
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultimodal hybrid 2D networks via the thiol-epoxide reaction on 1T/2H MoS2 polytypes†
Giulia
Tuci
*a,
Andrea
Rossin
 a,
Cuong
Pham-Huu
*b,
Dario
Mosconi
c,
Lapo
Luconi
a,
Stefano
Agnoli
c,
Gaetano
Granozzi
c and
Giuliano
Giambastiani
*ab
a,
Cuong
Pham-Huu
*b,
Dario
Mosconi
c,
Lapo
Luconi
a,
Stefano
Agnoli
c,
Gaetano
Granozzi
c and
Giuliano
Giambastiani
*ab
aInstitute of Chemistry of OrganoMetallic Compounds, ICCOM-CNR and Consorzio INSTM, Via Madonna del Piano, 10 – 50019, Sesto F.no, Florence, Italy. E-mail: giulia.tuci@iccom.cnr.it
bInstitute of Chemistry and Processes for Energy, Environment and Health (ICPEES), ECPM, UMR 7515 CNRS-University of Strasbourg, 25 rue Becquerel, 67087 Strasbourg Cedex 02, France. E-mail: cuong.pham-huu@unistra.fr; giambastiani@unistra.fr
cDepartment of Chemical Science of the University of Padua and INSTM Unit, Via Marzolo 1, 35131 Padova, Italy
First published on 9th March 2021
Abstract
Searching for new protocols aimed at the surface functionalization of TMDCs is the password for filling the gap between reactivity and versatility of 2D carbon-based networks and their inorganic counterparts. This contribution describes a straightforward but effective approach to the chemical decoration of chemically exfoliated MoS2 nanosheets with β-hydroxyl organic pendant arms. The adopted strategy is based on the thiol-epoxide “click” reaction and generates functional hybrids with a loading of dangling organic groups directly proportional to the epoxide electrophilic character. Most importantly, this grafted methodology occurs for the first time on both phases (1T and 2H) of the model chalcogenide and provides functional samples suitable for an additional functionalization step-forward (post-derivatization).
Introduction
The interest of the scientific community in organic or inorganic two-dimensional (2D) materials is growing exponentially year-by-year as their distinctive chemico-physical, optical and mechanical properties are often functional to address several technological issues.1–4 Layered 2D transition-metal dichalcogenides (TMDCs) have already shown their potentiality in various technological areas as energy storage materials, components for nano–optoelectronic, sensing and environmental protection devices up to their application in catalysis.5–11 Molybdenum disulfide (MoS2) is among the most studied material from the TMDCs series. It is available in several polymorphic structures classified on the basis of metal coordination, the most common ones being semiconducting 2H (trigonal prismatic) and metallic 1T (octahedral) phases.12,13 The former represents the natural and thermodynamically stable MoS2 polytype.14,15 It can be chemically exfoliated by metal ion intercalation (using mainly Li+ but also Na+ or K+ ions), thus inducing a material structural reorientation towards the metallic 1T polymorph.16,17Anyhow, a full exploitation of the unique features of these inorganic bidimensional networks implies a step forward in the direction of bridging the gap with their more celebrated and studied organic counterpart, graphene. In this regard, the surface tailoring of electronic and chemico-physical properties of MoS2, including material processability, can be conveniently achieved through chemical functionalization.18 Recent findings from the literature have unveiled how the covalent surface functionalization of TMDCs boosts the (photo)electrocatalytic properties of these materials in key processes at the heart of renewable energy technology such as the hydrogen evolution reaction (HER).19–21 Some of us have reported on the synthesis of an efficient hybrid TMDC sample with improved HER performance by anchoring newly designed porphyrins bearing different functional groups. We showed how mildly acidic hydroxyl groups in dangling organic fragments facilitate a local proton release, hence fostering hydrogen production.22 However, any step forward in the direction of tailored hybrid functional materials requires the development of effective and versatile functionalization protocols to provide multimodality to the nanomaterial surface.
In recent years, few protocols for the chemical grafting of organic species to TMDCs have been developed.23–26 The first example of MoS2 surface decoration was reported by Dravid and co-workers using organic thiols.27 These authors and related successive studies28–32 postulated a “ligand conjugation” of exogenous thiol molecules at the S-vacancies of coordinatively unsaturated Mo atoms. Later, Mc Donald and co-workers33 unveiled the real MoS2–thiols interaction nature and demonstrated that exogenous thiols were not covalently grafted to 2D nanosheets, but they rather underwent a formal oxidation step to give the corresponding disulfides that were in turn simply physisorbed at the material surface.
The first real covalent grafting of organic fragments to TMDC surface has been described by Chhowalla and collaborators in a seminal contribution that appeared in 2015.34 Their protocol relied on the generation of negatively charged 1T-MoS2 flakes (prepared by chemical exfoliation of a bulk 2H-MoS2 sample with nBuLi) to be reacted with organoiodide species, covalently binding organic dangling arms to the sulfur atoms of the inorganic network. In the same year, Backes and co-workers developed a similar approach for the covalent grafting of chemically-exfoliated 1T-MoS2 using reactive diazonium salts as electrophile species.16 More recently, Perez et al. have described a different strategy for the mild decoration of 2H-MoS2 obtained by liquid-phase exfoliation (LPE) in 2-propanol/water with an ultrasonic probe.35,36 These authors exploited the soft nucleophilicity of sulfur atoms for covalent bond formation with maleimide derivatives via Michael addition. The reaction occurred at room temperature in the presence of a base while preserving the 2H semiconducting polytype of the starting material. Despite these seminal outcomes, much work remains to be done for covering the existing gap with the functionalization of organic 1D and 2D networks and hence for boosting the applications of TMDCs. Moreover, the development of new functionalization strategies suitable for the chemical modification of both 1T and 2H phases still remains a challenging and highly desirable objective.
This paper describes an effective and straightforward covalent functionalization strategy for the TMDC nanosheets decoration under mild reaction conditions. Taking the advantage of the principle of the effective thiol-epoxide “click” protocol,37 we have reacted the negatively charged 1T-MoS2 with a series of epoxide derivatives generating β-hydroxyl nanohybrids upon anti-Markovnikov reactant's ring opening. The successful MoS2 nanosheet functionalization has been demonstrated by infrared (FT-IR) and X-ray photoelectron (XPS) spectroscopy, while elemental analysis (EA) crossed with TGA-MS provided a quantitative estimation of the grafted groups loading. The facile MoS2 surface engineering with different organic moieties along with the control on the dangling groups loading is a highly powerful tool for the fine-tuning of MoS2 chemico-physical properties. It is noteworthy that the proposed covalent grafting approach takes place on both metallic (1T) and semiconducting (2H) MoS2 phases. Finally, the hydroxyl moieties derived from the epoxides ring-opening have undergone further post-functionalization, thus conferring multimodality to MoS2 platforms and widening their applicability range.
Results and discussion
Chemical functionalization of CE-MoS2 by anti-Markovnikov epoxides ring-opening
Few-layer 1T-MoS2 nanosheets are prepared by Li-ion intercalation according to a literature procedure recently reported by some of us.38 In brief, a solid mixture of bulk 2H-MoS2 and LiBH4 was treated at 330 °C for 4 days and then transferred to degassed Milli-Q water. After sonication and careful purification, a stable and homogeneous aqueous dispersion of chemically-exfoliated MoS2 (CE-MoS2 ∼ 0.2 mg mL−1) was obtained. According to the literature, XPS analysis of the exfoliated material (Fig. S1, ESI†) revealed a predominant 1T phase38 (72% vs. 16% of 2H-MoS2) with only minor amounts of partially oxidized MoSxOy species (7%) and MoOx suboxides (5%).Our approach to chemical decoration of CE-MoS2 relies on the ability of the as-obtained negatively charged chalcogenide to react as a nucleophile towards epoxide derivatives, leading to their regioselective ring-opening via the so-called thiol-epoxide “click” reaction.37,39 The latter is a one-step, selective and 100% atom economy process that takes place with high yield under mild operative conditions.40 All these aspects make the protocol highly attractive for its exploitation as a new and efficient covalent functionalization method for the TMDC surface decoration with different chemical functionalities. Moreover, the hydroxyl moieties generated upon epoxide ring-opening are reactive sites that may undergo further post-functionalization, thus improving the versatility and widening the applicability range of this emerging class of 2D materials (vide infra).
A series of differently substituted epoxide derivatives (1–4) have been selected as model reactants and underwent nucleophilic attack from MoS2 nanosheets (Scheme 1). In a typical procedure, 200 mL of CE-MoS2 aqueous suspension reacted with a proper amount of epoxide (10 eq. respect to S) at 60 °C (see Experimental section for details).
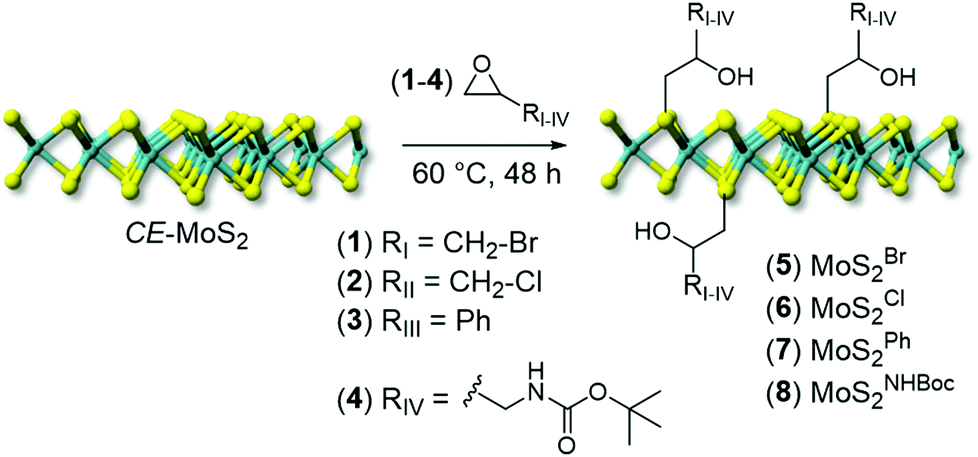 | ||
| Scheme 1 Chemical functionalization of CE-MoS2 with epoxide derivatives (1–4) to give decorated MoS2 nanohybrids (5–8). | ||
After the materials work-up, first and qualitative evidence of the occurred epoxide ring-opening is provided by FT-IR spectra of all functionalized samples (5–8, Fig. 1). In particular, distinctive signals ascribed to alkane C–H stretching (νC–H) and an intense broad band unambiguously assigned to the stretching of hydroxyl groups (νO–H) appear for all decorated materials at ≈2900 and ≈3400 cm−1, respectively.41 Noteworthy, 5 shows a remarkably less intense IR signal associated with –OH groups with respect to all other grafted materials (5vs.6–8, Fig. 1). Such evidence prompted us to invoke the occurrence of an alternative nucleophilic attack on epibromohydrin (1). Indeed, the presence of bromine as a good leaving group opens two different scenarios competitive with the plain anti-Markovnikov epoxide ring-opening: the direct S-nucleophilic displacement at the C–Br bond or a Payne rearrangement just after the initial epoxide ring-opening with the subsequent intramolecular ring-closure and oxirane regeneration.42–44 In both cases, the final product would give rise to a CE-MoS2 decorated sample bearing integral epoxide moieties instead of free-hydroxyl groups. Although these authors are aware of the qualitative character of the IR information, particularly for poorly distinctive signals like those arising from this region of the IR spectrum, the lower intensity of the IR band associated with –OH moieties for this sample (5) can be coherently rationalized as discussed above. Since the pioneering work by Chowalla and co-workers,34 alkyl iodide derivatives are known to undergo nucleophilic substitution (SN) in the TMDC surface functionalization. On the other hand, such reactivity was considered less feasible in the presence of alkyl bromides. As an example, McDonald and co-workers reported on the covalent grafting of 5-bromomethyl-2,2′-bipyridine to CE-1T-MoS2 in the presence of a catalytic amount of NaI in the reaction medium; the addition of NaI was necessary to allow the halogen scrambling on the organic reactant, with concomitant occurrence of the nucleophilic substitution reaction.45 However, more recently it has been demonstrated that alkyl bromide derivatives can also be exploited for the direct covalent decoration of CE-MoS2via the SN reaction,15,46 supporting our hypothesis of competing processes at work with epibromohydrin (1) as a substrate.
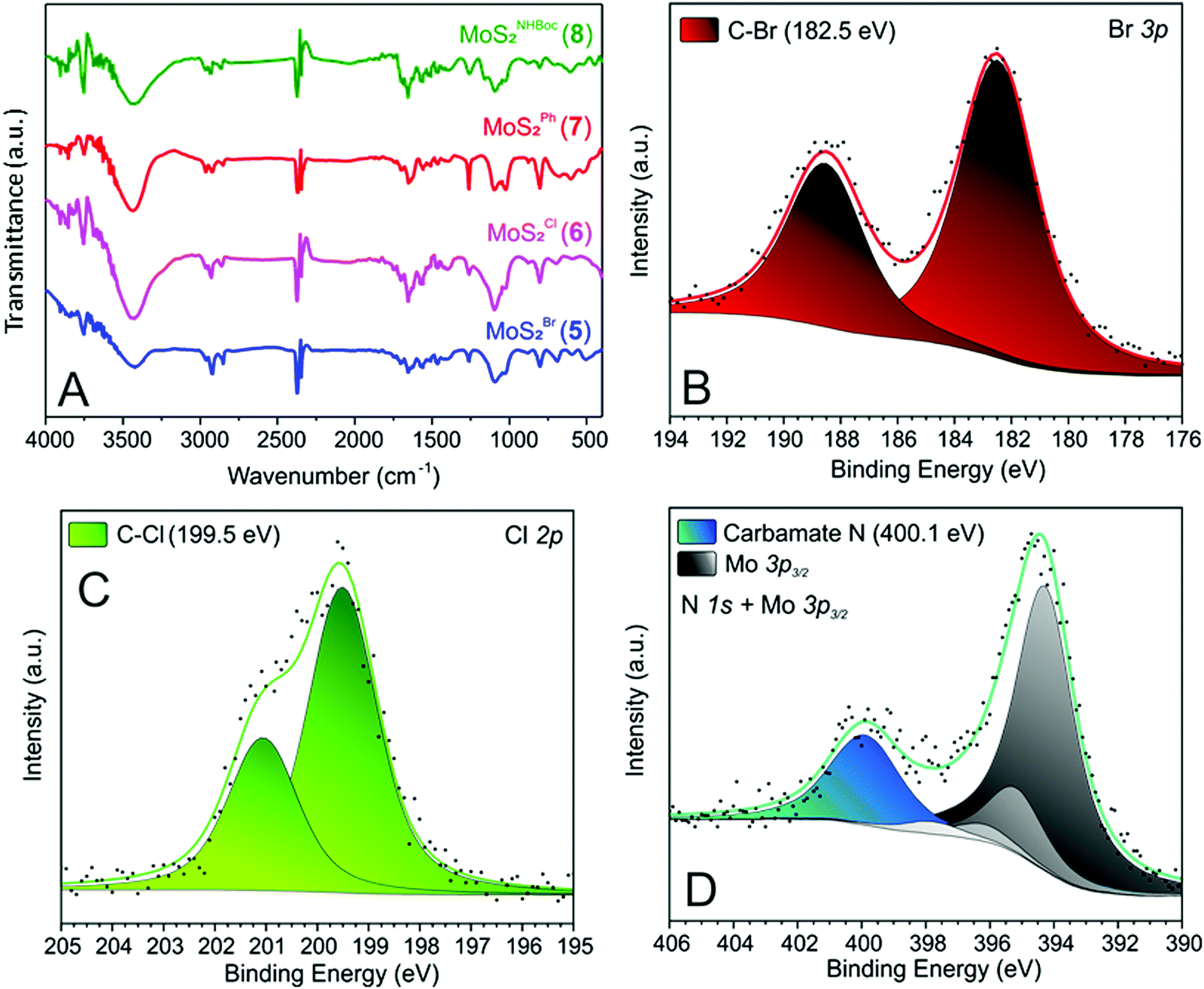 | ||
| Fig. 1 (A) IR spectra of MoS2 functionalized materials (5–8). (B) High-resolution Br 3p XPS core level region of 5. (C) High-resolution Cl 2p XPS core level region of 6. (D) High-resolution N 1s XPS core level region of 8. | ||
The signal around 3400 cm−1 in 5 (even if less intense with respect to the other hybrid materials 6–8, Fig. 1) suggests indeed that the epoxide ring-opening (partially) occurs also with 1. The presence of bromine in 5 (as evidenced by TGA-MS and XPS analyses – vide infra) provides additional evidence for the existence of two alternative reaction paths at work with 1 as a substrate.
Further proof of evidence of the occurred CE-MoS2 functionalization comes from XPS analyses. All MoS2 nanohybrids 5–8 show distinctive signals associated with heteroatoms at the respective grafted moieties. Binding energy (BE) signals at 182.5 and 199.5 eV for Br 3p and Cl 2p core regions of 5 and 6, respectively (Fig. 1B and C) account for the occurred nanomaterial grafting and epoxide ring-opening on oxiranes 1 and 2. Similarly, a distinctive N 1s signal ascribed to the carbamate moiety47 (400.1 eV, Fig. 1D) is observed in the XPS spectrum of 8.
As far as distinctive XPS signals of 7 are concerned, the high-resolution C 1s spectrum (Fig. S2C, ESI†) is in accordance with the styrene oxide (3) ring-opening as outlined in Scheme 1. Indeed, two components at 284.5 and 286.9 eV with a roughly 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 intensity ratio and ascribed to phenyl and C–OH/C–S moieties, respectively, are clearly observed in the C 1s photoemission spectrum of 7.
:1 intensity ratio and ascribed to phenyl and C–OH/C–S moieties, respectively, are clearly observed in the C 1s photoemission spectrum of 7.
Finally, the shift towards higher BEs of the S 2p XPS signal in all grafted 2D nanomaterials (indicating an electronic transfer from the inorganic MoS2 nanosheets to its covalently grafted organic arms) is an indirect evidence of the occurred surface MoS2 nanosheets decoration. Noteworthy, both 1T and 2H phases underwent a significant shift (1T: from 160.9 eV in the pristine CE-MoS2 to 161.1–161.5 eV in 5–8; 2H: from 161.9 eV in the pristine CE-MoS2 to 162.2–162.5 eV in 5–8, Fig. S1 vs. S2–S5, ESI†). Such evidence suggests that both metallic and semiconducting MoS2 polymorphs equally underwent covalent grafting. This aspect is of primary importance because the 2H phase cannot be directly functionalized by strong electrophiles48 such as alkyl halides34 or aryldiazonium salts16,38 (if not in the presence of pre-formed defects as nucleation sites).49 To the best of our knowledge, the only example of a direct 2H MoS2 functionalization reported so far, deals with the recent contribution by Perez and co-workers and it is limited to TMDCs decoration with maleimide derivatives only.35 Thereby, the thiol-epoxide functionalization offers a valid and practical alternative to existing methods for surface grafting of both metallic and semiconducting MoS2 phases using a wide variety of functional groups. A careful analysis of the S 2p photoemission spectra of 5–8 (Fig. S1 vs. S2–S5, ESI†) has finally unveiled that 5 and 6 present more pronounced signal shifts (∼0.5–0.6 eV vs. plain MoS2) towards higher BEs compared to those measured on the two other nanohybrids (7–8; ∼0.2–0.4 eV vs. plain MoS2). This trend mirrors the electronic properties of the material decorating groups: the higher the electron-withdrawing character of the pendant arms, the higher the electronic transfer from MoS2 nanosheets to its grafted species and thus the higher the BE values of the corresponding S 2p XPS signals. On the other hand, Mo 3d XPS photoemission spectra of all functionalized derivatives (5–8) did not change significantly with respect to the starting CE-MoS2 (Fig. S1 vs. S2–S5, ESI†). Such an effect indicates that electronic material properties are not altered (also in terms of 1T/2H phases ratio) by the thiol-epoxide “click” process. Overall, XPS analyses demonstrate that chemical functionalization occurs at negatively charged sulfur atoms, in line with their ability to react as nucleophiles towards epoxide derivatives.
On a more quantitative ground, thermogravimetric (TGA-MS, Fig. 2) and elemental analyses (Table 1) have been accomplished on samples 5–8 to better determine the functionalization loading of each nanohybrid. Weight losses measured on 5–8 have been compared with the TGA profile of pristine CE-MoS2 to provide direct and semi-quantitative evidence of the organic groups loading grafted on the inorganic 2D network. At the same time, the MS analysis of volatiles produced throughout the material's thermal decomposition has been used to confirm the identity of the chemically grafted functionalities.
 | ||
| Fig. 2 Thermogravimetric profiles of (A) MoSBr2 (5), (B) MoSCl2 (6), (C) MoSPh2 (7) and (D) MoSNHBoc2 (8) along with (whenever applicable) MS analysis of volatiles. Thermal program: 40–600 °C, 5 °C min−1, N2 atmosphere (50 mL min−1). | ||
| Samples | Elemental analysisa | S/RI–IV molar ratio | RI–IV-loading (mmol g−1) | |||
|---|---|---|---|---|---|---|
| N wt% | C wt% | S wt% | From EAb | From TGAc | ||
| a Calculated as average values over three independent runs. b Loading calculated from C wt%. c Calculated from TG weight losses (%) in the 60–300 °C temperature range. d Not determined because of secondary reaction paths at work (vide infra). e Loading calculated from N wt%. | ||||||
| CE-MoS2 | — | 1.63 | 23.19 | — | — | — |
| MoSBr2 (5) | — | 8.08 | 22.61 | 3.9 | 1.79 | —d |
| MoSCl2 (6) | — | 5.91 | 26.23 | 6.9 | 1.19 | 1.07 |
| MoSPh2 (7) | — | 8.99 | 23.41 | 9.5 | 0.77 | 0.49 |
| MoSNHBoc2 (8) | 1.31 | 9.57 | 27.39 | 10.3 | 0.83 (0.93)e | 0.57 |
As Fig. 2 shows, samples 5 and 7 display distinctive weight losses in the 60–300 °C range, whose net decrease reduced by that of the plain CE-MoS2 in the same temperature range (10.9 wt% for 5 and 5.9 wt% for 7) is roughly attributed to the complete thermal decomposition of organic moieties grafted to the material surface. MS analyses of low-molecular-weight volatiles produced throughout nanohybrids thermal decomposition additionally support the nature of the MoS2 grafted species.
While distinctive mass peaks attributed to HBr (m/z = 80 and 82 [M+] coming from 79Br and 81Br isotopes, respectively) together with less intense components due to CH2–Br species (m/z = 93 and 95 [M+]) were observed for 5 (Fig. 2A), mass signals ascribable to phenyl C6H5- (m/z = 77 [M+], m/z = 78 [M+ + 1]) and styrene Ph–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 moieties (m/z = 104 [M+], m/z = 105 [M+ + 1] and m/z = 103 [M+ − 1]) were detected for 7 (Fig. 2C). Similarly, a characteristic weight loss (12.9 wt%) was registered for sample 8 in the same 60–300 °C temperature range (∼5–52 min). The mass analysis of volatiles highlights the generation of isobutene (m/z = 56 [M+], m/z = 55 [M+ − 1] and m/z = 57 [M+ + 1]), typically ascribed to the thermal decomposition and rearrangement of tert-butoxyl groups of the N-Boc protected carbamate species (Fig. 2D).38,47 Finally, 6 shows a clear-cut weight loss of 13.0 wt% in the 60–300 °C temperature range (Fig. 2B) that accounts for 1.1 mmol g−1 of β-hydroxyl chloride-grafted moieties. In this case, the low molecular weight of volatiles evolved throughout the material thermal decomposition did not allow the detection of any characteristic mass signals.
CH2 moieties (m/z = 104 [M+], m/z = 105 [M+ + 1] and m/z = 103 [M+ − 1]) were detected for 7 (Fig. 2C). Similarly, a characteristic weight loss (12.9 wt%) was registered for sample 8 in the same 60–300 °C temperature range (∼5–52 min). The mass analysis of volatiles highlights the generation of isobutene (m/z = 56 [M+], m/z = 55 [M+ − 1] and m/z = 57 [M+ + 1]), typically ascribed to the thermal decomposition and rearrangement of tert-butoxyl groups of the N-Boc protected carbamate species (Fig. 2D).38,47 Finally, 6 shows a clear-cut weight loss of 13.0 wt% in the 60–300 °C temperature range (Fig. 2B) that accounts for 1.1 mmol g−1 of β-hydroxyl chloride-grafted moieties. In this case, the low molecular weight of volatiles evolved throughout the material thermal decomposition did not allow the detection of any characteristic mass signals.
Noteworthy, we found an excellent match between the functionalization degree extrapolated from TGA analyses and the organic fraction loading calculated from EA (Table 1).
The loading of grafted groups for nanohybrids 7 and 8 is quite similar with roughly one (1) decorating moiety every 9.5 and 10.3 sulfur atoms, respectively (as calculated from EA analyses, Table 1). Such a percentage slightly increases on 6 where a S/RII ratio close to 6.9 is calculated. This trend is in line with the higher electrophilic character of epichlorohydrin (2) that fosters the thiol-epoxide “click” reaction with respect to epoxides 3 and 4, thus enhancing the MoS2 surface coverage.19 On the other hand, the markedly higher functionalization degree measured on 5 (S/RII = 3.9) cannot simply be ascribed to the electron-withdrawing nature of bromine but rather to the occurrence of a competitive bromide displacement via direct SN (or Payne rearrangement – see above). In addition, the weight loss measured by TGA analyses on 5 (13.9%, Fig. 2A) is markedly lower with respect to what we expected for a pure thiol-epoxide ring-opening functionalization (≈28%). Such a discrepancy additionally supports the hypothesis for a competing SN process at work in the case of epoxide 1. Crossing EA outcomes with thermogravimetric weight loss measured for the nanohybrid 5, it can be inferred that such a secondary reaction path largely prevails on the thiol-epoxide “click” reaction (estimated SN: thiol-epoxide “click” ratio ∼75:25). Worthy of note, this ratio well-matches with that of 1T/2H phases of the starting CE-MoS2 (72/16, see Fig. S1, ESI†). The higher chemical inertness of the semiconducting 2H phase towards direct decoration in the presence of strong electrophiles (i.e. alkyl halides)16,34,38 has prompted us to postulate that the minor 2H MoS2 component is the only phase involved in the thiol-epoxide “click” reaction with epibromohydrin (1), while the major metallic 1T-MoS2 phase undergoes classical SN reaction with bromide ion elimination.
Post-synthetic modification of nanohybrids 7 and 8
One key challenge in the chemical functionalization of 2D organic/inorganic platforms is represented by the possibility to impart multimodality to these layered materials. The thiol-epoxide protocol is particularly relevant because the oxirane ring-opening provides at least one highly versatile hydroxyl group suitable to undergo tailored conversion/derivatization. On this ground, the nanohybrid 8 was treated with p-toluenesulfonyl chloride (CH3C6H4SO2Cl) and pyridine (C5H5N) as an acid scavenger in refluxing dichloroethane (DCE, C2H4Cl2) for the hydroxyl moiety conversion into an optimal leaving-group. All attempts to carry out the reaction at either room temperature or refluxing temperature of dichloromethane (CH2Cl2, bp 40 °C) failed and the nanohybrid 8 was entirely recovered with no alterations. As expected for the intermediate 9, the transiently formed tosyl-ester undergoes spontaneous intramolecular cyclization, thus leading to the oxazolidinones-decorated nanohybrid 10 (MoSoxaz2, Scheme 2).50,51 Such reactivity of the organyl pendant fragment is favored already at 0 °C in the case of N-alkyl substituted carbamates, while it occurs upon heating at about 60 °C on NH derivatives. After a careful material workup, sample 10 was analyzed first using TGA-MS. As Fig. 3A shows, MoSoxaz2 (10) shows a lower weight loss (10.1 wt%) if compared with the starting material (8) in the same temperature range. Moreover, the weight loss measured for the nanohybrid 10 is in excellent accord with that expected for a complete conversion of tosilated β-hydroxyl moieties in 8 (∼0.7 mmol g−1, Table 1) into the corresponding oxazolidinone species (10.0 wt%).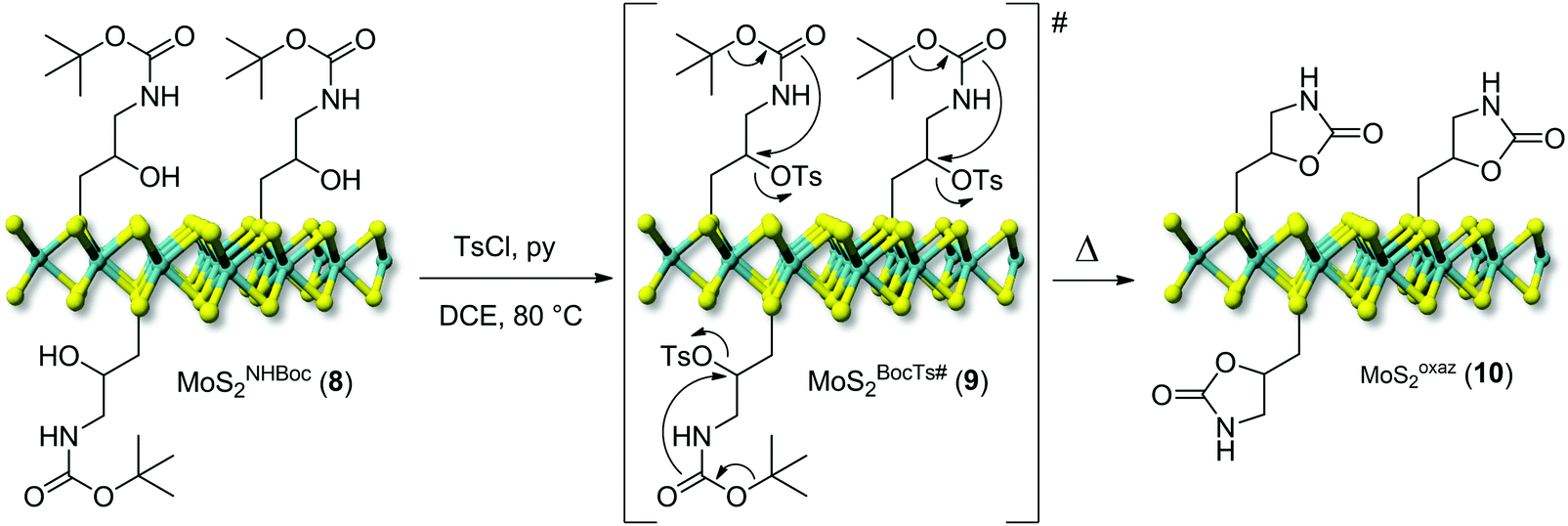 | ||
| Scheme 2 Post-derivatization of the nanohybrid MoSNHBoc2 (8) into the oxazolidinones-decorated MoSoxaz2 (10). | ||
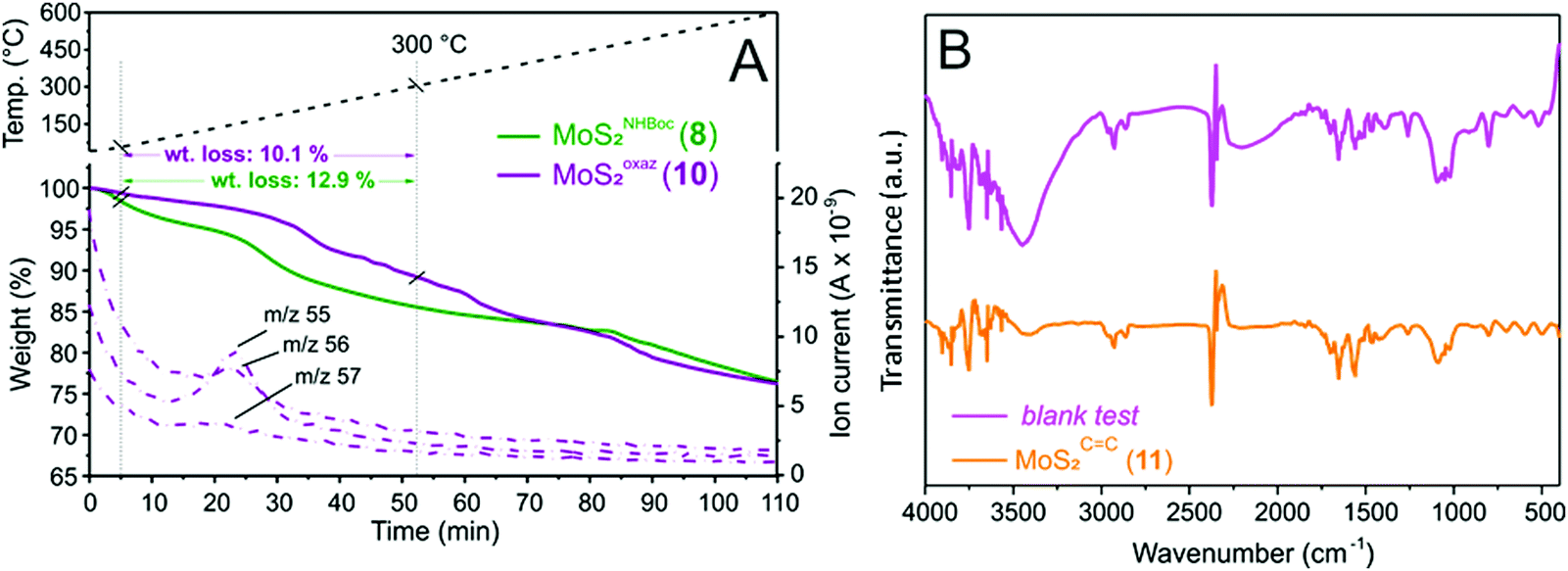 | ||
| Fig. 3 (A) Thermogravimetric profiles of MoSoxaz2 (10) along with mass analysis of volatile species. Thermal program: 40–600 °C, 5 °C min−1, N2 atmosphere (50 mL min−1). TG profile of MoSNHBoc2 (8) is reported for the sake of comparison. (B) IR spectrum of MoSCC2 (11) along with the relative blank test. | ||
Further proof of evidence of the occurred intramolecular cyclization is given from MS analysis of low-molecular-weight volatiles resulting from the thermal decomposition of 10 (Fig. 3A). MS signals ascribable to the oxazolidinone thermal rearrangement into 2-methyl-aziridine species (m/z = 56 [M+], m/z = 55 [M+ − 1])52 along with complete suppression of the mass peaks associated with the thermal rearrangement of tert-butoxyl groups in the Boc units of the starting material 8 (Fig. 2D), are useful additional tiles of a complex puzzle that validate the post-synthetic treatment of 8 as well as the supposed nature of the decorating fragments in 10.
Elemental analysis on MoSoxaz2 (10) has finally confirmed the occurred post-derivatization path (Table S1, ESI†). Indeed, a coherent reduction of C wt% was recorded from the nanohybrid 8 to its post-derivative 10, while the estimated functionalization degree remained almost unchanged (∼0.83 mmol g−1; 1 group every 9.7 sulfur atoms).
IR spectra of hybrids 8 and 10 at comparison show a marked decrease of the –OH groups stretching (νO–H; ≈3400 cm−1) on the latter (Fig. S6, ESI†vs.Fig. 1), while the N 1s photoemission spectrum of 10 presents a unique component centered at 400.0 eV (Fig. S7, ESI†) ascribed to a carbamate fragment. Both results are in line with the expected generation of oxazolidinone species on the 2D nanomaterial surface. At the same time, no appreciable variation of Mo 3d or S 2p photoemission lines in two nanohybrids at comparison (8vs.10) is observed (Fig. S5 vs. S7, ESI†), which confirms that the post-synthetic modification on 8 occurs without any appreciable alteration of the MoS2 pristine structure.
Finally, the reactivity of hydroxyl groups in the nanohybrid 7 was tested under similar post-derivatization conditions, using p-toluenesulfonic acid and pyridine as an acid scavenger. In this case, the treatment resulted in the formal dehydration of the tosylated secondary hydroxyl group with the subsequent generation of a conjugated CC bond (MoSCC211, Scheme 3).53,54
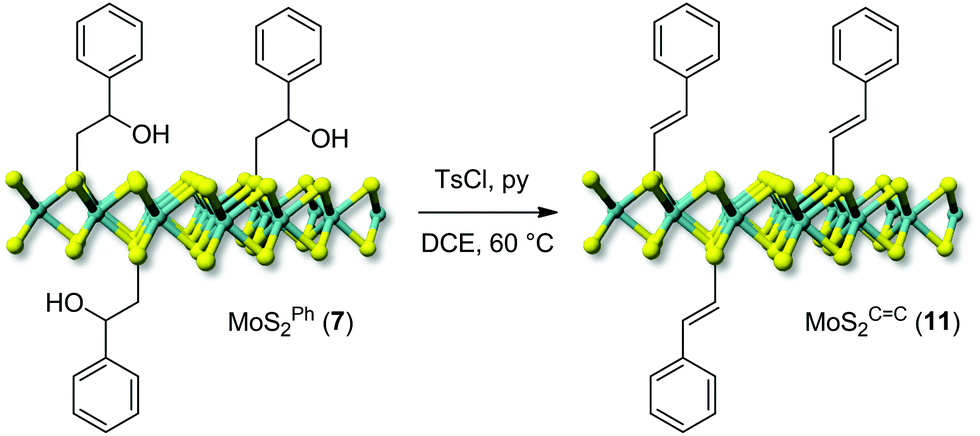 | ||
| Scheme 3 Post-derivatization of the nanohybrid MoSPh2 (7) into the styrene-decorated MoSCC2 (11). | ||
The first evidence of the occurred alcohol dehydration is given by IR analysis of derivative 11. As shown in Fig. 3B, the –OH stretching (νO–H ∼ 3400 cm−1) is markedly reduced on 11 compared to the starting precursor 7. To get further evidence of the occurred secondary alcohol dehydration, a blank trial has been carried out using 7 as a substrate to be reacted under identical conditions to those outlined on Scheme 3 except for the use of the p-toluenesulfonic acid. Noteworthy, the IR spectrum of the blank sample was largely superimposable on that of the starting hybrid 7 with no appreciable suppression of the stretching signal attributed to the secondary alcohol (Fig. 3B). All these data taken together strongly supported the hypothesis of an alcohol elimination path facilitated by the transient generation of the sulfonyl ester intermediate.
Thermogravimetric analysis conducted on MoSCC2 (11) was also consistent with the postulated alcohol dehydration path. Indeed, the nanohybrid 11 showed only a slightly lower weight loss with respect to the starting material 7 (8.4 wt% vs. 8.9 wt%) while keeping almost unchanged its TG profile all over the whole scanned temperature range (Fig. S8, ESI†). Similarly, elemental analysis of 11 (Table S1, ESI†) did not show any appreciable alteration with respect to 7, thus strengthening the supposed dehydration path. As a matter of fact, the functionalization degree calculated for hybrid 11 was in excellent accord with the loading measured on its starting material 7 (Table S1, ESI†vs.Table 1).
Finally, XPS analysis was used to corroborate the expected formal alcohol dehydration mechanism at work on 7. In particular, the C 1s core-level regions of the two nanohybrids in comparison (7vs.11; Fig. S2 vs. S9, ESI†) have unveiled a coherent increment in 11 of the component at 284.4 eV ascribed to C sp2 moieties along with a corresponding decrease of the peak at 286.9 eV attributed to C–OH groups.
Experimental
General considerations
All manipulations were carried out under dry nitrogen (≥99.999%; Rivoira) atmosphere using standard Schlenk-type techniques. Aqueous suspension of CE-MoS2 (∼0.2 mg mL−1) was obtained by following literature procedures.38,55,56 Unless otherwise stated, all chemicals were provided by commercial suppliers and used as received. Sample sonications were conducted on an Elma S15 Elmasonic sonicator (37 kHz) in a water/ice bath. Thermogravimetric analysis (TGA-MS) was run under N2 atmosphere (50 mL min−1) on an EXSTAR Seiko 6200 analyzer coupled with a ThermoStarTM GSD 301T for mass analysis of volatile species. All samples (from 10 to 40 mg) were stabilized to constant weight at 40 °C (from 30 min to 90 min) before running each measurement. Elemental analysis was carried out on a Thermo FlashEA 1112 series CHNS-O elemental analyzer with an accepted tolerance of ±2% on each element. N, C and S wt% were determined as mean values over three independent analyses. FT-IR spectroscopy was performed on a PerkinElmer Spectrum BX FT-IR spectrophotometer in the 400–4000 cm−1 range with a resolution of 1 cm−1. Samples for analyses were prepared by mixing spectroscopic-grade KBr with functionalized MoS2 hybrids. X-Ray photoemission spectroscopy (XPS) measurements were performed under UHV conditions (<10−8 mbar) at room temperature using a nonmonochromated Al Kα source (hν = 1486.7 eV) and an electron analyzer pass energy of 20 eV. The binding energy (BE) scale was calibrated using Au 4f7/2 core levels (BE = 83.8 eV). XPS multipeak analysis (KolXPD software)57 was performed using Voigt functions, keeping constant in all the peaks the full width at half maximum (FWHM) and the Gaussian–Lorentzian ratio.General procedure for the synthesis of MoS2 functionalized samples 5–8
200 mL of an aqueous suspension of CE-MoS2 (∼0.2 mg mL−1) is treated with a proper amount of epoxide derivative (5.5 mmol, 10 eq. vs. S) and sonicated for 20 minutes. Afterward, the suspension was stirred at 60 °C for 48 h, cooled to room temperature and centrifuged in order to recover the solid residue. The latter was washed/sonicated several times (3 × 30 mL of EtOH and 3 × 30 mL of AcOEt, 15 min sonication each) and recovered by centrifugation before being dried to constant weight and stored at room temperature.Post-derivatization of MoSPh2 (7) and MoSNHBoc2 (8)
20 mg of functionalized MoS2 [either MoSPh2 (7) or MoSNHBoc2 (8)] were suspended in 20 mL of dry and degassed dichloroethane (DCE) and sonicated for 20 minutes. Afterwards, an excess of tosyl chloride (TsCl, 5 eq. respect to hydroxyl group loading calculated on the basis of EA – 0.07 and 0.08 mmol for hybrids 7 and 8, respectively) and pyridine (25 eq.) was added to the suspension. After sonication for further 10 minutes, the mixture was maintained under stirring at 50 °C (80 °C for hybrid 8) for 3 days. After cooling to room temperature, the solid was recovered by centrifugation and subjected to several purification cycles (sonication/centrifugation) with ethyl acetate (3 × 30 mL) and EtOH (3 × 30 mL) before being dried to constant weight. For the blank test, sample 7 was treated exactly under the same reaction conditions except for the use of p-toluenesulfonic acid as a reagent.Conclusions
We described an efficient and general strategy for the covalent grafting of organic moieties to chemically exfoliated MoS2 nanosheets, taking advantage of the thiol-epoxide “click” protocol. To accomplish this aim, exfoliated MoS2 nanoflakes have been reacted under relatively mild conditions with a series of epoxide derivatives from a wide collection of commercially available compounds, giving rise to chemically decorated 2D MoS2 networks containing β-hydroxyl pendant arms. Besides, we have demonstrated the efficiency of the proposed functionalization technology for the material surface decoration that occurs on both semiconducting and metallic phases of CE-MoS2. To the best of our knowledge, this is the first example reported so far wherein covalent grafting of organic moieties at the inorganic surface of a model chalcogenide occurs at both 1T and 2H phases. Furthermore, the grafting approach generates reactive hydroxyl surface groups that may undergo further derivatization through classical post-functionalization procedures. Such a feature fills the existing gap between the reactivity of classical C-based 1D and 2D networks and that of 2D TMDCs.All materials were thoroughly characterized by different and complementary spectroscopic (IR, XPS) and analytical (Elemental analysis and TGA-MS) techniques, along with (whenever applicable) blank trials. The quantitative estimation of the grafted groups has unveiled a correlation between the material functionalization degree and electrophilic character of the epoxide derivatives dictated by the electron-withdrawing character of their substituents. Overall, these findings widen the toolkit of TMDC functionalization protocols, providing multimodality to the surface of the resulting materials. This aspect opens new horizons in the exploitation of hybrid TMDCs as (photo)electrocatalysts in processes where they have already shown excellent performances. Hybrid hetero-tagged TMDC derivatives of this type are currently being investigated in our labs as functional carriers for (photo)electrocatalytic active moieties in HER.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. G. and C. P.-H. thank the TRAINER project (Catalysts for Transition to Renewable Energy Future) of the “Make our Planet Great Again” program (Ref. ANR-17-MPGA-0017) for support. The Italian teams would also like to thank the Italian MIUR through the PRIN 2017 Project Multi-e (20179337R7) “Multielectron transfer for the conversion of small molecules: an enabling technology for the chemical use of renewable energy” for financial support to this work.References
- M. Xu, T. Liang, M. Shi and H. Chen, Graphene-Like Two-Dimensional Materials, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed.
- K. Khan, A. K. Tareen, M. Aslam, R. Wang, Y. Zhang, A. Mahmood, Z. Ouyang, H. Zhang and Z. Guo, Recent developments in emerging two dimensional materials and their applications, J. Mater. Chem. C, 2020, 8, 387–440 RSC.
- S. Das, J. A. Robinson, M. Dubey, H. Terrones and M. Terrones, Beyond Graphene: Progress in Novel Two-Dimensional Materials and van der Waals Solids, Annu. Rev. Mater. Res., 2015, 45, 1–27 CrossRef CAS.
- T.-H. Le, Y. Oh, H. Kim and H. Yoon, Exfoliation of 2D Materials for Energy and Environmental Applications, Chem. – Eur. J., 2020, 26, 6360–6401 CrossRef CAS PubMed.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutierrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, Progress, challenges, and opportunities in two-dimensional materials beyond graphene, ACS Nano, 2013, 7, 2898–2926 CrossRef CAS PubMed.
- Q. Jia, X. Huang, G. Wang, J. Diao and P. Jiang, MoS2 Nanosheet Superstructures Based Polymer Composites for High-Dielectric and Electrical Energy Storage Applications, J. Phys. Chem. C, 2016, 120, 10206–10214 CrossRef CAS.
- S. Rowley-Neale, C. Foster, G. Smith, D. Brownson and C. E. Banks, Mass-Producible, 2D-MoSe2 Bulk Modified Screen-Printed Electrodes Provide Significant Electrocatalytic Performances Towards the Hydrogen Evolution Reaction, Sustainable Energy Fuels, 2017, 1, 74–83 RSC.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Lithium Ion Battery Applications of Molybdenum Disulfide (MoS2) Nanocomposites, Energy Environ. Sci., 2014, 7, 209–231 RSC.
- X. Zhang, S. Y. Teng, A. C. M. Loy, B. S. How, W. D. Leong and X. Tao, Transition Metal Dichalcogenides for the Application of Pollution Reduction: A Review, Nanomaterials, 2020, 10, 1012 CrossRef CAS PubMed.
- R. Kumar, N. Goel, M. Hojamberdiev and M. Kumar, Transition metal dichalcogenides-based flexible gas sensors, Sens. Actuators, A, 2020, 303, 111875 CrossRef CAS.
- L. Ries, E. Petit, T. Michel, C. Coelho Diogo, C. Gervais, C. Salameh, M. Bechelany, S. Balme, P. Miele, N. Onofrio and D. Voiry, Enhanced sieving from exfoliated MoS2 membranes via covalent functionalization, Nat. Mater., 2019, 18, 1112–1117 CrossRef CAS PubMed.
- X. Zhao, S. Ning, W. Fu, S. J. Pennycook and K. P. Loh, Differentiating Polymorphs in Molybdenum Disulfide via Electron Microscopy, Adv. Mater., 2018, 30, 1802397 CrossRef PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- F. Wypych and R. Schollhorn, 1T-MoS2, a New Metallic Modification of Molybdenum Disulfide, J. Chem. Soc., Chem. Commun., 1992, 1386–1388 RSC.
- E. X. Yan, M. Cabán-Acevedo, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Reductant-Activated, High-Coverage, Covalent Functionalization of 1T′-MoS2, ACS Mater. Lett., 2020, 2, 133–139 CrossRef CAS.
- K. C. Knirsch, N. C. Berner, H. C. Nerl, C. S. Cucinotta, Z. Gholamvand, N. McEvoy, Z. Wang, I. Abramovic, P. Vecera, M. Halik, S. Sanvito, G. S. Duesberg, V. Nicolosi, F. Hauke, A. Hirsch, J. N. Coleman and C. Backes, Basal-plane functionalization of chemically exfoliated molybdenum disulfide by diazonium salts, ACS Nano, 2015, 9, 6018–6030 CrossRef CAS PubMed.
- E. Er, H.-L. Hou, A. Criado, J. Langer, M. Möller, N. Erk, L. M. Liz-Marzán and M. Prato, High-Yield Preparation of Exfoliated 1T-MoS2 with SERS Activity, Chem. Mater., 2019, 31, 5725–5734 CrossRef CAS.
- E. P. Nguyen, B. J. Carey, J. Z. Ou, J. van Embden, E. Della Gaspera, A. F. Chrimes, M. J. S. Spencer, S. Zhuiykov, K. Kalantar-zadeh and T. Daeneke, Electronic Tuning of 2D MoS2 through Surface Functionalization, Adv. Mater., 2015, 27, 6225–6229 CrossRef CAS PubMed.
- E. E. Benson, H. Zhang, S. A. Schuman, S. U. Nanayakkara, N. D. Bronstein, S. Ferrere, J. L. Blackburn and E. M. Miller, Balancing the Hydrogen Evolution Reaction, Surface Energetics, and Stability of Metallic MoS2 Nanosheets via Covalent Functionalization, J. Am. Chem. Soc., 2018, 140, 441–450 CrossRef CAS PubMed.
- M. Cai, F. Zhang, C. Zhang, C. Lu, Y. He, Y. Qu, H. Tian, X. Feng and X. Zhuang, Cobaloxime anchored MoS2 nanosheets as electrocatalysts for the hydrogen evolution reaction, J. Mater. Chem. A, 2018, 6, 138–144 RSC.
- R. Canton-Vitoria, Y. Sayed-Ahmad-Baraza, M. Pelaez-Fernandez, R. Arenal, C. Bittencourt, C. P. Ewels and N. Tagmatarchis, Functionalization of MoS2 with 1,2-dithiolanes: toward donor-acceptor nanohybrids for energy conversion, npj 2D Mater. Appl., 2017, 1, 13 CrossRef.
- M. Blanco, M. Lunardon, M. Bortoli, D. Mosconi, L. Girardi, L. Orian, S. Agnoli and G. Granozzi, Tuning on and off chemical- and photo-activity of exfoliated MoSe2 nanosheets through morphologically selective “soft” covalent functionalization with porphyrins, J. Mater. Chem. A, 2020, 8, 11019–11030 RSC.
- A. Stergiou and N. Tagmatarchis, Molecular Functionalization of Two-Dimensional MoS2 Nanosheets, Chem. – Eur. J., 2018, 24, 18246–18257 CrossRef CAS PubMed.
- L. Daukiya, J. Seibel and S. De, Feyter, Chemical modification of 2D materials using molecules and assemblies of molecules, Adv. Phys. X, 2019, 4, 1625723 CAS.
- A. Hirsch and F. Hauke, Post-Graphene 2D Chemistry: The Emerging Field of Molybdenum Disulfide and Black Phosphorus Functionalization, Angew. Chem., Int. Ed., 2018, 57, 4338–4354 CrossRef CAS PubMed.
- S. Bertolazzi, M. Gobbi, Y. Zhao, C. Backes and P. Samorì, Molecular chemistry approaches for tuning the properties of two-dimensional transition metal dichalcogenides, Chem. Soc. Rev., 2018, 47, 6845–6888 RSC.
- S. S. Chou, M. De, J. Kim, S. Byun, C. Dykstra, J. Yu, J. Huang and V. P. Dravid, Ligand conjugation of chemically exfoliated MoS2, J. Am. Chem. Soc., 2013, 135, 4584–4587 CrossRef CAS PubMed.
- K. Cho, M. Min, T.-Y. Kim, H. Jeong, J. Pak, J.-K. Kim, J. Jang, S. J. Yun, Y. H. Lee, W.-K. Hong and T. Lee, Electrical and Optical Characterization of MoS2 with Sulfur Vacancy Passivation by Treatment with Alkanethiol Molecules, ACS Nano, 2015, 9, 8044–8053 CrossRef CAS PubMed.
- R. Canton-Vitoria, C. Stangel and N. Tagmatarchis, Electrostatic Association of Ammonium-Functionalized Layered-Transition-Metal Dichalcogenides with an Anionic Porphyrin, ACS Appl. Mater. Interfaces, 2018, 10, 23476–23480 CrossRef CAS PubMed.
- S.-D. Jiang, G. Tang, Z.-M. Bai, Y.-Y. Wang, Y. Hu and L. Song, Surface functionalization of MoS2 with POSS for enhancing thermal, flame-retardant and mechanical properties in PVA composites, RSC Adv., 2014, 4, 3253–3262 RSC.
- S. Bertolazzi, S. Bonacchi, G. Nan, A. Pershin, D. Beljonne and P. Samorì, Engineering Chemically Active Defects in Monolayer MoS2 Transistors via Ion-Beam Irradiation and Their Healing via Vapor Deposition of Alkanethiols, Adv. Mater., 2017, 29, 1606760 CrossRef PubMed.
- S. Kumari, H. P. Mungse, R. Gusain, N. Kumar, H. Sugimura and O. P. Khatri, Octadecanethiol-grafted molybdenum disulfide nanosheets as oil-dispersible additive for reduction of friction and wear, FlatChem, 2017, 3, 16–25 CrossRef CAS.
- X. Chen, N. C. Berner, C. Backes, G. S. Duesberg and A. R. McDonald, Functionalization of Two-Dimensional MoS2: On the Reaction between MoS2 and organic thiols, Angew. Chem., Int. Ed., 2016, 55, 5803–5808 CrossRef CAS PubMed.
- D. Voiry, A. Goswami, R. Kappera, C. de Carvalho Castro, S. D. Kaplan, T. Fujita, M. Chen, T. Asefa and M. Chhowalla, Covalent functionalization of monolayered transition metal dichalcogenides by phase engineering, Nat. Chem., 2015, 7, 45–49 CrossRef CAS PubMed.
- M. Vera-Hidalgo, E. Giovanelli, C. Navío and E. M. Pérez, Mild Covalent Functionalization of Transition Metal Dichalcogenides with Maleimides: A “Click” Reaction for 2H-MoS2 and WS2, J. Am. Chem. Soc., 2019, 141, 3767–3771 CrossRef CAS PubMed.
- R. Quiros-Ovies, M. V. Sulleiro, M. Vera-Hidalgo, J. Prieto, I. J. Gomez, V. Sebastian, J. Santamaria and E. M. Perez, Controlled Covalent Functionalization of 2H-MoS2 with Molecular or Polymeric Adlayers, Chem. – Eur. J., 2020, 26, 6629–6634 CrossRef CAS PubMed.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Thiol-click chemistry: a multifaceted toolbox for small molecule and polymer synthesis, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- G. Tuci, D. Mosconi, A. Rossin, L. Luconi, S. Agnoli, M. Righetto, C. Pham-Huu, H. Ba, S. Cicchi, G. Granozzi and G. Giambastiani, Surface Engineering of Chemically Exfoliated MoS2 in a “Click”: How To Generate Versatile Multifunctional Transition Metal Dichalcogenides-Based Platforms, Chem. Mater., 2018, 30, 8257–8269 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Click Chemistry: Diverse Chemical Function from a Few Good Reactions, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. C. Stuparu and A. Khan, Thiol-Epoxy “Click” Chemistry: Application in Preparation and Postpolymerization Modification of Polymers, J. Polym. Sci. Pol. Chem., 2016, 54, 3057–3070 CrossRef CAS.
- R. M. Silverstein, F. X. Webster, D. J. Kiemle and D. L. Bryce, Spectrometric Identification of Organic Compounds, Wiley, 8th edn, 2014 Search PubMed.
- R. M. Hanson, in Organic Reactions, ed. L. E. Overman, et al., John Wiley & Sons Inc., 2002, ch. 1, vol. 60, pp. 1–156 Search PubMed.
- U. Albrecht, I. Freifeld, H. Reinke and P. Langer, One-pot cyclization of dilithiated nitriles with isothiocyanates and epibromohydrin. Synthesis of 2-cyano-1-(hydroxymethyl)-cyclopropanes and 2-cyanomethylidene-4-(hydroxymethyl)-thiazolidines, Tetrahedron, 2006, 62, 5775–5786 CrossRef CAS.
- G. S. Singh, K. Mollet, M. D’hooghe and N. De Kimpe, Epihalohydrins in Organic Synthesis, Chem. Rev., 2013, 113, 1441–1498 CrossRef CAS PubMed.
- X. Chen, D. McAteer, C. McGuinness, I. Ian Godwin, J. N. Coleman and A. R. McDonald, RuII Photosensitizer-Functionalized Two-Dimensional MoS2 for Light-Driven Hydrogen Evolution, Chem. – Eur. J., 2018, 24, 351–355 CrossRef CAS PubMed.
- M. Kaur, N. K. Singh, A. Sarkar, S. J. George and C. N. R. Rao, Supramolecular Hybrids of MoS2 and Graphene Nanosheets with Organic Chromophores for Optoelectronic Applications, ACS Appl. Nano Mater., 2018, 1, 5101–5107 CrossRef CAS.
- G. Tuci, L. Luconi, A. Rossin, E. Berretti, H. Ba, M. Innocenti, D. Yakhvarov, S. Caporali, C. Pham-Huu and G. Giambastiani, Aziridine-Functionalized Multiwalled Carbon Nanotubes: Robust and Versatile Catalysts for the Oxygen Reduction Reaction and Knoevenagel Condensation, ACS Appl. Mater. Interfaces, 2016, 8, 30099–30106 CrossRef CAS PubMed.
- X. Chen and A. R. McDonald, Functionalization of Two-Dimensional Transition-Metal Dichalcogenides, Adv. Mater., 2016, 28, 5738–5746 CrossRef CAS PubMed.
- X. S. Chu, A. Yousaf, D. O. Li, A. A. Tang, A. Debnath, D. Ma, A. A. Green, E. J. G. Santos and Q. H. Wang, Direct Covalent Chemical Functionalization of Unmodified Two-Dimensional Molybdenum Disulfide, Chem. Mater., 2018, 30, 2112–2128 CrossRef CAS.
- C. Agami, F. Couty, L. Hamon and O. Venier, Chiral Oxazolidinones from N-Boc Derivatives of b-Amino Alcohols. Effect of a N-Methyl Substituent on Reactivity and Stereoselectivity, Tetrahedron Lett., 1993, 34, 4509–4512 CrossRef CAS.
- C. Agami and F. Couty, The Reactivity of the N-Boc Protecting Group: an Underrated Feature, Tetrahedron, 2002, 58, 2701–2724 CrossRef CAS.
- C. Shimasaki, F. Hirata, H. Ohta, E. Tsukurimichi and T. Yoshimura, Thermal behavior and mass spectrometry studies of 2-oxazolidinone derivatives, J. Anal. Appl. Pyrolysis, 1993, 24, 291–300 CrossRef CAS.
- P. Hjerrild, T. Tørring and T. B. Poulsen, Dehydration reactions in polyfunctional natural products, Nat. Prod. Rep., 2020, 37, 1043–1064 RSC.
- Q. Meng, B. Du and A. Thibblin, Mechanisms of elimination and substitution reactions. Spontaneous solvolysis reactions via carbanion and carbocation intermediates, J. Phys. Org. Chem., 1999, 12, 116–122 CrossRef CAS.
- H.-L. Tsai, J. Heising, J. L. Schindler, C. R. Kannewurf and M. G. Kanatzidis, Exfoliated-restacked phase of WS2, Chem. Mater., 1997, 9, 879–8882 CrossRef CAS.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Conducting MoS2 nanosheets as catalysts for hydrogen evolution reaction, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- J. Libra, KolXPD: Software for spectroscopy data measurement and processing, http://www.kolibrik.net/science/kolxpd/, Kolibrik.net, s.r.o., Žďár nad Sázavou, Czech Republic.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qm00003a |
| This journal is © the Partner Organisations 2021 |