
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a cobalt(III)-based ponatinib prodrug system†
Marlene
Mathuber
a,
Michael
Gutmann
 b,
Mery
La Franca
bc,
Petra
Vician
b,
Anna
Laemmerer
bd,
Patrick
Moser
b,
Bernhard K.
Keppler
ad,
Walter
Berger
*bd and
Christian R.
Kowol
*ad
b,
Mery
La Franca
bc,
Petra
Vician
b,
Anna
Laemmerer
bd,
Patrick
Moser
b,
Bernhard K.
Keppler
ad,
Walter
Berger
*bd and
Christian R.
Kowol
*ad
aInstitute of Inorganic Chemistry, Faculty of Chemistry, University of Vienna, Waehringer Straße 42, 1090 Vienna, Austria
bInstitute of Cancer Research and Comprehensive Cancer Center, Medical University of Vienna, Borschkegasse 8A, 1090 Vienna, Austria
cDepartment of Biological, Chemical and Pharmaceutical Sciences and Technologies, University of Palermo, via Archirafi 32, 90123 Palermo, Italy
dResearch Cluster “Translational Cancer Therapy Research”, University of Vienna and Medical University of Vienna, 1090 Vienna, Austria. E-mail: christian.kowol@univie.ac.at; walter.berger@meduniwien.ac.at
First published on 30th March 2021
Abstract
Receptor tyrosine kinase inhibitors have become a central part of modern targeted cancer therapy. However, their curative potential is distinctly limited by both rapid resistance development and severe adverse effects. Consequently, tumor-specific drug activation based on prodrug designs, exploiting tumor-specific properties such as hypoxic oxygen conditions, is a feasible strategy to widen the therapeutic window. After proof-of-principal molecular docking studies, we have synthesized two cobalt(III) complexes using a derivative of the clinically approved Abelson (ABL) kinase and fibroblast growth factor receptor (FGFR) inhibitor ponatinib. Acetylacetone (acac) or methylacetylacetone (Meacac) have been used as ancillary ligands to modulate the reduction potential. The ponatinib derivative, characterized by an ethylenediamine moiety instead of the piperazine ring, exhibited comparable cell-free target kinase inhibition potency. Hypoxia-dependent release of the ligand from the cobalt(III) complexes was proven by changed fluorescence properties, enhanced downstream signaling inhibition and increased in vitro anticancer activity in BCR-ABL- and FGFR-driven cancer models. Respective tumor-inhibiting in vivo effects in the BCR-ABL-driven K-562 leukemia model were restricted to the cobalt(III) complex with the higher reduction potential and confirmed in a FGFR-driven urothelial carcinoma xenograft model. Summarizing, we here present for the first time hypoxia-activatable prodrugs of the clinically approved tyrosine kinase inhibitor ponatinib and a correlation of the in vivo activity with their reduction potential.
Introduction
Tyrosine kinases play an essential part in the signal transduction pathways of cells, by catalyzing the transfer of γ-phosphate groups of adenosine triphosphate (ATP).1 Thus, they fulfil a crucial role in the control of e.g. cell growth, proliferation and differentiation. Dysregulations of these signaling processes have been linked to several diseases, including cancer initiation and progression.2 The development of tyrosine kinase inhibitors (TKIs) is a promising targeted therapeutic approach and resulted in the approval of respective antibodies and small molecule inhibitors.3,4 The latter competitively block the ATP-binding pockets in the active sites of tyrosine kinases.3 One representative of this substance class is ponatinib, a multi-kinase inhibitor, which targets among others the Abelson kinase (ABL), fibroblast growth factor receptor (FGFR), and platelet-derived growth factor receptor (PDGFR) families.5–7 Ponatinib is clinically approved as second line treatment for Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML)/acute lymphoblastic leukemia (Ph+ ALL), especially in case of a BCR-ABLT315I mutation.8–10 Its anti-tumorigenic potential is also evaluated in several other cancer types,11 with the inhibition of FGFR being one major point of interest.12,13 However, despite the specific targeting of oncogene-dependent cancer cells, adverse effects, caused by the lack of distinction between healthy and cancerous tissue, could be frequently noticed.14 In a clinical phase III study severe arterial thrombotic events occured.15,16 As a result, the maximum applicable dose of ponatinib for clinical routine was reduced and further safety measures were included.15,17To optimize the therapeutic potential of ponatinib, a promising way could be the use of prodrug systems. In general, anticancer prodrugs consist of non-toxic, inactive compounds, which only release their active counterpart through specific activation in the tumor.18 Cancerous tissue exhibits certain features, such as a hypoxic environment, which can be exploited for this kind of approach.19,20 Hypoxia-activated prodrugs (HAPs) have been intensively explored over the past years for various substance classes with a focus on DNA alkylating agents.21–23 Although some of these compounds (e.g. tirapazamine, evofosfamide, apaziquone) have entered advanced clinical studies, none of them received approval so far.24,25 Surprisingly, until now this prodrug approach has been implemented only on a few TKI compounds,26–28 with TH-4000 as one of the first examples.29 Various chemical possibilities to exploit hypoxia are known with nitroimidazoles27–29 and cobalt(III) metal complexes as important representatives.26,30,31 The mechanism of the latter is based on distinct differences in ligand lability of the cobalt(III) (kinetically inert) and cobalt(II) (kinetically labile) oxidation state.32,33 Due to its bulkiness, the intact cobalt(III) complex should not be able to bind to the ATP binding pocket of the respective kinase (Fig. 1). In healthy tissue (normoxic conditions) the complex is stable, thus the release of the bioactive ligand is prevented.34
 | ||
| Fig. 1 Proposed mode of action of hypoxia-activated cobalt(III) prodrugs. Normoxic, healthy tissue (left side): The prodrug is inactive. Due to its bulkiness, the cobalt(III) complex does not fit into the ATP-binding pocket of the respective tyrosine kinase. Hypoxic cancer tissue (right side): The drug is active. In the hypoxic tissue of the tumor, the cobalt(III) complex is irreversibly reduced and the TKI ligand subsequently released able to inhibit the signaling processes of the target tyrosine kinases. | ||
In contrast, in the hypoxic environment of a tumor, the inactive prodrug will be irreversibly reduced. This results in the dissociation of the complex, with formation of [Co(II)(H2O)6]2+ and release of the bioactive ligand with subsequent binding to the kinase pocket (Fig. 1).
In such a prodrug approach, a well-balanced equilibrium is important: on the one hand the cobalt(III) complex needs to have a high stability in healthy tissue, on the other hand the bioactive ligand should be efficiently released after reduction under hypoxic conditions. Seminal work of Ware et al. demonstrated that monodentate aziridine ligands could not sufficiently stabilized and therefore bidentate chelating ligands are preferred.33
In a previous study of our group, we established the concept of cobalt(III) prodrugs for EGFR-inhibitors, based on Erlotinib derivatives (LEGFR; Fig. 2).26 We observed that, due to the direct connection of the ethylenediamine unit to the quinazoline ring system, the reduction potential of the respective cobalt(III) complex distinctly increased resulting in reduced stability in blood serum. In this study we focused on ponatinib, which is not only a highly active, clinically approved anticancer drug, but possesses also a –CH2-spacer between the solubilizing piperazine moiety and the aromatic ring system (LPon; Fig. 2). As ancillary ligands of the cobalt(III) complexes, acetylacetone (acac) or methylacetylaceton (Meacac) were used. The novel compounds were evaluated for their fluorescence properties, electrochemical potential, kinetic reduction behavior and serum stability. Additionally, their fluorescence properties and the biological activity were investigated under normoxic vs. hypoxic conditions in an FGFR- or ABL1-driven cancer cell background. Finally, the in vivo antitumor activity of the novel complexes was evaluated in the respective xenograft mouse models.
 | ||
| Fig. 2 (A) Chemical structure of the previously synthesized EGFR inhibitor ligand (LEGFR) with a direct attachment of the ethylenediamine moiety to the aromatic ring system.26 (B) The clinically approved ponatinib (Iclusig®). (C) Newly developed ponatinib derivative (LPon), where the metal-chelating ethylenediamine moiety is separated from the aromatic ring system. | ||
Results and discussion
Docking studies
To get a first indication if our proposed strategy is indeed promising, we performed docking studies of the novel ligand LPon and the two cobalt(III) complexes Co(acac)2LPon and Co(Meacac)2LPon (Scheme 1) compared to the approved parental compound ponatinib. In general, the anticancer effect of TKIs is based on their strong intermolecular, non-covalent interactions with target-specific residues mainly consisting of hydrogen bonds, van der Waals, and Coulomb interactions.35 Consequently, molecular docking is a well-established tool in the field of TKI development.36 As kinases, two of the main targets of ponatinib were chosen, namely FGFR1 (PDB ID: 4V04) and ABL1 (PDB ID: 4WA9). The results indicated that the replacement of the piperazine moiety of ponatinib by an ethylenediamine group (LPon) did not change the binding mode in the pockets of FGFR1 (Fig. 3A and B) as well as ABL1 (Fig. 3C and D). In addition, the data of the complexes Co(acac)2LPon and Co(Meacac)2LPon convincingly showed that LPon attached to the cobalt(III) core is unable to interact with the active sites of FGFR1 and ABL1. The steric effects of the complexation prohibited all crucial interactions with FGFR1 and ABL1 (Fig. 3 and S1†). Summarizing, the molecular docking studies showed that LPon is comparable to ponatinib in its kinase binding ability and that the cobalt(III) complexes can be considered as promising prodrugs.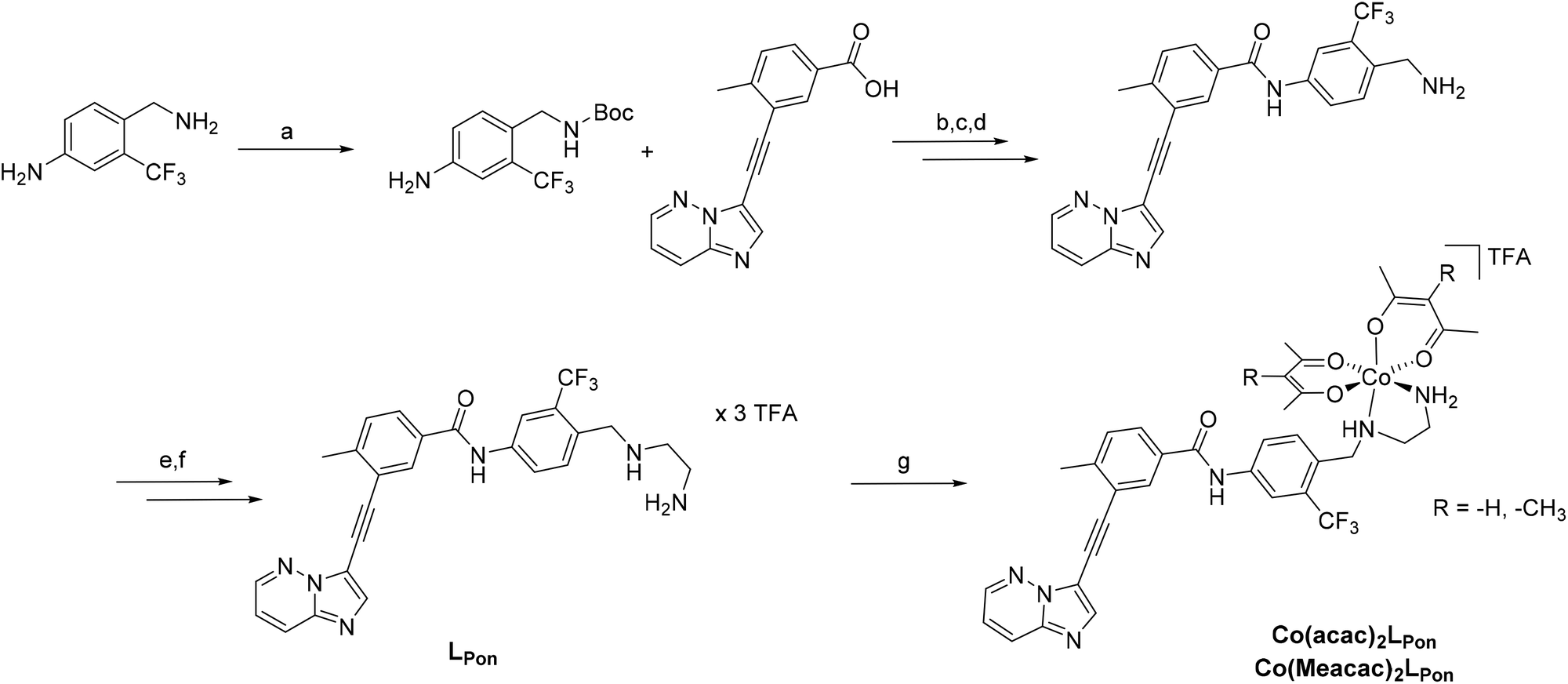 | ||
Scheme 1 Synthesis of LPon and its cobalt(III) complexes Co(acac)2LPon and Co(Meacac)2LPon. Reagents and conditions: (a) Di-tert-butyl dicarbonat, THF, 81%; (b) (COCl)2, abs. Toluol, NMM, DMAP, 68%; (c) 4 N HCl in dioxan/H2O, 95%; (d) NaHCO3 in EtOAc, 94%; (e) N-Boc-2-aminoacetaldehyde, sodium cyanoborhydride, abs. THF, molecular sieves (3–4 Å), 50%; (f) TFA in dichloromethane (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 40%; (g) in situ deprotonation with NaOH, Na[Co(acac)2(NO2)2] or Na[Co(Meacac)2(NO2)2], activated charcoal in MeOH, 36% for Co(acac)2LPon and 31% for Co(Meacac)2LPon. :1), 40%; (g) in situ deprotonation with NaOH, Na[Co(acac)2(NO2)2] or Na[Co(Meacac)2(NO2)2], activated charcoal in MeOH, 36% for Co(acac)2LPon and 31% for Co(Meacac)2LPon. | ||
 | ||
| Fig. 3 Visualizations of the best docking poses of LPon and Co(acac)2LPon in comparison to ponatinib with FGFR1 (A and B) (PDB ID: 4V04) as well as ABL1 (C and D) (PDB ID: 4WA9). Ponatinib is shown in red, LPon in blue and Co(acac)2LPon in pink. | ||
Synthesis and characterization
To obtain a suitable metal-binding moiety for the complexation with a cobalt precursor, the piperazine was exchanged with an ethylenediamine unit (Scheme 1). To this end, the aliphatic amino group of the commercially available 4-(aminomethyl)-3-(trifluoromethyl)aniline was protected using di-tert-butyl dicarbonate. For the subsequent coupling reaction between the free aromatic amino group and purchased 3-(2-{imidazo[1,2-b]pyridazin-3-yl}ethynyl)-4-methylbenzoic acid, the carboxylic acid was first converted into the corresponding acyl chloride using oxalylchloride (COCl)2 in toluene.37 Subsequently, the amino group was deprotected using HCl, followed by neutralization of the formed HCl salt. Reductive amination with N-boc-(metylamino)-acetaldehyde and sodium cyanoborhydride yielded in the introduction of the protected ethylenediamine moiety. Finally, a second deprotection step with trifluoroacetic acid (TFA) and purification by reversed-phase HPLC generated the ethylenediamine-bearing ponatinib derivative LPon as three-fold TFA salt.For the synthesis of the cobalt complexes, acetylacetone (acac) or methylacetylacetone (Meacac) were used as ancillary ligands, since these ligands can also distinctly influence the reduction potential of the complex.33,38 The cobalt(III) complexes were synthesized by reaction of Na[Co(acac)2(NO2)2] or Na[Co(Meacac)2(NO2)2] with LPon (after the in situ deprotonation of the ligand with NaOH) in methanol and the presence of activated charcoal following a slightly modified procedure of Denny et al.39 Afterwards, the crude products were directly purified by reversed-phase HPLC (with the addition of 0.1% TFA to the eluents) leading to the respective TFA salts of Co(acac)2LPon and Co(Meacac)2LPon. Due to the two stereogenic centers (one from the propeller chirality of the complex itself and the other from the aliphatic NH-group of the ethylenediamine bridge), two isomers of the cobalt(III) complexes are present. For Co(acac)2LPon we could separate the two isomers by adjusting the gradient of the preparative HPLC purification (Fig. S2†). All novel compounds were characterized by mass spectrometry as well as 1H and 13C one- and two-dimensional NMR spectroscopy and their purity was confirmed by elemental analysis. The amount of TFA was confirmed by 19F-NMR measurements via the ratio of the fluorine signals between LPon and TFA.
The 1H NMR spectra of the isomeric mixtures of the cobalt(III) complexes [∼60%:40% for Co(acac)2LPon and ∼70%:30% for Co(Meacac)2LPon] show only one signal set in the aromatic area (with the exception of H23; see Experimental part), but two in the aliphatic area and of the –NH groups (Fig. S3 and S4†). This is in contrast to our previously synthesized cobalt(III) complex with LEGFR, where all signals could be observed twice.26 An obvious explanation is that the conjugated system in the LPon ligand is much less extended compare to LEGFR (Fig. 2). A clear indication, that we were indeed able to lower the impact of the ring system on the chelating ethylenediamine moiety and vice versa.
Following the successful isolation of the two isomers of Co(acac)2LPon, we studied their interconversion behavior in phosphate buffered saline (PBS) (pH 7.4, 10 mM) incubated at 37 °C. An interconversion is possible due to the chiral N of the ethylenediamine moiety, which can detach, invert and bind again, resulting in the other diastereomer. The results showed that the conversion from isomer 1 into isomer 2 proceeded with ∼30% after 72 h (Fig. 4A). In contrast, the reverse reaction from pure isomer 2 into isomer 1 yielded in only ∼20% conversion after 72 h (Fig. 4B). A higher kinetic stability of isomer 2 is also supported by data after 9 days, which reveal that initial pure isomer 1 is converted to a racemic mixture with isomer 2 (50%:50%). In case of pure isomer 2 still a ratio of ∼60:40 is present after 9 days. At the same time, these experiments revealed that the complexes are highly stable in aqueous solution at pH 7.4 without any release of the ponatinib-like ligand. Because of the slow conversion rates the pure isomers of Co(acac)2LPon were also biologically investigated (vide infra).
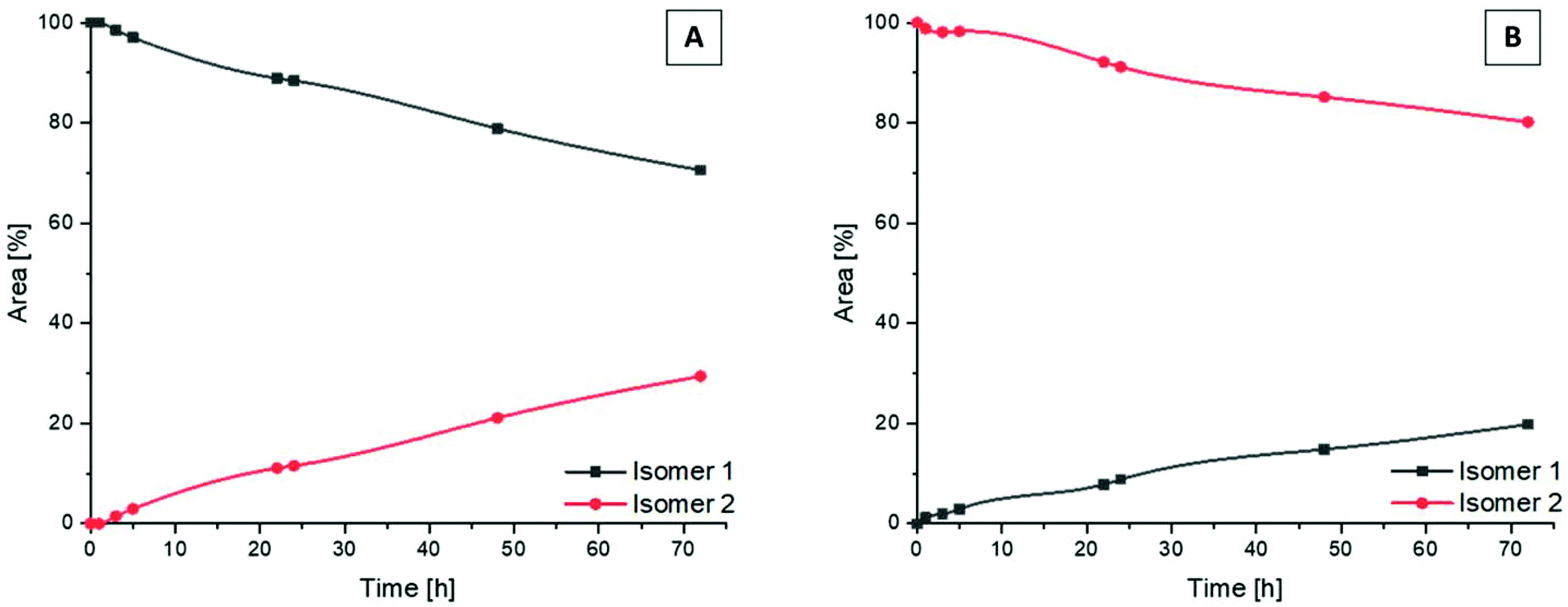 | ||
| Fig. 4 Kinetic behavior of the isomers of Co(acac)2LPon incubated in PBS at 37 °C (pH 7.4, 10 mM) and monitored by HPLC. Depicted is the conversion from pure isomer 1 (A) and pure isomer 2 (B) into the respective mixtures over a period of 72 h. | ||
Fluorescence properties
We could recently show that ponatinib possesses distinct fluorescence properties with an emission maximum at 470 nm when irradiated at 320 nm (measured in PBS pH = 7.40, concentration = 15 μM).40 Therefore, we also analyzed the fluorescence of the novel ligand LPon and its cobalt(III) complexes under the same conditions. The 3D spectrum of LPon showed a maximum and intensity similar to ponatinib (λem = 470 nm at λex = 320 nm) (Fig. 5A). In contrast, for the cobalt(III) complexes a strongly quenched fluorescence in PBS was observed (Fig. 5B). This phenomenon can be explained by the metal center, which influences the conversion of singlet-excited states to triplet-excited states with extremely fast intersystem crossing rates (typical lifetimes are in the fs scale). Hence, ligand-based fluorescence is often difficult to observe in a metal coordinated form.41 However, these distinct differences in the fluorescence properties between free ligand and cobalt(III) complex can be exploited for stability studies in cell culture (vide infra). | ||
| Fig. 5 Fluorescence properties of investigated ponatinib derivatives. (A) 3D full excitation–emission landscape of LPon (Rayleigh scattering of 1st and 2nd order appear as diagonal ridges). (B) Fluorescence emission spectra at λex = 320 nm of LPon, Co(acac)2LPon, and Co(Meacac)2LPon. Measurements were performed in PBS at pH = 7.40 [conc. ligand/complex = 15 μM; T = 25.0 °C]. | ||
Cyclic voltammetry
Since the redox potential is a crucial characteristic for the activation of cobalt(III)-based prodrugs systems, we investigated the novel complexes by cyclic voltammetry in comparison to Co(acac)2LEGFR. Measurements were performed in dimethylformamide (DMF) (+0.2 M [n-Bu4N][BF4]) at a scan speed of 100 mV s−1. The voltammograms revealed a single irreversible cathodic peak, which can be assigned to the reduction of cobalt(III) to cobalt(II) (Fig. 6A). Even at a higher scan speed of 1000 mV s−1, the redox processes were still completely irreversible for all complexes (Fig. S5†). | ||
| Fig. 6 (A) Cyclic voltammograms of Co(acac)2LPon, Co(Meacac)2LPon and Co(acac)2LEGFR in DMF (1.5 mM complex, I = 0.2 M [n-Bu4N][BF4], scan rate of 100 mV s−1, 25.0 °C). (B) Stability measurements of Co(acac)2LPon, Co(Meacac)2LPon and Co(acac)2LEGFR incubated in pure FCS at 37 °C (pH 7.4, 150 mM phosphate buffer) analyzed by HPLC-MS over a time period of 26 h. The y-axis shows the relative ratio of the integrated peak areas of the intact complex over time (in percent) compared to the area at the starting point (0 h). | ||
The data clearly showed that the “separation” of the chelating ethylenediamine moiety from the ring system in case of the ponatinib complex Co(acac)2LPon resulted in the desired strongly decreased cathodic peak potential at −315 mV vs. NHE, compared to the directly attached EGFR-inhibitor reference Co(acac)2LEGFR at −57 mV vs. NHE. Methylation of the ancillary acetylacetone ligands in Co(Meacac)2LPon further shifted the potential to −442 mV vs. NHE. This in line with literature data of other cobalt(III) prodrugs systems.33,38 Investigation of the same complexes in a DMF/H2O (7:3 v/v) mixture, resulted in similar shifts (Fig. S6†). In all measurements completely irreversible reduction processes were observed, regardless of the solvent or scan speed (100–1000 mV s−1). This is of interest, since the proposed mechanism for cobalt(III) prodrugs is often associated with a reversible redox behaviour of these complexes. The frequently proposed model suggests that under normoxic conditions in healthy tissue the reduced cobalt(II) complex will immediately be re-oxidized by oxygen to the inert cobalt(III) complex and thus the release of the bioactive ligand is prevented.34,42,43 Interestingly, for most cobalt(III) prodrugs reported in literature irreversible redox processes in aqueous solutions could be observed, which contradicts this hypothesis.44–47
Notably, pulse radiolysis studies of cobalt(III) nitrogen mustard complexes indeed suggested that the re-oxidation rate under normoxia is too slow and the cobalt(III) complexes rather compete with molecular oxygen for one-electron reductants.48 Nevertheless, different cobalt(III) complexes with irreversible electrochemical behavior exhibited strong hypoxia-dependent activity against cancer cells in vitro and in vivo, proving the potential of such complexes.26,45
Serum stability measurements
Recently, we could show that even the EGFR-inhibitor cobalt(III) complex Co(acac)2LEGFR (with the highest redox potential) is completely stable in the presence of the natural low-molecular weight reducing agents ascorbic acid, glutathione or reduced nicotinamide adenine dinucleotide (NADH).38 Therefore, we only investigated the stability of the novel complexes in fetal calf serum (FCS). As reference again Co(acac)2LEGFR26 was included. The complexes were dissolved in 50 mM phosphate puffer and mixed 1:10 with fetal calf serum (FCS; buffered with 150 mM phosphate buffer to keep a stable pH of 7.4) to a final concentration of 50 μM. The samples were incubated at 37 °C and after 0, 2, 4, 6, 24 and 26 h extracted with acetonitrile and measured by HPLC-MS. Both ponatinib-derived complexes Co(acac)2LPon and Co(Meacac)2LPon were highly stable in FCS with ∼90% intact complex after 26 h (Fig. 6B). In contrast, the reference Co(acac)2LEGFR showed less than 50% remaining complex after the same incubation time, in line with our previous results.38 This indeed confirms, that by avoiding the direct attachment of the ethylenediamine moiety to an aromatic ring system, complexes with much higher stability can be generated. Since no distinct difference in stability between Co(acac)2LPon and Co(Meacac)2LPon within 26 h could be observed, measurements were repeated up to 72 h. Now the slightly increased stability of Co(Meacac)2LPon at ∼80% of intact compound compared to Co(acac)2LPon at ∼70% could be uncovered (Fig. S7†).
Biological investigations
Hypoxia-inducible intracellular ligand release
Prior to the evaluation of the anticancer activity of our novel derivatives in cell culture models, we examined the stability of Co(acac)2LPon and Co(Meacac)2LPon in the presence of cells under normoxic conditions. The stability was determined by the appearance of LPon-associated fluorescence (vide supra) in BCR-ABL-driven K-562 leukemia cells via flow cytometry. As shown in Fig. 7, both ponatinib and LPon were taken up rapidly and efficiently by K-562 cells with no significant increase in cell-associated fluorescence from 1 h exposure onwards. In contrast, exposure of K-562 cells to equimolar concentrations of both cobalt(III) complexes resulted in distinctly reduced cell-associated fluorescence levels, proving the pronounced stability of the complexes even in the presence of cells. Despite significant LPon-release from the two complexes over time (up to 24 h), in case of Co(Meacac)2LPon cell-associated fluorescence remained more than 7.5-fold lower after 24 h as compared to the free ligand. Moreover, the fluorescence intensities of Co(Meacac)2LPon were significantly reduced as compared to Co(acac)2LPon, well in agreement with the higher reduction potential of the latter complex. | ||
| Fig. 7 Fluorescence-based evaluation of LPon release from Co(acac)2LPon and Co(Meacac)2LPon in K-562 cells under normoxic cell culture conditions by flow cytometry. Cells were incubated with 10 μM of the respective compounds for the indicated exposure times and mean fluorescence intensities were determined by flow cytometry (BD LSRFortessa X-20, pacific blue filter). Fluorescence intensities were normalized by subtracting the auto fluorescence of untreated cells. Data are given in arbitrary units (a.u.) as means ± SEM of five independent experiments. Statistical significance was calculated by two-way ANOVA with p < 0.05(*); <0.01 (**); <0.001 (***). | ||
Next, the impact of hypoxia on the stability of our novel derivatives was evaluated by monitoring intracellular fluorescence intensities under normoxic (21% O2) vs. hypoxic (0.1% O2) conditions. In general, only in case of the cobalt(III) complexes, but not the free ligand LPon and ponatinib, a significant impact of oxygen conditions on cell-associated fluorescence was observed (Fig. 8). Hypoxic fluorescence activation in case of both cobalt(III) complexes was time-dependent, reaching significance after 6 h of compound incubation and resulting in 2- to 4-fold higher intensities. The activation ability tended to be higher in the more stable Co(Meacac)2LPon complex, probably related to the lower spontaneous reduction under normoxic conditions (compare Fig. 7).
 | ||
| Fig. 8 Impact of hypoxia on cell-associated LPon-fluorescence from Co(acac)2LPon and Co(Meacac)2LPon. K-562 cells were incubated with 10 μM of compounds for the indicated time points under hypoxic and normoxic conditions and fluorescence intensities were determined by flow cytometry (BD LSRFortessa X-20, pacific blue filter). Fluorescence intensities were determined as described under Fig. 7 and mean fluorescence intensities under hypoxic cell culture conditions were normalized to the respective normoxic conditions. Data are given as means ± SEM of five independent experiments. Statistical significance was calculated by one-way ANOVA with Dunnett multiple comparison test with p < 0.05(*); <0.01 (**); <0.001 (***). | ||
The enhanced stability of Co(acac)2LPon under normoxia and its hypoxia-induced ligand release was subsequently confirmed by fluorescence microscopy. Fluorescence intensities indicated a ∼2.8-fold decreased stability of the cobalt(III) complex under hypoxic vs. normoxic cell culture conditions. In case of the free ligand LPon, fluorescence intensities remained unaffected by different oxygen levels (Fig. S9†). These findings are well in agreement with the flow cytometric analysis of the intracellular ligand release, demonstrating reduced stability, i.e. enhanced ligand release of the cobalt(III) complexes in hypoxic vs. normoxic environments.
Kinase screening
To investigate how the structural changes of the novel derivative affect its ABL1 and FGFR1 kinase-inhibitory potential in comparison to ponatinib, both substances were tested in cell-free kinase inhibition assays in the presence of a 10-fold excess of ATP (Fig. S8†). The IC50 values for ponatinib, which were 1.75 and 5.54 nM against ABL1 and FGFR1, are in the same range as reported in literature.10In comparison, the IC50 values of LPon against ABL1 and FGFR1 was 1.93 and 25.9 nM, respectively. Consequently, the ABL1 inhibition potential of our new ligand is comparable to ponatinib, whereas the FGFR inhibition is distinctly weaker by a factor of ∼5.
Anticancer activity against ABL1- and FGFR-driven cancer cell models
As a next step, the impacts of the novel derivatives on cell viability of the leukemia cancer cell line K-562 (BCR-ABL-dependent)49 and the urothelial cancer cell line UM-UC-14 (FGFR3-dependent)50 were evaluated under normoxic and hypoxic conditions. In general, a 72 h continuous cell exposure to all investigated compounds resulted in a dose-dependent reduction of cell vitality in the tested cell models under both oxygen conditions as determined by two independent methods, i.e. MTT assay (Fig. 9A) and ATP-based viability quantification (Fig. 9B). | ||
| Fig. 9 Anticancer activity of Co(acac)2LPon, Co(Meacac)2LPon, LPon and ponatinib against human cancer cell models under normoxic (N) and hypoxic (H) conditions. (A) BCR-ABL-positive K-562 leukemic cell viability was measured by MTT vitality assay and (B) by luminescence assay based on ATP quantification (CellTiter-Glo) after 72 h. (C) Clonogenic cell growth of the FGFR3-driven UM-UC-14 urothelial cancer cell model determined by colony formation assay after 5 d of incubation. Data are given as means ± SD of one representative experiment performed in triplicates. Statistical significance was calculated by two-way ANOVA with Sidak multiple comparison test with p < 0.05(*); <0.01 (**); <0.001 (***). | ||
Treatment of BCR-ABL-driven K-562 cells with ponatinib resulted in a pronounced reduction of cell vitality with IC50 in the nanomolar range. Notably, the anticancer activity of ponatinib was distinctly stronger as LPon. As in the above-mentioned cell-free kinase assay, IC50 values of both compounds against ABL1 were comparable (compare Fig. S8†), a reduced cellular uptake of LPon as compared to the more lipophilic ponatinib might be expected (calculated logP values51: ponatinib = 4.42; LPon = 3.39). Both cobalt(III) complexes were clearly less active as compared to the free ligand under normoxic conditions with Co(Meacac)2LPon showing the highest IC50 value. Anticancer activities of ponatinib and LPon were only marginally altered under hypoxia, reaching significance only at a concentration of 0.5 nM ponatinib with both assays (Fig. 9A). In contrast, reduction of oxygen to 0.1% massively increased the anticancer activity of Co(acac)2LPon and Co(Meacac)2LPon against K-562 cells at a concentration range between 5 and 10 nM.
In FGFR3-dependent UM-UC-14 cells a comparable activity pattern was observed but with IC50 values in the low micromolar range (Fig. S10†). Again, target cells could be strongly sensitized by oxygen reduction against both cobalt(III) complexes, but only marginally to ponatinib. The sensitizing effect in this cell model was more pronounced for Co(acac)2LPonvs.Co(Meacac)2LPon reflecting the differences in the reduction potentials. Purification of the novel compounds was performed in the presence of 0.1% TFA, leading to the respective TFA salts. MTT assays with TFA in UM-UC-14 cells elucidated, that TFA up to 25 μM had no impact on cell viability (Fig. S11†).
In order to investigate the impact of long-term oxygen restriction, clonogenic assays were performed in the FGFR3-dependent UM-UC-14 cell model (Fig. 9C). Under normoxic conditions both cobalt(III) complexes inhibited clonogenic cell growth by approximately 50% only, whereas ponatinib by 95%. In contrast, under hypoxic conditions all three compounds were comparably active with more than 80% inhibition of clone formation.
As mentioned above, two stereoisomers of Co(acac)2LPon could be isolated. To clarify, whether there are differences in their anticancer activity, they were analyzed (in addition to the isomeric mixture) in UM-UC-14 cells by MTT assay. The impact on cell viability was moderately stronger for isomer 1 as compared to isomer 2 at normoxic conditions. Accordingly, values for the isomeric mixture positioned intermediate. As expected, both stereoisomers and the isomeric mixture were distinctly activated by hypoxic conditions. The moderately higher activity of isomer 1 remained also under hypoxic conditions (Fig. S12A†). Comparable effects were found in K-562 by luminescence assay-based ATP quantification (Fig. S12B†). Since no remarkable difference in anticancer activity could be observed for the pure isomers, the isomeric mixture was used for all other experiments.
It is known from literature that cobalt(II) ions (which are released after reduction of the cobalt(III) complexes) can cause cytotoxicity52,53 and changes in the hypoxia inducible factor (HIF-1 alpha).54 In our previous work we therefore investigated the effect of CoCl2, and the two complexes [Co(II)(acac)2en] and [Co(III)(acac)2en]PF6 (“en” = ethylenediamine) without an EGFR-targeting moiety on two cancer cell lines (A431 and H1975).26 Notably, all compounds showed no significant cytotoxic activity under normoxic as well as hypoxic conditions. Consequently, a major contribution of cobalt(II) ions on the anticancer activity of our prodrugs can be widely excluded. Concerning upregulation of HIF-1 alpha signalling, it should be considered that our metal complexes are active in the nanomolar to the low micromolar range, while in most papers using CoCl2 as hypoxia-mimicry, concentrations of 100 μM or even higher are used.55
Impact of hypoxia on downstream signalling inhibition
Next, the impact of both cobalt(III) complexes, LPon and ponatinib on the phosphorylation of the downstream targets ERK1/2 and S6 was evaluated under normoxic vs. hypoxic conditions by western blot analysis in BCR-ABL-driven K-562 cells (Fig. 10). | ||
| Fig. 10 Impact of the cobalt(III) complexes on the ERK1/2 and S6 under normoxic and hypoxic conditions. K-562 cells were treated with the compounds, after 12 h cell lysates were prepared and protein expression as well as phosphorylation levels of downstream pathways (p-ERK1/2 and p-S6) analyzed by western blotting. One representative experiment out of three is shown. The ratio of phosphorylated to total protein is given between the respective lanes. | ||
Generally, ERK1/2 phosphorylation, as readout of the MAPK pathway, tended to be enhanced under hypoxic conditions, while S6, as PI3K/AKT pathway indicator, tended to be reduced. Ponatinib and the free ligand LPon were equally active under the two oxygen conditions. However, comparable to the viability assays, the efficacy of LPon was lower as compared to ponatinib. The cobalt(III) complexes were distinctly more active under hypoxic conditions, especially at the level of S6 phosphorylation. In this case, Co(acac)2LPon turned out to be equally active as compared to ponatinib. The activity of Co(Meacac)2LPon was generally weaker as compared to Co(acac)2LPon.
In vivo anticancer activity
Based on this promising in vitro data, the antitumor activity of our novel cobalt(III) complexes Co(acac)2LPon and Co(Meacac)2LPon was evaluated in xenograft mouse models. As a first in vivo experiment, BCR-ABL-driven K-562 cells were injected subcutaneously (s.c.) and treated intraperitoneally (i.p.) with the test compounds at 10 mg kg−1 or the respective amount of solvent three times a week, for two weeks. Treatment with our novel derivatives was well tolerated and even after repeated i.p. applications no substantial body weight loss was observed (Fig. S13A†). Administration of Co(acac)2LPon but not of the slower LPon-releasing Co(Meacac)2LPon complex significantly reduced tumor growth of K-562 xenografts as compared to solvent controls (Fig. 11). Consequently, anticancer activity of Co(acac)2LPon was also investigated against FGFR3-driven UM-UC-14 xenografts using the identical treatment regime. Again, administration of Co(acac)2LPon displayed significant reduction of tumor volumes compared to control, without significant impact on body weight (Fig. 11, S13B†). | ||
| Fig. 11 In vivo anticancer activity of the investigated cobalt(III) complexes. (A) BCR-ABL-driven leukemic K-562 cells or (B) FGFR3-driven urothelial UM-UC-14 cells were injected s.c. into the right flank of male CB17/SCID mice (n = 4 animals per experimental group). When tumors were measurable (day 5 and day 7, respectively) compounds (10 mg kg−1 i.p.) were applied as indicated (black arrows). Tumor sizes were evaluated by caliper measurement. Data are given as means ± SEM. Statistical significance was calculated by two-way ANOVA with Sidak multiple comparison test with p < 0.05(*); <0.01 (**). | ||
Conclusions
TKIs greatly improved cancer therapy since their first approval two decades ago. However, therapy resistance and severe side effects are limiting their clinical application. In this study, we developed the first ponatinib prodrugs with the aim to exploit the hypoxic environment present in the malignant tissue for cancer-specific drug activation. Molecular docking studies showed that both cobalt(III) prodrugs synthesized are unable to interact with the active sites of FGFR1 and ABL1 kinases and that the bioactive ligand LPon has comparable kinase inhibitory abilities as ponatinib. Cell-free kinase inhibition assays confirmed binding of the free ligand to the targeted kinases ABL1 and FGFR1, in the case of ABL1 with affinities comparable to ponatinib. Furthermore, investigation of the fluorescence properties demonstrated, that the intensities of LPon were comparable to ponatinib, whereas Co(acac)2LPon and Co(Meacac)2LPon exhibited a strongly quenched fluorescence. Both derivatives were highly stable in pure FCS with ∼90% intact complex after 26 h. Nevertheless, according to cyclic voltammetry, Co(Meacac)2LPon exhibited a clearly lower reduction potential as compared to Co(acac)2LPon, suggesting higher stability and slower ligand release. This was confirmed, exploiting the distinct differences in the fluorescence properties between free ligand and cobalt(III) complexes. Flow cytometry confirmed significantly enhanced spontaneous release of the bioactive ligand from Co(acac)2LPon as compared to Co(Meacac)2LPon under normoxic conditions, but potent release of both compounds under hypoxia. Moreover, reduced oxygen levels significantly increased inhibition of FGFR3-downstream signaling and improved the anticancer activity of both cobalt(III) complexes against ABL- and FGFR-dependent human cancer cell models. Finally, Co(acac)2LPon, but not Co(Meacac)2LPon significantly reduced in vivo xenograft growth of the leukemic K-562 model. The in vivo activity of Co(acac)2LPon was confirmed in an FGFR3 mutation-dependent urothelial carcinoma xenograft background. This suggests, that especially in the leukemic background hypoxic conditions might be too weak to activate Co(Meacac)2LPon based on the lower reduction potential and, hence, higher stability. However, it needs to be considered that in the current study relatively small tumors were treated, due to a rapid tumor growth. Therefore, we currently aim to compare these drugs in slow growing cancer xenografts at later stages and to identify other solid tumor models, characterized by massive tumor hypoxia and driven by ponatinib targets. This will allow to further elucidate the interplay between cobalt(III)-based prodrug systems and its reduction kinetics, molecular targets and cancer hypoxia.Experimental section
Docking studies
The 3D structures of the molecules (LPon, Co(acac)2LPon and Co(Meacac)2LPon) have been drawn and optimized with Avogadro version 1.2.0,56 using Force Field GAFF (General AMBER Force Field).57 The molecules obtained are those with minimum energy. The crystal structures of FGFR1 (PDB ID: 4V04) and ABL1 (PDB ID: 4WA9) have been obtained from the RCSB Protein Data Bank.58 The *.pdb files from proteins were manipulated by elimination of adsorbed water and sulfur dioxide molecules, addition of missing hydrogen atoms, protonation of histidine residues, conversion of selenomethionines in methionines, addition and optimization of the side chain atoms. New *.pdb files were created of the proteins without the co-crystallized ligands and of the native ligands without the proteins using Chimera UCFS version 1.11.2.59 The *.pdbqt files of drugs and proteins were generated using AutodockTools-1.5.7rc.60 Ligand based molecular docking was performed with Autodock Vina 1.5 using ponatinib coordinates for FGFR1 and Axitinib for ABL1 to create the grid box.61 The coordinates of the grid box are: FGFR1 (x-size: 30, x-center: 27.814, y-size: 46, y-center: 5.016, z-size: 54, z-center: 16.789); ABL1 (x-size: 48, x-center: 7.819, y-size: 46, y-center: 156.948, z-size: 46, z-center: 37.479). Docking studies were performed in triplicates and the calculated RMSD value was always less than 1.5 Å.62Materials and methods
All solvents and reagents were obtained from commercial suppliers. They were, unless stated otherwise, used without further purification. Anhydrous MeOH and THF were bought from Fisher Chemicals over molecular sieves. The cobalt(III) precursors Na[Co(acac)2(NO2)2] and Na[Co(Meacac)2(NO2)2] were obtained by following the protocol of Denny et al.33 For all HPLC measurements Milli-Q water (18.2 MΩ cm, Merck Milli-Q Advantage, Darmstadt, Germany) was used. Preparative RP-HPLC was performed on an Agilent 1200 Series system controlled by Chemstation software. As stationary phase a XBridge BEH C18 OBD Prep Column (130 Å, 5 μm, 19 mm × 250 mm) from Waters Corp., Massachusetts, USA, was used. The general procedure included a flow rate of 17.06 mL min−1, an injection volume of up to 10 mL and a column temperature of 25 °C. Milli-Q water and acetonitrile with addition of acids (0.1% TFA) were used as eluents unless stated otherwise. Elemental analyses were performed by the Microanalytical Laboratory of the University of Vienna on a PerkinElmer 2400 CHN Elemental Analyzer. The amount of TFA was also confirmed by 19F NMR spectra. Electrospray ionization (ESI) mass spectra were recorded on a Bruker Amazon SL ion trap mass spectrometer in positive and/or negative mode by direct infusion.High resolution mass spectra were measured on a Bruker maXis™ UHR ESI time of flight mass spectrometer. Expected and experimental isotope distributions were compared. 1H and 13C NMR, one- as well as two-dimensional, spectra were recorded in d6-DMSO with a Bruker FT-NMR Avance III 500 MHz spectrometer at 500.10 (1H) and 125.75 (13C) MHz at 298 K. Chemical shifts (ppm) were referenced internal to the solvent residual peaks. For the description of the spin multiplicities the following abbreviations were used: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet.
Synthesis
:ethyl acetate 3:2), resulting in yellow crystals. Yield: 2.51 g (81%). 1H NMR (500.1 MHz, DMSO-d6): δ 7.24 (t, 1H), 7.11 (d, J = 8.4 Hz, 1H), 6.85 (d, J = 2.3 Hz, 1H), 6.75 (d, J = 8.3 Hz, 1H), 5.44 (s, 2H), 4.12 (d, J = 5.8 Hz, 2H), 1.39 (s, 9H) ppm. MS: calcd for [C13H17F3N2O2–H]−: 289.12, found: 288.97.
1H NMR (500.1 MHz, DMSO-d6): δ 10.58 (s, 1H), 8.74 (dd, J = 4.5 Hz, J = 1.5 Hz, 1H), 8.28 (dd, J = 9.2 Hz, 1.6 Hz, 1H), 8.25 (s, 1H), 8.23 (d, J = 2.0 Hz, 1H), 8.22 (d, J = 2.0 Hz, 1H), 8.08 (dd, J = 8.5 Hz, 1.9 Hz, 1H), 7.96 (dd, J = 8.0 Hz, 1.9 Hz, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.56 (d, J = 8.1 Hz, 1H), 7.40 (dd, J = 8.9 Hz, 4.4 Hz, 1H), 6.80 (s, 1H), 3.84 (s, 2H), 3.09–3.02 (m, 2H), 2.62 (s, 3H), 2.60–2.55 (m, 2H), 1.38 (s, 9H) ppm. MS: calcd for [C31H31F3N6O3+H]+: 593.2482, found: 593.2485.
 :0.67.
:0.67.
Shifts of the main isomer; 1H NMR (500.1 MHz, DMSO-d6): δ 10.67 (s, 1H, H17), 8.73 (s, 1H, H6), 8.30–8.26 (m, 2H, H4, H19), 8.26–8.22 (m, 2H, H2, H14), 8.18–8.14 (m, 1H, H24), 7.97 (d, 1H, J = 9 Hz, H12), 7.78 (d, 1H, J = 8 Hz, H23), 7.58 (d, 1H, J = 9 Hz, H11), 7.43–7.39 (m, 1H, H5), 5.70 (s, 1H, CH, acac), 5.65 (s, 1H, CH, acac), 5.61 (s, 1H, H29), 5.26 (s, 1H, H26), 5.09 (s, 1H, H29), 3.89–3.79 (m, 2H, H25), 2.74–2.66 (m, 1H, H28), 2.63 (s, 3H, H15), 2.45–2.38 (m, 1H, H28), 2.55–2.45 (m, 1H, H27), 2.38–2.32 (m, 1H, H27), 2.15 (s, 3H, CH3, acac), 2.13–2.10 (s, 6H, CH3, acac), 2.06 (s, 3H, CH3, acac) ppm. 13C NMR (125.75 MHz, DMSO-d6): δ 189.9 (CQ, acac), 189.1 (CQ, acac), 188.9 (CQ, acac), 188.4 (CQ, acac), 164.7 (C16), 145.1 (C6), 143.7 (C10), 139.7 (C3), 139.3 (C18), 138.3 (C14), 133.7 (C23), 132.0 (C13), 130.2 (C11 + C19), 128.5 (C12), 128.4 (C22), 126.9 (C20), 126.1 (C4), 124.8 (C21), 123.2 (C24), 121.8 (C9), 119.1 (C5), 117.5 (C2), 111.6 (C1), 98.1 (CH, acac), 97.9 (CH, acac), 96.4 (C8), 81.2 (C7), 52.2 (C27), 47.1 (C25), 42.5 (C28), 26.5 (CH3, acac), 26.3 (CH3, acac), 26.1 (CH3, acac), 26.0 (CH3, acac), 20.4 (C15) ppm.
Shifts of the minor isomer; 1H NMR (500.1 MHz, DMSO-d6): δ 10.67 (s, 1H, H17), 8.73 (s, 1H, H6), 8.30–8.26 (m, 2H, H4, H19), 8.26–8.22 (m, 2H, H2, H14), 8.18–8.14 (m, 1H, H24), 7.97 (d, 1H, J = 9 Hz, H12), 7.91 (d, 1H, J = 8 Hz, H23), 7.58 (d, 1H, J = 9 Hz, H11), 7.43–7.39 (m, 1H, H5), 5.74 (s, 1H, CH, acac), 5.65 (s, 1H, CH, acac), 5.61 (s, 1H, H26), 5.48 (s, 1H, H29), 5.39 (s, 1H, H29), 3.63–3.58 (m, 1H, H25), 3.11–3.04 (m, 1H, H25), 2.63 (s, 3H, H15), 2.55–2.45 (m, 2H, H28), 2.45–2.38 (m, 2H, H28), 2.19 (s, 3H, CH3, acac), 2.13–2.10 (s, 6H, CH3, acac), 2.05 (s, 3H, CH3, acac) ppm. 13C NMR (125.75 MHz, DMSO-d6): δ 190.6 (CQ, acac), 189.9 (CQ, acac), 189.8 (CQ, acac), 189.5 (CQ, acac), 164.7 (C16), 145.1 (C6), 143.7 (C10), 139.7 (C3), 139.4 (C18), 138.3 (C14), 133.6 (C23), 132.0 (C13), 130.2 (C11 + C19), 128.5 (C12), 128.3 (C22), 126126.6 (C20), 126.1 (C4), 124.8 (C21), 123.2 (C24), 121.8 (C9), 119.1 (C5), 117.5 (C2), 111.6 (C1), 98.1 (CH, acac), 98.0 (CH, acac), 96.4 (C8), 81.2 (C7), 50.9 (C27), 48.1 (C25), 42.3 (C28), 26.4 (CH3, acac), 26.3 (CH3, acac), 26.1 (CH3, acac), 26.0 (CH3, acac), 20.4 (C15) ppm.
MS: calcd For [C36H37F3N6O5Co]+: 749.2104, found: 749.2093. Anal. calcd for C38H37F6N6O7Co·0.6TFA·1.5H2O (Mr = 958.10 g mol−1): C, 49.14; H, 4.27; N, 8.77. Found: C, 48.84; H, 3.96; N, 8.53.
:0.43.
Shifts of the main isomer; 1H NMR (500.1 MHz, DMSO-d6): δ 10.66 (s, 1H, H17), 8.73 (s, 1H, H6), 8.30–8.21 (m, 4H, H2, H4, H14, H19), 8.17–8.13 (m, 1H, H24), 7.97 (d, 1H, J = 9 Hz, H12), 7.75 (d, 1H, J = 8 Hz, H23), 7.58 (d, 1H, J = 9 Hz, H11), 7.43–7.39 (m, 1H, H5), 5.52–5.42 (m, 1H, H29), 4.99 (s, 1H, H29), 4.81 (s, 1H, H26), 3.94–3.84 (m, 2H, H25), 2.72–2.65 (m, 1H, H28), 2.63 (s, 3H, H15), 2.47–2.42 (m, 1H, H27), 2.44–2.38 (m, 1H, H28), 2.38–2.33 (m, 1H, H27), 2.24 (s, 3H, CH3, Meacac), 2.20 (s, 3H, CH3, Meacac), 2.15 (s, 3H, CH3, Meacac), 2.13 (s, 3H, CH3, Meacac), 1.92 (s, 3H, CH3, Meacac), 1.88 (s, 3H, CH3, Meacac) ppm. 13C NMR (125.75 MHz, DMSO-d6): δ 188.1 (2CQ, Meacac), 187.3 (CQ, Meacac), 186.9 (CQ, Meacac), 164.7 (C16), 145.1 (C6), 143.8 (C10), 139.7 (C3), 139.2 (C18), 138.3 (C14), 133.4 (C23), 132.0 (C13), 130.1 (C11 + C2), 128.5 (C12), 128.2 (C20), 127.3 (C22), 126.1 (C4), 124.8 (C21), 123.1 (C24), 121.9 (C9), 119.2 (C5), 117.5 (C19), 111.7 (C1), 100.8 (CQ, Meacac), 100.7 (CQ, Meacac), 96.4 (C8), 81.2 (C7), 52.1 (C27), 46.9 (C25), 42.4 (C28), 26.6 (CH3, Meacac), 26.5 (2CH3, Meacac), 26.2 (CH3, Meacac), 20.4(C15), 14.9 (CH3, Meacac), 14.8 (CH3, Meacac) ppm.
Shifts of the minor isomer; 1H NMR (500.1 MHz, DMSO-d6): δ 10.66 (s, 1H, H17), 8.73 (s, 1H, H6), 8.30–8.21 (m, 4H, H2, H4, H14, H19), 8.17–8.13 (m, 1H, H24), 7.97 (d, 1H, J = 9 Hz, H12), 7.89 (d, 1H, J = 8 Hz, H23), 7.58 (d, 1H, J = 9 Hz, H11), 7.43–7.39 (m, 1H, H5), 5.52–5.42 (m, 1H, H29), 5.32 (s, 1H, H29), 5.12 (s, 1H, H26), 3.54–3.49 (m, 1H, H25), 2.99–2.93 (m, 1H, H25), 2.72–2.65 (m, 1H, H28), 2.63 (s, 3H, H15), 2.47–2.42 (m, 1H, H27), 2.44–2.38 (m, 1H, H28), 2.38–2.33 (m, 1H, H27) 2.28 (s, 3H, CH3, Meacac), 2.23 (s, 3H, CH3, Meacac), 2.18 (s, 3H, CH3, Meacac), 2.11 (s, 3H, CH3, Meacac), 1.92 (s, 3H, CH3, Meacac), 1.87 (s, 3H, CH3, Meacac) ppm. 13C NMR (125.75 MHz, DMSO-d6): δ 188.7 (CQ, Meacac), 188.2 (CQ, Meacac), 187.3 (CQ, Meacac), 187.0 (CQ, Meacac), 164.7 (C16), 145.1 (C6), 143.8 (C10), 139.7 (C3), 139.3 (C18), 138.3 (C14), 133.3 (C23), 132.0 (C13), 130.1 (C11 + C2), 128.5 (C12), 127.0 (C22), 126.1 (C4), 128.4 (C20), 123.1 (C24), 121.9 (C9), 119.2 (C5), 117.5 (C19), 111.7 (C1), 101.0 (CQ, Meacac), 100.7 (CQ, Meacac), 96.4 (C8), 81.2 (C7), 50.7 (C27), 48.0 (C25), 42.0 (C28), 26.4 (CH3, Meacac), 26.3 (CH3, Meacac), 26.2 (2CH3, Meacac), 20.4 (C15), 14.9 (CH3, Meacac), 14.8 (CH3, Meacac) ppm.
MS: calcd for [C38H41F3N6O5Co]+: 777.2417, found: 777.2405. Anal. calcd for C40H41F6N6O7Co·TFA·H2O (Mr = 1022.75 g mol−1): C, 49.32; H, 4.34; N, 8.22. Found: C, 48.98; H, 4.19; N, 8.01.
Interconversion of the isomers
For the interconversion measurements, a stock solution of the two isomers of Co(acac)2LPon (5 mM in water) was prepared. The final concentration was reached by 1:100 dilution with PBS (pH 7.4, 10 mM). The samples were incubated at 37 °C. Measurements were performed on a Dionex UltiMate 3000 RS UPLC system with a Waters Acquity UPLC® BEH C18 column (3 × 50 mm, pore size 1.7 μm). Water and acetonitrile (both containing 0.1% TFA) were used as eluents, an isocratic gradient of 45% acetonitrile within 8 min was applied.
Fluorescence measurements
Fluorescence measurements were performed on a Horiba FluoroMax®-4 spectrofluorometer and the data was analyzed using the FluorEssence v3.5 software package. The tested solutions were dissolved immediately prior to analysis in PB (50 mM, pH 7.4) with a final concentration of 15 μM. Scans were run at room temperature with excitation and emission slit widths of 3 nm. Emission scans were run from 250–700 nm using an excitation wavelength of 320 nm. The 3D fluorescence spectra of LPon were determined at excitation wavelengths from 230–550 nm and emission was recorded within the range of 220–700 nm.Cyclic voltammetry
The compounds were dissolved in DMF (+0.2 M [n-Bu4N][BF4]) to obtain a final concentration of 1.5 mM. Electrochemical experiments were conducted on an EG&G PARC 273A potentiostat/galvanostat with a scan rate of 100–1000 mV s−1 at room temperature. Argon was bubbled through the solution before every measurement to remove oxygen. A three-electrode configuration cell was used with a glassy carbon electrode as working electrode, which was polished before every measurement. The reference electrode was Ag/Ag+/ACN (10 mM AgNO3) and the auxiliary electrode was a platinum wire. The potentials were referenced to an internal standard redox couple of ferrocenium/ferrocene and calculated to the NHE (E1/2 = +0.72 V vs. NHE).63 For the cyclic voltammetry measurements in DMF/H2O (7:3 v/v; aqueous phase was 10 mM phosphate buffer at pH 7.40 with 0.1 M KCl), a glassy carbon electrode as working electrode, a platinum auxiliary electrode, and an Ag/AgCl electrode containing 3.5 KCl were used. Redox potentials measured relative to the Ag/AgCl/KCl (3.5 M) reference electrode and converted to the NHE using +0.205 V. Measurements were at least repeated three times and the mean values were calculated.
Serum stability measurements
For serum stability measurements, 135 μL FCS, buffered with 150 mM phosphate (Na2HPO4/NaH2PO4) to maintain a pH value of 7.4, were mixed with 15 μL of a 500 μM stock solution of the respective complex in 50 mM phosphate buffer to reach a final concentration of 50 μM. The samples were incubated at 37 °C. At different time points to 20 μL serum 40 μL of ACN were added. After vigorously shaking for 2 min the suspension was centrifuged at 6000 rpm for 10 min. The supernatant was taken with a syringe and directly measured via HPLC-MS. The samples were analyzed on an Agilent 1260 Infinity system using a Waters Atlantis T3 column (150 mm × 4.6 mm) coupled to a Bruker Amazon SL ESI-IT mass spectrometer. Water and acetonitrile (both containing 0.1% formic acid) were used as eluents with a gradient of 1–99% acetonitrile within 29 min.Biological methods
Kinase screening
The ABL1 and FGFR1 kinase-inhibitory potentials of the novel derivative LPon in comparison to ponatinib were evaluated using the Select Screen® Biochemical Kinase Profiling Service at Life Technologies (ThermoFisher Scientific, Madison, USA). The test compounds were screened in a final concentration of 1% DMSO using the Z′-LYTE® Assay. Results are additionally depicted in Fig. S8.†Cell lines, cell culture conditions and drug treatments
All reagents were purchased from Sigma-Aldrich (St Louis, USA) unless specified otherwise. The human leukemia cell line K-562 (chronic myeloid leukemia; BCR-ABL-positive) and human urothelial cancer cell line UM-UC-14 (FGFR3-dependent), obtained from the American Type Culture Collection (ATCC) (Rockville, MD, USA), were cultured in RPMI 1640 (K-562) or MEM (Minimum Essential Medium Eagle) (UM-UC-14) supplemented with 10% fetal calf serum (PAA, Linz, Austria) at 37 °C, 5% CO2 in a humidified atmosphere. In case of hypoxic cell culture conditions, plated and treated cells were incubated at 37 °C, 5% CO2 and 0.1% O2 (ProOx Model C21 system, BioSpherix) in humidified incubators for the indicated time point before analysis. All investigated compounds were dissolved in dimethyl sulfoxide (DMSO) as 10 mM stock solutions and were stored at −20 °C. Dilutions in culture media supplemented with 10% fetal calf serum were made immediately before the experiments at the indicated concentrations. Corresponding dilutions of DMSO were used as untreated vehicle controls.Cytotoxicity assay
Cells were seeded (3–7 × 104 cells per well) in 96-well plates and after 24 h recovery treated with increasing concentrations of compounds in triplicates. After 72 h drug exposure under normoxic or hypoxic cell culture conditions (0.1% O2) (ProOx Model C21 system, BioSpherix), the proportion of viable cells was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-vitality assay (EZ4U, Biomedica, Vienna, Austria) or by luminescence assay based on adenosine triphosphate quantification (CellTiter-Glo, Promega, Madison, Wisconsin) following the manufacturer's recommendations. Absorbance was measured at 450 nm (at 620 nm as reference) (MTT) and luminescence after 1000 ms (CellTiter-Glo) at the Tecan infinite 200Pro (Zurich, Switzerland). MTT-derived cytotoxicity was expressed as IC50 values calculated from full dose–response curves using GraphPad Prism software (La Jolla, CA).Colony formation assay
UM-UC-14 cells were seeded at low densities of 3 × 103 cells per well in 24-well plates in 500 μL and after 24 h recovery treated with 100 μL of increasing concentrations of compounds in duplicates. Following drug exposure time of 5 days under normoxic or hypoxic (0.1% O2) culture conditions, cells were washed with phosphate-buffered saline (PBS), fixed with ice-cold methanol for 30 min at 4 °C and stained with crystal violet. Digital photographs were taken using a Nikon D3200 camera and processed with ImageJ software. For quantification, crystal violet was eluted using 2% sodium dodecyl sulfate (SDS) and color absorbance was measured at 560 nm at the Tecan infinite 200Pro (Zurich, Switzerland). Values are given in arbitrary units (a.u.) as mean ± SD normalized to untreated control.Flow cytometry
Cells were seeded (1 × 106 cells per well) in 96-well plates, dissolved compounds were added and cells were incubated under normoxic or hypoxic cell culture conditions. Samples were measured on a BD LSRFortessa X-20 cell analyzer high throughput sampler (HTS) (BD Biosciences, East Rutherford, NJ, USA). Compound fluorescence was detected using 405 nm excitation and pacific blue (450/50 nm) band pass emission filters. Data were analyzed using BD FACSDiva software and are given in arbitrary units (a.u.) as mean fluorescence intensities of cells normalized to the auto fluorescence of untreated control.Fluorescence microscopy
UM-UC-14 cells were seeded (2.5 × 105 cells per well) in 12-well plates and after 24 h recovery treated with 10 μM of compounds. After 24 h incubation, the drug solutions were removed, cells were washed with PBS and microphotographs of the different treatments were taken using UV fluorescence microscopy (Nikon Eclipse Ti2 microscope with a DAPI filter and a high-pressure mercury lamp) and a 20× objective. The level of cellular fluorescence was determined from fluorescence microscopy images using ImageJ software.Western blot analysis
K-562 cells were plated (5 × 105 cells per well) in 6-well plates and allowed to recover for 24 h. Subsequently, the cells were treated with the drugs in different concentrations. After 12 h cells were harvested, proteins were isolated, resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS/PAGE), and transferred onto a polyvinylidene difluoride membrane for western blotting. The following antibodies were used: ERK1/2 (p44/42) rabbit monoclonal antibody (mAb) (137F5, #4695), phospho-ERK1/2 rabbit mAb (Thr202/Tyr204) (20G11, #4376), phospho-S6 rabbit pAb (Ser240/244, #2215) (Cell Signaling Technology, Beverly, MA, USA), S6 mouse mAb (C-8. #sc-74459) (Santa Cruz Biotechnology, TX, USA) and β-actin mouse mAb (#A5441, Sigma). All primary antibodies were used in 1:1000 dilutions (in Tris-buffered saline containing 0.1% Tween20 (TBST) + 3% bovine serum albumin (BSA)). Secondary goat anti-mouse-IgG (Fc specific)-peroxidase antibody (#A0168, Sigma) and horseradish peroxidase(HRP)-labeled mouse anti-rabbit IgG (#sc-2357, Santa Cruz Biotechnology) were used at working dilutions of 1:10000 (in TBST + 1% BSA).
In vivo xenograft experiment
Eight-to nine-week-old male C.B.17/SCID mice were bread in-house (originally Harlan) and were kept in pathogen-free conditions and controlled environment with 12 h light–dark cycle. K-562 (5 × 106 in 100 μL serum-free RMPI medium, 20% Matrigel) or UM-UC-14 (1 × 106 in 100 μL serum-free RMPI medium) cells were injected into the right flank of male C.B.17/SCID mice. When all tumors were measurable (at day 5 and day 7, respectively), all animals were dosed i.p. with solutions (200 μL) containing Co(acac)2LPon or Co(Meacac)2LPon (10 mg kg−1, 200 μL per 20 g, dissolved in 50 mM PBS, 2% DMSO). The control groups received solvent alone (200 μL, 50 mM PBS, 2% DMSO). Compounds were dissolved in the respective amount of DMSO and then added to PBS buffer for the desired end concentration allowing application of 200 μL per 20 g and dosing using 30 gauze needles. Tumor growth was evaluated by daily recording of tumor size by caliper measurement. Animals were sacrificed by cervical dislocation. All procedures were performed in a laminar flow hood. The animal experiments were performed according to the regulations of the Ethics Committee for the Care and Use of Laboratory Animals at the Medical University Vienna (proposal number BMBWF-V/3b 2020-0.380.502).Statistical analysis
Data were analyzed using GraphPad Prism 8.0 software (GraphPad Software, Inc.) and are expressed as mean ± standard deviation (SD) of one independent experiment performed in triplicates, unless stated otherwise. One- or two-way analysis of variance (ANOVA) with multiple comparison test (Sidak, Bonferroni or Dunnett) was performed for statistical evaluation if not stated otherwise, p-values <0.05 were considered statistically significant [p < 0.05(*); <0.01 (**); <0.001 (***)].Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Austrian Science Fund (FWF) for financial support. Grant P28853 to CK and grant FG3 to WB and CK.References
- M. A. Lemmon and J. Schlessinger, Cell signaling by receptor tyrosine kinases, Cell, 2010, 141, 1117–1134 CrossRef CAS PubMed.
- Z. Du and C. M. Lovly, Mechanisms of receptor tyrosine kinase activation in cancer, Mol. Cancer, 2018, 17, 58 CrossRef PubMed.
- P. Wu, T. E. Nielsen and M. H. Clausen, FDA-approved small-molecule kinase inhibitors, Trends Pharmacol. Sci., 2015, 36, 422–439 CrossRef CAS PubMed.
- S. M. Chiavenna, J. P. Jaworski and A. Vendrell, State of the art in anti-cancer mAbs, J. Biomed. Sci., 2017, 24, 1–12 CrossRef.
- W.-S. Huang, C. A. Metcalf, R. Sundaramoorthi, Y. Wang, D. Zou, R. M. Thomas, X. Zhu, L. Cai, D. Wen and S. Liu, Discovery of 3-[2-(imidazo [1, 2-b] pyridazin-3-yl) ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl) methyl]-3-(trifluoromethyl) phenyl} benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant, J. Med. Chem., 2010, 53, 4701–4719 CrossRef CAS.
- S. M. Hoy, Ponatinib: a review of its use in adults with chronic myeloid leukaemia or Philadelphia chromosome-positive acute lymphoblastic leukaemia, Drugs, 2014, 74, 793–806 CrossRef CAS PubMed.
- F. H. Tan, T. L. Putoczki, S. S. Stylli and R. B. Luwor, Ponatinib: a novel multi-tyrosine kinase inhibitor against human malignancies, OncoTargets Ther., 2019, 12, 635–645 CrossRef CAS PubMed.
- J. E. Cortes, D.-W. Kim, J. l. Pinilla-Ibarz, P. Le Coutre, R. Paquette, C. Chuah, F. Nicolini, J. Apperley, H. Khoury and M. Talpaz, A phase 2 trial of ponatinib in Philadelphia chromosome–positive leukemias, New Eng. J. Med., 2013, 369, 1783–1796 CrossRef CAS PubMed.
- M. C. Mueller, F. Cervantes, H. Hjorth-Hansen, J. J. Janssen, D. Milojkovic, D. Rea and G. Rosti, Ponatinib in chronic myeloid leukemia (CML): Consensus on patient treatment and management from a European expert panel, Crit. Rev. Oncol. Hematol., 2017, 120, 52–59 CrossRef PubMed.
- T. O'Hare, W. C. Shakespeare, X. Zhu, C. A. Eide, V. M. Rivera, F. Wang, L. T. Adrian, T. Zhou, W.-S. Huang and Q. Xu, AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance, Cancer Cells, 2009, 16, 401–412 CrossRef PubMed.
- F. Musumeci, C. Greco, G. Grossi, A. Molinari and S. Schenone, Recent studies on ponatinib in cancers other than chronic myeloid leukemia, Cancers, 2018, 10, 430 CrossRef CAS PubMed.
- R. Roskoski Jr., The role of fibroblast growth factor receptor (FGFR) protein-tyrosine kinase inhibitors in the treatment of cancers including those of the urinary bladder, Pharmacol. Res., 2020, 151, 104567 CrossRef PubMed.
- J. M. Gozgit, M. J. Wong, L. Moran, S. Wardwell, Q. K. Mohemmad, N. I. Narasimhan, W. C. Shakespeare, F. Wang, T. Clackson and V. M. Rivera, Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models, Mol. Cancer Ther., 2012, 11, 690–699 CrossRef CAS PubMed.
- O. Chan, C. Talati, L. Isenalumhe, S. Shams, L. Nodzon, M. Fradley, K. Sweet and J. Pinilla-Ibarz, Side-effects profile and outcomes of ponatinib in the treatment of chronic myeloid leukemia, Blood Adv., 2020, 4, 530–538 CrossRef CAS PubMed.
- J. F. Gainor and B. A. Chabner, Ponatinib: accelerated disapproval, Oncologist, 2015, 20, 847 CrossRef PubMed.
- J. H. Lipton, C. Chuah, A. Guerci-Bresler, G. Rosti, D. Simpson, S. Assouline, G. Etienne, F. E. Nicolini, P. le Coutre and R. E. Clark, Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: an international, randomised, open-label, phase 3 trial, Lancet Oncol., 2016, 17, 612–621 CrossRef CAS PubMed.
- D. J. Dorer, R. K. Knickerbocker, M. Baccarani, J. E. Cortes, A. Hochhaus, M. Talpaz and F. G. Haluska, Impact of dose intensity of ponatinib on selected adverse events: multivariate analyses from a pooled population of clinical trial patients, Leuk. Res., 2016, 48, 84–91 CrossRef CAS PubMed.
- F. Kratz, I. A. Müller, C. Ryppa and A. Warnecke, Prodrug strategies in anticancer chemotherapy, ChemMedChem, 2008, 3, 20–53 CrossRef CAS PubMed.
- Y. A. Fouad and C. Aanei, Revisiting the hallmarks of cancer, Am. J. Cancer Res., 2017, 7, 1016 CAS.
- R. M. Phillips, Targeting the hypoxic fraction of tumours using hypoxia-activated prodrugs, Cancer Chemother. Pharmacol., 2016, 77, 441–457 CrossRef CAS PubMed.
- K. M. Johnson, Z. D. Parsons, C. L. Barnes and K. S. Gates, Toward hypoxia-selective DNA-alkylating agents built by grafting nitrogen mustards onto the bioreductively activated, hypoxia-selective DNA-oxidizing agent 3-amino-1, 2, 4-benzotriazine 1, 4-dioxide (tirapazamine), J. Org. Chem., 2014, 79, 7520–7531 CrossRef CAS PubMed.
- V. Liapis, I. Zinonos, A. Labrinidis, S. Hay, V. Ponomarev, V. Panagopoulos, A. Zysk, M. DeNichilo, W. Ingman and G. J. Atkins, Anticancer efficacy of the hypoxia–activated prodrug evofosfamide (TH–302) in osteolytic breast cancer murine models, Cancer Med., 2016, 5, 534–545 CrossRef CAS PubMed.
- R. M. Phillips, H. R. Hendriks, J. B. Sweeney, G. Reddy and G. J. Peters, Efficacy, pharmacokinetic and pharmacodynamic evaluation of apaziquone in the treatment of non-muscle invasive bladder cancer, Expert Opin. Drug Metab. Toxicol., 2017, 13, 783–791 CrossRef CAS PubMed.
- A. Sharma, J. F. Arambula, S. Koo, R. Kumar, H. Singh, J. L. Sessler and J. S. Kim, Hypoxia-targeted drug delivery, Chem. Soc. Rev., 2019, 48, 771–813 RSC.
- L. Spiegelberg, R. Houben, R. Niemans, D. de Ruysscher, A. Yaromina, J. Theys, C. P. Guise, J. B. Smaill, A. V. Patterson and P. Lambin, Hypoxia-activated prodrugs and (lack of) clinical progress: The need for hypoxia-based biomarker patient selection in phase III clinical trials, Clin. Transl. Radiat. Oncol., 2019, 15, 62–69 CrossRef PubMed.
- C. Karnthaler-Benbakka, D. Groza, K. Kryeziu, V. Pichler, A. Roller, W. Berger, P. Heffeter and C. R. Kowol, Tumor–Targeting of EGFR Inhibitors by Hypoxia–Mediated Activation, Angew. Chem., Int. Ed., 2014, 53, 12930–12935 CrossRef CAS PubMed.
- C. Karnthaler-Benbakka, D. Groza, B. Koblmüller, A. Terenzi, K. Holste, M. Haider, D. Baier, W. Berger, P. Heffeter and C. R. Kowol, Targeting a targeted drug: an approach toward hypoxia-activatable tyrosine kinase inhibitor prodrugs, ChemMedChem, 2016, 11, 2410–2421 CrossRef CAS PubMed.
- B. Bielec, H. Schueffl, A. Terenzi, W. Berger, P. Heffeter, B. K. Keppler and C. R. Kowol, Development and biological investigations of hypoxia-sensitive prodrugs of the tyrosine kinase inhibitor crizotinib, Bioorg. Chem., 2020, 103778 CrossRef CAS PubMed.
- A. V. Patterson, S. Silva, C. Guise, M. Bull, M. Abbattista, A. Hsu, J. D. Sun, C. P. Hart, T. E. Pearce and J. B. Smaill, TH-4000, a hypoxia-activated EGFR/Her2 inhibitor to treat EGFR-TKI resistant T790M-negative NSCLC, J. Clin. Oncol., 2015, 33(15 suppl) Search PubMed.
- C. R. Munteanu and K. Suntharalingam, Advances in cobalt complexes as anticancer agents, Dalton Trans., 2015, 44, 13796–13808 RSC.
- N. Yamamoto, A. K. Renfrew, B. J. Kim, N. S. Bryce and T. W. Hambley, Dual targeting of hypoxic and acidic tumor environments with a cobalt(III) chaperone complex, J. Med. Chem., 2012, 55, 11013–11021 CrossRef CAS PubMed.
- U. Jungwirth, C. R. Kowol, B. K. Keppler, C. G. Hartinger, W. Berger and P. Heffeter, Anticancer activity of metal complexes: involvement of redox processes, Antioxid. Redox Signal., 2011, 15, 1085–1127 CrossRef CAS PubMed.
- D. C. Ware, B. D. Palmer, W. R. Wilson and W. A. Denny, Hypoxia-selective antitumor agents. 7. Metal complexes of aliphatic mustards as a new class of hypoxia-selective cytotoxins. Synthesis and evaluation of cobalt(III) complexes of bidentate mustards, J. Med. Chem., 1993, 36, 1839–1846 CrossRef CAS PubMed.
- M. C. Heffern, N. Yamamoto, R. J. Holbrook, A. L. Eckermann and T. J. Meade, Cobalt derivatives as promising therapeutic agents, Curr. Opin. Chem. Biol., 2013, 17, 189–196 CrossRef CAS PubMed.
- X.-Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Molecular docking: a powerful approach for structure-based drug discovery, Curr. Comput.-Aided Drug Des., 2011, 7, 146–157 CrossRef CAS PubMed.
- Z. Gagic, D. Ruzic, N. Djokovic, T. Djikic and K. Nikolic, In silico methods for design of kinase inhibitors as anticancer drugs, Front. Chem., 2020, 7, 873 CrossRef PubMed.
- D. H. Zou, W.-S. Thomas, R. M. Romero, J. A. C. Qi, J. Wang, Y. Zhu, X. Shakespeare, W. C. Sundaramoorthi, R. Metcalf, C. A. Dalgarno III and D. C. Sawyer, T. K., Bicyclic heteroaryl compounds, W.O. Patent2007/075869A2, 2007 Search PubMed.
- M. Mathuber, H. Schueffl, O. Dömötör, C. Karnthaler, E. v. A. Enyedy, P. Heffeter, B. K. Keppler and C. R. Kowol, Improving the Stability of EGFR Inhibitor Cobalt(III) Prodrugs, Inorg. Chem., 2020, 59, 17794–17810 CrossRef CAS PubMed.
- D. C. Ware, H. R. Palmer, P. J. Brothers, C. E. Rickard, W. R. Wilson and W. A. Denny, Bis-tropolonato derivatives of cobalt(III) complexes of bidentate aliphatic nitrogen mustards as potential hypoxia-selective cytotoxins, J. Inorg. Biochem., 1997, 68, 215–224 CrossRef CAS PubMed.
- B. Englinger, A. Laemmerer, P. Moser, S. Kallus, C. Röhrl, C. Pirker, D. Baier, T. Mohr, L. Niederstaetter, S. M. Meier-Menches, C. Gerner, L. Gabler, J. Gojo, G. Timelthaler, J. Senkiv, W. Jäger, C. R. Kowol, P. Heffeter and W. Berger, Lipid droplet–mediated scavenging as novel intrinsic and adaptive resistance factor against the multikinase inhibitor ponatinib, Int. J. Cancer, 2020, 1680–1693 CrossRef CAS PubMed.
- Y. Chia and M. Tay, An insight into fluorescent transition metal complexes, Dalton Trans., 2014, 43, 13159–13168 RSC.
- N. Graf and S. J. Lippard, Redox activation of metal-based prodrugs as a strategy for drug delivery, Adv. Drug Delivery Rev., 2012, 64, 993–1004 CrossRef CAS PubMed.
- A. K. Renfrew, E. S. O'Neill, T. W. Hambley and E. J. New, Harnessing the properties of cobalt coordination complexes for biological application, Coord. Chem. Rev., 2018, 375, 221–233 CrossRef CAS.
- D. C. Ware, H. R. Palmer, F. B. Pruijn, R. F. Anderson, P. J. Brothers, W. A. Denny and W. R. Wilson, Bis (dialkyl) dithiocarbamato cobalt(III) complexes of bidentate nitrogen mustards: synthesis, reduction chemistry and biological evaluation as hypoxia-selective cytotoxins, Anticancer Drug Des., 1998, 13, 81–103 CAS.
- P. R. Craig, P. J. Brothers, G. R. Clark, W. R. Wilson, W. A. Denny and D. C. Ware, Anionic carbonato and oxalato cobalt(III) nitrogen mustard complexes, Dalton Trans., 2004, 611–618 RSC.
- T. W. Failes, C. Cullinane, C. I. Diakos, N. Yamamoto, J. G. Lyons and T. W. Hambley, Studies of a cobalt(III) complex of the MMP inhibitor marimastat: a potential hypoxia–activated prodrug, Chem. – Eur. J., 2007, 13, 2974–2982 CrossRef CAS PubMed.
- I. C. A. de Souza, S. de Souza Santana, J. G. Gómez, G. P. Guedes, J. Madureira, S. M. de Ornelas Quintal and M. Lanznaster, Investigation of cobalt(III)–phenylalanine complexes for hypoxia-activated drug delivery, Dalton Trans., 2020, 16425–16439 RSC.
- R. Anderson, W. Denny, D. Ware and W. Wilson, Pulse radiolysis studies on the hypoxia-selective toxicity of a colbalt-mustard complex, Br. J. Cancer, 1996, 27, S48 CAS.
- B. B. Lozzio and C. B. Lozzio, Properties and usefulness of the original K-562 human myelogenous leukemia cell line, Leuk. Res., 1979, 3, 363–370 CrossRef CAS.
- M. Miyake, M. Ishii, N. Koyama, K. Kawashima, T. Kodama, S. Anai, K. Fujimoto, Y. Hirao and K. Sugano, 1-tert-butyl-3-[6-(3, 5-dimethoxy-phenyl)-2-(4-diethylamino-butylamino)-pyrido [2, 3-d] pyrimidin-7-yl]-urea (PD173074), a selective tyrosine kinase inhibitor of fibroblast growth factor receptor-3 (FGFR3), inhibits cell proliferation of bladder cancer carrying the FGFR3 gene mutation along with up-regulation of p27/Kip1 and G1/G0 arrest, J. Pharmacol. Exp. Ther., 2010, 332, 795–802 CrossRef CAS PubMed.
- PerkinElmer, ChemBioDraw Ultra 13.0 Search PubMed.
- C. Bresson, C. Darolles, A. Carmona, C. Gautier, N. Sage, S. Roudeau, R. Ortega, E. Ansoborlo and V. Malard, Cobalt chloride speciation, mechanisms of cytotoxicity on human pulmonary cells, and synergistic toxicity with zinc, Metallomics, 2013, 5, 133–143 CrossRef CAS PubMed.
- J. N. Lee, J. Park, S.-G. Kim, M. S. Kim, J.-Y. Lim and S.-K. Choe, 3-Aminotriazole protects against cobalt(II) chloride-induced cytotoxicity by inhibiting reactive oxygen species formation and preventing mitochondrial damage in HepG2 cells, Mol. Cell. Toxicol., 2017, 13, 125–132 CrossRef CAS.
- Z.-J. Dai, J. Gao, X.-B. Ma, K. Yan, X.-X. Liu, H.-F. Kang, Z.-Z. Ji, H.-T. Guan and X.-J. Wang, Up-regulation of hypoxia inducible factor-1α by cobalt chloride correlates with proliferation and apoptosis in PC-2 cells, J. Exp. Clin. Cancer Res., 2012, 31, 28 CrossRef CAS PubMed.
- Y. Yuan, G. Hilliard, T. Ferguson and D. E. Millhorn, Cobalt inhibits the interaction between hypoxia-inducible factor-α and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-α, J. Biol. Chem., 2003, 278, 15911–15916 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 17 CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Erratum: Development and testing of a general amber force field (Journal of Computational Chemistry (2004) 25 (1157)), J. Comput. Chem., 2005, 26, 1157–1174 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The protein data bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera—a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- R. Harris, A. J. Olson and D. S. Goodsell, Automated prediction of ligand–binding sites in proteins, Proteins, 2008, 70, 1506–1517 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CAS.
- I. Kufareva and R. Abagyan, in Homology Modeling, Springer, 2011, pp. 231–257 Search PubMed.
- W. C. Barrette, H. Johnson and D. T. Sawyer, Voltammetric evaluation of the effective acidities (pKa’) for Broensted acids in aprotic solvents, Anal. Chem., 1984, 56, 1890–1898 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qi00211b |
| This journal is © the Partner Organisations 2021 |