
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAqueous assembly of a (pseudo)rotaxane with a donor–π–acceptor axis formed by a Knoevenagel condensation†
Laurent
Jucker
a,
Yves
Aeschi
a and
Marcel
Mayor
 *abc
*abc
aDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: marcel.mayor@unibas.ch
bInstitute for Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), P. O. Box 3640, 76021 Karlsruhe, Germany
cLehn Institute of Functional Materials (LIFM), School of Chemistry, Sun Yat-Sen University (SYSU), Guangzhou 510275, China
First published on 14th June 2021
Abstract
The incorporation of a linear D–π–A “push–pull” chromophore synthesized by a Knoevenagel condensation as axle of a rotaxane is reported. While the introduction of the mechanical stoppers by the reversible and thermodynamically controlled Knoevenagel reaction turned out to be challenging, mechanical fixation of the superstructure is achieved by “click” chemistry in aqueous solution. The isolated rotaxane with a red- to NIR-emitting dye as axle displays enhanced emission in aqueous buffered media compared to the unthreaded model chromophore.
Introduction
The influence of macrocyclic hosts on the optical properties of their guests (and vice versa) has been investigated in a wide range of supramolecular systems with various emerging applications. Depending on the particular implementation, fluorescence enhancement, electron and/or energy transfer mechanisms can be incorporated in optical triggered molecular switches, typically as a function of the proximity of the components involved.1–4 Commonly employed dyes include rylene diimides,5–7 naphthalene imides,8 azo dyes,9–11 squaraines,12,13 and poly-aromatic hydrocarbons like anthracene14,15 or pyrene.16 Despite the growing library of rotaxanes, only very few examples comprising linear donor–π–acceptor “push–pull” chromophores are reported.17–23 An even more surprising fact considering their synthetic accessibility and tunability of their optical properties.24The decoration of a pseudorotaxane with a bulky substituent as stopper to mechanically fix the macrocyclic host is a common assembly strategy for rotaxanes, often referred to as “capping” or “stoppering”. A variety of coupling reactions have been employed, with the copper(I)-catalysed azide–alkyne 1,3-cycloaddtion (CuAAC) as “click” reaction being particularly popular,10,16 combining mild conditions and the concomitant tolerance for a large variety of functional groups with high yields, especially in (metal-)templated assembly strategies.25–28 However, in contrast to the irreversible CuAAC, only stoppering by reversible transformations might provide thermodynamic control over the formed superstructures. So far mainly imine condensations29,30 and olefin metatheses31 have been used for this purpose. To the best of our knowledge, examples taking advantage of the dynamic nature of the Knoevenagel condensation32 are limited to the syntheses of two rotaxanes22,23 comprising cyclodextrin-encapsulated cyanine dyes under aqueous basic conditions. More frequent use of the Knoevenagel condensation is observed in the field of covalent organic frameworks33,34 and macrocyclizations.35
We thus explored the potential of the Knoevenagel condensation as stoppering reaction for pseudorotaxanes assembled in water (Scheme 1a). We not only hoped for the reversible, thermodynamically controlled assembly of rotaxanes, but also wanted to profit from the strong electron accepting nature of the formed olefin decorated with a pair of electron-withdrawing groups (EWG). Our focus was set on the formation of linear donor–π–acceptor (D–π–A) “push–pull” chromophores as axles with emission in the red to near-infrared region (NIR). This emission range is not only of interest for bioimaging,13,36 but also particularly appealing for spectroscopy experiments enhanced by gold nanostructures.37,38
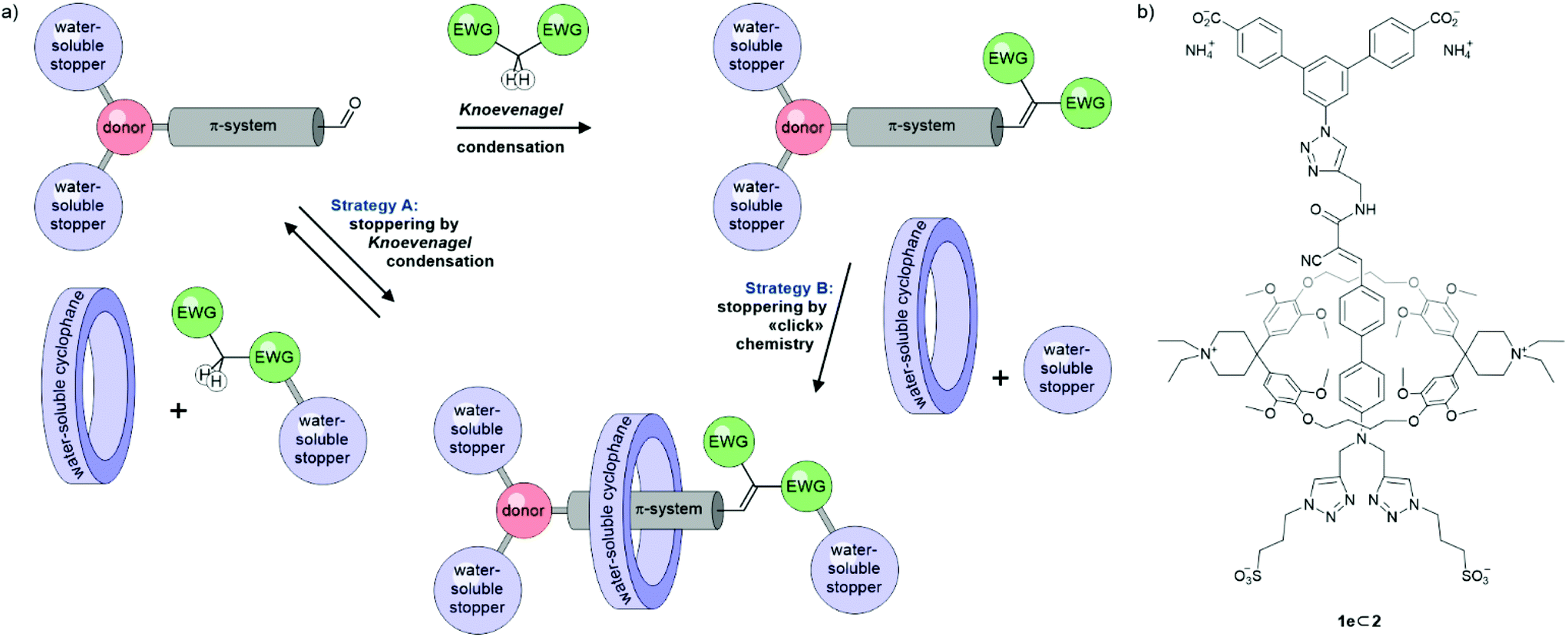 | ||
| Scheme 1 (a) Both strategies to assemble rotaxanes with a D–π-A chromophore as axis considered. (b) Rotaxane 1e⊂2 obtained via strategy B. | ||
As macrocycle forming (pseudo)rotaxanes with these axes, the Diederich-type cyclophane 239–41 combining strong complexation of hydrophobic guests with water-solubility was considered. Previously, 2 has been employed in rotaxanes comprising oligo(phenylene-ethynylene) (OPE) molecular wires,39,42,43 azo dyes,9 or naphthalenes44 as guests. We have previously used an OPE-functionalized modification of 2 to assemble dimeric, zwitterionic daisy chains in water45 and studied the slow association of 2 with carboxylate-terminated OPE structures forming kinetically inert pseudorotaxanes.46 Interestingly, the changes observed in the emission intensity upon complexation with 2 strongly depend on the nature of the guest and seem not to be fully understood yet. This further motivated the here reported approach, as axles assembled via Knoevenagel condensation might provide access to a series of (pseudo)rotaxanes incorporating “push–pull” D–π–A systems of various acceptor strengths.
Here we report our attempts to assemble water-soluble linear “push–pull” chromophores by Knoevenagel condensations as potential axes of rotaxanes. We present our initial struggles to balance reactivity, stability and solubility in water together with the first “proof-of-existence” rotaxane 1e⊂2 (Scheme 1b) with the desired axis structure. While this mechanically interlinked model compound enabled preliminary investigations of the physical properties of this class of rotaxanes, it was obtained applying a stepwise strategy, assembling first the axis with a Knoevenagel condensation and subsequently fixing the supermolecule by CuAAC “click” chemistry (strategy B in Scheme 1a).
Results and discussion
Precursor synthesis and pseudorotaxane formation
With the intention to profit from the Knoevenagel condensation as a stoppering reaction, the aldehyde decorated precursor 3 combining multiple key components was designed (Scheme 2). Its biphenyl backbone acts as both, π-system of the chromophore bridging donor and acceptor substituent and as hydrophobic station complexing the cyclophane. The electron-donating nitrogen is functionalized with two triazol units bearing sulfonates, which have a double role too, serving as mechanical stopper and as water-solubilizing groups at the same time.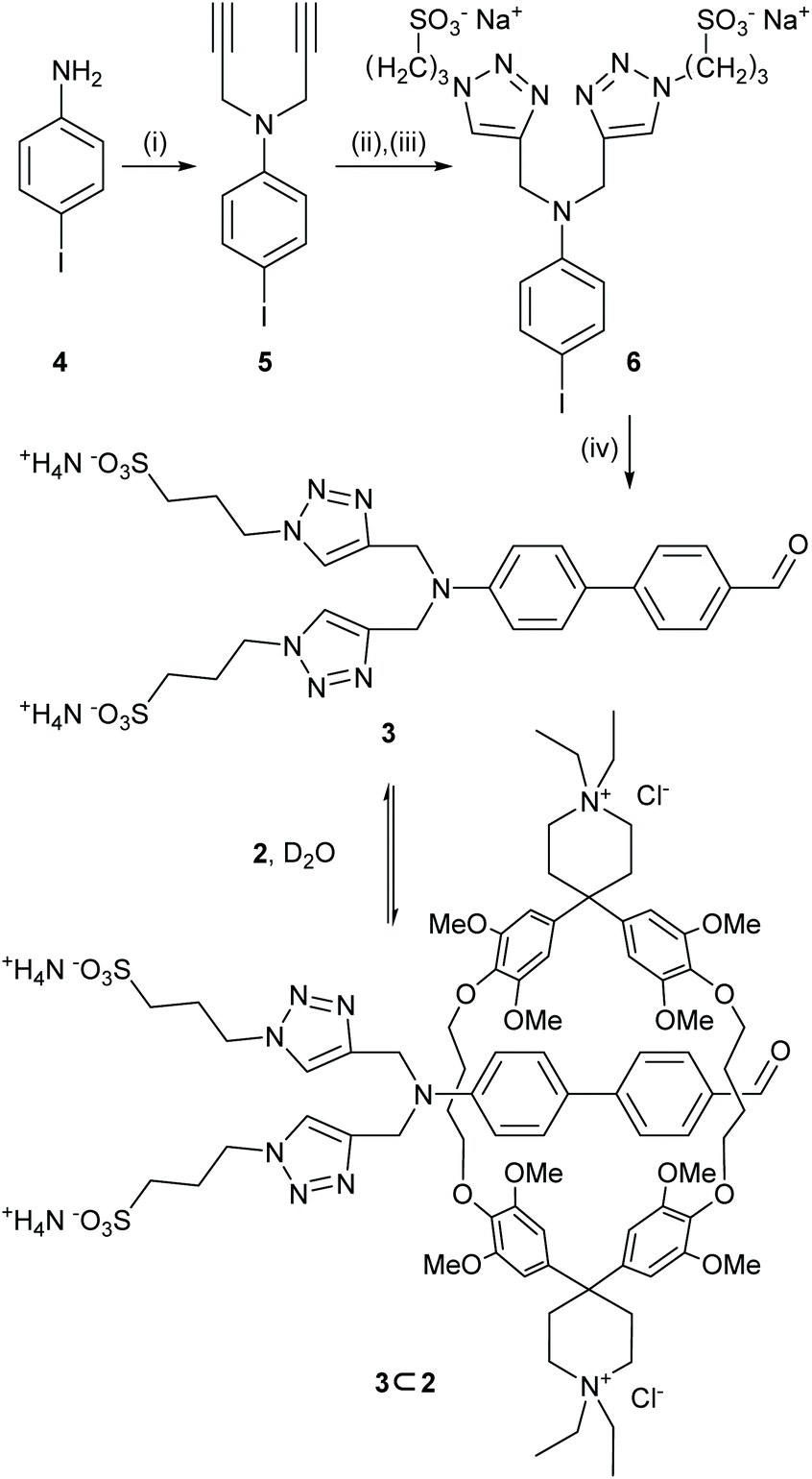 | ||
Scheme 2 Synthesis of 3 and pseudorotaxane formation with 2 in D2O. (i) Propargyl bromide (30 wt% in toluene), K2CO3, ACN, 60 °C, 3 d, 66%; (ii) NaN3, 1,3-propanesultone, DMF/H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), rt, 1 h; (iii) TBTA, Cu(CH3CN)4PF6, sodium ascorbate, 5, rt, 22 h, 45%; (iv) 4-formylphenylboronic acid, K3PO4, Pd(OAc)2, H2O, reflux, 1 h, 78%. :1), rt, 1 h; (iii) TBTA, Cu(CH3CN)4PF6, sodium ascorbate, 5, rt, 22 h, 45%; (iv) 4-formylphenylboronic acid, K3PO4, Pd(OAc)2, H2O, reflux, 1 h, 78%. | ||
Starting from 4-iodoaniline 4, two-fold alkylation with propargyl bromide in acetonitrile gave 5 in 66% yield. Formation of 3-azidopropane-1-sulfonate by SN2 of 1,3-propanesultone with sodium azide and subsequent CuAAC gave the water-soluble 6 as the disodium salt in 45% yield after decomposition of excess azide, purification by reverse-phase column chromatography, and precipitation from water with EtOH. After unsuccessful attempts in commonly employed mixed aqueous–organic media, we found suitable conditions47 for the Suzuki–Miyaura cross-coupling of 6 with 4-formylphenylboronic acid in water in absence of a ligand to give the desired aldehyde 3 as the ammonium salt in 78% yield after reverse-phase column chromatography.
The interaction between the one-sidedly stoppered water-soluble biphenyl 3 and the cyclophane 2 was investigated by 1H-NMR spectroscopy (Scheme S1†). Addition of an equimolar amount of 2 to a solution of 3 in D2O displayed the typical up-field shift of the protons encircled by the host expected for the formation of 3⊂2, along with a broadening of the signals. The aryl signals of the biphenyl system are shifted up-field by 0.5–2.2 ppm, with the central two protons experiencing the strongest shielding effect (as determined by NOESY, Fig. S1†). The aldehyde singlet only shifts up-field by 0.36 ppm and the triazole and propyl signals of the stopper are slightly deshielded. According to the extent of the experienced shielding we hypothesized the macrocyclic host in 3⊂2 to be mainly located around the centre of the biphenyl subunit, with the terminal aldehyde accessible for further reactions. A dilution series of 3⊂2 in D2O (Fig. S2†) in the range of 1 mM to 0.1 mM only showed a slight shift of the signals, indicating a high association constant (Ka) in aqueous solution (Ka > 105 M−1).48 This is in line with previously published pseudorotaxanes comprising 2.46
Aqueous Knoevenagel condensations
Having confirmed the formation of 3⊂2 by 1H-NMR spectroscopy, we investigated conditions for the Knoevenagel condensation in aqueous media (Scheme 3). Inspired by literature procedures,49,50 we tested the condensation of malononitrile with 3 in water at room temperature in absence of any catalyst and were delighted to obtain 1a in 69% yield after simple precipitation of the product by addition of acetonitrile. The “push–pull” dye 1a showed interesting optical properties (Fig. S14†) with an absorption maximum of λabs = 420 nm and emission extending into the NIR with a maximum of λem = 710 nm in H2O. The large Stokes’ shift of approx. 9700 cm−1 observed for 1a was particularly appealing for both, biological applications and the optimization of superstructures for single molecule experiments profiting from the field enhancement in gold nanostructures.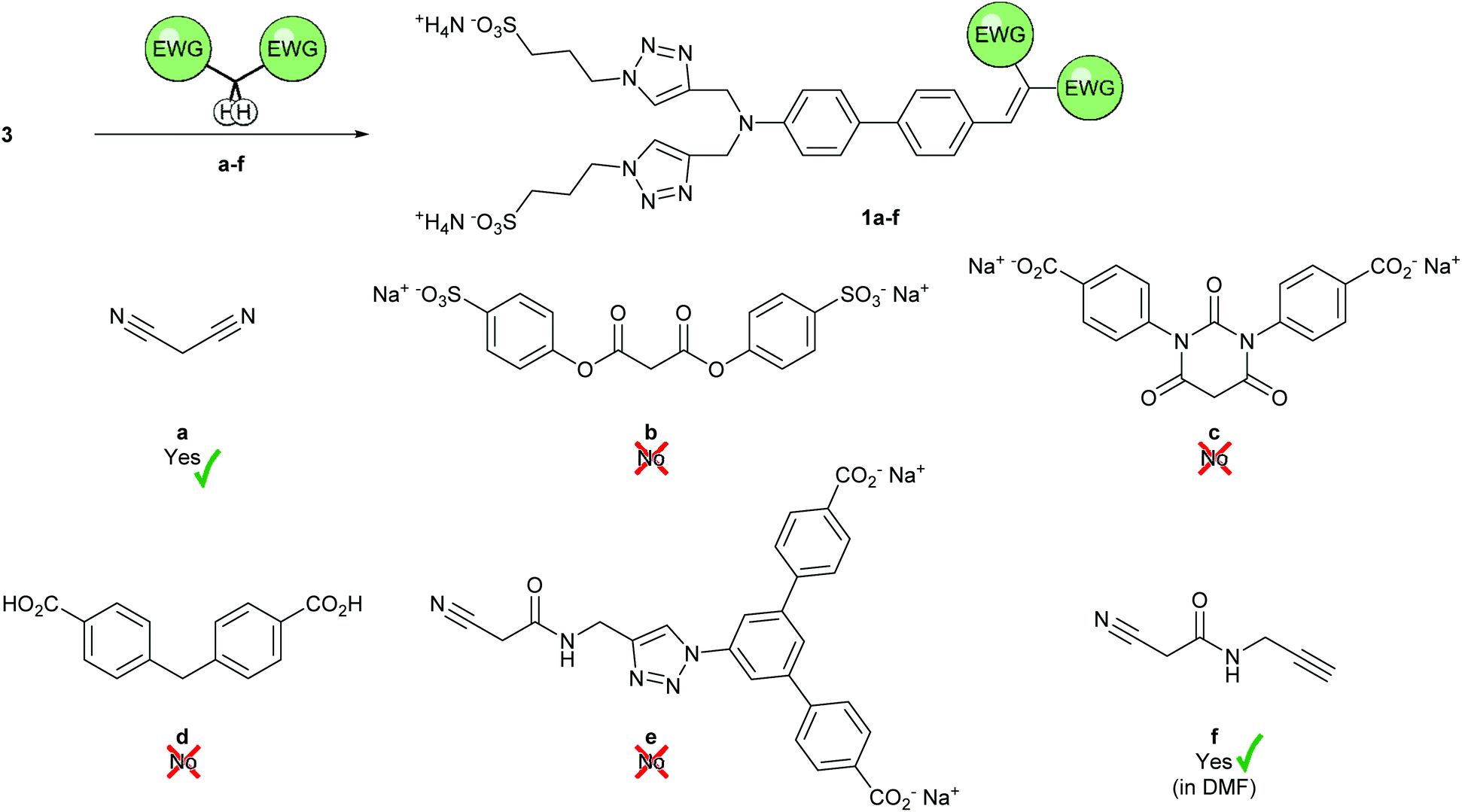 | ||
| Scheme 3 Series of methylene precursors a–f activated by electron-withdrawing groups (EWGs) for the Knoevenagel reaction with 3 with the intention to form 1a–f. Answer statements together with ticks and crosses show the success in the search for suitable conditions for the formation of the corresponding Knoevenagel products in isolable yields. | ||
Motivated by both, the encouraging preliminary synthesis and the exciting optical properties of the Knoevenagel product 1a, a large variety of sterically more demanding precursors were synthesized. From a molecular design view point, it seemed that the required methylene subunit trapped between a pair of EWGs was ideally suited to be decorated with bulky substituents, providing the option to profit from the Knoevenagel condensation to introduce the stopper required for the mechanically interlocked rotaxane structure. The overview and evaluation of considered methylene precursors comprising in part the stopper function is summarized in Scheme 3. Diester b was synthesized in an acid-catalyzed esterification of malonic acid; barbituric acid derivative c was obtained in two steps from a literature known urea derivative51 (Scheme S4†). Unfortunately, the diester b turned out to be unserviceable for the intended purpose in preliminary experiments. The compound was not reactive at room temperature and its ester moieties were prone to hydrolysis at elevated temperatures under all reaction conditions investigated (acidic, basic, and uncatalyzed aqueous conditions). At the first glance, preliminary experiments with the barbituric acid derivative c were more promising, as the colour of the reaction mixture changed immediately to red upon addition of acetic acid to an aqueous solution of the reactants, pointing at the successful product formation. However, reaction control by LC-MS and reverse-phase TLC merely showed the starting materials. Repeating the experiment in D2O, addition of CD3CO2D provided the expected red colour, yet it was not accompanied by any change in the 1H-NMR spectrum, likely due to the formation of only negligible amount of the intensely coloured desired product. Unfortunately, reaction conditions pushing the equilibrium towards the formation of 1c could not be found and we suspected the product to be of limited stability in aqueous medium.
We thus considered the less electron-withdrawing but likely more stable, commercially available 4,4′-methylenedibenzoic acid d next. Its reactivity was very poor even at elevated temperatures and reactions under more classic Knoevenagel conditions (DMF/piperidine) did not proceed either.
Puzzled by the refusal of the so far investigated methylene precursors to form the Knoevenagel product in aqueous media, we wondered whether their bulkiness or their charged state compromised either their reactivity or the stability of their condensation products. In a last attempt we developed the less symmetric precursor e, which features a bulky stopper on only one side of the active methylene component. In this compound the conjugation between the active methylene group and the stopper is broken by the methylene bridge between the amide and the triazole, thus the charged stopper is not participating in the electron withdrawal, unlike in molecules b–d. Of course we hoped that this variation in the molecular design will improve the reactivity of e and the stability of the resulting product 1e.
The synthesis of e was performed in three steps from the literature-reported52 aniline 7 (Scheme 4). First, a diazotation-azidation procedure gave 8 in 98% yield, followed by basic ester hydrolysis in THF/MeOH/H2O to produce the stopper 9 in 96% yield after centrifugation and removal of excess NaOH. In a final step, CuAAC with f led to the formation of e in 65% yield.
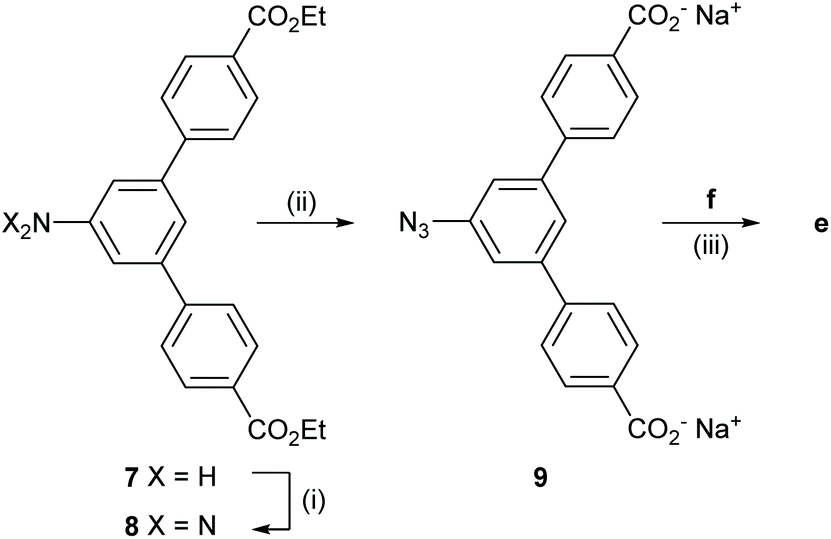 | ||
| Scheme 4 Synthesis of the stopper 9 in 2 steps and CuAAC “click” reaction providing e. Reagents and conditions: (i) TMSN3, tBuONO, DMF, 0 °C to rt, 6 h, 98%; (ii) NaOH, THF–MeOH–H2O (4:4:1), 50 °C, 1 h, 86%; (iii) CuSO4·5H2O, sodium ascorbate, H2O–MeOH (2:1), rt, 1 h, 65%. | ||
However, the Knoevenagel condensation of e and 3 to form 1e (Scheme 3) remained challenging in aqueous conditions. In absence of catalyst the reaction did not proceed towards the product even at elevated temperatures, while addition of Na2CO3 led to amide hydrolysis already at room temperature. By adjusting pH to ∼7–8 with a phosphate buffer, decomposition at elevated temperature was slowed down, however, the formation of the desired condensation product 1e was not observed. Acidic reaction conditions turned out to be even more challenging as the solubility of e decreased, substantially handicapping the formation of condensation product.
The reaction sequence forming the methylene precursor e with stopper potential by CuAAC “click” chemistry first and its subsequent engagement in the Knoevenagel condensation could also be inverted. The condensation could also be performed with the alkyne-bearing f and the obtained product exposed to the CuAAC “click” conditions to be further decorated by the bulky stopper. And indeed, the desired condensation product 1f was obtained, even so not in aqueous conditions. However, 1f profits from rather compact olefin substituents introduced by the Knoevenagel reaction, which should allow the subsequent rotaxane formation. As aqueous conditions are only required to unleash hydrophobic interaction to trigger the pseudorotaxane formation, the use of alternative solvents to obtain 1f is less an issue than in the case of bulkier methylene precursors. The formation of 1f was observed in DMF with piperidine as catalyst. However, under these conditions 1f was prone to react further in a Michael addition. To our delight, omission of piperidine led to the E-selective (as evidenced by the NOESY NMR spectrum, Fig. S13†) formation of 1f in 36% isolated yield.
The absorption and emission spectra of 1f in H2O (Fig. S15†) show a hypsochromic shift with respect to 1a, with maxima of 396 nm and 693 nm for the absorption and emission of 1f, respectively. The Stokes’ shift is, with a value of approximately 10800 cm−1, even larger than the one observed for 1a.
As already mentioned above, due to the relatively compact f, we anticipated that 2 should readily thread onto 1f. And indeed, the 1H-NMR analysis of a 1 mM 1:1 mixture in D2O displayed the expected shifts of the aromatic signals of 1f (Scheme S2†). In similarity to 3⊂2, the central protons of the biphenyl system in 1f⊂2 experienced the largest shift, suggesting the centre of the biphenyl subunit to be the most prevalent position of the host 2 in the superstructure. This is further corroborated by NOE signals of these central protons to the aromatic signal of 2 (Fig. S4†). 1H-NMR spectra of a dilution series (Fig. S5†) showed no significant changes in the chemical shift down to concentrations of 50 μM, indicating a Ka beyond the range suited for determination by NMR spectroscopy (>105 M−1). The association of 1f⊂2 in water was determined by recording the emission intensity during the titration of 1f against 2 (Fig. S11†). The obtained Ka of 1.5 × 107 M−1 is larger than previously reported association constants with the same cyclophane,42,46 which might be the result of supporting electrostatic attractions between the negatively charged sulfonate groups of the axis and the piperidinium moieties of the macrocycle.
With the successful syntheses of e and 1f, together with its strong association with 2, our focus moved away from the original idea of using a Knoevenagel condensation as stoppering reaction (strategy A in Scheme 1a) to the stepwise assembly strategy (strategy B in Scheme 1a). Finally accepting that aqueous conditions are a challenging environment to push a reversible condensation reaction to completion, we wondered if a rotaxane with an axle assembled by a Knoevenagel condensation might be stable in water at all. Furthermore, we hoped to access the model compound to be able to assess the influence of the integration of the D–π–A dye systems into the rotaxane superstructure on its optical properties.
[2]Rotaxane
With the confirmed formation of the pseudorotaxane 1f⊂2, the stoppering with 9via CuAAC “click” reaction was envisioned (Scheme 5). | ||
| Scheme 5 Mechanical fixation in the rotaxane 1e⊂2via CuAAC “click” reaction of 1f and 9 in presence of host 2 in aqueous environment. Reagents and conditions: (i) CuSO4·5H2O, sodium ascorbate, H2O, rt, 1.5 h, 0.9%. | ||
To simplify purification, classical conditions with CuSO4 and sodium ascorbate in absence of a ligand were chosen. The reaction was performed at 2 mM concentration of 2 in H2O, to prevent its self-aggregation.39 Higher host concentrations led to the formation of precipitate during the reaction. After 30 min, we were delighted to observe the formation of new species with masses of the desired [2]rotaxane 1e⊂2 and the unthreaded side product 1e by LC-MS. While the ratio of both products could not be determined, we proceeded with the challenging isolation of the [2]rotaxane.
The solubility of 1e⊂2 was lower in H2O than that of its precursors, however it turned out to be well soluble in mixed solvents of H2O/CH3CN and H2O/DMF, as well as in DMSO. While reverse-phase HPLC on C18-functionalized silica using H2O/CH3CN as eluent allowed to remove most impurities like 1e, 2, and 9 over multiple consecutive runs, some remaining impurities could also not be removed using H2O/MeOH eluent mixtures. Numerous alternative purification attempts like precipitations and crystallizations by vapour diffusion failed. Dialysis against water showed no significant improvement by 1H-NMR spectroscopy. Purification by gel permeation chromatography (GPC) using polystyrene beads (BioBeads SX-1) was considered, but the low solubility of 1e⊂2 in pure DMF and the poor swelling behaviour of the beads in polar protic media rendered the method ill-suited for this purpose. Finally, GPC using Sephadex LH-20 in a mixture of DMF and aqueous NH4OAc buffer, followed by a second dialysis, provided the rotaxane 1e⊂2 in 0.9% isolated yield with only minor impurities remaining. 1e⊂2 was characterized by 1H-NMR and 2D-NMR in D2O/CD3CN, and high-resolution mass spectrometry. The sparse availability of the mechanically interlinked model compound did neither allow to record 13C-NMR spectra showing all expected signals nor its characterization by elemental analysis, however, the chemical shifts of the missing 13C peaks were extracted from HMQC and HMBC measurements.
The low yield of the mechanically interlocked product led us to investigate the reaction further and 1H-NMR studies revealed the formation of undesired pseudorotaxane 9⊂2 when 9 and 2 were mixed in D2O (Fig. S6†). Furthermore, equimolar competition experiments of 1f, 9, and 2 in D2O (Fig. S9†) revealed the partial formation of both 1e⊂2 and 9⊂2, pointing towards similarly high Ka values. Unfortunately, higher ratios of 2 led to precipitation during the CuAAC reaction and formation of impurities which were inseparable by HPLC, GPC, or precipitation.
To exclude that deslipping of the macrocycle off the axle in the [2]rotaxane occurred during purification, we heated solutions of 1e⊂2 in acetonitrile and DMSO to 75 °C and 120 °C, respectively. Qualitative analysis by HPLC-MS (Fig. S27†) showed neither the formation of 1e nor 2 after 16 h of heating, confirming sufficient bulkiness of both stopper moieties.
The unthreaded reference compound 1e was prepared by CuAAC between 1f and 9, and obtained in 81% yield after precipitation from the reaction mixture with an excess of CH3CN (Scheme S6†). Interestingly, 1e turned out to be better soluble in water than the [2]rotaxane. However, 1H-NMR spectra of 1e displayed broad signals in both, D2O and D2O/CD3CN solutions, presumably due to the presence of different protonation states. Reasonably sharp NMR spectra were obtained in DMSO after addition of acetic acid.
To exclude solvent effects, the 1H-NMR spectra of 1e and 1e⊂2 were compared in DMSO-d6 with acetic acid as additive (Fig. 1). Akin to the NMR spectra of pseudorotaxanes 3⊂2 and 1f⊂2 measured in D2O, the aromatic protons of the biphenyl moiety are most affected by the cyclophane, showing a significant upfield shift. The protons of the stoppers remain largely unaffected, further corroborating the biphenyl bridge as the preferred resting position of the cyclophane in the superstructure.
 | ||
| Fig. 1 Comparison of 1H-NMR spectra of 1e (top, 400 MHz) and 1e⊂2 (bottom, 500 MHz) in DMSO-d6 with AcOH as additive. | ||
We are mainly interested in the effects the cyclophane exercises on the optical properties of the D–π–A chromophore, with the intention to develop model compounds for experiments investigating single molecule motions spectroscopically. Thus the optical properties of 1e and 1e⊂2 (Fig. 2a) were recorded in aqueous solutions containing 0.1 M Na2HPO4 adjusted to pH 6.4 with H3PO4 to guarantee similar ions and comparable protonation states. In the UV-Vis absorption spectra, the longest wavelength absorption maxima are 420 nm and 406 nm for 1e⊂2 and 1e, respectively, showing a bathochromic shift in the [2]rotaxane. Exciting both samples at their absorption maxima for the emission spectra, displayed a hypsochromically shifted emission for 1e⊂2 compared to 1e with maxima at 655 and 669 nm, respectively. More importantly, when corrected for the different absorptions, we noticed that the signal intensity at the emission maximum is significantly higher in the [2]rotaxane than in the unthreaded chromophore in the buffered aqueous solution. Intrigued by this result, we determined the absolute quantum yields (QY) in buffered aqueous solution to be below 1% for 1e, while an efficiency of 11.0% (±0.5%) was observed for 1e⊂2 under the same conditions (see ESI†). In DMSO, the effect seemed to persist (Fig. S16†) and indeed, both molecules showed higher QY in DMSO with values of 19.0% (±0.9%) and 58.5% (±1.5%) for the rod and [2]rotaxane, respectively. The mechanisms determining the extent of emission enhancement upon threading are not fully clarified yet and various factors might contribute like e.g., reduced solvent interaction or decreased rotational freedom. Aggregation-induced emission quenching of 1e seems unlikely as potential mechanism, as all the synthesised chromophores 3, 1a, 1e, 1f, and 1e⊂2 even emit in the solid state when irradiated with light of 366 nm (Fig. 2b and c). The emission spectra of the finely ground solids of 3, 1a, 1e, and 1f in between two glass slides were recorded in an integrating sphere, while 1e⊂2 was measured as a film on glass due to the lack of material. Interestingly, the emission spectra of dyes 1a, 1e, 1f, and 1e⊂2 (Fig. S21†) show no significant differences, sharing an emission maximum at roughly 600 nm. This hypsochromic shift can be explained by the absence of solvent interaction.
 | ||
| Fig. 2 (a) Absorption (solid lines) and emission (dashed lines) spectra of 1e⊂2 (black) and 1e (red) in aqueous 0.1 M Na2HPO4 solution adjusted to pH 6.4 with H3PO4. Absorption spectra are normalized. Emission spectra were corrected for differences in absorption at the excitation wavelength (420 nm for 1e⊂2; 406 nm for 1e). Photographs of 3, 1a, 1f, 1e, 1e⊂2 (left to right) under ambient (b) and 366 nm wavelength light (c). Photographs in (c) were slightly edited to decrease the blue discoloration caused by the lamp, unedited photographs can be found in the supporting information (Fig. S22–S26†). | ||
Knoevenagel reversibility
During experiments examining the pH-dependence of 1e in NMR spectroscopy, we observed the instability of 1e in D2O/CD3CN (1:1) in the presence of Na2CO3, as expected based on our previous results with e, which showed decomposition in Knoevenagel condensation attempts performed under basic conditions. Over time, an aldehyde signal arose, along with the characteristic peaks of the biphenyl moiety present in our chromophore design. To our delight, the 1H-NMR spectra of the formed species and 3 (in D2O/CD3CN, 1:1, Scheme S3†) share identical chemical shifts. Further examination by LC-MS corroborated 3 as the main species formed after 20 h (Fig. S28†). While e decomposed under these conditions, the experiment nonetheless indicated the reversibility of the Knoevenagel condensation in water, a prerequisite for its employment as a stoppering reaction for the formation of rotaxanes under thermodynamic control.
Conclusions
The challenges we faced during our ongoing journey studying the potential of the Knoevenagel condensation as stoppering reaction in water for the formation of rotaxanes are reported. The rigid-rod type water-soluble aldehyde precursor readily associated with the macrocyclic host in water and was suited for the synthesis of push–pull red to NIR-emitting dyes with solid-state emission. While we have not yet succeeded in finding a suitable reaction system enabling stoppering via Knoevenagel condensation, we synthesized and characterized a first “proof-of-existence” [2]rotaxane with a Knoevenagel product as axle. Significant emission enhancement was observed for the encapsulated dye in comparison to the unthreaded dye when measured in buffered aqueous solution and solid-state emission was preserved.We are currently working on mechanically interlocked model compounds for externally triggered single molecule experiments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous financial support by the Volkswagen-Stiftung (Az. 93438) is gratefully acknowledged. M. M. acknowledges support from the 111 project (Grant No. 90002-18011002).Notes and references
- X. Ma and H. Tian, Bright functional rotaxanes, Chem. Soc. Rev., 2010, 39, 70–80 Search PubMed.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Development of Pseudorotaxanes and Rotaxanes: From Synthesis to Stimuli-Responsive Motions to Applications, Chem. Rev., 2015, 115, 7398–7501 Search PubMed.
- P. Rajamalli, F. Rizzi, W. Li, M. A. Jinks, A. K. Gupta, B. A. Laidlaw, I. D. W. Samuel, T. J. Penfold, S. M. Goldup and E. Zysman-Colman, Using the Mechanical Bond to Tune the Performance of a Thermally Activated Delayed Fluorescence Emitter, Angew. Chem., Int. Ed., 2021, 60, 12066–12073 Search PubMed.
- J. E. M. Lewis, M. Galli and S. M. Goldup, Properties and emerging applications of mechanically interlocked ligands, Chem. Commun., 2017, 53, 298–312 Search PubMed.
- Z.-Q. Cao, H. Li, J. Yao, L. Zou, D.-H. Qu and H. Tian, A Perylene-Bridged Switchable [3]Rotaxane Molecular Shuttle with a Fluorescence Output, Asian J. Org. Chem., 2015, 4, 212–216 Search PubMed.
- B. J. Slater, E. S. Davies, S. P. Argent, H. Nowell, W. Lewis, A. J. Blake and N. R. Champness, A Perylene Diimide Rotaxane: Synthesis, Structure and Electrochemically Driven De-Threading, Chem. – Eur. J., 2011, 17, 14746–14751 Search PubMed.
- A. H. G. David, P. García–Cerezo, A. G. Campaña, F. Santoyo–González and V. Blanco, [2]Rotaxane End–Capping Synthesis by Click Michael–Type Addition to the Vinyl Sulfonyl Group, Chem. – Eur. J., 2019, 25, 6170–6179 Search PubMed.
- D. D. Günbaş, L. Zalewski and A. M. Brouwer, Solvatochromic rotaxane molecular shuttles, Chem. Commun., 2011, 47, 4977 Search PubMed.
- S. Anderson, T. D. W. Claridge and H. L. Anderson, Azo-Dye Rotaxanes, Angew. Chem., Int. Ed., 1997, 36, 1310–1313 Search PubMed.
- H. Yan, L. Zhu, X. Li, A. Kwok, X. Li, H. Ågren and Y. Zhao, Photothermal-responsive [2]rotaxanes, RSC Adv., 2013, 3, 2341 Search PubMed.
- M. R. Craig, T. D. W. Claridge, H. L. Anderson and M. G. Hutchings, Synthesis of a cyclodextrin azo dye [3]rotaxane as a single isomer, Chem. Commun., 1999, 1537–1538 Search PubMed.
- M. Xue, Y.-S. Su and C.-F. Chen, Isomeric Squaraine-Based [2]Pseudorotaxanes and [2]Rotaxanes: Synthesis, Optical Properties, and Their Tubular Structures in the Solid State, Chem. – Eur. J., 2010, 16, 8537–8544 Search PubMed.
- J. J. Gassensmith, J. M. Baumes and B. D. Smith, Discovery and early development of squaraine rotaxanes, Chem. Commun., 2009, 6329–6338 Search PubMed.
- W. Zhou, J. Li, X. He, C. Li, J. Lv, Y. Li, S. Wang, H. Liu and D. Zhu, A Molecular Shuttle for Driving a Multilevel Fluorescence Switch, Chem. – Eur. J., 2008, 14, 754–763 Search PubMed.
- D. A. Leigh, M. Á. F. Morales, E. M. Pérez, J. K. Y. Wong, C. G. Saiz, A. M. Z. Slawin, A. J. Carmichael, D. M. Haddleton, A. M. Brouwer, W. J. Buma, G. W. H. Wurpel, S. León and F. Zerbetto, Patterning through Controlled Submolecular Motion: Rotaxane-Based Switches and Logic Gates that Function in Solution and Polymer Films, Angew. Chem., Int. Ed., 2005, 44, 3062–3067 Search PubMed.
- T. Ogoshi, D. Yamafuji, T. Yamagishi and A. M. Brouwer, Förster resonance energy transfer by formation of a mechanically interlocked [2]rotaxane, Chem. Commun., 2013, 49, 5468 Search PubMed.
- W. Zhou, J. Xu, H. Zheng, H. Liu, Y. Li and D. Zhu, Charge Transfer Chromophore-Stopped [2]Rotaxane through [2+2] Cycloaddition, J. Org. Chem., 2008, 73, 7702–7709 Search PubMed.
- H. Zhang, Q. Wang, M. Liu, X. Ma and H. Tian, Switchable V-Type [2]Pseudorotaxanes, Org. Lett., 2009, 11, 3234–3237 Search PubMed.
- S. J. Vella, J. Tiburcio, J. W. Gauld and S. J. Loeb, Push−Pull [2]Pseudorotaxanes. Electronic Control of Threading by Switching ON/OFF an Intramolecular Charge Transfer, Org. Lett., 2006, 8, 3421–3424 Search PubMed.
- L. S. Atabekyan, A. I. Vedernikov, V. G. Avakyan, N. A. Lobova, S. P. Gromov and A. K. Chibisov, Photoprocesses in styryl dyes and their pseudorotaxane complexes with cucurbit[7]uril, J. Photochem. Photobiol. A. Chem., 2013, 253, 52–61 Search PubMed.
- L. Zhu, X. Li, F.-Y. Ji, X. Ma, Q.-C. Wang and H. Tian, Photolockable Ratiometric Viscosity Sensitivity of Cyclodextrin Polypseudorotaxane with Light-Active Rotor Graft, Langmuir, 2009, 25, 3482–3486 Search PubMed.
- J. E. H. Buston, J. R. Young and H. L. Anderson, Rotaxane-encapsulated cyanine dyes: enhanced fluorescence efficiency and photostability, Chem. Commun., 2000, 905–906 Search PubMed.
- J. E. H. Buston, H. L. Anderson and F. Marken, Enhanced chemical reversibility of redox processes in cyanine dye rotaxanes, Chem. Commun., 2001, 1046–1047 Search PubMed.
- F. Bureš, Fundamental aspects of property tuning in push–pull molecules, RSC Adv., 2014, 4, 58826–58851 Search PubMed.
- H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Macrocycle Size Matters: “Small” Functionalized Rotaxanes in Excellent Yield Using the CuAAC Active Template Approach, Angew. Chem., Int. Ed., 2011, 50, 4151–4155 Search PubMed.
- V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, Catalytic “Click” Rotaxanes: A Substoichiometric Metal-Template Pathway to Mechanically Interlocked Architectures, J. Am. Chem. Soc., 2006, 128, 2186–2187 Search PubMed.
- J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Metal ions in the synthesis of interlocked molecules and materials, Chem. Soc. Rev., 2017, 46, 2577–2591 Search PubMed.
- N. Hoyas Pérez and J. E. M. Lewis, Synthetic strategies towards mechanically interlocked oligomers and polymers, Org. Biomol. Chem., 2020, 18, 6757–6780 Search PubMed.
- S. J. Cantrill, S. J. Rowan and J. F. Stoddart, Rotaxane Formation under Thermodynamic Control, Org. Lett., 1999, 1, 1363–1366 Search PubMed.
- I. Neira, A. Blanco-Gómez, J. M. Quintela, C. Peinador and M. D. García, Adjusting the Dynamism of Covalent Imine Chemistry in the Aqueous Synthesis of Cucurbit[7]uril-based [2]Rotaxanes, Org. Lett., 2019, 21, 8976–8980 Search PubMed.
- Y. Suzaki and K. Osakada, Ferrocene-containing [2]- and [3]rotaxanes. Preparation via an end-capping cross-metathesis reaction and electrochemical properties, Dalton Trans., 2007, 2376 Search PubMed.
- E. Knoevenagel, Condensation von Malonsäure mit aromatischen Aldehyden durch Ammoniak und Amine, Berichte Dtsch. Chem. Ges., 1898, 31, 2596–2619 Search PubMed.
- R. Chen, J.-L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Designed Synthesis of a 2D Porphyrin-Based sp2 Carbon-Conjugated Covalent Organic Framework for Heterogeneous Photocatalysis, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 Search PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Two-dimensional sp2 carbon–conjugated covalent organic frameworks, Science, 2017, 357, 673–676 Search PubMed.
- S. Lee, C.-H. Chen and A. H. Flood, A pentagonal cyanostar macrocycle with cyanostilbene CH donors binds anions and forms dialkylphosphate [3]rotaxanes, Nat. Chem., 2013, 5, 704–710 Search PubMed.
- M. van Oosten, T. Schäfer, J. A. C. Gazendam, K. Ohlsen, E. Tsompanidou, M. C. de Goffau, H. J. M. Harmsen, L. M. A. Crane, E. Lim, K. P. Francis, L. Cheung, M. Olive, V. Ntziachristos, J. M. van Dijl and G. M. van Dam, Real-time in vivo imaging of invasive- and biomaterial-associated bacterial infections using fluorescently labelled vancomycin, Nat. Commun., 2013, 4, 2584 Search PubMed.
- R. Bardhan, N. K. Grady, J. R. Cole, A. Joshi and N. J. Halas, Fluorescence Enhancement by Au Nanostructures: Nanoshells and Nanorods, ACS Nano, 2009, 3, 744–752 Search PubMed.
- A. P. Francisco, D. Botequim, D. M. F. Prazeres, V. V. Serra, S. M. B. Costa, C. A. T. Laia and P. M. R. Paulo, Extreme Enhancement of Single-Molecule Fluorescence from Porphyrins Induced by Gold Nanodimer Antennas, J. Phys. Chem. Lett., 2019, 10, 1542–1549 Search PubMed.
- S. Anderson, R. T. Aplin, T. D. W. Claridge, T. Goodson III, A. C. Maciel, G. Rumbles, J. F. Ryan and H. L. Anderson, An approach to insulated molecular wires: synthesis of water-soluble conjugated rotaxanes, J. Chem. Soc., Perkin Trans. 1, 1998, 2383–2398 Search PubMed.
- S. B. Ferguson, E. M. Seward, F. Diederich, E. M. Sanford, A. Chou, P. Inocencio-Szweda and C. B. Knobler, Strong enthalpically driven complexation of neutral benzene guests in aqueous solution, J. Org. Chem., 1988, 53, 5593–5595 Search PubMed.
- F. Diederich, Complexation of Neutral Molecules by Cyclophane Hosts, Angew. Chem., Int. Ed. Engl., 1988, 27, 362–386 Search PubMed.
- P. N. Taylor, A. J. Hagan and H. L. Anderson, Hindered fluorescence quenching in an insulated molecular wire, Org. Biomol. Chem., 2003, 1, 3851 Search PubMed.
- S. Anderson and H. L. Anderson, Synthesis of a Water-Soluble Conjugated [3]Rotaxane, Angew. Chem., Int. Ed. Engl., 1996, 35, 1956–1959 Search PubMed.
- Y. Aeschi, S. Drayss-Orth, M. Valášek, F. Raps, D. Häussinger and M. Mayor, Assembly of [2]Rotaxanes in Water: Assembly of [2]Rotaxanes in Water, Eur. J. Org. Chem., 2017, 2017, 4091–4103 Search PubMed.
- Y. Aeschi, S. Drayss–Orth, M. Valášek, D. Häussinger and M. Mayor, Aqueous Assembly of Zwitterionic Daisy Chains, Chem. – Eur. J., 2019, 25, 285–295 Search PubMed.
- Y. Aeschi, L. Jucker, D. Häussinger and M. Mayor, Slow Formation of Pseudorotaxanes in Water: Slow Formation of Pseudorotaxanes in Water, Eur. J. Org. Chem., 2019, 2019, 3384–3390 Search PubMed.
- Z. Li, C. Gelbaum, W. L. Heaner, J. Fisk, A. Jaganathan, B. Holden, P. Pollet and C. L. Liotta, Palladium-Catalyzed Suzuki Reactions in Water with No Added Ligand: Effects of Reaction Scale, Temperature, pH of Aqueous Phase, and Substrate Structure, Org. Process Res. Dev., 2016, 20, 1489–1499 Search PubMed.
- P. Thordarson, Determining association constants from titration experiments in supramolecular chemistry, Chem. Soc. Rev., 2011, 40, 1305–1323 Search PubMed.
- F. Areias, M. Costa, M. Castro, J. Brea, E. Gregori-Puigjané, M. F. Proença, J. Mestres and M. I. Loza, New chromene scaffolds for adenosine A2A receptors: Synthesis, pharmacology and structure–activity relationships, Eur. J. Med. Chem., 2012, 54, 303–310 Search PubMed.
- F. Bigi, M. L. Conforti, R. Maggi, A. Piccinno and G. Sartori, Clean synthesis in water: uncatalysed preparation of ylidenemalononitriles, Green Chem., 2000, 2, 101–103 Search PubMed.
- A. Azhdari Tehrani, L. Esrafili, S. Abedi, A. Morsali, L. Carlucci, D. M. Proserpio, J. Wang, P. C. Junk and T. Liu, Urea Metal–Organic Frameworks for Nitro-Substituted Compounds Sensing, Inorg. Chem., 2017, 56, 1446–1454 Search PubMed.
- P. Martinez-Bulit, B. H. Wilson and S. J. Loeb, One-pot synthesis of porphyrin-based [5]rotaxanes, Org. Biomol. Chem., 2020, 18, 4395–4400 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qo00643f |
| This journal is © the Partner Organisations 2021 |