high conversion within 20 minutes {[rac-LA]:[M]:[BnOH] = 300:1:1} (Table 1). [Zn(2–3)]2 exhibited superior activity relative to [Zn(1)]2, presumably owing to a more Lewis acid Zn(II)-centre. [Zn(1)]2 exhibited excellent Mn control (Mn,theo = 29900 g mol−1, Mn = 28600 g mol−1; Table 1, entry 1), whilst reasonable Mn control was maintained for [Zn(2–3)]2, observing moderate dispersities (Đ = 1.24–1.37) in all cases. Reducing the catalyst loading to 0.033 mol% at 180 °C to simulate industrial conditions resulted in prolonged polymerisation times, achieving between 60–69% within 50 to 90 minutes {[rac-LA]:[M]:[BnOH] = 3000:1:10} (Table 1). The reactivity trend noted for [Zn(1–3)]2 was exacerbated under these conditions, likely due to an increase in temperature assisting catalyst dissociation and solubility. Interestingly, [Zn(1)]2 exhibited poorer Mn control (Mn,theo = 29500 g mol−1, Mn = 23500 g mol−1; Table 1, entry 2;), whilst comparable control was retained for [Zn(2–3)]2 under these conditions. Generally, a lower Mn value than expected (relative to theoretical values) was observed, possibly indicative of side transesterification reactions, consistent with the broader dispersities observed (Đ = 1.58–2.04). Reactivity trends discussed for [Zn(1–3)]2 could be extended to the Mg(II)-catalen series, observing comparable activity between [Zn(1)]2 and [Mg(1)]2 at 130 °C (Table 1). Both [Mg(1,3)]2 exhibited reduced activity relative to their Zn(II) counterparts at 180 °C, suggesting the active species to be inherently less active, despite [Mg(3)]2 (71%, 8 min; Table 1, entry 9;) outperforming [Zn(3)]2 (69%, 16 min; Table 1, entry 5;) at 130 °C. However, significantly lower Mn values relative to those reported for the Al(III)-catalens (Mn = 160500–252100 g mol−1) were observed, implying kp to be significantly slower relative to ki, possibly due to a mechanism shift. Under both sets of conditions, [Mg(1,3)]2 exhibited poor Mn control (Mn,theo = 27750–30800 g mol−1, Mn = 8750–17900 g mol−1). MALDI-ToF analysis confirmed the polymer (Table 1, entry 8) to be –OBn and –H end-capped with multiple series present indicating a high degree of transesterification (see ESI†), consistent with the broad dispersities observed (Đ = 1.50–2.00). Metal exchange was found to have no impact on stereocontrol, observing the production of atactic PLA in all instances (Pr = 0.49–0.57).
Under solution conditions {[rac-LA]:[M]:[BnOH] = 100:1:1} (Table 2), [Zn(1)]2 exhibited good polymerisation control, producing PLA of reasonably well-defined Mn (Mn,theo = 8750 g mol−1, Mn = 12400 g mol−1; Table 2, entry 1) and narrow dispersities (Đ = 1.08). GPC analysis revealed the Mn distribution to be bimodal in nature (see ESI†). Surprisingly, [Zn(2–3)]2 exhibited poorer activity relative to [Zn(1)]2, achieving between 21–31% conversion within 8 h, contrary to reactivity trends discussed for the melt (Table 1). This activity loss could potentially be due to greater catalyst aggregation under these conditions, which limits availability of the active species. Indeed, kinetically limited dissociation was previously reported for the Al(III)-catalens, resulting in a loss in polymerisation control.35 This trend was reflected in the Mg(II)-series but appeared less prevalent, consistent with observations in the melt. Promisingly, both [Mg(1,3)]2 outperformed their Zn(II) counterpart, achieving 94% and 77% conversion within 1.5 and 8 h respectively. MALDI-ToF analysis confirmed polymer produced by [Mg(3)]2 (Table 2, entry 5) to be –OBn and –H end-capped and transesterified, consistent with GPC analysis (Mn,theo = 12000 g mol−1, Mn = 9400 g mol−1, Đ = 1.63; Table 2, entry 5) (see ESI†). Interestingly, [Mg(1)]2 also facilitated intermolecular transesterification, consistent with a 72 g mol−1 peak separation in the MALDI-ToF spectra (see ESI†), but maintained excellent polymerisation control (Mn,theo = 13650 g mol−1, Mn = 13900 g mol−1, Đ = 1.17; Table 2, entry 4). It is possible transesterification was exacerbated during polymer work up in the presence of MeOH, consistent with the melt (Table 1, entries 7–10). [Mg(1,3)]2 produced atactic polymer (Pr = 0.46–0.48), whilst [Zn(1)]2 afforded PLA with a slight heterotactic bias (Pr = 0.61). In summary, all Zn(II)- and Mg(II)-complexes exhibited good activity in the production of biocompatible atactic PLA, although non-competitive with the industry standard; Sn(Oct)2.
Polymerisation kinetics
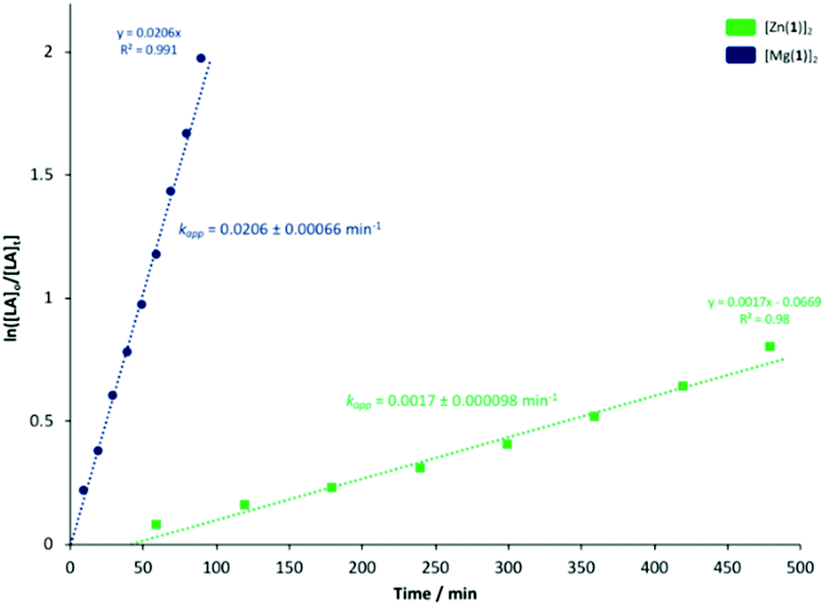
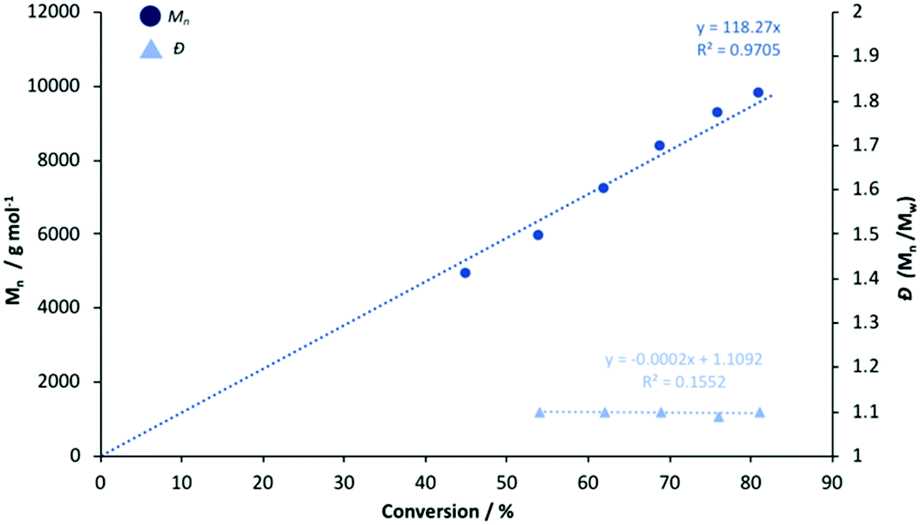
To ascertain a better understanding of metal-ligand cooperative effects on activity, a kinetic study was pursued using [Zn(1)]2 and [Mg(1)]2 as a model system. A plot of ln([LA]0/[LA]t) against time exhibited a linear relationship, indicating the reaction to be pseudo-first-order with respect to the consumption of rac-LA (Fig. 3). [Mg(1)]2 exhibited an apparent rate constant (kapp) of 0.0206 min−1, over 12 orders of magnitude higher relative to [Zn(1)]2 (kapp = 0.0017 min−1), consistent with solution results (Table 2). Indeed, [Zn(1)]2 also exhibited an induction period of ca. 40 minutes, potentially evidencing catalyst aggregation under these conditions. No induction period was observed for [Mg(1)]2, highlighting judicial choice of the metal can circumvent such limitations. It is tentatively suggested such aggregation is H-bonding in nature, although metal influence remains poorly understood. GPC analysis of the aliquots retained for [Mg(1)]2 confirmed the polymerisation to be well controlled and living (Fig. 4). This was demonstrated by a linear increase in Mn with conversion whilst maintaining narrow dispersities. A slightly lower Mr,monomer relative to ideal PLA was observed (Mr,theo = 144.12 g mol−1, Mr = 118.27 g mol−1), which could likely be attributed to minor transesterification, consistent with MALDI-ToF analysis (see ESI†).
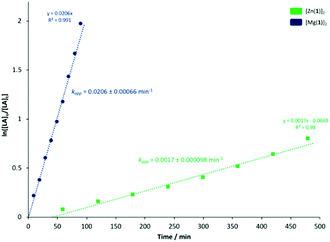 |
| | Fig. 3 Pseudo-first-order logarithmic plot for the polymerisation of rac-LA at 80 °C in toluene {[rac-LA]:[M]:[BnOH] = 100:1:1} using [Zn(1)]2 and [Mg(1)]2. N.B. [LA]0 = 0.69 mol dm−3. | |
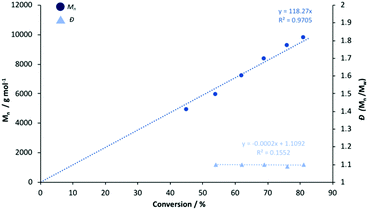 |
| | Fig. 4 Linear plot of Mn and Đ against conversion for the polymerisation of rac-LA at 80 °C in toluene {[rac-LA]:[M]:[BnOH] = 100:1:1} using [Mg(1)]2. | |
Polyester recycling
PLA degradation.
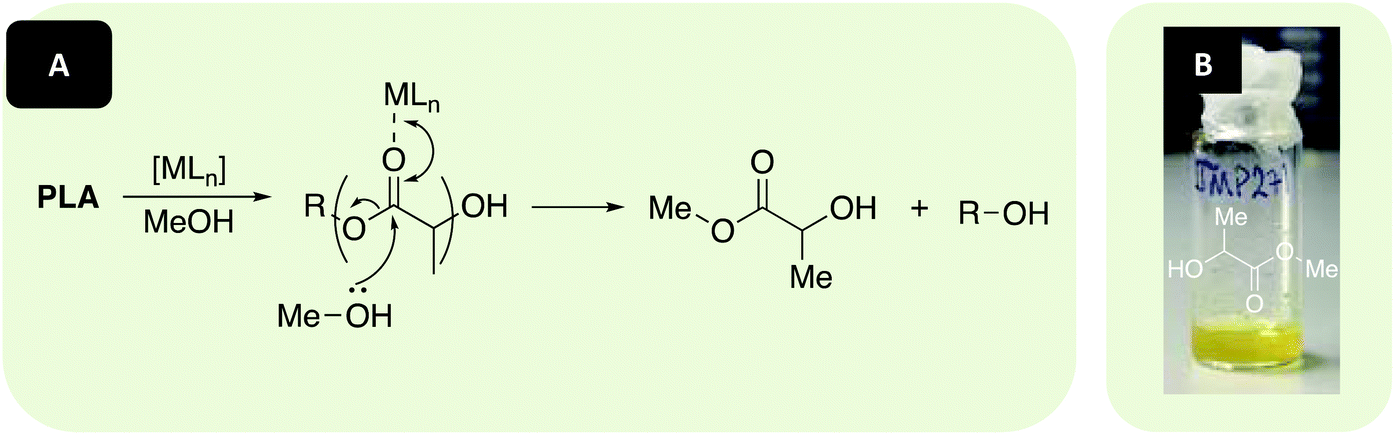
[Zn(1–3)]2 and [Mg(1,3)]2 were investigated in the metal-mediated degradation of PLA into Me-LA in solution at 80 °C (Fig. 5 and Table 3). Whilst Me-LA is a possible green solvent replacement, it is also a potentially valuable chemical to the PLA supply chain since it can be directly converted to lactide.3,91 Commercially available polymer (0.25 g, PLLA cup, Mn = 45510 g mol−1) and catalyst were dissolved in either THF or anhydrous toluene under Ar, with heat and stirring assisting dissolution. MeOH was then added and the conversion to Me-LA was determined via1H NMR analysis of the methine region (ca. δ = 4.2–5.2 ppm). The production of Me-LA has previously been shown to proceed via a two-step process through the intermediate formation of chain-end groups (see ESI†).109,123
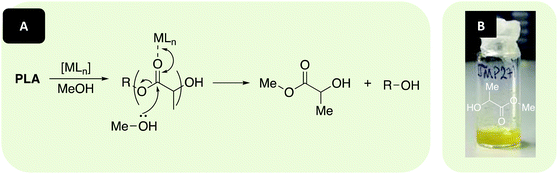 |
| | Fig. 5 (A) Metal-mediated degradation mechanism of PLA into methyl lactate (Me-LA) via transesterification with MeOH, where R denotes the growth polymer chain. (B) Example of recycled product (Me-LA). N.B. Discolouration in Me-LA product (B, liquid) originates from catalyst used ([Mg(1)]2 – yellow solid). | |
Table 3 Degradation of PLA into Me-LA using [Zn(1–3)]2 and [Mg(1,3)]2
| Catalyst |
Time/h |
T/°C |
Cat. loading/wt% |
Y
Me-LA/%
|
S
Me-LA/%
|
X
int/%
|
k
app/h−1 |
| Reaction conditions: 0.25 g of PLLA cup (Mn = 45510 g mol−1), VTHF:VMeOH = 4:1, nMeOH:nester = 7:1, [Zn(1–3)]2 = 4–8 wt% cat. loading (0.24–0.58 mol% relative to ester linkages), [Mg(1,3)]2 = 2–8 wt% cat. loading (0.13–0.57 mol% relative to ester linkages). YMe-LA, SMe-LA and Xint determined by 1H NMR upon solvent removal. Solvent: Anhydrous toluene, Vtoluene:VMeOH = 4:1. |
| [Zn(1)]2 |
8 |
80 |
8 |
19 |
28 |
68 |
— |
| 8 |
80 |
4 |
4 |
9 |
46 |
— |
| 8a |
80 |
8 |
51 |
53 |
96 |
— |
| 8a |
80 |
4 |
7 |
12 |
57 |
— |
| [Zn(2)]2 |
8 |
80 |
8 |
0 |
0 |
42 |
— |
| 8 |
80 |
4 |
0 |
0 |
20 |
— |
| 8a |
80 |
8 |
4 |
9 |
44 |
— |
| 8a |
80 |
4 |
0 |
0 |
28 |
— |
| [Zn(3)]2 |
8 |
80 |
8 |
0 |
0 |
36 |
— |
| 8a |
80 |
8 |
0 |
0 |
37 |
— |
| [Mg(1)]2 |
8 |
80 |
2 |
10 |
36 |
28 |
— |
| 8 |
80 |
4 |
64 |
66 |
97 |
0.628 |
| 8 |
80 |
8 |
31 |
56 |
55 |
0.0819 |
| [Mg(3)]2 |
8 |
80 |
2 |
13 |
38 |
34 |
— |
| 8 |
80 |
4 |
42 |
63 |
67 |
— |
| 8 |
80 |
8 |
64 |
77 |
83 |
0.265 |
Consequently, the methine groups can be categorised as internal (int), chain-end (CE) and those corresponding directly to the alkyl lactate (Me-LA). Conversion of internal methine units (Xint), methyl lactate selectivity (SMe-LA) and Me-LA yield (YMe-LA) are tabulated in Table 3 below. [Zn(1)]2 exhibited reasonably poor activity at 8 wt%, achieving 19% conversion to Me-LA within 8 h in THF with poor selectivity (Table 3, entry 1). Promisingly, superior activity and selectivity (YMe-LA = 51%, SMe-LA = 53%,) was observed upon shifting to a non-coordinating solvent, namely anhydrous toluene (Table 3, entry 3). This implies THF competes with the degrading polymeric chain with respect to coordination to the Zn(II)-centre, consistent with the near complete consumption of PLA (Xint = 96%). Interestingly, shifting to a more electron withdrawing catalen backbone in [Zn(2–3)]2 had a detrimental impact on YMe-LA, achieving 0% conversion to Me-LA under analogous conditions in THF. Whilst contrary to previous work by Payne et al.,110 this was consistent with solution polymerisation results (Table 2). Indeed, previously described solution reactivity trends were retained for PLA degradation. Since this behaviour was retained in anhydrous toluene, it is suggested the aforementioned activity loss due to possible catalyst aggregation (Table 2) likely persists under these conditions. It is possible bulky tBu substituents promote the dissociation of [Zn(1)]2 in solution, resulting in superior activity relative to [Zn(2–3)]2. [Mg(1)]2 significantly outperformed its Zn(II)-counterpart, achieving 31% conversion to Me-LA with good selectivity in THF at 8 wt% (Table 3, entry 13). Interestingly, significantly enhanced activity was realised upon decreasing the catalyst loading to 4 wt% (YMe-LA = 64%, SMe-LA = 66%, Xint = 97%), possibly evidencing a reduction in catalyst aggregation due to dilution. To improve industrial feasibility and investigate the limit of this effect, the catalyst loading was further reduced to 2 wt%. Significantly reduced YMe-LA relative to 4 wt% was observed (Table 3, entries 11 and 12), implying unavailability of the active species predominates, consistent with [Mg(3)]2 (Table 3, entry 14). In light of this, [Zn(1–3)]2 were investigated at 4 wt% in both THF and anhydrous toluene at 80 °C, although no activity enhancement was observed. [Mg(3)]2 exhibited superior activity relative to [Zn(3)]2, achieving 42% conversion to Me-LA within 8 h at 4 wt% (Table 3, entry 15). Whilst lower relative to [Mg(1)]2, enhanced YMe-LA and SMe-LA was observed at 8 wt% (Table 3, entry 16). This implies the liberated species to be inherently more active relative to [Mg(1)]2 and that possible catalyst aggregation dominates at 4 wt%. Judicial choice of the metal had previously been shown to circumvent such challenges in the solution polymerisation of rac-LA (Fig. 3). Degradation reactions using [Mg(1,3)]2 in anhydrous toluene were not pursued in alignment with the 12 principles of green chemistry.125 Overall, mass transfer limitations due to polymer particle size and stirring speeds were considered negligible based on previous work by Román-Ramírez et al.,109 which employed a homoleptic Zn(II)-complex bearing an ethylenediamine Schiff-base ligand.
PLA degradation kinetics.
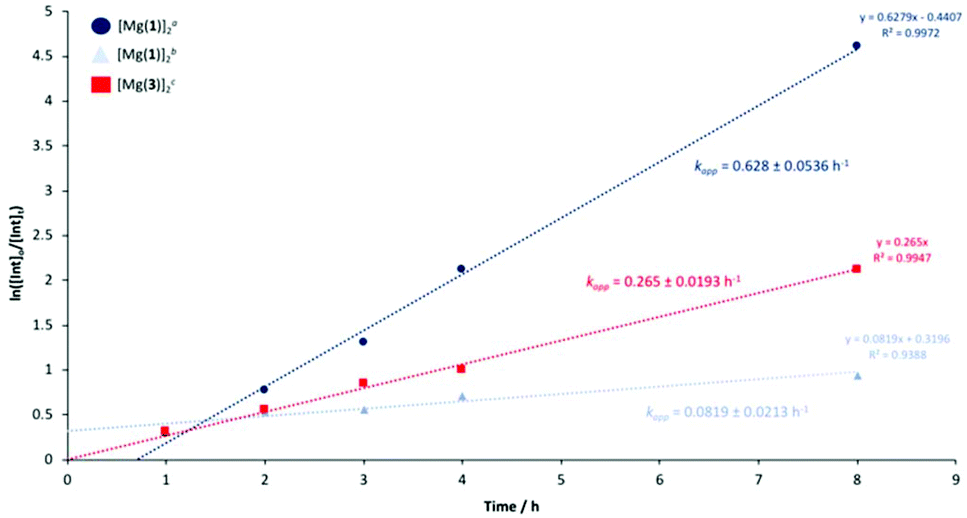
[Mg(1)]2 and [Mg(3)]2 were identified as the outstanding candidates and thus pursued for further kinetic analysis. Reaction progress was monitored hourly for the first 4 hours for 1H NMR (CDCl3) analysis of the methine region. A final aliquot was taken after 8 hours for analysis, totalling 5 data points (Fig. 6). PLA consumption was assumed to adopt pseudo-first-order kinetics in accordance to previous work by Román-Ramírez et al.109 Consequently, the gradient of the logarithmic plot is equivalent to the apparent rate constant, kapp (Table 3 and Fig. 6). [Mg(1)]2 exhibited a kapp value of 0.628 ± 0.0536 and 0.0819 ± 0.0213 h−1 at 4 and 8 wt% respectively in THF, indicating an increase in catalyst loading results in a statistically significant decrease in activity. [Mg(3)]2 was found to have a kapp value of 0.265 ± 0.0193 h−1, lower relative to [Mg(1)]2, consistent with preliminary methanolysis results (Table 3). Comparable YMe-LA values (Table 4) were observed relative to Table 3, indicating good reproducibility. Whilst promising, these kapp value remain lower compared to previously reported Zn(II)-complexes (kapp = 0.44–12.0 h−1) operating between 50 to 80 °C under analogous reaction conditions.109,110,123 However, to the best of our knowledge, [Mg(1,3)]2 represent the first example of PLA methanolysis mediated by a well-defined discrete Mg(II)-complex, operating under significantly milder conditions relative to Petrus et al.,111 who relied upon metallic Mg or Mg(nBu)2 as pre-catalysts. For [Mg(1,3)]2, inspection of the 1H NMR (Table 3, entries 12 and 16) following solvent removal revealed the formation of a new Mg(II)-species, although its identity remains unclear. It is suggested the dimeric framework dissociates in solution, affording a heteroleptic complex of the general formula Mg(1,3)L, where L could be methoxy, lactyl or higher chain oligomers, as previously described by Jones and co-workers.123 Consequently, [Mg(1,3)]2 should strictly be regarded as pre-catalysts and this can likely be extended to the remaining Zn(II)-complexes.
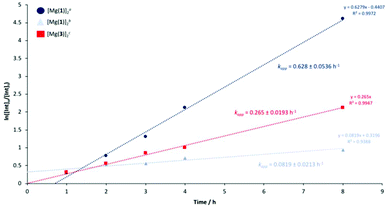 |
| | Fig. 6 Pseudo-first-order logarithmic plot for the degradation of PLLA cup using [Mg(1)]2 and [Mg(3)]2 in THF at 80 °C. a[Mg(1)]2 = 4 wt% cat. loading (0.29 mol% relative to ester linkages). b[Mg(1)]2 = 8 wt% cat. loading (0.57 mol% relative to ester linkages). c[Mg(3)]2 = 8 wt% cat. loading (0.53 mol% relative to ester linkages). | |
Table 4 PLLA cup degradation using [Mg(1,3)]2 in THF at 80 °C
| Catalyst |
Y
Me-LA/% |
k
app/h−1 |
| Reaction conditions: 0.25 g of PLLA cup (Mn = 45510 g mol−1), VTHF:VMeOH = 4:1, nMeOH:nester = 7:1. Error associated with kapp calculated using linear regression. [Mg(1)]2 = 4 wt% cat. loading (0.29 mol% relative to ester linkages). [Mg(1)]2 = 8 wt% cat. loading (0.57 mol% relative to ester linkages). [Mg(3)]2 = 8 wt% cat. loading (0.53 mol% relative to ester linkages). N.B. YMe-LA refers to maximum Me-LA conversion determined via1H NMR (CDCl3) after 8 h prior to solvent (THF) removal. |
| [Mg(1)]2a |
76 |
0.628 ± 0.0536 |
| [Mg(1)]2b |
38 |
0.0819 ± 0.0213 |
| [Mg(3)]2c |
63 |
0.265 ± 0.0193 |
PET degradation.
Presently, bio-based plastics account for ca. 1% of all processed plastics, with PLA accounting for just 13.9% of bioplastic production in 2019.126 Consequently, our attention shifted to PET, a commercial polyester widely exploited in the packaging industry, which consumed 38% of plastics produced globally in 2015, with PET accounting for 22.6% of plastic use in the sector.13 Glycolysis is the most widely used chemical recycling method for PET, characterised by cleavage of the ester bond via insertion of a glycol, commonly ethylene glycol (EG), to produce bis(2-hydroxyethyl) terephthalate (BHET) or higher alcohol derivatives. BHET can then be repolymerised to virgin PET or used as a precursor in the production of unsaturated polyester resins.127–131 In light of this, preliminary work sought to apply [Zn(1,3)]2 and [Mg(1)]2 to the glycolysis of PET. [Mg(3)]2 was not investigated due to insufficient yield. Typically, high temperatures (180–240 °C) and prolonged reaction times (0.5–8 h) in the presence of a transesterification catalyst, often a metal acetate, are required to achieve appreciable conversion. Whilst numerous metal acetate catalysts have been reported in the literature, zinc acetate is considered the benchmark.127,132
Consequently, Zn(OAc)2·2H2O (Sigma Aldrich) was chosen as an air-stable, commercially available reference. Additionally, high EG:PET (≥5:1) are used to mediate the formation of higher chain oligomers, thus favouring the formation of BHET.127 As such, a reaction temperature of 180 °C in the presence of 8 wt% catalyst and 27.5 equivalents of EG was chosen (Table 5). Two sources of PET were selected: 1. A carbonated drinks bottle (Mn ∼40000 g mol−1) and 2. Thin-films, representing waste from the manufacturing industry (Fig. 7).
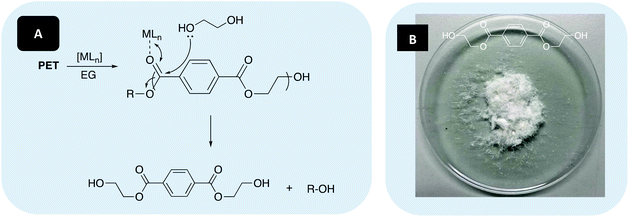 |
| | Fig. 7 (A) Metal mediated glycolysis of poly(ethylene terephthalate) (PET) into bis(2-hydroxyethyl) terephthalate (BHET) in the presence of ethylene glycol (EG), where R denotes the growth polymer chain. (B) Example of recycled product (BHET). | |
Table 5 PET degradation into BHET using selected Zn(II)- and Mg(II)-catalen complexes at 180 °C
| Catalyst |
Time/h |
T/°C |
Cat. loading/wt% |
EG/equiv. |
Y
BHET (wet) (g/%) |
H2O/equiv. |
Corrected YBHET (%) |
| Reaction conditions: 0.25 g of carbonated drinks bottle (Mn ∼40000 g mol−1), 27.5 equivalents of EG (relative to ester linkages), Ref: Zn(OAc)2·2H2O = 8 wt% cat. loading (0.02 g, 7 mol% relative to ester linkages), [Zn(1,3)]2 = 8 wt% cat. loading (0.02 g, 1.3–1.4 mol% relative to ester linkages), [Mg(1)]2 = 8 wt% cat. loading (0.02 g, 1.5 mol% relative to ester linkages). PET thin-film (0.25 g). PET (0.25 g, carbonated drinks bottle) + PVC (0.025 g, 10 wt%, Sigma Aldrich, Mn ∼22000 g mol−1). N.B. YBHET (wet) refers to the isolated yield of BHET recrystallised from deionised H2O, followed by drying at 90 °C for 3h in vacuo. Residual H2O (equiv.) was determined via1H NMR (D6-DMSO) analysis. A corrected YBHET is provided accounting for the complete removal of H2O. |
| Ref |
4 |
180 |
8 |
27.5 |
0.16 (48%) |
2.0 |
42 |
| 2a |
180 |
8 |
27.5 |
0.14 (42%) |
2.1 |
37 |
| [Zn(1)]2 |
4 |
180 |
8 |
27.5 |
0.20 (61%) |
3.3 |
49 |
| [Zn(3)]2 |
4 |
180 |
8 |
27.5 |
0.16 (48%) |
1.3 |
44 |
| [Mg(1)]2 |
3 |
180 |
8 |
27.5 |
0.20 (61%) |
2.7 |
51 |
| 0.75a |
180 |
8 |
27.5 |
0.14 (42%) |
3.5 |
34 |
| 3b |
180 |
8 |
27.5 |
0.18 (55%) |
2.4 |
46 |
Typically, a 6–12% reduction in YBHET (isolated yield) was observed on accounting for residual H2O (1.3–3.5 equivalents) after drying, and thus cannot be considered absolute. Consequently, the discussion of YBHET herein will refer to the corrected value. [Zn(1,3)]2 exhibited comparable performance to the reference, achieving between 44–49% YBHET within 4 h at 180 °C. Promisingly, [Mg(1)]2 exhibited superior activity, ascertaining the highest YBHET observed, achieving 51% within 3h under analogous conditions. For this system, tolerance stability was successfully demonstrated in the presence of 10 wt% PVC, retaining comparable YBHET (46%; Table 5, entry 7). PVC contamination as low as 100 ppm has previously been reported to adversely impact the quality of the final recycled product, owing to the production of acid catalysts that facilitate chain scission under melt reprocessing conditions (T = 160 °C) routinely employed in industry.127 Thus, this result is particularly promising from an industrial perspective. Both [Mg(1)]2 and Zn(OAc)2·2H2O exhibited superior activity on substituting the carbonated drinks bottle for thin-films, owing to superior sample dissolution. Remarkably, [Mg(1)]2 achieved 34% YBHET within 45 minutes, vastly outperforming the reference, which afforded comparable conversion within 2 h (YBHET = 37%; Table 5, entry 2). Whilst reaction times reflect the time taken to achieve complete PET dissolution, indicative of reaction completion, YBHET less than 50% were typically observed. It is possible the production of higher chain oligomers is partly responsible.127–132 In summary, [Zn(1,3)]2 and [Mg(1)]2 exhibited superior activity for PET glycolysis relative to Zn(OAc)2·2H2O, despite the wt% of the reference corresponding to a significantly higher zinc loading, highlighting the importance of structure-activity relationships.
Conclusions
A range of dimeric Zn(II)- and Mg(II)-catalen complexes were prepared and fully characterised. The production of biocompatible atactic PLA in solution and under industrially preferred solvent-free conditions was demonstrated, typically observing good activity and Mn control with a broad range of dispersities (Đ = 1.08–2.04). Mg(II)-Complexes were shown to facilitate the relatively mild methanolysis of PLA, achieving up to 64% conversion to Me-LA within 8 h at 80 °C in THF. Further kinetic analysis found [Mg(1,3)]2 to have kapp values of 0.628 ± 0.0536 {4 wt% cat. loading} and 0.265 ± 0.0193 h−1 {8 wt% cat. loading} respectively. For [Mg(1)]2, increasing the catalyst loading to 8 wt% resulted in a statistically significant reduction in activity (0.0819 ± 0.0213 h−1), potentially evidencing catalyst aggregation, which appeared prevalent in the Zn(II)-complexes. Preliminary work extended polymer scope to PET from various sources, demonstrating catalyst versatility.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We wish to thank the EPSRC for funding and the University of Bath and MC2 for use of their analysis facilities. We would like to thank the EPSRC for funding (EP/L016354/1) for a PhD studentship to J. P. and (EP/P016405/1) for P. M. We thank Avery Dennison for kindly providing waste PET thin-films used in this study.
References
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
-
PlasticsEurope, Plastics – the Facts 2019. An Analysis of European Plastics Production, Demand and Waste Data, 2019, https://www.plasticseurope.org/application/files/9715/7129/9584/FINAL_web_version_Plastics_the_facts2019_14102019.pdf, (Accessed: 13th October 2020) Search PubMed.
- J. Payne, P. McKeown and M. D. Jones, Polym. Degrad. Stab., 2019, 165, 170–181 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 1–5 Search PubMed.
- R. C. Thompson, C. J. Moore, F. S. vom Saal and S. H. Swan, Philos. Trans. R. Soc., B, 2009, 364, 2153–2166 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS.
-
Ellen MacArthur Foundation, The New Plastics Economy: Rethinking the future of plastics, 2016, https://www.ellenmacarthurfoundation.org/publications/the-new-plastics-economy-rethinking-the-future-of-plastics, (Accessed: 7th January 2019) Search PubMed.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady and R. Na, Science, 2015, 347, 768–771 CrossRef CAS.
- L. Lebreton, B. Slat, F. Ferrari, B. Sainte-Rose, J. Aitken, R. Marthouse, S. Hajbane, S. Cunsolo, A. Schwarz, A. Levivier, K. Noble, P. Debeljak, H. Maral, R. Schoeneich-Argent, R. Brambini and J. Reisser, Sci. Rep., 2018, 8, 4666 CrossRef CAS.
- E. T. H. Vink, K. R. Rábago, D. A. Glassner, B. Springs, R. P. O'Connor, J. Kolstad and P. R. Gruber, Macromol. Biosci., 2004, 4, 551–564 CrossRef CAS.
- E. T. H. Vink, D. A. Glassner, J. Kolstad, R. J. Wooley and R. P. O'Connor, Biotechnology, 2007, 3, 58–81 CAS.
- J. Lunt, Polym. Degrad. Stab., 1998, 59, 145–152 CrossRef CAS.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- H. R. Kricheldorf, I. Kreiser-Saunders and A. Stricker, Macromolecules, 2000, 33, 702–709 CrossRef CAS.
- N. Spassky, M. Wisniewski, C. Pluta and A. LeBorgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- Z. Y. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS.
- N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS.
- N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem. – Eur. J., 2007, 13, 4433–4451 CrossRef CAS.
- H.-L. Chen, S. Dutta, P.-Y. Huang and C.-C. Lin, Organometallics, 2012, 31, 2016–2025 CrossRef CAS.
- E. L. Whitelaw, G. Loraine, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 11469–11473 RSC.
- A. Pilone, K. Press, I. Goldberg, M. Kol, M. Mazzeo and M. Lamberti, J. Am. Chem. Soc., 2014, 136, 2940–2943 CrossRef CAS.
- P. McKeown, M. G. Davidson, G. Kociok-Köhn and M. D. Jones, Chem. Commun., 2016, 52, 10431–10434 RSC.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 CrossRef CAS.
- H. Du, A. H. Velders, P. J. Dijkstra, J. Sun, Z. Zhong, X. Chen and J. Feijen, Chem. – Eur. J., 2009, 15, 9836–9845 CrossRef CAS.
- J. Feijen, Chem. – Eur. J., 2009, 15, 9836–9845 CrossRef.
- K. Press, I. Goldberg and M. Kol, Angew. Chem., Int. Ed., 2015, 54, 14858–14861 CrossRef CAS.
- R. Hador, A. Botta, V. Venditto, S. Lipstman, I. Goldberg and M. Kol, Angew. Chem., Int. Ed., 2019, 58, 14679–14685 CrossRef CAS.
- S. Gesslbauer, H. Cheek, A. J. P. White and C. Romain, Dalton Trans., 2018, 47, 10410–10414 RSC.
- S. Gesslbauer, R. Savela, Y. Chen, A. J. P. White and C. Romain, ACS Catal., 2019, 9, 7912–7920 CrossRef CAS.
- J. Payne, P. McKeown, G. Kociok-Köhn and M. D. Jones, Chem. Commun., 2020, 56, 7163–7166 RSC.
- A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn, M. F. Mahon, A. F. Johnson, P. Khunkamchoo, S. L. Roberts and S. S. F. Wong, Macromolecules, 2006, 39, 7250–7257 CrossRef CAS.
- A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293–1295 RSC.
- A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293–1295 RSC.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
- E. Sergeeva, J. Kopilov, I. Goldberg and M. Kol, Inorg. Chem., 2010, 49, 3977–3979 CrossRef CAS.
- S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783–4790 CrossRef CAS.
- A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698–704 CrossRef CAS.
- J.-C. Buffet and J. Okuda, Polym. Chem., 2011, 2, 2758–2763 RSC.
- C. Romain, B. Heinrich, S. B. Laponnaz and S. Dagorne, Chem. Commun., 2012, 48, 2213–2215 RSC.
- X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS.
- M. D. Jones, S. L. Hancock, P. McKeown, P. M. Schafer, A. Buchard, L. H. Thomas, M. F. Mahon and J. P. Lowe, Chem. Commun., 2014, 50, 15967–15970 RSC.
- M. D. Jones, L. Brady, P. McKeown, A. Buchard, P. M. Schafer, L. H. Thomas, M. F. Mahon, T. J. Woodman and J. P. Lowe, Chem. Sci., 2015, 6, 5034–5039 RSC.
- E. L. Whitelaw, M. F. Mahon and M. D. Jones, Inorg. Chem., 2010, 49, 7176–7181 CrossRef CAS.
- E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004–10006 RSC.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS.
- M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2005, 44, 8004–8010 CrossRef CAS.
- H.-Y. Chen, H.-Y. Tang and C.-C. Lin, Macromolecules, 2006, 39, 3745–3752 CrossRef CAS.
- J. Borner, U. Florke, K. Huber, A. Doring, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2009, 15, 2362–2376 CrossRef.
- D. J. Darensbourg and O. Karroonnirun, Inorg. Chem., 2010, 49, 2360–2371 CrossRef CAS.
- J. Börner, I. dos Santos Vieira, A. Pawlis, A. Döring, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2011, 17, 4507–4512 CrossRef.
- H. Wang and H. Ma, Chem. Commun., 2013, 49, 8686–8688 RSC.
- C. Fliedel, D. Vila-Viçosa, M. J. Calhorda, S. Dagorne and T. Avilés, ChemCatChem, 2014, 6, 1357–1367 CrossRef CAS.
- H. Wang, Y. Yang and H. Ma, Macromolecules, 2014, 47, 7750–7764 CrossRef CAS.
- Y. Yang, H. Wang and H. Ma, Inorg. Chem., 2015, 54, 5839–5854 CrossRef CAS.
- T. Rosen, Y. Popowski, I. Goldberg and M. Kol, Chem. – Eur. J., 2016, 22, 11533–11536 CrossRef CAS.
- P. M. Schäfer, M. Fuchs, A. Ohligschläger, R. Rittinghaus, P. McKeown, E. Akin, M. Schmidt, A. Hoffmann, M. A. Liauw, M. D. Jones and S. Herres-Pawlis, ChemSusChem, 2017, 10, 3547–3556 CrossRef.
- D. E. Stasiw, A. M. Luke, T. Rosen, A. B. League, M. Mandal, B. D. Neisen, C. J. Cramer, M. Kol and W. B. Tolman, Inorg. Chem., 2017, 56, 14366–14372 CrossRef CAS.
- S. Abbina and G. Du, ACS Macro Lett., 2014, 3, 689–692 CrossRef CAS.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS.
- M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS.
- C. Kan, J. Hu, Y. Huang, H. Wang and H. Ma, Macromolecules, 2017, 50, 7911–7919 CrossRef CAS.
- P. McKeown, S. N. McCormick, M. F. Mahon and M. D. Jones, Polym. Chem., 2018, 9, 5339–5347 RSC.
- Al. Hermann, S. Hill, A. Metz, J. Heck, A. Hoffmann, L. Hartmann and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2020, 59(48), 21778–21784 CrossRef CAS.
- J. Kasperczyk and M. Bero, Polymer, 2000, 41, 391–395 CrossRef CAS.
- L. F. Sánchez-Barba, A. Garcés, M. Fajardo, C. Alonso-Moreno, J. Fernández-Baeza, A. Otero, A. Antiñolo, J. Tejeda, A. Lara-Sánchez and M. I. López-Solera, Organometallics, 2007, 26, 6403–6411 CrossRef.
- V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2009, 9820–9827 RSC.
- C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832–4846 RSC.
- L. Wang and H. Ma, Macromolecules, 2010, 43, 6535–6537 CrossRef CAS.
- L. F. Sánchez-Barba, A. Garcés, J. Fernández-Baeza, A. Otero, C. Alonso-Moreno, A. Lara-Sánchez and A. M. Rodríguez, Organometallics, 2011, 30, 2775–2789 CrossRef.
- H.-J. Chuang, H.-L. Chen, J.-L. Ye, Z.-Y. Chen, P.-L. Huang, T.-T. Liao, T.-E. Tsai and C.-C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 696–707 CrossRef CAS.
- A. Garcés, L. F. Sánchez-Barba, J. Fernández-Baeza, A. Otero, M. Honrado, A. Lara-Sánchez and A. M. Rodríguez, Inorg. Chem., 2013, 52, 12691–12701 CrossRef.
- W. Yi and H. Ma, Inorg. Chem., 2013, 52, 11821–11835 CrossRef CAS.
- M. J. Walton, S. J. Lancaster and C. Redshaw, ChemCatChem, 2014, 6, 1892–1898 CrossRef CAS.
- K. Devaine-Pressing, J. H. Lehr, M. E. Pratt, L. N. Dawe, A. A. Sarjeant and C. M. Kozak, Dalton Trans., 2015, 44, 12365–12375 RSC.
- H. Wang, J. Guo, Y. Yang and H. Ma, Dalton Trans., 2016, 45, 10942–10953 RSC.
- P. McKeown, J. Brown-Humes, M. G. Davidson, M. F. Mahon, T. J. Woodman and M. D. Jones, Dalton Trans., 2017, 46, 5048–5057 RSC.
- M. H. Chisholm, K. Choojun, A. S. Chow, G. Fraenkel and J. C. Gallucci, Inorg. Chem., 2013, 52, 11302–11310 CrossRef CAS.
- M. H. Chisholm, K. Choojun, J. C. Gallucci and P. M. Wambua, Chem. Sci., 2012, 3, 3445–3457 RSC.
- M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785–2794 CrossRef CAS.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS.
- M. Hong and E. X.-Y. Chen, Green Chem., 2017, 9, 3692–3706 RSC.
- P. McKeown and M. D. Jones, Sustainable Chem., 2020, 1, 1–22 CrossRef.
- C. T. Bowmer, R. N. Hooftman, A. O. Hanstveit, P. W. M. Venderbosch and N. van der Hoeven, Chemosphere, 1998, 37, 1317–1333 CrossRef CAS.
- C. S. M. Pereira, V. M. T. M. Silva and A. E. Rodrigues, Green Chem., 2011, 13, 2658–2671 RSC.
- M. Dusselier, P. V. Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- Y. Fan, C. Zhou and X. Zhu, Catal. Rev. Sci. Eng., 2009, 51, 293–324 CrossRef CAS.
- V. Piemonte, S. Sabatini and F. Gironi, J. Polym. Environ., 2013, 21, 640–647 CrossRef CAS.
- F. M. Lamberti, L. A. Román-Ramírez, P. Mckeown, M. D. Jones and J. Wood, Processes, 2020, 8, 738 CrossRef CAS.
- H. Tsuji, T. Saeki, T. Tsukegi, H. Daimon and K. Fujie, Polym. Degrad. Stab., 2008, 93, 1956–1963 CrossRef CAS.
- V. Piemonte and F. Gironi, J. Polym. Environ., 2013, 21, 313–318 CrossRef CAS.
-
P. Coszach, J.-C. Bogaert and J. Willocq, US Pat, 8431683B2, 2013 Search PubMed.
- C. F. VanNostrum, T. F. J. Veldhuis, G. W. Bos and W. E. Hennink, Polymer, 2004, 45, 6779–6787 CrossRef CAS.
- H. Tsuji, H. Daimon and K. Fujie, Biomacromolecules, 2003, 4, 835–840 CrossRef CAS.
- F. Codari, S. Lazzari, M. Soos, G. Storti, M. Morbidelli and D. Moscatelli, Polym. Degrad. Stab., 2012, 97, 2460–2466 CrossRef CAS.
- K. Odelius, A. Höglund, S. Kumar, M. Hakkarainen, A. K. Ghosh, N. Bhatnagar and A. C. Albertsson, Biomacromolecules, 2011, 12, 1250–1258 CrossRef CAS.
- K. Hirao, Y. Nakatsuchi and H. Ohara, Polym. Degrad. Stab., 2010, 95, 925–928 CrossRef CAS.
-
L. D. Brake, US Pat, 5264617, 1993 Search PubMed.
- X. Song, X. Zhang, H. Wang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2013, 98, 2760–2764 CrossRef CAS.
- X. Song, H. Wang, X. Zheng, F. Liu and S. Yu, J. Appl. Polym. Sci., 2014, 131, 40817–40822 CrossRef.
- C. Fliedel, D. Vila-Viçosa, M. J. Calhorda, S. Dagorne and T. Avilés, ChemCatChem, 2014, 6, 1357–1367 CrossRef CAS.
- E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004–10006 RSC.
- L. A. Román-Ramírez, P. Mckeown, M. D. Jones and J. Wood, ACS Catal., 2019, 9, 409–416 CrossRef.
- J. Payne, P. McKeown, M. F. Mahon, E. A. C. Emanuelsson and M. D. Jones, Polym. Chem., 2020, 11, 2381–2389 RSC.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- F. A. Leibfarth, N. Moreno, A. P. Hawker and J. D. Shand, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4814–4822 CrossRef CAS.
- F. Nederberg, E. F. Connor, T. Glausser and J. L. Hedrick, Chem. Commun., 2001, 2066–2067 RSC.
- P. McKeown, M. Kamran, M. G. Davidson, M. D. Jones, L. A. Román-Ramírez and J. Wood, Green Chem., 2020, 22, 3721–3726 RSC.
- A. C. Sanchéz and R. S. Collinson, Eur. Polym. J., 2011, 47, 1970–1976 CrossRef.
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 1–9 Search PubMed.
- E. M. Krall, T. W. Klein, R. J. Andersen, A. J. Nett, R. W. Glasgow, D. S. Reader, B. C. Dauphinais, S. P. Mc Ilrath, A. A. Fischer, M. J. Carney, D. J. Hudson and N. J. Robertson, Chem. Commun., 2014, 50, 4884–4887 RSC.
- T.-O. Kindler, C. Alberti, E. Fedorenko, N. Santangelo and S. Enthaler, ChemistryOpen, 2020, 9, 401–404 CrossRef CAS.
- L. Monsigny, J.-C. Berthet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS.
- H. Liu, X. Song, F. Liu, S. Liu and S. Yu, J. Polym. Res., 2015, 22, 135–141 CrossRef.
- C. Alberti, N. Damps, R. R. R. Meißner, M. Hofmann, D. Rijono and S. Enthaler, Adv. Sustainable Syst., 2020, 4, 1900081 CrossRef CAS.
- M. Hofmann, C. Alberti, F. Scheliga, R. R. R. Meißner and S. Enthaler, Polym. Chem., 2020, 11, 2625–2629 RSC.
- P. McKeown, L. A. Román-Ramírez, S. Bates, J. Wood and M. D. Jones, ChemSusChem, 2019, 12, 5233–5238 CrossRef CAS.
-
A. J. Hunt, T. J. Farmer and J. H. Clark, Chapter 1 Elemental Sustainability and the Importance of Scarce Element Recovery, in Element Recovery and Sustainability, ed. A. Hunt, RSC, 2013, ch. 1, pp. 1–28 Search PubMed.
-
ACS, 12 Principles of Green Chemistry, https://www.acs.org/content/acs/en/greenchemistry/principles/12-principles-of-green-chemistry.html, (Accessed: 19th October 2020) Search PubMed.
- European Bioplastics, Facts and Figures, https://docs.european-bioplastics.org/publications/EUBP_Facts_and_figures.pdf, (Accessed: 20th October 2020).
- F. M. Lamberti, L. A. Román-Ramírez and J. Wood, J. Polym. Environ., 2020, 28, 2551–2571 CrossRef CAS.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8–25 CrossRef CAS.
- S. M. Al-Salem, P. Lettieri and J. Baeyens, Waste Manage., 2009, 29, 2625–2643 CrossRef CAS.
- G. P. Karayannidis and D. S. Achilias, Macromol. Mater. Eng., 2007, 292, 128–146 CrossRef CAS.
- D. Paszun and T. Spychaj, Ind. Eng. Chem. Res., 1997, 36, 1373–1383 CrossRef CAS.
- K. R. Della Chiaie, F. R. McMahon, E. J. Williams, M. J. Price and A. P. Dove, Polym. Chem., 2020, 11, 1450–1453 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of the experimental protocols with selected spectra and raw data. CCDC 2041361–2041367. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0py01519a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Oliver
Driscoll
b,
Gabriele
Kociok-Köhn
b,
Oliver
Driscoll
b,
Gabriele
Kociok-Köhn