
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural and spectral properties of a non-classical C58 isomer and its fluorinated derivatives in theory†
Xiaoxi Song,
Renfeng Mao,
Ziwei Wang and
Jiayuan Qi *
*
College of Chemistry, Fuzhou University, Fuzhou, Fujian 350116, People's Republic of China. E-mail: jyqi@fzu.edu.cn
First published on 5th January 2021
Abstract
The traditional classical fullerene is only composed of pentagons and hexagons, with many different topologies, of which only a few structures conform to the isolated pentagon rule (IPR), which means all five-membered rings are separated by hexagons, whereas isomers that violate the rule are called non-IPR isomers. In contrast, the non-classical fullerene consists of other kinds of polygons such as squares and heptagons in addition to pentagons and hexagons. X-ray photoelectron spectra (XPS), near-edge X-ray absorption fine structure (NEXAFS) spectra and X-ray emission spectra (XES), as well as the ground-state electronic/geometrical structures of the important non-IPR isomers C3v-#1205C58 and C2-#1078C58, and the remarkable non-classical isomer Cs-C58(NC) with its two fluorides Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B), have been computed at the density functional theory (DFT) level. Significant differences in the electronic structures and simulated X-ray spectra have been observed after fluorination. Meanwhile, strong isomer dependence has been shown in these spectra, which means the “fingerprint” in the X-ray spectra can effectively identify the above-mentioned fullerene isomers. As a consequence, the work can provide useful information especially isomer identification for experimental and theoretical research in fullerene science.
1 Introduction
The fullerene family has drawn many researchers' extensive attention since the discovery of buckminsterfullerene in 1985.1 Many breakthroughs have been made in the years that followed. Ih-#1812C60 fullerene is the most typical IPR fullerene that obeys the isolated pentagon rule (IPR);2,3 in contrast, some fullerenes with fused pentagons which violate this rule are referred to as non-IPR fullerene. Generally speaking, isomers which satisfy the IPR rule are more stable than carbon cages with adjacent pentagons for higher fullerenes.4 Enhanced local steric strain and lack of resonance stabilization bring about the reactivity of non-IPR fullerene.5 IPR fullerene and non-IPR fullerene are collectively referred to as classical fullerenes for their carbon cages are both composed of pentagons and hexagons. However, non-classical fullerenes' carbon cages contain additional other kinds of polygons, such as the common square(s) or heptagon(s), or both exist at the same time.6 In the past, scientists proposed the possibility of the existence of non-classical fullerene,7 until the year of 2002, the first non-classical fullerene C2v-C62 was synthesized by Qian et al., which was also the first non-classical fullerene with a square.8 In 2005, Troshin et al. isolated two C58 fullerene derivatives C58F17CF3 and C58F18 whose parent moiety was the first non-classical fullerene to be found with a seven-membered ring.9 Since then, this field has attracted a lot of attention and more and more non-classical fullerenes have been discovered by researchers. In some previous studies, some non-classical fullerenes had been found to be more stable than the corresponding classical fullerenes. For instance, Ayuela et al.10 and Cui et al.11 found that C62 isomer with Cs symmetry, whose carbon cage contains a heptagon, was more stable than any classical C62 fullerene isomers. Furthermore, it was found that square(s) in carbon cage may lead to added strain and unusual kinetic stability; on the contrary, the incorporation of heptagon(s) may release the strain in fused pentagon–pentagon bonds.6C58 family, as the neighbour of significant C60 fullerene, has attracted attention from researchers. There are 1205 classical isomers of C58 fullerene, and all of them are non-IPR isomers, among which #1205C58 with C3v-symmetry (nomenclature follows Fowler's spiral algorithm,3 hereinafter simplicity referred to as C3v-#1205C58) is predicted to be the most stable classical fullerene.12 So far, no classical C58 fullerenes have been captured, because non-IPR fullerenes are difficult to synthesize which is a big challenge for researchers. But, as mentioned above, two stable non-classical fullerene derivatives C58F17CF3 and C58F18 containing a heptagonal ring had been separated and the same C58 moiety with a seven-membered ring in their structures was produced by removing a bond shared by a pentagon and a hexagon from Ih-C60 fullerene.9 The structure of resultant C58 with Cs symmetry (hereinafter labelled as Cs-C58(NC)) is hard to synthesize because it violates the strain-based nonadjacent pentagon rule. Nevertheless, as a result of the presence of sp3 carbon, its derivatives C58F17CF3 and C58F18 are less strained and they are isolated more easily. As early as 1991, Guo et al.13 discovered substituted fullerene C58B2, and then the electronic properties and binding energy of C58B2 and C58N2 were also studied theoretically. Gan's team synthesized C58 open cage derivatives in 2016.14 The foregoing results about C58 fullerene are rather finite experimentally, in contrast, it is more abundant in theory. For instance, Hu et al. used Hartree–Fork calculations to investigate the endohedral complexes of C58 cage with H2 and CO in 2004.15 Chen' group studied amply the structure, stability, electronic and optical properties of C58 fullerene isomers and two different fullerene derivatives C58X18(A) and C58X18(B), where X = H, F, and Cl in 2007, respectively.16 It is worth noting that C58X18(A) and C58X18(B) (named by Chen's team, so called in this paper) have the same parent carbon cage, which is the non-classical C58 isomer with a heptagon, but the X (X = H, F and Cl) atoms are connected in different ways on the cage. Similarly, Zhao et al. also calculated C58X18' structure and stability in 2010,17 and Tang et al.18 had computed derivatives C58F18 and C58F17CF3 at density functional theory level. Their results indicated that Cs-C58(NC) with a heptagon is more stable than the major non-IPR isomers and both C58X18(A) and C58X18(B) possess high stability, most of all, C58F18(A) had lower energy than C58F18(B) which was characterized experimentally by Troshin et al. in 2005.9 Although there are still many restrictions on the experimental and theoretical researches of C58 fullerene, its special structure and broad applications in materials, energy, medicine, biology, optics and magnetism19–28 have always inspired researchers to make unremitting efforts.
For Cs-C58(NC) and its fluorinated derivatives C58F18(A) and C58F18(B), only limited structures, energies and electronic properties were presented by theoretical calculations,12,16–18,29 further researches about electronic structures and spectroscopic properties for them are expected to contribute a deeper understanding. In this paper, to add insight into the electronic structures of C58 isomers as well as the fluorinated species, X-ray spectra have been computed at the density functional theory (DFT) level. X-ray spectroscopy not only has been proved to be effective for the electronic structure of many molecules or other complex systems, but also is a powerful tool for identifying fullerene isomers. Herein, a comparative and systematic study of near-edge absorption fine structure (NEXAFS) and X-ray photoelectron spectra (XPS) of C2-#1078C58 and C3v-#1205C58 which is regarded as the most stable classical C58 isomer, as well as the non-classical isomer Cs-C58(NC) and its fluorides C58F18(A) and C58F18(B) (denoted as Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B) hereafter, respectively) have been provided. What's more, X-ray emission spectra (XES) of C2-#1078C58, C3v-#1205C58 and Cs-C58(NC) have also been computed at the DFT level. NEXAFS is atom specific and sensitive to the chemical environment of atoms, which has been considered as an effective skill for accurate confirmation of electronic structure, especially the unoccupied orbitals.30,31 With regard to core orbits, XPS can provide useful information that is used to distinguish elements and their chemical states quantitatively, while XES was employed to describe the element specific valence band structures clearly.32,33 The paper aim to explore the electronic structures of C58 isomers and fluorinated derivatives, and provide a valid way for isomer characterization and identification because of X-ray spectra show strong isomer dependence in present study.34–38 It is expected that the results of this research will provide a further theoretical support for the progress of experimental and theoretical researches in C58 fullerene isomers and derivatives.
The framework of this article is as follows: the second part introduces the calculation methods of the XPS, NEXAFS and XES spectra, the third part discusses and analyzes the results in detail, and the final conclusion of this paper is given in the fourth part.
2 Computational details
Molecular mechanics, molecular graphics and quantum chemistry were well utilized in this study for geometrical structures of the non-IPR isomers C3v-#1205C58 and C2-#1078C58, as well as the non-classical isomer Cs-C58(NC) with its fluorinated derivatives Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B). At first, the three-dimensional models of Cs-C58(NC), Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B) were designed by GaussView39 software. The initial coordinates of C3v-#1205C58 and C2-#1078C58 were obtained by GaGe40 program. Then, the geometrical structures of the above-mentioned fullerenes and fluorinated derivatives were optimized by the Gaussian 09 program41 at the B3LYP42,43/6-31G** level. And single point energy was also calculated with a larger 6-311++G(3df,3pd) basis set in order to get more accurate results for further energy analysis. All energies we got were computed by ZPE correction. The natural atomic charges of the atoms within two fluorinated derivatives were analyzed by the natural bond orbital (NBO)44,45 at the B3LYP42,43/6-31G level. Besides, the nucleus-independent chemical shifts (NICS)46,47 which can estimate the aromatic properties of fullerene and derivatives were calculated by GIAO48-B3LYP/6-31G (d,p) method. The calculated variation tendency of energies and the relative stability among the non-IPR isomers C3v-#1205C58 and C2-#1078C58, as well as the non-classical isomer Cs-C58(NC) conform to the previous researches.12,16 Furthermore, we use a larger basis set and thus the results are more reasonable and reliable to provide a more accurate prediction.The NEXAFS and XPS spectra are computed by the StoBe49 program at the density functional theory level and the gradient corrected Becke (BE88) exchange50 functional and Perdew (PD86) correlation functional51 have been used. On the one hand, the excited carbon atoms have used the triple-ζ quality individual gauge for the localized orbital (IGLO-III) basis set of Kutzelnigg et al.,52 on the other hand, model core potentials have been applied to the other non-excited carbon atoms and fluorine atoms, which can facilitate the convergence of the core-hole state. Furthermore, the theoretical NEXAFS spectra have been simulated by using a full core-hole (FCH) potential method in combination with a double basis set technique, where a normal orbital basis set for the minimization of the energy and an added augmented diffuse basis set (19s, 19p, 19d) has been used for the excited carbon atoms to obtain a proper expression of the relaxation effects.
It is confirmed that FCH method have provided excellent transition moments as well as good relative energy positions when evaluating the NEXAFS spectra of fullerenes and the derivatives.53,54 For the sake of determining the absolute energy position of the spectrum, the ΔKohn–Sham (ΔKS) calculations have been performed, which can give more accurate ionization potential (IP) values compared with other methods. In the ΔKS scheme, the total energies of the system in the ground state and the N − 1 electron system after the core ionization have been optimized.55 Then, the IP is obtained by the energy difference between the ground state and the fully optimized core-ionized state. In order that the first spectral feature corresponding to the transition [1s → the lowest unoccupied molecular orbital (LUMO)] coincides with the same one obtained from the ΔKohn–Sham approach,55,56 all the raw spectra have been calibrated. As a result of introduction of the core hole,53 all spectra have congruously used a shift of +0.2 eV to explicate the differential relativistic effect. The final NEXAFS spectra are obtained by convoluting the discrete intensities, a Gaussian function with full width at half maximum (FWHM) set at 0.3 eV below the IP as well as a Stieltjes imaging approach57,58 in the region above has been used. The final XPS spectra are generated by broadening the IP bars with a Lorentzian line shape that FWHM set at 0.15 eV, each IP bar weighted with the portion of equivalent centers among all centers of the studied element. The simulated X-ray spectra also have been demonstrated to be effective for the determination of the electronic structures of many molecules, surfaces, clusters and bulk materials, such as DNAs, carbon nanotubes, fullerenes, etc.59–63
The XES spectra have been performed by using the tool package BIONANO LEGO64 at the B3LYP42,43/6-31G level with the group theory formulation developed by Luo et al.32,65 X-ray emission spectroscopy, which can provide information about the valence occupied orbitals, is composed of the radiative decay of valence electron into a core hole. Central insertion scheme (CIS) method32,66 and a category of Krylov subspace methods67 are applied to the BIONANO LEGO software. SCF iterations and computational time are reduced obviously by a better initial guess of above methods. The CIS method can add the identical units in the middle of the system continuously to elongate the initial system and obtain the initial Hamiltonian in the site representation which possess identical central parts.67–69 Meanwhile, the category of Krylov subspace methods can effectively reduce the storage and computational requirements so that large-scale characteristic value problems are well solved. In the XES spectrum, the transition moment between valence and core orbital is computed, and the ground state electronic structure is utilized to calculate the adiabatic approximation intensities.70 Further convolution by a Lorentzian function with FWHM = 0.5 eV has been applied to generate the final XES spectra.
3 Results and discussion
3.1 Geometrical and electronic structures
The optimized structures of the non-IPR isomers C3v-#1205C58 and C2-#1078C58, as well as the non-classical isomer Cs-C58(NC) with its fluorinated derivatives Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B) are demonstrated in Fig. 1. The distinct geometrical features of five different optimized structures in Fig. 1 have been individually marked. There are three pairs of pentagon–pentagon ring fusions (denoted as DFP) and two pairs of triple sequentially fused pentagons (referred to as TSFP) which are both colored in purple in the structures of C3v-#1205C58 and C2-#1078C58, respectively. And there is a heptagon colored in gold and two TSFPs that are also colored in purple in the carbon cages of Cs-C58(NC) and its two fluorides. As illustrated in the Fig. 1, the fluorine atoms which colored in green are attached to carbon atoms within two fluorinated derivatives in different ways. Compared with Cs-C58(NC)F18(A), which has three fluorine atoms connected with heptagon sites, only two fluorine atoms of Cs-C58(NC)F18(B) are located at the heptagon sites, most of the remaining fluorine atoms are attached to the triple sequentially fused pentagons as shown in Fig. 1. Addition of fluorine atoms contributes to the release of local strain and hybridization changes from sp2 to sp3, nevertheless the presence of them did not change the symmetry of the carbon cage which still maintains the Cs symmetry. The result is consistent with the results of previous theoretical studies.12,16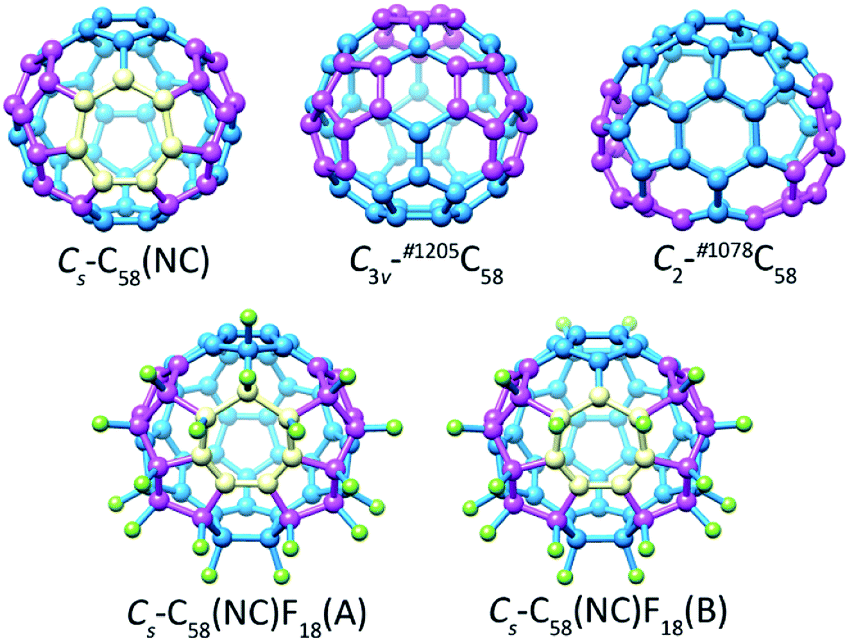 | ||
| Fig. 1 Optimized structures of the non-IPR isomers C3v-#1205C58 and C2-#1078C58, as well as the non-classical isomer Cs-C58(NC) with its fluorinated derivatives Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B). | ||
Table 1 manifests the statistics of relative energies, bond lengths as well as HOMO (highest occupied molecular orbital)–LUMO (lowest unoccupied molecular orbital) gaps of three C58 isomers and the fluorinated derivatives Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B). The singlet and triplet states of three C58 isomers have been separately considered. Based on the results of the Table 1, although the shortest and longest bond lengths of three C58 isomers are different, their average values are almost the same (1.435 Å −1.436 Å). Average C–C bond lengths of both two fluorinated derivatives are 1.473 Å which is longer than the average bond length of the pristine Cs-C58(NC). Due to fluorination, the C–C bond lengths have been elongated in general which reflects the considerable changes in the C58 backbone. The energies of singlet Cs-C58(NC) and C2-#1078C58 are 8.61 kcal mol−1 and 5.75 kcal mol−1 lower than their triplet isomers, respectively. In contrast, the triplet C3v-#1205C58 is 6.13 kcal mol−1 lower than its singlet one in energy, which agrees well with the previous results.12,71,72 Hence, we focus on the singlet Cs-C58(NC) and C2-#1078C58, as well as the triplet electronic state of C3v-#1205C58 in the following discussion. The results indicate that the stability of class fullerene C3v-#1205C58 is higher than non-classical fullerene Cs-C58(NC) and another classical isomer C2-#1078C58. According to our calculations, the energy of fluoride Cs-C58(NC)F18(B) is 5.567 kcal mol−1 higher than Cs-C58(NC)F18(A) which indicates that Cs-C58(NC)F18(A) is more popular isomer in energy than the former. In the previous report,12 Chen et al. stated that the singlet C2-#1078C58 and Cs-C58(NC) are also more stable than their triplet electronic state at the B3LYP42,43/6-31G level, and the triplet C3v-#1205C58 is also the most popular isomer in energy, whose energy is 9.15 and 18.99 kcal mol−1 lower than that of singlet Cs-C58(NC) and C2-#1078C58, respectively. Meanwhile, Chen's work16 also indicated the fluoride Cs-C58(NC)F18(A) is more stable than the B-former fluoride. In summary, our results of variation tendency of energies and the relative stability among three C58 isomers conform to the previous researches.12,16 Furthermore, we use a larger basis set, thus the results are more reasonable and reliable to provide a more accurate prediction. The non-classical isomer Cs-C58(NC) shows a larger HOMO–LUMO gap, that does not provide direct information on the relative chemical stability compared with the non-IPR isomers C3v-#1205C58 and C2-#1078C58. Meanwhile, Cs-C58(NC)F18(A) has a larger HOMO–LUMO gap than another fluoride C58(NC)F18(B), and both their HOMO–LUMO gaps are much larger than their parent fullerene.
| Molecule | State | Shortest Rcc | Longest Rcc | Average Rcc | HOMO–LUMO gap | Relative energy | NICS | NBO(F) |
|---|---|---|---|---|---|---|---|---|
| C3v-#1205C58 | 1A′ | 1.385 | 1.495 | 1.435 | — | 6.13 | — | — |
| 3A1 | 1.388 | 1.468 | 1.436 | 1.382 | 0.00 | −7.520 | — | |
| C2-#1078C58 | 1A′ | 1.374 | 1.492 | 1.436 | 1.376 | 18.25 | 1.865 | — |
| 3A1 | 1.388 | 1.476 | 1.436 | — | 24.00 | — | — | |
| Cs-C58(NC) | 1A′ | 1.375 | 1.489 | 1.436 | 1.516 | 8.65 | −5.254 | — |
| 3A′′ | 1.385 | 1.471 | 1.436 | — | 17.26 | — | — | |
| Cs-C58(NC)F18(A) | 1A′ | 1.335 | 1.629 | 1.473 | 3.320 | — | −14.83 | −0.323 |
| Cs-C58(NC)F18 (B) | 1A′ | 1.339 | 1.632 | 1.473 | 2.732 | — | −11.21 | −0.326 |
To better understand the interaction between C58 backbone and fluorine atoms, the natural bond orbital (NBO) analyses for Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B) have also been performed. The charge transfer during the fluorination process can qualitatively represented by the average of NBO charges on fluorine atoms for two fluorides. As shown in Table 1, the average natural atomic charge of fluorine atoms is −0.323 (−0.314 to −0.334) in fluorinated derivatives Cs-C58(NC)F18(A), and that is −0.326 (−0.318 to −0.336) in another fluorides, which is consist with the previous studies.16 It is worth mentioned that the natural atomic charges of C (sp3) atoms in two fluorides are from 0.309 to 0.35, while that of C (sp2) atoms are from −0.039 to 0.026. Such results indicate strong electron acceptor and donor characteristics of fluorine atoms and the C58 backbone, respectively. What's more, it can be inferred that the addition of fluorine atoms to the C58 mainly affects the charges of C (sp3) atoms and the influence on C (sp2) atoms is negligible. The strong electron transfer and electrostatic interaction reveal large changes have taken place in electronic and geometrical structures from fullerene to the fluorinated derivative. The change in structure has been confirmed by the growth of the C–C bond length, while the change in electronic structure has been exhibited by the XPS and NEXAFS spectra, as will be discussed below.
Furthermore, we also calculated the nucleus-independent chemical shifts (NICS) values at the center of the fullerene based on the GIAO-B3LYP/6-31G (d,p) method to get information on the aromatic character. According to the NICS characterization, a negative NICS value indicates that it has the aromaticity, the antiaromaticity is characterized by a positive NICS value, and nonaromaticity by a value close to zero. As displayed in the Table 1, Cs-C58(NC) is aromatic with a NICS value of −5.25 ppm and the triplet C3v-#1205C58 possesses a more negative NICS value of −7.52 ppm, while the singlet C2-#1078C58 shows antiaromaticity by a positive NICS value of 1.865 ppm. Fluorofullerene Cs-C58(NC)F18(A) not only has lower energy than the synthesized Cs-C58(NC)F18(B), but also owns more negative NICS value of −14.83 ppm, which suggests that Cs-C58(NC)F18(A) is stable and may be isolated experimentally. In addition, both the NICS values of two fluorinated derivatives are more negative than their parent fullerene, this phenomenon could be reasonably explained by that the addition of fluorine atoms has changed the C–C bond lengths of the C58 backbone, and thus hybridization of some carbon atoms changed from sp2 to sp3, finally, the loss of electron density in the skeleton greatly affects the aromatic character of the cage center.
In order to discuss the spectra of fullerene isomers more conveniently, it is necessary to classify the carbon atoms of fullerene through their local environment in which they are located. There are six distinguished types of carbon sites displayed in Fig. 2. (1) The pyracylene site C1, in which the carbon atom lying in a pentagon is connected through an exo bond to another pentagon. (2) The corannulene site C2, in which the carbon atom lying in a pentagon is connected through an exo bond to a hexagon. (3) The pyrene site C3, in which the carbon atom is part of three hexagons. (4) The DFP site C4, in which the carbon atom lies in a pentagon–pentagon ring fusion. (5) The TSFP site C5, in which the carbon atom lies in the fusion of triple sequentially fused pentagons. (6) The heptagon site C6, in which the carbon atom lies in the proximate hexagon–hexagon–heptagon vertexes and pentagon–hexagon–heptagon vertexes. Different combinations of these six carbon sites will produce different isomers. For example, there are 45 pyracylene sites, 8 TSFP sites and 5 heptagon sites both for Cs-C58(NC) and its fluorinated derivatives; 36 pyracylene sites, 12 corannulene sites, 6 DFP sites and 4 pyrene sites for C3v-#1205C58; 34 pyracylene sites, 10 corannulene sites, 8 TSFP sites and 6 pyrene sites for C2-#1078C58.
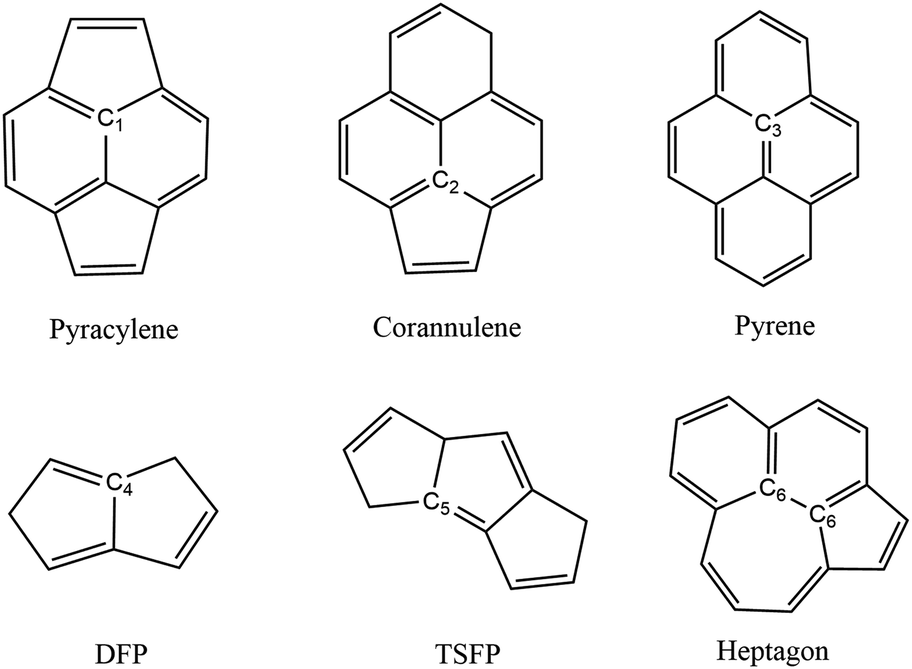 | ||
| Fig. 2 Schematic illustration of the local environment of different types of carbons. | ||
In addition, the Fowler–Manolopoulos spiral algorithm mentioned above is one of the most widely used algorithms for the generation and nomenclature of fullerene isomers, which advantageously solves the topological problem of fullerene isomerism. Fowler and Manolopoulos developed the algorithm in 1992 (ref. 73) and demonstrated its limitations in 1993.74 The algorithm is not guaranteed to yield all possible isomers, for instance the non-classical fullerene isomers, but it is reliable and suitable for constructing polyhedrons with lower symmetries, especially for the fullerenes with atoms within 380. In the present work, the C3v- and C2-symmetric C58 isomers are, according to the Fowler's spiral algorithm,3 corresponding to the fullerenes58 numbered as 1205 and 1078.
3.2 XPS
The calculated C 1s XPS spectra of different non-equivalent carbon atoms in the non-IPR isomers C3v-#1205C58 and C2-#1078C58, as well as the non-classical isomer Cs-C58(NC) are displayed in Fig. 3(a). It is easily observed that the three C58 isomers all show a strong peak at around 289.9–290 eV and two weak peaks at lower energy about 289.5 eV and higher energy about 290.3 eV, respectively. All carbon atoms of non-IPR isomer C3v-#1205C58 are classified as only 13 symmetry-independent atoms and four types: DFP, pyracylene, corannulene and pyrene. While compared with C3v-#1205C58, the symmetries of another non-IPR isomer C2-#1078C58 and the non-classical isomer Cs-C58(NC) are reduced and the non-equivalent carbon atoms of them are increased to 29 and 31, respectively. Particularly worth mentioning is that the spectrum of non-classical isomer have an un-conspicuous wave-packet at around 290.6 eV, which is not observed in that of two non-IPR isomers. The clear differences among the three isomers could be attributed to the presence of heptagon sites in Cs-C58(NC). Such a particular characteristic can provide useful evidence for the identification of the three isomers. In addition, there are similar IP values for the same or similar types of carbon atoms in different molecules. According to the Fig. 3(b)–(d), the pyrene sites in these two non-IPR molecules have the same IP values at high-energy regions around 290.3 eV, while there are no pyrene sites in the non-classical isomer, which are replaced by the heptagon sites with high-energy range of 290.2–290.6 eV. Besides, carbons at the pentagon–pentagon ring fusions (DFP/TSFP) of the three C58 isomers exhibit at the lower-energy regions. This phenomenon could be reasonably inferred by that the stability of the different types of carbon atoms. The presence of adjacent pentagons leads to the enhanced local steric strain and lack of resonance stabilization, thus the increased chemical active sites result in the decrease of ionization energy. In contrast, the incorporation of heptagon may release the strain of carbon skeleton, resulting in the higher ionization energy. All of the above results are well consist with the previous theoretical conclusions of C54,36 C60,34 C72,35 C74,37 and C78.38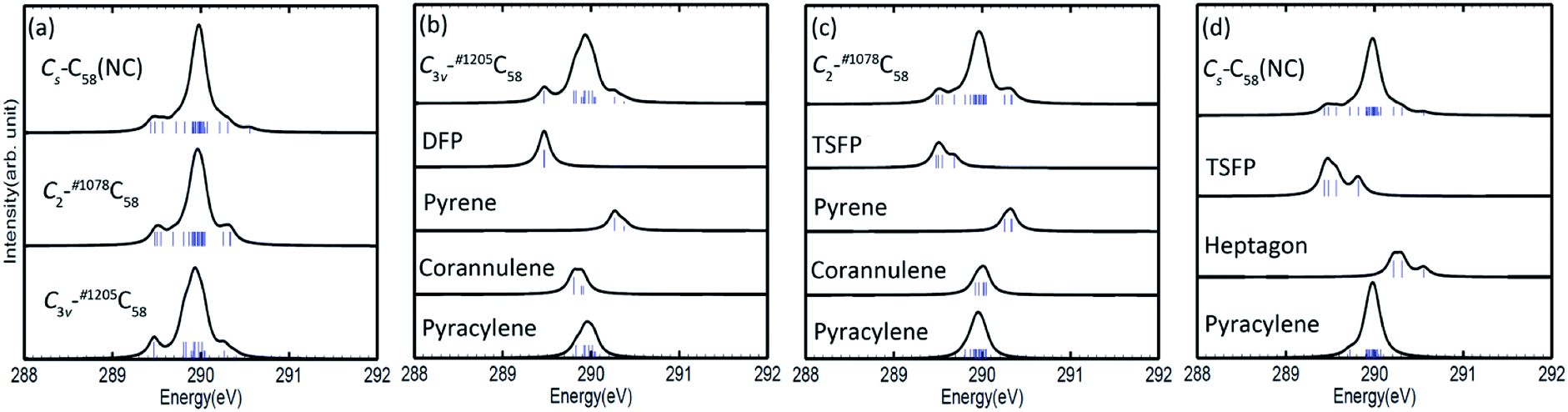 | ||
| Fig. 3 Calculated 1s ionization potentials (IPs, denoted as bars) of different symmetry-independent carbon atoms in each C58 isomer (a) and type-specific contributions for the non-IPR isomers C3v-#1205C58 (b) and C2-#1078C58 (c), as well as the non-classical isomer Cs-C58(NC) (d). The XPS spectra are generated from these IPs through a Lorentzian convolution with FWHM = 0.15 eV. | ||
The calculated C 1s XPS spectra of the two fluorinated species compared with the corresponding C58 backbone are presented in Fig. 4(a). The spectra of Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B) both demonstrate a strong peak at around 291 eV and a weak shoulder in the range of 291.4–291.6 eV, as well as a wave-pocket at higher energy range of 293.4–293.8 eV, which is not observed in that of the pristine fullerene Cs-C58(NC). In addition, significant differences in the XPS spectra have been observed after fluorination, the whole features of the fluorinated species exhibit an obvious blue shift in a sense after fluorination. Meanwhile, there are slight differences between the two fluorides. For a deeper insight, the contributions from the three types of carbons have been displayed in Fig. 4(b) and (c). One can observe that the carbon atoms belong to the same types of the two fluorides have similar IP values. Compared with the parent fullerene, the TSFP sites of two fluorides both exhibit an obvious blue shift, especially for that directly bonded to fluorine atoms, which have the higher IP values at 293.4–293.8 eV. Since the carbon atoms which are connected to the fluorine atoms have the characteristic of an electron donor, the energy level position of the inner 1s orbital of them is reduced, that is, the peaks in XPS spectra of two fluorides appear in the higher-energy regions.36 This is accordant with previously reported results on halogenation of carbon compounds that the binding energy of carbon 1s electrons will increase differently depending on the bond type of carbon atoms.75,76 Such observation again confirms the effective changes in the geometrical and electronic structures of the C58 backbone after fluorination. Compared with the previous studies of C54,36 C60,34 C72,35 C74,37 C78,38 and their chlorinated derivatives, the fluorides still have an obvious blue shift after fluorination. It is worth mentioning that the blue shift after fluorination is 0.2 eV more than that produced by chlorination.
 | ||
| Fig. 4 Calculated 1s ionization potentials (IPs, denoted as bars) of different symmetry-independent carbon atoms in (a) the non-classical fullerene derivatives Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B) as well as type-specific contributes for the fluorides (b) Cs-C58(NC)F18(A) and (c) Cs-C58(NC)F18(B). The XPS spectra are generated from these IPs through a Lorentzian convolution with FWHM = 0.15 eV. | ||
Accordingly, the XPS spectra can in principle be considered as a useful technique for identifying isomers with different symmetries and detecting changes in their geometrical and electronic structures after fluorination.
3.3 NEXAFS
The calculated NEXAFS spectra of the three C58 isomers are shown in Fig. 5(a), which are obtained by the weighted (according to their relative abundance) summation of all type-specific spectra. The non-IPR isomers C3v-#1205C58 and C2-#1078C58, as well as the non-classical isomer Cs-C58(NC) exhibit seven major peaks, which are labeled as A–G (marked in the ascending order of energy and hereafter). Distinct differences have been shown by the spectra of these three isomers in Fig. 5(a), especially the energy range of 283–285 eV. For example, both an obvious peak labeled as A exists in the spectra of Cs-C58(NC) and C2-#1078C58, while the same position of the spectrum of C3v-#1205C58 shows a weak shoulder that is not easily detectable, which is on the left of the peak A. In addition, the non-IPR isomer C3v-#1205C58 possesses a broad wave-packet which consists of two peaks A and B, while there is a much stronger peak B with a weak left shoulder in the spectrum of the Cs-C58(NC), besides, a weak peak B and a sharp peak C appear in the similar energy range of C2-#1078C58. Slight differences also could been observed in the high energy regions, especially the clear peak G in the spectrum of C3v-#1205C58, which is hardly found in the same regions of Cs- and C2-symmetric isomers.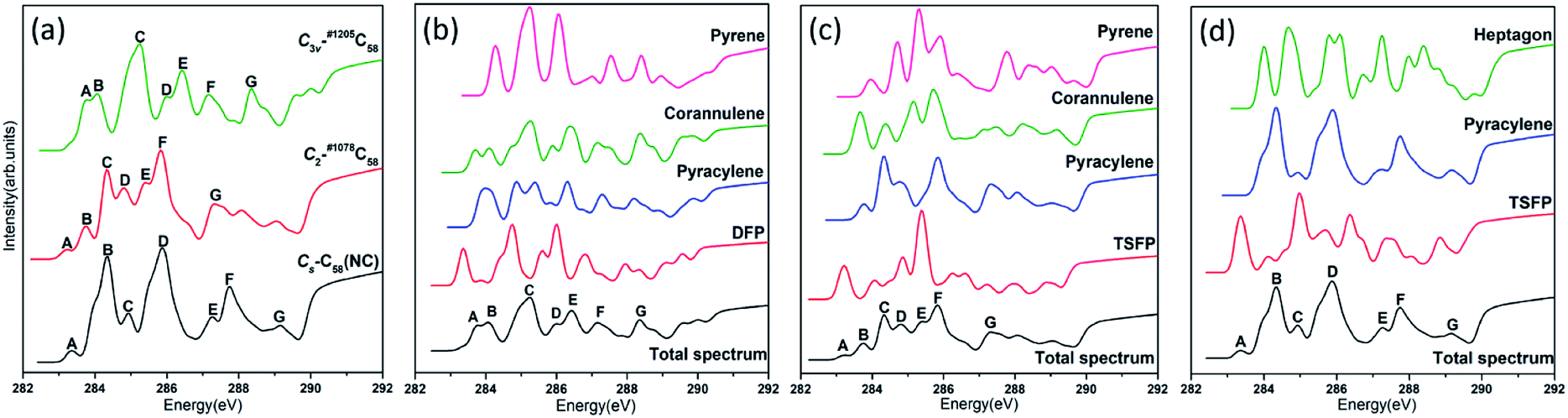 | ||
| Fig. 5 Calculated (a) total NEXAFS spectra of the three C58 isomers as well as type-specific contributions for (b) C3v-#1205C58, (c) C2-#1078C58 and (d) Cs-C58(NC). Each total spectrum is obtained by weighted (according to their relative abundance) summation of all type-specific spectra. | ||
For a deeper understanding, Fig. 5(b), (c) and (d) show the contributions of different carbon types to C3v-#1205C58, C2-#1078C58 and Cs-C58(NC), respectively. As illustrated in the Fig. 5(b), the weak shoulder that is on the left of the peak A only stems from the DFP sites, which is special to C3v-#1205C58. The double wave-packet A and B only originate from the excitation of pyracylene and corannulene sites. As for the peaks C, D, E, F and G, they all originate from all four types of carbons. As depicted in Fig. 5(c), the peak A only arises from the TSFP sites, which is regarded as the “fingerprints”. The peak B arises from the excitation of pyracylene and corannulene sites, and all four types of carbon atoms contribute to the remaining peaks. In Fig. 5(d), there is a clear peak A which is only contributed by the TSFP sites, and it is the “fingerprint” of the spectrum of the Cs-C58(NC). The remaining peaks arise from all three types of carbons.
The comparative total NEXAFS spectra of Cs-C58(NC) and its two fluorinated derivatives Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B) are displayed in Fig. 6(a). There are significant differences among the non-classical isomer and its fluorinated derivatives. For example, the two fluorides both have a double peak (both marked as G and H, respectively) in the high-energy regions of 290–292 eV, which is hardly found in their parent fullerene. In summary, an obvious blue shift exhibits in the NEXAFS spectrum after fluorination. Slight differences between the two fluorides also can be observed, the double peak (G and H) in Cs-C58(NC)F18(A) is sharper than that in Cs-C58(NC)F18(B). Based on the above characteristics, we can easily distinguish the three species. The contributions from the three types of carbons for two fluorides are displayed in Fig. 6(b) and (c), respectively. It is worth mentioning that both the weak peaks A of two fluorides only originate from the excitation of the TSFP sites, which are regarded as “fingerprints”. Besides, the peaks B of the two derivatives both arise from the excitation of the pyracylene and heptagon sites. All the three different types of carbons contribute to the remaining peaks of two fluorides, except for the peak C of Cs-C58(NC)F18(A) only stems from the pyracylene sites. According to a further comparison of the type-specific contributions for Cs-C58(NC) and its two fluorinated derivatives which are displayed in Fig. 5(d), 6(b) and (c), respectively, the features of TSFP carbon sites in the high-energy regions have greatly enhanced after the substitution of the fluorine atoms, resulting in the blue shift of the spectral peaks. All these differences in the NEXAFS spectra among non-classical isomer and its two derivatives demonstrate that fluorination has effectively changed the electronic structure of the C58 backbone.
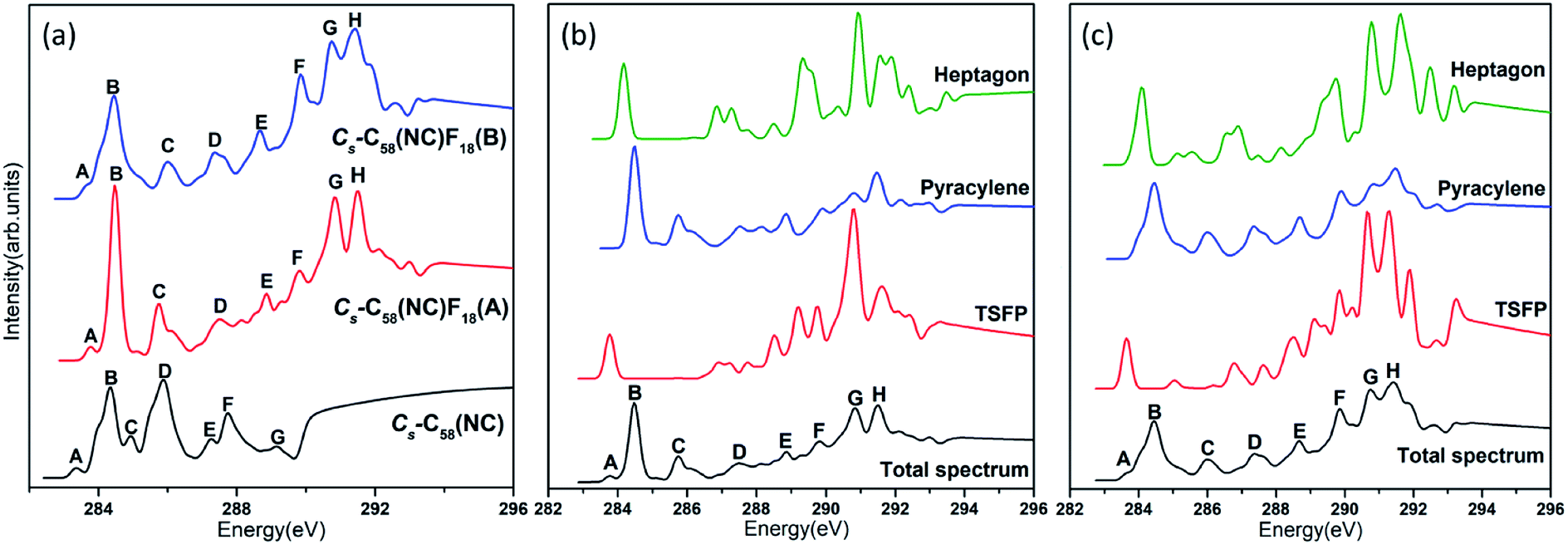 | ||
| Fig. 6 Calculated (a) total NEXAFS spectra of Cs-C58(NC) and two corresponding fluorinated derivatives as well as type-specific contributions for the fluorides (b) Cs-C58(NC)F18(A) and (c) Cs-C58(NC)F18(B). Each total spectrum is obtained by weighted (according to their relative abundance) summation of all type-specific spectra. | ||
In conclusion, notable differences have been observed in the NEXAFS spectra of the three C58 fullerene isomers and two fluorinated derivatives, which result from and directly reflect the isomer difference and the effects of fluorination on the geometrical and electronic structures. The presences of TSFP and DFP carbon sites are the most distinct feature of C3v-, C2- and Cs-C58 isomers and two fluorinated species. As a consequence, the fingerprints of the NEXAFS spectra of three C58 isomers may be reasonably deemed as the peak A, while the fingerprints could be considered as peaks A, G and H for two fluorinated derivatives. NEXAFS spectra of the fluorides have the same variation rule as that of chlorides. The similar blue shift exists in the NEXAFS spectra of fluorides in this work compared with that after chlorination in the previous studies of C54,36 C60,34 C72,35 C74,37 and C78.38 Hence, the NEXAFS spectra can provide effective identification information for different isomers.
3.4 XES
The calculated XES spectra of the non-IPR isomers C3v-#1205C58 and C2-#1078C58, as well as the non-classical isomer Cs-C58(NC) are presented in Fig. 7. XES spectra aim to provide the emitted energy and intensity during the relaxation process of electron in the inner layer of a specific element after being excited to obtain more useful information. It is sensitive to different elements, thus, the general spectral shapes of C 1s emission spectra of three fullerene isomers are quite similar. The XES spectra of the three isomers are all characterized by two main peaks at the range of 265–270 eV and a lower double peak at around 272–273 eV. Although the general spectra are similar, some slightly differences still could be observed among them. For instance, the non-IPR isomer C3v-#1205C58 has a broad left shoulder and a slim strong peak in the range of 265–267 eV, while a double peak appears in the spectra of C2-#1078C58 and Cs-C58(NC), respectively. Besides, a broader peak at around 272 eV exists in the spectrum of the non-IPR isomer C2-#1078C58 compared with another two C58 isomers. It could be speculated that different types of carbon atoms change the electronic structure of the carbon cage, resulting in the differences among the spectra of the three isomers. In summary, the C 1s emission spectra can also provide effective evidences for distinguishing isomers. | ||
| Fig. 7 Calculated XES spectra at C 1s edges of the non-IPR isomers (a) C3v-#1205C58 and (b) C2-#1078C58, as well as the non-classical isomer (c) Cs-C58(NC). | ||
4 Conclusions
The electronic structures, XPS, NEXAFS and XES spectra of the non-IPR isomers C3v-#1205C58 and C2-#1078C58, as well as the non-classical isomer Cs-C58(NC) with its two non-classical fluorinated derivatives Cs-C58(NC)F18(A) and Cs-C58(NC)F18(B) have been investigated at the DFP level. Both the XPS and NEXAFS spectra show strong isomer dependence and the distinct effect of the fluorination process on the carbon cages have been demonstrated. In particular, the carbon atoms at the fusions of the pentagon–pentagon rings (DFP or TSFP) demonstrate special signals which are particular in the non-IPR isomers C3v-#1205C58 and C2-#1078C58, as well as the non-classical isomer Cs-C58(NC) with its two fluorides, respectively. The fingerprints originated from the DFP and TSFP sites in the XPS and NEXAFS spectra of the two non-IPR isomers and the non-classical isomer appear in the lower-energy regions, while that in higher energy regions in spectra of the two fluorides. In other words, there is a large blue shift in the XPS and NEXAFS spectra after fluorination. The different spectral features in X-ray spectra of the non-IPR isomers C3v-#1205C58 and C2-#1078C58, as well as the non-classical isomer Cs-C58(NC) can provide effective evidence for distinguishing the non-IPR and non-classical isomers. In conclusion, these results indicate X-ray spectra can provide a valuable method for further experimental and theoretical researches in fullerene science.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21503042, No. 21973014), the Research Fund for the Doctoral Program of Higher Education of China (20123514120003), Natural Science Foundation of Fujian Province (2014J05015), Foundation of Educational Commission of Fujian Province (JA14036) and Fuzhou University (2012-XQ-12).Notes and references
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- H. W. Kroto, Nature, 1987, 329, 529–531 CrossRef CAS.
- P. W. Fowler and D. E. Manolopoulos, An atlas of fullerenes, Oxford Univ. Press, Oxford, 1995 Search PubMed.
- M. Bühl and A. Hirsch, Chem. Rev., 2001, 101, 1153–1184 CrossRef.
- Y. Z. Tan, S. Y. Xie, R. B. Huang and L. S. Zheng, Nat. Chem., 2009, 1, 450–460 CrossRef CAS.
- L. H. Gan, J. Q. Zhao and F. S. Pan, J. Mol. Struct. Theochem., 2010, 953, 24–27 CrossRef CAS.
- R. Taylor, Interdiscip. Sci. Rev., 1992, 17, 161–170 CrossRef.
- W. Qian, S. C. Chuang, R. B. Amador, T. Jarrosson, M. Sander, S. Pieniazek, S. I. Khan and Y. Rubin, J. Am. Chem. Soc., 2003, 125, 2066–2067 CrossRef CAS.
- P. A. Troshin, A. G. Avent, A. D. Darwish, N. Martsinovich, A. K. Abdul-Sada, J. M. Street and R. Taylor, Science, 2005, 309, 278 CrossRef CAS.
- A. Ayuela, P. W. Fowler, D. Mitchell, R. Schmidt, G. Seifert and F. Zerbetto, J. Phys. Chem., 1996, 100, 15634–15636 CrossRef CAS.
- Y. H. Cui, D. L. Chen, W. Q. Tian and J. K. Feng, J. Phys. Chem. A, 2007, 111, 7933–7939 CrossRef CAS.
- D. L. Chen, W. Q. Tian, J. K. Feng and C. C. Sun, ChemPhysChem, 2007, 8, 1029–1036 CrossRef CAS.
- T. Guo, C. M. Jin and R. E. Smalley, J. Phys. Chem., 1991, 95, 4948–4950 CrossRef CAS.
- Y. M. Yu, L. Xu, X. C. Huang, S. S. Liang and L. B. Gan, Synlett, 2016, 27, 2123–2127 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Chem. Phys. Lett., 2004, 390, 472–474 CrossRef CAS.
- D. L. Chen, W. Q. Tian, J. K. Feng and C. C. Sun, J. Phys. Chem. B, 2007, 111, 5167–5173 CrossRef CAS.
- H. L. Zhao, F. S. Pan, Z. H. Liu, C. Y. Tao and L. H. Gan, Comput. Theor. Chem., 2011, 963, 115–118 CrossRef CAS.
- C. M. Tang, W. H. Zhu and K. M. Deng, Chin. J. Chem., 2010, 28, 1355–1358 CrossRef CAS.
- N. Gharbi, M. Pressac, M. Hadchouel, H. Szwarc, S. R. Wilson and F. Moussa, Nano Lett., 2005, 5, 2578–2585 CrossRef CAS.
- C. M. Sayes, A. A. Marchione, K. L. Reed and D. B. Warheit, Nano Lett., 2007, 7, 2399–2406 CrossRef CAS.
- R. Charvet, S. Acharya, J. P. Hill, M. Akada, M. Liao, S. Seki, Y. Honsho, A. Saeki and K. Ariga, J. Am. Chem. Soc., 2009, 131, 18030–18031 CrossRef CAS.
- B. J. Li and Z. Xu, J. Am. Chem. Soc., 2009, 131, 16380–16382 CrossRef CAS.
- Y. F. Shen, J. B. Wang, U. Kuhlmann, P. Hildebrandt, K. Ariga, H. Möhwald, D. G. Kurth and T. Nakanishi, Chem.–Eur. J, 2009, 15, 2763–2767 CrossRef CAS.
- J. B. Wang, Y. F. Shen, S. Kessel, P. Fernandes, K. Yoshida, S. Yagai, D. G. Kurth, H. Möhwald and T. Nakanishi, Angew. Chem., Int. Ed., 2009, 48, 2166–2170 CrossRef CAS.
- T. Baati, F. Bourasset, N. Gharbi, L. Njim, M. Abderrabba, A. Kerkeni, H. Szwarc and F. Moussa, Biomaterials, 2012, 33, 4936–4946 CrossRef CAS.
- N. Wang, L. Sun, X. N. Zhang, X. C. Bao, W. Zheng and R. Q. Yang, RSC Adv., 2014, 4, 25886–25891 RSC.
- Z. Y. Zhang, L. Wei, X. J. Qin and Y. Li, Nano Energy, 2015, 15, 490–522 CrossRef CAS.
- K. N. Semenov, N. A. Charykov, V. N. Postnov, V. V. Sharoyko, I. V. Vorotyntsev, M. M. Galagudza and I. V. Murin, Prog. Solid State Chem., 2016, 44, 59–74 CrossRef CAS.
- W. Q. Tian, D. L. Chen, Y. H. Cui and J. K. Feng, J. Comput. Theor. Nanosci., 2009, 6, 239–256 CrossRef CAS.
- J. Stöhr, NEXAFS spectroscopy, Springer-Verlag, Berlin, 1992 Search PubMed.
- F. Gel'mukhanov and H. Ågren, Phys. Rep., 1999, 312, 87–330 CrossRef.
- Y. Luo, H. Ågren, F. Gel'mukhanov, J. Guo, P. Skytt, N. Wassdahl and J. Nordgren, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 14479–14496 CrossRef CAS.
- M. Nyberg, Y. Luo, L. Qian, J. E. Rubensson, C. Såthe, D. Ding, J. H. Guo, T. Käämbre and J. Nordgren, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 115117 CrossRef.
- J. Y. Qi, W. J. Hua and B. Gao, Chem. Phys. Lett., 2012, 539–540, 222–228 CrossRef CAS.
- J. Y. Qi, X. Y. Hu, H. H. Zhu and M. Zheng, Phys. Chem. Chem. Phys., 2016, 18, 8049–8058 RSC.
- X. T. Song, X. Q. Li and J. Y. Qi, RSC Adv., 2018, 8, 32731–32739 RSC.
- M. Zheng, X. T. Song, X. Q. Li and J. Y. Qi, Mol. Phys., 2018, 116, 1772–1781 CrossRef CAS.
- X. Q. Li, X. X. Song, X. T. Song and J. Y. Qi, J. Phys. Chem. C, 2019, 123, 13837–13845 CrossRef CAS.
- R. Dennington, T. Keith and J. Millam, GaussView 5.0.8, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- G. Brinkmann, O. D. Friedrichs, S. Lisken, A. Peeters and N. V. Cleemput, Match, 2010, 63, 533–552 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. M. Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, J. N. R. Kobayashi, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision D.01), Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CAS.
- F. Weinhold, C. R. Landis and E. D. Glendening, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- Z. F. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- K. Hermann, L. Pettersson, M. Casida, C. Daul, A. Goursot, A. Koester, E. Proynov, A. St-Amant and D. Salahub, StoBe-deMon (Version 3.0) and Documentation for STOBE2007, StoBe Software, Stockholm, 2007 Search PubMed.
- A. D. Becke, Phys. Rev. A: At. Mol. Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- W. Kutzelnigg, U. Fleischer and M. Schindler, NMR-basic principles and progress, Springer-Verlag, Heidelberg, 1990 Search PubMed.
- P. J. W. Weijs, M. T. Czyżyk, J. F. van Acker, W. Speier, J. B. Goedkoop, H. van Leuken, H. J. M. Hendrix, R. A. de Groot, G. van der Laan, K. H. J. Buschow, G. Wiech and J. C. Fuggle, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 11899–11910 CrossRef CAS.
- A. Bassan, M. Nyberg and Y. Luo, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 165402 CrossRef.
- L. Triguero, O. Plashkevych, L. G. M. Pettersson and H. Ågren, J. Electron Spectrosc. Relat. Phenom., 1999, 104, 195–207 CrossRef CAS.
- C. Kolczewski, R. Püttner, O. Plashkevych, H. Ågren, V. Staemmler, M. Martins, G. Snell, A. S. Schlachter, M. Sant'Anna, G. Kaindl and L. G. M. Pettersson, J. Chem. Phys., 2001, 115, 6426–6437 CrossRef CAS.
- P. W. Langhoff, Electron-molecule and photon-molecule collisions, Plenum, New York, 1979 Search PubMed.
- P. W. Langhoff, Theory and applications of moment methods in many-fermion systems, Plenum, New York, 1980 Search PubMed.
- W. J. Hua, Y. J. Ai, B. Gao, H. B. Li, H. Ågren and Y. Luo, Phys. Chem. Chem. Phys., 2012, 14, 9666–9675 RSC.
- B. Gao, Z. Y. Wu, H. Ågren and Y. Luo, J. Chem. Phys., 2009, 131, 034704 CrossRef.
- W. J. Hua, B. Gao, S. H. Li, H. Ågren and Y. Luo, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 155433 CrossRef.
- J. Jiang, B. Gao, T. T. Han and Y. Fu, Appl. Phys. Lett., 2009, 94, 092110 CrossRef.
- J. Jiang, K. Liu, W. Lu and Y. Luo, J. Chem. Phys., 2006, 124, 214711 CrossRef.
- B. Gao, J. Jiang, K. Liu and Y. Luo, Bionano Lego, Version 2.0, Royal Institute of Technology, Sweden, 2008 Search PubMed.
- Y. Luo, H. Ågren and F. Gel'mukhanov, J. Phys. B: At., Mol. Opt. Phys., 1994, 27, 4169–4180 CrossRef CAS.
- B. Gao, J. Jiang, Z. Wu and Y. Luo, J. Chem. Phys., 2008, 128, 084707 CrossRef.
- B. Gao, Doctoral thesis, comprehensive summary, KTH, Royal Institute of Technology, 2008 Search PubMed.
- B. Gao, J. Jiang, K. Liu, Z. Wu, W. Lu and Y. Luo, J. Comput. Chem., 2008, 29, 434–444 CrossRef CAS.
- W. J. Hua, Doctoral thesis, comprehensive summary, KTH, Royal Institute of Technology, 2011 Search PubMed.
- Y. F. Deng, B. Gao, M. S. Deng and Y. Luo, J. Chem. Phys., 2014, 140, 124304 CrossRef.
- L. H. Gan, R. Wu, J. L. Tian, J. Clarke, C. Gibson and P. W. Fowler, J. Comput. Chem., 2017, 38, 144–151 CrossRef CAS.
- S. Díaz Tendero, M. Alcamí and F. Martín, J. Chem. Phys., 2005, 123, 184306 CrossRef.
- P. W. Fowler, D. E. Manolopoulos and R. P. Ryan, Carbon, 1992, 30, 1235–1250 CrossRef CAS.
- D. E. Manolopoulos and P. W. Fowler, Chem. Phys. Lett., 1993, 204, 1–7 CrossRef CAS.
- E. Papirer, R. Lacroix, J. B. Donnet, G. Nanse and P. Fioux, Carbon, 1994, 32, 1341–1358 CrossRef CAS.
- E. Papirer, R. Lacroix, J. B. Donnet, G. Nansé and P. Fioux, Carbon, 1995, 33, 63–72 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08215e |
| This journal is © The Royal Society of Chemistry 2021 |