
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling properties of thermogels by tuning critical solution behaviour of ternary copolymers†
Anton A. A.
Smith‡
 a,
Caitlin L.
Maikawa
b,
Hector
Lopez Hernandez
a and
Eric A.
Appel
*ab
a,
Caitlin L.
Maikawa
b,
Hector
Lopez Hernandez
a and
Eric A.
Appel
*ab
aDepartment of Materials Science & Engineering, Stanford University, Stanford, CA 94305, USA. E-mail: eappel@stanford.edu
bDepartment of Bioengineering, Stanford University, Stanford, CA 94305, USA
First published on 26th March 2021
Abstract
Thermoresponsive hydrogel materials show promise as biomaterials as their properties can be widely tuned to fit engineering requirements for an array of important applications. Here we show that the properties of thermogelling tri-block copolymers consisting of a central poly(ethylene glycol) block and pendant N-substituted polyacrylamides can be tuned by altering the random terpolymer compositions. The heterogeneity of the pendant terpolymer blocks is reflected in the concentration dependence of their critical solution behavior. The lower critical solution temperature (LCST) of the pendant blocks can be finely tuned, enabling controlled modulation of thermogel properties. Altering terpolymer composition to control LCST behaviour, therefore, provides a facile approach to design thermogel properties.
Introduction
Thermoresponsive gelators, or thermogels, are materials that undergo a transition from sol to gel in aqueous solution at elevated temperatures. A class of these materials are multi-block copolymers wherein one or more blocks undergo phase separation when the temperature is elevated above the lower critical solution temperature (LCST). Pluronics, which are triblock copolymers comprising poly(ethylene oxide) (PEO) and poly(propylene oxide), are an example of thermogel materials that have found broad use as biomaterials in both tissue engineering and drug delivery.1–5 Acrylic based polymers with thermogelation properties are often based on poly(N-isopropyl acrylamide) (PNIPAM), since PNIPAM exhibits an LCST at 32 °C, conveniently between ambient and physiological temperatures.6 When ABA or ABC triblock copolymers containing terminal PNIPAM blocks are raised above their LCST, the terminal blocks phase separate and form micelles that are connected into a 3D network by a hydrophilic block.7–14 As the LCST of these PNIPAM blocks are the fundamental feature enabling thermogel formulation, modulation of this critical solution behaviour provides a unique opportunity to tune thermogel properties. It is well known that the LCST of PNIPAM can be modulated through copolymerization, whereby inclusion of hydrophobic monomers, such as tertbutyl acrylamide (TBAM), decreases the LCST and inclusion of hydrophilic monomers, such as hydroxyethyl acrylamide (HEAM), increases the LCST.15,16 In addition to changing the LCST, thermogel properties can be modulated through the composition and lengths of either the hydrophobic or hydrophilic blocks within the copolymer.7,10 In this study, we perform a systematic variation of terpolymer composition of terminal thermally-responsive blocks to ascert greater control over critical solution behaviour, and correspondingly modulate crosslink strength and dynamics in triblock copolymer thermogelling systems. Towards this goal, NIPAM is copolymerized with TBAM and/or HEAM and the critical solution behaviour of the resulting copolymers are measured. We hypothesize that thermogelation temperature and mechanical properties of triblock copolymers can be modulated through alterations to the ternary composition of the phase-transitioning block. The terpolymer compositions are used to extend a 24 kDa PEG bifunctional RAFT agent to create triblock copolymers (Fig. 1), and the gel transition temperatures and mechanical properties are measured for the resulting gels.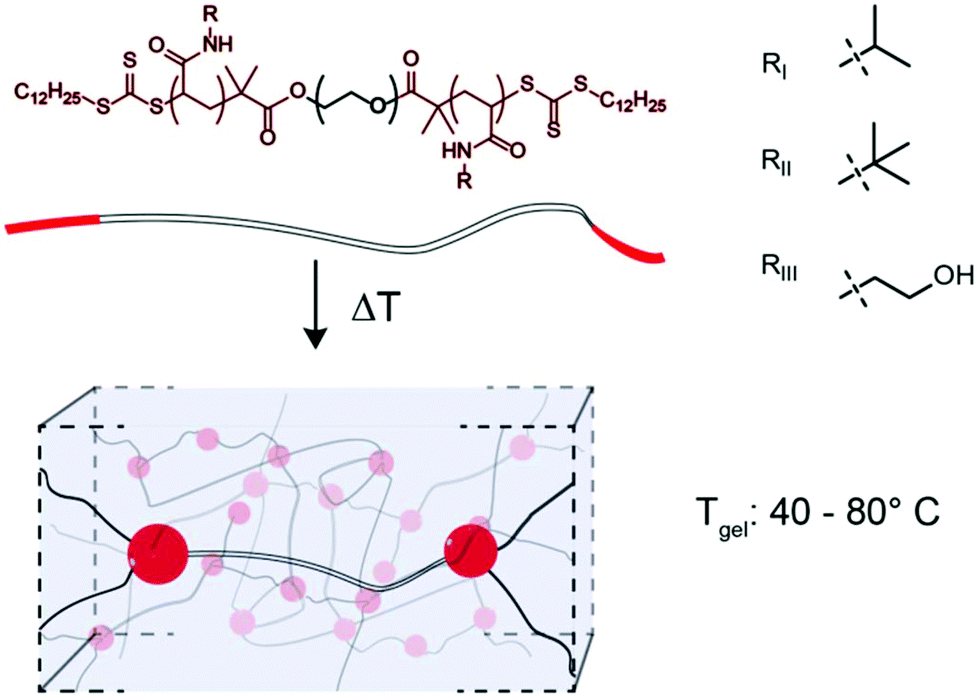 | ||
| Fig. 1 A PEG based bifunctional RAFT agent extended with terpolymer compositions of N-substituted acrylamides form 3D physically crosslinked networks upon heating due to LCST transition of terminal blocks. Gelation temperature is controlled by monomer composition of terminal blocks. | ||
Experimental
Materials and methods
All chemicals were obtained through Millipore-Sigma. N-Hydroxyethyl acrylamide was passed through basic alumina prior to use. N-Isopropyl acrylamide was recrystallized from hexane and dried in vacuo prior to use. Commercial 20 kDa PEG was measured to be 24 kDa by PEG callibrated DMF GPC, and was used as such. Molecular weights and dispersity indexes were determined by GPC callibrated with PEG standards using two size exclusion chromatography columns in sequence, Resolve Mixed Bed Low DVB, ID 7.8 mm, Mw range 200–600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (Jordi Labs) in a mobile phase of N,N-dimethylformamide (DMF) with 0.1 M LiBr at 35 °C and a flow rate of 1.0 mL min−1. The GPC was a Dionex Ultimate 3000 pump, degasser, and autosampler (Thermo Fisher Scientific), equipped with an ERC RefractoMax520 RI refractometer. Fluorescense determination of LCST was performed with dansyl hydroxypropyl suphonamide as the probe at 40 μg mL−1, using a BioTek Synergy H1 microplate reader, exciting at 350 nm and recording a full excitation spectrum.
000 g mol−1 (Jordi Labs) in a mobile phase of N,N-dimethylformamide (DMF) with 0.1 M LiBr at 35 °C and a flow rate of 1.0 mL min−1. The GPC was a Dionex Ultimate 3000 pump, degasser, and autosampler (Thermo Fisher Scientific), equipped with an ERC RefractoMax520 RI refractometer. Fluorescense determination of LCST was performed with dansyl hydroxypropyl suphonamide as the probe at 40 μg mL−1, using a BioTek Synergy H1 microplate reader, exciting at 350 nm and recording a full excitation spectrum.
Rheological characterization
All rheometry experiments were performed on a torque-controlled Discovery HR2 Rheometer (TA Instruments). Small amplitude oscillatory shear (SAOS) measurements were performed using a 8 mm parallel plate geometry (Peltier plate steel) from 0.6 rad s−1 to 10 rad s−1 at a strain amplitude within the linear viscoelastic regime. Polymer solutions were tested at 5 wt% polymer for rheological experiments. SAOS measurements below the LCST of all polymers were performed at 25 °C. Measurements above the LCST of all polymers were performed at 78 °C unless otherwise specified. SAOS temperature ramps were performed at a frequency of 6 rad s−1 with a ramp rate of 2 °C min−1. All experiments were performed in triplicate, with the mean being reported, except #13, which was performed in duplicate.Synthetic procedures
For the synthesis of PEG-DOPAT RAFT agent (Scheme 1A), PEG 24 kDa (3 g, 0.125 mmol), 2-(dodecylthiocarbonothioylthio)propionic acid (DOPAT) (0.26 g, 0.75 mmol), and 4-(dimethylamino)pyridine (5.5 mg, 0.05 mmol) was dissolved in 50 mL DCM. N,N′-Diisopropylcarbodiimide (DIC) (0.95 g, 0.75 mmol) in 5 mL DCM was added, and the reaction mixture stirred at room temperature overnight. The solution was precipitated into diethyl ether, and the product was recovered by filtration and dried in vacuo. 1H-NMR characterization shown in Fig. S1.† Polymerizations with PEG-DOPAT RAFT agent (Scheme 1C) were conducted in a total reaction volume of 4 mL of water:dioxane (1:1 v/v), containing PEG-DOPAT (0.45 g, 0.0186 mmol), 120 eq. monomers (2.25 mmol), and 0.48 eq. VA-044 thermal initiator (0.3 mg, 0.009 mmol). The reaction mixtures were heated to 100 °C for 3 minutes. Following polymerization, all volatiles were removed in vacuo by a GeneVac evaporator to recover the polymers. 1H-NMR characterization shown in Fig. S2.† Characteristics shown for all polymers in Table S1.†
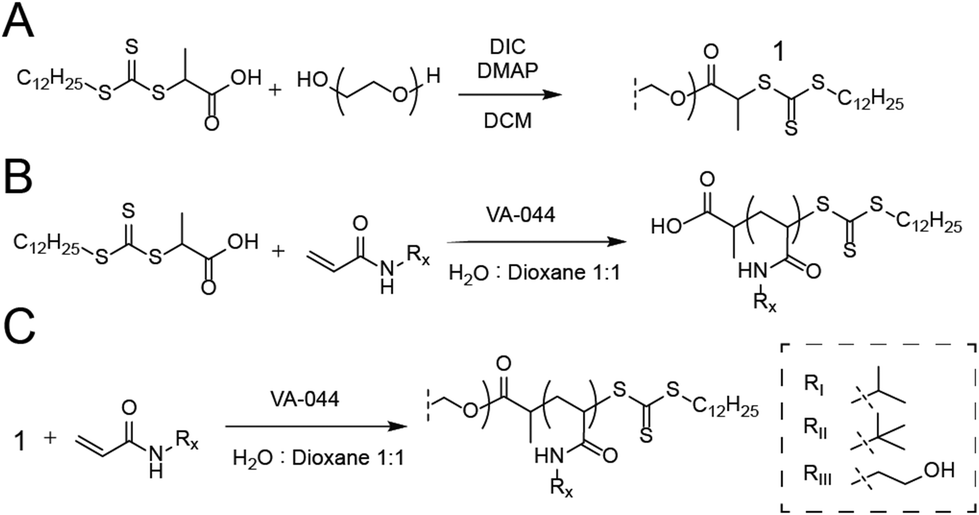 | ||
| Scheme 1 Synthesis of NIPAM-based copolymers. (A) PEG-DOPAT RAFT agent was made with DIC coupling to DOPAT. (B and C) RAFT polymerizations using either PEG-DOPAT RAFT agent or DOPAT varying acrylamide ratios. | ||
Polymerizations with DOPAT (Scheme 1B) were conducted in a total reaction volume of 1 mL of water:dioxane (1:1 v/v) containing DOPAT (10 mg, 0.029 mmol), 100 eq. of monomers (0.29 mmol), and 0.2 eq. VA-044 thermal initiator (1. mg, 5.7 μmol). Following polymerization, volatiles were removed in vacuo, and the polymers washed with a 1:1 mixture of diethyl ether and acetone. 1H-NMR characterization shown in Fig. S3.† Characteristics shown for all polymers in Table S1.† Dansyl hydroxypropyl suphonamide was synthesized as described in literature.17
Results and discussion
LCST of terpolymers
We first synthesized a series of NIPAM-based copolymers through NIPAM copolymerization with TBAM and/or HEAM. The compositions were varied in mol% of each of the three monomers of interest as shown in Scheme 1 and Table 1. For all polymerizations we used the ultrafast oxygen tolerant conditions developed by Perrier et al. for Reverible Addition–Fragmentation chain-Transfer (RAFT) polymerization, enabling rapid generation of the desired collection of acrylamide-derived copolymers.18 The DOPAT polymers exhibited higher dispersities than expected, which we attribute to a low solubility of DOPAT at low temperatures. All DOPAT was dissolved in the reaction vessel by gentle heating, prior to reaching the targeted reaction temperature, though some unintended initiation likely occurred in some samples. The same issue was not observed for the PEG-DOPAT RAFT agent polymerizations. PNIPAM retains a relatively high water content after phase separation from aqueous solutions at temperatures above the LCST, previously characterized to be approximately 20–50 wt%.19| Polymer # | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| References to polymers in text is given by the polymer number followed by the composition in mol % (NIPAM:TBAM:HEAM). |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I NiPAM % | 100 | 95 | 90 | 95 | 90 | 85 | 85 | 80 | 80 | 80 | 75 | 75 | 75 | 75 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| II TBAM % | 5 | 10 | 5 | 10 | 5 | 15 | 10 | 5 | 20 | 15 | 10 | 5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| III HEAM % | 5 | 5 | 5 | 10 | 5 | 10 | 15 | 5 | 10 | 15 | 20 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
We hypothesized that in the context of thermogelling block copolymers, the entrapped water effectively functions as a plasticizer within the phase-segregated micellar domains that act as physical crosslinks in the thermogelling networks. For thermogels, the chain dynamics of the PNIPAM polymers within these phase segregated domains is expected to correlate to the strength and dynamics of the networks at a given temperature. Following on this rationale, we hypothesized that a lowering of the water content within these micellar domains should produce stronger crosslinks, and that such a decrease in entrapped water can be achieved by introducing more hydrophobic monomers into the terpolymer to alter the critical solution behavior of these materials. For measuring LCST, an amphiphilic dansyl derived fluorescent probe was chosen in favour of the traditional cloud point measurement. When in a hydrophobic environment, the fluorescence emission of dansyl derivatives increases markedly,20 making it an efficient probe for LCST transitions at very low concentrations of the polymer. This allowed for a rapid high-throughput characterization of LCST behavior, as well as a qualitative assessment of overall hydrophobicity of the polymers, due to the overall increased fluorescent emission caused by more hydrophobic polymers (Fig. 2, Fig. S4 and Table S2†). Polymer 1 (100:0:0), which is a PNIPAM homopolymer, exhibited an onset of fluorescense at the same temperature at all signal producing concentrations, whereas polymer 2 (95:5:0), which is a P(NIPAM-co-TBAM) copolymer, showed a concentration dependent onset temperature for probe fluorescence. This distinct behaviour likely reflects the heterogeneity of the copolymer, where some chains exhibit different critical solution behavior and commensurate LCSTs than others, depending on the exact TBAM content in individual chains. With more TBAM, the concentration dependence of the LCST is even further increased, again suggesting an impact on measured LCSTs from heterogeneous polymer composition. Polymer 4 (95:0:5), which is a P(NIPAM-co-HEAM) copolymer, a homogeneous onset of signal is observed, albeit at a higher temperature than for the PNIPAM homopolymer, consistent with literature reports.21
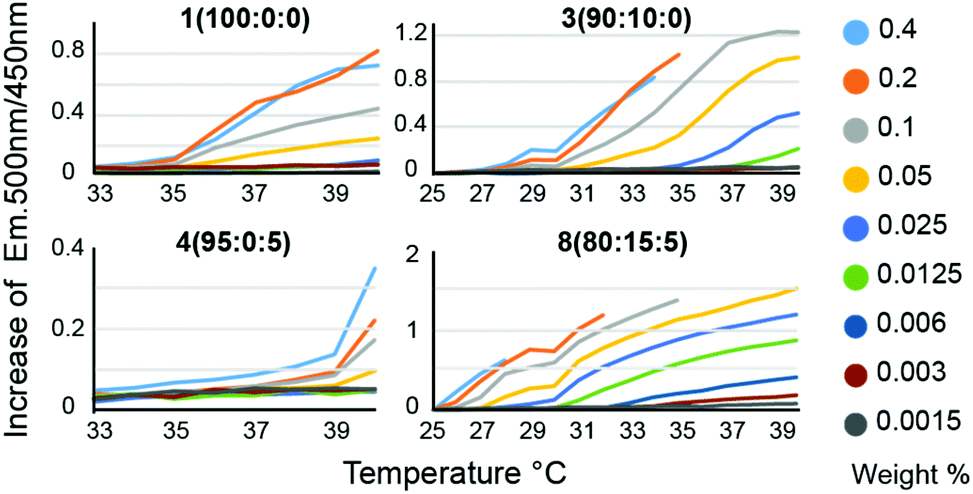 | ||
| Fig. 2 LCST determination for PNIPAM copolymers at various concentrations. LCSTs were determined for all copolymer compositions in the temperature range of 25–40 °C across different concentrations using dansyl hydroxypropyl suphonamide as a fluorescent probe. Onset of LCST is defined as an increase in slope of the 500 nm/450 nm emission ratio. A pure PNIPAM polymer 1 (100:0:0) displayed an LCST at 35–36 °C, whereas a copolymer containing 10% TBAM, polymer 3 (90:0:10), had a broad onset spanning 27–39 °C. A PNIPAM copolymer containing 5% HEAM, polymer 4 (95:0:5), exhibited homogeneous onset at 39 °C, whereas terpolymers containing NIPAM, TBAM and HEAM, polymer 8 (80:15:5), exhibited LCST onset spanning 25–37 °C. | ||
We expect the onset from polymer 4 (95:0:5) arises from the subset of polymers containing little to no HEAM. The greatest concentration dependence is observed for copolymers containing 10–20% TBAM and 5% HEAM, where the sporadic incorporation of HEAM increases the heterogeneity of the copolymers. In all samples, it is likely that the dodecyl Z-group has contributed to the LCST. Altogether, the concentration dependence of the LCSTs observed for these copolymers is heavily influenced by the heterogeneity in monomer composition encorporated into the chains for short polymers. The LCST for all polymers for all concentrations are listed in Table S2.†
We then employed the program ‘Compositional Drift’ to simulate the incorporation distribution of HEAM and TBAM comonomers into PNIPAM polymers at feed ratios of 5%, 10%, 15%, and 20% mol% for copolymers with a degree of polymerization 50 and dispersities of Đ = 1.02 and Đ = 1.32 (Fig. 3A).22 Even with Đ = 1.02, the distribution of comonomers across all mol% feed ratios is quite broad, and this breadth is exascerbated further when Đ = 1.32. This inherent heterogeneity of comonomer incorporation, even for low dispersity polymers, can be expected to translate into broad temperature dependence of the mechanical properties for thermogels using relatively short terpolymers as a thermally-responsive phase-transitioning block. This distribution narrows as DP increases, and simulated DP 1000 show a narrow compositional variation (Fig. 3B).
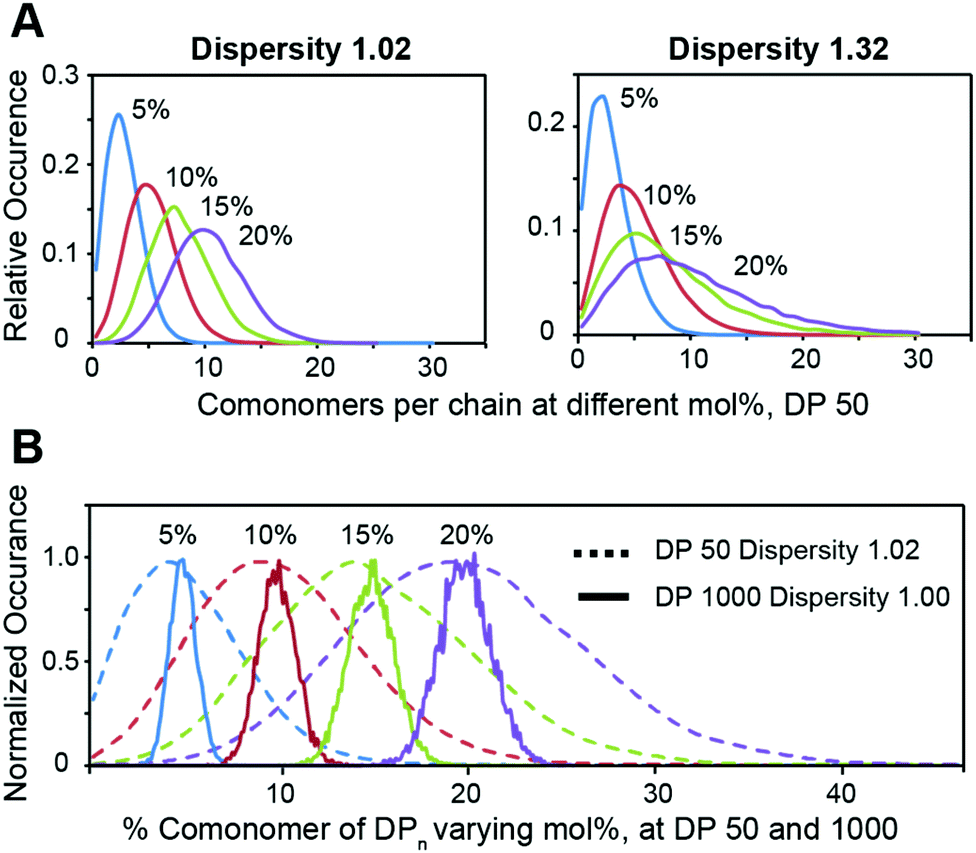 | ||
| Fig. 3 Simulated composition variation for various copolymers. (A) Variation in absolute number of comonomers per chain at different mol% for copolymers with a degree of polymerization of 50, for two different polymer dispersity indexes. While the number average number of comonomers reflects the monomer ratio, the distribution is wide regardless of polymer dispersity index. Correspondingly, any short copolymer will always have a wide distribution of polymers with critical solution behaviours, which is reflected in the wide onset of LCST temperatures reported for some copolymers above. (B) Simulated variation in % co-monomer incorporation, at DP 50 and DP 1000. At DP 1000, the variation in composition has decreased significantly for all compositions. All simulations are done assuming reactivity ratios of 1 for all monomers and complete conversion. | ||
A bifunctional PEG RAFT agent (PEG-DOPAT) was synthesized and RAFT polymerization was employed to generate ABA triblock copolymers with various (ter)polymer compositions for the phase-transitioning A blocks (Scheme 1B). We explored the thermogelling behaviour using a combination of dynamic frequency and temperature sweep rheological measurements. The frequency sweeps (Fig. S5 and S6†) provided valuable information about the viscoelasticity of the fluids or gels below and above the LCST for the phase-transitioning blocks. The temperature ramps (Fig. S7†) provided information about the temperature responsiveness of the formulations, including the LCST and gelation temperature. We hypothesized that the phase separation behaviour above the LCST would alter the amount of viscous dissipation (G′′) observed within these triblock copolymer hydrogels and that the appearance of interchain crosslinks above the LCST of each formulation would significantly increase the elastic response (G′). All NIPAM-based (co)polymer formulations showed liquid-like behaviour at 25 °C (Fig. S6†), demonstrating loss moduli greater than storage moduli at all frequencies measured. Indeed, for every formulation the loss modulus (G′′) scaled linearly with frequency throughout the entire frequency regime evaluated, and the storage modulus (G′) scaled with the frequency squared, characteristic of viscous fluid-like behaviour.23 Temperature ramps were performed at a frequency of 1 s−1 from 25 °C to 78 °C (unless otherwise specified) at 2 °C min−1. For a typical (co)polymer solution, increases in the temperature are analogous to decreasing measurement frequencies,24 resulting in a decrease in the storage and loss modulus and an increase in tanδ. For these LCST block copolymers, behaviour similar to that of a viscous solution was observed up to the LCST temperature, beyond which the (co)polymers start to phase separate. Temperature ramps (Fig. 4Ai and Bi) reveal peaks in tanδ, indicating temperatures at which the viscoelasticity stops scaling as a simple viscous fluid.
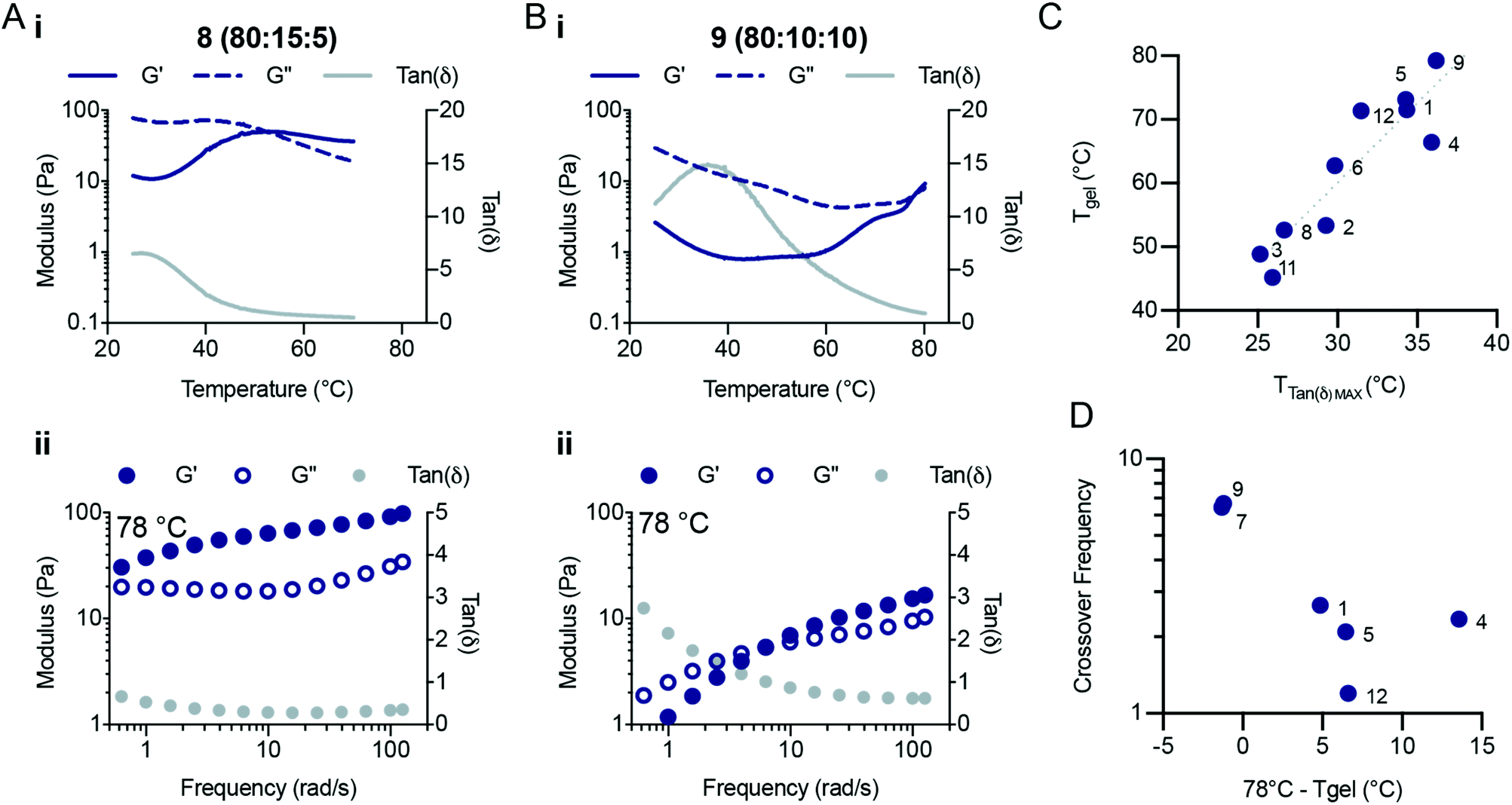 | ||
| Fig. 4 Thermogelling behaviour of NIPAM-based (co)polymer formulations. Temperature responsive hydrogel formation for a representative (A) low LCST polymer and (B) high LCST polymer. (i) Temperature ramps show the transition from a primarily viscous material response to the formation of a hydrogel with a primarily elastic response. The onset of phase separation was observed as tanδ decreases after surpassing the LCST. Network formation is observed by the crossover of the storage and loss moduli at increased temperatures. (C) Scaling of the gelation temperature vs. the temperature at the peak of tanδ. The gelation temperature is approximately double the LCST temperature for all formulations that gelled. (D) Crossover frequency vs. the distance from the gel point. The crossover frequency measured for each formulation decreased as the distance from the gel point increased, indicating the continued formation of an elastic network after the LCST. | ||
The terpolymer compositions tested showed peaks in tanδ ranging from 25 °C to 36 °C, indicative of the LCST temperature, where the polymers begin to phase separate and stop contributing to viscous dissipation within the solution The LCST measured for the triblock copolymer compositions by fluorescense was in good agreement with the temperature that corresponds to the peak value in tanδ (Fig. S7;† Fluorescence LCST vs. Peak TanDelta LCST). The peak in tanδ preceded a crossover of the storage and loss modulus, which is indicative of gelation and occurred at a temperature (Tgel) approximately double the LCST (Fig. 4C and Fig. S8, S9†). At this temperature, sufficient network formation results in an elastically dominant mechanical response. Gelation temperatures were measured in the range of 45 °C and 80 °C and gelation was observed for every formulation tested except those with HEAM compositions exceeding 15 mol%. The storage modulus of the resulting gels increases with temperature, as water is excluded from the terpolymer rich mesoglobular phase, to the point of forming an elastic network.
The viscoelasticity of each formulation was measured at elevated temperatures to probe their frequency-dependent mechanical response after gelation (Fig. 4 and Fig. S5†). This analysis was particularly important for formulations which demonstrated higher LCSTs and no crossover during the temperature ramp at a frequency of 1 s−1. Fig. 4Aii and Bii show characteristic responses for a lower LCST polymer and a higher LCST polymer, respectively. The frequency sweeps at 78 °C showed complex viscoelastic behaviours with a rubbery plateau and a crossover frequency. We found that as the temperature is increased past the Tgel the crossover frequency of the hydrogels continues to decrease. The crossover frequency for each formulation shown as a function of (78 °C – Tgel) highlights the decrease in crossover frequency as the distance from Tgel increases (Fig. 4D). The crossover frequency is the inverse of the relaxation time, so as the crossover frequency decreases the relaxation time of the network increases. Interestingly, these observations suggest that the NIPAM-based copolymers continue to phase separate as the temperature of the system is increased, retarding the relaxation processes in the hydrogels. Several of the compositions tested did not demonstrate crossover frequencies at 78 °C. In contrast, polymers 2 (95:5:0), 3 (90:10:0), 6 (85:10:5), 8 (80:15:5), and 11 (75:20:5) showed solid-like behavior throughout the entire frequency range measured (0.5–100 rad s−1). All compositions demonstrated similar plateau values for both the storage and loss moduli (with the exception of polymers 8 and 9), demonstrating that these (co)polymers generate a similar network structure after gelation. Therefore, modification of the copolymer LCST through alteration of terpolymer compositions affords tunability over the temperature transitions and viscoelastic responses of triblock thermogelling materials.
It is curious that the Tgel for these ABA triblock copolymers does not coincide with the LCST of the NIPAM-based copolymer A blocks. We hypothesize that this transition occurs at higher temperatures due to local rearrangements of the polymers near the LCST, whereby higher temperatures are required to expel sufficient water from the micellar crosslinks to produce sufficiently strong crosslinks for gellation. Similar above-LCST behaviour has been observed for NIPAM-based hydrogels,25 and the observations made here corroborates that transitions above the LCST can be exploited in the design of the mechanical responses of thermoresponsive materials.
Conclusions
In this work we show that the critical solution behavior of PNIPAM heteropolymers can be modulated through the copolymerization of both hydrophilic and hydrophobic monomers to create terpolymers. For short polymers, copolymerization introduces a broad dispersity in polymer composition, where the LCST of a specific subpopulation of polymer chains will be determined by its specific composition. This dispersity of copolymer composition translates to different thermoresponsive behaviours when used as the temperature-dependent phase transitioning block in thermogelling heteropolymers. The storage and loss moduli plateau values determined well above the LCST of the phase transitioning blocks are similar for all polymers evaluated, regardless of composition, suggesting these polymers eventually assume a similar network structure. The minor differences in mechanical behaviours observed across different copolymer compositions likely reflect variation in the strength and dynamics of the physical crosslinks that correlated to the LCST, whereby copolymers with lower LCST values produced slightly stiffer and less dynamic gels. All together, heteropolymer LCST can be fine-tuned by their exact monomer composition, and the resulting phase segregation behaviour can translate into control of material properties of the ABA triblock thermogels.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was financially supported by the Center for Human Systems Immunology with Bill and Melinda Gates Foundation (OPP1113682) and the NIDDK (R01DK119254). A. A. A. S. was funded by grant NNF18OC0030896 from the Novo Nordisk Foundation and the Stanford Bio-X Program. C. L. M. was supported by the NSERC Postgraduate Scholarship and the Stanford BioX Bowes Graduate Student Fellowship. Hector Lopez Hernandez was also partially supported by the NSF AGEP postdoctoral fellowship.References
- L. Bromberg, J. Controlled Release, 2008, 128, 99–112 CrossRef CAS PubMed.
- C. P. Fu, F. Ren, Q. Zhang, G. J. Lao and L. M. Zhang, Colloid Polym. Sci., 2015, 293, 2191–2200 CrossRef CAS.
- M. Gori, S. M. Giannitelli, M. Torre, P. Mozetic, F. Abbruzzese, M. Trombetta, E. Traversa, L. Moroni and A. Rainer, Adv. Healthcare Mater., 2020, 9, 2001163 CrossRef CAS PubMed.
- A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, J. Controlled Release, 2002, 82, 189–212 CrossRef CAS PubMed.
- Z. P. Liu, X. Y. Su, S. S. Liow, M. J. Tan, Z. B. Li, X. J. Loh, C. Chee and G. Lingam, Invest. Ophthalmol. Visual Sci., 2018, 59, 5923 Search PubMed.
- S. Fujishige, K. Kubota and I. Ando, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef CAS.
- L. Despax, J. Fitremann, M. Destarac and S. Harrisson, Polym. Chem., 2016, 7, 3375–3377 RSC.
- M. K. Gupta, J. R. Martin, T. A. Werfel, T. W. Shen, J. M. Page and C. L. Duvall, J. Am. Chem. Soc., 2014, 136, 14896–14902 CrossRef CAS PubMed.
- B. Hu, W. X. Fu and B. Zhao, Macromolecules, 2016, 49, 5502–5513 CrossRef CAS.
- Z. F. Lin, S. Q. Cao, X. Y. Chen, W. Wu and J. S. Li, Biomacromolecules, 2013, 14, 2206–2214 CrossRef CAS PubMed.
- C. H. Luo, N. Wei, X. F. Luo and F. L. Luo, Macromol. Chem. Phys., 2018, 219, 1800124 CrossRef.
- M. Onoda, T. Ueki, R. Tamate, A. M. Akimoto, C. C. Hall, T. P. Lodge and R. Yoshida, ACS Macro Lett., 2018, 7, 950–955 CrossRef CAS.
- C. Zhou, M. A. Hillmyer and T. P. Lodge, J. Am. Chem. Soc., 2012, 134, 10365–10368 CrossRef CAS PubMed.
- C. Zhou, G. E. S. Toombes, M. J. Wasbrough, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2015, 48, 5934–5943 CrossRef CAS.
- N. Bayat, Y. Zhang, P. Falabella, R. Menefee, J. J. Whalen, M. S. Humayun and M. E. Thompson, Sci. Transl. Med., 2017, 9, eaan3879 CrossRef PubMed.
- H. Therien-Aubin, Z. L. Wu, Z. H. Nie and E. Kumacheva, J. Am. Chem. Soc., 2013, 135, 4834–4839 CrossRef CAS PubMed.
- E. A. Archer and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 5074–5083 CrossRef CAS PubMed.
- G. Gody, R. Barbey, M. Danial and S. Perrier, Polym. Chem., 2015, 6, 1502–1511 RSC.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS PubMed.
- R. F. Chen, Arch. Biochem. Biophys., 1967, 120, 609–620 CrossRef CAS.
- E. A. Appel, J. del Barrio, X. J. Loh, J. Dyson and O. A. Scherman, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 181–186 CrossRef CAS.
- A. A. A. Smith, A. Hall, V. Wu and T. Xu, ACS Macro Lett., 2019, 8, 36–40 CrossRef CAS.
- R. G. Larson, The structure and rheology of complex fluids, Oxford university Press, New York (N.Y.), 1999 Search PubMed.
- N. G. Gaylord and J. R. Van Wazer, J. Polym. Sci., 1961, 51, S77–S78 Search PubMed.
- I. Bischofberger and V. Trappe, Sci. Rep., 2015, 5, 15520 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01696a |
| ‡ Current address: Technical University of Denmark, Department of Health Technology, 2800 Kgs. Lyngby, Denmark. |
| This journal is © The Royal Society of Chemistry 2021 |