
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWell-defined cyclic polymer synthesis via an efficient etherification-based bimolecular ring-closure strategy†
Sandeep
Sharma
 ,
Konstantinos
Ntetsikas
,
Viko
Ladelta
,
Saibal
Bhaumik
and
Nikos
Hadjichristidis
*
,
Konstantinos
Ntetsikas
,
Viko
Ladelta
,
Saibal
Bhaumik
and
Nikos
Hadjichristidis
*
Physical Sciences and Engineering Division, KAUST Catalysis Center, Polymer Synthesis Laboratory, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia. E-mail: nikolaos.hadjichristidis@kaust.edu.sa
First published on 28th October 2021
Abstract
The synthesis of cyclic polymers on a large scale is a challenging task for polymer scientists due to the requirement of ultra-high dilution conditions. In this paper, we demonstrate an alternative method to prepare cyclic polymers with moderate dilution and up to 1 gram scale. We employed a simple Williamson etherification reaction to prepare cyclic polymers with a good solvent/non-solvent combination. In this way, various polystyrene (PS) and polyethylene glycol (PEG) cyclic homopolymers were synthesized. Anionic polymerization using high vacuum techniques combined with the postpolymerization reaction was used to generate linear dihydroxy PS precursors. The synthesized linear and cyclic homopolymers were fully characterized using various spectroscopic and analytical techniques, such as size exclusion chromatography (SEC), matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF-MS), and differential scanning calorimetry (DSC). Detailed nuclear magnetic resonance (NMR) spectroscopic studies were also performed to obtain the complete structural information of the synthesized polymers.
Introduction
In recent years, cyclic polymers have gained significant attention from the scientific community due to their unique properties.1,2 Cyclic polymers possess different properties from their linear counterparts in terms of the glass transition temperature, crystallinity, and melt-flow dynamics.1,3 In general, there are two major strategies to synthesize cyclic polymers: (a) ring-expansion, where the cyclic topology of the growing polymer chain is preserved throughout, and (b) ring-closure of a linear polymer.4,5 The ring-expansion approach is induced by the insertion of monomer units into an activated (pseudo)cyclic chain.6,7 This method avoids high dilution and precursors with functionalized end groups; however, it has limitations towards different monomers. Moreover, high molecular weight polymers can be easily prepared without entropic penalties or the formation of linear by-products, but the control over molecular weight and dispersity is limited.1On the other hand, the ring-closure method comprises the coupling reaction of an α,ω-functionalized linear polymer, generally performed under high dilution conditions on a small scale and using a catalyst.8 This strategy can further be divided into unimolecular and bimolecular coupling reactions. The unimolecular ring-closure technique involves the cyclization of homo-difunctional9,10 or hetero-difunctional linear polymers.11–13 Unimolecular cyclization is not affected by the stoichiometric ratio; however, it requires high dilution to suppress the oligomerization. The bimolecular ring-closure strategy involves the reaction between an α,ω-homodifunctional linear polymer and a difunctional coupling agent in a dilute solution. The exact stoichiometric ratio of all reagents is required to generate pure cyclic polymers.
Initially, Rempp et al. used the bimolecular coupling method to synthesize cyclic polymers.14 They reported the synthesis of cyclic polystyrene (PS) by the coupling reaction of a living difunctional PS, prepared via anionic polymerization, with a difunctional electrophilic compound (dibromo-p-xylene) in a stoichiometric ratio under high dilution. In the same year, Höcker et al. reported the synthesis of cyclic PS by the slow addition of living difunctional PS to α,α′-dichloro-p-xylene solution in tetrahydropyran (THP), but still the yield of cyclization was below 50%.15 Roovers et al.16 reported the synthesis of high molecular weight ring PS (molecular weights in the range of 5000 to 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) using sodium naphthalene as a difunctional initiator and dichlorodimethylsilane as the linking agent. Similarly, various cyclic homopolymers and block copolymers were synthesized using anionic polymerization and coupling agents with different functionalities.17–21
000 g mol−1) using sodium naphthalene as a difunctional initiator and dichlorodimethylsilane as the linking agent. Similarly, various cyclic homopolymers and block copolymers were synthesized using anionic polymerization and coupling agents with different functionalities.17–21
Apart from anionic polymerization, other polymerization methods have also been used to synthesize cyclic polymers combined with different bimolecular and unimolecular coupling methods, such as the self-accelerating click reaction22 and the electrostatic self-assembly and covalent fixation (ESA-CF) method.23 However, the upgrading of these processes to a larger scale (grams) is still a very challenging task. Some efforts have been made to synthesize cyclic polymers by the continuous-flow technique24 and the self-accelerating double strain-promoted azide–alkyne click reaction (DSPAAC) for cyclization.25 Nonetheless, these reactions require a large amount of solvent to prepare cyclic polymers on a large scale.
The Williamson etherification is a well-known and straightforward reaction in organic chemistry. The synthesis of cyclic polyethers via the Williamson etherification reaction has been reported, where a homodifunctional polyethylene glycol (PEG) bearing hydroxyl end groups was cyclized by reacting with dichloromethane (DCM) in the presence of potassium hydroxide.26 Recently, the scale-up of cyclic PEG polymers was also attempted via the Williamson etherification.27 In the latter, cyclic PEG polymers were generated through the reaction of dihydroxyl PEG with tosyl chloride (Ts-Cl) and purified through multi-step procedures. A combination of a good solvent and non-solvent was used for the synthesis of cyclic PEG to minimize the chain distance. The influence of good solvent/non-solvent mixtures on the preparation of cyclic PS was also studied in radical trap assisted atom transfer radical coupling (RTA-ATRC).28
Cyclic polymers, which are synthesized by click and other coupling reactions, are generally prepared by atom transfer radical polymerization (ATRP)29 and reversible addition–fragmentation chain-transfer (RAFT) polymerization.30 These techniques do not always lead to well-defined polymers with controlled molecular characteristics and a narrow polydispersity index (PDI < 1.1). On the other hand, living anionic polymerization leads to well-defined polymers having the highest molecular weight, structural and compositional homogeneity. Hence, we designed a bimolecular ring-closing reaction between telechelic polymers and coupling agents [1,4-bis(bromomethyl)benzene and/or 2,6-bis(bromomethyl)pyridine] to generate cyclic polymers in a good solvent/non-solvent mixture. Benzylic bromo compounds as linking agents were chosen due to their higher reactivity compared to the aliphatic ones.
Herein, we report a traditional Williamson etherification-based efficient cyclization reaction to prepare cyclic PS and PEG homopolymers up to 1 gram scale in moderate dilution. The linear α,ω-dihydroxy PS precursors (HO-PS-OH) were synthesized via anionic polymerization using high vacuum techniques and the postpolymerization reaction (deprotection of the α-tert-butyldimethylsilyl group). Overall, seven different cyclic polymers (four PS and three PEG) have been prepared with different molecular weights ranging from 4000 to 23000 g mol−1 for the PS and 2000 and 6000 g mol−1 for the PEG samples, respectively.
Experimental
Materials
1,4-Bis(bromomethyl)benzene (98%), 2,6-bis(bromomethyl)pyridine (98%), potassium hydroxide (KOH) and tetra-n-butylammonium fluoride (TBAF, 1 M in THF) were purchased from Sigma-Aldrich and used without further purification. Tetrahydrofuran (THF) was purchased from VWR and dried over sodium and benzophenone. Dichloromethane (DCM) was purchased from VWR and used without further purification. Benzene (VWR 99%) was purified over CaH2 and distilled under high vacuum in a round bottom flask containing PS(−)Li(+), exhibiting the characteristic yellow-orange color. The protected hydroxyl-functional initiator, 3-(t-butyldimethylsiloxy)-1-propyllithium (Gelest, 0.5 M in cyclohexane), was diluted to the appropriate concentration using purified benzene in custom made glass apparatus. Styrene (Sigma Aldrich, 99%) was purified by distillation over CaH2 and subsequently over di-n-butylmagnesium (Sigma-Aldrich, 1 M solution in heptane) and stored at −20 °C in pre-calibrated ampoules. N,N,N′,N′-Tetramethylethylenediamine (TMEDA) was purified over CaH2 overnight and distilled twice over a sodium mirror, followed by distillation in custom-made glass apparatus and diluted in the appropriate concentration using purified benzene. Ethylene oxide (EO, Sigma-Aldrich, 99.5%) was used as the end-capping agent, purified by successive distillations over CaH2 and n-BuLi at 0 °C, and finally stored in ampoules under high vacuum. Methanol (terminating agent) was purified by successive distillations over CaH2 and stored in ampoules under high vacuum. All the above purification methods were performed in custom-made glass apparatus by employing high-vacuum techniques (HVT) and standard procedures, based on the requirements of anionic polymerization, described in more detail elsewhere.31,32 Dithranol (≥98.0%) and sodium trifluoroacetate (NaTFA, 98%) were used as received. Poly(ethylene glycol) samples (PEG-2k, PEG-6k) were purchased from Sigma-Aldrich and purified by dissolving in 1,4-dioxane (anhydrous, 99.8%, Sigma-Aldrich) and cryoevaporating 1,4-dioxane, followed by drying under vacuum overnight.Instrumentation
Size exclusion chromatography (SEC) measurements were carried out at 35 °C on a Viscotek GPC max VE-2001 with THF as the eluent at a flow rate of 1.0 mL min−1, equipped with an isocratic pump, Styragel HR2, and HR4 columns in series (300 mm × 8 mm) and a differential refractive index detector (DRI). The system was calibrated with polystyrene (PS) standards (Mp: 370 to 4220000 g mol−1). Proton nuclear magnetic resonance spectra (1H-NMR) and quantitative 13C-NMR spectra were recorded on a Bruker AVANCE III spectrometer operating at 500 MHz. 1H–1H correlation spectra (COSY) and total correlation spectra (TOCSY) were recorded on a Bruker AVANCE III-600 MHz spectrometer. Chloroform-d (CDCl3) was used as the solvent in all cases, and all the measurements were conducted at 25 °C. Matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS) experiments were carried out using dithranol as the matrix in THF and sodium trifluoroacetate (NaTFA) as the ionizing agent on a Bruker Ultraflex III MALDI-TOF mass spectrometer (Bruker Daltonik, Bremen, Germany). Differential scanning calorimetry (DSC) measurements were performed on a Mettler Toledo DSC1/TC 100 system. The samples were heated from room temperature to 150 °C to erase the thermal/solvent history, cooled to −80 °C, and heated again to 150 °C, with a heating/cooling rate of 10 °C min−1.
Synthesis of α-tert-butyldimethylsilyloxy-ω-PS-OH (Pr-l-PS) homopolymers
All synthetic procedures were conducted using conventional high-vacuum techniques. The anionic polymerization was carried out in evacuated, n-BuLi washed custom-made glass reactors at room temperature. Reagents were added via break-seals, and aliquots for characterization were taken by heat-sealing the proper constrictions in all steps. More details concerning the techniques and the apparatus used are given in previous reports.31–33 For example, the synthesis of the α-protected tert-butyldimethylsilyloxy-ω-functionalized with hydroxyl group PS (labeled as Pr-l-PS-4k) is described in detail. Styrene (5 g) was added to 80 mL of benzene, followed by the addition of TMEDA (2.50 mmol) and 1.25 mmol of the protected functional initiator 3-(t-butyldimethylsilyloxy)-1-propyllithium, and kept for 24 h at room temperature. After completion of the polymerization, an aliquot was taken by heat-sealing the proper constriction to verify the molecular characteristics of the synthesized PS. Subsequently, ethylene oxide (∼1 mL) was added to the reaction mixture and left for 12 h at room temperature, and the reaction was terminated by the addition of degassed methanol (∼0.5 mL) (Scheme 1). The resultant reaction mixture was precipitated into a large excess of methanol, and the final polymer was dried in a vacuum oven for 24 h at 40 °C. The number-average molecular weight and dispersity index of Pr-l-PS-4k were calculated by SEC (Mn = 4100 g mol−1 and Đ = 1.04) calibrated with PS standards. Other polymers (Pr-l-PS-7.5k, Pr-l-PS-13k, and Pr-l-PS-23k) were synthesized in a similar way. | ||
| Scheme 1 General synthetic strategy for the preparation of HO-PS-OH homopolymers via anionic polymerization and the postpolymerization reaction. | ||
Synthesis of α,ω-dihydroxy PS (l-PS) difunctional homopolymers
In a round bottom flask, 3 g of Pr-l-PS-4k (0.73 mmol) homopolymer were dissolved in 50 mL of dry THF. The solution was purged with argon for 15 minutes, followed by the slow addition of TBAF (7.31 mmol) at room temperature. The reaction mixture was stirred for 48 h at room temperature. The deprotected telechelic α,ω-dihydroxy PS (l-PS-4k) homopolymer was precipitated in methanol three times and dried overnight in a vacuum oven at 40 °C. The successful deprotection of the α-t-butyldimethylsiloxy-ω-PS-OH sample was confirmed by 1H-NMR spectroscopy. Other polymers (Pr-l-PS-7.5k, Pr-l-PS-13k, and Pr-l-PS-23k) were deprotected by the same method. For simplicity reasons, all α,ω-dihydroxy PS were labeled as l-PS-4k, l-PS-7.5k, l-PS-13k, and l-PS-23k.Cyclization procedure of telechelic α,ω-dihydroxy PS homopolymers
A typical example is as follows. An excess amount of KOH powder (2 g) was dispersed in a 100 mL mixture of THF and n-hexane (10%) in a 500 mL Schlenk flask. In another round bottom flask, an equimolar mixture of l-PS-4k (1 g, 0.24 mmol) and 2,6-bis(bromomethyl)pyridine (64.6 mg, 0.24 mmol) was dissolved in 100 mL of dry THF. The polymer mixture was added to the Schlenk flask dropwise using a syringe pump in 24 h at 55 °C (4.2 mL h−1). After the complete addition, the mixture was left under stirring for 12 h at 55 °C. Afterward, the excess KOH was removed via filtration, and all solvents were evaporated under reduced pressure. Subsequently, the obtained crude reaction mixture was dissolved in DCM (100 mL), washed with water (3 × 50 mL), and dried over magnesium sulfate (MgSO4). The final cyclic polymer was purified by repeated good solvent/non-solvent (toluene/methanol) fractionations to remove the high molecular weight linear condensation by-product. The l-PS-7.5k polymer was cyclized with 2,6-bis(bromomethyl)pyridine, while l-PS-13k and l-PS-23k polymers were cyclized with 1,4-bis(bromomethyl)benzene. All cyclic PS polymers were labeled as c-PS-4k, c-PS-7.5k, c-PS-13k, and c-PS-23k.Cyclization procedure of α,ω-dihydroxy PEG homopolymers
A typical example is as follows. An excess amount of KOH powder (2 g) was dispersed in a 100 mL mixture of THF and n-hexane (10%) in a 500 mL Schlenk flask. In another round bottom flask, an equimolar mixture of l-PEG-2k (1 g, 0.5 mmol) and 2,6-bis(bromomethyl)pyridine (132.5 mg, 0.5 mmol) was dissolved in 100 mL of dry THF. The polymer mixture was added to the Schlenk flask dropwise using a syringe pump in 24 h at 55 °C (4.2 mL h−1). After the complete addition, the mixture was left under stirring for 12 h at 55 °C. Afterward, the excess KOH was removed via filtration, and all solvents were evaporated under reduced pressure. Subsequently, the obtained crude reaction mixture was dissolved in DCM and precipitated in hexane. The final cyclic PEG polymer was purified by repeated good solvent/non-solvent (toluene/hexane) fractionations to remove the high molecular weight linear condensation by-product. All cyclic PEG polymers were labeled as c-PEG-2k (linking agent: 2,6-bis(bromomethyl)pyridine), c-PEG-2k-b (linking agent: 1,4-bis(bromomethyl)benzene) and c-PEG-6k (linking agent: 2,6-bis(bromomethyl)pyridine).Results and discussion
Synthesis and molecular characterization results of linear and cyclic homopolymers
The synthesis of hydroxyl (–OH) functionalized telechelic polymers with a well-defined structure and low polydispersity via free-radical or other controlled/living polymerization techniques is not feasible.21 The living anionic polymerization method is well known for generating well-defined polymers with controlled molecular characteristics. 3-(t-Butyldimethylsiloxy)-1-propyllithium was used as the initiator in the polymerization of styrene in a hydrocarbon solvent (benzene), followed by end-capping with EO and termination with degassed methanol, to generate α-tert-butyldimethylsilyloxy-ω-PS-OH functionalized (Pr-l-PS) homopolymers. As the next step, a deprotection reaction took place by reacting Pr-l-PS with an excess of TBAF solution in THF to generate α,ω-dihydroxyl PS (l-PS) homopolymers. The use of functionalized alkyllithium initiators provides a simple and general methodology for the synthesis of telechelic and heterotelechelic polymers.34,35 The t-butyldimethylsiloxy protecting group is readily removed using a variety of reagents, such as hydrochloric acid or fluoride sources.36 On the other hand, end-capping of the carbanionic chain end with EO and termination with methanol lead to OH-functionalized polymers with a high degree of functionalization (>99%).37 In general, epoxides are reactive toward nucleophilic attack by living carbanions, due to their high ring strain. When lithium is used as a counterion in anionic polymerization, it does not proceed to further propagation of epoxides due to the strong oxygen–lithium aggregation, leading to the insertion of only one terminal unit.38 A series of dihydroxyl functionalized PS were synthesized via the combination of living anionic polymerization using high vacuum techniques and the postpolymerization reaction (deprotection) as described above (Scheme 1). Four different telechelic PS exhibiting different molecular weights (4k, 7.5k, 13k, and 23k) were prepared.The telechelic polymers were cyclized through a bimolecular ring-closure strategy by reacting an equimolar quantity of 2,6-bis(bromomethyl)pyridine or 1,4-bis(bromomethyl) benzene with a mixture of solvents (THF and n-hexane) (Schemes 2A and B). The purpose of using a non-solvent (n-hexane) was to improve the ring-closure reaction by reducing (on average) the end-to-end distance of the polymer chains. This approach helped to perform the cyclization process in higher concentrations due to the presence of the non-solvent. Thus, a series of cyclic polymers up to 1 g scale were synthesized in 80–85% yield.
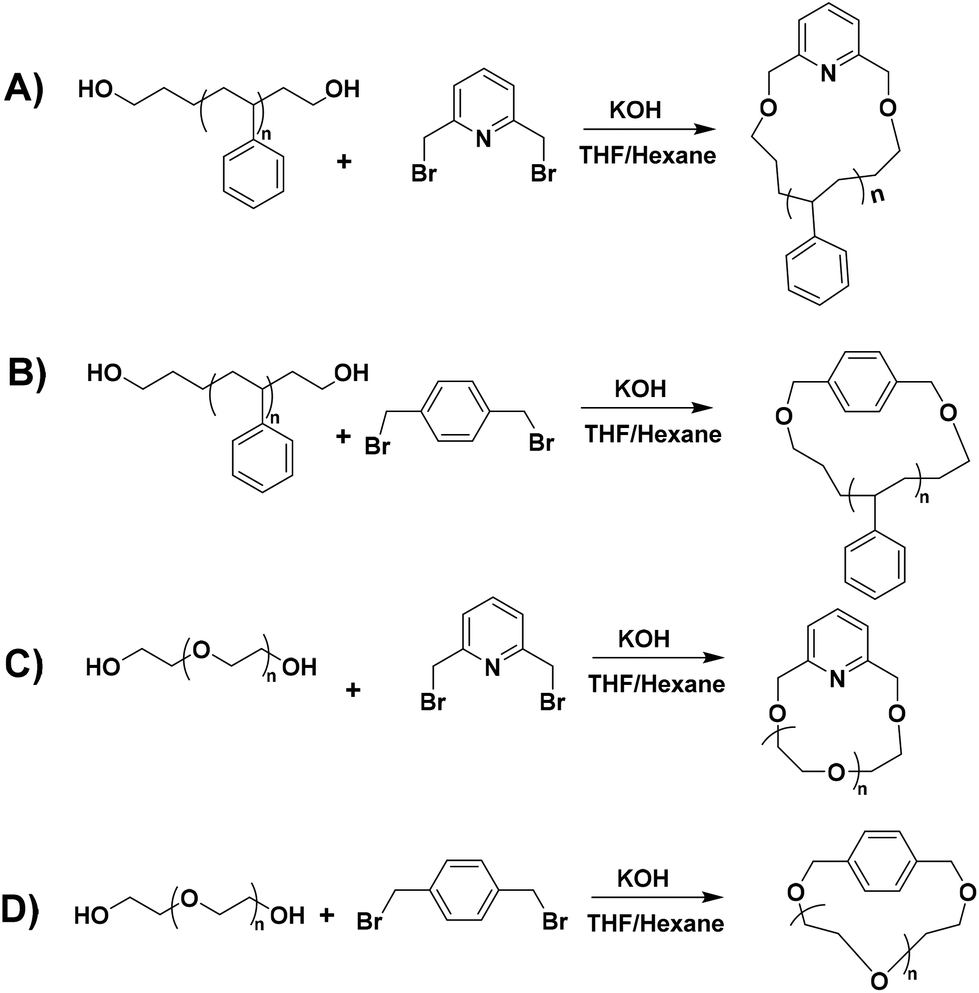 | ||
| Scheme 2 General synthetic strategy for the preparation of cyclic polymers: (A) reaction of the HO-PS-OH (l-PS) homopolymer with 2,6-bis(bromomethyl)pyridine, (B) reaction of the HO-PS-OH (l-PS) homopolymer with 1,4-bis(bromomethyl) benzene, (C) reaction of l-PEG with 2,6-bis(bromomethyl)pyridine and (D) reaction of l-PEG with 1,4-bis(bromomethyl) benzene. | ||
In the case of cyclic PEG, commercial linear dihydroxyl l-PEG-2k and l-PEG-6k were used for the bimolecular coupling reaction with 2,6-bis(bromomethyl)pyridine (Scheme 2C) and 1,4-bis(bromomethyl) benzene as linking agents (Scheme 2D).
To confirm the effect of the non-solvent (hexane) on the cyclization of l-PEG-2k with 1,4-bis(bromomethyl) benzene, we performed the reaction in the presence and the absence of a non-solvent and the synthesized polymers were characterized using SEC and 1H-NMR spectroscopy.
The molecular characteristics along with the corresponding glass transition temperatures (Tg) of all synthesized linear and cyclic homopolymers are given in Table 1. As a representative example, SEC chromatographs of the linear and cyclic PS-4k polymers are given in Fig. 1. The SEC chromatograph of the linear PS-4k (l-PS-4k) showed a peak molecular weight of 4800 g mol−1 (Mp), while the cyclic PS-4k (c-PS-4k) showed a lower Mp value (3700 g mol−1), leading to a higher retention time compared to the linear precursor. It is well known that cyclic polymers exhibit a lower hydrodynamic volume than linear ones due to the absence of free chain ends.39 Therefore, the SEC analysis indicated the first evidence for the successful synthesis of the cyclic polymer. The SEC chromatograph of the crude cyclic polymer revealed some additional peaks (mainly the condensation product), which were removed by fractionation in a toluene/methanol (good solvent/non-solvent) system. The SEC chromatographs of the crude and the purified c-PS-4k are shown in Fig. S1.† The ratio of the cyclic and the high molecular weight condensation products (linear) was calculated from each SEC chromatograph (Table S1†). For the c-PS-4k-crude sample, the ratio of the cyclic and linear products is 79:21 (Fig. S1†). The SEC chromatographs of all cyclic crude samples, and the respective linear and purified cyclic polymers are shown in the ESI (Fig. S2–S7†). The PDI of the cyclic polymer and its linear counterpart was found to be nearly the same. The number-average molecular weight (Mn) of all linear PS was determined by SEC since the instrument was calibrated with PS standards, and in all cases, the PDI was found in the range of 1.02 to 1.05.
 | ||
| Fig. 1 SEC chromatographs of l-PS-4K and the purified c-PS-4k (THF, 35 °C, PS standards). | ||
| Sample |
M
na (g mol−1) |
M
pa (g mol−1) |
PDIa (Đ) |
T
gb (°C) |
|---|---|---|---|---|
| a M n, Mp, and Đ were determined by SEC in THF at 35 °C. b T g was measured by DSC. | ||||
| l-PS-4k | 4100 | 4800 | 1.04 | 79.5 |
| c-PS-4k | 3500 | 3700 | 1.05 | 86.9 |
| l-PS-7.5k | 7600 | 7900 | 1.02 | 89.7 |
| c-PS-7.5k | 6500 | 6800 | 1.03 | 94.2 |
| l-PS-13k | 13200 |
14200 |
1.02 | 92.8 |
| c-PS-13k | 10200 |
10900 |
1.03 | 96.6 |
| l-PS-23k | 23100 |
25200 |
1.02 | 94.2 |
| c-PS-23k | 19100 |
19800 |
1.03 | 103.2 |
The molecular characteristics along with the corresponding melting and crystallization transition temperatures (Tm and Tc) of l-PEG and c-PEG are given in Table 2. The corresponding SEC chromatographs of l-PEG-2k and c-PEG-2k are shown in Fig. 2. The l-PEG-2k polymer showed a peak molecular weight (Mp) of 2400 g mol−1, while c-PEG-2k showed a lower value (1800 g mol−1), leading to a higher retention time than the linear polymer. The ratio of the cyclic and linear products was calculated from the SEC chromatographs of the crude cyclic polymers (Fig. S8 and S9†), given in Table S2.† The SEC chromatographs of l-PEG-2k and the purified c-PEG-2k-b are presented in Fig. S10.† Similarly, c-PEG-2k-b showed a lower value of peak molecular weight (Mp) at 1750 g mol−1, compared to l-PEG-2k. Furthermore, c-PEG-6k also followed a similar trend confirming the successful cyclization (Fig. S11†).
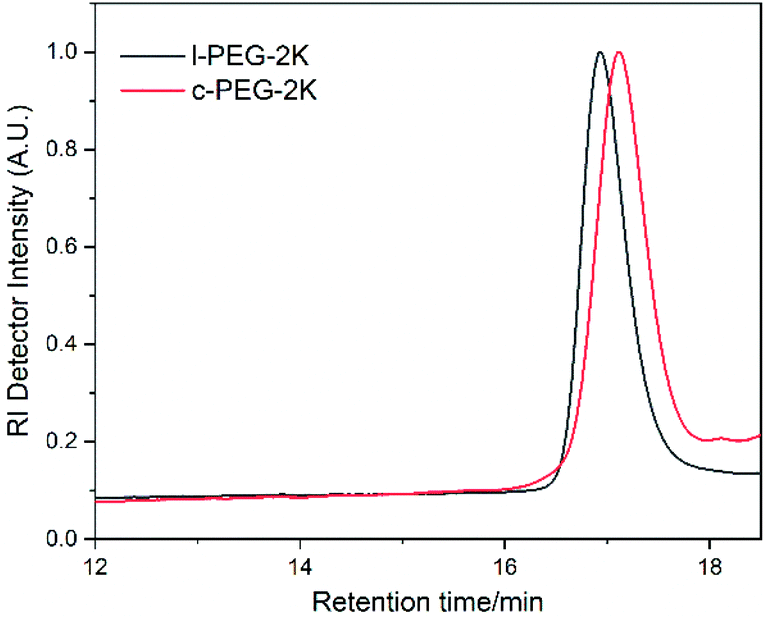 | ||
| Fig. 2 SEC chromatographs of l-PEG-2k and the corresponding purified c-PEG-2k (THF, 35 °C, PS standards). | ||
To further investigate the role of the good solvent/non-solvent system, cyclization reactions were carried out in a good solvent, i.e. THF and in a mixture of good and non-solvents (THF/hexane) for the case of l-PEG-2k using 1,4-bis(bromomethyl) benzene as the linking agent. As is evident from the SEC chromatographs (Fig. S12†), a mixture of good and non-solvents is an effective medium to promote cyclization (only 13% of the condensation product); however when only THF was used, the proportion of high molecular condensation products significantly increased (13% to 49%).
The synthesized (linear and cyclic) polymers were further characterized by one-dimensional (1D) and two-dimensional (2D) NMR spectroscopy, and the complete structural information is reported. In all cases, the successful deprotection reaction for PS and subsequently the cyclization reaction were confirmed by 1H-NMR spectroscopy. Out of all the synthesized polymers, the PS-4k case was selected as an example for detailed NMR studies.
The 1H-NMR spectra of Pr-l-PS-4k and l-PS-4k (after the deprotection) are given in Fig. 3A and B, respectively. The quantitative deprotection of the t-butyldimethyl-silyl groups was accomplished using TBAF solution. The progress of the reaction was monitored by 1H-NMR spectroscopy and specifically by the observation of the characteristic peak of the t-butylsilyl moiety at 0.9 ppm (protons d in Fig. 3A). Treatment of the protected PS homopolymer with TBAF led to the disappearance of the chemical shift at 0.9 ppm (Fig. 3B), indicating the complete removal of the t-butyldimethyl-silyl groups and resulting in the formation of an α,ω-dihydroxy-PS homopolymer. Interestingly, the –OCH2 protons, corresponding to both chain ends, presented two different peaks at 3.51 (proton a in Fig. 3B corresponds to the propyl chain end) and 3.34 ppm (proton b in Fig. 3B corresponds to the ethyl chain end). To further confirm this assumption, a monofunctionalized-OH PS of similar molecular weight was synthesized via anionic polymerization (details of the synthetic procedure in the ESI†) for comparison. The corresponding 1H-NMR spectrum was compared with that of the l-PS-4k polymer (Fig. S13†). The peak position and shape confirmed that the 3.34 ppm peak corresponds to the –OCH2 protons for the chain end group and is located near the phenyl ring of PS. Due to the effect of the phenyl ring of PS, these protons were shielded and showed a peak around 3.34 ppm, while the protons of the –OCH2 group of the α-chain end are located far from the phenyl ring and appeared at 3.51 ppm. However, the integration of the area of both peaks was calculated to be almost equal (Fig. S13†).
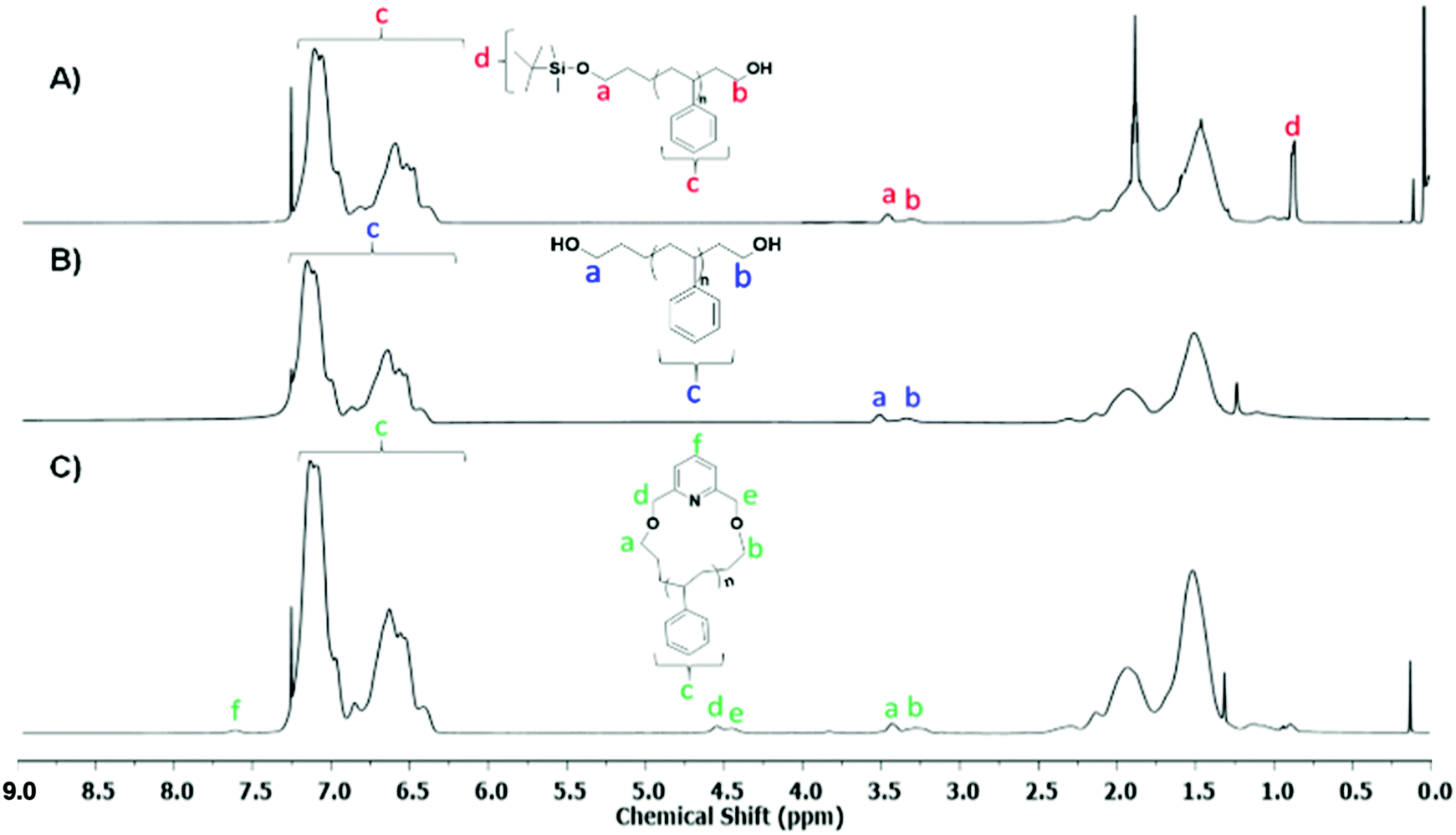 | ||
| Fig. 3 1H-NMR spectra of (A) Pr-PS-4k, (B) l-PS-4k, and (C) the corresponding purified c-PS-4k (CDCl3, 25 °C, 500 MHz). | ||
Additionally, the quantitative 13C-NMR spectrum of the l-PS-4k polymer also indicated two different peaks at 62.86 and 61.33 ppm (Fig. S14†). The peak at 62.86 ppm corresponds to the propyl carbon denoted as b in Fig. S14,† while the peak at 61.33 ppm corresponds to the ethyl carbon (represented as a in Fig. S14†). A 1:1 integration ratio in the quantitative 13C-NMR analysis also verified that these signals belong to the protons of the –OCH2 end groups. The complete structural information of the l-PS-4k polymer was established by 1H–1H correlation spectroscopy (COSY) and total correlation spectroscopy (TOCSY), as presented in Fig. S15† and Fig. 4 respectively.
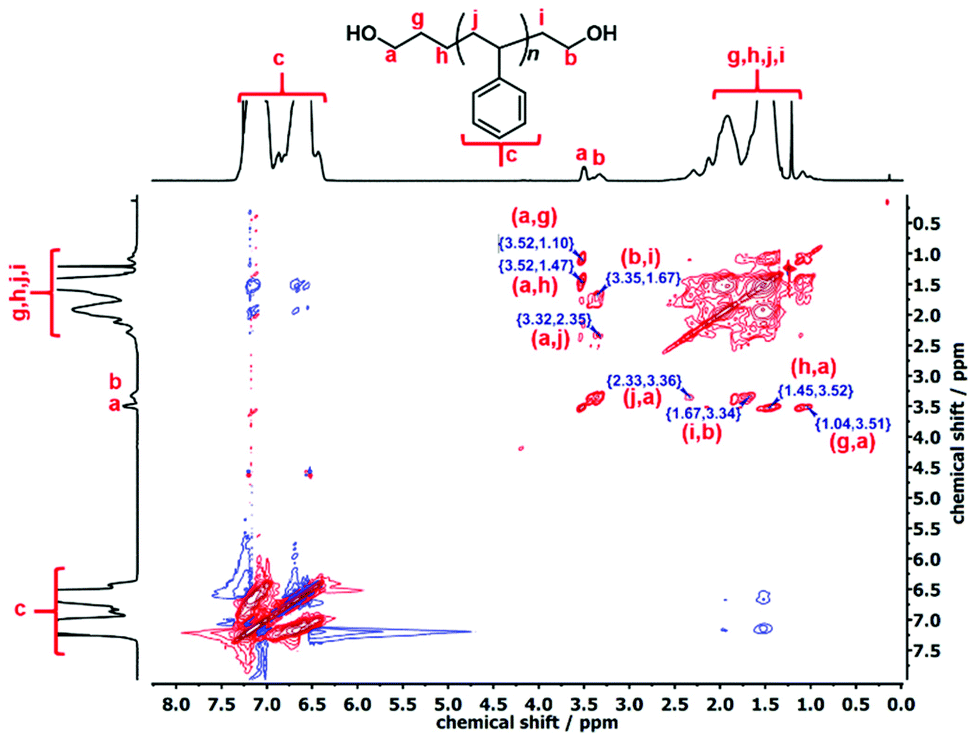 | ||
| Fig. 4 TOCSY NMR spectrum of l-PS-4k (CDCl3, 25 °C, 600 MHz). | ||
The COSY spectrum of the l-PS-4k polymer revealed the correlation between neighboring protons separated by one C–C bond and provided useful information regarding the end-group analysis (Fig. S15†). Protons (b) at 3.33 ppm correlate with protons (i) at 1.67 ppm and protons (a) at 3.51 ppm correlate with protons (g) at 1.38 ppm. Furthermore, long-distance 1H–1H correlation analysis by TOCSY (Fig. 3) revealed that protons (a) correlate with the protons (g), (h), and (j) at 1.10, 1.47, and 2.05 ppm, respectively. These protons [(g) and (h)] can be assigned as the –CH2–CH2– protons of the propyl end-group (α-chain end group) and (j) protons from the main chain. On the other hand, protons (b) correlate with protons (i), indicating that they correspond to the ω-chain end group. Thus it was confirmed that the two distinct peaks are assigned to the end groups: the propyl end group (–OCH2) signal indicated at 3.51 ppm and the ethyl end group (–OCH2) signal at 3.34 ppm.
The cyclization reaction of l-PS-4k was carried out using 2,6-bis(bromomethyl)pyridine as a linking agent in the presence of KOH. The 1H-NMR spectrum of the c-PS-4k polymer revealed three new peaks at 4.46, 4.53, and 7.63 ppm (Fig. 3C). The peaks at 4.46 and 4.53 ppm (d and e) correspond to the –OCH2 groups near to the pyridine ring and the signal at 7.63 ppm (protons f in Fig. 3C) to the proton of the pyridine ring. Along with this, the upfield shift of protons at 3.51 (protons a in Fig. 3B) to 3.42 (protons b in Fig. 3B) and 3.34 (protons a in Fig. 3C) to 3.27 (protons b in Fig. 3C) ppm confirmed the successful cyclization (transformation of the –OH group to the –OCH2 group). Unfortunately, the corresponding carbons could not be detected in the quantitative 13C-NMR spectrum (Fig. S16†) after the cyclization reaction. In the cyclic polymer, the –CH2 group near the pyridine ring (protons d and e, at 4.46 and 4.53 ppm) is surrounded by a tertiary carbon and oxygen as heteroatoms. Therefore, these protons did not show any short- and long-distance correlation as confirmed by the TOCSY spectrum of the c-PS-4k polymer in Fig. 5. Other long-distance correlations from propyl and ethyl end-groups remained intact.
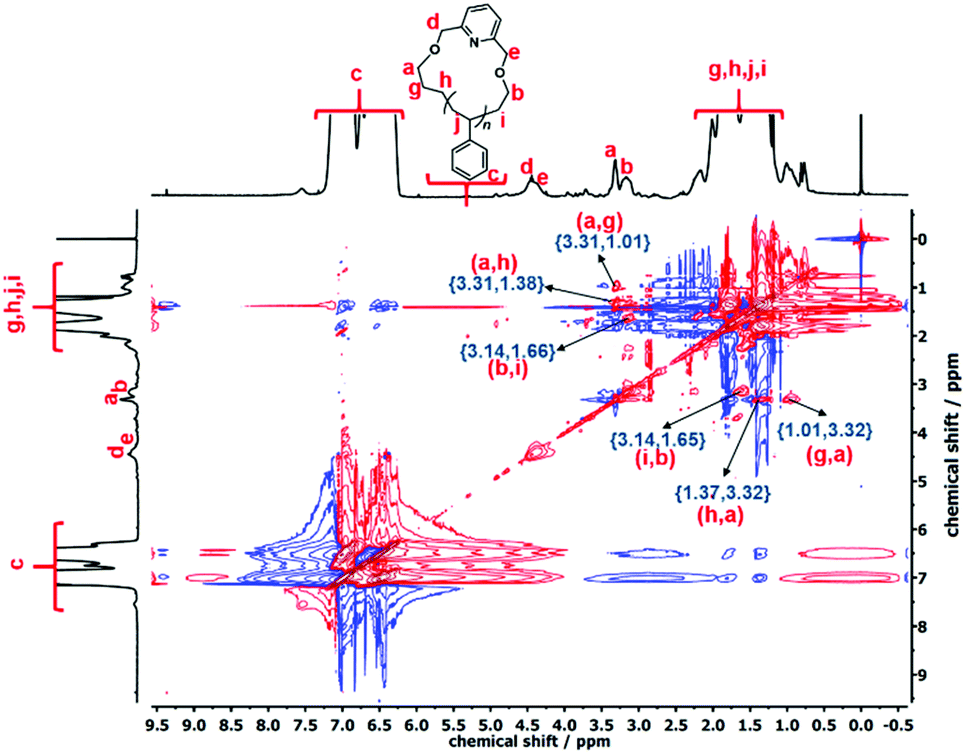 | ||
| Fig. 5 TOCSY NMR spectrum of c-PS-4k (CDCl3, 25 °C, 600 MHz). | ||
Likewise, all the other linear and cyclic PS samples (l-PS-7.5k, l-PS-13k, l-PS-23k, and the corresponding cyclic ones) were also characterized by 1H-NMR spectroscopy, and the cyclization was confirmed in all cases (Fig. S17–S19†). Furthermore, l-PEG and c-PEG samples were also characterized by 1H-NMR spectroscopy (Fig. S20–S22†). The 1H-NMR spectra of l-PEG-2k showed peaks around 3.65 ppm (protons a, Fig. S20†), referring to the ethylene glycol repeating unit (–OCH2CH2–). The protons of the end groups (–OCH2) of l-PEG-2k were merged with the –OCH2CH2– protons of the main chain at 3.65 ppm. The molecular weight of the commercial PEG polymers was confirmed by the end-group analysis technique using trifluoroacetic anhydride (TFA) (Fig. S23 and S24†).40 To distinguish the chain end group, a few drops of TFA were added into the NMR sample tubes of the commercial l-PEG samples. TFA can interact with the hydroxyl end-group (–CH2–OH), resulting in the downfield shift of the adjacent methylene protons from 3.56 ppm to 4.49 ppm. Thus, the obtained molecular weights were in good agreement with the reported ones; i.e., l-PEG-2k showed 2000 g mol−1 and l-PEG-6k showed 6200 g mol−1. The 1H-NMR spectrum of c-PEG-2k was compared with that of l-PEG-2k, and new peaks were observed at 4.67, 7.38, and 7.70 ppm (protons c, e, and d, respectively) in c-PEG-2k (Fig. S20†). Also, in the case where 1,4-bis(bromomethyl)benzene was used as the linking agent for the cyclization, the 1H-NMR spectrum of c-PEG-2k-b (Fig. S21†) showed new peaks at 7.29, 4.53 and 3.93 ppm (protons a, b and c, respectively) compared to the linear precursor.
Similarly, in c-PEG-6k also new peaks were observed, which confirm the successful cyclization of the linear precursor (Fig. S22†).
The synthesized linear and cyclic polymers (l-PS-4k and c-PS-4k) were also analyzed by MALDI-TOF MS. The MALDI-TOF MS spectra of l-PS-4k and c-PS-4k are depicted in Fig. 6. According to the structure of l-PS-4k, the spectrum should show mass distribution: M + Na+ = [104.08 (chain ends) + 104.15n + 23 (Na+)] and the corresponding cyclic: M + Na+ = [207.13 + 104.15n + 23 (Na+)]. The peak distribution of l-PS-4k could be accurately assigned to the linear l-PS-4k ionized with Na+. Similarly, for c-PS-4k, the peak distribution was precisely ascribed to the cyclic structure ionized with Na+. A regular m/z difference of 104.1 was observed between the neighboring peaks in the distribution of both the linear and cyclic polymers, which corresponds to the molar mass of the monomeric styrene unit. The presence of linear and higher molecular weight impurities such as macrocyclic dimers was not observed. Thus, MALDI-TOF MS confirmed the high purity of the final c-PS-4k.
 | ||
| Fig. 6 MALDI-TOF MS of (A) l-PS-4k and (B) c-PS-4k (matrix: dithranol, ionizing agent: NaTFA). | ||
The MALDI-TOF MS spectra for l-PEG-2k and c-PEG-2k are presented in Fig. 7. According to the structure of l-PEG-2k, the spectrum should show mass distribution: M + Na+ = [18.02 (chain ends) + 44.05n + 23 (Na+)] and the corresponding cyclic: M + Na+ = [121.05 + 44.05n + 23 (Na+)]. The peak distribution of l-PEG-2k and c-PEG-2k could be accurately assigned to the corresponding polymer ionized with Na+. The m/z difference of 44.05 was observed between the neighboring peaks in the distribution of both linear and cyclic polymers, which corresponds to the molar mass of the ethylene glycol repeating unit (–OCH2CH2–). The presence of linear and higher molecular weight products like macrocyclic dimers and condensation products was also not observed in this case.
 | ||
| Fig. 7 MALDI TOF MS of (A) l-PEG-2k and (B) c-PEG-2k (matrix: dithranol, ionizing agent: NaTFA). | ||
DSC analysis of the linear and cyclic polymers
DSC is an essential tool to distinguish the differences between cyclic and linear polymers by comparing their glass transition temperatures (Tg). Generally, Tg of a linear polymer increases with molecular weight up to an asymptotic value corresponding to a chain of infinite molecular weight.1 Although cyclic polymers have shown a similar trend with molecular weight, the Tg values observed for any given molecular weight are higher than those of linear analogs. The reason for the higher Tg in cyclic polymers is the absence of free chain ends that affect the mobility and the inter- and intra-chain interactions of a cyclic polymer.1 DSC traces were recorded for all the linear and cyclic PS polymers, and their Tg values are listed in Table 1. l-PS-4k showed a Tg around 79.5 °C, while the cyclic counterpart (c-PS-4k) showed a higher Tg value, at 86.9 °C (Fig. 8). All PS showed a similar trend, where cyclic polymers presented higher Tg values than the linear ones (Fig. S25–S27†) and an increase in Tg with the increase of molecular weight. | ||
| Fig. 8 DSC traces of l-PS-4K and purified c-PS-4k. | ||
In the DSC traces of the l-PEG and c-PEG samples, melting and crystallization transitions were observed, as presented in Fig. 9 and S28.† The thermal behavior of the cyclic and linear crystalline polymers is still debatable.41,42 In some cases, cyclic polymers show lower melting point (Tm) and crystallization temperature (Tc) values compared to linear analogs.43 The DSC trace of l-PEG-2k revealed transitions at 42.3 °C and 16.3 °C (attributed to Tm and Tc, respectively), while the DSC trace of c-PEG-2k showed transitions at 38.5 °C and 9.6 °C (Fig. 9), respectively. The c-PEG-6k polymer also followed similar behavior regarding Tm and Tc (Table 2 and Fig. S28†).
 | ||
| Fig. 9 DSC traces of l-PEG-2k (heating and cooling) and c-PEG-2k (heating and cooling). | ||
Conclusions
An alternative method for the cyclization of linear dihydroxy-PS and -PEG has been developed. The PS polymers were synthesized by the living anionic polymerization technique and show low PDI values (<1.1). The cyclization was carried out through a Williamson etherification reaction among the dihydroxy functionalized polymers and two different linking agents [1,4-bis(bromomethyl)benzene and 2,6-bis(bromomethyl)pyridine] in moderate dilution. This procedure led to the formation of cyclic polymers in a relatively high yield and up to 1 gram scale. All the synthesized polymers were fully characterized by various analytical techniques such as 1D and 2D NMR, SEC, MALDI-TOF MS, and DSC. Comparison of the 1D and 2D NMR spectra of the linear and cyclic polymers further confirmed that cyclization was successful. Furthermore, the linking agent 2,6-bis(bromomethyl)pyridine can form an active metal complex which further can lead to the formation of complex macromolecular architectures such as rotaxanes44 and catenanes.45Author contributions
Nikos Hadjichristidis designed and supervised the project. Sandeep Sharma and Konstantinos Ntetsikas designed (with N. H.) the project and performed synthesis, characterization, and data analysis. Saibal Bhaumik assisted in the synthesis and Viko Ladelta performed the 2D NMR measurements. All authors assisted in the preparation of the manuscript/ESI and approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this publication was supported by King Abdullah University of Science and Technology (KAUST).References
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS PubMed.
- Z. Jia and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2085–2097 CrossRef CAS.
- T. Josse, J. De Winter, P. Gerbaux and O. Coulembier, Angew. Chem., Int. Ed., 2016, 55, 13944–13958 CrossRef CAS PubMed.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202–2213 RSC.
- J. P. Edwards, W. J. Wolf and R. H. Grubbs, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 228–242 CrossRef CAS.
- Y. A. Chang and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2892–2902 CrossRef CAS.
- N. Kusuyama, Y. Daito, H. Kubota, Y. Kametani and M. Ouchi, Polym. Chem., 2021, 12, 2532–2541 RSC.
- Z. Jia and M. J. Monteiro, in Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize II, ed. V. Percec, Springer International Publishing, Cham, 2013, vol. 238, pp. 295–327 Search PubMed.
- M. Schappacher and A. Deffieux, J. Am. Chem. Soc., 2011, 133, 1630–1633 CrossRef CAS PubMed.
- Q. He, A. M. Yol, S.-F. Wang, H. Ma, K. Guo, F. Zhang, C. Wesdemiotis, R. P. Quirk and M. D. Foster, Polym. Chem., 2016, 7, 5840–5848 RSC.
- M. Glassner, J. P. Blinco and C. Barner-Kowollik, Macromol. Rapid Commun., 2011, 32, 724–728 CrossRef CAS PubMed.
- T. Yamamoto, S. Yagyu and Y. Tezuka, J. Am. Chem. Soc., 2016, 138, 3904–3911 CrossRef CAS PubMed.
- M. Schappacher and A. Deffieux, Macromolecules, 2001, 34, 5827–5832 CrossRef CAS.
- G. Hild, A. Kohler and P. Rempp, Eur. Polym. J., 1980, 16, 525–527 CrossRef CAS.
- D. Geiser and H. Höcker, Macromolecules, 1980, 13, 653–656 CrossRef CAS.
- J. Roovers and P. M. Toporowski, Macromolecules, 1983, 16, 843–849 CrossRef CAS.
- J.-M. Boutillier, B. Lepoittevin, J.-C. Favier, M. Masure, P. Hémery and P. Sigwalt, Eur. Polym. J., 2002, 38, 243–250 CrossRef CAS.
- R. Yin and T. E. Hogen-Esch, Macromolecules, 1993, 26, 6952–6957 CrossRef CAS.
- H. Iatrou, N. Hadjichristidis, G. Meier, H. Frielinghaus and M. Monkenbusch, Macromolecules, 2002, 35, 5426–5437 CrossRef CAS.
- A. E. Madani, J.-C. Favier, P. Hémery and P. Sigwalt, Polym. Int., 1992, 27, 353–357 CrossRef.
- G. Polymeropoulos, G. Zapsas, K. Ntetsikas, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2017, 50, 1253–1290 CrossRef CAS.
- P. Sun, J. Chen, J. A. Liu and K. Zhang, Macromolecules, 2017, 50, 1463–1472 CrossRef CAS.
- Y. Tezuka, Acc. Chem. Res., 2017, 50, 2661–2672 CrossRef CAS PubMed.
- P. Sun, J. A. Liu, Z. Zhang and K. Zhang, Polym. Chem., 2016, 7, 2239–2244 RSC.
- L. Zhang, Y. Wu, S. Li, Y. Zhang and K. Zhang, Macromolecules, 2020, 53, 8621–8630 CrossRef CAS.
- Z.-G. Yan, Z. Yang, C. Price and C. Booth, Makromol. Chem., Rapid Commun., 1993, 14, 725–732 CrossRef CAS.
- C. H. Hövelmann, S. Gooßen and J. Allgaier, Macromolecules, 2017, 50, 4169–4179 CrossRef.
- C. W. P. Maya, M. Arce, M. M. Thursby, J. P. Wu, E. M. Carnicom and E. S. Tillman, Macromolecules, 2016, 49, 7804–7813 CrossRef.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS PubMed.
- J. Zhao, Y. Zhou, Y. Zhou, N. Zhou, X. Pan, Z. Zhang and X. Zhu, Polym. Chem., 2016, 7, 1782–1791 RSC.
- N. Hadjichristidis, H. Iatrou, S. Pispas and M. Pitsikalis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3211–3234 CrossRef CAS.
- D. Uhrig and J. W. Mays, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6179–6222 CrossRef CAS.
- S. Bhaumik, K. Ntetsikas and N. Hadjichristidis, Macromolecules, 2020, 53, 6682–6689 CrossRef CAS PubMed.
- A. Touris, S. Lee, M. A. Hillmyer and F. S. Bates, ACS Macro Lett., 2012, 1, 768–771 CrossRef CAS.
- R. P. Quirk, S. H. Jang, H. Yang and Y. Lee, Macromol. Symp., 1998, 132, 281–291 CrossRef CAS.
- A. J. Meuler, M. K. Mahanthappa, M. A. Hillmyer and F. S. Bates, Macromolecules, 2007, 40, 760–762 CrossRef CAS.
- R. P. Quirk and J.-J. Ma, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 2031–2037 CrossRef CAS.
- C. Tonhauser and H. Frey, Macromol. Rapid Commun., 2010, 31, 1938–1947 CrossRef CAS PubMed.
- Y. Shi, S.-P. R. Chen, Z. Jia and M. J. Monteiro, Polym. Chem., 2020, 11, 7354–7361 RSC.
- V. Ladelta, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2017, 8, 511–515 RSC.
- L. Haiying, J. Robert and L. Philippe, Polymer, 2006, 47, 8406–8413 CrossRef.
- Y. Ga-Er, S. Tao, Y. Ze-Gui, P. Colin, B. Colin, C. Jennifer, J. R. Anthony and V. Kyriakos, Polymer, 1997, 38, 35–42 CrossRef.
- R. Watanabe, M. Noba, T. Uno, T. Itoh and M. Kubo, J. Polym. Sci., 2020, 58, 1982–1988 CrossRef CAS.
- V. Aucagne, J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS PubMed.
- S. M. Goldup, D. A. Leigh, T. Long, P. R. McGonigal, M. D. Symes and J. Wu, J. Am. Chem. Soc., 2009, 131, 15924–15929 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01337h |
| This journal is © The Royal Society of Chemistry 2021 |