
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of polarity on the molecular exchange dynamics in imine-based covalent adaptable networks†
Sybren K.
Schoustra
 ,
Timo
Groeneveld
and
Maarten M. J.
Smulders
*
,
Timo
Groeneveld
and
Maarten M. J.
Smulders
*
Laboratory of Organic Chemistry, Wageningen University, Stippeneng 4, 6708 WE Wageningen, The Netherlands. E-mail: maarten.smulders@wur.nl
First published on 9th December 2020
Abstract
Covalent adaptable networks (CANs) are a rising type of polymeric materials that consist of covalently crosslinked polymer chains, but with the inclusion of dynamic covalent bonds, and that can perform bond exchange reactions under equilibrium control. The dynamic behaviour of these exchange reactions within a polymer matrix has been established to be a key parameter in the control of the material properties. Therefore, in order to fully control the macroscopic material properties of CANs, understanding the underlying molecular exchange processes of these dynamic covalent bonds is essential. In this work, we studied the effect of polarity in polyimine-based CANs, and considered not only the network response itself, but also the – so far often overlooked – effect on the exchange dynamics. By combining results from kinetic studies and material analysis we were firstly able to show a distinct correlation between the presence of polar domains in the molecular structure and the thermal and dynamic mechanical properties of the materials. More importantly, the presence of polar domains also greatly affected the exchange kinetics of the dynamic imine bonds. On the molecular level, we showed that the imine exchange could be greatly enhanced (up to 20 times) when polar groups were present near the reactive imine species. As a result, in our polymer materials we established a tuneable range of phase transition temperatures from glass-to-rubber and rubber-to-liquid over roughly 100 °C as a result of either presence or absence of polar groups in the polymer matrix. Furthermore, detailed analysis in the stress relaxation behaviour of the polyimine materials revealed three relaxation processes, which we could attribute to the relaxation in network topology, to the imine exchange on a local level, and to the imine exchange as a result of diffusion through the polymer network. From this analysis we were also able to illustrate the effect of polarity on the polymer network to each of the three relaxation mechanisms.
Introduction
Vitrimer-like materials are a relatively new type of polymer that exhibit the strength of thermosets via a crosslinked network structure, but are still able to be recycled, reshaped, and even self-healed, as a result of dynamic covalent bonds within their molecular structure.1–5 These dynamic covalent bonds can perform bond exchange reactions under equilibrium control6,7 to construct covalent adaptable networks (CANs) when built into a polymer matrix.8–10 By stimulating the exchange reactions of the dynamic covalent bonds, for example by introduction of heat or light, a molecular flow is introduced in the material.11 Several types of bond exchange reactions have been studied over the last decade, such as transesterifications,1,12 boronic ester exchange,13–16 1,2,3-triazolium exchange,17,18 diketoenamine exchange,19,20 (alkyl)urea exchange,21–23 urethane exchange,24–28 thiol–ene/yne exchange,29,30 and imine exchange.31–34 Individually, each type of bond exchange reaction possesses different dynamics according to the mechanism of the exchange reaction.35 Therefore, CANs can be synthesised with selective properties based on the type of bond exchange reaction. However, not only the type of bond exchange reaction is determinative for the dynamic (mechanical) response of the material, but also the composition of the polymer matrix plays a definitive role,36 which includes for example the crosslinking density37 or steric effects.22 For many polymeric materials, the effect of such network characteristics has been evaluated for their influence on the material properties at the macroscopic level.21,24,38 However, as the dynamic mechanical properties of these materials are directly influenced by the underlying molecular exchange reaction,22,39,40 further understanding of the specific effects of polymer composition on the molecular bond exchange of dynamic covalent bonds in CANs is still essential.36,41Tailoring the flexibility of polymer chains is a classic example of a commonly applied parameter to tune material properties, and a conventional approach for enhancing this flexibility is by incorporating ethylene oxide (EO) moieties in the polymer,42 as the energy barrier for rotation of the C–O bond in ethers is considerably lower than for C–C bond rotation in alkanes. For this reason, ethylene oxides and similar ether moieties are frequently found in CANs.36,43–47 One factor that is however mainly overlooked when it comes to these ether moieties is the concomitant induced polar effect on the polymer network (dynamics), which was previously reported for materials that rely on ion transport.48,49 For example, in the study by Zhao and co-workers, the differences between polar EO linkers and apolar alkyl linkers were noticeable as a significant decrease in the glass transition temperature (Tg), as well as an increased conductivity for the polar EO-containing materials.49 Furthermore, in a study by Polgar and co-workers on Diels–Alder-based CANs, polarity induced effects as a result of introduced acetate groups were shown to affect cluster formation within the material.50 Also in recent work on vitrimer-like anilinium salt CANs, solvation effects as a result of polar groups have been proposed.51 These works thus suggest that polarity differences in CANs can have greater consequences to the dynamic behaviour on both macroscopic and molecular level than is typically assumed. Additionally, such polar effects could play a distinct role in the kinetics of the bond exchange reactions in CANs, which is a key parameter to their dynamic features.22,34
To investigate the role of polarity-induced effects by EO moieties, we made use of a CAN consisting of dynamic imine bonds, as we recently reported that the imine exchange was very sensitive to electronic effects and charge distribution at the imine bond.34 Furthermore, imines are favourable materials as they can be readily synthesised from a condensation reaction between an aldehyde and amine at room temperature, without a catalyst, and with water as the only side product.52,53 While earlier work on polyimine CANs already described the effect of solvent polarity,54,55 and moisture sensitivity,56 the quantification of polar effects in the polymer matrix itself on the imine exchange has not been addressed in great detail yet. Therefore, in this study we synthesised different polyimine materials with varying polar, EO-based moieties (also in different components) of the polymer network (Scheme 1). With a combination of kinetic studies using small-molecule analogues by NMR analysis, and temperature-dependent rheology experiments on the network polymers, we were able to dissect the effect of the polarity in the material on both the macroscopic level, and on the molecular level of the bond exchange. Furthermore, we obtained a better understanding in the relaxation behaviour of CANs, as we found the stress relaxation process to proceed in three individual phases, which we could attribute to (1) chain distribution within the polymer network, (2) imine exchange at a local level, and (3) imine exchange as a result of diffusion through the polymer matrix.
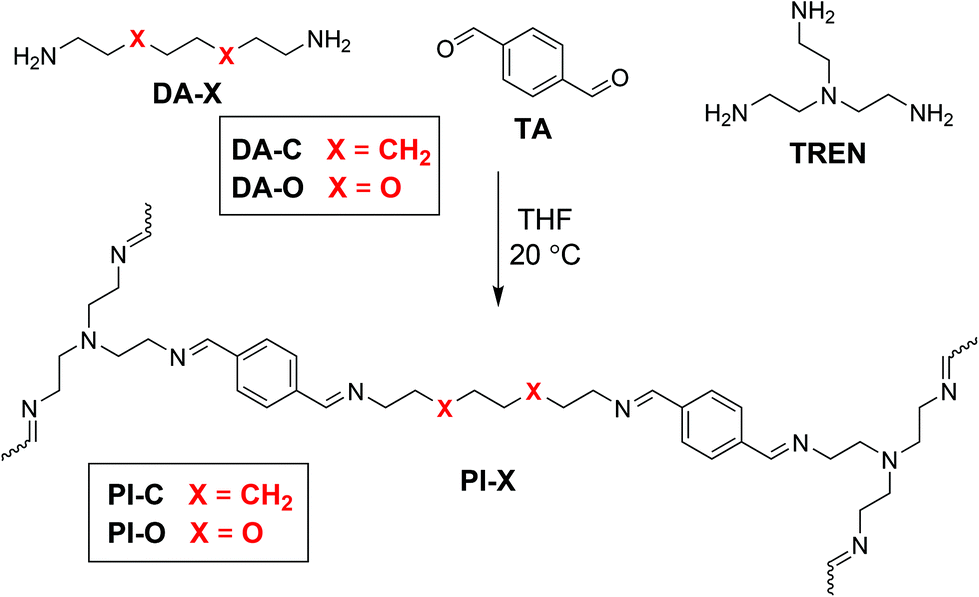 | ||
| Scheme 1 Reaction scheme for the formation of dynamic polyimine CANs from terephthalaldehyde (TA), tris(2-aminoethyl)amine (TREN) and a tuneable dianiline (DA-X) with built-in domains being either polar (X = O) or apolar (X = CH2) of nature. All materials were synthesised at room temperature, followed by drying in vacuo at 50 °C. | ||
Results & discussion
Small-molecule bond exchange studies
For the initial kinetic evaluation of the bond exchange reaction, two same-sized diamine monomers (DA-X) were selected (Scheme 1). The first consisted of a fully carbon octyl chain (DA-C), and the second included two oxygen atoms on the 3′ and 6′ position of the chain (DA-O) to represent the EO moiety. Once the amines have reacted with an appropriate aldehyde, imines are formed. These imines can then perform bond exchange reactions via either a transimination reaction with a primary amine, or via imine metathesis with another imine.53 In this section, the study of the kinetics of both these exchange reactions will be divided in four parts: (1) transimination for which the tuneable monomer acts as the nucleophile, (2) transimination for which the tuneable monomer acts as the electrophile, (3) metathesis for which the tuneable monomer acts as the nucleophile, and (4) metathesis for which the tuneable monomer acts as the electrophile (Scheme 2). The material that is added in excess acts as the attacking moiety. The excess is required to obtain a pseudo first-order reaction towards formation of the new imine, as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio would result in an equilibrium between starting compound and product. In the transimination reactions (Scheme 2A and B) the primary amine acts as the nucleophile, which will attack on the C end of the imine bond.57 For the imine metathesis reactions (Scheme 2C and D) the material that is added in excess is considered as the attacking party (nucleophile), as the exchange reaction of the starting material with the excess material will be the most prominent until new equilibrium is reached.
:1 ratio would result in an equilibrium between starting compound and product. In the transimination reactions (Scheme 2A and B) the primary amine acts as the nucleophile, which will attack on the C end of the imine bond.57 For the imine metathesis reactions (Scheme 2C and D) the material that is added in excess is considered as the attacking party (nucleophile), as the exchange reaction of the starting material with the excess material will be the most prominent until new equilibrium is reached.
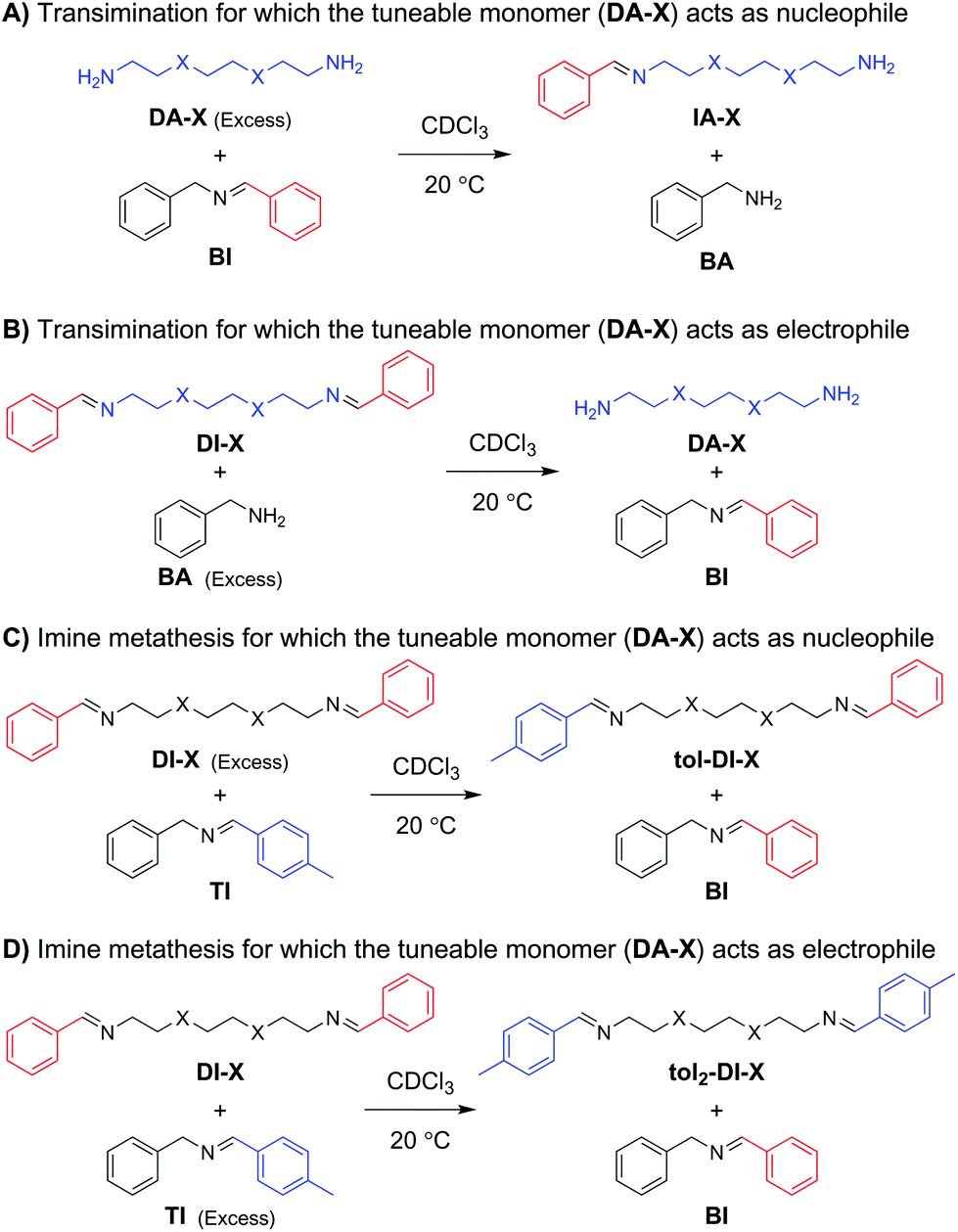 | ||
| Scheme 2 Overview of imine exchange reactions. (A) Transimination with DA-X as nucleophile, (B) transimination with DI-X as electrophile, (C) imine metathesis with DI-X as nucleophile, and (D) imine metathesis with DI-X as electrophile. | ||
For the first transimination reaction, in which DA-X is the nucleophile (Scheme 2A), a benzyl imine (BI) was synthesised and dissolved in CDCl3. An excess of DA-X was then added to push the reaction towards formation of the imine on the tuneable diamine chain (IA-X) and freeing the benzylamine (BA) from the starting benzyl imine (BI) in a pseudo first-order fashion. The conversion over time was followed using 1H NMR by integration of the imine signals of BI and IA-X. The data (Fig. 1A) were fitted using the equation for first order reaction kinetics to obtain the reaction rate constant (k) for the exchange reaction (see ESI†). The determined k values for all exchange reactions have been combined in Table 1.
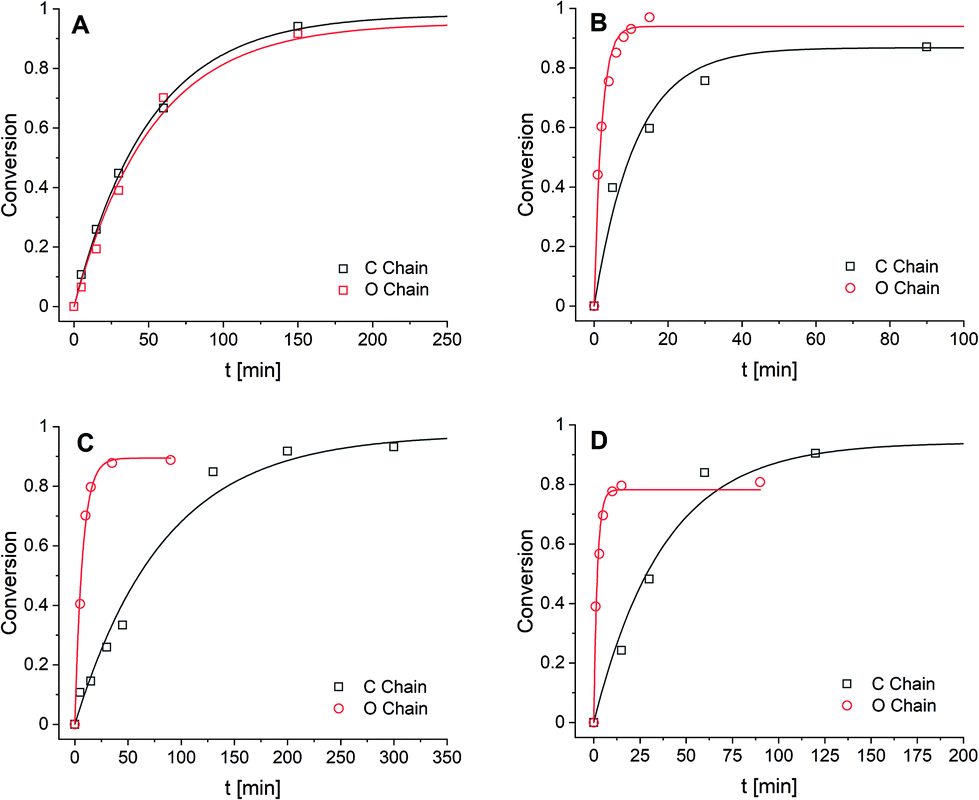 | ||
| Fig. 1 Conversion as function of time for (A) transimination with DA-X as nucleophile, (B) transimination with DI-X as electrophile, (C) imine metathesis with DI-X as nucleophile, and (D) imine metathesis with DI-X as electrophile. Data points were fitted according to first-order kinetic model. Note the different timescale on the x-axis for each plot. | ||
| X | Transimination (nucleophilic) | Transimination (electrophilic) | Metathesis (nucleophilic) | Metathesis (electrophilic) |
|---|---|---|---|---|
| CH2 | 0.33 ± 0.01 | 1.68 ± 0.33 | 0.20 ± 0.02 | 0.44 ± 0.07 |
| O | 0.32 ± 0.03 | 8.88 ± 0.92 | 2.30 ± 0.17 | 8.69 ± 1.20 |
For the second transimination reaction, in which DI-X is the electrophile (Scheme 2B), imines DI-C and DI-O were first synthesised. The imines were then dissolved in CDCl3 and an excess of benzylamine (BA) was added to push the reaction towards the formation of the benzyl imine (BI) and transformation of the imines of the tuneable chains back to amine monomer (DA-X). The conversion over time (Fig. 1B) was followed using 1H NMR by integration of the imine signals of DI-X and BI. Reaction rate constants (k) were obtained using the same procedure as before and are also presented in Table 1.
For the first metathesis reaction, in which DI-X acts as nucleophile (Scheme 2C), a toluene imine (TI) was first synthesised and dissolved in CDCl3. An excess of the tuneable imine chain (DI-X) derived from benzaldehyde was then added to push the reaction towards the formation of the benzyl imine (BI). The conversion was followed over time (Fig. 1C), and k (Table 1) was determined following the same procedure as before.
For the second metathesis reaction, in which DI-X acts as electrophile (Scheme 2D), DI-X was dissolved in CDCl3 and reacted with an excess of toluene imine TI to push the reaction towards the formation of the bis-toluene imine chain (tol2-DI-X). The conversion was followed over time (Fig. 1D), and k (Table 1) was determined following the same procedure as before.
From the combined data (in Table 1) we observed that for the transimination reaction in which the DA-X acts as a nucleophile, that the k values were similar for the apolar and polar compound, which thus implies that the presence of the more polar EO moiety in the material has no observable effect on the reaction rate. However, for the transimination in which DI-X acts as electrophile, we observed that the ether-containing material yielded a five-fold higher reaction rate. These results together suggest that the reactivity of the amine is not affected by the presence or absence of the polar EO moiety in the material, but the reactivity of the imine is affected: the presence of the ether moiety enhances the reaction rate five-fold compared to the alkyl chain. The observation that imines are more affected by the polar effect than the amines might be explained by their prominent charge separation via the (conjugated) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond. The transimination reaction can also be catalysed by trace amount of acid (which could even be water) via protonation of the imine nitrogen before formation of the aminal intermediate, and can therefore result in faster exchange.57 As this charged species would be stabilised better in polar media, in addition to the availability of oxygen lone pairs to transfer the H+, this could explain the enhanced reaction rate for the EO-containing material. This acid-catalysed reaction would also only affect the reactivity of the imine, which could explain why no difference in reaction rate was observed for different amines. For the metathesis reactions we observed that the reaction rate increased significantly (more than 10-fold) for the polar, EO-containing linker for both the nucleophilic and the electrophilic reaction. Furthermore, we observed that the electrophilic effect (∼20-fold rate enhancement) was slightly more prominent than the nucleophilic effect (∼10-fold rate enhancement), which suggests that the imines of the more polar EO-containing molecules are somewhat more prone to being attacked relative to performing the attack. Just as for the transimination reaction, trace amounts of acid could catalyse the metathesis, and the same effects regarding polarity in the materials could be envisioned. Additionally, trace amounts of (unreacted) primary amine could enhance the reaction rate via a series of fast, uncatalysed transimination reactions producing metathetic products.58,59
N double bond. The transimination reaction can also be catalysed by trace amount of acid (which could even be water) via protonation of the imine nitrogen before formation of the aminal intermediate, and can therefore result in faster exchange.57 As this charged species would be stabilised better in polar media, in addition to the availability of oxygen lone pairs to transfer the H+, this could explain the enhanced reaction rate for the EO-containing material. This acid-catalysed reaction would also only affect the reactivity of the imine, which could explain why no difference in reaction rate was observed for different amines. For the metathesis reactions we observed that the reaction rate increased significantly (more than 10-fold) for the polar, EO-containing linker for both the nucleophilic and the electrophilic reaction. Furthermore, we observed that the electrophilic effect (∼20-fold rate enhancement) was slightly more prominent than the nucleophilic effect (∼10-fold rate enhancement), which suggests that the imines of the more polar EO-containing molecules are somewhat more prone to being attacked relative to performing the attack. Just as for the transimination reaction, trace amounts of acid could catalyse the metathesis, and the same effects regarding polarity in the materials could be envisioned. Additionally, trace amounts of (unreacted) primary amine could enhance the reaction rate via a series of fast, uncatalysed transimination reactions producing metathetic products.58,59
In conclusion, from the small-molecule kinetic studies we could clearly observe that imines of the more polar, EO-containing molecules showed significantly higher imine exchange rates, compared to the apolar, alkyl-linked molecules. This prompted us to investigate whether these differences in molecular exchange kinetics would also manifest themselves when these exchangeable imine groups are incorporated into a polymer matrix, by revealing different dynamic and/or thermal material properties dependent on the nature (i.e. polarity) of the used linker.
Thermal properties of polyimine networks
Initially the thermal properties of the polyimine materials were studied. To this end, the polymers were synthesised using the DA-X monomer, in combination with terephthalaldehyde (TA) and tris(2-aminoethyl)amine (TREN) (Scheme 1). The synthesis was performed in THF at room temperature, followed by drying in vacuo at 50 °C, according to our previously reported polymerisation procedure for polyimine materials.34 Two polyimine materials were synthesised: PI-C, containing the alkyl-linked diamine monomer DA-C, and PI-O, containing the EO-linked diamine monomer DA-O (Scheme 1). Before the materials were analysed with the use of rheology, they were hot-pressed into flat discs with a 10 mm diameter and 0.4 mm thickness. As the monomer ratios were constant, and the molecular lengths of DA-C and DA-O are similar, the crosslinking density was assumed to be identical for both PI-X materials.Using a rheology setup, a temperature sweep experiment was performed in which the storage (G′) and loss (G′′) modulus were measured as a function of the temperature (Fig. 2). Over the entire duration of the experiment, the materials were exposed to a stress of 0.1% at a constant frequency of 1 Hz on a temperature ramp with steps of 1 °C per 10 s. From the G′ and G′′ curves, typically two phase transitions of the materials were observed: from the glass to the rubber state and from the rubber to the liquid state. The glass-to-rubber phase transition can typically be observed as a steep decrease in the G′ when the glass-like material is heated.22 The phase transition from rubber to liquid can be noticed from a second steep drop in G′ at increasing temperature, or can sometimes be better visualised at the crossover point of G′ and G′′ (tan(δ) = 1).9
 | ||
| Fig. 2 Representative temperature sweep curves of (A) PI-C and (B) PI-O, where the vertical blue line indicates the glass-rubber transition and the vertical red line indicates the rubber-liquid transition. (C) Visual representation of the glass (blue), rubber (yellow) and liquid (red) phases of PI-O (top) and PI-C (bottom) as a function of the temperature. | ||
From the temperature sweep curves of PI-O and PI-C we observed that both phase transitions for PI-O occurred at significantly lower temperature than those of PI-C. These observations of decreasing phase transition temperatures for the polar materials are in agreement with the results from the kinetic experiments. As we discussed above, the more polar EO-containing molecules showed a faster bond exchange reaction, which would result in faster dynamic processes in the materials. As a result, the corresponding phase transition temperatures decreased. Furthermore, the rubber domain of the PI-C materials is relatively small (temperature range of 19 °C) compared to PI-O (temperature range of 62 °C). This could be attributed to a lack of chain mobility in the polymer network of PI-C due to poor chain diffusion.
Apart from the polar effect in the diamine monomer, we also considered the effect of more polar EO-containing chains in the dialdehyde monomer. Therefore, we synthesised two dialdehyde monomers (AL-X) that differ in their linker, similar to those of the previously used diamines, where one monomer included an octyl chain, and the other included the EO moiety (Fig. 3).
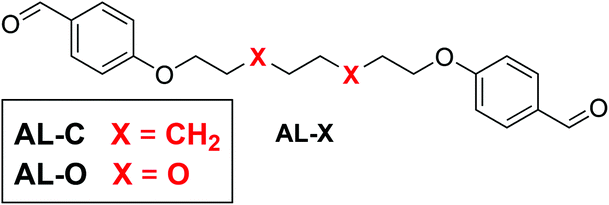 | ||
| Fig. 3 Molecular structure of the tuneable dialdehyde monomer, for which X is either CH2 (AL-C, apolar chain) or O (AL-O, polar chain). | ||
With two different aldehyde (AL-X) and two diamine (DA-X) monomers we could synthesise four different polyimines (crosslinked with TRENvia the same procedure as before): both diamine and dialdehyde are polar (i.e. EO-containing linker, PI-OO), both diamine an dialdehyde are apolar (i.e. alkyl linker, PI-CC), the diamine is polar and the dialdehyde is apolar (PI-OC), the diamine is apolar and the dialdehyde is polar (PI-CO).
A temperature sweep experiment was performed on the four polyimine samples using the same rheology setup as before to determine the phase transition temperatures (Fig. 4). From the results we can draw a clear conclusion that the increasing polarity lowers both phase transition temperatures. In the extreme case where both the aldehyde and amine chains are polar (PI-OO) the phase transition of glass to rubber was observed at a temperature as low as 1 °C and the phase transition from rubber to liquid at 37 °C. If we compare this to the other extreme where both monomers were apolar (PI-CC), we observed a phase transition from glass to rubber at 119 °C, and a phase transition from rubber to liquid above 150 °C. This means that the phase transition temperatures were increased by more than 100 °C by simply replacing the polar linkers for apolar ones (that vary only in two of the eight atoms that make up the linker). When only one of the monomers – either de dialdehyde or diamine – was polar, we observed that the phase transition temperatures were found in between the two extremes. Comparing the PI-OC and PI-CO individually shows that the phase transition from glass to rubber is similar (57 °C for PI-OC and 59 °C for PI-CO), but the phase transition from rubber to liquid occurred at slightly higher temperatures for PI-OC (83 °C) compared to PI-CO (68 °C). This means that the presence of polar domains in either the diamine or dialdehyde monomer decreases the phase transition temperatures of the polyimine materials, with the addition that the presence of EO moieties in the dialdehyde monomer is slightly more effective in lowering the rubber-to-glass phase transition, and therefore shortening the rubber phase of the material. This slight difference between the effect of polar groups in either the aldehyde or amine monomer could possibly be explained by other, secondary interactions within the network, e.g. in relation to the aromatic moieties being present next to the EO moiety.
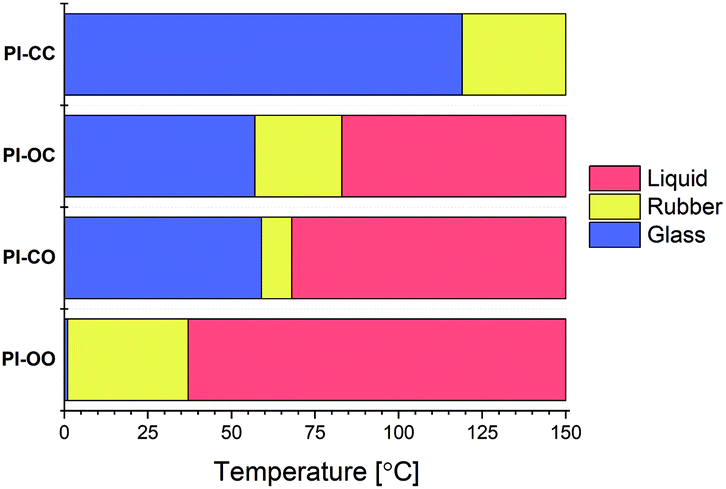 | ||
| Fig. 4 Visual representation of the glass (blue), rubber (yellow) and liquid (red) phases of the PI-XX materials as a function of the temperature. | ||
Stress relaxation of polyimine networks
Next to the thermal properties, the stress relaxation behaviour of PI-C and PI-O was also investigated by applying a 1% deformation to the materials and following the relaxation modulus (G(t)) over time until it reached zero again. In this relaxation process, we observed multiple relaxation phases, as can be seen in the normalised stress relaxation plots (Fig. 5). Please note that for PI-C and PI-O different temperatures needed to be selected in order to study the relaxation behaviour. The relaxation curves could not be accurately fitted to a one-component Maxwell model that is commonly used for the determination of relaxation times (τ) in CANs.60 However, the data could be fitted with a three-component function (see ESI†), revealing that three individual modes of relaxation (with corresponding relaxation times) occur within the material: a fast, intermediate and slow process. The relaxation curves for PI-O and PI-C were measured at several temperatures within their rubber state (Fig. S14†) and at every given temperature the three relaxation times were determined (Table 2 for PI-O and Table 3 for PI-C).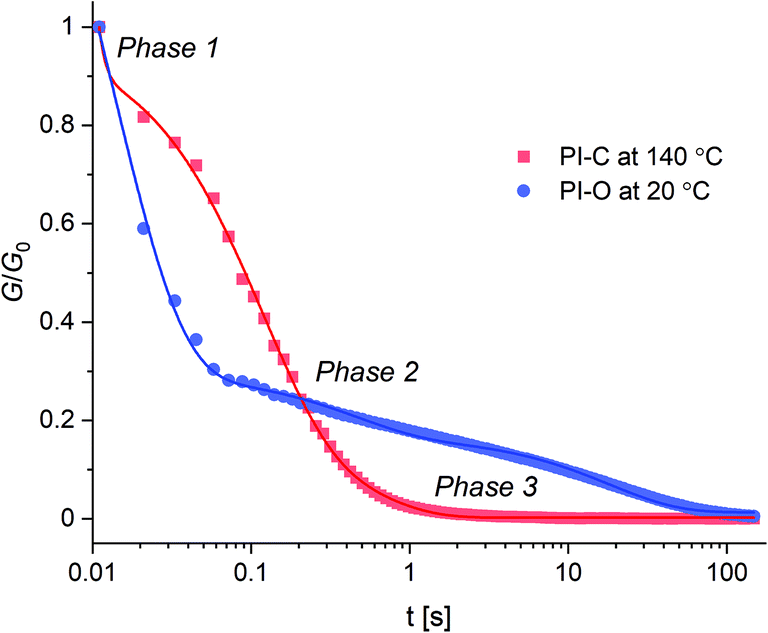 | ||
| Fig. 5 Normalised three-step relaxation curves for PI-C at 140 °C (red) and PI-O at 20 °C (blue). | ||
| T (°C) | τ 1 (ms) | τ 2 (ms) | τ 3 (ms) |
|---|---|---|---|
| 20 | 13.7 | 4.4 × 102 | 18.4 × 103 |
| 25 | 13.2 | 3.3 × 102 | 9.6 × 103 |
| 30 | 12.1 | 2.3 × 102 | 5.1 × 103 |
| 35 | 9.0 | 1.4 × 102 | 2.8 × 103 |
| 40 | 7.3 | 1.0 × 102 | 1.6 × 103 |
| 45 | 5.5 | 0.74 × 102 | 1.0 × 103 |
| 50 | 5.0 | 0.63 × 102 | 0.67 × 103 |
| 55 | 4.1 | 0.56 × 102 | 0.46 × 103 |
| T (°C) | τ 1 (ms) | τ 2 (ms) | τ 3 (ms) |
|---|---|---|---|
| 140 | 1.0 | 1.12 × 102 | 49 × 103 |
| 145 | 1.0 | 0.94 × 102 | 37 × 103 |
| 150 | 1.0 | 0.83 × 102 | 32 × 103 |
| 155 | 1.0 | 0.77 × 102 | 36 × 103 |
| 160 | 1.0 | 0.70 × 102 | 38 × 103 |
| 165 | 1.0 | 0.65 × 102 | 48 × 103 |
| 170 | 1.1 | 0.59 × 102 | 47 × 103 |
| 175 | 1.1 | 0.56 × 102 | 48 × 103 |
From the relaxation times of PI-O (Table 2) we observed a clear temperature dependency for each of the three relaxation processes: τ1 changed by a factor ∼3.5 over a temperature range of 35 °C, τ2 by a factor ∼8, and τ3 by a factor ∼40. When we compared these observations to the relaxation times of PI-C (Table 3), we found distinct differences. Firstly, we observed that both τ1 and τ3 seem to be independent of the temperature for PI-C. However, τ2 was dependent on the temperature, as the relaxation time changed by a factor ∼2 over a temperature range of 35 °C.
Based on the observed three-step relaxation process and determined τ values as a function of the temperature, we propose three modes of relaxation to each of the relaxation phases, with corresponding τ. The initial response (τ1) of the material when it is exposed to a stress was attributed to chain rearrangements in the material.61,62 This process is generally very fast, but quickly reaches it maximum potential due to the crosslinked structure of the polymer network.8,63 Therefore, in order to fully relax the materials, additional relaxation through bond exchange is required.64 We do however see that this additional relaxation through bond exchange proceeds on two different timescales (τ2 and τ3). This separation in two timescales could be attributed to imine exchange on a local level (τ2), meaning exchange between imines groups that are in close proximity, and secondly to imine exchange through diffusion (τ3). The latter occurs on a longer timeframe, as this mechanism requires the imine groups to diffuse through the network as well as find partnering imine groups to perform the bond exchange. The concept of diffusion and flow in the materials could be provided by reptation theory,65,66 which applies to random Brownian movement of polymer strains in entangled macromolecular structures,67 and could be extrapolated to dynamic polymer networks as well.68 More flexible chains would show an enhanced reptation process (better “slithering” of chains),68 which in turn promotes the availability of imines as they move better through the medium.
Regarding the mentioned three relaxation mechanisms, we can then further discuss the observations on the stress relaxation behaviour between the polar PI-O and apolar PI-C material. As can be seen in Table 2, each of the relaxation phases for PI-O was significantly influenced by the temperature, whereas for PI-C (Table 3) only τ2 showed a clear temperature response. The temperature response of the imine exchange is expected, although the absence of a clear temperature response for the imine exchange through diffusion (τ3) is peculiar. An explanation for this absence might be found in earlier work on cluster formation of exchanging polar groups in apolar polymer networks.50 The diffusion of imine groups in the apolar PI-C material might be limited as the polar imine groups tend to cluster together. This would mean that the local imine exchange becomes the main exchange pathway as the imine groups are already in close reach to each other, and the diffusion of imine groups through the polymer matrix is limited. The observation that τ2 showed stronger temperature response for the polar PI-O material (factor ∼8 over 35 °C range) over the apolar PI-C material (factor ∼2 over 35 °C range) suggests that the increasing polarity in the polymer network also enhances the thermal response of the imine exchange. As τ1 was found to be independent of the temperature for the apolar PI-C, this suggests that the glass transition of the material is mainly caused by the imine exchange, whereas for the polar PI-O material the flexibility within the material might also contribute to the softening of the material. Lastly, this could also add to the observation that the τ3 in PI-O showed a significant temperature response (factor ∼40 over 35 °C) as both the imine exchange and network flexibility are enhanced by increasing the temperature.
Overall, we can conclude that τ1 can be linked to reptation and is affected by the flexibility of the polymer chains.68 Additionally, the ease of reptation alters the diffusion and availability of exchanging imine groups, affecting τ3. Cluster formation of exchanging groups as a result of polarity in the polymer network might cause additional alterations to τ3, as well as to τ2.50 The temperature dependency of the imine exchange further applies to both τ2 and τ3. Lastly, our supporting kinetic data from the NMR experiments showed a clear correlation between the rate of imine exchange and presence of polar EO groups, which applies again to both τ2 and τ3.
Determination of the relaxation behaviour of the PI-XX materials was harder to achieve as the materials were generally very brittle, with the exception of PI-OO, and would break easily at the required applied stress. Measurements in the liquid phase of the materials were also unsuccessful as relaxation times were too short (<1 ms) to properly analyse. Therefore, no in-depth stress relaxation analysis could be performed on these materials. Only for the PI-OO material (Table 4) we were able to analyse the relaxation behaviour, for which we observed a relatively similar trend in the three relaxation phases as the PI-O material (Table 2). This resemblance may result from the fact that both materials contain the polar EO linkers. Note, however, that the two materials have different crosslinking densities and relative polarity effects, as the PI-O material contains the small TA dialdehyde monomer, and PI-OO contains the longer AL-O dialdehyde monomer that also contains an additional polar EO linker.
| T (°C) | τ 1 (ms) | τ 2 (ms) | τ 3 (ms) |
|---|---|---|---|
| 15 | 11.0 | 3.7 × 102 | 12.2 × 103 |
| 20 | 10.0 | 2.7 × 102 | 7.3 × 103 |
| 25 | 8.0 | 1.7 × 102 | 4.0 × 103 |
| 30 | 5.7 | 1.1 × 102 | 2.2 × 103 |
| 35 | 4.1 | 0.76 × 102 | 1.3 × 103 |
| 40 | 2.7 | 0.58 × 102 | 8.6 × 102 |
| 45 | 2.3 | 0.52 × 102 | 6.4 × 102 |
| 50 | 1.4 | 0.50 × 102 | 4.8 × 102 |
Conclusions
Ethylene oxide and similar ether groups are commonly applied in (dynamic) polymer materials to increase flexibility of the material. What should, however, not be overlooked in the case of CANs, is the polarity-induced effect of these ethylene oxide groups on the kinetics of the dynamic covalent bond exchange, which in turn results in alteration of the physical and thermal properties of the material. In this study, we showed that the dynamic exchange behaviour in polyimine CANs could be directly related to polarity effects in the polymer matrix. Small-molecule kinetic studies revealed that the reaction rate of imine exchange could be enhanced by introduction of more polar ethylene oxide groups when compared to apolar purely aliphatic carbon chains in the monomer. Additionally, from rheology studies on the crosslinked polymer materials we concluded that the addition of the more polar ethylene oxide groups resulted in a higher dynamic response in the materials, which was reflected in a decrease in the phase transitions from glass to rubber, and from rubber to liquid. Furthermore, extensive investigations into the stress relaxation behaviour of the materials revealed a gradual three-phase relaxation process. These were attributed to relaxation by chain redistribution within the polymer network, relaxation via imine exchange on local level, and imine exchange after diffusion through the network. This division in three relaxation processes within one material offers a better physical–chemical understanding of the dynamic behaviour of CANs and how molecular interactions within the polymer matrix relate to macroscopic properties of the material. These results firstly reveal the subtle, sometimes unexpected, effect that small changes in molecular structure can have on material properties, as reflected here in the particular changes in the multi-step relaxation processes. Moreover, we envision that this multi-step relaxation could also be observed in other CANs to achieve a more precise insight in, and ultimately control over, the molecular dynamics of the material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr. Joshua Dijksman and Prof. Han Zuilhof for their help and involvement in this project. The Netherlands Organisation for Scientific Research (NWO) is acknowledged for funding (NWO Vidi grant 016.Vidi.189.031 to MMJS).Notes and references
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- M. Podgórski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 32, 1906876 CrossRef.
- Y. Jin, Z. Lei, P. Taynton, S. Huang and W. Zhang, Matter, 2019, 1, 1456–1493 CrossRef.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS.
- A. Gablier, M. O. Saed and E. M. Terentjev, Soft Matter, 2020, 16, 5195–5202 RSC.
- S. Wu, H. Yang, S. Huang and Q. Chen, Macromolecules, 2020, 53, 1180–1190 CrossRef CAS.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef.
- B. Marco-Dufort, R. Iten and M. W. Tibbitt, J. Am. Chem. Soc., 2020, 142, 15371–15385 CrossRef CAS.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS.
- C. He, P. R. Christensen, T. J. Seguin, E. A. Dailing, B. M. Wood, R. K. Walde, K. A. Persson, T. P. Russell and B. A. Helms, Angew. Chem., Int. Ed., 2020, 59, 735–739 CrossRef CAS.
- L. Chen, L. Zhang, P. J. Griffin and S. J. Rowan, Macromol. Chem. Phys., 2019, 221, 1900440 CrossRef.
- L. Zhang and S. J. Rowan, Macromolecules, 2017, 50, 5051–5060 CrossRef.
- Z. Wang, S. Gangarapu, J. Escorihuela, G. Fei, H. Zuilhof and H. Xia, J. Mater. Chem. A, 2019, 7, 15933–15943 RSC.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps and B. S. Sumerlin, J. Am. Chem. Soc., 2020, 142, 283–289 CrossRef CAS.
- W. Denissen, I. De Baere, W. Van Paepegem, L. Leibler, J. Winne and F. E. Du Prez, Macromolecules, 2018, 51, 2054–2064 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS.
- J. P. Brutman, D. J. Fortman, G. X. De Hoe, W. R. Dichtel and M. A. Hillmyer, J. Phys. Chem. B, 2019, 123, 1432–1441 CrossRef CAS.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2020, 59, 3609–3617 CrossRef CAS.
- B. M. El-Zaatari, J. S. A. Ishibashi and J. A. Kalow, Polym. Chem., 2020, 11, 5339–5345 RSC.
- P. Taynton, K. Yu, R. K. Shoemaker, Y. Jin, H. J. Qi and W. Zhang, Adv. Mater., 2014, 26, 3938–3942 CrossRef CAS.
- F. García, J. Pelss, H. Zuilhof and M. M. J. Smulders, Chem. Commun., 2016, 52, 9059–9062 RSC.
- Z. Q. Lei, P. Xie, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2015, 3, 19662–19668 RSC.
- S. K. Schoustra, J. A. Dijksman, H. Zuilhof and M. M. J. Smulders, Chem. Sci., 2020 10.1039/d0sc05458e.
- F. García and M. M. J. Smulders, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3551–3577 CrossRef.
- Y. Spiesschaert, C. Taplan, L. Stricker, M. Guerre, J. M. Winne and F. E. Du Prez, Polym. Chem., 2020, 11, 5377–5385 RSC.
- Y. Liu, Z. Tang, J. Chen, J. Xiong, D. Wang, S. Wang, S. Wu and B. Guo, Polym. Chem., 2020, 11, 1348–1355 RSC.
- R. Mo, J. Hu, H. Huang, X. Sheng and X. Zhang, J. Mater. Chem. A, 2019, 7, 3031–3038 RSC.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2010, 10, 14–27 CrossRef.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- F. Sugiyama, A. T. Kleinschmidt, L. V. Kayser, D. Rodriquez, M. Finn, M. A. Alkhadra, J. M. H. Wan, J. Ramírez, A. S. C. Chiang, S. E. Root, S. Savagatrup and D. J. Lipomi, Polym. Chem., 2018, 9, 4354–4363 RSC.
- R. L. Snyder, C. A. L. Lidston, G. X. De Hoe, M. J. S. Parvulescu, M. A. Hillmyer and G. W. Coates, Polym. Chem., 2020, 11, 5346–5355 RSC.
- P. Yan, W. Zhao, X. Fu, Z. Liu, W. Kong, C. Zhou and J. Lei, RSC Adv., 2017, 7, 26858–26866 RSC.
- N. Zheng, Z. Fang, W. Zou, Q. Zhao and T. Xie, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS.
- H. Li, J. Bai, Z. Shi and J. Yin, Polymer, 2016, 85, 106–113 CrossRef CAS.
- D. J. Fortman, D. T. Sheppard and W. R. Dichtel, Macromolecules, 2019, 52, 6330–6335 CrossRef CAS.
- B. K. Wheatle, N. A. Lynd and V. Ganesan, ACS Macro Lett., 2018, 7, 1149–1154 CrossRef CAS.
- Q. Zhao, C. Shen, K. P. Halloran and C. M. Evans, ACS Macro Lett., 2019, 8, 658–663 CrossRef.
- L. M. Polgar, E. Hagting, P. Raffa, M. Mauri, R. Simonutti, F. Picchioni and M. van Duin, Macromolecules, 2017, 50, 8955–8964 CrossRef CAS.
- P. Chakma, C. N. Morley, J. L. Sparks and D. Konkolewicz, Macromolecules, 2020, 53, 1233–1244 CrossRef CAS.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC.
- A. Chao, I. Negulescu and D. Zhang, Macromolecules, 2016, 49, 6277–6284 CrossRef CAS.
- C. Zhu, C. Xi, W. Doro, T. Wang, X. Zhang, Y. Jin and W. Zhang, RSC Adv., 2017, 7, 48303–48307 RSC.
- P. Taynton, C. Zhu, S. Loob, R. Shoemaker, J. Pritchard, Y. Jin and W. Zhang, Polym. Chem., 2016, 7, 7052–7056 RSC.
- M. Ciaccia and S. Di Stefano, Org. Biomol. Chem., 2015, 13, 646–654 RSC.
- M. Ciaccia, R. Cacciapaglia, P. Mencarelli, L. Mandolini and S. Di Stefano, Chem. Sci., 2013, 4, 2253–2261 RSC.
- M. Ciaccia, S. Pilati, R. Cacciapaglia, L. Mandolini and S. Di Stefano, Org. Biomol. Chem., 2014, 12, 3282–3287 RSC.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS.
- S. Ciarella, F. Sciortino and W. G. Ellenbroek, Phys. Rev. Lett., 2018, 121, 058003 CrossRef CAS.
- P. E. Rouse, J. Chem. Phys., 1953, 21, 1272–1280 CrossRef CAS.
- R. P. Wool, Soft Matter, 2008, 4, 400–418 RSC.
- E. B. Stukalin, L. Cai, N. A. Kumar, L. Leibler and M. Rubinstein, Macromolecules, 2013, 46, 7525–7541 CrossRef CAS.
- P.-G. de Gennes, Phys. Today, 1983, 36, 33–39 CrossRef CAS.
- P.-G. de Gennes, J. Chem. Phys., 1971, 55, 572–579 CrossRef.
- M. Doi and S. F. Edwards, J. Chem. Soc., Faraday Trans. 2, 1978, 74, 1789–1801 RSC.
- P. C. Cai, B. A. Krajina and A. J. Spakowitz, Phys. Rev. E, 2020, 102, 020501 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01555e |
| This journal is © The Royal Society of Chemistry 2021 |