
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mass spectral and theoretical investigations of the transient proton-bound dimers on the cleavage processes of the peptide GHK and its analogues†
Jinhu Wang *a,
Cheng Wangb,
Han Zhanga,
Yang Liua and
Tiesheng Shia
*a,
Cheng Wangb,
Han Zhanga,
Yang Liua and
Tiesheng Shia
aCollege of Chemistry, Chemical Engineering and Materials Science, Zaozhuang University, Zaozhuang 277160, Shandong Province, P. R. China
bDepartment of Traditional Chinese Medicine, Zaozhuang Municipal Hospital, Zaozhuang 277102, Shandong Province, P. R. China. E-mail: wangjinhu@uzz.edu.cn
First published on 20th January 2021
Abstract
Fragmentation mechanisms of the singly protonated peptides GHK, GHKH and HGHK have been investigated by mass spectrometry and theoretical calculations. Fragmentation behavior of the protonated H–K amide bond in GHK was changed completely when a histidinyl residue was introduced into the C-terminus of GHK. The H–K amide bond breaking was a predominant pathway in the case of GHK and GHKH. For HGHK, the histidinyl residue at the N-terminus hampered significantly breaking of the H–K amide bond resulting in a high potential energy barrier; calculations indicated that this histidinyl effect played a vital role for the H–K amide bond fragmentation. Subsequent analysis of the fragmentation mechanism revealed that recombination processes of the hydrogen bonding for the intermediate products were all exergonic. Formation of a proton-bound dimer (PBD) lowering the energy barriers from a thermodynamic perspective for all the designed fragmentation pathways was demonstrated to be feasible by our systematic calculations. Moreover, the involvement of different PBDs was further confirmed by analyses of the reduced density gradient (RDG) isosurfaces and scatter maps. A dynamically favored pathway was likely via six-membered ring or nine-membered ring structures generated by the diketopiperazine as revealed by atom-in-molecules (AIM) analyses, since the steric interaction energies in the newly formed ring were estimated to be relatively small when compared to the products generated from a lactam and/or an oxazolone pathway. This is the first feasibility investigation from a dynamic viewpoint for formation of different rings involved in the lactam, oxazolone or diketopiperazine pathways in the fragmentation mechanisms proposed.
1. Introduction
Histidine (His) is an essential amino acid that belongs to the group of aromatic and heterocyclic amino acids.1 His-containing peptide GHK is known as a wound healing factor.2 It is normally present in human plasma, saliva, and urine, and it stimulates collagen, dermatan sulfate, chondroitin sulfate, and the small proteoglycan, decorin.3 GHK also enhances osteogenesis of cord blood mesenchymal stem cells, induces stem cell osteogenic differentiation,4 and participates actively in the processes of wound healing and tissue repair.5 The wound healing and antiaging properties of GHK are due probably to its strong affinity for copper ions (Cu2+) to form GHK–Cu complexes.6–10Except for formation of the GHK–Cu complexes, protonation is also favored for GHK since the proton affinity of the His side chain is high.11–13 Experimentally and theoretically, there is compelling evidence for formation of nondirectly sequential ions via cyclization/reopening chemistry in the collision-induced dissociation (CID) spectra of the bn ions when the His residue is near the C-terminus of peptides.14 The MS/MS spectra of the protonated peptides (HAAAAA, AHAAAA, AAHAAA, AAAHAA, and AAAAHA) show a series of bn ions with a enhanced cleavage at the amide bond of the C-terminus to His, rendering a histidinyl effect.14
Histidinyl effect originates from a mobilization of the proton located at the His side chain to the nitrogen of the C-terminally neighboured amide bond.11–13 A subsequent nucleophilic attack of the imidazolyl nitrogen on the carbon of the protonated amide bond can generate a transient lactam structure, which is a non-classical oxazolone derivative (Path a in Scheme 1). Moreover, an isomerization from the transient lactam to an oxazolone is potentially possible.15 Formation of the well-known oxazolone involves a nucleophilic attack of backbone amide oxygen (the N-terminally neighboured amide bond) on the C-terminally adjacent nitrogen of the protonated amide bond (Path b in Scheme 1).16 Several studies have suggested that the oxazolone structure is likely a b2 isomer.17–20 Moreover, the possibility for an isomerization between an oxazolone and a diketopiperazine form has been exploited.21 In the diketopiperazine pathway, the N-terminal amino group acts as a nucleophile and attacks the carbonyl carbon of the second amino acid residue, forming a more stable six-membered ring structure (Path c in Scheme 1).22 Such a cyclized ion involves a trans–cis isomerization of the amide bond.13 The fragmentation patterns of His-containing b2 ions from the protonated HG–OMe, GH–OMe, HGG–OMe, and GHG–OMe were compared with the diketopiperazine structure generated from dipeptide GH and very similar fragmentation spectra were found, supporting the formation of a diketopiperazine (cyclic dipeptide) b2 ion.12
 | ||
| Scheme 1 Possible mechanisms for the protonated GHK leading to amide bond fragmentation: (a) lactam pathway; (b) oxazolone pathway; (c) diketopiperazine pathway. | ||
A spectroscopic study of protonated tripeptide AAA and a cyclo dipeptide (cyclo-AA) by infrared multiple photon dissociation (IRMPD) technique indicated that the product is not a diketopiperazine, rather an oxazolone protonated at the oxazolone N-atom.23 Another IRMPD study of the protonated peptide HAAAA and cyclic dipeptide cyclo (HA) provided the first spectroscopic evidence for the presence of a mixture of oxazolone and diketopiperazine.24 A jointly multistage mass spectral and ab initio study demonstrated that the cyclic structures involving the side chains of Arg, His, and Lys, are more stable than the oxazolone ones.25
A study of His-containing peptides including FH–OMe, FH–NH2, FHA, HF–OMe, HF–NH2 and HFA by energy-resolved electrospray/Quadrupole/Time-of-Flight (QqToF) and by Density Functional Theory (DFT) calculations showed that the position of the His residue indeed influences the identity of the subsequent b2 ion, where the leaving group significantly affects the transition state energies and structures of the b2 ion.21 Under the conditions of low-energy collisions for the mass spectral measurements, the loose complex of the protonated oxazolone derivative and the leaving C-terminal fragment could undergo a rearrangement, resulting in formation of a proton-bound dimer (PBD) (see Scheme 1).26 Furthermore, under the low-energy conditions, the lifetime of the dimer was long enough so that various PBD isomers could interconvert27 and proton transfers took place between the proton donors and acceptors.28,29
The separation of PBD was the final step in most of the fragmentation mechanisms proposed for the protonated peptides.15 However, most investigations were focusing only on the products of charged ions (such as lactam, oxazolone or diketopiperazine ions), where the leaving groups (neutral counterparts) were not investigated systematically. The structure of neutral counterparts affects the energies of the dissociation reaction (as does the structure of the product ion), which in turn has a major impact on the preferred dissociation pathway and the structure of the product ion. Occasionally, neutral counterparts of charged ions could attract a proton and form a new charged ion.13,30 When only a PBD was formed, the followed dissociation reactions possibly generated bx or yz ions.15
In this work, the fragmentation reactions were investigated involving some leaving groups and formation of PBDs which are labeled by Pn′ in the text. The fragmentations of protonated GHK and of its analogues GHKH and HGHK were analyzed for examinations of the influence of His residue. All the three pathways described in Scheme 1 contain different dissociation channels, leading to formation of a mixture of three isomeric forms of the N-terminus fragments. Since the PBD was vital in the formation of bx or yz ions, cleavage processes of the peptide GHK and its analogues (GHKH and HGHK) were studied. As the Lac pathway only took place on the amide bond of H–K for our subjects, the cleavage of H–K amide bond was selected as the cleavage amide bond for the experimental and theoretical study in the following Lac, Dik and Oxa pathways. The identification of the intermediates in these pathways bolsters the dissociation mechanisms, enabling us to have a deeper understanding of the cleavage behavior of H–K amide bond in these peptides.
2. Experimental and theoretical methods
2.1 Experimental section
The peptides GHK, GHKH and HGHK with a purity ≥ 95% were purchased from Guoping Pharmaceutical Inc. (Hefei, China); they were used without further purification. Stock solutions of 0.1 mM peptides were prepared by dissolving desired amounts of the peptides in a solvent mixture of water and methanol (in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) containing 0.5% formic acid); HPLC-grade methanol and ultrapure deionized water (>18.2 M Ω cm−1) were used as the solvents.
:1 (v/v) containing 0.5% formic acid); HPLC-grade methanol and ultrapure deionized water (>18.2 M Ω cm−1) were used as the solvents.
The stock solutions were diluted to 20 μM by water, and then directly infused, at a rate of 15 μL min−1, into the capillary of an electrospray ionization (ESI) tandem mass spectrometer (LTQ Orbitrap Velos, Thermo Scientific, San Jose, CA, USA); methanol was used as the mobile phase. Electrospray and ion focusing conditions were adjusted to maximize the relative abundance of the singly protonated peptides and fragmented ions. Full scan mass spectra with a mass window of 1000 Da were acquired after an accumulation of about 1 × 106 counts in the positive mode. Helium was used as the collision gas for the ion trap at a pressure of 2.05 × 10−5 torr. Protonated peptides subjected to low energies of CID yield the complementary bn and yn series of sequence ions in the trap cell, where the ESI parameters were optimized to be: electrode needle voltage of 3500 V, sample cone voltage of 55 V, extraction cone voltage of 0.50 V, source temperature of 110 °C, and cone nitrogen gas flow rate of 30 L h−1. Mass number deviation was set at 100 ppm, where the delta of retention time was adopted the default value of 0.5 min.
2.2 Theoretical methods
Since the electron densities in the non-covalent bonding regions are relatively low, examination of the non-covalent bonding interactions by evaluating the reduced density gradient (RDG) functions is a reliable approach.41 Secondary Hessian eigenvalue (λ2) varies in sign (sign(λ2)ρ) depending on a bonding nature, thus sign(λ2)ρ can be used as an indicator to distinguish the interactions of non-bonding (when sign(λ2)ρ > 0) from bonding (when sign(λ2)ρ < 0).42 Molecular structures and non-covalent bonding interactions were directly visualized and quantified from the analysis of 3-D RDG isosurfaces when the sign(λ2)ρ values were encoded with different colours.41 The analysis of 3-D RDG isosurfaces was also based on the optimized chemical structures38 resembling the AIM approach.
3. Results and discussion
3.1 Mass spectra
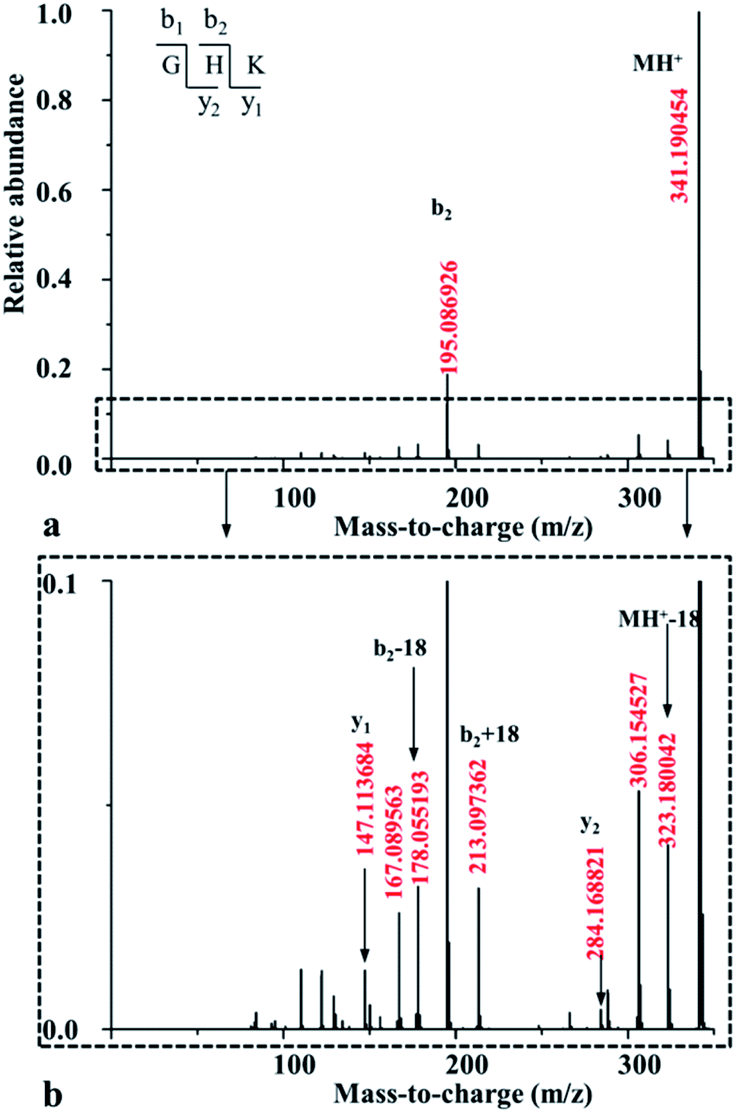 | ||
| Fig. 1 MS/MS spectrum acquired for the singly protonated GHK at the potential of 5.0 eV (a), together with its enlarged one in y-axis (b). | ||
| Precursor | Fragment | m/z | Relative abundance (%) |
|---|---|---|---|
| a (1) MH+ represents the precursor ion for singly protonated peptides; (2) relative abundances presented here were obtained in relative to the abundance of precursor ion at CID 5 eV. | |||
| GHKH | b2 | 195.085 | 15.6 |
| b3 | 323.178 | 13.5 | |
| y1 | 156.070 | 6.6 | |
| MH+-18-17 | 443.216 | 6.2 | |
| MH+ | 478.248 | 99.9 | |
| HGHK | a1 | 110.073 | 7.5 |
| b2 | 195.084 | 7.9 | |
| b3 | 323.150 | 2.1 | |
| y2 | 284.167 | 7.2 | |
| y3 | 341.190 | 3.0 | |
| MH+-18 | 460.235 | 4.3 | |
| MH+ | 478.251 | 99.9 | |
For both GHKH and HGHK, the precursor ions (MH+ in Table 1) are all at a relative abundance of 99.9%; only a small percentage of precursor ions fragments at low energies, base on which we can judge which amide bond is easily cleavage. Apart from the loss of a water and/or an ammonia (MH+-18-17 for GHKH and MH-18 for HGHK), other site cleavages occurred entirely at the backbone amide bonds at CID potential of 5 eV. The mass peak at m/z 478.248 is a precursor ion for the protonated GHKH. Similar to protonated GHK, the relative abundance of the b2-type fragment ion (m/z 195.085) is increased to 15.6%, indicating that a cleavage of the H–K amide bond is preferred. Obviously, the addition of an His residue to the C-terminus of GHK does not hamper the H–K amide bond cleavage. Thus, all the three fragmentation pathways described in Scheme 1 are all possible to occur rendering a formation of the b2 ion similar to the case of the protonated GHK. Another peak at m/z 323.178 is assigned to the truncated GHK fragment as the b3-type ion, being generated by a cleavage of the K–H amide bond. The y1-type fragment ion derived from the K–H amide bond cleavage is also detectable. Comparing with that of b3 ion, the strength of this ion is relatively lower, suggesting that b3 is the dominant ion for the K–H amide bond cleavage. On the other hand, the cleavage at the N-terminal side of His from the G–H amide bond is not detectable.
The mass peak at m/z 478.251 in Table 1 corresponds to the protonated ion of HGHK. A dominant peak at m/z 110.073, differentiating from those of the protonated GHK and GHKH, is a secondary fragmentation product from an amide cleavage by loss of a CO (a b1 ion), being assigned to an a1-type fragment ion. Fragmentation efficiencies versus the applied collision energies for H–G amide bond are given in Fig. S5 in ESI.† Clearly, the collision energies have a significant influence on the H–G amide bond fragmentations, i.e., the higher of the energies, the stronger of the relative abundance of the a1-type fragment ions. Hence, the His effect determines if the cleavage at the C-terminus of His and the a1 ion is generated. The protonated GHK and GHKH did not produce a1 ions since GHK and GHKH possess the same N-terminal Gly and the charge cannot be stabilized by N-terminal fragment after cleavage. However, when an His residue is at the N-terminus, and the charge can be fixed by the side chain of the His.
Another major peak at m/z 195.085 is formed from the G–H amide bond cleavage which is assigned to a b2-type fragment ion being produced by the cleavage at the N-terminal His. Mass peak at m/z 284.167 is assigned to the truncated HK fragment as an y2-type ion, being generated from the cleavage of the G–H amide bond. A b3-type fragment ion derived from the H–K amide bond cleavage is also detectable. The strength of b3 ion is relatively lower compared with that of the b2 ion. It is thus concluded that the His residue at N-terminus can prohibit cleavage of the backbone H–K amide bond.
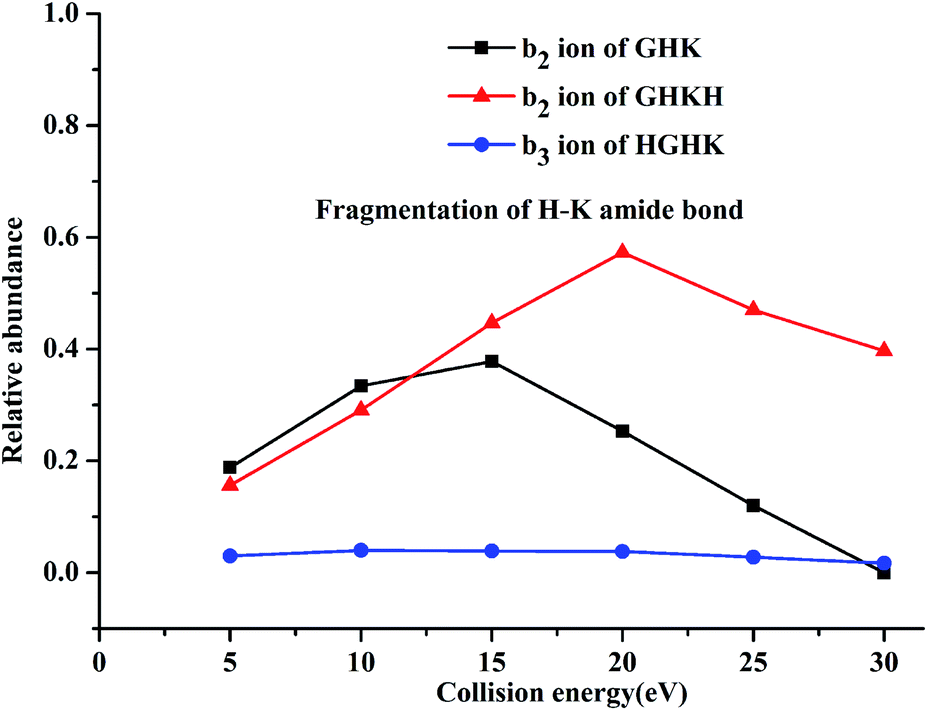 | ||
| Fig. 2 Relative abundances for the H–K amide bond fragmentations for the singly protonated peptidyl ions of GHK, GHKH and HGHK. | ||
3.2 Theoretical calculations
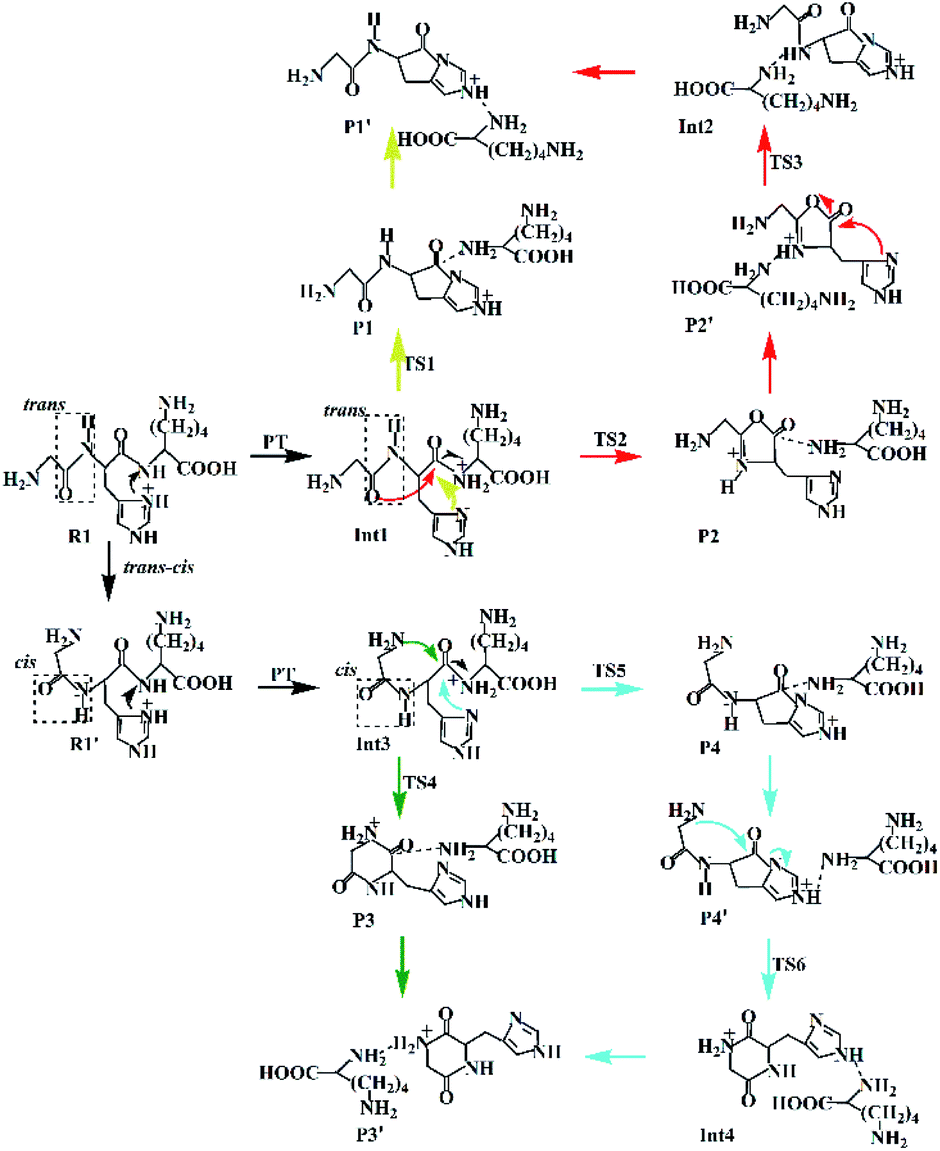 | ||
| Scheme 2 Proposed fragmentation mechanism for the protonated GHK. | ||
Reaction coordinates of all the optimized structures for the protonated GHK and its analogues are listed in Fig. S6–S16 in the ESI† while energies of all the optimized structures are given in Tables S1–S3 in the ESI.† Energy profiles of the designed reaction pathways are shown in Fig. S17–S19 in the ESI,† and the major optimized transition states for the H–K amide bond cleavage are given in Fig. S20–S22 in the ESI.† These major transition states are further validated by IRC calculations and are shown in Fig. S23–S25 in the ESI.†
(a) The lactam direct pathway. In the lactam pathway, reactant R1 firstly undergoes a proton transfer from the His side chain to the H–K amide bond oxygen, where the intermediate Int1 is possibly formed. The cleavage of the carbonyl carbon to the H–K amide nitrogen occurs consecutively. The C–N amide bond cleavage directly affords the product P1 via the transition state TS1. This product can generate a PBD (P1′). The barrier to the H–K amide bond breaking from Int1 to P1 is calculated to be 29.6 kcal mol−1 (yellow line in Fig. 3). The formation of P1′ for the N- and C-terminal fragments (P1→P1′) readily takes place since P1′ is 7.8 kcal mol−1 lower in energy than that of the product P1.
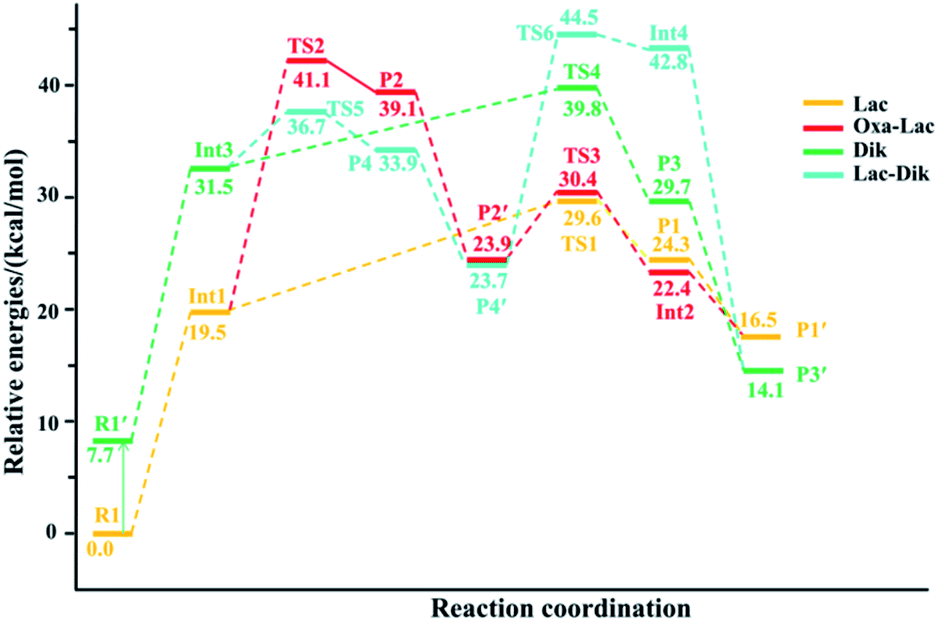 | ||
| Fig. 3 Energy profile for the H–K amide bond cleavage of the protonated GHK. | ||
Two nucleophilic attacks may occur sequentially for Int1 in the combined Oxa–Lac pathway, cf. Scheme 2. One attack takes place by the N-terminally neighboured amide oxygen on the carbon centre of the protonated amide bond, breaking the C-terminally neighboured amide bond and affording the oxazolone product P2 via the transition state TS2. This needs to overcome an energy barrier of 41.1 kcal mol−1 (the red line in Fig. 3). After the recombination of the hydrogen bonding model, a favoured product of P2′ is generated, and it is exergonic by 15.2 kcal mol−1 relative to P2. The other attack occurs by the imidazolyl nitrogen of the His on the amide carbonyl carbon, giving rise to the lactam intermediate Int2 via the transition state TS3 and requiring to overcome an energy barrier of 30.4 kcal mol−1.
(b) The diketopiperazine direct pathway. Starting from R1, the trans–cis isomerization of the N-terminal G–H amide bond gives the potential intermediate R1′ (green line in Scheme 2) with an endothermic energy of 7.7 kcal mol−1 (green line in Fig. 3). Thus, structures generated from the diketopiperazine (green line) and the lactam-diketopiperazine (cyan line) pathways all have the cis G–H amide bond. The initial part of the diketopiperazine pathway also involves a proton transfer from R1′ to the intermediate Int3 requiring an energy of 23.8 kcal mol−1 (Fig. 3). Int3 then undergoes a cyclization by a nucleophilic attack of the N-terminal amine group on the amide carbonyl carbon (green line in Scheme 2) via the transition state TS4, yielding the diketopiperazine product P3 with an energy barrier of 39.8 kcal mol−1 (TS4 in Fig. 3). Subsequently, P3 recombines the hydrogen bonding model, and the favourable product P3′ is generated with an exergonic energy of 15.6 kcal mol−1 relative to P3.
(c) The combined lactam–diketopiperazine direct pathway. Starting from Int3, two nucleophilic attacks are proposed to occur sequentially in the combined Lac–Dik pathway shown in Scheme 2 (cyan line). A concerted nucleophilic attack of the imidazolyl nitrogen of the His on the carbon centre of the protonated amide bond proceeds firstly to afford the lactam product P4 via the transition state TS5 by overcoming an energy barrier of 36.7 kcal mol−1 (the cyan line in Fig. 3). After the recombination of the hydrogen bonding model, the favourable product P4′ is generated with an exergonic energy of 10.2 kcal mol−1 relative to P4. This is followed by another nucleophilic attack of the N-terminal amine group on the amide carbonyl carbon, giving rise to the diketopiperazine intermediate Int4 via the transition state TS6 with an energy barrier of 44.5 kcal mol−1.
The optimized transition states for protonated GHK are shown in Fig. S20 in the ESI.† The transition state TS1 is generated from the Lac pathway. The nucleophilic attack of the imidazolyl nitrogen of the His leads to an elongatation of the H–K amide bond to 2.00 Å. The electron rich imidazolyl nitrogen atom is about 1.56 Å away from the carbon of the protonated H–K amide bond. The optimized structures of TS2 and TS3 are obtained from the Oxa–Lac pathway. The nucleophilic attack makes the N-terminally adjacent carbonyl oxygen to be 1.59 Å away from the protonated amide carbon atom in TS2, where this kind of distance is 1.73 Å in TS3. The protonated amide bond is elongated to 2.35 Å in TS2, which is 0.35 Å longer than that in TS1. The transition state TS4 is achieved from the Dik pathway when the N-terminal amine group is 1.80 Å away from the amide carbonyl carbon, being accompanied with an elongation of the H–K amide bond to 1.77 Å. Another Lac–Dik pathway gives two transition states (TS5 and TS6). TS5 is obtained by an approach of the imidazolyl nitrogen of His onto the carbon centre of the protonated amide bond (distance of 1.53 Å), while TS6 is achieved by a nucleophilic attack of the N-terminal amine group on the amide carbonyl carbon (1.70 Å). Undoubtedly, the above nucleophilic attacks largely weaken the covalent bonds linking to the carbon centre of the protonated amide bond as shown by the bond lengths (2.21 and 1.94 Å for TS5 and TS6, respectively).
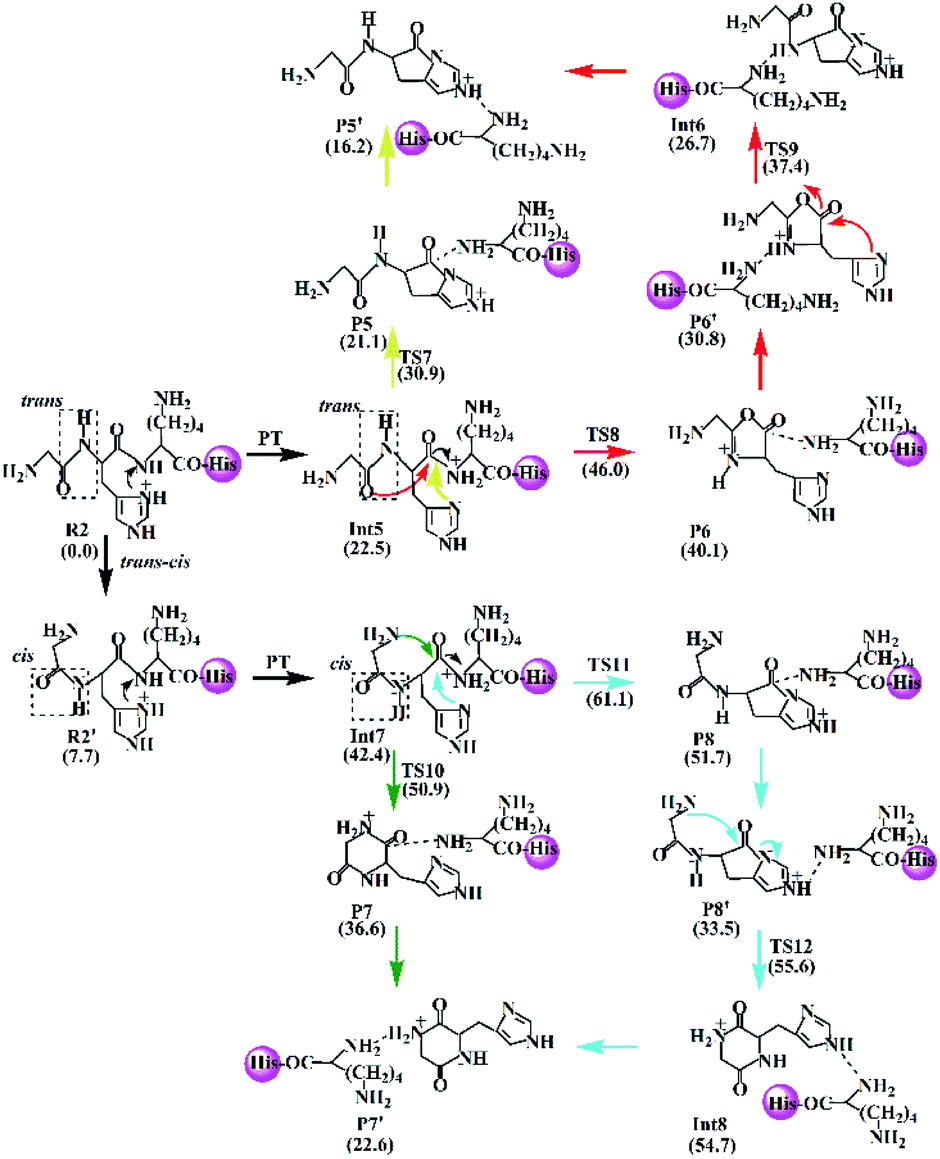 | ||
| Scheme 3 Proposed fragmentation mechanism for the protonated GHKH. | ||
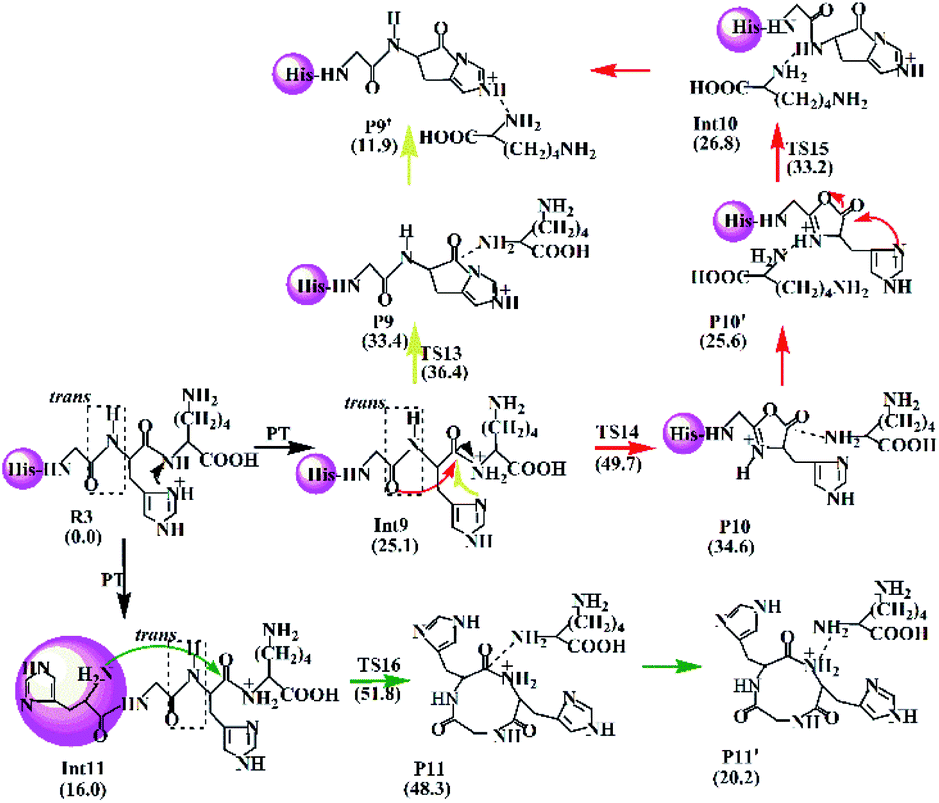 | ||
| Scheme 4 Proposed fragmentation mechanism for the protonated HGHK. | ||
The Lac pathway involves the reactant R3, the intermediate Int9, the product P9, the transition state TS13 and the PBD P9′; the combined Oxa–Lac pathway encompasses the intermediate Int11, the product P11, the transition state TS15 and the PBD P10′; the Dik direct pathway consists of the intermediates Int9 and Int10, the product P10, the transition state TS16 and the PBD P11′. The energy barriers are also shown in Scheme 4. The optimized transition states for protonated HGHK are shown in Fig. S22 in the ESI.† In TS13, the nucleophilic attack of the imidazolyl nitrogen of the His leads to an elongation of the H–K amide bond to 1.73 Å. The electron-rich imidazolyl nitrogen atom is about 1.69 Å away from the carbon of the protonated H–K amide bond. In TS14, the nucleophilic attack makes the N-terminally adjacent carbonyl oxygen at 1.63 Å away from the protonated amide carbon atom whereas this distance is 1.53 Å in TS15. The protonated amide bond is elongated to 2.13 Å in TS14, which is 0.40 Å longer than that in TS13. In TS16, the N-terminal amine group is 1.82 Å away from the amide carbonyl carbon and an elongation of the H–K amide bond is up to 2.01 Å.
Analyses in terms of relative energy in Fig. 3 and in Schemes 3 and 4 indicate that the Lac pathway is a low energy route for all the protonated peptides, which is confirmed by the energy profile (red line) along scan coordinates of protonated amide bond (r1) in Fig. 4. The range of the scan coordinates (r1) increases from about 1.5 to 3.0 Å, where variations of energy profiles and possible reaction pathways (Lac, Oxa and Dik) are investigated. For intermediates Int3, Int7, and Int11, two possible nucleophilic pathways are proposed. Differences of the two pathways lie in that the distance of r4 decreases firstly in the Dik pathway, where the length of r2 shortens preferentially in the Lac–Dik pathway. Fig. 4 shows that only distances of r2 become short quickly along with the cleavage of the protonated bond (r1), revealing that the nucleophilic attack of the imidazolyl nitrogen of the His takes place preferentially along with the elongation of the H–K amide bond for GHK analogues. This accounts for the lower energy barrier for the Lac pathway.
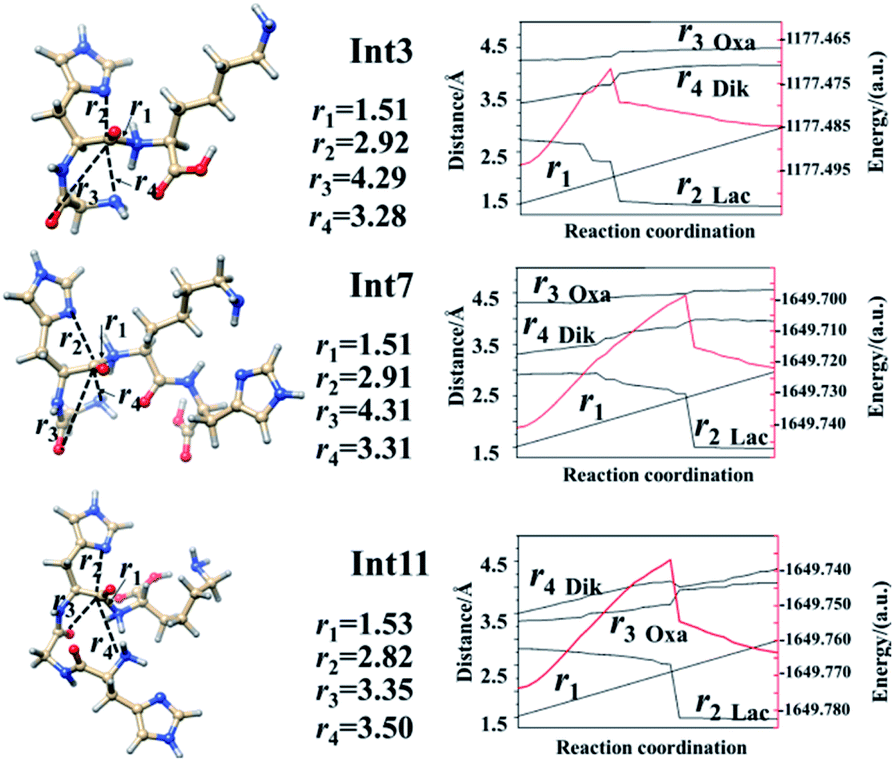 | ||
| Fig. 4 Energy profiles along the scan coordinate of protonated amide bonds (r1 in images, unit in Å). Left lines are optimized structures while right lines correspond to the energy profiles (unit in a.u.) of the left side structure. | ||
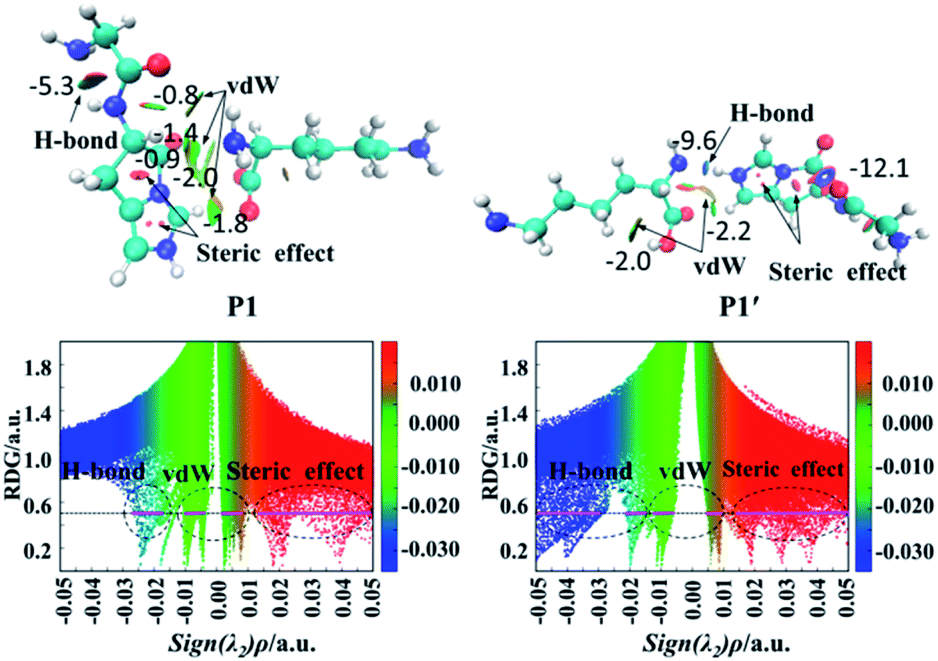 | ||
| Fig. 5 Graphic noncovalent interaction for products (P1 and P1′) obtained from the lactam pathway of the protonated GHK. (Upper panels): Isosurfaces (RDG = 0.5 a.u.). (Lower panels): Scatter maps of RDG versus sign(λ2)ρ value, where the RDG isosurface of the horizontal line is 0.5 a.u. The colour scale bar ranges from −0.035 to 0.02. | ||
Fig. 5 also gives graphically the noncovalent interaction for products P1 and P1′ by RDG isosurfaces and scatter maps. The upper panels correspond to the RDG isosurfaces at RDG = 0.5 a.u. Each colour-filled region is called a slab. For the RDG isosurface of P1, the slab between N-terminal amine group and hydrogen atom of the N-terminally neighboured amide bond shows a deeply green colour, implying that the interaction there is a weak hydrogen bond (H-bond). The lower panels correspond to scatter maps of RDG versus the value of sign(λ2)ρ. There are several shapes with the points at their peaks approaching the horizontal axis, which correspond to different types of interactions. A horizontal line was drawn on the graph of RDG = 0.5 a.u, the segments crossing these colour regions corresponded to the points was then used to construct the RDG isosurfaces in the upper panels of this figure. The horizontal line corresponding H-bond segment (lower panels in Fig. 5) is the shortest among the three segments. The slab between carbonyl group of the breaking amide bond and the leaving group is marked by the green region in which a van der Waals (vdW) contact interaction is identified.
Analysis of the scatter maps endows that there are two vdW segments crossing green regions (lower panels in Fig. 5) which are relatively longer than that of an H-bond segment. The vdW region is dominated apparently by the main interactions between the binding of the N-terminal and C-terminal fragments. Besides, regions at the centre of the imidazolyl ring and at the newly formed ring generated by the nucleophilic attack were filled by the red colour, corresponding to steric interactions. The electronic density in these regions is high since the mapped colour is red. The horizontal line crossing the red region is the longest, suggesting strong steric interactions.
The RDG isosurface and scatter map of P1′ are given on right side of Fig. 5. The segment of horizontal line crossing the blue region at RDG = 0.5 a.u is much longer than that of P1, which is also true for other PBDs. The large segment crossing the blue region contributes to a blue H-bond slab between the amine group of truncated C-terminal fragment and the imidazolyl ring of N-terminal fragment.
The AIM plots of interacting energies for products P1 and P1′ based on critical points (CPs) are shown in Fig. 6, while similar plots for other products are given in Fig. S36–S46 in the ESI.† For electron density analysis, the bond critical point (BCP) (3, −1) coloured in the orange sphere reflects the atomic interactions, and it has higher electron density if the calculated EHB values are more negative. For P1, no EHB values are more than 2.0 kcal mol−1 between the N-terminal and C-terminal fragments, even the sum of the EHB values is only about −6.9 kcal mol−1. Thus, the truncated C-terminal fragment interacts with the protonated N-terminal fragment weakly. These small values are consistent with the green slabs between carbonyl group of the breaking amide bond and the truncated department shown in Fig. 5. Hence, the vdW interactions are confirmed by these small EHB values. Structural analyses indicate that the truncated C-terminus only forms a weak H-bond (C–H⋯N) with a distance of 2.61 Å (upper panels of Fig. 6). As to P1′, the recombination of the hydrogen bonding model generates strong interactions by hydrogen bond with the bond distance of 1.85 Å (the upper panels of Fig. 6). The amine group of the truncated C-terminal fragment can form a strong hydrogen bond with the positively charged imidazolyl ring of the His, the calculated EHB value is up to −9.6 kcal mol−1. The largely negative value of EHB certainly contributes to the blue H-bond slab in the RDG isosurface (Fig. 5).
 | ||
| Fig. 6 The AIM plots of interaction energies for products P1 and P1′. CPs (3, −1) based on critical points (CPs) with orange spheres (upper panels), CPs (3, +1) with yellow spheres (lower panels), and the bond paths connecting (3, −1) with brown lines. | ||
For electron density analysis, CP (3, +1) is often named as ring critical point (RCP), generally displaying the steric interactions. Interaction energies based on CPs (3, +1) for products P1 and P1′ are also shown in Fig. 6 with yellow sphere. Those energies obtained from the CPs (3, +1) between carbonyl group of the breaking amide bond and the truncated department are all small. On the other hand, the interaction energies generated from the CPs (3, +1) in the centre of ring are much larger. For P1 and P1′, each contains two rings which are the imidazolyl ring and the newly formed ring generated by the nucleophilic attack of the His side chain. The estimated interaction energy in the centre of the imidazolyl ring is almost constant (−27.3 kcal mol−1 for P1 versus −27.7 kcal mol−1 for P1′), where the energy in the centre of newly formed five-membered ring changes slightly (−16.1 kcal mol−1 for P1 versus −15.1 kcal mol−1 for P1′).
Examination of readiness of formation of the five-membered rings in the Lac and Oxa pathways and of six/nine-membered rings in the Dik pathway is of critical importance. Consequently, the relations of interaction energies in the centre of formed rings are depicted in Fig. 7, demonstrating that all estimated steric interaction energies in the Oxa pathway (P2 and P2′; P6 and P6′; P10 and P10′) are the largest among all the pathways. Those derived in the Dik pathway (P3 and P3′; P7 and P7′; P11 and P11′) are the smallest. Indeed, our results support the six-membered ring structure in the cases of GHK and GHKH and the nine-membered ring structure in the case of HGHK in the Dik pathway.12,13,22 The statistic values of attractive and steric interaction energies in the N-terminal and C-terminally truncated fragments are summarized in Table 2. An overall analysis unveils that the steric interactions are much larger than attractive interactions. Furthermore, the total attractive interactions become much big when the PBD is formed, i.e., the attractive interaction energy in P1 is −12.4 kcal mol−1, while it adds up to −25.9 kcal mol−1 in P1′. Clearly, the formation of the PBD enhances the stability.
 | ||
| Fig. 7 Relations of steric interaction energies obtained on CPs (3, +1); unit in kcal mol−1. | ||
| Products | Total of attractive interaction/(kcal mol−1) | Total of steric effect/(kcal mol−1) |
|---|---|---|
| P1 | −12.4 | −52.9 |
| P1′ | −25.9 | −60.8 |
| P2 | −14.2 | −62.3 |
| P2′ | −21.2 | −53.6 |
| P3 | −26.3 | −37.5 |
| P3′ | −48.4 | −43.5 |
| P4 | −16.7 | −48.3 |
| P4′ | −21.3 | −50.9 |
| P5 | −27.8 | −78.8 |
| P5′ | −51.6 | −90.5 |
| P6 | −11.4 | −70.8 |
| P6′ | −48.9 | −87.5 |
| P7 | −36.3 | −72.6 |
| P7′ | −59.8 | −72.1 |
| P8 | −18.8 | −79.9 |
| P8′ | −23.6 | −83.3 |
| P9 | −34.5 | −84.3 |
| P9′ | −30.1 | −85.2 |
| P10 | −20.6 | −88.3 |
| P10′ | −26.8 | −90.4 |
| P11 | −38.2 | −75.9 |
| P11′ | −46.7 | −72.8 |
4. Conclusions
Fragmenting mechanisms of the singly protonated GHK and its synthetic analogues GHKH and HGHK have been investigated by electrospray ionization tandem mass spectrometry, whereas the H–K amide bond breaking mechanisms are further investigated by theoretical methods. Fragmentations at the H–K amide bond of the protonated GHK and GHKH are the dominant cleavages observed experimentally. In the case of HGHK, the N-terminal His hampers significantly the breaking of H–K amide bond due to a high energy barrier. Theoretical calculations indicate that the His effect plays an important role for the H–K amide bond fragmentation. The energy barriers of the lactam pathway are lower than those of the classical Oxa and the Dik pathways. The recombination processes of the intermediate products being transferred to the PBDs are favoured thermodynamically since these transformations are all exergonic. We further demonstrated that formation of PBDs is definitely feasible which can reduce the energy barriers significantly for all the reaction pathways. A comparison of the RDG isosurface with the scatter map suggests that hydrogen bonding contributes largely to the stabilities of PBDs. An AIM analysis of the interaction energies in the newly formed ring at critical points (3, +1) indicate that a six-membered and a nine-membered ring structures generated in the Dik pathway is favoured dynamically when compared with products generated from the Lac or the Oxa pathways. This is the first feasibility investigation for the formation of different rings generated in the Lac, Oxa or Dik pathways from a dynamic perspective.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support of this work by grants from Natural Science Foundation of Shandong Province (ZR201702200464) and from the Critical R&D Plans of Shandong Province (Major Scientific Innovation Projects, 2019JZZY010516) is gratefully acknowledged. The calculations were performed on CNGrid (China National Grid).Notes and references
- F. Turecek, C. Yao, Y. E. Fung, S. Hayakawa, M. Hashimoto and H. Matsubara, Histidine-containing radicals in the gas phase, J. Phys. Chem. B, 2009, 113, 7347 CrossRef CAS.
- P. Gonzalez, K. Bossak, E. Stefaniak, C. Hureau, L. Raibaut, W. Bal and P. Faller, N-terminus Cu-binding motifs (Xxx-Zzz-His, Xxx-His) and their derivatives: chemistry, biology and medicinal applications, Chem.–Eur. J., 2018, 24, 8029 CrossRef CAS.
- L. Pickart, J. M. Vasquez-Soltero and A. Margolina, GHK peptide as a natural modulator of multiple cellular pathways in skin regeneration, BioMed Res. Int., 2015, 648048 Search PubMed.
- M. E. Klontzas, S. Reakasame, R. Silva, J. C. F. Morais, S. Vernardis, R. J. MacFarlane, M. Heliotis, E. Tsiridis, N. Panoskaltsis, A. R. Boccaccini and A. Mantalaris, Oxidized alginate hydrogels with the GHK peptide enhance cord blood mesenchymal stem cell osteogenesis: a paradigm for metabolomics-based evaluation of biomaterial design, Acta Biomater., 2019, 88, 224 CrossRef CAS.
- L. Sun, A. Li, Y. Hu, Y. Li, L. Shang and L. Zhang, Self-assembled fluorescent and antibacterial GHK–Cu nanoparticles for wound healing applications, Part. Part. Syst. Charact., 2019, 36, 1800420 CrossRef.
- L. Pickart and S. Lovejoy, Biological activity of human plasma copper-binding growth factor glycyl-L-histidyl-L-lysine, Methods Enzymol., 1987, 147, 314 CAS.
- T. Badenhorst, D. Svirskis and Z. Wu, Physicochemical characterization of native glycyl-L-histidyl-L-lysine tripeptide for wound healing and anti-aging: a preformulation study for dermal delivery, Pharm. Dev. Technol., 2016, 21, 152 CrossRef CAS.
- L. Pickart, J. M. Vasquez-Soltero and A. Margolina, The human tripeptide GHK–Cu in prevention of oxidative stress and degenerative conditions of aging: implications for cognitive health, Oxid. Med. Cell. Longevity, 2012, ID324832 Search PubMed.
- S. Sharma, A. Dua and A. Malik, Biocompatible stimuli responsive superabsorbent polymer for controlled release of GHK–Cu peptide for wound dressing application, J. Polym. Res., 2017, 24, 104 CrossRef.
- Z. Y. Liu, S. Y. Chen, F. F. Qiao and X. H. Zhang, Interaction of peptide backbones and transition metal ions: 1. an IM-MS and DFT study of the binding pattern, structure and fragmentation of Pd(II)/Ni(II)–polyalanine complexes, Int. J. Mass Spectrom., 2019, 438, 87 CrossRef CAS.
- C. Gu, G. Tsaprailis, L. Breci and V. H. Wysocki, Selective gas-phase cleavage at the peptide bond C-terminus to aspartic acid in fixed-charge derivatives of Asp-containing peptides, Anal. Chem., 2000, 72, 5804 CrossRef CAS.
- J. M. Farrugia, T. Taverner and R. A. J. O'Hair, Side-chain involvement in the fragmentation reactions of the protonated methyl esters of histidine and its peptides, Int. J. Mass Spectrom., 2001, 209, 99 CrossRef CAS.
- B. Paizs and S. Suhai, Fragmentation pathways of protonated peptides, Mass Spectrom. Rev., 2005, 24, 508 CrossRef CAS.
- B. J. Bythell, M. Knapp-Mohammady, B. Paizs and A. G. Harrison, Effect of the His residue on the cyclization of b ions, J. Am. Soc. Mass Spectrom., 2010, 21, 1352 CrossRef CAS.
- B. J. Bythell, Comment on: “Quantum chemical mass spectrometry: verification and extension of the mobile proton model for histidine” by Julie Cautereels and Frank Blockhuys, J. Am. Soc. Mass Spectrom. 28, 1227–1235 (2017), J. Am. Soc. Mass Spectrom., 2017, 28, 2728 CrossRef CAS.
- T. Yalcin, C. Khouw, I. G. Csizmadia, M. R. Peterson and A. G. Harrison, Why are b ions stable species in peptide spectra?, J. Am. Soc. Mass Spectrom., 1995, 6, 1165 CrossRef CAS.
- A. G. Harrison, I. G. Csizmadia and T. H. Tang, Structure and fragmentation of b2 ions in peptide mass spectra, J. Am. Soc. Mass Spectrom., 2000, 11, 427 CrossRef CAS.
- D. Y. J Huo, T. Qin and L. L. Zu, Energetic switch of the proline effect in collision-induced dissociation of singly and doubly protonated peptide Ala–Ala–Arg–Pro–Ala–Ala, J. Mass Spectrom., 2019, 54(1), 55 CrossRef.
- H. E. Aribi, C. F. Rodriquez, D. R. Almeida, Y. Ling, W. W. N. Mak, A. C. Hopkinson and K. M. Siu, Elucidation of fragmentation mechanisms of protonated peptide ions and their products: a case study on glycylglycylglycine using density functional theory and threshold collision-induced dissociation, J. Am. Chem. Soc., 2003, 125, 9229 CrossRef.
- B. J. Bythell, D. F. Barofsky, F. Pingitore, M. J. Polce, P. Wang, C. Wesdemiotis and B. Paizs, Backbone cleavages and sequential loss of carbon monoxide and ammonia from protonated AGG: a combined tandem mass spectrometry, isotope labeling, and theoretical study, J. Am. Soc. Mass Spectrom., 2007, 18, 1291 CrossRef CAS.
- C. R. Nelson, M. T. Abutokaikah, A. G. Harrison and B. J. Bythell, Proton mobility in b2 ion formation and fragmentation reactions of histidine containing peptides, J. Am. Soc. Mass Spectrom., 2016, 27, 487 CrossRef CAS.
- M. M. Cordero, J. J. Houser and C. Wesdemiotis, Neutral products formed during backbone fragmentations of protonated peptides in tandem mass spectrometry, Anal. Chem., 1993, 65, 1594 CrossRef CAS.
- J. Oomens, S. Young, S. Molesworth and M. van Stipdonk, Spectroscopic evidence for an oxazolone structure of the b2 fragment ion from protonated tri-alanine, J. Am. Soc. Mass Spectrom., 2009, 20, 334 CrossRef CAS.
- B. R. Perkins, J. Chamot-Rooke, S. H. Yoon, A. C. Gucinski, A. Somogyi and V. H. Wysocki, Evidence of diketopiperazine and oxazolone structures for HA b2+ ion, J. Am. Chem. Soc., 2009, 131, 17528 CrossRef CAS.
- J. M. Farrugia, A. J. Richard and G. E. Reid, Do all b2 ions have oxazolone structures? Multistage mass spectrometry and ab initio studies on protonated N-acyl amino acid methyl ester model systems, Int. J. Mass Spectrom., 2001, 210–211, 71 CrossRef.
- B. Paizs and S. Suhai, Combined quantum chemical and RRKM modeling of the main fragmentation pathways of protonated GGG. II. Formation of b(2), y(1), and y(2) ions, Rapid Commun. Mass Spectrom., 2002, 16, 375 CrossRef CAS.
- B. Paizs and S. Suhai, Towards understanding some ion intensity relationships for the tandem mass spectra of protonated peptides, Rapid Commun. Mass Spectrom., 2002, 16, 1699 CrossRef CAS.
- C. Bleiholder and B. Paizs, Competing gas-phase fragmentation pathways of asparagine-, glutamine-, and lysine-containing protonated dipeptides, Theor. Chem. Acc., 2010, 125, 387 Search PubMed.
- R. Wu and T. B. McMahon, Infrared multiple photon dissociation spectra of proline and glycine proton-bound homodimers. evidence for zwitterionic structure, J. Am. Chem. Soc., 2007, 129, 4864 CrossRef CAS.
- E. Halin, S. Hoyas, V. Lemaur, J. D. Winter, S. Laurent, J. Cornil, J. Roithová and P. Gerbaux, Side-chain loss reactions of collisionally activated protonated peptoids: a mechanistic insight, Int. J. Mass Spectrom., 2019, 435, 217 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Electronic structure calculations on workstation computers: the program system turbomole, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, All-atom empirical potential for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B, 1998, 102, 3586 CrossRef CAS.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, Charmm: a program for macromolecular energy, minimization, and dynamics calculations, J. Comput. Chem., 1983, 4, 187 CrossRef CAS.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French and S. Billeter, QUASI: a general purpose implementation of the QM/MM approach and its application to problems in catalysis, J. Mol. Struct.: THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- D. K. Papayannis and A. M. Kosmas, The catalytic role of the water or acidic zeolite in the oxidation of BrCH2OH. A theoretical study, Chem. Phys., 2016, 479, 53 CrossRef CAS.
- J. Ghosh and A. Bhattacharya, Prediction of electronically nonadiabatic decomposition mechanisms of isolated gas phase nitrogen-rich energetic salt: guanidium-triazolate, Chem. Phys., 2016, 464, 26 CrossRef CAS.
- X. L. Cheng, Cyclization mechanisms of the cyclic dimer of aziridine aldehyde with vinyl aldehyde, Comput. Theor. Chem., 2017, 1113, 105 CrossRef CAS.
- R. W. F. Bader, Atoms in molecules: a quantum theory, Oxford University Press, New York, USA, 1990 Search PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities, Chem. Phys. Lett., 1998, 285, 170 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyser, J. Comput. Chem., 2012, 33, 580 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS.
- R. Chaudret, B. De Courcy, J. Contreras-Garcia, E. Gloaguen, A. Zehnacker-Rentien, M. Mons and J. P. Piquemal, Unraveling non-covalent interactions within flexible biomolecules: from electron density topology to gas phase spectroscopy, Phys. Chem. Chem. Phys., 2014, 16, 9876 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07600g |
| This journal is © The Royal Society of Chemistry 2021 |