
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater-assisted synthesis of highly stable CsPbX3 perovskite quantum dots embedded in zeolite-Y†
Hui Chengab,
Yanfeng Yinb,
Jianbo Tangb,
Donghua Fan*a,
Jan J. Huang*a and
Shengye Jin *b
*b
aSchool of Applied Physics and Materials, Wuyi University, Jiangmen, 529020, China
bNational Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China 116023
First published on 13th January 2021
Abstract
All-inorganic perovskite materials have emerged as highly promising materials for solar cells and photoelectronic applications. However, the poor stability of perovskites in ambient conditions significantly hampers their practical applications. In this work, we report a three-step synthesis of size tunable CsPbX3 (X = Br, Cl, or I) quantum dots (QDs) embedded in zeolite-Y (CsPbX3-Y), which involves efficient chemical transformation of non-luminescent Cs4PbX6 to highly luminescent CsPbX3 by stripping CsX through an interfacial reaction with water. We show that the size and the emission of CsPbX3 in CsPbX3-Y can be tuned by the amount of water added as well as the halide composition. More importantly, the as-prepared CsPbX3-Y show significantly enhanced stability against moisture upon protection by zeolite-Y. This work not only reports a new pathway for the preparation of highly luminescent CsPbX3 but also provided new insights into the chemical transformation behavior and stabilization mechanism of these emerging perovskites.
Introduction
Benefiting from their compositional tunability and outstanding charge transport properties, all inorganic perovskites and hybrid lead-halide perovskites have attracted significant attention in recent years in the materials research community,1–4 particularly in solar cells,5–7 solid-state light-emitting diodes,8,9 photodetectors and lasers.10,11 However, the sensitivity of perovskites to moisture and their apparent instability upon contact with water significantly impede their practical applications.12–14 Recent work focusing on investigating the interaction mechanism between perovskites and water uncovered the existence of weak interactions between the organic and inorganic constituents,15,16 particularly at the surface of the perovskite, which is believed to be responsible for the rapid decomposition of perovskite upon exposure to H2O.17–19 As a result, efforts have been devoted to modify the surface of perovskite in attempt to improve their stability against moisture.20–24 For example, organic ligands or polymers such as tri-n-octylphosphine,25 polyhedral oligomeric silsesquioxane and titanium butoxide have been used to modify the surface to enhance the stability of perovskite quantum dots (QDs).26,27 In addition to organic components,28 many inorganic materials which exhibit good thermal resistance such as SiO2, TiO2 and Al2O3 have been used to protect perovskite QDs.4,27,29–32 More recently, porous zeolites have drawn increasing attention as hosts to encapsulate and stabilize perovskite QDs.33 Zeolites are porous aluminosilicates, in which an open framework structure of [AlO4]5− and [SiO4]4− tetrahedra forms a regular 3D structure of cages and channels. As a result, they can accommodate a variety of charge balancing cations that are readily exchangeable and have been demonstrated to improve and stabilize emission from various types of luminescent centers that were introduced into the porous zeolite structure by ion exchange.34In this work, we report a successful three-step synthesis method for perovskite QDs embedded in zeolite-Y. The zeolite-Y is first exchanged with Cs+ ions by an ion-exchange reaction from aqueous solution. Cs+ ion in zeolite then reacts with Pb2+ and X− (with X = Cl, Br, or I) ions to form non-luminescent Cs4PbX6. In the final step, Cs4PbX6 is converted into CsPbX3 by stripping CsX through an interfacial reaction with water. We not only show that the size and the emission property of CsPbX3 in CsPbX3-Y can be controlled by the amount of water absorbed and the halide composition but also demonstrate that the stability of CsPbX3-Y is significantly enhanced against moisture.
Experimental section
Materials
Caesium bromide (CsBr, Alfa Aesar, 99.999%), oleic acid (OA, Sigma-Aldrich, 90%), ODE (Sigma-Aldrich, 90%), oleylamine (OAm, Aladdin, 80–90%), lead chloride (PbCl2, Alfa Aesar, 99.999%), lead bromide (PbBr2, Alfa Aesar, 99.999%), lead iodide (PbI2, Alfa Aesar, 99.999%), n-trioctylphosphine (TOP, Strem, 97%), n-hexane (Aladdin, AR, 97%), isopropanol (AR, Fuyu Chemicals), Na+–zeolite-Y (SiO2/Al2O3 = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Alfa Aesar). All solvents and chemicals were purchased from a commercial company and directly used without further purification.
:1, Alfa Aesar). All solvents and chemicals were purchased from a commercial company and directly used without further purification.
Preparation of Cs+-Y
1.0 g of zeolite-Y and 0.01 mol CsBr were dispersed in 10 mL aqueous solution in single neck flask. The mixture was stirred in a water bath at 60 °C for 24 h. The products were centrifuged and washed with deionized water, and then dried in air at 80 °C for 12 h. The formation of Cs+-Y can be represented by the following chemical equation (eqn (1)).| Na+-Y + Cs+ → Cs+-Y + Na+ | (1) |
Synthesis of Cs4PbX6-Y
PbX2 solution was prepared by mixing 0.188 mmol of PbX2, 1.0 mL of OA, 1.0 mL of OAm in a 50 mL three-necked bottle along with 5.0 mL ODE. The mixture was dried for 30 min at 120 °C under vacuum with stirring. After that, the mixture was purged under N2 flow until PbX2 was completely dissolved. For preparing Cs4PbCl6-Y, PbCl2 is used. PbCl2 was dissolved at 150 °C with TOP (1 mL). The mixed PbX2 with PbCl2:PbBr2 = 0.4:0.6 and PbI2:PbBr2 = 0.4:0.6 were used to prepare Cs4Pb(Br0.6Cl0.4)6-Y and Cs4Pb(Br0.6I0.4)6, respectively. Then Cs+-Y (0.5 g) was loaded into a 50 mL three-necked bottle along with 5.0 mL ODE. The mixture was dried for 30 min under vacuum at 120 °C. The reaction temperature was raised to 150 °C under N2 atmosphere and maintained for another 30 min. The as-prepared PbX2 solution was swiftly injected into the hot reaction mixture. The mixture was stirred about 10 min. For the larger halide ion I−, a longer reaction time (∼15 min) was needed for PbX2 to diffuse into the zeolite and react with the previously exchanged Cs+. The mixture was then cooled down using an ice-water bath. Finally, the obtained product was washed with n-hexane and isopropanol twice followed by centrifugation. The Cs4PbX6 composites were dried at 80 °C for 5 h. The chemical equation corresponding to this reaction can be represented by (eqn (2)):| 4Cs+-Y + Pb2++ 6X− → Cs4PbX6-Y | (2) |
Synthesis of CsPbX3-Y
A small amount of water was added into the Cs4PbX6-Y composites, which results into the conversion of Cs4PbX6-Y to CsPbX3-Y (eqn (3)). The size of CsPbX3 in CsPbX3-Y can be controlled by the amount of water added.
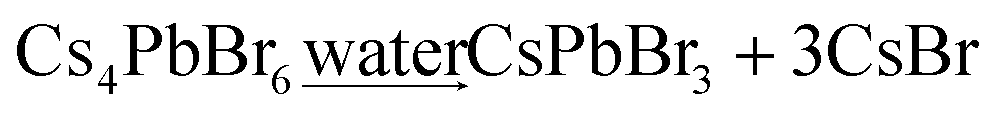 | (3) |
Synthesis of CsPbBr3 quantum dots (QDs)
In a typical synthesis of CsPbBr3 QDs, the Cs-precursor was prepared according to the literature protocol.35 Briefly, 0.188 mmol of PbBr2, 1.0 mL of OA, and 1.0 mL of OAm were loaded into a 50 mL three-necked bottle along with 5.0 mL ODE. The mixture was dried for 30 min at 120 °C under vacuum and stirring. Then the reaction temperature was raised to 150 °C under N2 atmosphere and maintained for another 30 min. The pre-prepared Cs-precursor solution (0.4 mL) was swiftly injected into the hot reaction mixture. The system was cooled down with an ice-water bath after reaction about 5 seconds. Finally, the obtained product was dispersed in n-hexane after centrifugation.Characterization and general procedure
The powder X-ray diffraction (PXRD) patterns were collected using Empyrean-100 diffractometer with Cu Kα radiation (λ = 1.5406 Å) at 293 K. The data were collected at 5°–40° range and scan speed is 5.0 degree per min. A Bruker TENSOR27 spectrometer was applied to measure FT-IR (Fourier transform infrared) spectra (as KBr pressed pellets) in the 4000–400 cm−1 range. The surface morphology of the powder was observed using a JSN-7800F scanning electron microscope (SEM) with an accelerating voltage of 1.0 kV. Diffuse reflectance UV-visible spectra were obtained using Agilent Technologies Carry 60 UV-vis spectrophotometer. For the luminescence measurements, a laser source at 405 nm was employed. The luminescence signals were collected and directed into a fiber integrated spectrograph with spectra resolution < 1.0 nm. The fluorescence lifetime measurement setup used in this study was based on the time-correlated single photon counting technology (TCSPC). The excitation beam was picosecond pulse diode laser with 405 nm output wavelength. The optical detector was single photon counting module.Results and discussion
Fig. 1 schematically illustrated the three-step synthesis of CsPbBr3-Y. The first step is the exchange of Na+ in zeolite-Y (Na+-Y) by Cs+ to form Cs+-Y (see details of procedure in Experimental section).33 In the second step, PbX2 was added into the suspension of Cs+-Y in ODE to form non-luminescent Cs4PbX6-Y. In the last step, non-luminescent Cs4PbX6-Y is converted to luminescent CsPbX3-Y upon the addition of water. The formation of CsPbX3 is believed to undergo stripping CsX from Cs4PbX6 via interfacial reaction with water.36 The products formed during each step in Fig. 1 were confirmed by powder X-ray diffraction (XRD), Fourier transform infrared (FTIR) spectroscopy, and scanning electron microscope (SEM).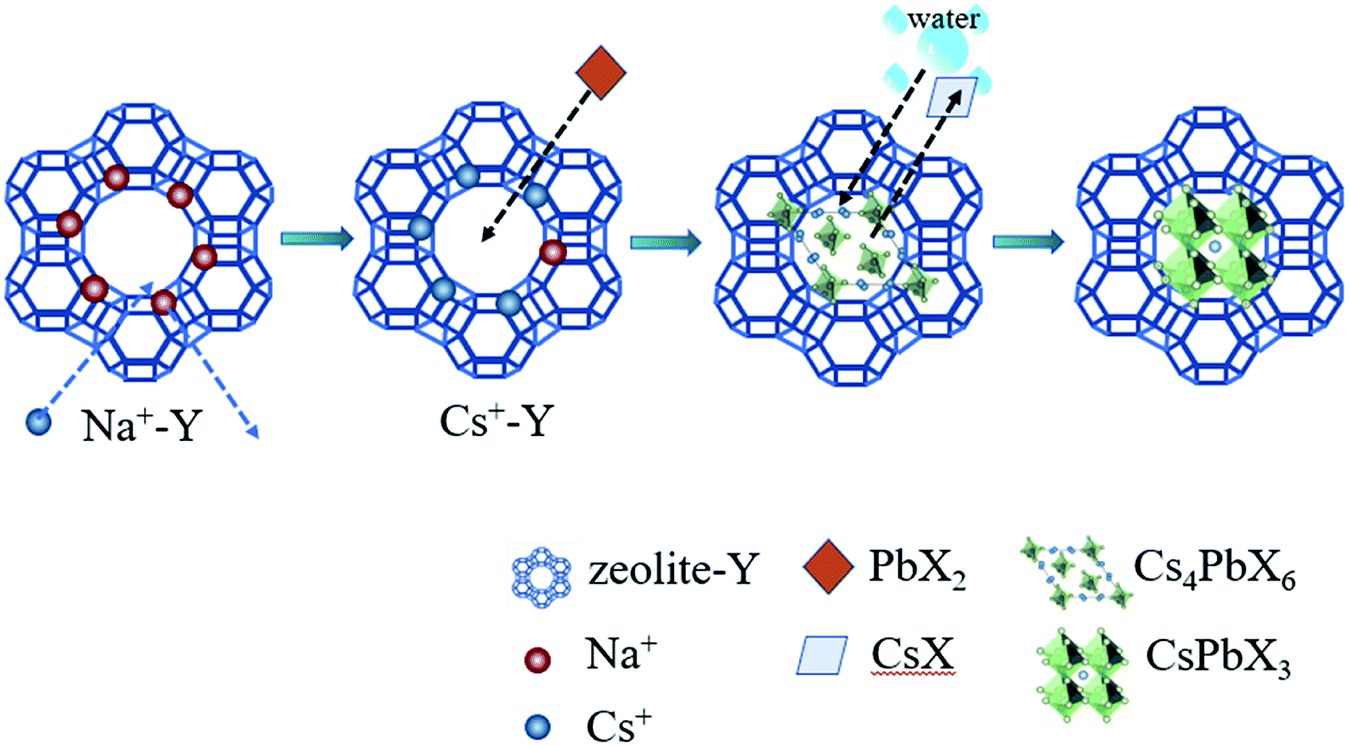 | ||
| Fig. 1 Schematic illustration of the three-step synthesis of CsPbBr3-Y composites. | ||
Fig. 2a shows the X-ray diffraction (XRD) patterns of the zeolite-Y, Cs+-Y, Cs4PbBr6-Y and CsPbBr3-Y. While the majority of the diffraction angles of Cs+-Y, Cs4PbBr6-Y and CsPbBr3-Y match that of zeolite-Y, the relative intensity of several diffraction peaks change in Cs+-Y, Cs4PbBr6-Y and CsPbBr3-Y with respect to that in zeolite-Y. For example, compared to zeolite-Y, the intensity of the peaks at 12.41°, 14.38°, 18.66°, and 21.61° in Cs+-Y and CsPbBr3-Y increases, while the intensity of peaks at 15.78° and 20.27° decreases significantly. The similar results have been observed in previous literatures,33 which can be attributed to the change of the unit cell of zeolite-Y while zeolite crystal structure remains fixed. These results suggest that the variations during ion-exchange reaction occur inside the pore of zeolite-Y, i.e. the formation of QDs occurs in mesopores of zeolite-Y although we cannot exclude that some QDs form on the surface of zeolite-Y. No XRD peaks originating from the CsPbBr3 perovskite crystal structure are observed, which is likely due to the much smaller volume of embedded QDs (3–10 nm) than zeolite-Y (2 μm), resulting in much weaker diffraction signals in the former than the latter.
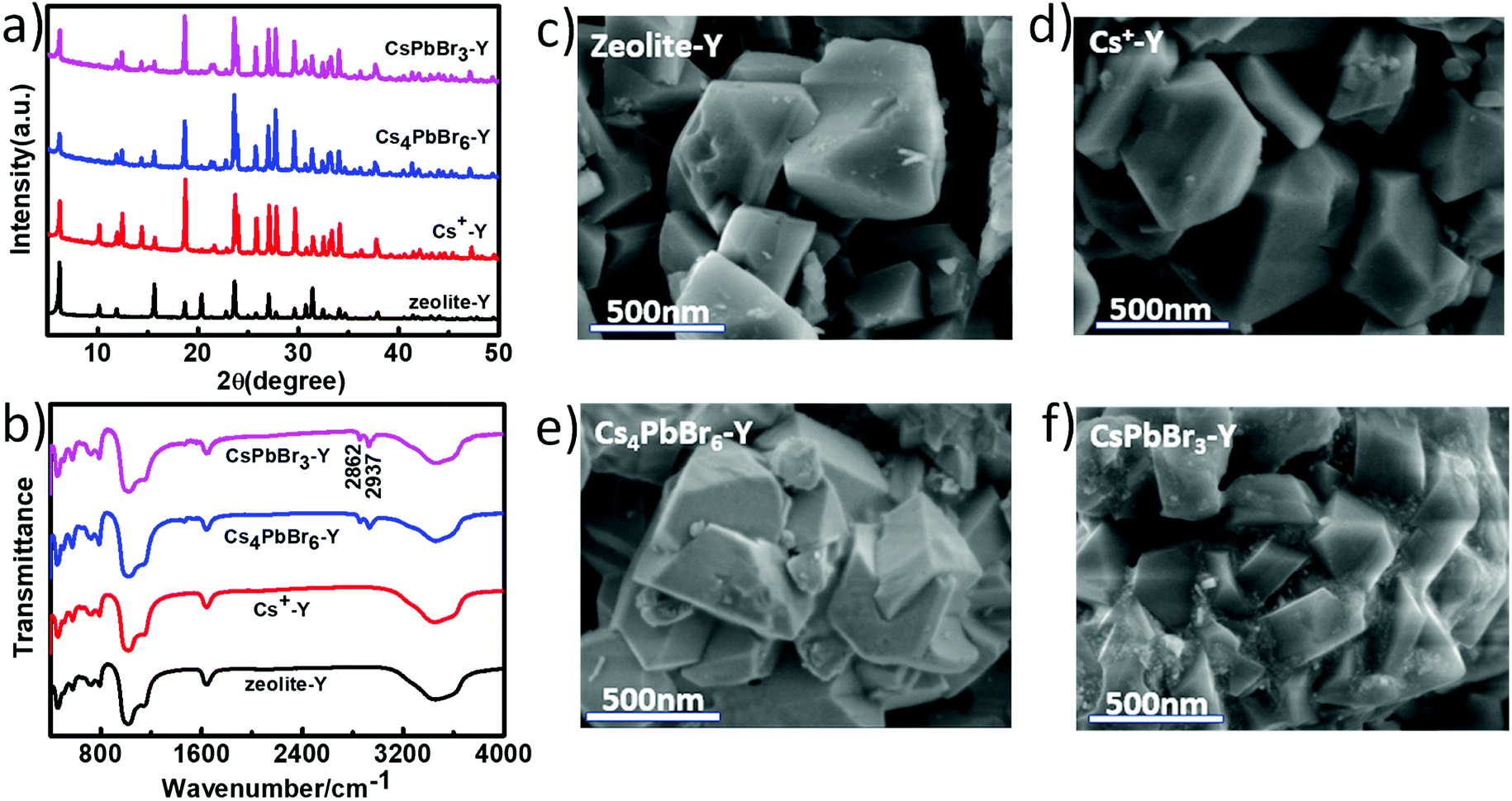 | ||
| Fig. 2 PXRD patterns (a) and FTIR spectra (b) of zeolite-Y, Cs+-Y, Cs4PbBr6-Y, CsPbBr3-Y; the SEM images of Zeolite-Y (c), Cs+-Y (d), Cs4PbBr6-Y (e), and CsPbBr3-Y (f). | ||
Fig. 2b compares the FTIR spectra of Zeolite-Y, Cs+-Y, Cs4PbBr6-Y and CsPbBr3-Y. The peak at 1642 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O symmetric stretching vibration of the deprotonated carboxylate group and the broad band at 3300 cm−1 can be assigned to the OH stretching vibration of the water molecule. The strong peaks at 2862 and 2937 cm−1 can be assigned to C–H bending that results from the hydrocarbon chains in organic species. The bands at 1140 cm−1 and 1021 cm−1 can be attributed to the vibration of Si–O–Si and Si–O–C, respectively.37 Fig. 2c–f show scanning electron microscopy (SEM) images of zeolite-Y, Cs+-Y, Cs4PbBr6-Y and CsPbBr3-Y composites, respectively. The Cs+-Y, Cs4PbBr6-Y, and CsPbBr3-Y all show similar morphology as that of zeolite-Y, suggesting the retain of zeolite structure among these samples, which is consistent with XRD results.
O symmetric stretching vibration of the deprotonated carboxylate group and the broad band at 3300 cm−1 can be assigned to the OH stretching vibration of the water molecule. The strong peaks at 2862 and 2937 cm−1 can be assigned to C–H bending that results from the hydrocarbon chains in organic species. The bands at 1140 cm−1 and 1021 cm−1 can be attributed to the vibration of Si–O–Si and Si–O–C, respectively.37 Fig. 2c–f show scanning electron microscopy (SEM) images of zeolite-Y, Cs+-Y, Cs4PbBr6-Y and CsPbBr3-Y composites, respectively. The Cs+-Y, Cs4PbBr6-Y, and CsPbBr3-Y all show similar morphology as that of zeolite-Y, suggesting the retain of zeolite structure among these samples, which is consistent with XRD results.
Fig. 3a shows the diffuse reflectance UV-visible spectra of as prepared CsPbBr3-Y. Immediately after the formation of CsPbBr3-Y, the first exciton peak of CsPbBr3 QDs is centered at 470 nm (black plot). With increasing time of exposure to water, the first exciton peak of CsPbBr3-Y shows a bathochromic shift, suggesting the gradual increase of QD size in CsPbBr3-Y.29,38,39 The spectrum stops shifting when exciton peak reaches 520 nm (blue plot), which suggests that the QDs strop growing. These results are further supported from the steady state emission spectrum, where the emission peak for the above CsPbBr3-Y samples also show red shift, ranging from 460 nm to 520 nm (Fig. 3b) upon exposure to water and stops shifting afterwards. These results are consistent with the diffuse reflectance UV-visible data above and suggest the growth of QDs in CsPbBr3-Y with the amount of water absorbed. The sample when the size stops growing (i.e. the exciton peak at 520 nm) was used for further study and is denoted as CsPbBr3-Y (520 nm).
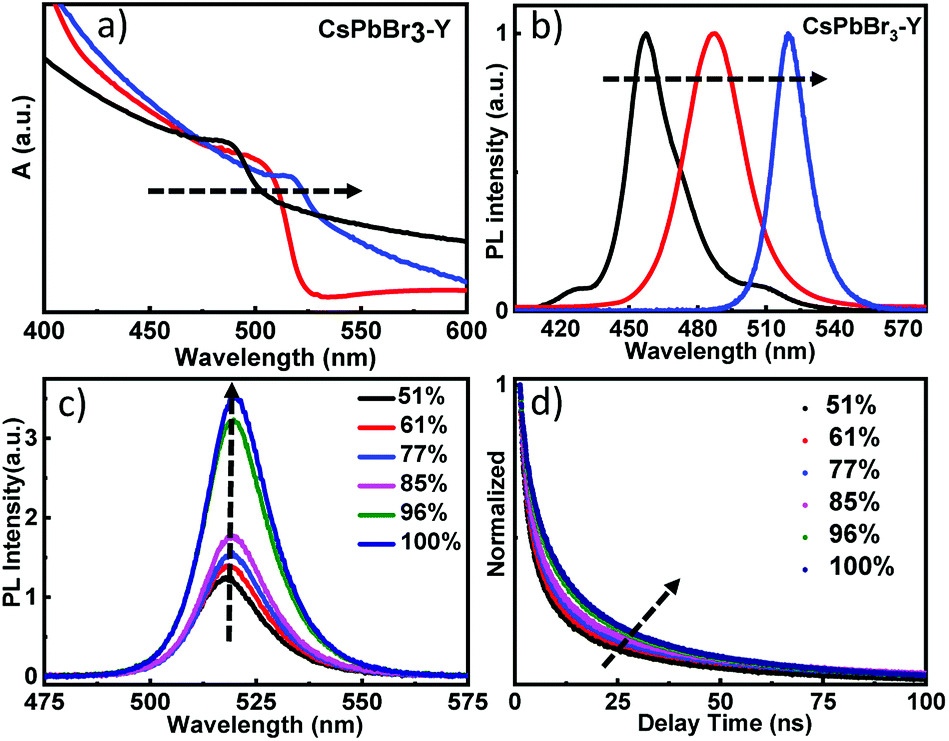 | ||
| Fig. 3 The diffuse reflectance spectra (a) and the corresponding emission spectra (b) of CsPbBr3-Y with different sizes of CsPbBr3; (c) the emission spectra of CsPbBr3-Y (520 nm) in different relative humidity; (d) the emission decay curves of CsPbBr3-Y (520 nm) in different relative humidity. | ||
The impact of relative humidity on the emission properties of CsPbBr3-Y (520 nm) was examined under different humidity. As shown in Fig. 3c, the emission intensity of CsPbBr3-Y (520 nm) gradually increases with increasing humidity.36 Furthermore, the emission lifetime of CsPbBr3-Y (520 nm) measured by time-resolved emission spectroscopy using time correlated single photon counting (TCSPC) technique becomes longer with increasing humidity (Fig. 3d). These results, consistent with the description in the previous literature,40 can be attributed to that the nonradiative recombination has been inhibited with increasing humidity, consistent with the steady state emission results above.
In addition to the relative humidity, we also evaluated the stability of CsPbBr3-Y (520 nm) by measuring the impact of environment on its emission intensity. As shown in Fig. 4a, the emission intensity of CsPbBr3 QDs with similar size without zeolite-Y decreases more than 51.3% after 30 days and the emission is almost completely quenched after 60 days. In contrast, the emission intensity in CsPbBr3-Y (520 nm) only decays 15.4% by 30 days and 21.8% after 60 days (Fig. 4b). These results together suggest that embedding CsPbBr3 in zeolite can significantly increase its stability in environment. The stability of CsPbBr3-Y (520 nm) was further evaluated by exposing CsPbBr3-Y (520 nm) to 100% relative humidity, where the intensity of CsPbBr3 QDs is completely quenched after 6 hours (Fig. 4c), whereas the emission of CsPbBr3-Y (520 nm) only decrease by 18.4% after 240 hours (Fig. 4d). Moreover, the emission lifetime of CsPbBr3-Y (520 nm) is much longer than CsPbBr3 QDs (Fig. S1a, S1b, Tables S1 and S2†). Based on these results, we can conclude that the stability of CsPbBr3 in CsPbBr3-Y composites is significantly improved.
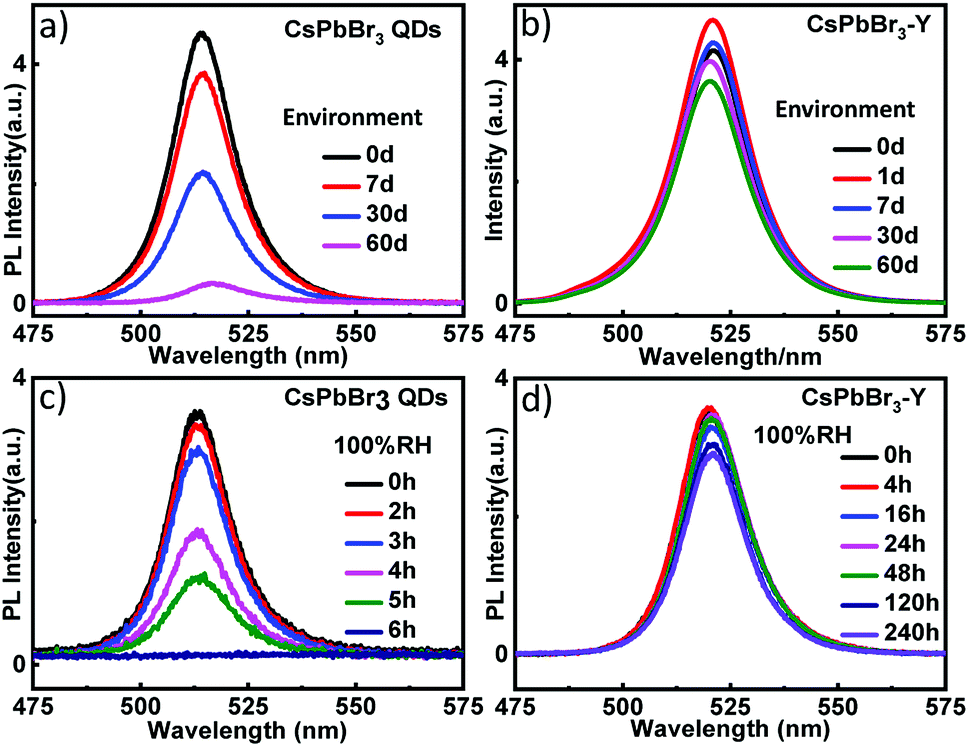 | ||
| Fig. 4 The emission spectra of CsPbBr3 QDs (a) and CsPbBr3-Y (520 nm) (b) upon exposure to environment; the emission spectra of CsPbBr3 QDs (c) and CsPbBr3-Y (520 nm) (d) under 100% relative humidity. | ||
It has been shown previously that the luminescence properties of CsPbX3 are strongly dependent on their halide composition. In this work, we also examined the impact of halide composition to the emission property of CsPbX3-Y. We first incorporated X (X = Cl or I) to Cs4PbBr6-Y with X:Br = 0.4:0.6, which forms Cs4Pb(Br0.6Cl0.4)6-Y and Cs4Pb(Br0.6I0.4)6-Y, respectively. After exposing Cs4Pb(Br0.6Cl0.4)6-Y and Cs4Pb(Br0.6I0.4)6-Y to water, the corresponding CsPb(Br0.6Cl0.4)3-Y and CsPb(Br0.6I0.4)3-Y were formed. Both Cs4Pb(Br0.6Cl0.4)6-Y (i and iii in Fig. 5a) and Cs4Pb(Br0.6I0.4)6-Y (i and iii in Fig. 5b) do not emit under sun light and ultraviolet light. After they absorb water, the color of CsPb(Br0.6Cl0.4)3-Y changes from white to yellow under sun light (ii in Fig. 5a) and cyan under ultraviolet (iv in Fig. 5a), while the color of CsPb(Br0.6I0.4)3-Y, change from white to red under sunlight (ii in Fig. 5b) and yellow under ultraviolet (iv in Fig. 5b). These results suggest that non-luminescent Cs4Pb(Br0.6X0.4)6-Y (X = Cl or I) turn into luminescent CsPb(Br0.6X0.4)3-Y. It is worth mentioning that the reaction rate of the transformation process depends on the halide composition. The transformation process of Cs4Pb(Br0.6Cl0.4)6-Y to CsPb(Br0.6Cl0.4)3-Y started immediately upon the addition of water, while it took several minutes to gradually see the color change for Cs4Pb(Br0.6I0.4)6-Y. These phenomena can be attributed to the higher solubility of CsCl than that of CsI in water, resulting into the faster CsX-stripping process in the former than the latter. As shown in Fig. 5c, the emission spectra of these perovskite samples shifts significantly, where CsPb(Br0.6Cl0.4)3-Y displays an emission peak at 480 nm, CsPb(Br0.6I0.4)3-Y shows a relatively broad emission spectrum centered at 560 nm, both of which distinct from that of CsPbBr3– Y, suggesting that the emission of CsPbX3-Y can be tuned by controlling the halide component.
 | ||
| Fig. 5 (a) Cs4Pb(Br0.6Cl0.4)6-Y under sun light (i), CsPb(Br0.6Cl0.4)3 under sun light (ii), Cs4Pb(Br0.6Cl0.4)6-Y under UV (iii), CsPb(Br0.6Cl0.4)3 under UV (iv); (b) Cs4Pb(Br0.6I0.4)6-Y under sun light (i), CsPb(Br0.6I0.4)3-Y under sun light (ii), Cs4Pb(Br0.6I0.4)6-Y under UV (iii), CsPb(Br0.6I0.4)3-Y under UV (iv); (c) the emission spectra of CsPb(Br0.6Cl0.4)3-Y, CsPbBr3-Y and CsPb(Br0.6I0.4)3-Y. | ||
Conclusions
In this work, we report a successful three-step method to synthesize perovskite QDs embedded in zeolite-Y, through which the efficient chemical transformation of non-luminescent Cs4PbX6 to highly luminescent CsPbX3 occurs by stripping CsX from the former through an interfacial reaction with water. We show that the size and the emission of CsPbX3 in CsPbX3-Y can be tuned by the amount of water added as well as the halide composition. More importantly, the as-prepared CsPbX3-Y show significantly enhanced stability against moisture upon protection by zeolite-Y. This work not only reports a new pathway for the preparation of highly luminescent CsPbX3 but also provides important insights into the chemical transformation behavior and stabilization mechanism of these emerging perovskites.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the Natural Science Foundation of Guangdong Province, 2018A030313460, Science and Technology Projects of Jiangmen (No. (2017) 307, (2017) 149, (2018) 352), Key Laboratory of Optoelectronic materials and Applications in Guangdong Higher Education (No: 2017KSYS011).Notes and references
- L. N. Quan, B. P. Rand, R. H. Friend, S. G. Mhaisalkar, T. W. Lee and E. H. Sargent, Chem. Rev., 2019, 119, 7444–7477 CrossRef CAS.
- S. Lee, J. H. Park, Y. S. Nam, B. R. Lee, B. Zhao, D. Di Nuzzo, E. D. Jung, H. Jeon, J. Y. Kim, H. Y. Jeong, R. H. Friend and M. H. Song, ACS Nano, 2018, 12, 3417–3423 CrossRef CAS.
- W. Chen, H. Liu, R. Fan, P. Wang, T. Sun and Y. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 9851–9857 CrossRef CAS.
- L. Polavarapu, B. Nickel, J. Feldmann and A. S. Urban, Adv. Energy Mater., 2017, 7, 1700267 CrossRef.
- X. Wang, L. Wang, T. Shan, S. Leng, H. Zhong, Q. Bao, Z. H. Lu, L. L. Deng and C. C. Chen, Nano-Micro Lett., 2020, 12, 84 CrossRef CAS.
- N. J. Jeon, H. Na, E. H. Jung, T. Y. Yang, Y. G. Lee, G. Kim, H. W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
- M. Anaya, G. Lozano, M. E. Calvo and H. Míguez, Joule, 2017, 1, 769–793 CrossRef CAS.
- H. Cho, S. Jeong, M. Park, Y. Kim, C. Wolf, C. Lee and T. Lee, Science, 2015, 350, 1222–1225 CrossRef CAS.
- Z. K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS.
- X. He, Y. Qiu and S. Yang, Adv. Mater., 2017, 29, 201700775 Search PubMed.
- G. Xing, N. Mathews, S. S. Lim, N. Yantara, X. Liu, D. Sabba, M. Gratzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS.
- Y. Wei, X. Deng, Z. Xie, X. Cai, S. Liang, P. a. Ma, Z. Hou, Z. Cheng and J. Lin, Adv. Funct. Mater., 2017, 27, 1703535 CrossRef.
- C. Sun, Y. Zhang, C. Ruan, C. Yin, X. Wang, Y. Wang and W. W. Yu, Adv. Mater., 2016, 28, 10088–10094 CrossRef CAS.
- J. Hai, H. Li, Y. Zhao, F. Chen, Y. Peng and B. Wang, Chem. Commun., 2017, 53, 5400–5403 RSC.
- K. K. Liu, Q. Liu, D. W. Yang, Y. C. Liang, L. Z. Sui, J. Y. Wei, G. W. Xue, W. B. Zhao, X. Y. Wu, L. Dong and C. X. Shan, Light: Sci. Appl., 2020, 9, 44 CrossRef CAS.
- C. Wu, Y. Zou, T. Wu, M. Ban, V. Pecunia, Y. Han, Q. Liu, T. Song, S. Duhm and B. Sun, Adv. Funct. Mater., 2017, 27, 1700338 CrossRef.
- G. Niu, X. Guo and L. Wang, J. Mater. Chem. A, 2015, 3, 8970–8980 RSC.
- D. Zhang, Y. Xu, Q. Liu and Z. Xia, Inorg. Chem., 2018, 57, 4613–4619 CrossRef CAS.
- J. Ren, T. Li, X. Zhou, X. Dong, A. V. Shorokhov, M. B. Semenov, V. D. Krevchik and Y. Wang, Chem. Eng. J., 2019, 358, 30–39 CrossRef CAS.
- V. Malgras, J. Henzie, T. Takei and Y. Yamauchi, Chem. Commun., 2017, 53, 2359–2362 RSC.
- Z. C. Kong, J. F. Liao, Y. J. Dong, Y. F. Xu, H. Y. Chen, D. B. Kuang and C. Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- Y. Wang, T. Wu and J. Barbaud, et al., Science, 2019, 365, 687–691 CrossRef CAS.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. Cacovich, C. Stavrakas, B. Philippe, J. M. Richter, M. Alsari, E. P. Booker, E. M. Hutter, A. J. Pearson, S. Lilliu, T. J. Savenije, H. Rensmo, G. Divitini, C. Ducati, R. H. Friend and S. D. Stranks, Nature, 2018, 555, 497–501 CrossRef CAS.
- L. Yang, T. Wang, Q. Min, B. Liu, Z. Liu, X. Fan, J. Qiu, X. Xu, J. Yu and X. Yu, ACS Omega, 2019, 4, 6084–6091 CrossRef CAS.
- C. Lu, H. Li, K. Kolodziejski, C. Dun, W. Huang, D. Carroll and S. M. Geyer, Nano Res., 2017, 11, 762–768 CrossRef.
- H. Huang, B. Chen, Z. Wang, T. F. Hung, A. S. Susha, H. Zhong and A. L. Rogach, Chem. Sci., 2016, 7, 5699–5703 RSC.
- Z. J. Li, E. Hofman, J. Li, A. H. Davis, C. H. Tung, L. Z. Wu and W. Zheng, Adv. Funct. Mater., 2018, 28, 1704288 CrossRef.
- Y. Cai, L. Wang, T. Zhou, P. Zheng, Y. Li and R. J. Xie, Nanoscale, 2018, 10, 21441–21450 RSC.
- V. Malgras, S. Tominaka, J. W. Ryan, J. Henzie, T. Takei, K. Ohara and Y. Yamauchi, J. Am. Chem. Soc., 2016, 138, 13874–13881 CrossRef CAS.
- H. C. Yoon, S. Lee, J. K. Song, H. Yang and Y. R. Do, ACS Appl. Mater. Interfaces, 2018, 10, 11756–11767 CrossRef CAS.
- H. C. Wang, S. Y. Lin, A. C. Tang, B. P. Singh, H. C. Tong, C. Y. Chen, Y. C. Lee, T. L. Tsai and R. S. Liu, Angew. Chem., Int. Ed. Engl., 2016, 55, 7924–7929 CrossRef CAS.
- S. Guarnera, A. Abate, W. Zhang, J. M. Foster, G. Richardson, A. Petrozza and H. J. Snaith, J. Phys. Chem. Lett., 2015, 6, 432–437 CrossRef CAS.
- J. Y. Sun, F. T. Rabouw, X. F. Yang, X. Y. Huang, X. P. Jing, S. Ye and Q. Y. Zhang, Adv. Funct. Mater., 2017, 27, 1704371 CrossRef.
- T. Altantzis, E. Coutino-Gonzalez, W. Baekelant, G. T. Martinez, A. M. Abakumov, G. V. Tendeloo, M. B. Roeffaers, S. Bals and J. Hofkens, ACS Nano, 2016, 10, 7604–7611 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS.
- W. Lv, L. Li, M. Xu, J. Hong, X. Tang, L. Xu, Y. Wu, R. Zhu, R. Chen and W. Huang, Adv. Mater., 2019, 31, e1900682 CrossRef.
- X. Zhang, X. Bai, H. Wu, X. Zhang, C. Sun, Y. Zhang, W. Zhang, W. Zheng, W. W. Yu and A. L. Rogach, Angew. Chem., Int. Ed. Engl., 2018, 57, 3337–3342 CrossRef CAS.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- X. Gong, M. Li, X. B. Shi, H. Ma, Z. K. Wang and L. S. Liao, Adv. Funct. Mater., 2015, 25, 6671–6678 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD of the CsPb(Cl0.4Br0.6)3-Yand CsPb(Br0.6I0.4)3-Y, the emission spectra and decay curves of different size CsPbBr3-Y, the emission decay curves and lifetime of CsPbBr3 ODs and CsPbBr3-Y in environment and 100% RH, respectively, the emission spectra of CsPbBr3-Y immersion in water, the images of Cs4PbBr6-Y and CsPbBr3-Y in sun light and under UV. See DOI: 10.1039/d0ra08311a |
| This journal is © The Royal Society of Chemistry 2021 |