
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe power of architecture – cage-shaped PEO and its application as a polymer electrolyte†
Andreas Johannes
Butzelaar‡
 a,
Martin
Gauthier-Jaques‡
b,
Kun Ling
Liu
c,
Gunther
Brunklaus
cd,
Martin
Winter
cd and
Patrick
Theato
*ab
a,
Martin
Gauthier-Jaques‡
b,
Kun Ling
Liu
c,
Gunther
Brunklaus
cd,
Martin
Winter
cd and
Patrick
Theato
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstraße 18, 76131 Karlsruhe, Germany. E-mail: patrick.theato@kit.edu
bSoft Matter Synthesis Laboratory – Institute for Biological Interfaces III (IBG-3), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cIEK-12/Forschungszentrum Jülich GmbH, Helmholtz-Institute Münster, Corrensstraße 46, 48149, Münster, Germany
dMEET Battery Research Center/Institute of Physical Chemistry, University of Münster, Corrensstraße 46, 48149 Münster, Germany
First published on 8th July 2021
Abstract
Herein we report for the first time on the gram-scale synthesis of a four-arm cage-shaped poly(ethylene oxide) (PEO) and its pioneering application as polymer electrolyte. The well-supressed crystallization by the cage architecture proves the great toolbox of polymer chemists to overcome crystallization issues in PEO-based polymer electrolytes.
Solid-state electrolytes (SSEs) are supposed to supersede organic liquid electrolytes because of their high mechanical strength, electrochemical stability, thermal tolerance, overall low toxicity and safety.1 Among the different SSEs, polymer electrolytes (PEs) are of particular interest due to their intrinsic set of properties, including a high flexibility, thin-film forming ability, easy processability and wide electrochemical windows.2 Within this material class, the first studied PE was reported nearly 45 years ago by P. Wright and was comprised of alkali metal ions and poly(ethylene oxide) (PEO).3 Although numerous reports of PEs displaying a wide variety of structures were reported over the past decades, the interest for PEO-based materials never decreased due to their remarkable flexibility, low glass transition temperature (Tg), electrochemical stability against lithium metal as well as unmatched solubility for conductive lithium salts.4 Furthermore, Bollorés lithium metal polymer (LMP) battery technology, which was introduced to the market in 2011,5 is based on PEO and led to a fleet of more than 8000 electric vehicles until today. Despite its market success, the fairly low ionic conductivities of PEO-based materials at lower temperatures, i.e. below its melting point (∼65 °C (ref. 6)) constitute a major drawback to the commercialization on a larger scale.4 Considering that ion transport is expected to only occur through the free volume provided by an amorphous PEO phase,7 various approaches such as cross-linking,8 plasticizer implementation,9 blending with other polymers10 or additional composite manufacturing9a,d,11 were investigated in order to reduce crystallinity within PEO-based materials and thus increase the resulting ionic conductivity at more convenient working temperatures. In this context, also architectural variations of PEO have been studied with the aim to improving the Li+-ion conductivity. Herein, one of the most applied approaches features the grafting of PEO side chains onto a polymer backbone and thus enabling reduction of crystallization.12 We recently highlighted the coupled influence of different parameters such as side chain length, grafting density and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) loading content on the thermal properties and the resulting ionic conductivity of such a series of comb-shaped PEO side chain copolymers, successfully showing an effective reduction of the degree of crystallinity within the PE due to the systematic architectural variations.13
Noteworthy, the interest in complex polymer architectures in materials science increased over the past decades.14 Among the different polymer architectures, cage polymers stand out particularly because of the absence of chain end-groups and their multi-cyclic structures, thus lowering their hydrodynamic radius in solution as well as their propensity to crystallize, which is of interest within the present study. However, the synthesis of covalently-closed cage polymers still remain a great challenge resulting in only a small number of reports so far and thus limiting their application. Pioneering studies in the last 20 years by the groups of Tezuka,15 Shea,16 Pan,17 Paik,18 and Satoh19 explored different approaches for the synthesis of cage polymers. Yet only in 2020, the first example of cage-shaped PEO synthesis was published by Matsushita and co-workers.20 Recently, we reported on a novel cage polymer synthesized via a closing reaction based on an intramolecular tetramerization of end-functionalized m-azidoethynylbenzene into [34]-triazolophane macrocycles,21 which was demonstrated for a series of four-arm cage-shaped poly(ε-caprolactone)s (ε-PCLs), all exhibiting a reduced crystallinity induced by the topological conversion in accordance with former reports of similar ε-PCL structures.19b
Despite being studied for 20 years, it is interesting to note that cage polymers hardly found any broader application so far. Their unique properties are indisputable, yet studies on cage polymers were so far rather limited to academic curiosities because they could commonly only be synthesized on the milligram scale. In order to overcome this current limitation and subsequently explore potential applications, the optimization of the cage polymer synthesis on a gram-scale is necessary. In this regard, we opted for a semi-batch process by adapting our recently developed synthetic protocol, allowing the production of sufficient amounts of polymer in a single reaction. Having all these considerations in mind, we planned to move our research interest to the synthesis of cage-shaped PEO in order (a) to demonstrate the universality of our intramolecular tetramerization process for the efficient synthesis of cage polymers, (b) to enable the synthesis of cage polymers at the gram-scale, and (c) to investigate the topological effect that a reduction in PEO crystallinity might induce on the ionic conductivity of PEO-based polymer electrolytes.
As starting material, a commercially available four-arm star-shaped PEO polymer (PEOstar), claiming a number average molar mass (Mn) of 5.0 kg mol−1 and a dispersity (Đ) below 1.05 was purchased and carefully characterized prior the following synthesis. While the size-exclusion chromatography (SEC) confirmed the presence of a single Gaussian distribution with a low dispersity of Đ = 1.04, proton nuclear magnetic resonance (1H-NMR) and Fourier-transform infrared spectroscopy (FT-IR) ensured the chemical purity of the material. In addition, 1H-NMR spectroscopy allowed to determine Mn,1H-NMR of 5.7 kg mol−1 from the integral ratio between the singlet located at 3.40 ppm corresponding to the central CH2 hydrogens and the remaining CH2 signals (Table 1, ESI II/1†). The functional end-group 3-azido-5-ethynylbenzoic acid was synthetized beforehand in accordance with our previous study.21 The PEOstar esterification was successfully conducted under mild conditions by EDC coupling, yielding end-functionalized star-shaped PEO (PEOend-func) in 94% yield after purification by chromatography. Again, the quantitative end-functionalization and the polymer purity were ensured by 1H-NMR analysis. Thus, the three aromatic protons, the terminal alkyne proton at 3.19 ppm, as well as the four protons of the terminal ethylene oxide units located between 4.52 and 3.77 ppm were successfully assigned. Furthermore, a Mn,SEC shift to higher molar mass was observed by SEC analysis (from 7.2 to 8.0 kg mol−1) while retaining a single Gaussian distribution and a low dispersity of Đ = 1.05. Further, the introduction of the end-group functionalities was also proven by FT-IR with the azide double bond stretching and alkyne proton stretching signals located at 1724 cm−1 and 2881 cm−1, respectively, as well as the introduction of aromatic proton stretching signals (Table 1, ESI II/2†).
The topological conversion of the star-shaped polymer into the cage-shaped PEO was adapted and expanded to the gram-scale in order to obtain a sufficient quantity of material. To do so, the synthetic closing step was performed in a semi-batch process guaranteeing a steady-state concentration of the reactive species throughout the reaction, thereby efficiently suppressing undesired intermolecular reactions that would yield a cross-linked material (Scheme 1). In detail, the topological conversion of the star-shaped PEOend-func into its cage-shaped counterpart (PEOcage) was achieved by copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) and isolated in 43% yield (i.e. 1.548 g) after subsequent chromatography column purification. The [34]-triazolophane structure formation was confirmed by 1H-NMR spectroscopy, via the complete disappearance of the alkyne proton signal previously located at 3.19 ppm and the appearance of the triazole proton signal at 10.69 ppm as well as the downfield shifting and broadening of the three aromatic proton signals from 7.29–7.92 ppm to 8.89–9.24 ppm (Fig. 1a). According to the SEC analysis, the Mn,SEC value of the PEOcage decreased from 8.0 to 4.7 kg mol−1 due to the topological conversion (Fig. 1b). Noteworthy, a single Gaussian curve shape was retained while maintaining a low dispersity of Đ = 1.14, indicating the absence of PEOend-func or PEOstar. This is in accordance with the results of the FT-IR spectra, in which the alkyne proton stretching signal, the broadening of the aromatic protons stretching signals and the strong attenuation of the azide double bond stretching signal at 1723 cm−1 were also clearly observed (Table 1, ESI II/3†). Finally, PEOcage was characterized by ESI-MS analysis. It is worth noting that no polymer distribution could be recorded in positive mode, but was acquired in negative mode with chloride anions, as a result of the acidic extraction step during the purification process. Considering the high affinity of [34]-triazolophane macrocycles and PEO toward chloride anions,22 the results previously obtained for cage-shaped ε-PCL,21 and the natural propensity of PEO to stabilize numerous ionic species, the presence of the main [M + 2Cl]2− distribution and of the additional minor distributions are very plausible (ESI II/4†).
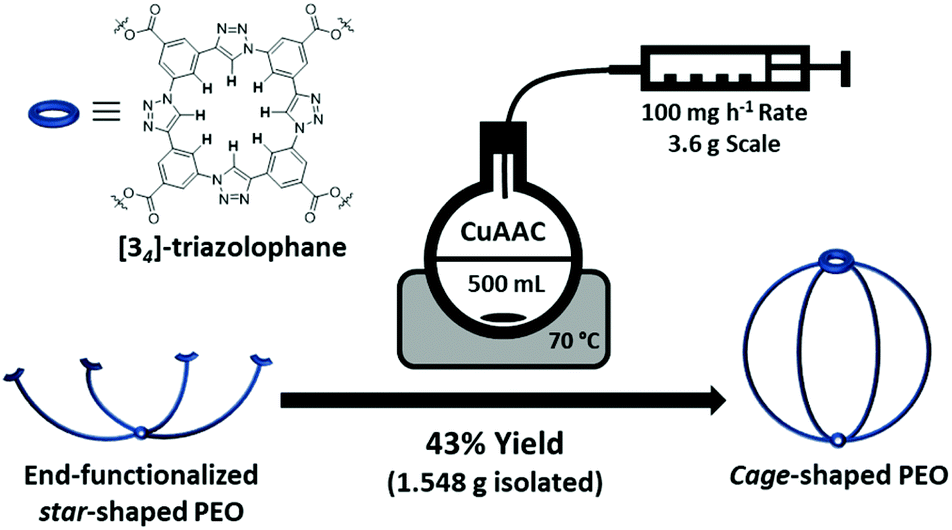 | ||
| Scheme 1 Schematic summary of the gram-scale synthesis of PEOcage by semi-batch CuAAC reaction. | ||
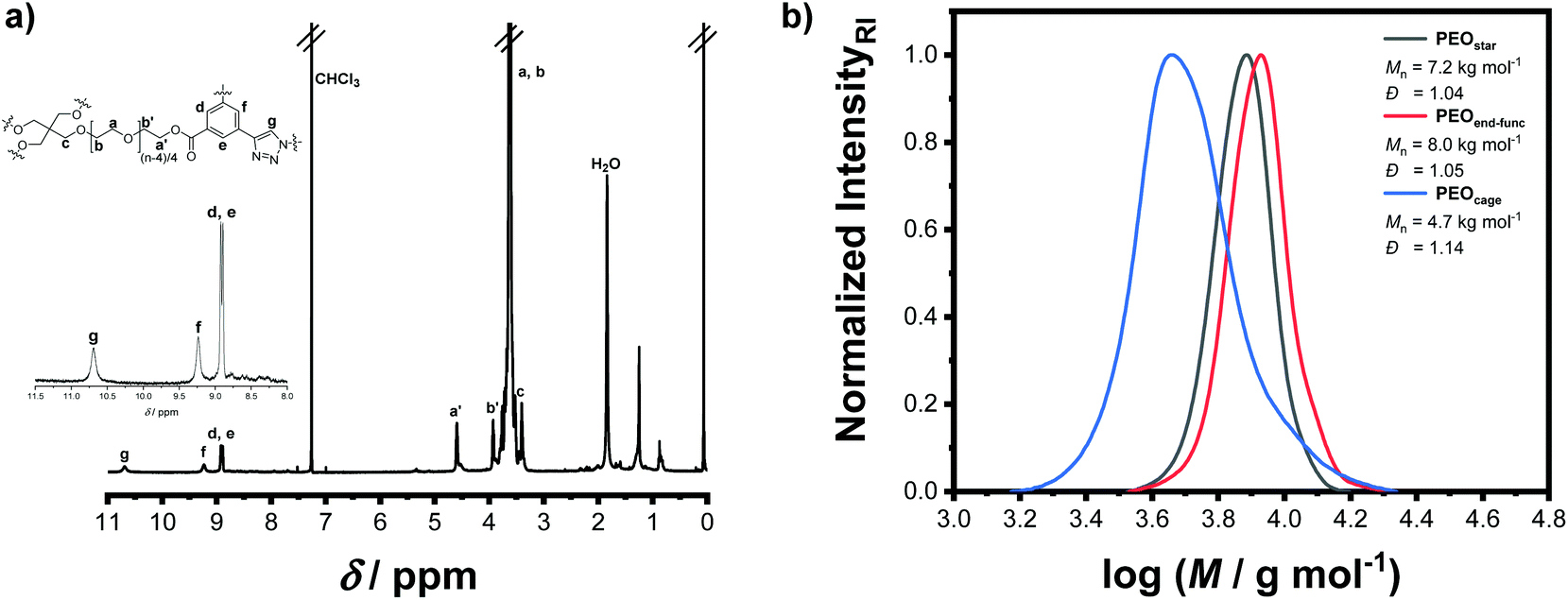 | ||
| Fig. 1 (a) 1H-NMR spectrum of the final PEOcage architecture. (b) SEC traces of PEOstar, PEOend-func and PEOcage. | ||
Now with sufficient material in hand, the thermal properties of the cage-shaped PEO were characterized by differential scanning calorimetry (DSC) and compared to the star-shaped PEO. As PEO-based materials are commonly comprised of both crystalline and amorphous domains in variable ratios, their semi-crystalline nature can be characterized by their melting temperature (Tm) and glass transition temperature (Tg) as well as the related enthalpies.23 Within this study, PEO-based PEs are of primary interest, which should preferably exhibit a completely amorphous phase, i.e a non-existing crystallization, in order to enable ionic conductivity in the created free volume.24 Additionally, a low Tg ensures a maximal chain mobility and an optimal ion transport by segmental motion.25 The predominant crystalline nature of PEOstar was highlighted by the prominent Tm located at 47.5 °C, the high fusion enthalpy 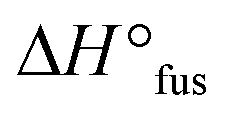 of 121.0 J g−1 and the absence of a noticeable Tg (Fig. 2a, Table 2). In comparison, literature values for high molar mass linear PEO are reported as Tm ∼ 65 °C,26
of 121.0 J g−1 and the absence of a noticeable Tg (Fig. 2a, Table 2). In comparison, literature values for high molar mass linear PEO are reported as Tm ∼ 65 °C,26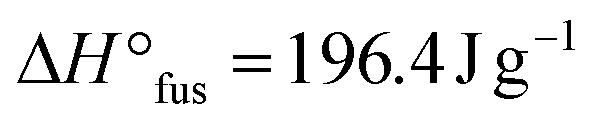 ,27 indicating an influence of the PEO architecture on the crystallization, yet not suppressing it completely. A further suppression of the crystallization was observed after the topological conversion into PEOcage, with Tm and
,27 indicating an influence of the PEO architecture on the crystallization, yet not suppressing it completely. A further suppression of the crystallization was observed after the topological conversion into PEOcage, with Tm and 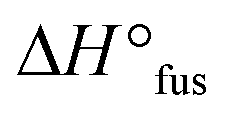 reduced to 30.3 °C and 51.6 J g−1, respectively, and a clearly detectable Tg of −45.9 °C (Fig. 2a, Table 2). These values correspond to a reduction of crystalline domains of 58% or 74% in comparison to PEOstar or pure PEO,28 respectively and represent an impressive reduction of crystallinity induced via a topological conversion into a cage-shaped architecture. In comparison, the reduction of crystallinity observed in a previously reported comb-shaped polymer architecture, consisting of comparable 24 and 54 ethylene oxide (EO) repeating units per side chain, was only 51% and 39%, respectively, relative to pure PEO.13 Furthermore, the thermal properties of different PEs prepared from PEOcage (PEcage) as well as PEOstar by addition of Li+-salt (PEstar) for comparison were examined considering that the addition of LiTFSI salt impacts the crystallinity substantially due to its plasticizing character. While lithium-salt loadings of [Li+]
reduced to 30.3 °C and 51.6 J g−1, respectively, and a clearly detectable Tg of −45.9 °C (Fig. 2a, Table 2). These values correspond to a reduction of crystalline domains of 58% or 74% in comparison to PEOstar or pure PEO,28 respectively and represent an impressive reduction of crystallinity induced via a topological conversion into a cage-shaped architecture. In comparison, the reduction of crystallinity observed in a previously reported comb-shaped polymer architecture, consisting of comparable 24 and 54 ethylene oxide (EO) repeating units per side chain, was only 51% and 39%, respectively, relative to pure PEO.13 Furthermore, the thermal properties of different PEs prepared from PEOcage (PEcage) as well as PEOstar by addition of Li+-salt (PEstar) for comparison were examined considering that the addition of LiTFSI salt impacts the crystallinity substantially due to its plasticizing character. While lithium-salt loadings of [Li+]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[EO] of 1:20 and 1:25 completely suppressed crystallization of the PEOcage, these loading values were not sufficient to suppress the crystallization for the comparable PEstar samples with
:[EO] of 1:20 and 1:25 completely suppressed crystallization of the PEOcage, these loading values were not sufficient to suppress the crystallization for the comparable PEstar samples with 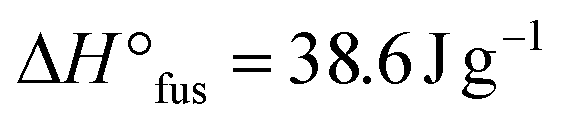 and 61.6 J g−1 for [Li+]:[EO] of 1:20 and 1:25, respectively, as shown in Table 2. Notably, usually significantly higher LiTFSI loadings comprised of [Li+]:[EO] between 1:6–1:12 are necessary to ensure a completely amorphous PEO.28 Moreover, in both PEstar and PEcage samples, the Tg values decreased when the [Li+]:[EO] ratio was reduced from 1:20 to 1:25, due to a lower amount of quasi-ionic cross-linking between the PEO chain segments.13,29 On the contrary, higher Tg values were systematically observed for PEcage in comparison with PEstar arising from the architecture-induced restriction of the polymers segmental motion. Nonetheless, the thermal characterization showed quite impressively that the topological conversion into a cage-based architecture not only reduces crystallinity, but also allows for low LiTFSI loadings. In addition, the thermal stabilities of PEOstar, PEOcage and PEcage1:25 were examined by thermogravimetric analysis (TGA) measurements.
and 61.6 J g−1 for [Li+]:[EO] of 1:20 and 1:25, respectively, as shown in Table 2. Notably, usually significantly higher LiTFSI loadings comprised of [Li+]:[EO] between 1:6–1:12 are necessary to ensure a completely amorphous PEO.28 Moreover, in both PEstar and PEcage samples, the Tg values decreased when the [Li+]:[EO] ratio was reduced from 1:20 to 1:25, due to a lower amount of quasi-ionic cross-linking between the PEO chain segments.13,29 On the contrary, higher Tg values were systematically observed for PEcage in comparison with PEstar arising from the architecture-induced restriction of the polymers segmental motion. Nonetheless, the thermal characterization showed quite impressively that the topological conversion into a cage-based architecture not only reduces crystallinity, but also allows for low LiTFSI loadings. In addition, the thermal stabilities of PEOstar, PEOcage and PEcage1:25 were examined by thermogravimetric analysis (TGA) measurements.
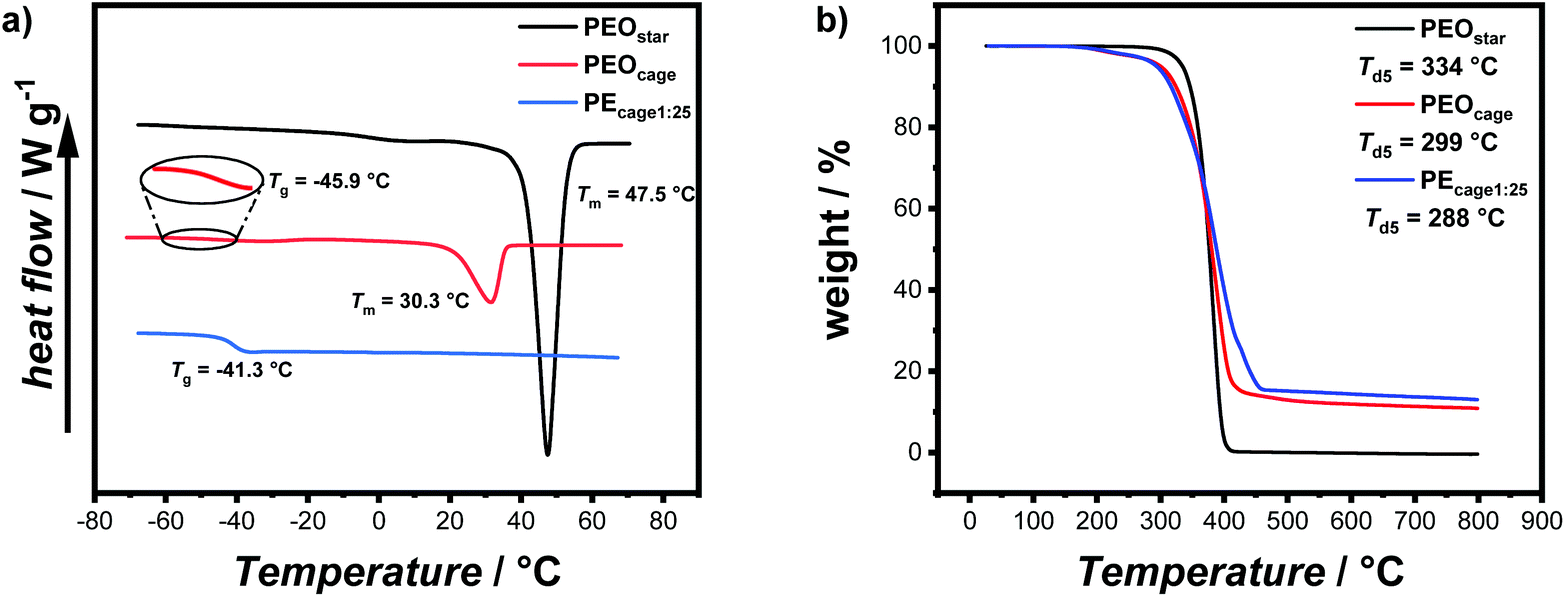 | ||
| Fig. 2 (a) DSC thermogram of PEOstar, PEOcage and PEcage1:25 samples, showing the effect of architecture change and lithium salt addition. (b) TGA thermogram displaying the degradation profile of PEOstar, PEOcage and PEcage1:25. Subscripted numbers correspond to [LiTFSI]:[EO] ratio. | ||
| Entry | Polymer/PE | [Li+]:[EO] ratio |
T m/°C |
|
T g/°C |
|---|---|---|---|---|---|
| 1 | PEOstar | n/a | 47.5 | 121.0 | n/a |
| 2 | PEstar1:20 | 1:20 |
35.0 | 38.6 | −42.5 |
| 3 | PEstar1:25 | 1:25 |
39.0 | 61.6 | −44.2 |
| 4 | PEOcage | n/a | 30.3 | 51.6 | −45.9 |
| 5 | PEcage1:20 | 1:20 |
n/a | n/a | −40.8 |
| 6 | PEcage1:25 | 1:25 |
n/a | n/a | −41.3 |
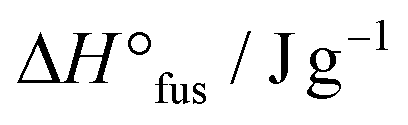
All materials showed a good thermal stability up to over 280 °C with a decomposition temperature at 5% weight loss Td5 of around 334 °C for PEOstar, 299 °C for PEOcage and 288 °C for PEcage1:25 (Fig. 2b). Here, the presence of thermally more labile ester moieties might induce a small reduction in thermal stability when comparing PEOstar with both other samples. Further, the remaining char above 500 °C of ∼12% (for PEOcage) and ∼15% (for PEcage1:25), could be correlated to the theoretical content of [34]-triazolophane within PEOcage (11.8%) and the remaining lithium species within PEcage1:25.
Last but not least, the ionic conductivity of both PEcage samples were measured within a temperature range of 70 °C to 0 °C via electrochemical impedance spectroscopy (EIS) and compared to the values obtained from PEstar and a linear PEO-based electrolyte (PElinear with 5 Mg mol−1) (Fig. 3). In accordance with the observation by DSC analysis, the complete crystallization suppression of the PEcage samples led to a typical Vogel–Tammann–Fulcher behavior regarding their ionic conductivity. In addition, PEcage1:25 performed slightly better than PEcage1:20 over the whole temperature range, as predicted by the difference in Tg of 0.5 °C noticed between their respective DSC thermograms resulting from the lower LiTFSI salt loading, which reduced the quasi-ionic cross-linking and thus increased the segmental motion. Instead, EIS analyses of PEstar and PElinear showed the known and eminent drop in ionic conductivity as soon as PEO crystallization occurred in the range from 50 °C to 30 °C depending of the respective topology and on the LiTFSI content. The somewhat higher chain mobility of PEstar and PElinear above their melting points resulted in a slightly higher ionic conductivity than the PEcage. Yet, PEcage exhibited a superior ionic conductivity below 40 °C, resulting in ionic conductivity values of 1 × 10−5 S cm−1 at 20 °C, outperforming the PEstar and PElinear by a factor of 10. Nonetheless, it has to be stated that these ionic conductivities are still relatively low from a practical point of view, though, they clear show the capability of architectural approaches taken by polymer chemists.
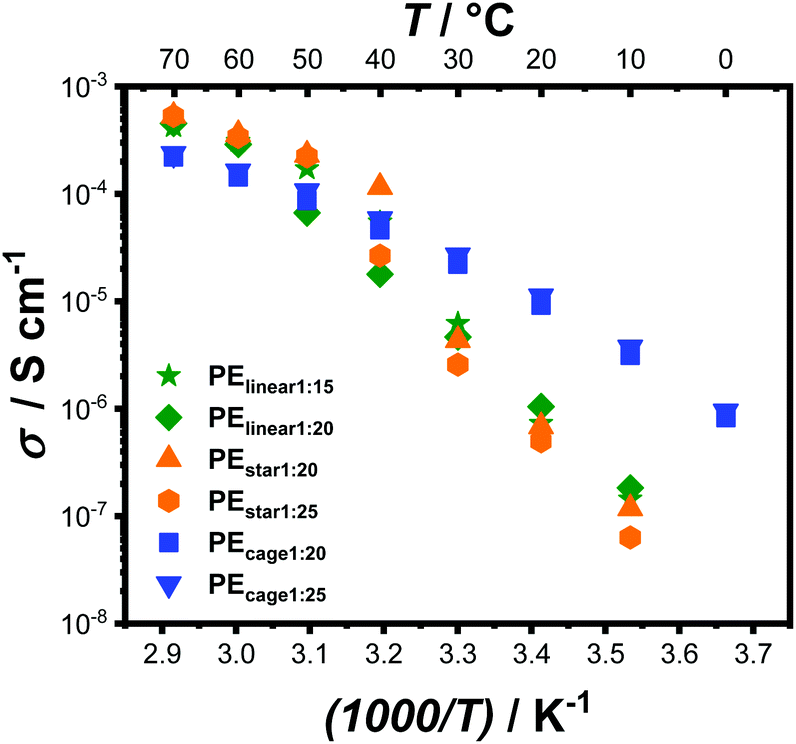 | ||
| Fig. 3 Temperature-dependent ionic conductivity of PEcage compared to PEstar and PElinear samples with different LiTFSI loading ratios. Subscripted numbers correspond to [LiTFSI]:[EO] ratio. | ||
Conclusions
In conclusion, the gram-scale synthesis of a four-arm cage-shaped PEO was successfully accomplished, opening up the possibility for applicational studies. For this, PEOcage was investigated as a potential polymer electrolyte for lithium-ion batteries. In this regard, addition of a reduced amount of lithium-salt to the polymer electrolyte resulted in purely amorphous samples with superior ionic conductivity below 40 °C. Notably, the ionic conductivity gap recorded at 20 °C exceeded the values of the polymer electrolyte control samples by 10 times. Beyond being a significant step ahead in the research of applications for cage polymers, the present study clearly underlines the importance of topology and architecture when designing applications of polymer materials. Lastly, the exciting opportunities offered by architectural approaches might contribute to the conception of next generation polymer electrolytes to advance electric energy storage.Author contributions
A. J. Butzelaar: conceptualization, investigation, writing – original draft. M. Gauthier-Jaques: conceptualization, investigation, writing – original draft. K. L. Liu: investigation. Gunther Brunklaus: writing – review & editing, supervision, funding acquisition. Martin Winter: supervision, funding acquisition. Patrick Theato: conceptualization, writing – review & editing, supervision, funding acquisition. A. J. Butzelaar and M. Gauthier-Jaques contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the German Federal Ministry of Education and Research (BMBF) within ‘FestBatt’ (13XP0175A and 13XP0175C) and the Helmholtz Association is gratefully acknowledged.Notes and references
- Q. Zhao, S. Stalin, C. Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS
.
- K. S. Ngai, S. Ramesh, K. Ramesh and J. C. Juan, Ionics, 2016, 22, 1259–1279 CrossRef CAS
- P. V. Wright, Br. Poly. J., 1975, 7, 319–327 CrossRef CAS
-
(a) Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC
- Batteries LMP®|Blue Solutions. https://www.blue-solutions.com/en/blue-solutions/technology/batteries-lmp/ (accessed 2021-04-09).
-
(a) P. J. Sánchez-Soto, J. M. Ginés, M. J. Arias, C. Novák and A. Ruiz-Conde, J. Therm. Anal. Calorim., 2002, 67, 189–197 CrossRef
-
(a) D. Bamford, A. Reiche, G. Dlubek, F. Alloin, J. Y. Sanchez and M. A. Alam, J. Chem. Phys., 2003, 118, 9420–9432 CrossRef CAS
-
(a) M. Falco, C. Simari, C. Ferrara, J. R. Nair, J. G. Meligrana, F. Bella, I. Nicotera, P. Mustarelli, M. Winter and C. Gerbaldi, Langmuir, 2019, 35, 8210–8219 CAS
-
(a) X. Qian, N. Gu, Z. Cheng, X. Yang, E. Wang and S. Dong, Mater. Chem. Phys., 2002, 74, 98–103 CrossRef CAS
-
(a) J. Xi, X. Qiu, J. Li, X. Tang, W. Zhu and L. Chen, J. Power Sources, 2006, 157, 501–506 CrossRef CAS
- K. Liu, R. Zhang, J. Sun, M. Wu and T. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 46930–46937 CrossRef CAS PubMed
-
(a) J. Rolland, J. Brassinne, J. P. Bourgeois, E. Poggi, A. Vlad and J. F. Gohy, J. Mater. Chem. A, 2014, 2, 11839 RSC
- A. J. Butzelaar, K. L. Liu, P. Röring, G. Brunklaus, M. Winter and P. Theato, ACS Appl. Polym. Mater., 2021, 3, 1573–1582 CrossRef CAS
-
(a) L. Y. Qiu and Y. H. Bae, Pharm. Res., 2006, 23, 1–30 CrossRef CAS PubMed
-
(a) H. Oike, H. Imaizumi, T. Mouri, Y. Yoshioka, A. Uchibori and Y. Tezuka, J. Am. Chem. Soc., 2000, 122, 9592–9599 CrossRef CAS
- C. E. Wagner, J. S. Kim and K. J. Shea, J. Am. Chem. Soc., 2003, 125, 12179–12195 CrossRef CAS PubMed
- G. Y. Shi and C. Y. Pan, J. Polym. Sci. Polym. Chem., 2009, 47, 2620–2630 CrossRef CAS
-
(a) J. Jeong, K. Kim, R. Lee, S. Lee, H. Kim, H. Jung, M. A. Kadir, Y. Jang, H. B. Jeon, K. Matyjaszewski, T. Chang and H. J. Paik, Macromolecules, 2014, 47, 3791–3796 CrossRef CAS
-
(a) Y. Satoh, H. Matsuno, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, Macromolecules, 2017, 50, 97–106 CrossRef CAS
- T. Noda, Y. Doi, Y. Ohta, S. I. Takata, A. Takano and Y. Matsushita, J. Polym. Sci., 2020, 58, 2098–2107 CrossRef CAS
- M. Gauthier-Jaques and P. Theato, ACS Macro Lett., 2020, 9, 700–705 CrossRef CAS
-
(a) Y. Hua, R. O. Ramabhadran, E. O. Uduehi, J. A. Karty, K. Raghavachari and A. H. Flood, Chem. – Eur. J., 2011, 17, 312–321 CrossRef CAS PubMed
- X. Li, S. Cheng, Y. Zheng and C. Y. Li, Mol. Syst. Des. Eng., 2019, 4, 793–803 RSC
- D. E. Martínez-Tong, L. A. Miccio and A. Alegria, Soft Matter, 2017, 13, 5597–5603 RSC
-
(a) S. Das and A. Ghosh, AIP Adv., 2015, 5, 27125 CrossRef
-
(a) S. A. Madbouly and B. A. Wolf, J. Chem. Phys., 2002, 117, 7357–7363 CrossRef CAS
- G. Dreezen, M. H. J. Koch, H. Reynaers and G. Groeninckx, Polymer, 1999, 40, 6451–6463 CrossRef CAS
- S. Lascaud, M. Perrier, A. Vallée, S. Besner, J. Prud'homme and M. Armand, Macromolecules, 1994, 27, 7469–7477 CrossRef CAS
- A. Vallée, S. Besner and J. Prud'homme, Electrochim. Acta, 1992, 37, 1579–1583 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, instrumentation, polymers and polymer electrolytes preparation and characterization procedures. See DOI: 10.1039/d1py00490e |
| ‡ These authors contributed equally to this publication. |
| This journal is © The Royal Society of Chemistry 2021 |