
From a 1,2-azaborinine to large BN-PAHs via electrophilic cyclization: synthesis, characterization and promising optical properties†
Yannik
Appiarius
 ab,
Tim
Stauch
bcd,
Enno
Lork
ce,
Pascal
Rusch
dfg,
Nadja C.
Bigall
dfg and
Anne
Staubitz
*ab
ab,
Tim
Stauch
bcd,
Enno
Lork
ce,
Pascal
Rusch
dfg,
Nadja C.
Bigall
dfg and
Anne
Staubitz
*ab
aUniversity of Bremen, Institute for Analytical and Organic Chemistry, Leobener Straße 7, D-28359 Bremen, Germany
bMAPEX Center for Materials and Processes, Bibliothekstraße 1, D-28359 Bremen, Germany. E-mail: staubitz@uni-bremen.de
cUniversity of Bremen, Institute for Physical and Theoretical Chemistry, Leobener Straße 7, D-28359 Bremen, Germany
dBremen Center for Computational Materials Science, Am Fallturm 1, D-28359 Bremen, Germany
eUniversity of Bremen, Institute of Inorganic Chemistry and Crystallography, Leobener Straße 7, D-28359 Bremen, Germany
fLeibniz University Hannover, Institute for Physical Chemistry and Electrochemistry, Callinstraße 3a, D-30167 Hannover, Germany
gCluster of Excellence PhoenixD (Photonics, Optics, and Engineering – Innovation Across Disciplines), Hannover, Germany
First published on 20th August 2020
Abstract
We present a convergent synthetic route towards boron–nitrogen containing polycyclic aromatic hydrocarbons (BN-PAHs) that allowed us to synthesize six derivatives. Starting from the conjunction of a 1,2-azaborinine nucleophile and various aryl electrophiles, the key step was the extension of the aromatic system via an electrophilic ring closure of the respective alkyne precursors. Our route allows the use of substituted PAH precursors to be circumvented, which are often unavailable. Instead, it builds up the BN-PAHs solely from easily accessible monocycles. All derivatives were emissive in solution and solid state with quantum yields up to Φlum = 0.40 and small Stokes shifts. The emission wavelengths in solid state were notably dependent on the connectivity of the rings. Due to excimer formation in one derivative, its emission was significantly redshifted with a comparatively slow secondary photoluminescence (PL) decay.
Introduction
1,2-Azaborinines represent a class of six-membered heteroarenes, in which a CC unit is formally replaced by a dipolar BN unit, making them the isosteric and isoelectronic1,2 but less aromatic3–6 congeners of benzene. However, the imbalanced distribution of π-electrons7,8 causes notably divergent optoelectronic properties, which was demonstrated for several BN containing materials including polymers.9–14PAHs are characterized by planar, rigid and thermally stable backbones.15–17 Regarding their optoelectronic properties, large delocalized π-systems18,19 and π–π-stacking20,21 often lead to a high charge carrier mobility.22,23 Moreover, fluorescence in the visible spectrum24,25 in PAHs such as pyrenes26,27 or perylene diimides28,29 made them widely used, semiconducting materials in organic electronics like organic light emitting diodes (OLEDs).30,31 Over the past decade, the incorporation of one or more 1,2-azaborinine units into PAHs has gained increased interest because the resulting BN-PAHs feature several beneficial qualities: compared to their CC analogs, narrowed frontier molecular orbital gaps with particularly stabilized highest occupied molecular orbitals (HOMOs)32–35 as well as redshifted emission spectra36 could be observed. Moreover, quantum yields up to unity37,38 and hole mobilities up to 0.15 cm2 V−1 s−1 (ref. 39) emphasize the application potential of BN-PAHs. At the same time, the BN substitution did not induce significant thermal destabilization.35,40–42
The synthetic approach towards BN-PAHs highly depends on the desired position of the BN unit in the target molecule. Most frequently, electrophilic borylations were performed with suitable (poly)aromatic amines, either with neighboring vinyl- or aryl-groups.32,43–46 This approach is generally simple to follow, high yielding, and it forms the BN bond at a late stage. Moreover, metathesis reactions can be employed to build up new six-membered rings that include B and N heteroatoms.47,48
An innovative methodology was developed by Piers and co-workers, involving consecutive BN bond formation and cycloisomerization of a boracycle and various ethynylpyridine precursors49 (Scheme 1). Both reaction steps typically proceed at ambient temperature without the requirement for catalysts due to a strongly nucleophilic carbon atom α to boron. Only in case of R = TMS, the addition of platinum(II) chloride, a commonly used soft metal catalyst for electrophilic cyclizations,50 was required.
 | ||
| Scheme 1 Examples of recent approaches towards BN phenanthrenes. *Also involves an electrophilic borylation. | ||
This work and later also other reports44,51 impressively clarified the high impact of the position of the BN heteroatoms on the optical properties of BN-PAHs. The influence of internally implemented BN units was throughout much higher than the influence of BN moieties in peripheral positions.37,52
Most of the discussed routes follow a linear strategy: the entire series of precursors is fixed at the outset, depending on the desired product. However, the requirement for large and appropriately substituted PAH precursors can be difficult to meet, as the introduction of reactive groups into PAHs often favors only one regiochemistry and others can be impossible to access.53–55 Consequently, a lack of purchasable or preparable precursors leads to the methods’ limitation to relatively small targets like phenanthrenes. However, the major benefits of extended PAHs are often not observable in these small systems.
Results and discussion
We envisioned a convergent synthetic pathway that allows the construction of much larger systems, using the same 1,2-azaborinine precursor and conditions for all desired molecules. Herein, we present a three-step synthesis of three- and five-membered PAHs via a cross-coupling and subsequent electrophilic cyclization sequence (Table 1). This represents the first bottom-up BN-PAH synthesis starting from a monocyclic 1,2-azaborinine.|
|
||||
|---|---|---|---|---|
| Entry | Starting material | Electrophilic arene | Cross-coupled product | Ring-closed product |
| 1 |

|

|
|
|
| 2 |
|

|

|

|
| 3 |

|

|

|

|
| 4 |

|
|
|

|
| 5 |

|
|

|
|
| 6 |

|
|

|
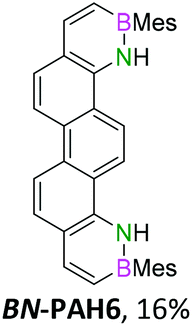
|

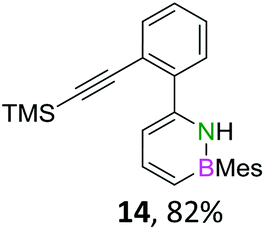
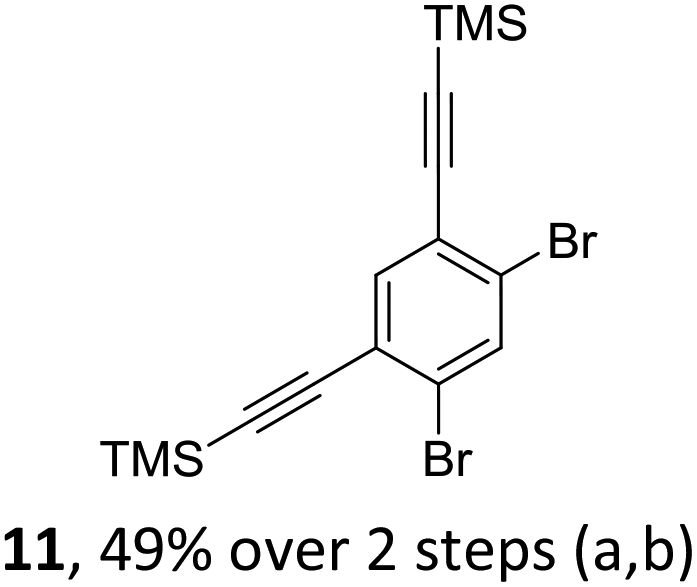
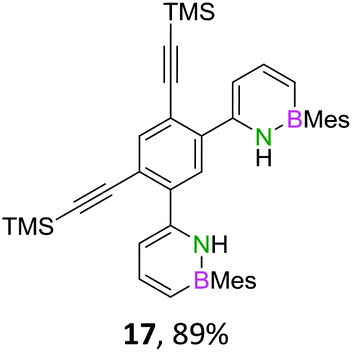
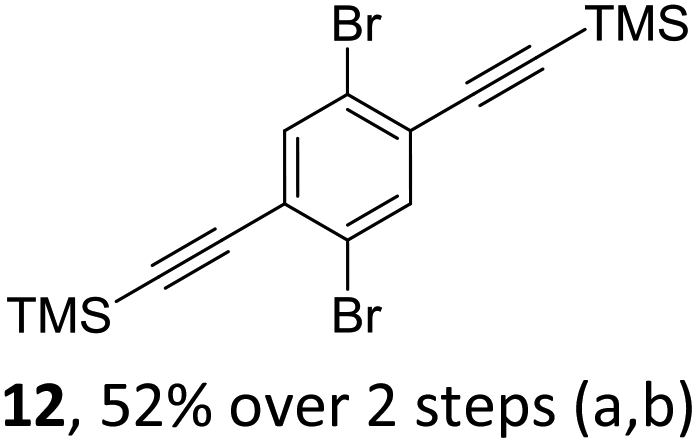
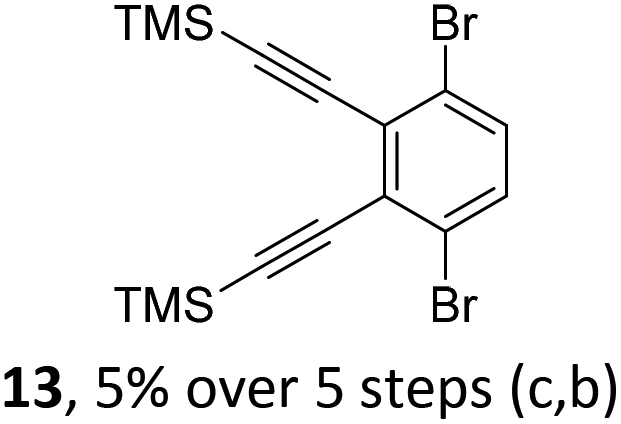
The initial key step of our approach was a Suzuki–Miyaura cross-coupling of azaborinine nucleophile 1 with six different alkynylaryl halides, using slightly modified conditions from Liu et al.56 While 1 was synthesized over seven steps in 5.3% total yield,56 the electrophiles were mostly prepared by electrophilic halogenations and subsequent, iodo-selective Sonogashira cross-coupling reactions with trimethylsilylacetylene. Besides bromo(trimethylsilylethynyl)benzene 8, -thiophene 9 and -pyridine 10, three regioisomers of dibromobis(trimethylsilylethynyl)benzenes 11–13 were used as electrophilic components (Table 1). All Suzuki–Miyaura cross-coupling reactions were complete after 6 h at 80 °C in a microwave reactor, giving biphenylyl or terphenylyl alkynes 14–19 in good yields (Table 1). A recurring by-product was the homo-coupled bis(azaborinine) 20, which we could isolate and characterize (Scheme 2). Subsequent deprotection of the TMS-protected alkynes with KOH furnished the corresponding alkynes, which were directly converted in an electrophilic ring closure by heating them with 30 mol% platinum(II) chloride in toluene for 4 h.49 Without any exceptions, the main components were the desired products with newly formed six-membered rings (Table 1). However, some syntheses suffered from the formation of side-products with equal molecular mass and very similar retardation factors, which explains the divergent yields. Both the formation of new five-membered rings via a competing 5-exo-dig cyclization and the formation of new six-membered rings via the azaborinine nitrogen atom are plausible mechanisms (see ESI†).
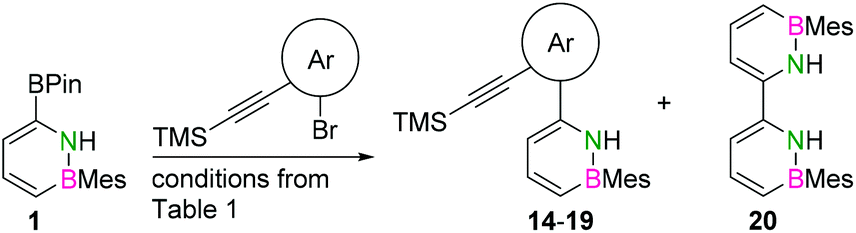 | ||
| Scheme 2 Illustration of by-product 20, which arose in many Suzuki–Miyaura cross-coupling reactions involving azaborinine nucleophile 1 and different electrophiles 14–19. | ||
In the subsequent discussion of the molecules’ properties, we compare the newly synthesized BN-PAH1–6 with their CC analogs. However, as the exact CC counterparts have not been reported, the data of their unsubstituted analogs phenanthrene 21, naphtho[1,2-b]thiophene 22, benzo[h]quinoline 23, dibenz[a,j]anthracene 24, dibenz[a,h]anthracene 25 and picene 26 are used.
All BN-PAHs were chemically stable and soluble in common organic solvents (CHCl3, THF, slightly in n-pentane and methanol), which can be attributed to the solubilizing BN units, inducing strong dipole moments. The BN-PAHs were characterized by NMR, HR-MS, IR, UV/vis and PL spectroscopy.
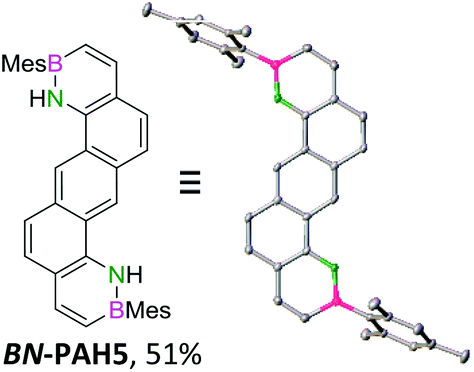
For small BN-PAH1–3, melting points of 100–150 °C were measured. Large BN-PAH4 and BN-PAH5, containing anthracene units, did not melt but decomposed above 300 °C, in contrast to CC analogs 24 and 25, whose melting points are below 300 °C.57 The comparatively low melting point of BN-PAH6 (<125 °C) was presumably caused by the unfavorably oriented mesityl groups, which prevent intense overlapping of the PAH backbones by inducing steric repulsion, in contrast to BN-PAH4 and BN-PAH5 (see next paragraphs). Furthermore these values mirror the notably lower melting point of phenanthrene compared to anthracene.58
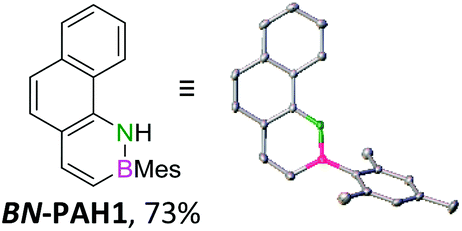
Single crystals suitable for X-ray diffraction analysis were obtained for BN-PAH1, 2, 4 and 5 by slow evaporation of the solvents (see Table 1 and ESI†). BN-PAH4 crystallized with one molecule CDCl3 per asymmetric unit, which can be attributed to its cavity. The results showed significantly distorted mesityl-groups (φmax between 61.7° and 81.2°), while the backbones were almost planar with the highest torsion angle found in BN-PAH4 (φmax = 5.2°, due to slight proton–proton repulsion). Bond lengths in the azaborinine unit match reported values, showing a significant BN double bond character.51
Except for BN-PAH1, which showed a herringbone packing arrangement with a plane to plane angle of 44.9°, all derivatives revealed parallel-displaced packing of the PAH scaffolds (see ESI†). Low plane distances (3.40–3.60 Å) support the presence of classical π–π-interactions. As Hirshfeld surface analysis clarified, the dipolar BN units were uninvolved in the packing formations of all derivatives, most likely due to their peripheral locations (see ESI†). Likewise, the mesityl groups apparently did not impede stacking, different from a previously described BN-PAH with more central mesityl units.59 On the contrary, BN-PAH4 and BN-PAH5 showed large overlaps of the PAH scaffolds.
The electronic properties of the target molecules were analyzed by performing nucleus-independent chemical shift (NICS)60 calculations at the MP2/cc-pVDZ level.61,62 The azaborinine rings were moderately aromatic, displaying NICS(0) values of −3.0 to −3.4 ppm. All other rings were highly aromatic (NICS(0) = −7.0 to −12.1 ppm), in accordance with the common differences between anthracene and phenanthrene cores63 (see ESI†).
The photophysical properties were determined by means of UV/vis absorption and PL spectroscopy (Table 2). Except for BN-PAH6, all compounds showed absorption spectra of similar shapes in solution (see ESI†): one high energetic global maximum band was followed by three local maximum bands at lower energies (315–355 nm for small BN-PAH1–3, 365–425 nm for large BN-PAH4–5), representing vibrational sub-levels. For all derivatives, the molar extinction coefficients at the intensity maxima were particularly high.
| Compound | λ abs (CHCl3) a [nm] | ε [mol−1 L cm−1] | λ lum (CHCl3) a [nm] | Stokes shift (CHCl3) [cm−1] | Φ lum (CHCl3) | λ lum (solid) a [nm] | Stokes shift (solid) [cm−1] | Φ lum (solid) |
|---|---|---|---|---|---|---|---|---|
| a Bold values represent intensity maxima. c = 10−6–10−4 mol L−1. b The highest energy PL band was not identified due to a lack of vibronic resolution. | ||||||||
| BN-PAH1 | 275, 322, 338, 354 | 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 931 931 |
361, 379, 398 | 548 | 0.40 | 368, 409 | 1401 | 0.03 |
| BN-PAH2 | 258, 317, 331, 347 | 40303 |
356, 372 | 729 | 0.09 | 378, 394 | 1323 | <0.01 |
| BN-PAH3 | 283, 331, 348 | 42583 |
389 | —b | 0.06 | 380 | 1774 | 0.05 |
| BN-PAH4 | 317, 365, 384, 406 | 58811 |
423, 451, 480 | 990 | 0.17 | 574 | 5943 | 0.06 |
| BN-PAH5 | 316, 378, 399, 422 | 122343 |
427, 456, 486 | 278 | 0.24 | 460, 487, 521 | 1249 | 0.02 |
| BN-PAH6 | 285, 341, 371, 387 | 51736 |
392, 412, 437 | 330 | 0.16 | 399, 417, 441 | —b | 0.02 |
CC analogs 21, 24, 25 and 26 revealed very similar spectra, though slightly hypsochromically shifted maxima and more closely adjoined bands.57,64
Except for BN-PAH3, the emission spectra in solution resembled each other and consisted of three main bands and one or two less intense bands at high wavelengths (Fig. 1 and Table 2). While three-membered BN-PAH1–3 showed emission maxima between 355 and 400 nm, the emission maxima of their five-membered congeners BN-PAH4–5 were located between 420 and 490 nm, which demonstrates the powerful effect that the addition of rings to a small PAH structure can have. BN-PAH6 also exhibited five emission bands, albeit at higher energies (390–440 nm). The emission band of BN-PAH3 was significantly broadened with the loss of vibronic resolution. We assume that this arises from an increased dipole moment in its excited state geometry. This feature also makes BN-PAH3 slightly solvatochromic if compared to all other BN-PAHs (see ESI†).
 | ||
| Fig. 1 Normalized PL spectra of BN-PAH1–6 in chloroform solution. | ||
For all other BN-PAHs, the observed Stokes shifts were small (278–990 cm−1), suggesting small structural changes in the stiff backbone upon excitation. Along with the vibrationally well-resolved shapes of the spectra, this implies the presence of rigid, delocalized structures. Our results approximately reflect reported spectra for CC analogs 21,6523,662567 and 26,64 however these were shifted hypsochromically to BN-PAH1–6 by 13–40 nm. Especially the large BN-PAHs almost exactly mirrored the spectral shapes of common PAHs like anthracene,68 which indicates that they behave like substituted PAHs rather than like PAHs, in which the heteroatoms predominate the electronic structures.
The PL quantum yield of BN-PAH1 was good (Φlum = 0.40), while the other three-membered PAHs showed low values (Φlum < 0.10). Large BN-PAH4–6 displayed values of Φlum = 0.16–0.24. In contrast, CC analogs 21 (Φlum = 0.09)37 and 25 (Φlum = 0.035)67 were significantly less emissive.
The emission bands in solid state were mostly comparable to these in solution, though broadened. Often, the emission curve mirrored the shape of the absorption curve, with Stokes shifts of typically <2000 cm−1 and noticeable fine structuring. Most remarkably, the spectra of regioisomers BN-PAH4 and BN-PAH5, which reveal very similar emission spectra in solution (Fig. 1), differed dramatically in solid state (Fig. 2), giving rise to very different emission colors (Fig. 3). While BN-PAH5 showed a fine-structured spectrum with three main bands between 460 and 560 nm, BN-PAH4 exhibited a single broad band with a maximum of 574 nm.
 | ||
| Fig. 2 Normalized PL spectra of BN-PAH4 and BN-PAH5 in solid state and corresponding PL lifetime curves. | ||
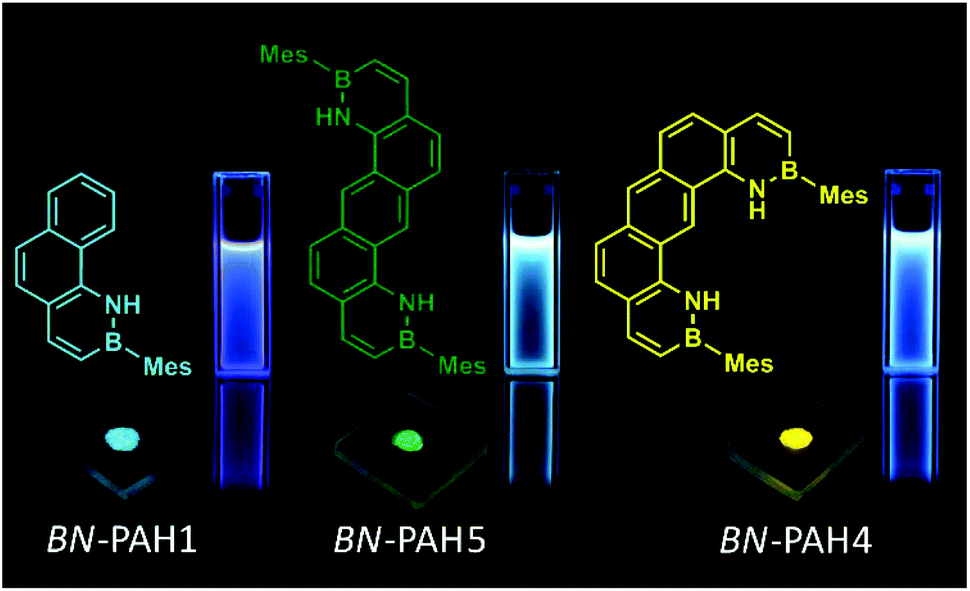 | ||
| Fig. 3 Photographs of dissolved samples of BN-PAH1, BN-PAH5 and BN-PAH4 in glass cuvettes (solvent: chloroform) and in solid state on glass slides after dropcasting from highly concentrated DCM solutions. Samples were irradiated at λ = 365 nm. | ||
Furthermore, the Stokes shift increased from 1249 cm−1 in BN-PAH5 to 5943 cm−1 in BN-PAH4. It is likely that this particularity arises from the formation of excimers in the solid state. This is supported by PL lifetime measurements of BN-PAH4 that showed a second decay that was much slower than the first one (100 ns vs. 2.91 ns), and the absence of fine-structuring. In contrast, none of the other compounds displayed lifetimes >10 ns. The phenomenon of considerable extension of PL lifetimes in excimers has been reported for other PAHs, especially pyrene.69 The quantum yields in the solid state were generally low (Φlum < 0.01–0.06), which indicates that the molecules undergo strong aggregation-caused quenching due to their close intermolecular π–π-interactions.
To improve our understanding of the measured emission spectra, we performed time-dependent Density Functional Theory (TD-DFT)70 calculations at the B3LYP/cc-pVDZ level of theory.62,71–73 All calculated fluorescence wavelengths were in good agreement with the maximum intensities of the measurements, with deviations of λmax between −31 and +22 nm (see ESI†). For large BN-PAH4–6, natural transition orbital (NTO) calculations revealed a π–π* character of the S1 states, from which fluorescence takes place, with extensive delocalization of the orbitals over the PAH scaffold (Fig. 4).
 | ||
| Fig. 4 Highest occupied (HONTO) and lowest unoccupied (LUNTO) NTO for the S1 state of BN-PAH5. | ||
Conclusions
In conclusion, we have developed a convergent synthetic method towards BN-PAHs via a cross-coupling and electrocyclization protocol. Besides the benefit of employing the same nucleophilic 1,2-azaborinine precursor for all derivatives, this flexible approach allows the introduction of a broad scope of electrophilic cross-coupling partners and does not require substituted PAH precursors.The optical measurements disclosed that all derivatives were emissive both in solution and in the solid state. The quantum yields in solution were low to moderate and comprised a broad range of values of Φlum = 0.06–0.40, however, these values were still superior compared to the CC analogs. Due to aggregation-caused quenching, the quantum yields in the solid state were consistently low. As an unexpected attribute, BN-PAH4 revealed the presence of excimers in solid state. This additional intermolecular interaction induces a large bathochromic shift, compared to its regioisomers, and notably enhances the PL lifetime.
This dependency of the emission wavelength on the connectivity of the rings along with the potential of introducing substituents to expand the π-system37 could make our methodology the starting point for synthesizing a number of novel BN-PAHs with precisely tuned emission wavelengths in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A.S. and Y.A. thank the German Research Foundation (DFG) for the Emmy-Noether-Grant STA1195/2-1. N.B. and P.R. thank the DFG for partial funding under Germany's Excellence Strategy within the Cluster of Excellence PhoenixD (EXC 2122, Project ID 390833453) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 714429). We thank Philipp J. Gliese for his support in the optimization of the azaborinine precursor syntheses and Daniel Duvinage for his support in the cyclic voltammetry measurements.Notes and references
- A. J. Ashe and X. Fang, A Synthesis of Aromatic Five- and Six-Membered B−N Heterocycles via Ring Closing Metathesis, Org. Lett., 2000, 2, 2089–2091 CrossRef CAS.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Recent Advances in Azaborine Chemistry, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- E. R. Abbey, L. N. Zakharov and S.-Y. Liu, Crystal Clear Structural Evidence for Electron Delocalization in 1,2-Dihydro-1,2-azaborines, J. Am. Chem. Soc., 2008, 130, 7250–7252 CrossRef CAS PubMed.
- P. G. Campbell, E. R. Abbey, D. Neiner, D. J. Grant, D. A. Dixon and S.-Y. Liu, Resonance Stabilization Energy of 1,2-Azaborines: A Quantitative Experimental Study by Reaction Calorimetry, J. Am. Chem. Soc., 2010, 132, 18048–18050 CrossRef CAS.
- M. Baranac-Stojanović, Aromaticity and Stability of Azaborines, Chem. – Eur. J., 2014, 20, 16558–16565 CrossRef.
- R. Carion, V. Liégeois, B. Champagne, D. Bonifazi, S. Pelloni and P. Lazzeretti, On the Aromatic Character of 1,2-Dihydro-1,2-azaborine According to Magnetic Criteria, J. Phys. Chem. Lett., 2010, 1, 1563–1568 CrossRef CAS.
- A. M. Daly, C. Tanjaroon, A. J. V. Marwitz, S.-Y. Liu and S. G. Kukolich, Microwave Spectrum, Structural Parameters, and Quadrupole Coupling for 1,2-Dihydro-1,2-azaborine, J. Am. Chem. Soc., 2010, 132, 5501–5506 CrossRef CAS PubMed.
- M. J. D. Bosdet and W. E. Piers, B-N as a C-C substitute in aromatic systems, Can. J. Chem., 2009, 87, 8–29 CrossRef.
- B. Thiedemann, P. J. Gliese, J. Hoffmann, P. G. Lawrence, F. D. Sönnichsen and A. Staubitz, High molecular weight poly(N-methyl-B-vinylazaborine) - a semi-inorganic B-N polystyrene analogue, Chem. Commun., 2017, 53, 7258–7261 RSC.
- A. W. Baggett, F. Guo, B. Li, S.-Y. Liu and F. Jäkle, Regioregular Synthesis of Azaborine Oligomers and a Polymer with a syn Conformation Stabilized by NH⋯π Interactions, Angew. Chem., Int. Ed., 2015, 54, 11191–11195 CrossRef CAS PubMed.
- H. Lin, C. R. McConnell, B. Jilus, S.-Y. Liu and F. Jäkle, Changing up BN-Polystyrene: Effect of Substitution Pattern on the Free-Radical Polymerization and Polymer Properties, Macromolecules, 2019, 52, 4500–4509 CrossRef CAS.
- H. L. van de Wouw, J. Y. Lee and R. S. Klausen, Gram-scale free radical polymerization of an azaborine vinyl monomer, Chem. Commun., 2017, 53, 7262–7265 RSC.
- H. L. van de Wouw, J. Y. Lee, E. C. Awuyah and R. S. Klausen, A BN Aromatic Ring Strategy for Tunable Hydroxy Content in Polystyrene, Angew. Chem., Int. Ed., 2018, 57, 1673–1677 CrossRef CAS.
- H. L. van de Wouw and R. S. Klausen, BN Polystyrenes: Emerging Optical Materials and Versatile Intermediates, J. Org. Chem., 2019, 84, 1117–1125 CrossRef CAS.
- R. Rieger and K. Müllen, Forever young: polycyclic aromatic hydrocarbons as model cases for structural and optical studies, J. Phys. Org. Chem., 2010, 23, 315–325 CAS.
- Y.-H. Chung, L. Sheng, X. Xing, L. Zheng, M. Bian, Z. Chen, L. Xiao and Q. Gong, A pure blue emitter (CIEy ≈ 0.08) of chrysene derivative with high thermal stability for OLED, J. Mater. Chem. C, 2015, 3, 1794–1798 RSC.
- S. W. Slayden and J. F. Liebman, The Energetics of Aromatic Hydrocarbons: An Experimental Thermochemical Perspective, Chem. Rev., 2001, 101, 1541–1566 CrossRef CAS PubMed.
- J. E. Anthony, Functionalized Acenes and Heteroacenes for Organic Electronics, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS.
- C. F. Matta and J. Hernández-Trujillo, Bonding in Polycyclic Aromatic Hydrocarbons in Terms of the Electron Density and of Electron Delocalization, J. Phys. Chem. A, 2003, 107, 7496–7504 CrossRef CAS.
- K. Balakrishnan, A. Datar, R. Oitker, H. Chen, J. Zuo and L. Zang, Nanobelt Self-Assembly from an Organic n-Type Semiconductor: Propoxyethyl-PTCDI, J. Am. Chem. Soc., 2005, 127, 10496–10497 CrossRef CAS PubMed.
- F. Liu, C. Tang, Q.-Q. Chen, F.-F. Shi, H.-B. Wu, L.-H. Xie, B. Peng, W. Wei, Y. Cao and W. Huang, Supramolecular π−π Stacking Pyrene-Functioned Fluorenes: Toward Efficient Solution-Processable Small Molecule Blue and White Organic Light Emitting Diodes, J. Phys. Chem. C, 2009, 113, 4641–4647 CrossRef CAS.
- A. M. v. d. Craats, J. M. Warman, A. Fechtenkötter, J. D. Brand and M. A, Harbison and K. Müllen, Record Charge Carrier Mobility in a Room-Temperature Discotic Liquid-Crystalline Derivative of Hexabenzocoronene, Adv. Mater., 1999, 11, 1469–1472 CrossRef.
- H.-Y. Oh, C. Lee and S. Lee, Efficient blue organic light-emitting diodes using newly-developed pyrene-based electron transport materials, Org. Electron., 2009, 10, 163–169 CrossRef CAS.
- Y. Li, T. Yu, W. Su, Y. Wang, Y. Zhao and H. Zhang, Polycyclic aromatic hydrocarbon-bridged coumarin derivatives for organic light-emitting devices, Arabian J. Chem., 2020, 13, 4126–4133 CrossRef CAS.
- E. Heyer and R. Ziessel, Mono-functionalized dibenzo[g,p]-chrysenes for cascade energy transfer, Tetrahedron Lett., 2013, 54, 3388–3393 CrossRef CAS.
- Y. Liu, Q. Bai, J. Li, S. Zhang, C. Zhang, F. Lu, B. Yang and P. Lu, Efficient pyrene-imidazole derivatives for organic light-emitting diodes, RSC Adv., 2016, 6, 17239–17245 RSC.
- S. Chidirala, H. Ulla, A. Valaboju, M. R. Kiran, M. E. Mohanty, M. N. Satyanarayan, G. Umesh, K. Bhanuprakash and V. J. Rao, Pyrene–Oxadiazoles for Organic Light-Emitting Diodes: Triplet to Singlet Energy Transfer and Role of Hole-Injection/Hole-Blocking Materials, J. Org. Chem., 2016, 81, 603–614 CrossRef CAS PubMed.
- E. Kozma, W. Mróz, F. Villafiorita-Monteleone, F. Galeotti, A. Andicsová-Eckstein, M. Catellani and C. Botta, Perylene diimide derivatives as red and deep red-emitters for fully solution processable OLEDs, RSC Adv., 2016, 6, 61175–61179 RSC.
- F. Zhang, Y. Ma, Y. Chi, H. Yu, Y. Li, T. Jiang, X. Wei and J. Shi, Self-assembly, optical and electrical properties of perylene diimide dyes bearing unsymmetrical substituents at bay position, Sci. Rep., 2018, 8, 8208 CrossRef.
- T. M. Figueira-Duarte and K. Müllen, Pyrene-Based Materials for Organic Electronics, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS.
- M. Chen, L. Yan, Y. Zhao, I. Murtaza, H. Meng and W. Huang, Anthracene-based semiconductors for organic field-effect transistors, J. Mater. Chem. C, 2018, 6, 7416–7444 RSC.
- J. S. A. Ishibashi, J. L. Marshall, A. Mazière, G. J. Lovinger, B. Li, L. N. Zakharov, A. Dargelos, A. Graciaa, A. Chrostowska and S.-Y. Liu, Two BN Isosteres of Anthracene: Synthesis and Characterization, J. Am. Chem. Soc., 2014, 136, 15414–15421 CrossRef CAS.
- X.-Y. Wang, F.-D. Zhuang, J.-Y. Wang and J. Pei, Incorporation of polycyclic azaborine compounds into polythiophene-type conjugated polymers for organic field-effect transistors, Chem. Commun., 2015, 51, 17532–17535 RSC.
- X.-Y. Wang, H.-R. Lin, T. Lei, D.-C. Yang, F.-D. Zhuang, J.-Y. Wang, S.-C. Yuan and J. Pei, Azaborine Compounds for Organic Field-Effect Transistors: Efficient Synthesis, Remarkable Stability, and BN Dipole Interactions, Angew. Chem., Int. Ed., 2013, 52, 3117–3120 CrossRef CAS.
- F.-D. Zhuang, Z.-H. Sun, Z.-F. Yao, Q.-R. Chen, Z. Huang, J.-H. Yang, J.-Y. Wang and J. Pei, BN-Embedded Tetrabenzopentacene: A Pentacene Derivative with Improved Stability, Angew. Chem., Int. Ed., 2019, 58, 10708–10712 CrossRef CAS.
- E. R. Abbey, L. N. Zakharov and S.-Y. Liu, Boron in Disguise: The Parent “Fused” BN Indole, J. Am. Chem. Soc., 2011, 133, 11508–11511 CrossRef CAS.
- A. Abengózar, D. Sucunza, P. García-García, D. Sampedro, A. Pérez-Redondo and J. J. Vaquero, A New Member of the BN-Phenanthrene Family: Understanding the Role of the B—N Bond Position, J. Org. Chem., 2019, 84, 7113–7122 CrossRef PubMed.
- C. J. Saint-Louis, L. L. Magill, J. A. Wilson, A. R. Schroeder, S. E. Harrell, N. S. Jackson, J. A. Trindell, S. Kim, A. R. Fisch, L. Munro, V. J. Catalano, C. E. Webster, P. P. Vaughan, K. S. Molek, A. K. Schrock and M. T. Huggins, The Synthesis and Characterization of Highly Fluorescent Polycyclic Azaborine Chromophores, J. Org. Chem., 2016, 81, 10955–10963 CrossRef CAS.
- T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, Synthesis of BN-Fused Polycyclic Aromatics via Tandem Intramolecular Electrophilic Arene Borylation, J. Am. Chem. Soc., 2011, 133, 18614–18617 CrossRef CAS.
- X. Wang, F. Zhang, J. Liu, R. Tang, Y. Fu, D. Wu, Q. Xu, X. Zhuang, G. He and X. Feng, Ladder-Type BN-Embedded Heteroacenes with Blue Emission, Org. Lett., 2013, 15, 5714–5717 CrossRef CAS.
- G. Li, Y. Zhao, J. Li, J. Cao, J. Zhu, X. W. Sun and Q. Zhang, Synthesis, Characterization, Physical Properties, and OLED Application of Single BN-Fused Perylene Diimide, J. Org. Chem., 2015, 80, 196–203 CrossRef CAS.
- M. Lepeltier, O. Lukoyanova, A. Jacobson, S. Jeeva and D. F. Perepichka, New azaborine-thiophene heteroacenes, Chem. Commun., 2010, 46, 7007–7009 RSC.
- S. R. Wisniewski, C. L. Guenther, O. A. Argintaru and G. A. Molander, A Convergent, Modular Approach to Functionalized 2,1-Borazaronaphthalenes from 2-Aminostyrenes and Potassium Organotrifluoroborates, J. Org. Chem., 2014, 79, 365–378 CrossRef CAS.
- C. Zhang, BN-Phenanthrenes: Synthesis, Reactivity, and Optical Properties, Org. Lett., 2019, 21, 3476–3480 CrossRef CAS PubMed.
- A. Mazzanti, M. Boffa, E. Marotta and M. Mancinelli, Axial Chirality at the Boron–Carbon Bond: Synthesis, Stereodynamic Analysis, and Atropisomeric Resolution of 6-Aryl-5,6-dihydrodibenzo[c,e][1,2]azaborinines, J. Org. Chem., 2019, 84, 12253–12258 CrossRef CAS.
- S. Hashimoto, T. Ikuta, K. Shiren, S. Nakatsuka, J. Ni, M. Nakamura and T. Hatakeyama, Triplet-Energy Control of Polycyclic Aromatic Hydrocarbons by BN Replacement: Development of Ambipolar Host Materials for Phosphorescent Organic Light-Emitting Diodes, Chem. Mater., 2014, 26, 6265–6271 CrossRef CAS.
- A. Abengózar, P. García-García, D. Sucunza, A. Pérez-Redondo and J. J. Vaquero, Synthesis of functionalized helical BN-benzo[c]phenanthrenes, Chem. Commun., 2018, 54, 2467–2470 RSC.
- X. Fang, H. Yang, J. W. Kampf, M. M. Banaszak Holl and A. J. Ashe, Syntheses of Ring-Fused B−N Heteroaromatic Compounds, Organometallics, 2006, 25, 513–518 CrossRef CAS.
- M. J. D. Bosdet, C. A. Jaska, W. E. Piers, T. S. Sorensen and M. Parvez, Blue Fluorescent 4a-Aza-4b-boraphenanthrenes, Org. Lett., 2007, 9, 1395–1398 CrossRef CAS.
- A. Fürstner and V. Mamane, Flexible Synthesis of Phenanthrenes by a PtCl2-Catalyzed Cycloisomerization Reaction, J. Org. Chem., 2002, 67, 6264–6267 CrossRef.
- L. Zi, J. Zhang, C. Li, Y. Qu, B. Zhen, X. Liu and L. Zhang, Synthesis, Properties, and Reactivity of Bis-BN Phenanthrenes: Stepwise Bromination of the Main Scaffold, Org. Lett., 2020, 22, 1499–1503 CrossRef CAS.
- M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, 10a-Aza-10b-borapyrenes: Heterocyclic Analogues of Pyrene with Internalized BN Moieties, Angew. Chem., Int. Ed., 2007, 46, 4940–4943 CrossRef CAS PubMed.
- J. M. Casas-Solvas, J. D. Howgego and A. P. Davis, Synthesis of substituted pyrenes by indirect methods, Org. Biomol. Chem., 2014, 12, 212–232 RSC.
- S. Duan, J. Turk, J. Speigle, J. Corbin, J. Masnovi and R. J. Baker, Halogenations of Anthracenes and Dibenz[a,c]anthracene with N-Bromosuccinimide and N-Chlorosuccinimide, J. Org. Chem., 2000, 65, 3005–3009 CrossRef CAS PubMed.
- P. H. Gore, Abnormal Substitution Reactions of Anthracene and Phenanthrene, J. Org. Chem., 1957, 22, 135–138 CrossRef CAS.
- A. W. Baggett, M. Vasiliu, B. Li, D. A. Dixon and S.-Y. Liu, Late-Stage Functionalization of 1,2-Dihydro-1,2-azaborines via Regioselective Iridium-Catalyzed C–H Borylation: The Development of a New N,N-Bidentate Ligand Scaffold, J. Am. Chem. Soc., 2015, 137, 5536–5541 CrossRef CAS.
- Photoelectric Spectrometry Group England Staff, UV Atlas of Organic Compounds, Springer, Boston, 1967 Search PubMed.
- J. L. Goldfarb and I. Külaots, Melting points and enthalpies of fusion of anthracene and its heteroatomic counterparts, J. Therm. Anal. Calorim., 2010, 102, 1063 CrossRef CAS.
- T. Agou, J. Kobayashi and T. Kawashima, Syntheses, Structure, and Optical Properties of Ladder-Type Fused Azaborines, Org. Lett., 2006, 8, 2241–2244 CrossRef CAS.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- C. Møller and M. S. Plesset, Note on an Approximation Treatment for Many-Electron Systems, Phys. Rev., 1934, 46, 618–622 CrossRef.
- T. H. Dunning Jr., Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- J. Poater, M. Duran and M. Solà, Aromaticity Determines the Relative Stability of Kinked vs. Straight Topologies in Polycyclic Aromatic Hydrocarbons, Front. Chem., 2018, 6, 561 CrossRef PubMed.
- A. Verdelli and A. Girlando, Vibronic structure of picene electronic transitions, Chem. Phys. Lett., 2014, 591, 47–51 CrossRef CAS.
- X.-Y. Wang, J.-Y. Wang and J. Pei, BN Heterosuperbenzenes: Synthesis and Properties, Chem. – Eur. J., 2015, 21, 3528–3539 CrossRef CAS PubMed.
- M. Norek, J. Dresner and J. Prochorow, Spectroscopy and Photophysics of Monoazaphenanthrenes I. Absorption and Fluorescence Spectra of Phenanthridine and 7,8-Benzoquinoline, Acta Phys. Pol., A, 2002, 104, 425–439 CrossRef.
- R. Umeda, S. Miyake and Y. Nishiyama, Synthesis of Dibenz[a,h]anthracenes by Pd-Catalyzed Intramolecular Double-cyclization of (Z,Z)-p-Styrylstilbenes, Chem. Lett., 2012, 41, 215–217 CrossRef CAS.
- K. Sudha, S. Sundharamurthi, S. Karthikaikumar, K. Abinaya and P. Kalimuthu, Switching of Förster to Dexter Mechanism of Short-Range Energy Transfer in meso-Anthrylporphyrin, J. Phys. Chem. C, 2017, 121, 5941–5948 CrossRef CAS.
- R. Katoh, S. Sinha, S. Murata and M. Tachiya, Origin of the stabilization energy of perylene excimer as studied by fluorescence and near-IR transient absorption spectroscopy, J. Photochem. Photobiol., A, 2001, 145, 23–34 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, A new mixing of Hartree–Fock and local density–functional theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, photophysical data, NMR spectra, single crystal data and additional calculations. CCDC 1998251–1998254. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qo00723d |
| This journal is © the Partner Organisations 2021 |