
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porous hollow TiO2 microparticles for photocatalysis: exploiting novel ABC triblock terpolymer templates synthesised in supercritical CO2†
Ryan R.
Larder
 a,
Thomas M.
Bennett
a,
L. Scott
Blankenship
a,
Jesum A.
Fernandes
a,
Bethany K.
Husband
a,
Rachel L.
Atkinson
a,
Matthew J.
Derry
b,
Daniel T. W.
Toolan
c,
Higor A.
Centurion
d,
Paul D.
Topham
b,
Renato V.
Gonçalves
d,
Vincenzo
Taresco
a and
Steven M.
Howdle
*a
a,
Thomas M.
Bennett
a,
L. Scott
Blankenship
a,
Jesum A.
Fernandes
a,
Bethany K.
Husband
a,
Rachel L.
Atkinson
a,
Matthew J.
Derry
b,
Daniel T. W.
Toolan
c,
Higor A.
Centurion
d,
Paul D.
Topham
b,
Renato V.
Gonçalves
d,
Vincenzo
Taresco
a and
Steven M.
Howdle
*a
aSchool of Chemistry, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: steve.howdle@nottingham.ac.uk
bAston Institute of Materials Research, Aston University, Aston Triangle, Birmingham, B7 4ET, UK
cDepartment of Chemistry, University of Sheffield, South Yorkshire, S3 7HF, UK
dSão Carlos Institute of Physics, University of São Paulo, PO Box 369, 13560-970 São Carlos, SP, Brazil
First published on 22nd April 2021
Abstract
Reversible addition–fragmentation chain transfer (RAFT) mediated dispersion polymerisation in supercritical carbon dioxide (scCO2) is an efficient and green method for synthesising block copolymer microparticles with internal nanostructures. Here we report for the first time the synthesis of phase separated poly(methyl methacrylate-block-styrene-block-4-vinylpyridine) (PMMA-b-PS-b-P4VP) triblock terpolymer microparticles using a simple two-pot sequential synthesis procedure in scCO2, with high monomer conversions and no purification steps. The microparticles, produced directly and without further processing, show a complex internal nanostructure, appearing as a “lamellar with spheres” [L + S(II)] type morphology. The P4VP block is then exploited as a structure-directing agent for the fabrication of TiO2 microparticles. Through a simple and scalable sol–gel and calcination process we produce hollow TiO2 microparticles with a mesoporous outer shell. When directly compared to porous TiO2 particles fabricated using an equivalent PMMA-b-P4VP diblock copolymer, increased surface area and enhanced photocatalytic efficiencies are observed.
Introduction
The ability of block copolymers to self-assemble into various periodic nanostructures has long been of interest in both theoretical and experimental capacities.1–3 This interest has brought to light a plethora of possible applications in electronics, photonics and drug delivery.4–8 The application of block copolymers as structure-directing agents for inorganic nanofabrication has also been of particular interest in recent years.9–12 The main advantage being the flexibility in both the size and morphology of the polymer templates used to control the final structure.A large portion of work on structure direction focuses on using block copolymer thin films as masks for nanolithography.13 Alternatively, various approaches have instead exploited a selective chemical association of one polymer block to an inorganic species in order to direct assembly of polymer-inorganic hybrids.14,15 A prominent polymer used in this method is poly(4-vinyl pyridine) (P4VP), as the coordinative ability of the pyridinyl nitrogen offers great scope for inorganic complexation.16 This bottom-up approach to inorganic nanofabrication has provided the means to create intricate 3D architectures that would be impossible to make via lithography methods.9,17–19
We have recently shown that diblock copolymers of poly(methyl methacrylate) (PMMA) and P4VP can be used as templates to fabricate hierarchically structured metal oxides, in both a versatile and scalable manner.20 Reversible addition–fragmentation chain transfer (RAFT) controlled dispersion polymerisation in supercritical carbon dioxide (scCO2) was used to synthesise nanostructured block copolymer microparticles.21 The method yielded block copolymers with high blocking efficiencies and minimal block tapering, due to almost complete conversion of monomers at each stage of the polymerisation.22 As a heterogeneous polymerisation process, the dispersion method affords well-defined microparticles, while the plasticising nature of scCO2 enables block copolymer phase separation within the particles during the synthesis. The use of scCO2 as the solvent not only eliminates the need for annealing steps to induce self-assembly, but also makes this a green synthetic route, avoiding the use of potentially toxic non-renewable solvents and energy intensive drying steps.23 The resulting PMMA-b-P4VP microparticles were then used as sacrificial templates in the inorganic sol–gel synthesis of TiO2 and LiFePO4 with structural features on both the micro and nano-length scales.20
However, our studies thus far have only focused on the use of simple AB-type diblock copolymers for application in structure directing. While it is well-established that AB copolymers predominantly form four distinct morphologies in the bulk (spheres, cylinders, bicontinuous gyroid and lamellar), addition of a third unique block has the potential to produce a wealth of additional nanostructures.24
When transitioning from an AB to an ABC-type system, the available parameter space is greatly expanded. The self-assembly depends on three unique interaction parameters (χAB, χAC and χBC) as well as an additional block fraction parameter.25 Various studies have already identified a multitude of stable and metastable morphologies including core–shell versions of the four common diblock morphologies, knitting patterns and complex interconnected networks.26–29 The confinement of these polymers in microparticles, like that demonstrated in our scCO2 dispersion method, could be expected to further increase the abundance of possible ABC morphologies.30
The complex structural morphologies formed by ABC triblock terpolymers are desirable as templates for inorganic materials. The wide variety of possible assemblies, particularly the many additional network structures, offer a route to enhance the properties of functional materials to an extent not possible using simple AB diblock copolymers.31–33 Also, the chemistries of each individual block can be tailored to interact with different chemical species, opening up the possibility of directing the structure of multiple compounds simultaneously.34
Here we report the synthesis of the first ABC triblock terpolymer via RAFT-mediated dispersion in scCO2, where the polymer self-assembly occurs in situ within the confines of the sterically stabilised microparticles. We have focused upon the structure-directing properties of the included P4VP block to selectively incorporate TiO2 into the new terpolymer morphology, before removal of the polymer template by calcination. The resulting TiO2 product is compared directly to the equivalent mesoporous TiO2 microparticles we have produced previously using an equivalent PMMA-b-P4VP microparticulate structure-directing agent.20 Specifically, we report on the enhanced functionality in photocatalytic activity for hydrogen generation.
Experimental
Materials
Methyl methacrylate (MMA, ProSciTech, 99%), styrene (Sigma Aldrich, >99%) and 4-vinylpyridine (4VP, Acros Organics, 95%) were purified by eluting through a basic alumina column to remove inhibitor. 2,2′-Azobis(2-methylpropionitrile) (AIBN, Sigma Aldrich, 98%) was purified by recrystallisation from methanol. 2-(Dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT, Sigma Aldrich, 98%), Poly(dimethylsiloxane)monomethyl methacrylate (PDMS-MA, Fluorochem, Mn ∼ 10 kg mol−1) and titanium tetraisopropoxide (TTIP, Sigma Aldrich, 97%) were all used as received.Triblock terpolymer synthesis
Triblock terpolymer synthesis was achieved using a two-pot seeded dispersion method, modified from previous publications. Firstly, a 25 kg mol−1 PMMA macro-RAFT agent (Scheme S1†) was synthesised. MMA (10 g, 99.9 mmol), AIBN (32.8 mg, 0.20 mmol), DDMAT (145.7 mg, 0.40 mmol) and PDMS-MA polymeric stabiliser (0.5 g, Mn ∼ 10 kg mol−1) were mixed in a sealed vial and degassed by purging with argon for 30 minutes. Meanwhile, a 60 mL autoclave was degassed by flushing with CO2 at 1–2 bar for 30 minutes. The monomer solution was added to the autoclave via syringe against a positive pressure of CO2 to prevent the ingress of air. The autoclave was then sealed, pressurised to 50 bar and heated to 65 °C before further addition of CO2 to the reaction pressure of 241 bar. After stirring at 300 rpm for 24 hours, the reaction was cooled to room temperature, slowly vented and the PMMA product collected as a fine free-flowing powder.The target triblock terpolymer (Scheme S2†) was synthesised by loading the autoclave with the PMMA macro-RAFT agent (3 g, Mn ∼ 25 kg mol−1) and additional PDMS-MA (0.45 g, Mn ∼ 10 kg mol−1), before degassing by flushing with CO2 at 1–2 bar for 30 minutes. The autoclave was then sealed, pressurised to 50 bar and heated to 65 °C before further addition of CO2 to 207 bar. A solution of styrene (6 g, 57.6 mmol) and AIBN (9.8 mg, 0.06 mmol) was degassed by purging with argon for 30 minutes then added to the autoclave via high-performance liquid chromatography (HPLC) pump (Jasco) at a flow rate of 0.5 mL min−1. After 72 hours a small sample of the diblock copolymer was collected through the outlet tap before a degassed solution of 4VP (3 g, 28.5 mmol) and AIBN (7.5 mg, 0.05 mmol) was added to the autoclave in the same manner as the styrene. After a further 24 hours, the reaction was cooled to room temperature, slowly vented and the triblock terpolymer product collected as an off-white, dry free-flowing powder (9.21 g).
Porous hollow TiO2 microparticle synthesis
The polymer-TiO2 composite was synthesised via a sol–gel procedure, identical to the one described in a previous publication using a PMMA-b-P4VP template, for direct comparison of the terpolymer's structure-directing capability.20 The composite was synthesised targeting a final mass of 25 wt% TiO2 relative to triblock terpolymer. The sol–gel solution was created by adding TTIP (0.062 mL, 0.21 mmol) to a solution of concentrated hydrochloric acid (0.031 mL, 37 wt% HCl in water) in ethanol (7.92 mL) for a final volume of 8 mL. The solution was stirred for 1 hour before addition of a slurry of the triblock terpolymer microparticles in ethanol (2 mL, 25 mg mL−1). The sol–gel mixture was then stirred for a further 24 hours before being evaporated in a Petri dish for approximately 48 hours at room temperature. The polymer-TiO2 composite material was obtained as a fine white powder (77 mg) that was loosely settled on the dish surface. The TiO2 hollow microparticles were obtained by calcining the composite material at 500 °C for 2 hours (10 °C min−1 ramp) in air to completely remove the polymer. The product was obtained as a fine white powder (19 mg).Nuclear magnetic resonance (1H NMR) spectroscopy was used to measure conversion values and polymer fractions. Spectra were obtained on a Bruker AV-III 400 MHz spectrometer on samples dissolved in CDCl3. Data was analysed using MestReNova software.
Gel permeation chromatography (GPC) was used to measure the molecular weight and dispersity of the polymers. An Agilent 1260 infinity SEC system was used with a Wyatt Optilab dRI detector. The mobile phase was a solvent mixture of chloroform and ethanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with triethyl amine stabiliser (1% v/v). Samples were injected at a flow rate of 0.5 mL min−1 and passed through a guard column followed by two separation columns (2 × Agilent PLgel 5 μm mixed C). The system was calibrated using PMMA narrow standards (Mn range: 0.5–2000 kg mol−1).
:1) with triethyl amine stabiliser (1% v/v). Samples were injected at a flow rate of 0.5 mL min−1 and passed through a guard column followed by two separation columns (2 × Agilent PLgel 5 μm mixed C). The system was calibrated using PMMA narrow standards (Mn range: 0.5–2000 kg mol−1).
Differential scanning calorimetry (DSC) was used to measure polymer glass transition temperatures (Tg). Using a TA Instruments Q2000 DSC, a 10 mg sample was heated to 200 °C (10 °C min−1 ramp), cooled to −100 °C then reheated to 200 °C. Data was collected during the second heating run. Tg values were determined using TA universal analysis software.
Thermal gravimetric analysis (TGA) was performed using a TA Instruments Q500 TGA with platinum crucibles over a temperature range of 30 to 500 °C (10 °C min−1 ramp) in air. Data was analysed using TA universal analysis software.
Scanning electron microscopy (SEM) was used to characterise the microstructure of polymer, TiO2 and composite products. Samples were imaged using a JEOL 6490LV SEM at an accelerating voltage of 10 kV after first being coated with a 6 nm layer of platinum. The average particle diameters reported from SEM are a mean average of 100 measured particles (ImageJ processing software), as described by Richez et al.35
Transmission electron microscopy (TEM) was used to characterise the internal nanostructure of all products. Samples were imaged using a JEOL 2100+ TEM at an accelerating voltage of 80 kV. Samples were prepared by embedding in an epoxy resin (Agar 100) at 50 °C for 48 hours before being cut into 100 nm thick sections using an RMC Powertome microtome equipped with a Diatome diamond knife. Polymer samples were then stained with I2 vapour for 2 hours prior to imaging. STEM/EDXS maps of the elemental composition were acquired during the imaging process using an Oxford Instruments XMax 100TLE detector controlled through the Aztec software package.
Small angle X-ray scattering (SAXS) analysis was performed on the terpolymer powder. SAXS patterns were recorded at a synchrotron source (Diamond Light Source, station I22, Didcot, UK) using monochromatic X-ray radiation (wavelength λ = 0.999 Å, with scattering vector q ranging from 0.003 to 0.25 Å−1, where q = 4πsinθ/λ and θ is one-half of the scattering angle) and a 2D Pilatus 2 M pixel detector (Dectris, Switzerland). Scattering data were reduced and normalised with glassy carbon being used for the absolute intensity calibration utilising standard routines available at the beamline.36
Surface area measurements were performed by measuring nitrogen sorption using a Micrometric 3FLEX Sorptometer at −196 °C. Samples were degassed at 200 °C for 12 hours prior to measurement. The Brunauer–Emmett–Teller (BET) method was used to calculate the surface area from the isotherm plots in the relative pressure range of 0.05–0.30, while the Barrett–Joyner–Halenda (BJH) method was used to calculate pore size distributions. BJH calculations were performed using the Harkins–Jura thickness curve on the isotherm's adsorption branch and employed the Faas correction.
Powder X-ray diffraction (PXRD) measurements were obtained on a PANalytical X′pert pro MPD diffractometer using a Cu-Kα radiation source (λ = 1.5406 Å). Samples were placed on a silicon zero background holder and analysed over a 2θ range from 10° to 80°.
Hydrogen evolution was assessed by preparing a suspension of 25 mg of the TiO2 photocatalyst in 50 ml of an aqueous solution with 20 vol% of methanol. The suspension was sonicated for 15 min and placed in a 90 mL quartz reactor. The reactor was purged with vacuum and argon to remove the residual atmosphere gases. The H2 produced was extracted by a continuous flux of Ar, to a Gas Chromatograph Agilent 7890B equipped with a Thermal Conductivity Detector (TCD). A Cermax Xe lamp (300 W) was used as the irradiation source, equipped with AM 1.5G filter and a water column to attenuate the thermal effect induced by the IR radiation. The irradiation power was adjusted to 300 mW cm−2, calibrated using a photometer (Gentec XLP 12-3S-H2-D0).
Results and discussion
The PMMA-b-PS-b-P4VP triblock terpolymer template was synthesised via RAFT-mediated dispersion polymerisation in scCO2, in a two-step procedure similar to that described previously.37 The first step creates a microparticulate PMMA macro-RAFT agent with a specific molecular weight which is then re-dispersed in the scCO2 solvent. Subsequent monomers are then added to grow the desired block terpolymer. Due to the pseudo-living nature of this polymerisation, specific polymer block fractions can be targeted by simply varying the quantity of monomers added with respect to the PMMA macro-RAFT agent.This two-pot approach was specifically chosen over a one-pot method developed previously.21 By splitting the polymerisation over two stages, the total mass of polymer at any one reaction stage can be kept reasonably constant. This becomes a necessity when adding additional monomer into a fixed volume high pressure autoclave, to prevent over-filling and hence overpressure.
This method was used to synthesise a triblock terpolymer with target weight fractions of 25% PMMA, 50% poly(styrene) (PS) and 25% P4VP blocks with a total molecular weight of 100 kg mol−1. This composition was chosen because of its similarity to the PMMA-b-P4VP diblock copolymer used in the previous study (75:25 weight ratio).20 Shifts in the measured GPC peak to lower retention time after addition of each block (Fig. 1a) demonstrate successful growth of a block terpolymer. Calculated molecular weights for the PMMA macro-RAFT, intermediate diblock copolymer and final triblock product are also presented (Table 1).
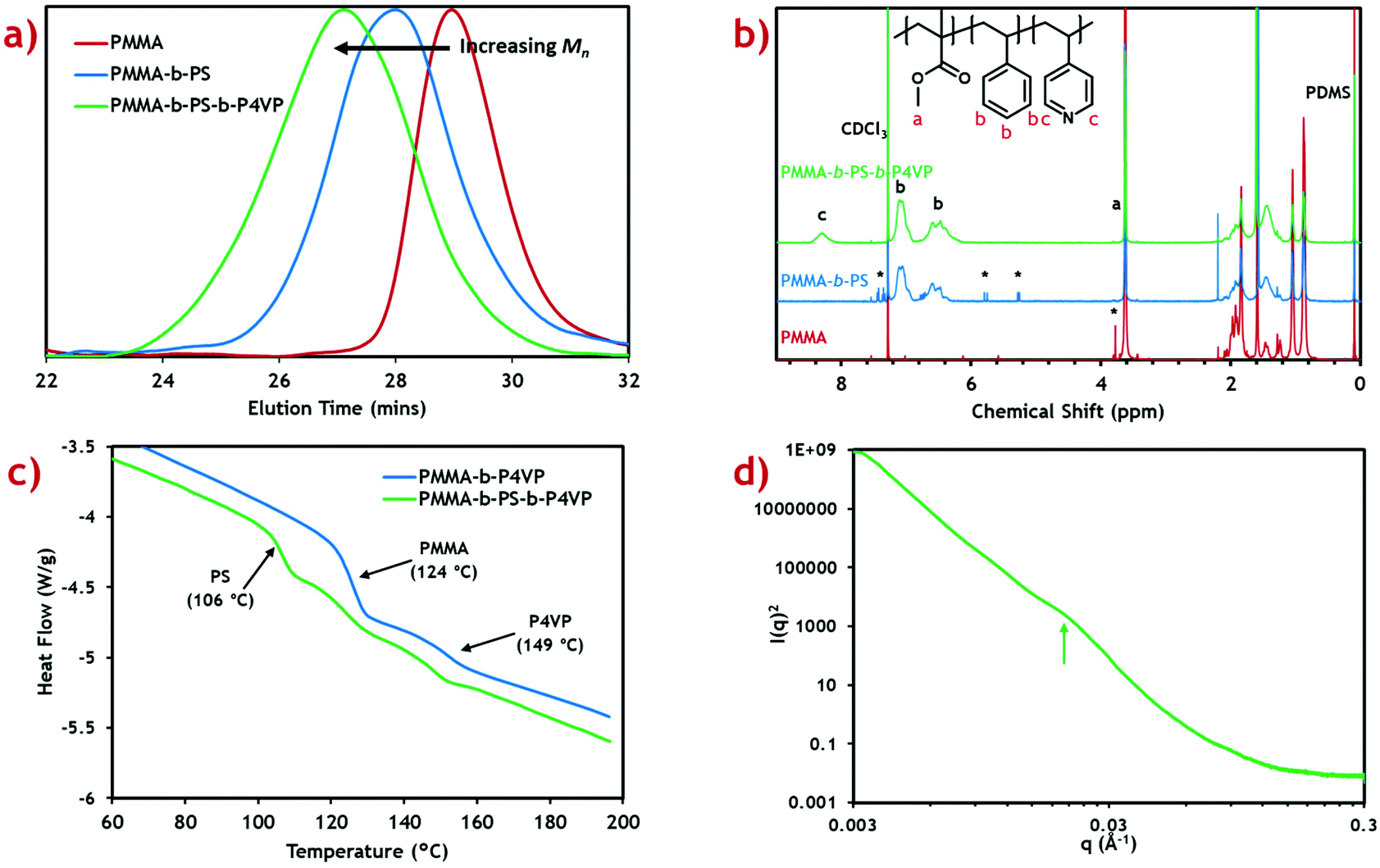 | ||
| Fig. 1 Characterisation data for the PMMA-b-PS-b-P4VP triblock terpolymer. (a) GPC chromatograms at each stage of the polymer growth. (b) 1H NMR assignment for samples at each stage of the polymer growth, peaks used for calculation of block fractions are labelled. (c) DSC plot for the triblock terpolymer product (green) and previously synthesised PMMA-b-P4VP diblock copolymer (blue). Tg values for each polymer block are labelled at the mid-point of step transitions. (d) 1D SAXS data for the triblock terpolymer, with the peak indicated at q ≈ 0.021 Å−1. | ||
| Polymer | M n Target (kg mol−1) |
M
n expa (kg mol−1) |
Đ | Block fractionsb (wt%) | Approximate block ratiosc (vol%) |
|---|---|---|---|---|---|
|
a Obtained via GPC eluting in a CHCl3:EtOH:TEA solvent mixture with dRI detector calibrated against PMMA standards.
b Obtained via1H NMR.
c Calculated using polymer densities (PMMA: 1.16 g cm−3, PS: 1.05 g cm−3, P4VP: 1.11 g cm−3), factoring in approximate volume swelling caused by scCO2 (PMMA: 24.8 ΔV/V%,38 PS: 9.4 ΔV/V%,39 P4VP: 15 ΔV/V%).40
|
|||||
| PMMA | 25 | 25.3 | 1.26 | 100 | 100 |
| PMMA-b-PS | 75 | 49.7 | 1.49 | 34:66 |
35:65 |
| PMMA-b-PS-b-P4VP | 100 | 74.8 | 1.59 | 27:52:21 |
28:52:20 |
Molecular weight for the PMMA macro-RAFT agent (25.3 kg mol−1) shows very good agreement with the target value (25 kg mol−1) with reasonably low dispersity (Đ = 1.26). This indicates, along with the 1H NMR, that the reaction proceeds to almost complete monomer conversion with good control of chain growth. The molecular weight for the diblock intermediate product (49.7 kg mol−1) is considerably lower than the target value of 75 kg mol−1. A reasonable error is expected since the GPC detector is calibrated against PMMA standards.41 The same effect is seen in the final triblock terpolymer product (74.8 kg mol−1). Dispersity for these block copolymers is reasonably low and comparable to those of previous diblock copolymers synthesised in scCO2.21 Some PMMA and PMMA-b-PS impurities are expected in the final triblock terpolymer product, however this is expected to be low due to the high blocking efficiencies reported in scCO2 RAFT dispersion polymerisations.22
To confirm the correct polymer block fractions had been achieved, the triblock product was analysed by 1H NMR (Fig. 1b). Integration of distinctive peaks for each polymer block (PMMA: 3.62 ppm, PS: 7.11 ppm, P4VP: 8.31 ppm) gave the molar ratios of each polymer, which were then converted into weight fractions. Calculated weight fractions showed good agreement with the target ratios (Table 1).
Weight fractions were converted to block volume ratios using the polymer densities and swelling factors for each polymer in scCO2. The scCO2 solvent is well-known to induce swelling in polymers which varies based on the “CO2-philicity” of the polymer.42 This effect has previously been shown to drastically alter the expected morphology in diblock copolymer systems, forming kinetically trapped morphologies.43 Swelling factors are only approximations based on limited data available at particular solvent temperatures and pressures.
Thermal characterisation of the triblock terpolymer product was performed using differential scanning calorimetry (DSC) (Fig. 1c). From the heat flow plot, three clear glass transitions (Tg) can be seen, each coinciding with the known Tg for each homopolymer constituent block.44 This indicates self-assembly has occurred within the microparticles to form a morphology with three distinct polymer phases. Any polymer blending would be indicated by intermediate Tg values.45
SEM analysis of the triblock terpolymer product confirmed the sample consists of well-defined microparticles (Fig. 2a) formed through use of the CO2-soluble steric stabiliser poly(dimethyl siloxane) (PDMS). This microparticle formation is typical for a heterogeneous dispersion-type polymerisation in scCO2.46 The average particle diameter and size dispersity are shown as inserts in the SEM image and are also summarised (Table S1†). The internal morphology of the particles was analysed by imaging thin sections (∼100 nm) of the particles via TEM (Fig. 2d). Contrast between the polymer phases was enhanced by staining the polymer sections with iodine vapor, which selectively binds to the nitrogen of the P4VP polymer causing it to appear darker in the TEM image. The PMMA and PS blocks remain lighter and indistinguishable from one another.
 | ||
| Fig. 2 SEM and TEM micrographs of (a and d) the triblock terpolymer template, (b and e) the polymer-TiO2 composite and (c and f) the calcined TiO2 product. Average microparticle sizes and standard deviation (SD) in measurements are indicated in the SEM micrographs. The scale bar in high magnification SEM insets is 2 μm. In the TEM micrographs, contrast is achieved (d) by I2 staining of the P4VP block or (e) due to infusion of TiO2. | ||
The P4VP block is seen arranged in lamellar rings running through the microparticles, akin to the “onion-like” morphology seen for lamellar-forming diblock copolymers in microparticle confinement.47 However, the morphology presents an added degree of complexity as the P4VP forms distinct spherical domains in the lamellar sheets, rather than forming continuous unbroken rings. The morphology also appears to break down slightly towards the particle centre, leading to a more randomly disordered distribution of P4VP domains. This is likely caused by the increased spatial constraint imposed towards the centre of the microparticle. Based on previous studies of ABC-type triblock materials, this morphology is predicted to be a “lamellar with spheres inside a domain” [L + S(II)] type morphology.26
In this system, the PMMA-b-PS diblock copolymer is already known to microphase separate to form the simple lamellar morphology, based on our previously constructed phase diagram.21 We propose that addition of the P4VP block then causes spherical domains to form in the PMMA lamellar sheets, growing to form interspersed columns of P4VP running throughout the PMMA lamellae, connecting the adjacent PS sheets (Fig. 3). P4VP domains are expected to run through the PMMA lamellae based on the lower volume fraction and the high χN value between the PS and P4VP blocks.
 | ||
| Fig. 3 Schematic representations and TEM cross-sectional images of (a) lamellar morphology of the PMMA-b-PS diblock copolymer, from our previous publications21,43 and (b) the proposed lamellar with spheres [L + S(II)] morphology of the synthesised PMMA-b-PS-b-P4VP triblock terpolymer. Approximate volume fractions and χN values for each block pair in the synthesised triblock terpolymer are shown below the schematic. χN values were calculated using χ values from the literature16,48 and a monomer reference volume of 118 Å3. Values do not account for scCO2 swelling. | ||
Based on TEM analysis (Fig. 2d) and calculated interaction parameters (χN), we tentatively assign the self-assembled morphology of this PMMA-b-PS-b-P4VP triblock terpolymer as L + S(II), which has been previously shown for other ABC triblock terpolymer systems.25,26 However, this morphology was only reported previously for a system with χACN ≪ χABN ≈ χBCN, while in this system χBCN ≫ χABN ≈ χACN, therefore making this previous study not directly comparable. Additionally, we cannot accurately predict the role that scCO2-swelling will play on these estimated χN values. Because of the influence of CO2 on the polymer self-assembly, morphological assignment based solely on the polymer composition or its post-processed bulk film state, is not possible. The kinetically trapped scCO2 morphology we observe within the microparticles is unlikely to represent the thermodynamically stable arrangement for the terpolymer. Thus, definitive assignment of this triblock terpolymer morphology is not possible, especially when considering that this assignment is made using only TEM images.
Small angle X-ray scattering (SAXS) was performed in an attempt to provide further evidence of the existence of this proposed morphology (Fig. 1d). A subtle, broad feature can be seen in the scattering profile at q ≈ 0.021 Å−1, when plotting I(q)2vs. q to enhance features at low q. This corresponds to a length scale of d ≈ 30 nm, which is in good agreement with the TEM analysis. Though this is indicative of nanoscale self-assembly, a greater scattering resolution is required to assign the morphology. Poor scattering resolution in this sample can be attributed to the presence of large microparticles, which dominate the scattering profile, since I(q) ∝ Vp2 (where Vp is the microparticle volume).49 Nevertheless, internal nanoscale assembly is evident in both TEM and SAXS analyses, suggesting the formation of a complex polymer morphology.
Inclusion of the P4VP block in the triblock terpolymer allows us to exploit the pyridine groups to incorporate inorganic species into select areas of the polymer morphology. This is achieved through integration of the polymer into the sol–gel synthesis of the desired metal oxide.20 The inclusion of TiO2 into the triblock terpolymer was achieved by dispersing the terpolymer microparticles in an acid-stabilised solution of titanium tetraisopropoxide (TTIP) in ethanol. After mixing, the solvent was evaporated to yield the polymer-TiO2 composite particles.
SEM analysis of the composite material shows that the microparticle structure of the polymer is retained when dispersing in the ethanol sol–gel, with only a minor increase (+0.16 μm) in the average particle size (Fig. 2b). Close inspection of the particle surfaces shows the formation of additional surface porosity not observed in the pure polymer material. This new topography is the result of the so-called “swelling-induced porosity effect” applied during the sol–gel process.50 The P4VP domains are selectively swollen by the sol–gel solvent, which then collapse to form pores once the solvent is evaporated.
TEM analysis of microparticle cross-sections (Fig. 2e) shows that the TiO2 has been incorporated into the polymer in layers near the surface. The concentration of TiO2 then diminishes towards the particle centre. This is apparent from the dark contrast formed by the dense TiO2 material in the TEM image. Outer layers of the composite appear as almost perfect lamellar rings, instead of being split into distinct spherical domains as observed in the original triblock terpolymer. This apparent overlap is likely a result of some swelling of the P4VP regions upon addition of the inorganic component. As the contrast in this image is generated solely from incorporation of the heavier TiO2, this is not necessarily a reflection of a change in the P4VP morphology.
Elemental mapping of the composite was also performed using the STEM/EDX mode of the TEM to confirm the absence of TiO2 from the centre of the microparticles. Areas of intense Ti characteristic X-rays were highlighted in coloured pixels (Fig. S1†). Ti X-ray signals where shown to match the position of the dark contrast in the TEM images.
Reduced infusion of the inorganic components throughout the triblock particles is thought to be due to the restrictive nature of the lamellar structure. This is in contrast to the full infusion of inorganic precursor seen previously when using the spherically structured PMMA-b-P4VP diblock copolymer microparticles.20 Therefore, we propose that the addition of hydrophobic PS lamellae in these triblock terpolymer microparticles, directly hinders diffusion of the TiO2 into the central P4VP domains.
The block terpolymer template was fully removed by calcining the material at 500 °C in air to leave only the TiO2 product. Thermal gravimetric analysis (TGA) of the calcination process shows 25.2 wt% of the composite material remained after 2 hours (Fig. S2†). This is in good agreement with the target value of 25 wt% TiO2 incorporation into the block terpolymer during the sol–gel synthesis, indicating complete removal of the polymer template was successful under these conditions.
The calcined product was analysed by SEM (Fig. 2c) to confirm that the microparticle structure had been retained after removal of the template. High magnification images also show the surface porosity present in the composite material was also retained. The average particle size was measured to be approximately half that of the original triblock terpolymer (1.60 μm). This is as anticipated and represents the expected contraction of the TiO2 regions in the composite to fill voids left by the combusted terpolymer.
TEM analysis of the internal structure (Fig. 2f) reveals these particles contain large hollow cores. These voids correlate to the central area of the composite material into which the TiO2 precursor was unable to infuse during the sol–gel synthesis. Thus, the solid polymer core of the composite is fully removed, leading to a central void surrounded by an outer TiO2 shell. The TEM image also confirms that the porosity seen on the particle surfaces persists throughout the shell. The overall result is formation of mesoporous hollow-cored microparticles. A lack of solid ring structures in the final TiO2 material also suggests no full lamellar structures were formed in the composite material.
Hollow TiO2 microparticles have already proven to be valuable in applications such as dye-sensitised solar cells, photocatalysis and energy storage.51–53 In many cases the hollow particle structure is more desirable than TiO2 nanoparticles or structures with higher surface areas. The internal interface of the hollow particle can lead to enhanced scattering of light for increased performance.54 Various studies have also explored the application of hollow TiO2 microparticles with mesoporous shells.51,55,56 Though, the structures are shown to be beneficial, the synthesis routes employed to achieve this specific structure can be difficult and demanding.55 Our approach using the triblock terpolymer as a template represents an improvement on these methods, due to its simplicity and low environmental impact through the use of scCO2. Both the scCO2 polymer synthesis and sol–gel process have already proven to be scalable synthetic methods, granting our approach greater viability on the industrial scale.20,57
This new TiO2 morphology produced using the triblock terpolymer template appears to be more structurally complex than mesostructured TiO2 produced using the equivalent PMMA-b-P4VP diblock template (Fig. 4b).20 The surface area of both TiO2 materials was measured through nitrogen sorption isotherms (Fig. 4d) and the calculated surface areas and average pore sizes are summarised (Table 2). The hollow-cored TiO2 was shown to have a significantly larger available surface area, making it more attractive for functional applications, and photocatalysis in particular.
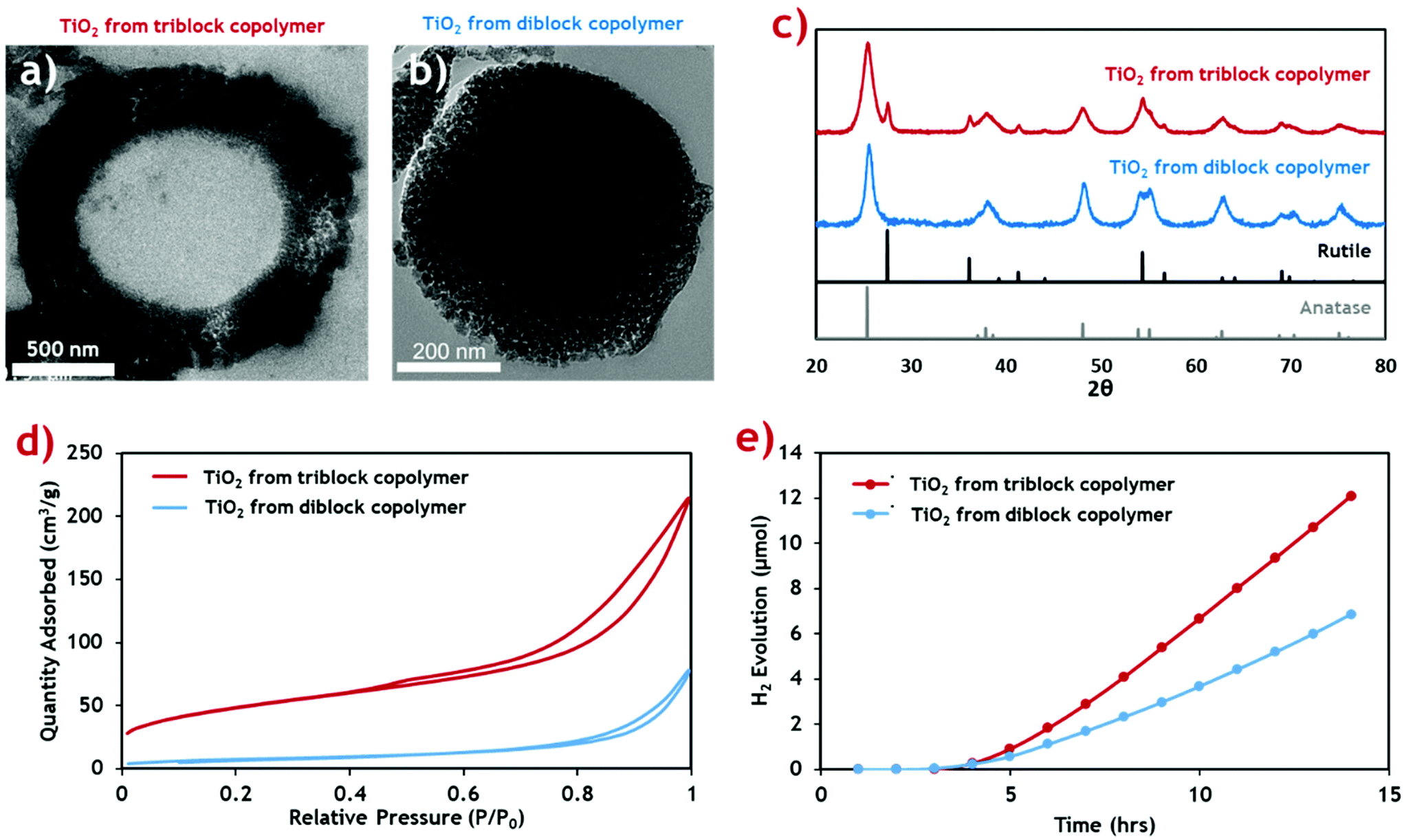 | ||
| Fig. 4 Characterisation data for the porous hollow-cored TiO2 product (red) compared with the mesoporous TiO2 produced with a diblock copolymer (blue), from our previous publication.20 (a and b) Cross-sectional TEM micrographs of the triblock TiO2 and diblock TiO2 products respectively. (c) PXRD diffractograms of the TiO2 materials showing their respective crystal phase compositions. Rutile and anatase reference patterns are shown below. (d) N2 sorption isotherms for the TiO2 materials. (e) Cumulative H2 evolution profiles for the TiO2 products when exposed to simulated solar radiation. | ||
| Polymer template used | Specific surface area (BET)a (m2 g−1) | Average pore size (BJH)a (nm) | Anatase:rutile phase compositionb (wt%) |
H2 evolution (μmol g−1 h−1) |
|---|---|---|---|---|
| a Calculated using nitrogen sorption measurements. b Calculated from PXRD measurements. | ||||
| Diblock | 26.4 | 17.9 | 100:0 |
30.3 |
| Triblock | 170.9 | 9.1 | 70.5:29.5 |
53.4 |
Both samples exhibit type-II adsorption isotherms, with hysteresis loops indicative of adsorption metastability in the multilayer. In the case of the isotherm for the triblock TiO2, its higher surface area is indicated by the greater overall N2 uptake. Additionally, higher uptakes in the low-pressure region suggest the presence of smaller pores in the triblock TiO2 than in its diblock counterpart. These two assertions are confirmed quantitatively using BET and BJH theories, respectively.58 The contribution of the additional smaller pores and the added internal surface of the hollow core ensure that the triblock TiO2 has a much greater surface area.
The crystal phase composition was determined by PXRD analysis and compared against the common anatase and rutile TiO2 patterns (Fig. 4c). The weight distribution of each phase was quantified using the empirical method of Spurr and Myers.59 The calculated phase compositions for both TiO2 samples (Table 2) show that while the diblock template yielded a pure anatase phase TiO2 material upon calcination, the new triblock template encouraged formation of a sizable quantity of rutile phase impurities. We envisage that this is a result of the more uniform morphology of the diblock copolymer composite, with an even distribution of inorganic material throughout the composite material. By comparison, the triblock TiO2 composite shows a gradual reduction in inorganic content towards the centre of the microparticles which likely led to inconsistencies in the crystallisation of the TiO2 during the calcination process.
The photocatalytic activity of the two TiO2 materials was assessed by measuring H2 generation under exposure to simulated solar radiation (Fig. 4e). The rate of H2 production was calculated from the gradient of the linear portion of the evolution plot (Table 2). The photocatalytic activity of the triblock TiO2 was found to be considerably higher than the diblock counterpart, likely resulting from the combination of higher active surface area and the mixed phase composition. Previous studies identify a possible synergistic effect between the anatase and rutile phases, leading to increased photocatalytic activity.60 The H2 evolution rates of both TiO2 materials are higher than those reported in the literature for common commercial P25 TiO2 nanoparticulate materials, under near identical conditions (∼20 μmol g−1 h−1).61 P25 is a mixed phase TiO2 material (∼25% rutile) making it highly comparable to our hollow particles.62
Conclusions
To summarise, we have demonstrated for the first time a robust two-pot method for synthesis of PMMA-b-PS-b-P4VP terpolymers in scCO2, via RAFT-mediated dispersion. The controlled radical dispersion polymerisation affords microparticles with well-defined internal nanostructures, assigned as a L + S(II) morphology. This unique structural hierarchy represents an extension over the four classical diblock morphologies previously reported in microparticle confinement.63 Moreover, the synthesis via scCO2 dispersion negates the need for additional drying, purification and polymer annealing steps, while also being considered to be an environmentally benign polymerisation route.The included P4VP block enabled the terpolymer to be used as a structure-directing agent for the templating of nanostructured TiO2. The unique triblock morphology within microparticle confinement was used to produce mesoporous TiO2 microparticles (∼1.6 μm) containing a large hollow core. The facile and scalable synthesis of this complex 3D architecture is useful for creating significant quantities of material with enhanced properties. Furthermore, this new triblock terpolymer was found to be a superior template when compared to the equivalent PMMA-b-P4VP diblock copolymer, in terms of the resulting active surface area and photocatalytic activity of the TiO2. Using identical synthetic procedures, the triblock TiO2 product was found to have a ∼6-fold greater surface area and ∼2-fold increase in the rate of hydrogen production.
In the future such new polymeric templates have the potential to facilitate the fabrication of a very wide range of useful inorganic materials, largely because of the vast array of possible sol–gel syntheses.64 We believe that this new, more complex, 3D polymer template will provide further opportunities to control the surface area and other properties of a variety of functional materials, for a broad range of applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Engineering and Physical Science Research Council (EPSRC) for their support of this project, as part of the Low Dimensional Materials and Interfaces (LDMI) doctoral training programme (EP/N50970X/1). The authors would also like to thank the Nanoscale and Microscale Research Centre (nmRC) at the University of Nottingham for access to electron microscope instruments and sample preparation equipment. Small angle X-ray scattering (SAXS) data were obtained on the I22 beamline at the Diamond Light Source (proposal number SM23501). We acknowledge the technical assistance provided by Dr Michael Fay, Dr Elizabeth Steer, Dr Julia Watts and Ms Nicola Western. Finally, we would like to thank Mr Richard Wilson and Mr Mark Guyler for their technical support with the high-pressure equipment.References
- M. W. Matsen, Macromolecules, 1995, 28, 5765–5773 CrossRef CAS.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 7641–7644 CrossRef CAS.
- N. A. Lynd, A. J. Meuler and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef CAS.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS PubMed.
- M. Stefik, S. Guldin, S. Vignolini, U. Wiesner and U. Steiner, Chem. Soc. Rev., 2015, 44, 5076–5091 RSC.
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- Y.-C. Tseng and S. B. Darling, Polymers, 2010, 2, 470–489 CrossRef CAS.
- P. D. Topham, A. J. Parnell and R. C. Hiorns, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1131–1156 CrossRef CAS.
- H.-Y. Hsueh, C.-T. Yao and R.-M. Ho, Chem. Soc. Rev., 2015, 44, 1974–2018 RSC.
- X. Li, J. Iocozzia, Y. Chen, S. Zhao, X. Cui, W. Wang, H. Yu, S. Lin and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 2046–2070 CrossRef CAS PubMed.
- S. Greil, A. Rahman, M. Liu and C. T. Black, Chem. Mater., 2017, 29, 9572–9578 CrossRef CAS.
- E. C. Giraud, P. Mokarian-Tabari, D. T. W. Toolan, T. Arnold, A. J. Smith, J. R. Howse, P. D. Topham and M. A. Morris, ACS Appl. Nano Mater., 2018, 1, 3426–3434 CrossRef CAS.
- S. Ji, L. Wan, C.-C. Liu and P. F. Nealey, Prog. Polym. Sci., 2016, 54–55, 76–127 CrossRef CAS.
- P. F. W. Simon, R. Ulrich, H. W. Spiess and U. Wiesner, Chem. Mater., 2001, 13, 3464–3486 CrossRef CAS.
- M. C. Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520–535 RSC.
- J. G. Kennemur, Macromolecules, 2019, 52, 1354–1370 CrossRef CAS.
- L. A. Connal, N. A. Lynd, M. J. Robb, K. A. See, S. G. Jang, J. M. Spruell and C. J. Hawker, Chem. Mater., 2012, 24, 4036–4042 CrossRef CAS PubMed.
- M. G. Fischer, X. Hua, B. D. Wilts, I. Gunkel, T. M. Bennett and U. Steiner, ACS Appl. Mater. Interfaces, 2017, 9, 22388–22397 CrossRef CAS PubMed.
- J. Hwang, C. Jo, K. Hur, J. Lim, S. Kim and J. Lee, J. Am. Chem. Soc., 2014, 136, 16066–16072 CrossRef CAS PubMed.
- T. M. Bennett, G. He, R. R. Larder, M. G. Fischer, G. A. Rance, M. W. Fay, A. K. Pearce, C. D. J. Parmenter, U. Steiner and S. M. Howdle, Nano Lett., 2018, 18, 7560–7569 CrossRef CAS PubMed.
- J. Jennings, M. Beija, A. P. Richez, S. D. Cooper, P. E. Mignot, K. J. Thurecht, K. S. Jack and S. M. Howdle, J. Am. Chem. Soc., 2012, 134, 4772–4781 CrossRef CAS PubMed.
- J. Jennings, M. Beija, J. T. Kennon, H. Willcock, R. K. O'Reilly, S. Rimmer and S. M. Howdle, Macromolecules, 2013, 46, 6843–6851 CrossRef CAS.
- E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121–191 CrossRef CAS.
- F. S. Bates, Science, 1991, 251, 898–905 CrossRef CAS PubMed.
- W. Zheng and Z.-G. Wang, Macromolecules, 1995, 28, 7215–7223 CrossRef CAS.
- Z. Guo, G. Zhang, F. Qiu, H. Zhang, Y. Yang and A.-C. Shi, Phys. Rev. Lett., 2008, 101, 028301 CrossRef PubMed.
- T. S. Bailey, H. D. Pham and F. S. Bates, Macromolecules, 2001, 34, 6994–7008 CrossRef CAS.
- F. S. Bates, MRS Bull., 2011, 30, 525–532 CrossRef.
- C. Auschra and R. Stadler, Macromolecules, 1993, 26, 2171–2174 CrossRef CAS.
- Z. Xu, J. Lin, Q. Zhang, L. Wang and X. Tian, Polym. Chem., 2016, 7, 3783–3811 RSC.
- M. Stefik, S. Mahajan, H. Sai, T. H. Epps, F. S. Bates, S. M. Gruner, F. J. Disalvo and U. Wiesner, Chem. Mater., 2009, 21, 5466–5473 CrossRef CAS PubMed.
- Z. Li, K. Hur, H. Sai, T. Higuchi, A. Takahara, H. Jinnai, S. M. Gruner and U. Wiesner, Nat. Commun., 2014, 5, 3247 CrossRef PubMed.
- M. Stefik, S. Wang, R. Hovden, H. Sai, M. W. Tate, D. A. Muller, U. Steiner, S. M. Gruner and U. Wiesner, J. Mater. Chem., 2012, 22, 1078–1087 RSC.
- C. D. Cowman, E. Padgett, K. W. Tan, R. Hovden, Y. Gu, N. Andrejevic, D. Muller, G. W. Coates and U. Wiesner, J. Am. Chem. Soc., 2015, 137, 6026–6033 CrossRef CAS PubMed.
- A. P. Richez, L. Farrand, M. Goulding, J. H. Wilson, S. Lawson, S. Biggs and O. J. Cayre, Langmuir, 2014, 30, 1220–1228 CrossRef CAS PubMed.
- B. R. Pauw, A. J. Smith, T. Snow, N. J. Terrill and A. F. Thünemann, J. Appl. Crystallogr., 2017, 50, 1800–1811 CrossRef CAS PubMed.
- M. Alauhdin, T. M. Bennett, G. He, S. P. Bassett, G. Portale, W. Bras, D. Hermida-Merino and S. M. Howdle, Polym. Chem., 2019, 10, 860–871 RSC.
- A. Rajendran, B. Bonavoglia, N. Forrer, G. Storti, M. Mazzotti and M. Morbidelli, Ind. Eng. Chem. Res., 2005, 44, 2549–2560 CrossRef CAS.
- S. Hilic, S. A. E. Boyer, A. A. H. Pádua and J.-P. E. Grolier, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 2063–2070 CrossRef CAS.
- Y. Zhang, K. K. Gangwani and R. M. Lemert, J. Supercrit. Fluids, 1997, 11, 115–134 CrossRef CAS.
- P. J. Docherty, C. Girou, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 3332–3339 RSC.
- R. G. Wissinger and M. E. Paulaitis, J. Polym. Sci., Part B: Polym. Phys., 1987, 25, 2497–2510 CrossRef CAS.
- J. Jennings, S. P. Bassett, D. Hermida-Merino, G. Portale, W. Bras, L. Knight, J. J. Titman, T. Higuchi, H. Jinnai and S. M. Howdle, Polym. Chem., 2016, 7, 905–916 RSC.
- J. E. C. Mark, Polymer data handbook, Oxford University Press, Oxford, UK, 2009 Search PubMed.
- I. M. Kalogeras and W. Brostow, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 80–95 CrossRef CAS.
- J. M. DeSimone, E. E. Maury, Y. Z. Menceloglu, J. B. McClain, T. J. Romack and J. R. Combes, Science, 1994, 265, 356–359 CrossRef CAS PubMed.
- M. P. Kim and G.-R. Yi, Front. Mater., 2015, 2, 1–12 Search PubMed.
- T. P. Russell, R. P. Hjelm and P. A. Seeger, Macromolecules, 1990, 23, 890–893 CrossRef CAS.
- L. Boldon, F. Laliberte and L. Liu, Nano Rev., 2015, 6, 25661 CrossRef PubMed.
- G. He, T. M. Bennett, M. Alauhdin, M. W. Fay, X. Liu, S. T. Schwab, C.-g. Sun and S. M. Howdle, Polym. Chem., 2018, 9, 3808–3819 RSC.
- S. Dadgostar, F. Tajabadi and N. Taghavinia, ACS Appl. Mater. Interfaces, 2012, 4, 2964–2968 CrossRef CAS PubMed.
- H. Li, Z. Bian, J. Zhu, D. Zhang, G. Li, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 8406–8407 CrossRef CAS PubMed.
- Z. Wang, L. Zhou and X. W. Lou, Adv. Mater., 2012, 24, 1903–1911 CrossRef CAS PubMed.
- H.-J. Koo, Y. J. Kim, Y. H. Lee, W. I. Lee, K. Kim and N.-G. Park, Adv. Mater., 2008, 20, 195–199 CrossRef CAS.
- W. H. Suh, A. R. Jang, Y.-H. Suh and K. S. Suslick, Adv. Mater., 2006, 18, 1832–1837 CrossRef CAS.
- T. Yanagishita, M. Imaizumi, K. Toshiaki and H. Masuda, RSC Adv., 2015, 5, 41830–41834 RSC.
- A. J. Haddleton, S. P. Bassett and S. M. Howdle, J. Supercrit. Fluids, 2020, 160, 104785 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- R. A. Spurr and H. Myers, Anal. Chem., 1957, 29, 760–762 CrossRef CAS.
- W. R. Siah, H. O. Lintang, M. Shamsuddin and L. Yuliati, IOP Conf. Ser.: Mater. Sci. Eng., 2016, 107, 012005 Search PubMed.
- V. Kumaravel, M. D. Imam, A. Badreldin, R. K. Chava, J. Y. Do, M. Kang and A. Abdel-Wahab, Catalysts, 2019, 9, 276 CrossRef CAS.
- T. Ohno, K. Sarukawa, K. Tokieda and M. Matsumura, J. Catal., 2001, 203, 82–86 CrossRef CAS.
- J. Jennings, G. He, S. M. Howdle and P. B. Zetterlund, Chem. Soc. Rev., 2016, 45, 5055–5084 RSC.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00334h |
| This journal is © The Royal Society of Chemistry 2021 |