
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiobased polymers derived from itaconic acid bearing clickable groups with potent antibacterial activity and negligible hemolytic activity†
A.
Chiloeches
ab,
A.
Funes
c,
R.
Cuervo-Rodríguez
c,
F.
López-Fabal
de,
M.
Fernández-García
af,
C.
Echeverría
 *af and
A.
Muñoz-Bonilla
*af
*af and
A.
Muñoz-Bonilla
*af
aInstituto de Ciencia y Tecnología de Polímeros (ICTP-CSIC), C/Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: sbonilla@ictp.csic.es; cecheverria@ictp.csic.es
bUniversidad Nacional de Educación a Distancia (UNED), C/Bravo Murillo, 38, 28015 Madrid, Spain
cFacultad de Ciencias Químicas, Universidad Complutense de Madrid, Avenida Complutense s/n, Ciudad Universitaria, 28040 Madrid, Spain
dHospital Universitario de Móstoles C/Dr. Luis Montes, s/n, 28935 Móstoles, Madrid, Spain
eFacultad de Ciencias Experimentales, Universidad Francisco de Vitoria, Carretera Pozuelo a Majadahonda, Km 1.800, 28223, Madrid, Spain
fInterdisciplinary Platform for Sustainable Plastics towards a Circular Economy-Spanish National Research Council (SusPlast-CSIC), Madrid, Spain
First published on 5th May 2021
Abstract
Herein, we report, for the first time, the synthesis of clickable polymers derived from biobased itaconic acid, which was then used for the preparation of novel cationic polymers with antibacterial properties and low hemotoxicity via click chemistry. Itaconic acid (IA) was subjected to chemical modification by incorporating clickable alkyne groups on the carboxylic acids. The resulting monomer with pendant alkyne groups was easily polymerized and copolymerized with dimethyl itaconate (DMI) by radical polymerization. The feed molar ratio of comonomers was varied to precisely tune the content of alkyne groups in the copolymers and the amphiphilic balance. Subsequently, an azide with a thiazole group, which is a component of the vitamin thiamine (B1), was attached onto the polymers by copper-catalyzed azide-alkyne cycloaddition (CuAAC) click chemistry leading to triazole linkages. N-Alkylation reactions of the thiazole and triazole groups with methyl and butyl iodides provide the corresponding itaconate derivatives with pendant azolium groups. The copolymers with variable cationic charge densities and hydrophobic/hydrophilic balances, depending on the comonomer feed ratio, display potent antibacterial activity against Gram-positive bacteria, whereas the activity was almost null against Gram-negative bacteria. Hemotoxicity assays demonstrated that the copolymers exhibited negligible hemolysis and excellent selectivity, more than 1000-fold, for Gram-positive bacteria over human red blood cells.
Introduction
In the last few years, antimicrobial peptides (AMPs) have inspired the synthesis of novel antimicrobial polymers with potent efficiency for the treatment of microbial infections also caused by antibiotic-resistant bacteria.1–3 These polymers are typically amphiphilic structures able to attach to negatively charged bacterial membranes with cationic hydrophilic segments, and, then, insert into them through the hydrophobic parts disrupting the cytoplasmic membrane.3,4 This action is rapid and makes it relatively difficult for the bacteria to develop resistance. As an additional advantage, in the majority of the reported synthetic polymers, the problems associated with AMPs such as high cost and poor pharmacokinetic properties have been overcome. However, most synthetic antimicrobial polymers are based on non-degradable backbones,5–8 which limit their application in clinical uses as they can be accumulated in the body and exert long term toxicity. Biodegradability is also an important and desired property for many biomedical applications including bioresorbable stents and prosthesis, food packaging and agricultural uses, which also contributes to sustainability by reducing the waste impact of fossil-based polymers. Although little research has been performed until now on the synthesis of biobased/biodegradable antimicrobial polymers,9 recently, a few examples have been reported that include functionalized polycarbonates10,11 polycaprolactone12 and polylactides.13 Biobased polymers such as polylactides also possess properties such as biocompatibility, environmental safety and sustainability, which could be essential in biomedical devices, wound dressing, food packaging, textiles and cosmetic applications. Therefore, research on sustainable antimicrobial materials is necessary and remains a challenge in the fields of polymer chemistry and materials science. On this basis, itaconic acid (IA) is a very promising biorenewable building block and one of the top chemicals obtained from biomass, whose annual production is estimated to be more than 80 kilotons.14 Itaconic acid is produced on a large scale by fermentation of biomass such as corn or rice, and also from lignocellulosic feedstocks.15 Due to the different functionalities of itaconic acid, polymeric derivatives can be synthesized either through radical polymerization16,17 of the itaconic acid via the α,β-unsaturated double bonds or through subjecting the double carboxylic groups to polycondensation.18,19 Thanks to these structural characteristics and its similarity to acrylic acid, IA and its derivatives such as dimethyl itaconate (DMI) have been extensively investigated as alternative monomers to prepare acrylic and methacrylic polymers. Also, the possibility of modifying the remaining functional groups via post polymerization reactions20,21 extends even more the potential of developing new materials with tunable properties. Most of these modification reactions reported in the literature involve the double bond functionalities rather than the carboxylic acid groups.Here, we proposed a new versatile method to modify the carboxylic acids of the itaconic acid monomer by incorporating pendant alkyne groups leading to clickable itaconic acid derivatives that can be polymerized via radical polymerization. This new approach can be used to further functionalize the IA-biobased polymers by copper-catalyzed azide–alkyne cycloaddition (CuAAC) click chemistry via triazole linkages. The facile and efficient reaction will allow the synthesis of new functional polymers. Specifically, we focused on incorporating antimicrobial azolium functionalities derived from vitamin thiamine (B1) to render biobased antimicrobial polymers.
Experimental section
Materials
For the preparation of polymers, the following chemicals were obtained. 2-(4-Methylthiazol-5-yl)ethanol azide was synthesized as previously described.22 Itaconic acid (IA, ≥99%), propargyl alcohol (≥99%), 4-(dimethylamino)pyridine (DMAP, ≥99%), N,N′-dicyclohexylcarbodiimide (DCC, 99%), hydroquinone (99%), copper(I) chloride (CuCl, ≥99.995%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), dimethyl itaconate (DMI, 99%), iodomethane (MeI, 99.5%), 1-iodobutane (BuI, 99%), neutral aluminum oxide, sodium bicarbonate (NaHCO3, ≥99.7%), magnesium sulfate anhydrous (MgSO4, ≥99.5%), ammonium persulfate (APS, 98%), anhydrous tetrahydrofuran (THF, 99.9%), and anhydrous N,N-dimethylformamide (DMF, 99.8%) were purchased from Sigma-Aldrich and used as received. The radical initiator 2,2′-azobisisobutyronitrile (AIBN, 98%) was purchased from Acros and was recrystallized twice from methanol. All the organic solvents were of AR grade, and tetrahydrofuran (THF), N,N-dimethylformamide (DMF), ethanol (EtOH), isopropyl alcohol (iPrOH), hexane and chloroform (CHCl3) were obtained from Scharlau. Ethyl acetate (EtOAc) was obtained from Cor Química S.L., toluene from Merck and sulfuric acid (H2SO4) from Panreac. Deuterated chloroform (CDCl3), water (D2O) and dimethyl sulfoxide (DMSO-d6) were acquired from Sigma-Aldrich. Cellulose dialysis membranes (CelluSep T1) were purchased from Membrane Filtration Products, Inc.For the antibacterial assay, the following were obtained: sodium chloride solution (NaCl suitable for cell culture, BioXtra) and phosphate buffered saline powder (pH 7.4) were obtained from Sigma-Aldrich. BBL Mueller–Hinton broth used as a microbial growth medium was purchased from Becton, Dickinson and Company and 96 well microplates were purchased from BD Biosciences. Columbia agar (5% sheep blood) plates were obtained from BioMérieux. American Type Culture Collection (ATCC): Pseudomonas aeruginosa (P. aeruginosa, ATCC 27853), Escherichia coli (E. coli, ATCC 25922), Staphylococcus epidermidis (S. epidermidis, ATCC 12228) and Staphylococcus aureus (S. aureus, ATCC 29213), used as bacterial strains, were purchased from Oxoid.
Characterization
1H and 13C NMR spectra were recorded on a Bruker Avance III HD-400AVIII spectrometer at room temperature using CDCl3, DMSO-d6 and D2O as solvents (purchased from Sigma-Aldrich). Fourier transform infrared (FTIR) spectra were recorded on a PerkinElmer Spectrum Two instrument, equipped with an attenuated total reflection (ATR) module. Mass spectrometry (MS) analysis was performed for high resolution ESI measurements using an Agilent 6500 Series Accurate-Mass Q-TOF LC/MS System, equipped with an Agilent 1200 LC. Size exclusion chromatography (SEC) measurements were performed using a Waters Division Millipore system equipped with a Waters 2414 refractive index detector. DMF (Scharlau) stabilized with 0.1 M LiBr (Sigma Aldrich, >99.9%) was used as an eluent at a flow rate of 1 mL min−1 at 50 °C. Calibration was made with poly(methyl methacrylate) standards (Polymer Laboratories Ltd). Zeta potential measurements of the cationic copolymers in distilled water at 25 °C were performed with a Zetasizer Nano series ZS (Malvern Instruments Ltd) using the Smoluchowski equation.Synthesis of di(prop-2-yn-1-yl) itaconate (PrI)
The monomer di(prop-2-yn-1-yl)itaconate bearing clickable alkyne groups was synthesized via the condensation reaction of itaconic acid with propargyl alcohol according to Scheme 1.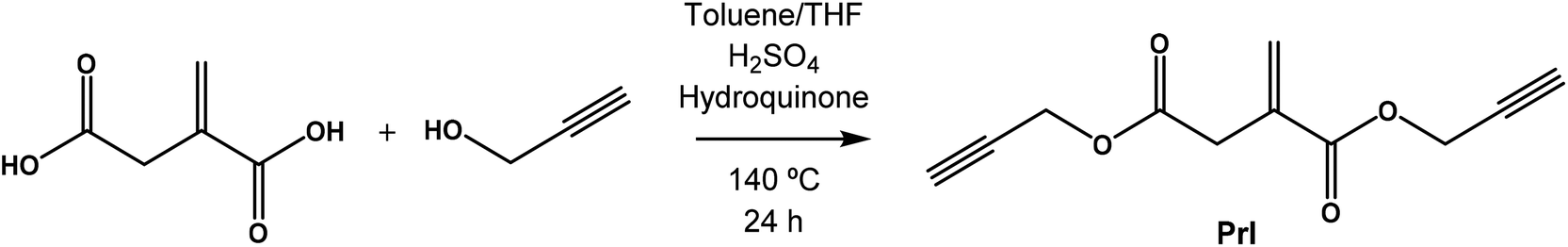 | ||
| Scheme 1 Synthesis of di(prop-2-yn-1-yl) itaconate (PrI). | ||
Briefly, itaconic acid (15.0 g, 115 mmol), propargyl alcohol (32.3 g, 576 mmol) and hydroquinone (1.26 g, 11.5 mmol) were placed in a three neck flask equipped with a Dean–Stark trap and the mixture was dissolved in THF (50 mL) at 60 °C. Then, toluene (250 mL) and H2SO4 (300 μL, 5.75 mmol) were added and the reaction mixture was heated under reflux for 24 h, during which period 4 mL of water was collected. After that, the solvents were partially removed under reduced pressure using a rotary evaporator, and the mixture was washed repeatedly with saturated NaHCO3 aqueous solution. The organic extract was dried over anhydrous MgSO4 and then filtered. The residual reaction mixture was finally purified by passing through a neutral alumina column using hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc (1:1) as a solvent. After solvent evaporation under reduced pressure, a yellow oil was obtained (22.998 g, 97% yield). HR-MS (ESI): m/z required for C11H10O4, 206.05863; found, 206.05791.
:EtOAc (1:1) as a solvent. After solvent evaporation under reduced pressure, a yellow oil was obtained (22.998 g, 97% yield). HR-MS (ESI): m/z required for C11H10O4, 206.05863; found, 206.05791.
1
H-NMR (400MHz, CDCl
3
), δ(ppm): 6.40 (d, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.80 (d, 1H, CH2), 4.76 (d, 2H, –CH2C
CH2), 5.80 (d, 1H, CH2), 4.76 (d, 2H, –CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 4.70 (d, J = 2.45, 2H, –CH2CCH), 3.40 (s, 2H, –CH2), 2.48 (m, J = 2.45, 2H, –CH2CCH).
CH), 4.70 (d, J = 2.45, 2H, –CH2CCH), 3.40 (s, 2H, –CH2), 2.48 (m, J = 2.45, 2H, –CH2CCH).
13
C-NMR (100MHz, CDCl
3
), δ(ppm): 169.80 (CO), 165.75 (CO), 132.78 (–CCH2), 130.03 (–CCH2), 75.22 (2C, –CH2CCH), 52.72 (–CH2CCH), 52.56 (–CH2CCH), 37.38 (–CH2).
Synthesis of bis((1-(2-(4-methylthiazol-5-yl)ethyl)-1H-1,2,3-triazol-4-yl)methyl) itaconate (TTI)
The monomer TTI bearing thiazol and triazol moieties was synthesized by the alkyne–azide click cycloaddition reaction between the PrI monomer previously prepared and 2-(4-methylthiazol-5-yl)ethanol azide (see Scheme S1 in the ESI†). In a typical experiment, PrI (0.612 g, 2.97 mmol), 2-(4-methylthiazol-5-yl)ethanol azide (1.025 g, 6.10 mmol), PMDETA (208 μL, 1 mmol) and CuCl (0.030 g, 0.30 mmol) were dissolved in 30 mL of CHCl3. The reaction mixture was stirred at room temperature for 24 h. Then, the reaction mixture was passed through a neutral alumina column to remove the copper compounds. The monomer was purified by column chromatography using first hexane:EtOAc (from 1:1 to 1:3) as a solvent and then ethanol. Afterwards, the solvent was removed by rotary evaporation to obtain a yellow liquid (0.822 g, 51% yield).
1
H-NMR (400 MHz, CDCl
3
), δ(ppm): 8.57 (s, 1H, H-thiazole), 8.56 (s, 1H, H-thiazole), 7.42 (s, 1H, H-triazole), 7.40 (s, 1H, H-triazole), 6.32 (s, 1H, CH2), 5.70 (s, 1H, CH2), 5.22 (s, 2H, O–CH2-triazole), 5.15 (s, 2H, O–CH2-triazole), 4.55 (t, J = 6.8, 4H, CH2–N), 3.39 (t, J = 6.8, 4H, CH2-thiazole), 3.32 (s, 2H, –CH2–), 2.21 (s, 6H, CH3-thiazole).
13
C-NMR (100 MHz, CDCl
3
), δ(ppm): 170.50 (CO), 165.90 (CO), 150.85 (2C, thiazole C–CH3), 150.50 (2C, thiazole C–H), 142.77 (2C, triazole Cquat), 133.23 (–CCH2), 129.77 (–CCH2), 125.85 (2C, thiazole Cquat), 124.5 (triazole C–H), 124.4 (triazole C–H), 58.26 (O–CH2–), 58.14 (O–CH2–), 51.18 (2C, CH2–N), 37.94 (–CH2–), 27.45 (2C, –CH2 thiazole), 14.71 (2C, CH3-thiazole).
Synthesis of poly(di(prop-2-yn-1-yl)itaconate-co-dimethyl itaconate) copolymers (P100, P75, P50, P25, and P0)
A series of copolymers and homopolymers with different chemical compositions were prepared by conventional radical polymerization of PrI and DMI comonomers using different feed molar ratios (PrI/DMI = 100/0, 75/25, 50/50, 25/75 and 0/100), at a total concentration of 2 M in anhydrous DMF, at 70 °C. The copolymers were named P100, P75, P50, P25 and P0 for the feed molar ratios PrI/DMI = 100/0, 75/25, 50/50, 25/75 and 0/100, respectively. Briefly (e.g. for the sample PrI/DMI = 50/50, copolymer P50), both monomers, PrI (2.06 g, 10.0 mmol) and DMI (1.59 g, 10.0 mmol), at a total concentration of 2 M in anhydrous DMF, were added into a glass tube. Subsequently, the initiator AIBN (0.16 g, 1.0 mmol) was added and the mixture was deoxygenated by purging argon for 15 min. The polymerization reaction mixture was stirred at 70 °C for 24 h. The copolymer was isolated by precipitation in ethanol, yielding a white solid, which was collected and dried overnight under vacuum at room temperature (1.654 g, 45%).
Copolymer P50:
1
H-NMR (400 MHz, CDCl
3
), δ(ppm): 4.67 (4H, –CH2CCH), 3.58 (6H, O–CH3), 2.49 (2H, –CH2CCH), 1.99–1.00 (8H, CH2–CO and –CH2-chain).
Copolymer P50:
1
H-NMR (400 MHz, DMSO-d
6
), δ(ppm): 4.67 (4H, –CH2CCH), 3.58 (6H, O–CH3 and 2H, –CH2CCH), 3.00–1.50 (8H, –CH2CO and CH2-chain–).
Synthesis of poly(bis((1-(2-(4-methylthiazol-5-yl)ethyl)-1H-1,2,3-triazol-4-yl)methyl)itaconate-co-dimethyl itaconate) copolymers (P100T, P75T, P50T and P25T)
The incorporation of the thiazole and triazole moieties into the homopolymer (P100) and copolymers (P75, P50, and P25) was performed by Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) click chemistry using 2-(4-methylthiazol-5-yl)ethanol azide. In a typical procedure, the copolymer (e.g. for the sample P50) (1.60 g, 8.8 meq. of alkyne groups), 2-(4-methylthiazol-5-yl)ethanol azide (1.51 g, 9.0 mmol), PMDETA (312 μL, 1.5 mmol) and CuCl (0.05 g, 0.5 mmol) were dissolved in 40 mL of CHCl3. The mixture was stirred at room temperature for 24 h and then it was passed through a neutral alumina column. The resulting copolymer was isolated by precipitation in hexane, and the degree of modification was almost quantitative. The copolymers were named P100T, P75T, P50T, and P25T for the molar ratios TTI/DMI = 100/0, 75/25, 50/50 and 25/75, respectively.Copolymer P50T: 1 H-NMR (400 MHz, CDCl 3 ), δ(ppm): 8.61 (2H, H-thiazole), 7.71 (2H, H-triazole), 5.15 (4H, O–CH2-triazole), 4.62 (4H, CH2–N), 3.64 (6H, O–CH3) 3.45 (4H, CH2-thiazole), 2.21 (6H, CH3-thiazole), 2.00–1.00 (8H, CH2–CO and –CH2-chain).
Copolymer P50T: 1 H-NMR (400 MHz, DMSO-d 6 ), δ(ppm): 8.74 (2H, H-thiazole), 8.00 (2H, H-triazole), 5.01 (4H, O–CH2-triazole), 4.52 (4H, CH2–N), 3.50 (6H, O–CH3) 3.26 (4H, CH2-thiazole), 2.10 (6H, CH3-thiazole), 3.00–1.50 (8H, CH2CO and CH2-chain–).
Quaternization reactions: synthesis of cationic polymers (P100T-Q, P75T-Q, P50T-Q, and P25T-Q)
The homopolymer (P100T) and the copolymers (P75T, P50T and P25T) were modified by the N-alkylation reaction with either iodomethane (MeI) or 1-iodobutane (BuI) leading to the corresponding cationic polymers. A typical quaternization reaction with MeI is described below for the P50T copolymer as an example. The copolymer (1.50 g, 4.3 meq. of thiazole and 4.3 meq. of triazole groups) was dissolved in 25 mL of anhydrous DMF and then a large excess of MeI was added (2.7 mL, 43.0 mmol; the ratio of thiazole and triazole groupstoalkyl iodide is ≈ 1:5). The mixture was deoxygenated with argon for 15 min, sealed, and then stirred at 70 °C for one week to achieve a high degree of modification. The resulting cationic copolymer was purified by precipitation in n-hexane followed by dialysis against distilled water and finally was isolated by freeze-drying. The degree of quaternization was almost quantitative. The copolymers quaternized with methyl iodide were named P100T-Me, P75T-Me, P50T-Me, and P25T-Me and those quaternized with butyl iodide were named P100T-Bu, P75T-Bu, P50T-Bu, and P25T-Bu.
Homopolymer P100T-Me: 1 H-NMR (400 MHz, D 2 O), δ(ppm): 8.89 (2H, H-thiazole), 7.96 (2H, H-triazole), 5.44 (4H, O–CH2-triazole), 5.02 (4H, CH2–N), 4.37 (6H, N+CH3 triazole), 4.10 (6H, N+CH3 thiazole), 3.77 (4H, CH2thiazole), 2.51 (6H, CH3-thiazole).
Homopolymer P100T-Bu: 1 H-NMR (400 MHz, D 2 O), δ(ppm): 8.93 (2H, H-thiazole), 8.20 (2H, H-triazole), 5.46 (4H, O–CH2-triazole), 5.05 (4H, CH2–N), 4.65 (4H, N+CH2 triazole), 3.45 (4H, N+CH2 thiazole), 3.9–3.50 (4H, CH2-thiazole), 2.54 (6H, CH3-thiazole), 1.90 (8H, CH2–CH2–CH3), 1.37 (8H, CH2–CH2–CH3), 0.96 (12H, CH2–CH2–CH3).
Copolymer P50T-Me: 1 H-NMR (400 MHz, D 2 O), δ(ppm): 8.92 (2H, H-thiazole), 8.07 (2H, H-triazole), 5.48 (4H, O–CH2-triazole), 5.05 (4H, CH2–N), 4.41 (6H, N+CH3 triazole), 4.13 (6H, N+CH3 thiazole), 3.80 (4H, CH2-thiazole), 3.68 (6H, –O–CH3), 2.53 (6H, CH3-thiazole).
Copolymer P150T-Bu: 1 H-NMR (400 MHz, D 2 O), δ(ppm): 8.96 (2H, H-thiazole), 8.26 (2H, H-triazole), 5.47 (4H, O–CH2-triazole), 5.10 (4H, CH2–N), 4.68 (4H, N+CH2 triazole), 4.47 (4H, N+CH2 thiazole), 3.82 (4H, CH2-thiazole), 3.68 (6H, –O–CH3), 2.54 (6H, CH3-thiazole), 1.93 (8H, CH2–CH2–CH3), 1.40 (8H, CH2–CH2–CH3), 0.95 (12H, CH2–CH2–CH3).
Antibacterial assays
The antibacterial activities of the cationic polymers were tested following a standard broth dilution method according to the Clinical Laboratory Standards Institute (CLSI) to determine the minimum inhibition concentrations (MICs).23 Bacterial cells were grown on 5% sheep blood Columbia agar plates for 24 h at 37 °C. Subsequently, the bacterial concentration was adjusted with saline to 108 colony-forming units (CFU) mL−1 (turbidity equivalent to ca. 0.5 McFarland turbidity standard). These suspensions were further diluted to 106 CFU mL−1 with fresh Mueller–Hinton broth. Stock solutions of the polymers at a concentration of 20000 μg mL−1 were prepared in the Mueller–Hinton broth medium using a minimum amount of DMSO (less than 6% v/v; higher DMSO content was found to be toxic for these bacterial strains22,24). Then, 100 μL from each polymer solution were placed in the first column of a 96-well round-bottom microplate, and 50 μL of broth was added into the rest of the wells. From the first column, polymer solution (50 μL) was diluted by 2-fold serial dilutions in the rest of the wells, followed by the addition of 50 μL of the bacterial to yield a total volume of 100 μL and a bacterial concentration of 5 × 105 CFU mL−1. A positive control without the polymer and a negative control without bacteria were also prepared. The plates were incubated at 37 °C for 24 h, and the MIC values were determined by checking the absence of bacterial growth visually. All the tests were performed in triplicate.
Hemotoxicity assays
Hemolysis studies were carried out as described previously.24,25 Fresh human blood was collected from healthy donors and used within the same day. Blood was drawn directly into the blood collecting tubes containing EDTA to prevent coagulation. The tubes were centrifuged at 3500 rpm for 20 min. Afterwards, the supernatant (plasma) and the buffy coat at the middle (white blood cells) were discarded and the red blood cells (RBCs) at the bottom were collected. The RBCs were washed with fresh sterile PBS and centrifuged three times. Subsequently, the RBCs were resuspended to a final concentration of 5% (v/v) in PBS. Polymer solutions were prepared in a mixture of PBS and a minimum amount of DMSO (up to 5% v/v, nontoxic under the experimental conditions used in this study) at a concentration of 40000 μg mL−1. Then, 100 μL of these polymer solutions were added in the first column of 96-well round-bottom microplates, while in the rest of the wells, 50 μL of PBS were added. Two-fold sequential dilutions of the polymer solutions were performed to obtain a series of concentrations, and finally, 150 μL of the RBC suspension were added to each well. Equally, Triton X-114 (1% v/v solution in PBS) was used as a positive control for 100% of hemolysis, whereas PBS was used as a negative control for 0% hemolysis. The microplates were incubated for 1 h at 37 °C. After this period, the plates were centrifuged at 1000 rpm for 10 min and the resulting supernatant in each well was transferred to a new 96-well microplate. Hemolysis was monitored by measuring the absorbance of the released hemoglobin at 550 nm using a microplate reader (Synergy HTX Multi-Mode Reader spectrophotometer, Bio-Tek). The percentage of hemolyzed erythrocytes was calculated according to the following equation: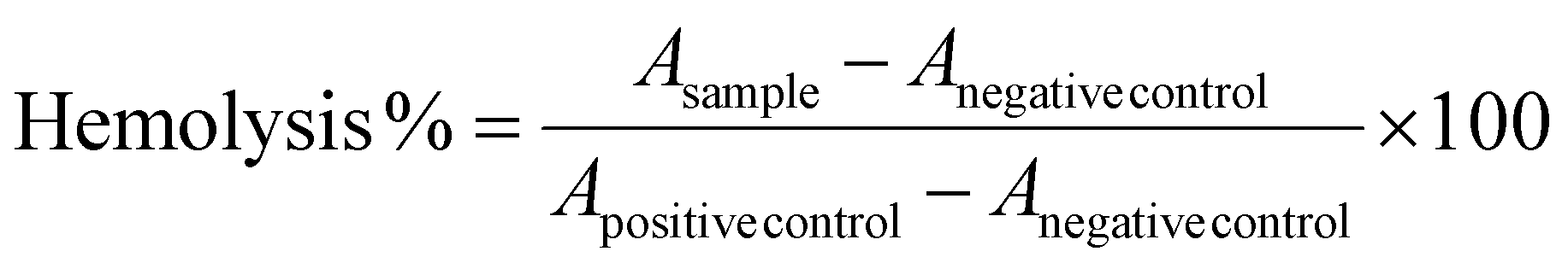
An absolutely achromatic supernatant solution indicates no hemolysis (Anegative control) while a red solution indicates hemolysis (Apositive control). All experiments were performed in triplicate, and the data were expressed as mean ± SD (n = 3).
Results and discussion
Synthesis of antibacterial polymers derived from itaconic acid
Antimicrobial biobased polymers derived from itaconic acid were prepared following a novel strategy consisting of the incorporation of clickable alkyne groups into the structure, which enables the posterior inclusion of antimicrobial groups by click chemistry. For this purpose, three different synthetic approaches were proposed (Scheme S1 in the ESI† and Scheme 2). | ||
| Scheme 2 Synthesis of the antibacterial cationic copolymers derived from itaconic acid. | ||
In the first approach, presented in Scheme S1A,† we attempted the radical polymerization of IA through the α,β-unsaturated double bond using an APS initiator in aqueous media at 70 °C for 24 h. Subsequently, the obtained poly(itaconic acid) (PIA) was subjected to post-modification of the two carboxylic acid functionalities by the condensation reaction with propargyl alcohol, using EDC/NHS chemistry in aqueous media. However, this approach was discarded because, in addition to the problems associated with the polymerization of PIA such as low conversion and slow rate, the poly(itaconic acid) was insoluble in most organic solvents. Then, the incorporation of propargyl alcohol into the PIA led to a polymer insoluble in an aqueous reaction medium, which hindered the complete modification of PIA.
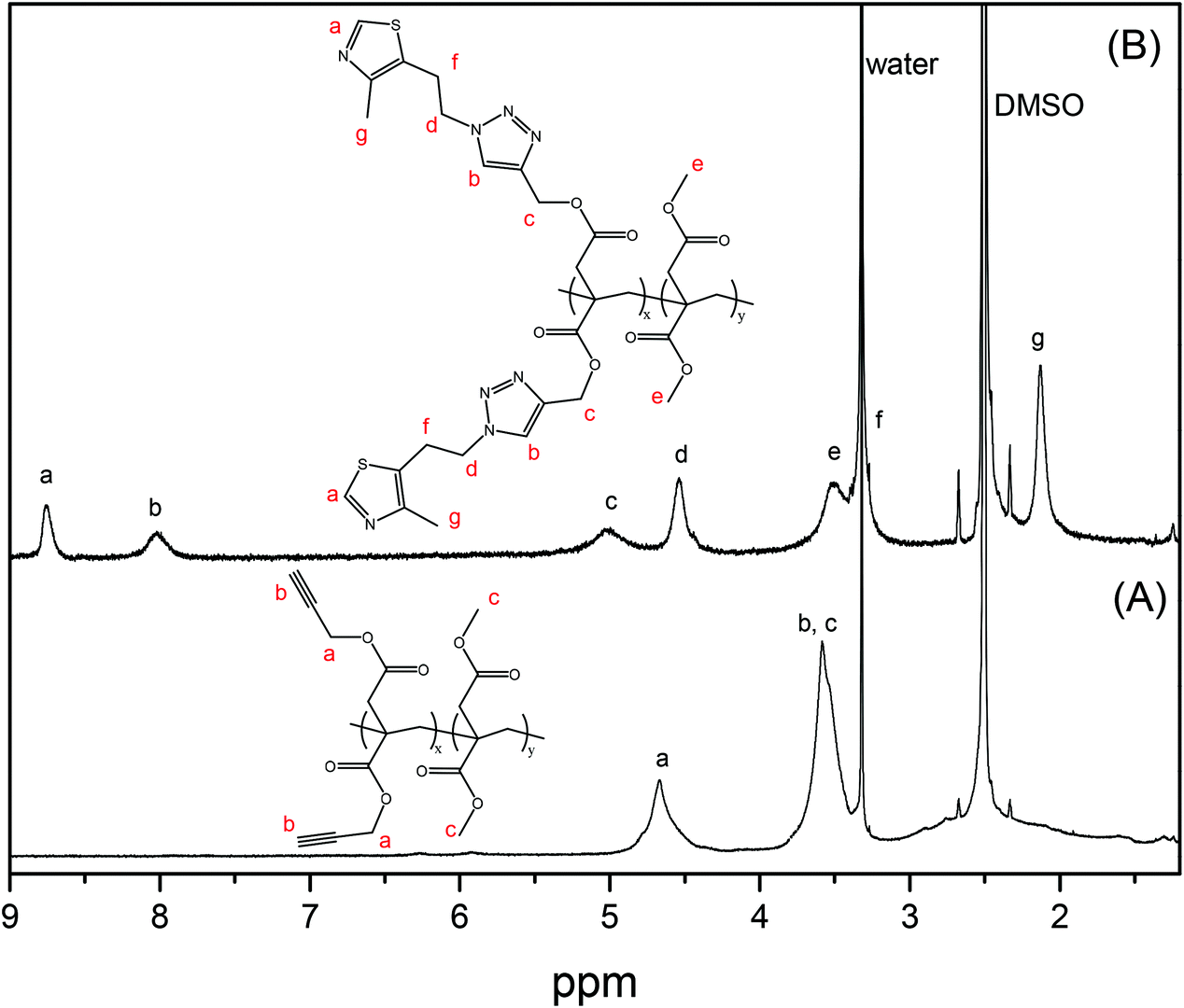
The second approach that we considered was the synthesis of a clickable monomer derived from IA, di(prop-2-yn-1-yl)itaconate (PrI) (see the Experimental section and Scheme S1B†). In this approach, the solubility issues were solved as the IA monomer has a higher solubility than PIA. The PrI monomer was successfully synthesized by the reaction between IA and propargyl alcohol in a mixture of toluene/THF and H2SO4, reaching a yield of 97%. Fig. 1A shows the 1H-NMR spectrum of the obtained PrI monomer, which confirms the complete functionalization of the carboxylic acid groups and the presence of the vinyl signals at 6.40 and 5.80 ppm. The 13C-NMR spectrum displayed in Fig. S1A in the ESI† also corroborated the chemical structure of the PrI monomer. The second step of this approach consisted in the incorporation of thiazole groups by Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) click chemistry between the alkyne groups of PrI and 2-(4-methylthiazol-5-yl)ethanol azide forming a 1,2,3-triazole group, which is also susceptible to quaternization. Likewise, 1H-NMR (Fig. 1B) and 13C-NMR spectra show typical peaks of the introduced functional groups, confirming the success of the reaction and the formation of the monomer derivative with thiazole and triazole groups in its structure (TTI). See the ESI, Fig. S1B,† for the 13C NMR spectrum of this monomer.
 | ||
| Fig. 1 1H-NMR spectra of (A) clickable monomer PrI and (B) monomer derivative TTI with thiazole and triazole groups in deuterated chloroform. | ||
In spite of the satisfactory synthesis of the biobased monomer TTI, this monomer did not easily polymerize or copolymerize with a comonomer such as dimethyl itaconate (DMI). We tried different polymerization conditions, both in bulk and in DMF solution. However, after 48 h of reaction, we were not able to obtain any polymer under all conditions tested, probably due to high steric hindrance among other reasons.
Then, a third approach was proposed (Scheme 2), the development of a clickable polymer derived from itaconic acid instead of using a clickable monomer. In this strategy, the clickable monomer PrI was successfully radical homopolymerized and copolymerized with dimethyl itaconate in DMF solution, at a total monomer concentration of 2 M, at 70 °C, with AIBN as a radical initiator. Therefore, homopolymers and a series of copolymers with different chemical compositions were obtained using different feed molar ratios (PrI/DMI = 100/0, 75/25, 50/50, 25/75 and 0/100).
Fig. 2 and 3 show the FTIR and 1H-NMR spectra, respectively, of the clickable copolymer P(PrI-co-DMI) for a feed chemical composition of PrI/DMI = 50/50 (named P50). In the FTIR spectrum, the characteristic peak of the alkyne C–H stretching band at 3283 cm−1 and the bands assigned to the CC bond at 2128 cm−1 and the CC–H bond at 642 cm−1 clearly demonstrate the successful synthesis of the alkyne-functionalized polymers. Likewise, in the 1H-NMR spectrum (Fig. 3A), the terminal methyne proton of the alkyne groups appears at 3.58 ppm and the rest of the signals are consistent with the chemical structure of the copolymer. The chemical compositions of the obtained polymers, thus the molar content of PrI in the copolymer, were calculated by integration of the 1H NMR spectral signals at 3.50 ppm (6H, O–CH3 from DMI) compared to the signal at 4.67 ppm (4H) from PrI. Table 1 summarizes the molecular characteristics of the synthesized copolymers and homopolymers including the obtained chemical compositions. The molar ratios of the comonomers in the copolymers were found to be very similar to the feed molar ratios, and therefore, polymers will be referred to by their feed compositions to maintain uniformity. This fact is important because the copolymer composition can be easily modulated by varying the comonomer feed ratios.
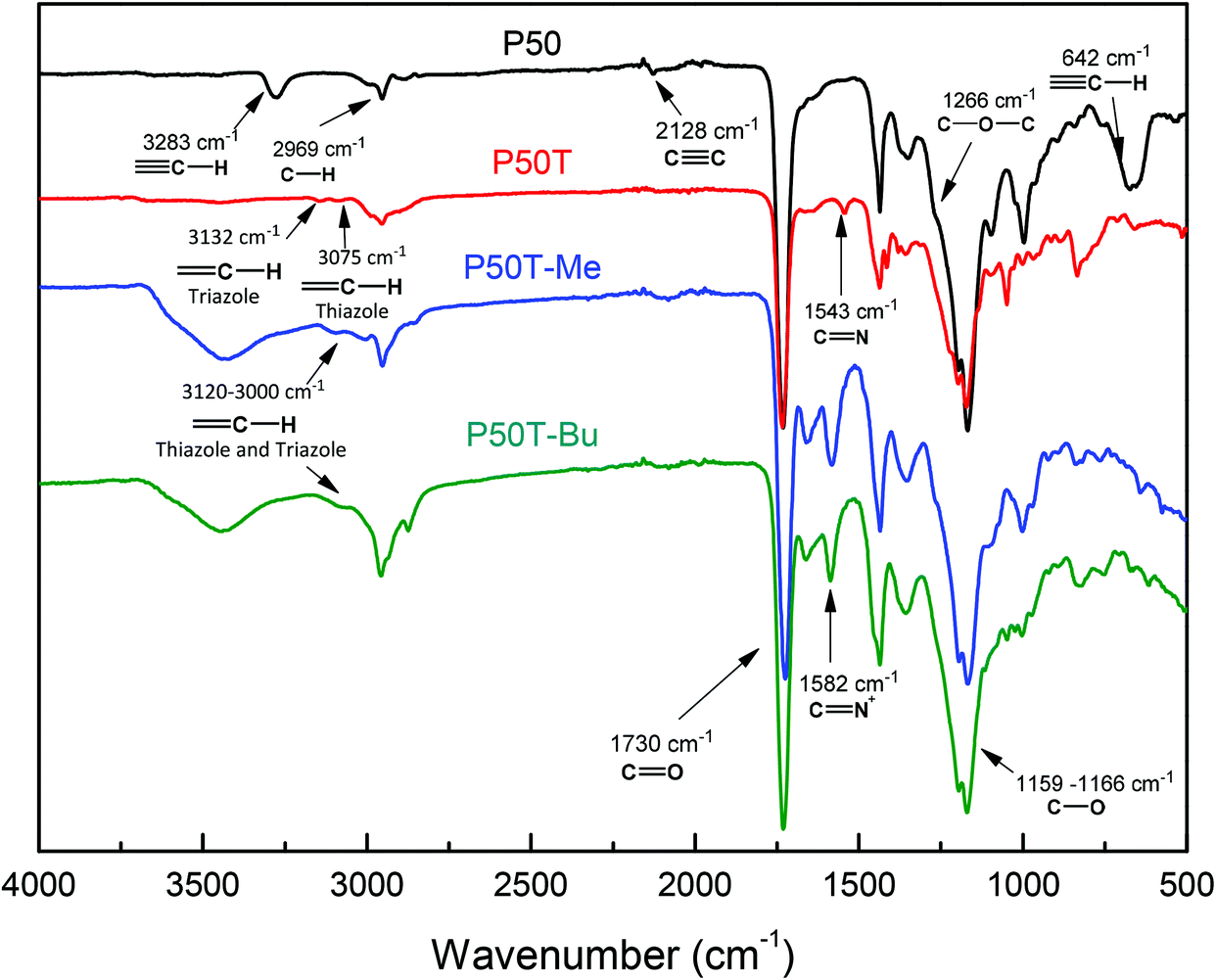 | ||
| Fig. 2 FTIR spectra of the copolymers P50, P50T, P50T-Me and P50T-Bu. | ||
 | ||
| Fig. 3 1H NMR spectra of the copolymers (A) P50 and (B) P50T in DMSO-d6. | ||
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n) and molecular weight dispersity (Đ) determined by SEC in DMF as an eluent. n and Đ values of the polymers after click reactions leading to P(TTI-co-DMI) are also included
n) and molecular weight dispersity (Đ) determined by SEC in DMF as an eluent. n and Đ values of the polymers after click reactions leading to P(TTI-co-DMI) are also included
| Sample P(PrI-co-DMI) | Feed ratio [PrI]/[DMI] | Polymer ratio [PrI]/[DMI]a |
n (g mol−1) |
Đ | Sample P(TTI-co-DMI) |
nb (g mol−1) |
Đ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Polymer ratio [PrI]/[DMI] was determined by 1H-NMR.
b
n and Đ values of the polymers after click reactions leading to P(TTI-co-DMI) polymers.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P100 | 100.0/0 | 100.0/0 | 6700 | 1.58 | P100T | 7100 | 1.59 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P75 | 75.0/25.0 | 72.5/27.5 | 8100 | 1.35 | P75T | 11100 |
1.27 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P50 | 50.0/50.0 | 50.1/49.9 | 7500 | 1.31 | P50T | 11700 |
1.25 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P25 | 25.0/75.0 | 23.1/76.9 | 6300 | 1.36 | P25T | 6800 | 1.49 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P0 | 0/100.0 | 0/100.0 | 4800 | 1.27 | P0 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The molecular weights of the polymers were determined using SEC (Table 1) and were found to be low for all polymers, with values in the range of n = 4800–8100 g mol−1, presenting low molecular weight dispersity (Đ) (1.58–1.27).
Once clickable polymers were synthesized with different contents of alkyne groups, the next step consists in the CuAAC click chemistry with 2-(4-methylthiazol-5-yl)ethanol azide, which led to polymers with thiazole and 1,2,3-triazole groups, P(TTI-co-DMI). The post-polymerization reaction was carried out under mild conditions and the degree of modification was almost quantitative. After the click reaction, the molecular weights of the polymers detected by SEC increased as a result of the incorporation of the azide molecules (Table 1), while the polydispersity indexes practically did not change. Equally, the new thiazole and triazole groups attached to the polymer structures were detected by FTIR. Fig. 2 shows the FTIR spectrum of P50T. The signals at 3132 and 3075 cm−1 corresponding to the C–H stretching vibrations of the triazole and thiazole groups, the signal at 1543 cm−1 attributed to the CN bond of thiazole, and the absence of the band associated with the CN bond clearly indicate the successful coupling. The NMR spectra also confirm the completion of the click reaction and the formation of the P(TTI-co-DMI) copolymers. All the signals of the 1H NMR spectra were consistent with the expected structures. The 1H NMR spectrum of P50T displayed in Fig. 3B, as an example, shows the characteristic peaks at 8.74 ppm and 8.00, corresponding to the thiazole and triazole protons, respectively, concomitant with the disappearance of propargyl methylene signal at 4.67 ppm.
The last step of the synthesis procedure consists in the incorporation of permanent positive charges into the polymers to provide them with antibacterial activities. Both the pendant nucleophilic azole groups, triazole and thiazole, can be modified using very reactive alkylating reagents such as alkyl iodides. In this study, two alkylating agents with different chain lengths were used, methyl and butyl iodide, to tune the final hydrophobic/hydrophilic balance of the copolymer. This hydrophobic/hydrophilic balance is well known to have a strong influence on the antimicrobial activity and toxicity of the resulting polymers,2,26–28 and, in this work, it was also controlled by the content of the hydrophobic comonomer DMI. The synthesized P(TTI-co-DMI) (P100T, P75T, P50T and P25T) copolymers were reacted with a large excess of either methyl iodide or butyl iodide in DMF at 70 °C under an argon atmosphere. The reaction was performed for one week to ensure quantitative modification, which was confirmed by NMR and FTIR spectroscopy. Fig. 2 shows the comparison of the FTIR spectra of the quaternized copolymers with either methyl or butyl iodide of the samples with a molar ratio [PrI]/[DMI] = 50 (P50T-Me and P50T-Bu, respectively) with their unquaternized copolymer precursor (P50T). In both cases, the band at 1543 cm−1 assigned to the ν(CN) disappears and a new band corresponding to the ν(CN)+ clearly emerges due to the formed thiazolium and triazolium groups, after the quaternization reactions. Similarly, the 1H NMR spectrum demonstrates the success of the quaternization reaction and almost quantitative modifications of the thiazole and triazole groups (Fig. 4). The two peaks assigned to the aromatic protons of the 1,2,3-thiazole (∼8.6 ppm) and 1,3-triazole (8.0–7.6 ppm) rings shift to lower field regions (∼8.90 and ∼8.3–8.0 ppm, respectively) as a result of the quaternization reaction due to the higher polarity resulting from the formation of azolium groups. Fig. S2, ESI†, displays the 1H-NMR spectrum of P50T-Me in DMSO-d6, which supports the signal assignments. Likewise, Figs. S3 and S4 in the ESI† display the COSY-NMR spectra of the P50T-Me and P50T-Bu copolymers, which were also obtained to support the signal assignments in the 1H-NMR spectra of the Hf and Hg protons at 3.80 ppm (4H, CH2-thiazole) and 3.68 ppm (6H, –O–CH3), respectively. These assignments were verified through the analysis of the COSY-NMR spectra based on the correlation of the Hf protons with the Hd protons.
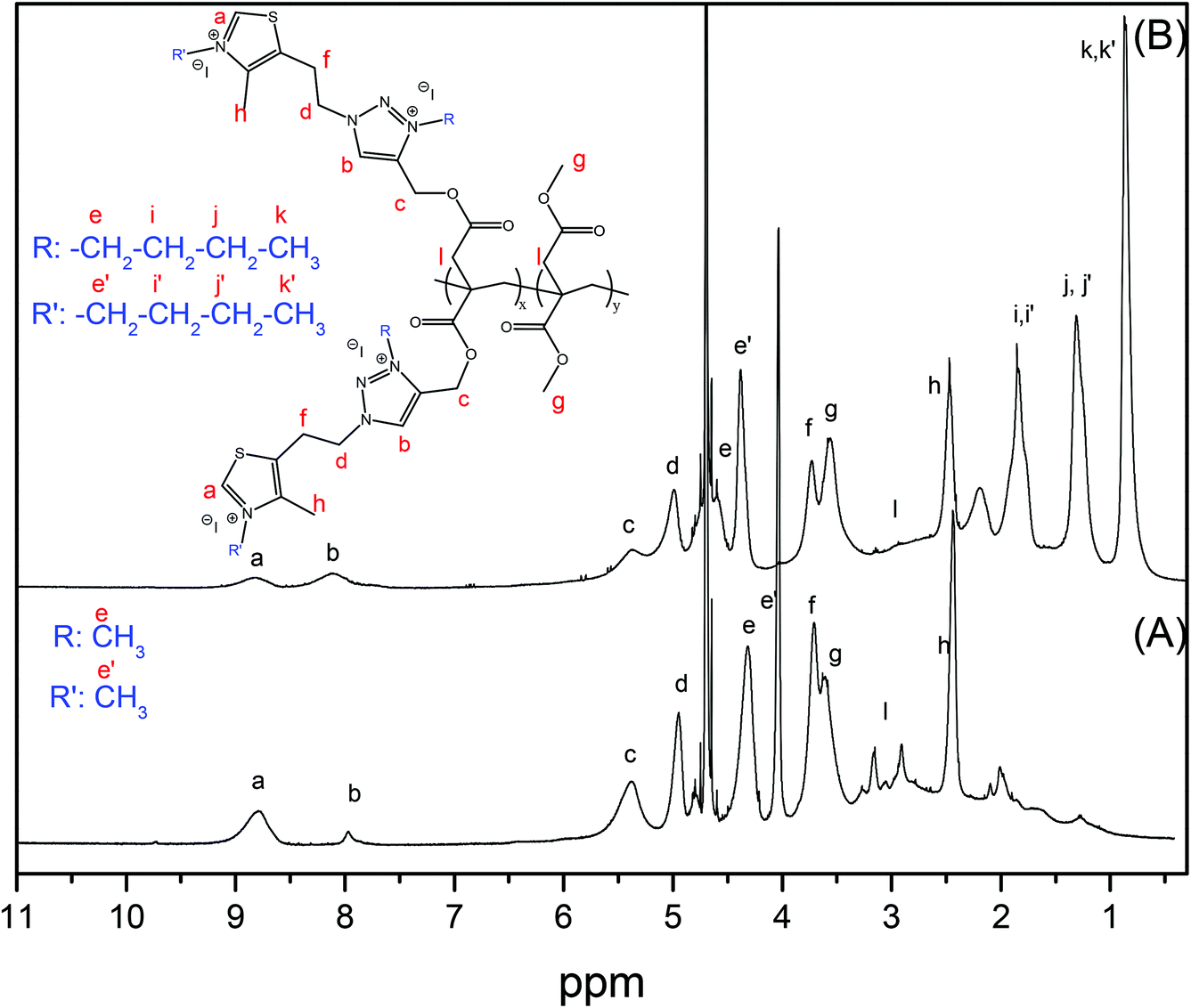 | ||
| Fig. 4 1H-NMR spectra of the copolymers (A) P75T-Me and (B) P75T-Bu in deuterated water. | ||
For the estimation of the positive charge density of the synthesized copolymers, zeta potential measurements were performed in distilled water. Table 2 shows the zeta potential values (ζ) obtained for all the synthesized copolymers. As confirmed by NMR and FTIR studies, the quaternization reactions result in cationic copolymers with high positive ζ values, around +50 mV for both methylated and butylated copolymers. Only the copolymers with a low content of cationic units, P50T-Me and P25T-Me, present reduced charge density.
| ζ (mV) | ζ (mV) | ||||||
|---|---|---|---|---|---|---|---|
| P100T-Me | P75T-Me | P50T-Me | P25T-Me | P100T-Bu | P75T-Bu | P50T-Bu | P25T-Bu |
| 56 ± 1 | 49 ± 3 | 38 ± 2 | 40 ± 3 | 48 ± 1 | 48 ± 2 | 49 ± 2 | 51 ± 2 |
Antibacterial activities of cationic polymers
The antibacterial efficiency of the obtained cationic polymers derived from biobased itaconic acid was evaluated for both Gram-negative (P. aeruginosa and E. coli) and Gram-positive (S. epidermidis and S. aureus) opportunistic bacteria. The minimum inhibitory concentration (MIC) of the polymer derivatives, which is the lowest concentration of a polymer needed to prevent the visible growth of bacteria, was measured by the CLSI microbroth dilution reference method23 against the different bacterial strains. The MIC values determined for all the polymers tested are summarized in Table 3.| Copolymer | MIC (μg mL−1) | HC50 (μg mL−1) | HC50/MICa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | S. epidermidis | S. aureus | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Calculated based on the MIC values of S. aureus. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P100T-Me | >10000 |
10000 |
31 | 10 | >10000 |
>1000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P75T-Me | 10000 |
10000 |
31 | 10 | >10000 |
>1000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P50T-Me | >10000 |
10000 |
312 | 312 | >10000 |
>32 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P25T-Me | >10000 |
10000 |
312 | 312 | >10000 |
>32 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P100T-Bu | >10000 |
5000 | 8 | 10 | >10000 |
>1000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P75T-Bu | 5000 | 5000 | 8 | 10 | >10000 |
>1000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P50T-Bu | 5000 | 10000 |
16 | 10 | >10000 |
>1000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P25T-Bu | >10000 |
10000 |
31 | 10 | >10000 |
>1000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Remarkably, huge differences in the activities of the polymers against Gram-positive and Gram-negative bacteria are clearly appreciated. Whereas the biobased cationic polymers present excellent activity against Gram-positive bacteria, they are totally inefficient against Gram-negative bacteria. Typically, Gram-negative bacteria are found to be less susceptible to cationic polymers than Gram-positive due to the additional outer membrane that provides a tough barrier to be overcome.29 The nature of the membrane also varies; in Gram-negative bacteria, the membranes are made of two negative phospholipidic membranes and lipopolysaccharides within the outer bilayer, while Gram-positive bacteria have thick cell walls, which consist of a large multilayer region of peptidoglycan with wall teichoic acid and lipoteichoic acid. Although this better behavior against Gram-positive bacteria was also revealed in our previous investigations with cationic methacrylic polymers bearing thiazole and triazole groups, such polymers also presented significant activity against Gram-negative bacteria.22,25,30 In the current work, the polymers derived from IA present four positive charges per monomeric unit, thus, a very high positive charge. In this case, with a high charge density, even when high content of the hydrophobic monomer DMI is used, the activity is almost nullified against Gram-negative bacteria. It seems that an excessive positive charge is detrimental to the disruptive action of polymers on Gram-negative bacterial membranes. On the other hand, against Gram-positive cells, the polymers were able to completely inhibit the bacterial growth at a low concentration, with MIC values that depend on the chemical composition, [TTI]/[DMI] ratio, and the length of the alkyl group. The activity was increased as the content of the active TTI units augments in the copolymers. The butylated polymers with the highest contents of TTI, P100T-Bu and P75T-Bu, have a MIC value as low as 8 μg mL−1. When the influence of the length of the alkyl group on activity is compared, the copolymers quaternized with butyl iodide exhibit the lowest MIC values, which indicates that longer hydrophobic alkyl chains impart stronger antibacterial activity, as previously discussed in the literature.4,6 Then, it appears that increasing the hydrophobicity of the alkylating chains in the antimicrobial polymers enhances their activity, whereas the incorporation of the hydrophobic comonomer DMI into the copolymer structure not only did not improve the antibacterial potential, but also decrease the activity. Apparently the activity against Gram-positive bacteria strongly depends on the cationic charge in the polymers, which is diluted by incorporating the DMI units. In fact, the copolymers with higher content of DMI units exhibited lower charge density as determined by zeta potential measurements. Then, modifying the hydrophobic/hydrophilic balance by varying the length of the alkylating agent seems to be an effective way to improve the activity, as the density of cationic charge is maintained by increasing the hydrophobicity.
Hemotoxicity of cationic polymers
To evaluate the toxicity of the cationic polymers derived from itaconic acid, the hemolysis test was performed on human eukaryotic cells. In this study, the hemoglobin released from the human red blood cells (RBCs) was measured after one hour of incubation with each of the polymers at various concentrations. The hemolysis percentage as a function of the polymer concentration is shown in Fig. 5. The HC50 values, summarized in Table 3, refer to the polymer concentration that triggers 50% lysis of RBCs31,32 and the selectivity values for bacterial cells over mammalian cells that were estimated by the ratio of HC50 to MIC values for S. aureus ATCC 29213. Nevertheless, the high selectivity can also be applied to S. epidermidis bacteria. | ||
| Fig. 5 Hemolytic activity of the copolymers quaternized with either (A) methyl iodide or (B) butyl iodide. | ||
As shown in Fig. 5, all the copolymers exhibit very low hemolysis, with hemolysis percentages well below 50% for the highest concentration tested, 10000 μg mL−1. In fact, most of them present values even below 5%, in particular copolymers quaternized with butyl iodide. With regard to selectivity against bacteria over red blood cells, calculated by the ratio of HC50 and MIC values (Table 3; herein, the MIC values against S. aureus were used), all the itaconic acid derivatives demonstrate excellent selectivity values, with most copolymers showing more than 1000-fold selectivity toward bacteria over RBCs. This series of copolymers derived from biobased itaconic acid are promising antibacterial polymers as they exhibit excellent activity against Gram-positive bacteria and negligible hemolysis. It is well established that the hydrophobic/hydrophilic balance of polymers plays a crucial role in the selective attachment to a bacterial cell membrane.33–35 Typically, polymers with high hydrophobicity show high hemolysis activity due to the strong interaction with the mammalian cell membrane.36 The polymers developed in the current work are very hydrophilic with high charge density, demonstrating null toxicity while maintaining the antibacterial activity.
Conclusions
We described a facile approach to functionalize biobased polymers derived from itaconic acid. Using this strategy we successfully developed potent and highly selective antibacterial polymers containing azolium groups. This strategy consists in the modification of the carboxylic groups of poly(itaconic acid) by incorporating alkyne clickable groups, which can be further functionalized through the CuAAC click reaction. Then, click chemistry allows the coupling of 1.3-thiazole groups, a component of vitamin B1, concomitantly with the formation of 1,2,3-triazole linkages under mild conditions, reaching almost the quantitative degree of modification. The N-alkylation reaction of the azole groups provides cationic azolium groups to the polymers with consequent potent antibacterial activity against Gram-positive bacteria and very low toxicities against human red blood cells. Although the copolymers were not active against Gram-negative bacteria, the approach reported herein provides the basis to develop polymers with a broad spectrum of antibacterial activity in the future. Biobased polymers functionalized with clickable alkyne groups could be easily post modified by attachment of a variety of antimicrobial components, extending the activity to other microbial strains.Ethical statement
All experiments were performed in accordance with the Guidelines of Microbiology and Parasitology Service, and the experiments were approved by the ethics committee at Hospital Universitario de Móstoles. Informed consent was obtained from the human participants of this study.Author contributions
Conceptualization: MFG, CE, and AMB; Investigation: AC, AF, and RCR; Formal analysis: AC, RCR, and FLF; Validation: FLF and CE; Supervision: MFG, CE, and AMB; Funding acquisition: MFG and AMB; Writing-original draft: AMB; and Writing-review & editing: AC, AF, RCR, FLF, MFG, CE, and AMB.Conflicts of interest
Authors declare that they do not have conflict of interest.Acknowledgements
This work was funded by the MICINN (PID2019-104600RB-I00), the Agencia Estatal de Investigación (AEI, Spain) and Fondo Europeo de Desarrollo Regional (FEDER, EU). A. Chiloeches acknowledges MICIU for his FPU fellowship FPU18/01776.References
- A. Al-Ahmad, D. Laird, P. Zou, P. Tomakidi, T. Steinberg and K. Lienkamp, PLoS One, 2013, 8, e73812 CrossRef CAS PubMed.
- H. Takahashi, G. A. Caputo, S. Vemparala and K. Kuroda, Bioconjugate Chem., 2017, 28, 1340–1350 CrossRef CAS PubMed.
- M. M. Konai, B. Bhattacharjee, S. Ghosh and J. Haldar, Biomacromolecules, 2018, 19, 1888–1917 CrossRef CAS PubMed.
- A. Muñoz-Bonilla and M. Fernández-García, Prog. Polym. Sci., 2012, 37, 281–339 CrossRef.
- A. Nimmagadda, X. Liu, P. Teng, M. Su, Y. Li, Q. Qiao, N. K. Khadka, X. Sun, J. Pan, H. Xu, Q. Li and J. Cai, Biomacromolecules, 2017, 18, 87–95 CrossRef CAS PubMed.
- I. Sovadinova, E. F. Palermo, M. Urban, P. Mpiga, G. A. Caputo and K. Kuroda, Polymers, 2011, 3, 1512–1532 CrossRef CAS.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. Degrado, Acc. Chem. Res., 2009, 43, 30–39 CrossRef PubMed.
- S. Laroque, M. Reifarth, M. Sperling, S. Kersting, S. Klopzig, P. Budach, J. Storsberg and M. Hartlieb, ACS Appl. Mater. Interfaces, 2020, 12, 30052–30065 CrossRef CAS PubMed.
- A. Munoz-Bonilla, C. Echeverria, A. Sonseca, M. P. Arrieta and M. Fernandez-Garcia, Materials, 2019, 12, 641 CrossRef CAS PubMed.
- W. Chin, G. Zhong, Q. Pu, C. Yang, W. Lou, P. F. De Sessions, B. Periaswamy, A. Lee, Z. C. Liang, X. Ding, S. Gao, C. W. Chu, S. Bianco, C. Bao, Y. W. Tong, W. Fan, M. Wu, J. L. Hedrick and Y. Y. Yang, Nat. Commun., 2018, 9, 917 CrossRef PubMed.
- W. Chin, C. Yang, V. W. L. Ng, Y. Huang, J. Cheng, Y. W. Tong, D. J. Coady, W. Fan, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2013, 46, 8797–8807 CrossRef CAS.
- Y. Xu, K. Zhang, S. Reghu, Y. Lin, M. B. Chan-Park and X. W. Liu, Biomacromolecules, 2019, 20, 949–958 CrossRef CAS PubMed.
- P. P. Kalelkar, Z. Geng, M. G. Finn and D. M. Collard, Biomacromolecules, 2019, 20, 3366–3374 CrossRef CAS PubMed.
- A. I. Magalhaes Jr., J. C. de Carvalho, J. D. C. Medina and C. R. Soccol, Appl. Microbiol. Biotechnol., 2017, 101, 1–12 CrossRef PubMed.
- J. Cunha da Cruz, A. Machado de Castro and E. F. Camporese Servulo, 3 Biotech, 2018, 8, 138 CrossRef PubMed.
- S. Bednarz, A. Wesołowska-Piętak, R. Konefał and T. Świergosz, Eur. Polym. J., 2018, 106, 63–71 CrossRef CAS.
- K. Satoh, D. H. Lee, K. Nagai and M. Kamigaito, Macromol. Rapid Commun., 2014, 35, 161–167 CrossRef CAS PubMed.
- T. Robert and S. Friebel, Green Chem., 2016, 18, 2922–2934 RSC.
- G. J. Noordzij, Y. J. G. van den Boomen, C. Gilbert, D. J. P. van Elk, M. Roy, C. H. R. M. Wilsens and S. Rastogi, Polym. Chem., 2019, 10, 4049–4058 RSC.
- A. Lv, Z.-L. Li, F.-S. Du and Z.-C. Li, Macromolecules, 2014, 47, 7707–7716 CrossRef CAS.
- S. Chanda and S. Ramakrishnan, Polym. Chem., 2015, 6, 2108–2114 RSC.
- R. Tejero, D. López, F. López-Fabal, J. L. Gómez-Garcés and M. Fernández-García, Polym. Chem., 2015, 6, 3449–3459 RSC.
- CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard-Ninth Edition, CLSI document M07-A9, Clinical and Laboratory Standards Institute, Wayne, PA, 2012 Search PubMed.
- M. Alvarez-Paino, A. Munoz-Bonilla, F. Lopez-Fabal, J. L. Gomez-Garces, J. P. Heuts and M. Fernandez-Garcia, Biomacromolecules, 2015, 16, 295–303 CrossRef CAS PubMed.
- R. Cuervo-Rodriguez, A. Munoz-Bonilla, F. Lopez-Fabal and M. Fernandez-Garcia, Polymers, 2020, 12, 972 CrossRef CAS PubMed.
- E. F. Palermo, I. Sovadinova and K. Kuroda, Biomacromolecules, 2009, 10, 3098–3107 CrossRef CAS PubMed.
- C. Ergene, K. Yasuhara and E. F. Palermo, Polym. Chem., 2018, 9, 2407–2427 RSC.
- E. F. Palermo, K. Lienkamp, E. R. Gillies and P. J. Ragogna, Angew. Chem., Int. Ed., 2019, 58, 3690–3693 CrossRef CAS PubMed.
- K. Lienkamp, K. N. Kumar, A. Som, K. Nusslein and G. N. Tew, Chemistry, 2009, 15, 11710–11714 CrossRef CAS PubMed.
- A. Chiloeches, C. Echeverría, R. Cuervo-Rodríguez, D. Plachà, F. López-Fabal, M. Fernández-García and A. Muñoz-Bonilla, Prog. Org. Coat., 2019, 136, 105272 CrossRef CAS.
- C. Krumm, S. Harmuth, M. Hijazi, B. Neugebauer, A.-L. Kampmann, H. Geltenpoth, A. Sickmann and J. C. Tiller, Angew. Chem., Int. Ed., 2014, 53, 3830–3834 CrossRef CAS PubMed.
- K. Lienkamp and G. N. Tew, Chem. – Eur. J., 2009, 15, 11784–11800 CrossRef CAS PubMed.
- M. Singh, A. Singh, S. Kundu, S. Bansal and A. Bajaj, Biochim. Biophys. Acta, 2013, 1828, 1926–1937 CrossRef CAS PubMed.
- M. A. Rahman, M. Bam, E. Luat, M. S. Jui, M. S. Ganewatta, T. Shokfai, M. Nagarkatti, A. W. Decho and C. Tang, Nat. Commun., 2018, 9, 5231 CrossRef CAS PubMed.
- Z. Zhu, G. Jeong, S.-J. Kim, I. Gadwal, Y. Choe, J. Bang, M.-K. Oh, A. Khan and J. Rao, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2391–2396 CrossRef CAS.
- S. Venkataraman, Y. Zhang, L. Liu and Y. Y. Yang, Biomaterials, 2010, 31, 1751–1756 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00098e |
| This journal is © The Royal Society of Chemistry 2021 |