
Radical heteroarylation of unactivated remote C(sp3)–H bonds via intramolecular heteroaryl migration†
Zhu
Cao‡
a,
Huihui
Zhang‡
a,
Xinxin
Wu
a,
Yahong
Li
 *a and
Chen
Zhu
*ab
*a and
Chen
Zhu
*ab
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, 199 Ren-Ai Road, Suzhou, Jiangsu 215123, China. E-mail: chzhu@suda.edu.cn; liyahong@suda.edu.cn
bKey Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 20th September 2021
Abstract
The radical-mediated heteroarylation of unactivated remote C(sp3)–H bonds via intramolecular heteroaryl migration is achieved, leading to a variety of heteroaryl-substituted aliphatic ketones. A library of O-/S-/N-containing heteroaryls such as benzofuryl, benzothiazolyl, benzothienyl, benzoxazolyl, oxazolyl, and thiazolyl are amenable to the migration approach. The heteroaryl migration is triggered by an azido radical-mediated hydrogen atom abstraction from unactivated aliphatic C(sp3)–H bonds. The transformation features mild C–C bond cleavage, good selectivity for tertiary C(sp3)–H bonds, and broad functional group compatibility.
Introduction
Heteroarenes are important structural motifs widely existing in natural and pharmaceutical products, and materials science.1 Over the past century, tremendous efforts from organic chemistry communities have been devoted to construct heteroarenes and investigate their transformations. Notwithstanding the great progress achieved in heterocyclic chemistry, our group has a long-term interest in developing ingenious approaches to incorporate heteroarenes to target molecules by radical-mediated functional group migration.2–4 For instance, radical-mediated difunctionalization of alkenes via intramolecular heteroaryl migration provides an efficient tactic for the concomitant installation of a heteroaryl and another functional group in an alkene.3 An upgraded docking-migration strategy significantly extends the compatibility of substrates, allowing the radical heteroarylation of more general alkenes.4Direct functionalization of inert C(sp3)–H bonds represents a powerful and atom-economical synthetic strategy.5 As a complement to transition metal-catalyzed C(sp3)–H activation, radical-mediated site-selective functionalization of C(sp3)–H bonds has received rapidly increasing attention,6,7 and many new reaction modes have been unveiled during the last decade. It should be noted that although remote radical C–H-functionalization has become a heavily investigated field of the chemical community, the remote C(sp3)–H arylation as addressed herein is particularly challenging and only very few examples have been reported complementing transition-metal-catalyzed C(sp3)–H arylation that only work for primary and secondary alkyl sites.6h,i In 2018, we disclosed a tertiary alcohol-directed radical heteroarylation of unactivated C(sp3)–H bonds via remote heteroaryl migration under photochemical conditions (Scheme 1a).8 In the presence of an Ir complex as a photocatalyst and potassium persulfate as an oxidant, the nascent alkoxy radical enabled the hydrogen atom transfer (HAT) and the ensuing heteroaryl migration. Although (benzo)thiazolyl and pyridyl were readily incorporated at the δ-position of alcohols, the substrates were limited to alcohols bearing N-containing heteroaryls. Despite the fact that O/S-containing heteroaryls have also showcased the migratory aptitude,3h,i they were not tolerated under strong oxidation conditions due to their high electron density, and thus failed to generate the desired products. To address this issue, we conceive an intermolecular HAT instead of the alkoxy radical-mediated intramolecular HAT to generate the alkyl radical, and meanwhile avoid the use of harsh conditions. Considering that the azido radical is a common HAT species easily accessible under mild conditions,9 we envision that the azido radical-mediated HAT might be compatible with the following O-/S-containing heteroaryl migration, thus realizing the unprecedented O-/S-heteroarylation of unactivated C(sp3)–H bonds by a functional group migration strategy (Scheme 1b).
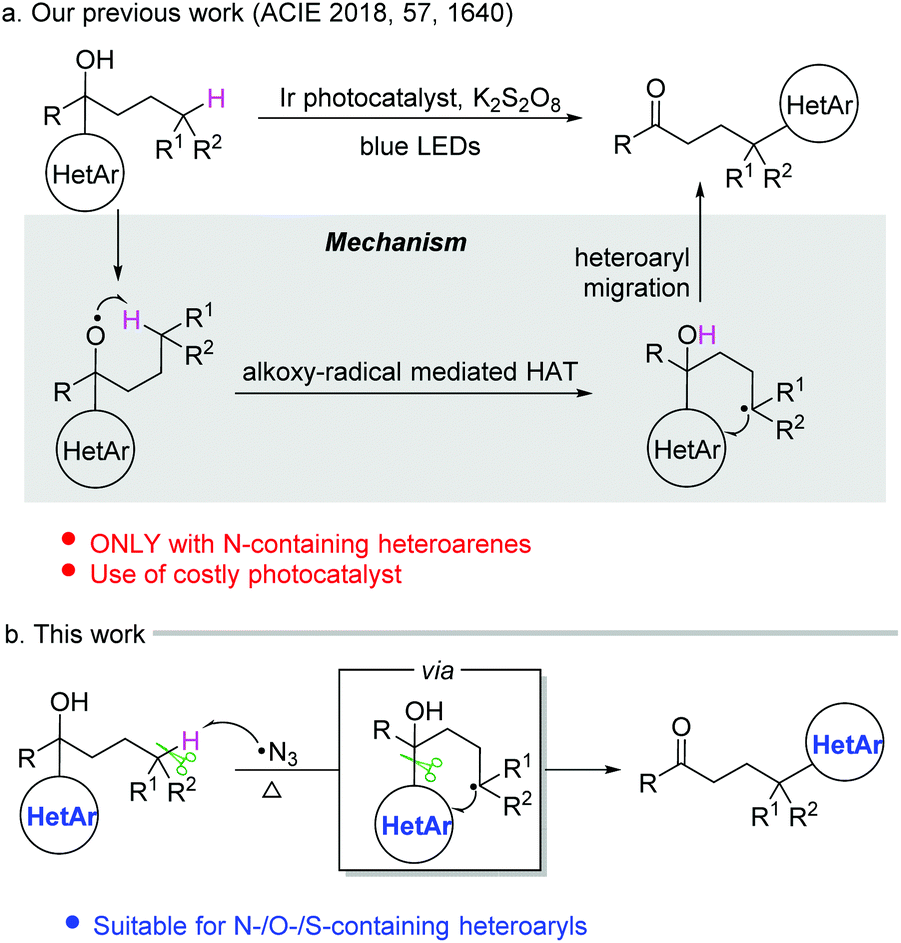 | ||
| Scheme 1 Radical-mediated heteroarylation of unactivated C(sp3)–H bonds by means of functional group migration. | ||
Herein, we provide the proof of principle for the hypothesis. Compared to the previous protocol,8 the current one provides a broader substrate scope for the azido radical-mediated heteroarylation of C(sp3)–H bonds. In addition to N-containing heteroaryls, O- and S-containing heteroaryls also readily migrate in the reaction, leading to heteroaryl-substituted aliphatic ketones in synthetically useful yields. The transformation features mild C–C bond cleavage, good selectivity for tertiary C(sp3)–H bonds, and wide functional group compatibility.
Results and discussion
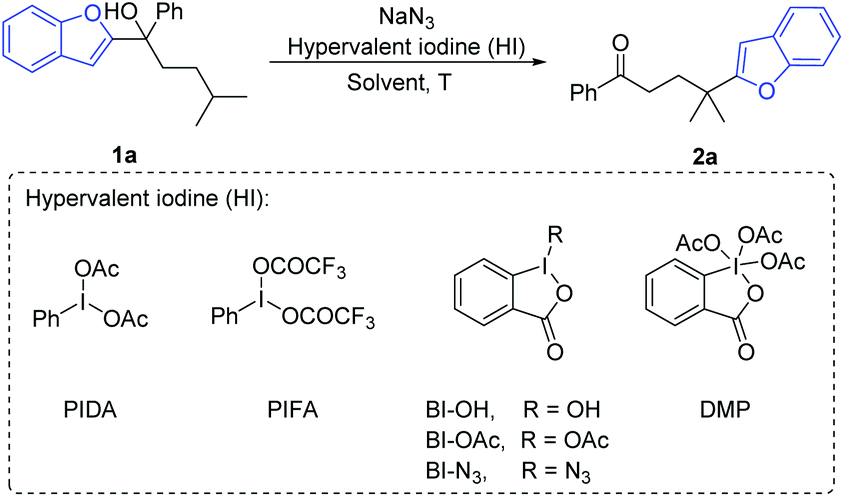
We commenced the investigation with the benzofuryl-substituted tertiary alcohol 1a as a model substrate and the combination of (diacetoxyiodo)benzene (PIDA) and sodium azide as the azido radical source (Table 1). Among the screened organic solvents, MeCN delivered the highest yield of the desired product 2a (entries 1–7). Elevating the reaction temperature to 80 °C improved the isolated yield of 2a to 53% (entry 8). It was found that changing the temperature to be higher or lower did not further increase the yield (entries 9 and 10). The amounts of PIDA and NaN3 were then examined (entries 11–15), showing that the use of excess PIDA and NaN3 was indeed beneficial for the reaction (entry 11). Slightly increasing the volume of MeCN to 2.5 mL resulted in a better yield (entry 16). The survey of hypervalent iodine (HI) reagents indicated that PIDA is still the most efficient reagent (entry 16, entries 18–22). Surprisingly, BI-N3 capable of serving both as an oxidant and azido radical source did not provide the expected reactivity in the absence of NaN3 (entry 23).|
|
||||
|---|---|---|---|---|
| Entry | HI | Solvent (mL) | T (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), HI (4.0 equiv.), and NaN3 (4.0 equiv.) in MeCN (2.5 mL), 80 °C for 12 h. b Isolated yield. c PIDA (2.5 equiv.), NaN3 (2.5 equiv.). d PIDA (3.5 equiv.), NaN3 (3.5 equiv.). e PIDA (4.5 equiv.), NaN3 (4.5 equiv.). f PIDA (4.0 equiv.), NaN3 (2.0 equiv.). g PIDA (2.0 equiv.), NaN3 (4.0 equiv.). h Without NaN3. | ||||
| 1c | PIDA | MeCN (2.0) | 30 | 37 |
| 2c | PIDA | THF (2.0) | 30 | <10 |
| 3c | PIDA | PhCF3 (2.0) | 30 | 25 |
| 4c | PIDA | DMF (2.0) | 30 | <10 |
| 5c | PIDA | EA (2.0) | 30 | 31 |
| 6c | PIDA | DCM (2.0) | 30 | 35 |
| 7c | PIDA | t BuCN (2.0) | 30 | <10 |
| 8c | PIDA | MeCN (2.0) | 80 | 53 |
| 9c | PIDA | MeCN (2.0) | 70 | 47 |
| 10c | PIDA | MeCN (2.0) | 90 | 46 |
| 11 | PIDA | MeCN (2.0) | 80 | 60 |
| 12d | PIDA | MeCN (2.0) | 80 | 58 |
| 13e | PIDA | MeCN (2.0) | 80 | 58 |
| 14f | PIDA | MeCN (2.0) | 80 | 44 |
| 15g | PIDA | MeCN (2.0) | 80 | 36 |
| 16 | PIDA | MeCN (2.5) | 80 | 70 |
| 17 | PIDA | MeCN (3.0) | 80 | 65 |
| 18 | PIFA | MeCN (2.5) | 80 | 30 |
| 19 | BI-OH | MeCN (2.5) | 80 | Trace |
| 20 | BI-OAc | MeCN (2.5) | 80 | 53 |
| 21 | BI-N3 | MeCN (2.5) | 80 | 57 |
| 22 | DMP | MeCN (2.5) | 80 | <10 |
| 23h | BI-N3 | MeCN (2.5) | 80 | Trace |
With the optimized reaction conditions in hand, the generality of the protocol was assessed (Scheme 2). A variety of tertiary alcohols bearing either an electron-rich or electron-deficient substituent were converted into the corresponding ketone products (2a–2t). The use of substrates with para-, meta-, or ortho-substitution resulted in comparable yields (2b–2d, 2e–2g, and 2k–2m), probably suggesting that the steric hindrance of tertiary alcohols did not affect the reaction yield much. The moderate yields of products 2b–2d might be attributed to the competitive HAT with the benzylic C–H bonds of substrates.10 Other tertiary C(sp3)–H bonds, such as the cyclohexyl C–H bond (2u), and the C–H bond adjacent to the O-atom (2v) were also regioselectively functionalized. Not only aryl ketones but also some dialkyl ketones were generated, albeit with decreased yields (2w–2y). In addition to benzofuryl, other heteroaryls such as benzothienyl, benzothiazolyl, thiazolyl, benzoxazolyl, and oxazolyl also readily migrated in the reaction, leading to the desired products in synthetically useful yields (2z–2af).
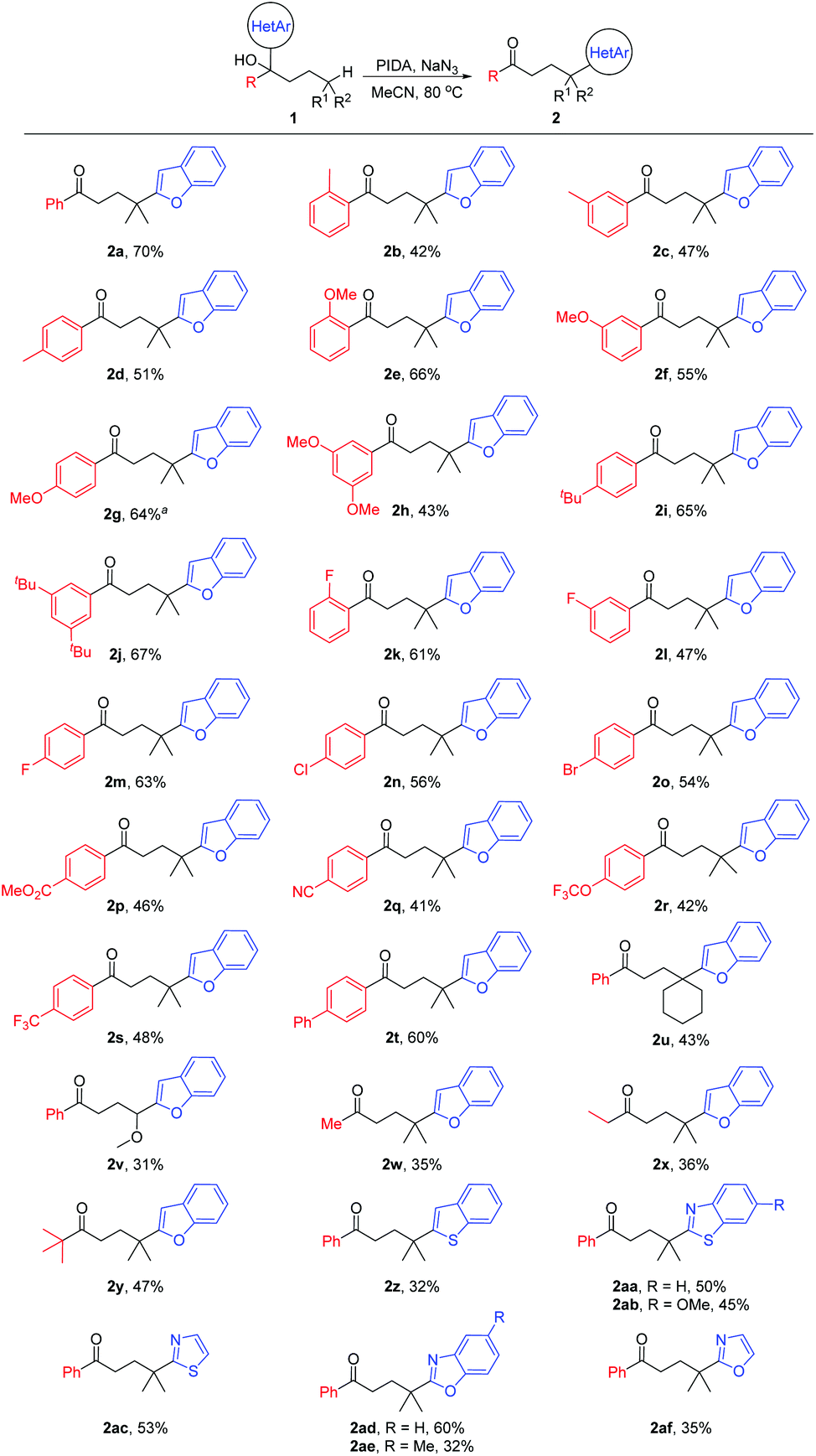 | ||
| Scheme 2 Substrate scope. Reaction conditions: 1 (0.2 mmol), PIDA (4.0 equiv.), and NaN3 (4.0 equiv.) in MeCN (2.5 mL), 80 °C for 12 h. Yields of the isolated products are given. aPIDA (6.0 equiv.) and NaN3 (6.0 equiv.). | ||
In order to illustrate the utility of the product, the transformations of 2a were performed. Upon treating 2a with methyl lithium, the tertiary alcohol 3 was generated, which was then subjected to intramolecular annulation via dehydration in the presence of formic acid, leading to the valuable ring-fused heterocyclic product 4 in a high yield (Scheme 3a). Treating 2a with hydroxylamine hydrochloride and sodium acetate generated the oxime 5, which served as the precursor of Beckmann rearrangement to afford the corresponding amide 6 (Scheme 3b).
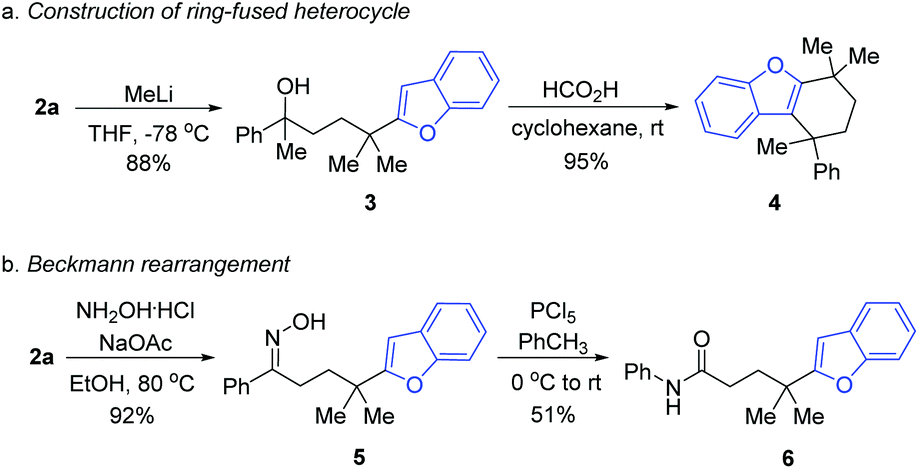 | ||
| Scheme 3 Synthetic applications. | ||
Afterwards, the migration distance was varied in the substrates to probe the transition states that might influence the heteroaryl migration (Scheme 4). In Scheme 2, all products were obtained via 1,4-heteroaryl migration, facilitated by a kinetically favoured five-membered cyclic intermediate. The 1,2-/1,5-benzofuryl migration via a three- or six-membered cyclic intermediate also proceeded, however, affording the corresponding products 2ag and 2ai in low yields (Scheme 4a and c). In sharp contrast, the 1,3-benzofuryl migration did not proceed owing to the disfavoured four-membered cyclic intermediate (Scheme 4b).
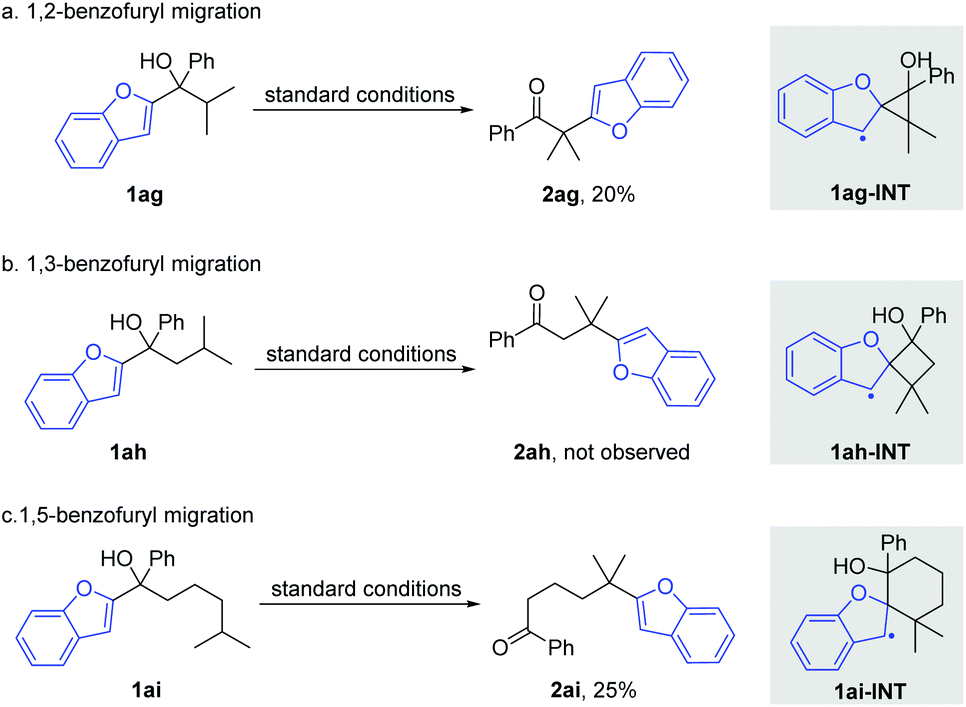 | ||
| Scheme 4 Investigations on the preference of cyclic intermediates. | ||
Based on the experimental results, a proposed mechanism is shown in Scheme 5. The interaction of PIDA with NaN3 generates an azido radical that regioselectively abstracts a hydrogen atom from the tertiary C(sp3)–H bond with the lowest bond dissociation energy. The resulting alkyl radical intermediate A undergoes intramolecular cyclization to generate the spiro radical intermediate B. The subsequent 1,4-heteroaryl migration via a five-membered cyclic intermediate results in the metastable ketyl radical C. Finally, single-electron oxidation of C with PIDA followed by deprotonation furnishes the desired product 2a.
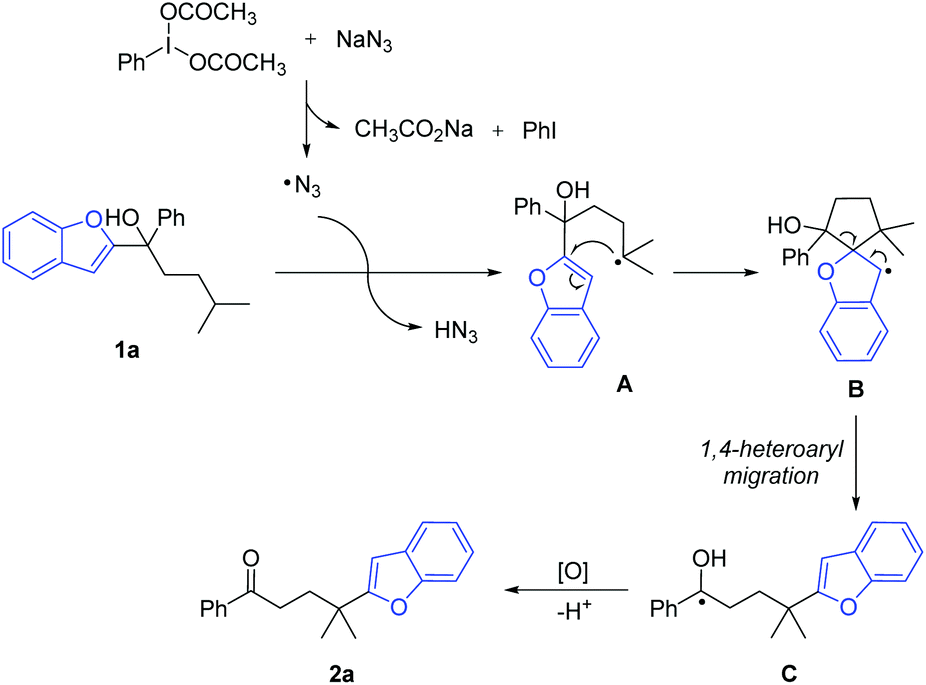 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
In summary, a radical-mediated remote heteroarylation of unactivated C(sp3)–H bonds of tertiary alcohols has been disclosed, leading to a variety of useful heteroaryl-substituted aliphatic ketones. The reaction proceeds via a cascade of azido radical-mediated HAT and intramolecular heteroaryl migration, in which an inert C–H bond and C–C bond are consecutively cleaved under mild conditions. A library of O-/S-/N-containing heteroaryls, such as benzofuryl, benzothiazolyl, benzothienyl, benzoxazolyl, oxazolyl, and thiazolyl, readily migrate in the reactions. Mechanistic studies reveal that the heteroaryl migration prefers a five-membered cyclic intermediate. Moreover, the transformation features broad functional group tolerance and good regioselectivity. This protocol offers an ingenious approach for selective functionalization of inert C(sp3)–H bonds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (grant no. 21971173, 22001185, 21772140), the Project of Scientific and Technologic Infrastructure of Suzhou (SZS201905), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- (a) N. A. McGrath, M. Brichacek and J. T. Njardarson, A graphical journey of innovative organic architectures that have improved our lives, J. Chem. Educ., 2010, 87, 1348 CrossRef CAS; (b) A. J. Waldman, T. L. Ng, P. Wang and E. P. Balskus, Heteroatom−heteroatom bond formation in natural product biosynthesis, Chem. Rev., 2017, 117, 5784 CrossRef CAS PubMed; (c) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Heterocyclic nanographenes and other polycyclic heteroaromatic compounds: synthetic routes, properties, and applications, Chem. Rev., 2017, 117, 3479 CrossRef PubMed; (d) Z. Cai, M. A. Awais, N. Zhang and L. Yu, Exploration of syntheses and functions of higher ladder-type π-conjugated heteroacenes, Chem, 2018, 4, 2538 CrossRef CAS; (e) M. N. Peerzada, E. Hamel, R. Bai, C. T. Supuran and A. Azam, Deciphering the key heterocyclic scaffolds in targeting microtubules, kinases and carbonic anhydrases for cancer drug development, Pharmacol. Ther., 2021, 225, 107860 CrossRef CAS PubMed.
- For reviews on radical-mediated heteroaryl migration, see: (a) X. Wu and C. Zhu, Radical-mediated remote functional group migration, Acc. Chem. Res., 2020, 53, 1620 CrossRef CAS PubMed; (b) X. Wu, S. Wu and C. Zhu, Radical-mediated difunctionalization of unactivated alkenes through distal migration of functional groups, Tetrahedron Lett., 2018, 59, 1328 CrossRef CAS; (c) X. Wu, Z. Ma, T. Feng and C. Zhu, Radical-mediated rearrangements: past, present, and future, Chem. Soc. Rev., 2021 10.1039/d1cs00529d.
- (a) Z. Wu, D. Wang, Y. Liu, L. Huan and C. Zhu, Chemo- and regioselective distal heteroaryl ipso-migration: a general protocol for heteroarylation of unactivated alkenes, J. Am. Chem. Soc., 2017, 139, 1388 CrossRef CAS PubMed; (b) H. Zhang, X. Wu, Q. Zhao and C. Zhu, Copper-catalyzed heteroarylsilylation of unactivated olefins through distal heteroaryl migration, Chem. – Asian J., 2018, 13, 2453 CrossRef CAS PubMed; (c) D. Chen, Z. Wu, Y. Yao and C. Zhu, Phosphinoyl-functionalization of unactivated alkenes through phosphinoyl radical-triggered distal functional group migration, Org. Chem. Front., 2018, 5, 2370 RSC; (d) M. Wang, Z. Wu, B. Zhang and C. Zhu, Azidoheteroarylation of unactivated olefins through distal heteroaryl migration, Org. Chem. Front., 2018, 5, 1896 RSC; (e) D. Chen, M. Ji, Y. Yao and C. Zhu, Difunctionalization of unactivated alkenes through SCF3 radical-triggered distal functional group migration, Acta Chim. Sin., 2018, 76, 951 CrossRef CAS; (f) J. Yu, D. Wang, Y. Xu, Z. Wu and C. Zhu, Distal functional group migration for visible-light induced carbo-difluoroalkylation/monofluoroalkylation of unactivated alkenes, Adv. Synth. Catal., 2018, 360, 744 CrossRef CAS; (g) N. Tang, S. Yang, X. Wu and C. Zhu, Visible-light-induced carbosulfonylation of unactivated alkenes via remote heteroaryl and oximino migration, Tetrahedron, 2019, 75, 1639 CrossRef CAS; (h) H. Zhang, L. Kou, D. Chen, M. Ji, X. Bao, X. Wu and C. Zhu, Radical-mediated distal ipso-migration of O/S-containing heteroaryls and DFT studies for migratory aptitude, Org. Lett., 2020, 22, 5947 CrossRef CAS PubMed; (i) H. Zhang, M. Ji, Y. Wei, H. Chen, X. Wu and C. Zhu, Radical-mediated hetaryl functionalization of nonactivated alkenes through distal ipso-migration of O- or S-hetaryls, Synlett, 2021, 32, 401 CrossRef CAS.
- (a) J. Yu, Z. Wu and C. Zhu, Efficient docking-migration strategy for selective radical difluoromethylation of alkenes, Angew. Chem., Int. Ed., 2018, 57, 17156 CrossRef CAS PubMed; (b) J. Liu, S. Wu, J. Yu, C. Lu, Z. Wu, X. Wu, X.-S. Xue and C. Zhu, Polarity umpolung strategy for the radical alkylation of alkenes, Angew. Chem., 2020, 59, 8195 CrossRef CAS PubMed; (c) T. Niu, J. Liu, X. Wu and C. Zhu, Radical heteroarylalkylation of alkenes via three-component docking-migration thioetheri-fication cascade, Chin. J. Chem., 2020, 38, 803 CrossRef CAS; (d) H. Zhang, M. Wang, X. Wu and C. Zhu, Designed heterocyclizing reagents for rapid assembly of N-fused heteroarenes from alkenes, Angew. Chem., 2021, 60, 3714 CrossRef CAS PubMed; (e) M. Ji, X. Wang, J. Liu, X. Wu and C. Zhu, Catalyst-free, radical-mediated intermolecular 1,2-arylheteroarylation of alkenes by cleaving inert C-C bond, Sci. China: Chem., 2021 DOI:10.1007/s11426-021-1077-4; (f) M. Ji, C. Chang, X. Wu and C. Zhu, Photocatalytic intermolecular carboarylation of alkenes by selective C−O bond cleavage of diarylethers, Chem. Commun., 2021, 57, 9240 RSC.
- For selected reviews, see: (a) X. Chen, K. M. Engle, D. H. Wang and J.-Q. Yu, Palladium(II)-catalyzed C-H activation/C-C cross-coupling reactions: versatility and practicality, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (b) J. Yamaguchi, A. D. Yamaguchi and K. Itami, C-H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS; (c) S. A. Girard, T. Knauber and C.-J. Li, The cross-dehydrogenative coupling of C(sp3)-H bonds: a versatile strategy for C-C bond formations, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (d) L. Yang and H. Huang, Transition-metal-catalyzed direct addition of unactivated C-H bonds to polar unsaturated bonds, Chem. Rev., 2015, 115, 3468 CrossRef CAS PubMed; (e) K. Yang, M. Song, H. Liu and H. Ge, Palladium-catalyzed direct asymmetric C-H bond functionalization enabled by the directing group strategy, Chem. Sci., 2020, 11, 12616 RSC; (f) N. Y. S. Lam, K. Wu and J.-Q. Yu, Advancing the logic of chemical synthesis: C-H activation as strategic and tactical disconnections for C-C bond construction, Angew. Chem., 2021, 60, 15767 CrossRef CAS PubMed; (g) D. Aynetdinova, M. C. Callens, H. B. Hicks, C. Y. X. Poh, B. D. A. Shennan, A. M. Boyd, Z. H. Lim, J. A. Leitch and D. J. Dixon, Installing the “magic methyl” - C-H methylation in synthesis, Chem. Soc. Rev., 2021, 50, 5517 RSC.
- For selected examples, see: (a) J. C. K. Chu and T. Rovis, Complementary strategies for directed C(sp3)-H functionalization: a comparison of transition-metal-catalyzed activation, hydrogen atom transfer, and carbene/nitrene transfer, Angew. Chem., Int. Ed., 2018, 57, 62 CrossRef CAS; (b) S. Sarkar, K. P. S. Cheung and V. Gevorgyan, C-H functionalization reactions enabled by hydrogen atom transfer to carbon-centered radicals, Chem. Sci., 2020, 11, 12974 RSC; (c) W. Guo, Q. Wang and J. Zhu, Visible light photoredox-catalysed remote C–H functionalisation enabled by 1,5-hydrogen atom transfer (1,5-HAT), Chem. Soc. Rev., 2021, 50, 7359 RSC; (d) L. Capaldo, D. Ravelli and M. Fagnoni, Direct photocatalyzed hydrogen atom transfer (HAT) for aliphatic C-H bonds elaboration, Chem. Rev., 2021 DOI:10.1021/acs.chemrev.1c00263; (e) X.-Q. Hu, J.-R. Chen and W.-J. Xiao, Controllable remote C−H bond functionalization by visible-light photocatalysis, Angew. Chem., Int. Ed., 2017, 56, 1960 CrossRef CAS; (f) X. Wu and C. Zhu, Radical functionalization of remote C(sp3)-H bonds mediated by unprotected alcohols and amides, CCS Chem., 2020, 2, 813 CrossRef CAS; (g) X. Wu and C. Zhu, Recent advances in alkoxy radical-promoted C-C and C-H bond functionalization starting from free alcohols, Chem. Commun., 2019, 55, 9747 RSC; (h) F. W. Friese, C. Mück-Lichtenfeld and A. Studer, Remote C-H functionalization using radical translocating arylating groups, Nat. Commun., 2018, 9, 2808 CrossRef PubMed; (i) N. Radhoff and A. Studer, Functionalization of α-C(sp3)-H bonds in amides using radical translocating arylating groups, Angew. Chem., 2021, 60, 3561 CrossRef CAS PubMed.
- For selected examples from our group, see: (a) X. Wu, H. Zhang, N. Tang, Z. Wu, D. Wang, M. Ji, Y. Xu, M. Wang and C. Zhu, Metal-free alcohol-directed regioselective heteroarylation of remote unactivated C(sp3)-H bonds, Nat. Commun., 2018, 9, 3343 CrossRef PubMed; (b) S. Wu, X. Wu, D. Wang and C. Zhu, Regioselective vinylation of remote unactivated C(sp3)-H bonds: Access to complex fluoroalkylated alkenes, Angew. Chem., Int. Ed., 2019, 58, 1499 CrossRef CAS PubMed; (c) N. Tang, X. Wu and C. Zhu, Practical, metal-free remote heteroarylation of amides via unactivated C(sp3)-H bond functionalization, Chem. Sci., 2019, 10, 6915 RSC; (d) S. Yang, X. Wu, S. Wu and C. Zhu, Regioselective sulfonylvinylation of the unactivated C(sp3)-H bond via a C-centered radical-mediated hydrogen atom transfer (HAT) process, Org. Lett., 2019, 21, 4837 CrossRef CAS PubMed; (e) M. Wang, L. Huan and C. Zhu, Cyanohydrin-mediated cyanation of remote unactivated C(sp3)-H bonds, Org. Lett., 2019, 21, 821 CrossRef CAS PubMed; (f) S. Wu, X. Wu, Z. Wu and C. Zhu, Regioselective introduction of vinyl trifluoromethylthioether to remote unactivated C(sp3)-H bonds via radical translocation cascade, Sci. China: Chem., 2019, 62, 1507 CrossRef CAS; (g) X. Shao, X. Wu, S. Wu and C. Zhu, Metal-free radical-mediated C(sp3)-H heteroarylation of alkanes, Org. Lett., 2020, 22, 7450 CrossRef CAS PubMed; (h) T. Niu, S. Yang, X. Wu and C. Zhu, Remote C(sp3)-H vinylation via radical-mediated consecutive fission of C-H and C-C bonds, Org. Chem. Front., 2020, 7, 2981 RSC.
- X. Wu, M. Wang, L. Huan, D. Wang, J. Wang and C. Zhu, Tertiary-alcohol-directed functionalization of remote C(sp3)-H bonds by sequential hydrogen atom and heteroaryl migrations, Angew. Chem., Int. Ed., 2018, 57, 1640 CrossRef CAS PubMed.
- For selected examples, see: (a) A. P. Antonchick and L. Burgmann, Direct selective oxidative cross-coupling of simple alkanes with heteroarenes, Angew. Chem., Int. Ed., 2013, 52, 3267 CrossRef CAS PubMed; (b) Y. Wang, X. Hu, C. A. Morales-Rivera, G.-X. Li, X. Huang, G. He, P. Liu and G. Chen, Epimerization of tertiary carbon centers via reversible radical cleavage of unactivated C(sp3)-H bonds, J. Am. Chem. Soc., 2018, 140, 9678 CrossRef CAS PubMed; (c) A. S. H. Ryder, W. B. Cunningham, G. Ballantyne, T. Mules, A. G. Kinsella, J. Turner-Dore, C. M. Alder, L. J. Edwards, B. S. J. McKay, M. N. Grayson and A. J. Cresswell, Photocatalytic α-tertiary amine synthesis via C-H alkylation of unmasked primary amines, Angew. Chem., 2020, 59, 14986 CrossRef CAS PubMed; (d) L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, Manganese-catalyzed oxidative azidation of C(sp3)-H bonds under electrophotocatalytic conditions, J. Am. Chem. Soc., 2020, 142, 17693 CrossRef CAS PubMed.
- (a) Y. Zhang and C.-J. Li, DDQ-mediated direct cross-dehydrogenative coupling (CDC) between benzyl ethers and simple ketones, J. Am. Chem. Soc., 2006, 128, 4242 CrossRef CAS PubMed; (b) Z. Li, Q. Wang and J. Zhu, Copper-catalyzed remote C(sp3)-H arylation of carboxamides and sulfonamides, Angew. Chem., Int. Ed., 2018, 57, 13288 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qo01209f |
| ‡ These authors have contributed equally to this paper. |
| This journal is © the Partner Organisations 2021 |