
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnisotropic microparticles for differential drug release in nerve block anesthesia†
Shivakumar B. S,
Vignesh Gopalakrishnan-Prema,
Gayathri Raju,
Sumi E. Mathew,
Neeraj Katiyar,
Deepthy Menon and
Sahadev A. Shankarappa *
*
Center for Nanosciences & Molecular Medicine, Amrita Vishwa Vidyapeetham, Kochi 682041, Kerala, India. E-mail: sahadevs@icloud.com; sahadevs@aims.amrita.edu; Tel: +91 4842 801234 (extn 8705)
First published on 22nd January 2021
Abstract
Microparticle shape, as a tunable design parameter, holds much promise for controlling drug-release kinetics from polymeric microparticulate systems. In this study we hypothesized that the intensity and duration of a local nerve block can be controlled by administration of bupivacaine-loaded stretch-induced anisotropic poly(lactic-co-glycolic acid) microparticles (MPs). MPs of size 27.3 ± 8.5 μm were synthesized by single emulsion method and subjected to controlled stretching force. The aspect ratio of the anisotropic–bupivacaine MPs was quantified, and bupivacaine release was measured in vitro. The anisotropic MPs were administered as local nerve block injections in rats, and the intensity and duration of local anesthesia was measured. Bupivacaine-loaded anisotropic MPs used in this study were ellipsoid in shape and exhibited increased surface pores in comparison to spherical MPs. Anisotropic MPs exhibited a higher rate of bupivacaine release in vitro, and showed significantly (P < 0.05) stronger sensory nerve blocking as compared to spherical bupivacaine MPs, even though the duration of the nerve block remained similar. This study demonstrates the utility of stretch-induced anisotropic MPs in controlling drug release profiles from polymeric MPs, under both in vitro and in vivo conditions. We show that shape, as a tunable design parameter, could play an important role in engineering drug-delivery systems.
Introduction
Anisotropic, non-spherical polymeric particles have been recently gaining much importance for their rather interesting ability to avoid non-specific uptake by certain blood,1 tumor,2 and stem cells.3 Most of these reported findings are in particles that range in size from 500 nm to 5 μm.4,5 Larger, non-spherical shaped microparticles (MPs) in the size range beyond 10 μm, though not suitable for intracellular uptake, may have a role in controlled drug delivery.6,7 Shape as a tunable design parameter for regulating drug-release from polymeric MPs could be utilized as an additional strategy in drug-delivery.8,9 However, actual proof of utilization of shape and anisotropy as design parameters for regulating drug-release from polymeric MPs is lacking.The internal structural arrangement of polymeric chains is fairly uniform within spherical shaped MPs synthesized by conventional methods.10 However, altering the shape and inducing anisotropy in MPs changes the internal and surface architecture of the polymeric matrix,11 which can affect particle degradation. Passive drug efflux from drug-loaded polymeric delivery systems occurs via a combination of diffusion, hydrolytic degradation, and bulk erosion.12,13 Even though the rate of drug diffusion from polymeric MPs is closely linked to drug hydrophilicity, the ingress of water molecules into the MP matrix determines to a large extent, the efflux of drug from the delivery system.14–16 Key factors that allow water molecules to enter and percolate through polymeric MPs are the sub-micron sized defects on the surface of MPs, along with the structural arrangement of the polymeric matrix. Incorporation of ‘porogens’ such as pluronic into the polymeric emulsion during MP synthesis, or addition of organic solvents to induce partial dissolution of polymer monomers, are few well-known strategies to alter the rate of drug efflux from the microparticulate matrix.17,18
Prolonged, controlled release of encapsulated drug molecules from polymeric microparticulate systems is a well-proven strategy to regulate intensity and duration of drug action and is widely used in several therapeutic applications.19,20 In this study, we wanted to examine if altering the shape of drug-loaded MPs by controlled morphological deformation could change drug release patterns. We hypothesized that stretch-induced anisotropic poly(lactic-co-glycolic acid) (PLGA) particles loaded with bupivacaine, a local anesthetic, can be utilized to produce functional nerve blocks. Bupivacaine-induced nerve blocks are widely used in clinical practice to perform minor surgical procedures. However, bupivacaine is extremely short-acting and hence limits its use. Delivery systems using spherical polymeric MPs have successfully demonstrated extended release of bupivacaine for prolonged nerve blocks.21,22 In this proof-of-concept study, by altering the shape and inducing anisotropy in bupivacaine-loaded PLGA microparticles, we have attempted to alter the dynamics of drug release and thereby affecting bupivacaine-induced nerve blocks.
Results
Morphology of stretched PLGA microparticles
To determine the effect of controlled stretching on microparticle topography, integrity, and shape, we subjected poly vinyl alcohol (PVA) films embedded with spherical PLGA microparticles to a continuous stretching force using a customized gear-based puller. We determined the effect of various stretching parameters on the aspect ratio (AR) (maximum length/maximum width) of the resulting anisotropic particles. PVA films containing naïve PLGA MPs (polymer ratios 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 and 75:25) were subjected to varying temperature, stretching rate, and extent of stretch (Table 1, Fig. S1†). Expectedly, the extent of film stretching had the most effect on the overall MP aspect ratio, while the rate of stretching had the least effect. Increasing the temperature of the PVA film during stretching only had a moderate effect, while the different lactide to glycolide ratio of PLGA used in this study did not seem to affect the overall MP shape. Similar to what was observed in naïve MPs, the overall aspect ratio of bupivacaine-loaded MPs was also strongly determined by the extent of applied stretch (Table 1).
:50 microparticles was 27.3 ± 8.5 μm, and that of PLGA 75:25 was 30.6 ± 8.8 μm. While varying each of the three stretching parameters, the remaining parameters were fixed at 50 mm min−1 (rate of stretching), 2.4 (length of stretching), or 80 °C (temperature)
:50 and 75:25) were subjected to varying temperature, stretching rate, and extent of stretch (Table 1, Fig. S1†). Expectedly, the extent of film stretching had the most effect on the overall MP aspect ratio, while the rate of stretching had the least effect. Increasing the temperature of the PVA film during stretching only had a moderate effect, while the different lactide to glycolide ratio of PLGA used in this study did not seem to affect the overall MP shape. Similar to what was observed in naïve MPs, the overall aspect ratio of bupivacaine-loaded MPs was also strongly determined by the extent of applied stretch (Table 1).
:50 microparticles was 27.3 ± 8.5 μm, and that of PLGA 75:25 was 30.6 ± 8.8 μm. While varying each of the three stretching parameters, the remaining parameters were fixed at 50 mm min−1 (rate of stretching), 2.4 (length of stretching), or 80 °C (temperature)
| Stretching parameters (PVA film) | PLGA 50:50 MPs |
PLGA 75:25 MPs |
|
|---|---|---|---|
| Temperature | 60 °C | 2.8 ± 0.9 | 3.8 ± 1.1 |
| 70 °C | 2.8 ± 0.7 | 4.1 ± 1.3 | |
| 80 °C | 3.3 ± 1.0 | 3.7 ± 0.8 | |
| Rate of stretching | 25 mm min−1 | 2.7 ± 0.7 | 1.9 ± 0.4 |
| 50 mm min−1 | 3.0 ± 1.0 | 3.3 ± 1.1 | |
| 82 mm min−1 | 2.9 ± 1.0 | 3.3 ± 1.0 | |
| Length of stretching | Film AR 1.5 | 1.9 ± 0.4 | 1.8 ± 0.4 |
| Film AR 2.0 | 2.3 ± 0.7 | 2.5 ± 0.5 | |
| Film AR 3.5 | 3.8 ± 0.9 | 3.7 ± 1.0 | |
| Bupivacaine loaded MPs | Film AR 1.5 | 1.9 ± 0.5 | — |
| Length of stretching | Film AR 3.5 | 4.0 ± 1.5 | — |
Controlled stretching of the PVA film resulted in the deformation of embedded spherical microparticles (Fig. 1a) into oblong and ellipsoid anisotropic particles (Fig. 1b), as visualized under SEM. Despite change in shape, stretched naïve MPs maintained their smooth and even surface topography with uniform taper at the poles (Fig. 1b), which was distinctly different from bupivacaine-loaded particles. Stretched microparticles containing bupivacaine demonstrated an uneven surface topography with slightly jagged tapering (Fig. 1c and d). Interestingly, bupivacaine particles subjected to a larger stretching force demonstrated visibly higher number of surface pores measuring 0.5–1 μm in diameter, as compared to non-stretched and moderately stretched particles (Fig. 1e–h).
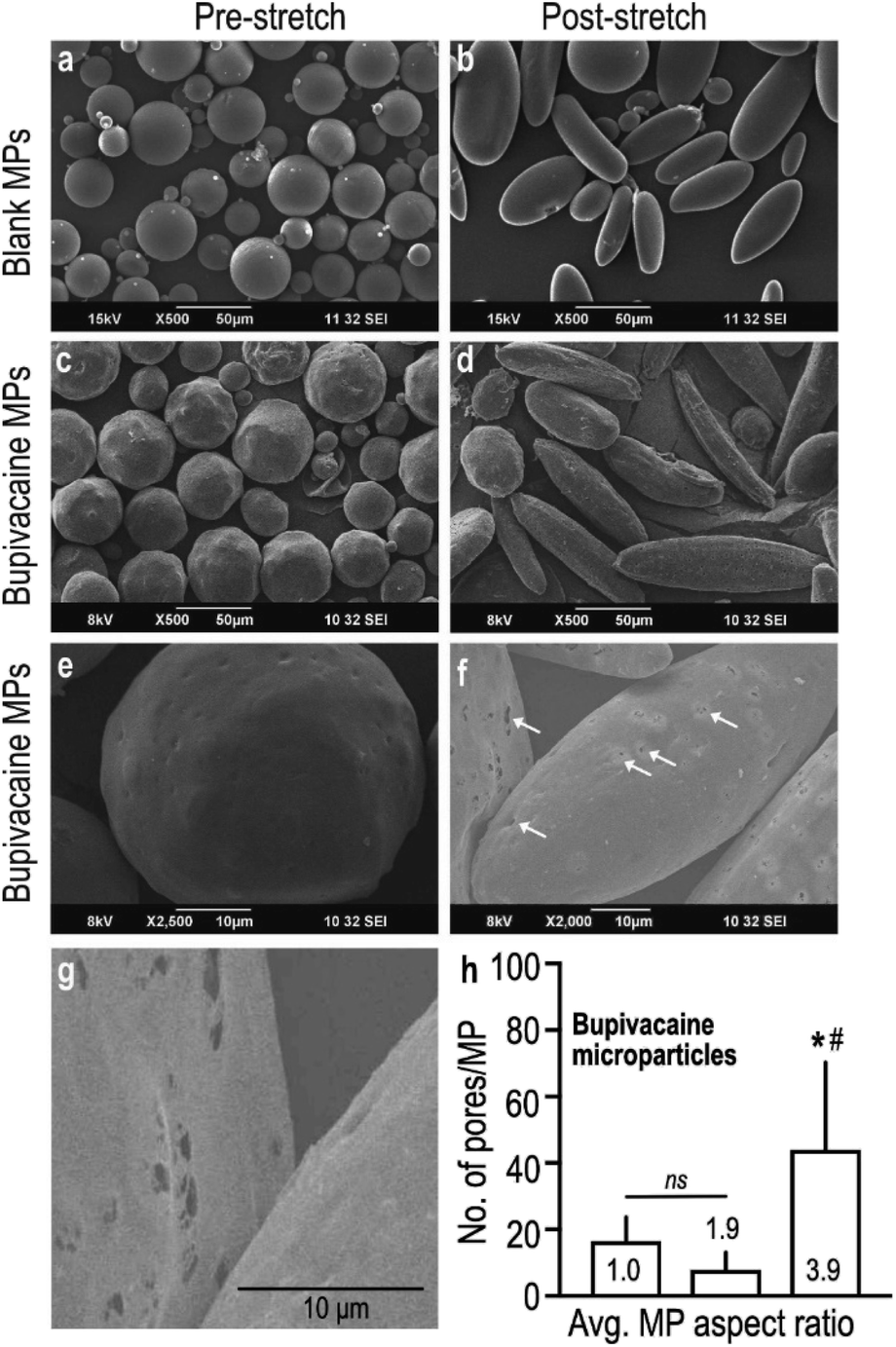 | ||
| Fig. 1 Morpho-physical characteristics of stretched polymeric microparticles. Representative SEM images of blank (a), or drug loaded spherical microparticles (c and e), that were subjected to a controlled stretching force, resulting in oblong and ellipsoid shaped microparticles (b, d and f). Magnified images of bupivacaine microparticles (e, f and g) showed small irregularly distributed surface pores (arrows) that were not visualized in other microparticle groups. Number of surface pores on bupivacaine microparticles were counted from magnified images (n = 10–15 particles) and plotted in (h) Data shown are mean ± SD, *, #P < 0.05, where * indicates statistical comparison between the indicated and non-stretched group (AR 1.0), while # indicates comparison between the indicated and AR 1.9 group. | ||
In a parallel experiment, to further determine the role of incorporated drug in overall MP morphology, an unrelated hydrophobic drug (ketoprofen) was loaded within PLGA MPs and subjected to similar stretching conditions as described before. Curiously, stretched ketoprofen-loaded particles demonstrated a large central hollow defect that was observed in 20–40% of MPs (Fig. S2a–c†), that was morphologically very different from stretched bupivacaine-loaded MPs. Partly similar to what was observed with stretched bupivacaine particles, the number of defect-carrying ketoprofen-loaded MPs increased with increase in stretching force. These observations confirm that encapsulated drugs indeed play a role in determining the overall architecture of the MP matrix, in addition to the stretching parameters.
Characterization of drug-loaded post-stretched microparticles
To further confirm the nature of interaction between encapsulated bupivacaine and the PLGA matrix, bupivacaine-PLGA MPs were subjected to Fourier transform infrared spectroscopy (FTIR). Characteristic PLGA peaks at 2949–3000 cm−1, and 1756 cm−1 corresponding to C–H, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching23 respectively, were distinctly observed in all anisotropic bupivacaine-loaded PLGA particles (Fig. 2a). Bupivacaine associated peaks at 1651 cm−1 corresponding to CO stretching vibration of amide I group, and the peak at 1534 cm−1 corresponding to classic amide II vibration,24,25 were relatively unchanged in the stretched particles. The seemingly unchanged bupivacaine FTIR spectra within the anisotropic MPs suggests physical entrapment of bupivacaine within the PLGA matrix.
O stretching23 respectively, were distinctly observed in all anisotropic bupivacaine-loaded PLGA particles (Fig. 2a). Bupivacaine associated peaks at 1651 cm−1 corresponding to CO stretching vibration of amide I group, and the peak at 1534 cm−1 corresponding to classic amide II vibration,24,25 were relatively unchanged in the stretched particles. The seemingly unchanged bupivacaine FTIR spectra within the anisotropic MPs suggests physical entrapment of bupivacaine within the PLGA matrix.
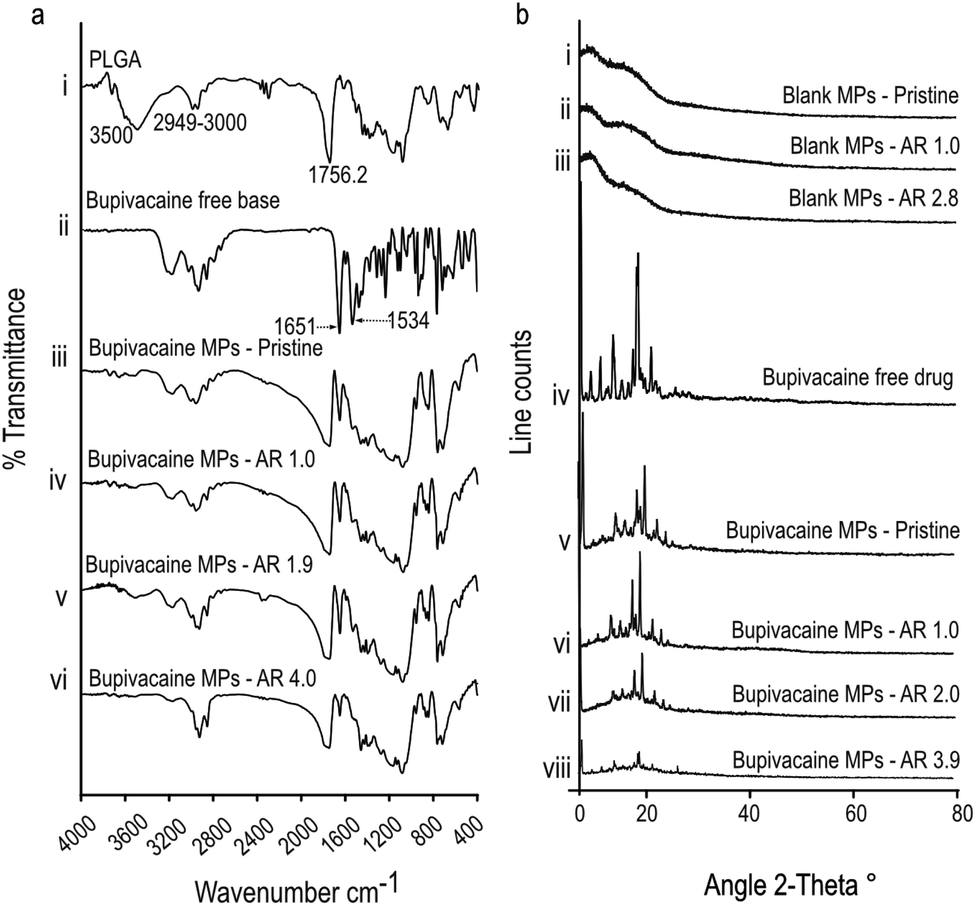 | ||
| Fig. 2 Physio-chemical analysis of drug-loaded stretched microparticles. Representative FTIR peaks (a) corresponding to the chemical bonds at indicated regions obtained from bupivacaine loaded PLGA microparticles of varying aspect ratio. Pristine drug loaded MPs refer to ‘as-synthesized’ spherical particles. Representative XRD peaks (b) obtained from drug-free (i, ii and ii), bupivacaine (v–viii), loaded microparticles. Pure drug form of bupivacaine (iv) were used as comparative reference. Pristine drug loaded MPs refer to ‘as-synthesized’ spherical particles. | ||
Further, X-ray diffraction (XRD) data of bupivacaine was assessed to determine its crystallinity and distribution within the deformed PLGA matrix. Absence of sharp peaks in all drug-free MP groups suggests the amorphous nature of the PLGA matrix. The sharp peaks associated with the crystalline nature of bupivacaine was distinctly noticeable in all stretched bupivacaine loaded MPs. This could be due to initial high-bupivacaine loading in MPs, which was essential to offset the drug loss due to the stretching process. However, there was a noticeable decrease in the intensity of bupivacaine associated peaks with increase in the aspect ratio of the stretched MPs (Fig. 2b), possibly due to loss of drug after the stretching process.
Drug release from stretched microparticles
To determine drug-release characteristics of stretched PLGA MPs, particles loaded with bupivacaine (Table 2), were suspended in a dialysis buffer with infinite sink conditions, and samples were collected at regular time intervals. Buffer conditions were optimized to mimic physiological temperature (37 °C) and pH (7.4). The total amount of drug in stretched MPs was lower, compared to unstretched particles (Table 2), partly due to the inevitable drug-loss that occurs during the process of stretching. Cumulative drug-release patterns from bupivacaine-loaded stretched MPs demonstrated faster release, compared to unstretched particles (Fig. 3). In addition, stretched particles retained a much smaller amount of bupivacaine after one-week, compared to non-stretched MPs. Interestingly, moderately stretched bupivacaine particles (AR 1.9) demonstrated comparable release with longer stretched (AR 3.9) particles. To further confirm if the observed bupivacaine release pattern was mainly due to MP stretching, the release profile of an unrelated drug (ketoprofen) from stretched MPs was measured (Fig. S2†). Similar to stretched-bupivacaine MPs, anisotropic ketoprofen-loaded MPs also demonstrated enhanced ketoprofen release. However, unlike bupivacaine MPs, longer-stretched ketoprofen particles (AR 3.9) released ketoprofen much quicker than moderately stretched MPs (AR 2.0). This strongly suggests that the drug itself partially contributes to its release in anisotropic MPs.:50 microparticles. MPs were stretched at 80 °C, with a stretching rate of 50 mm min−1 at variable extent of stretch to obtain microparticle AR's as depicted in the table. Data shown are from a single batch of microparticles that were subjected to 2–3 separate stretching sessions
| MP Aspect ratio | Bupivacaine content (wt%) |
|---|---|
| 1.0 | 44.9 |
| 1.9 | 33.6 |
| 3.9 | 19 |
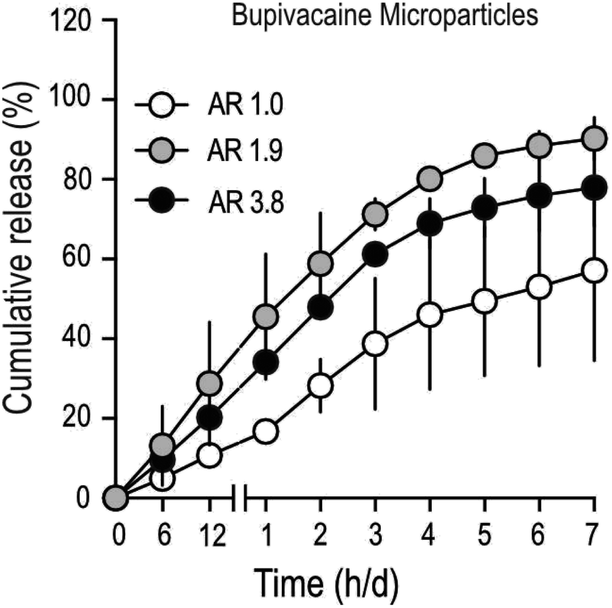 | ||
| Fig. 3 Differential drug release from stretched microparticles. Percent cumulative drug release from bupivacaine loaded microparticles of varying aspect ratio. Data shown are mean ± SD from 2–3 separate experiments conducted in triplicates. | ||
Differential nerve blocks with stretched and unstretched bupivacaine microparticles
To further assess the in vivo utility of stretched bupivacaine particles, sensory and motor nerve functions of the sciatic nerve were blocked by administering bupivacaine MPs as a nerve-block injection around the sciatic nerve in rats (Fig. 4a). Care was taken to ensure that the total amount of bupivacaine in the MP injectate in both the groups was similar (27.5 mg). The paw-withdrawal latency, an indirect measure of sensory function, was strongly blocked (9.8 ± 1.2 s) in stretched, compared to unstretched MPs (5.1 ± 0.8 s) (Fig. 4b and c), while there was no difference in the total duration of the nerve block between the two groups. Motor nerve block, usually considered as an unavoidable secondary effect in nerve-block anesthesia, was reversed much quicker in rats injected with stretched microparticles (Fig. 4d). Overall, stretched microparticles induced a strong sensory block, with a shorter-duration motor block. All animals had recovered their gait, and movement appeared normal within 12 hours of MP administration in both the groups. In this study, the total amount of bupivacaine (loaded in PLGA MPs) injected per animal was optimized to 27.5 mg, mainly because previous studies26 have shown that MPs containing ≥40 mg of bupivacaine tend to produce strong sensory blocks with paw-withdrawal latency values touching the upper limit (12 s) of the behavioral tests. Hence to allow room for possible detection of any difference in nerve block intensity between stretched and non-stretched groups, a lower dose of 27.5 mg was chosen. In addition, variability observed in the stretching process was reflected in the utilization of MPs of AR 2.8 for nerve block injection compared to those utilized in in vitro studies. Because there was minimal difference in bupivacaine release from MPs with AR 1.9 and 3.8 (Fig. 3), MPs with AR 2.8 were still considered relevant and used for nerve block injections.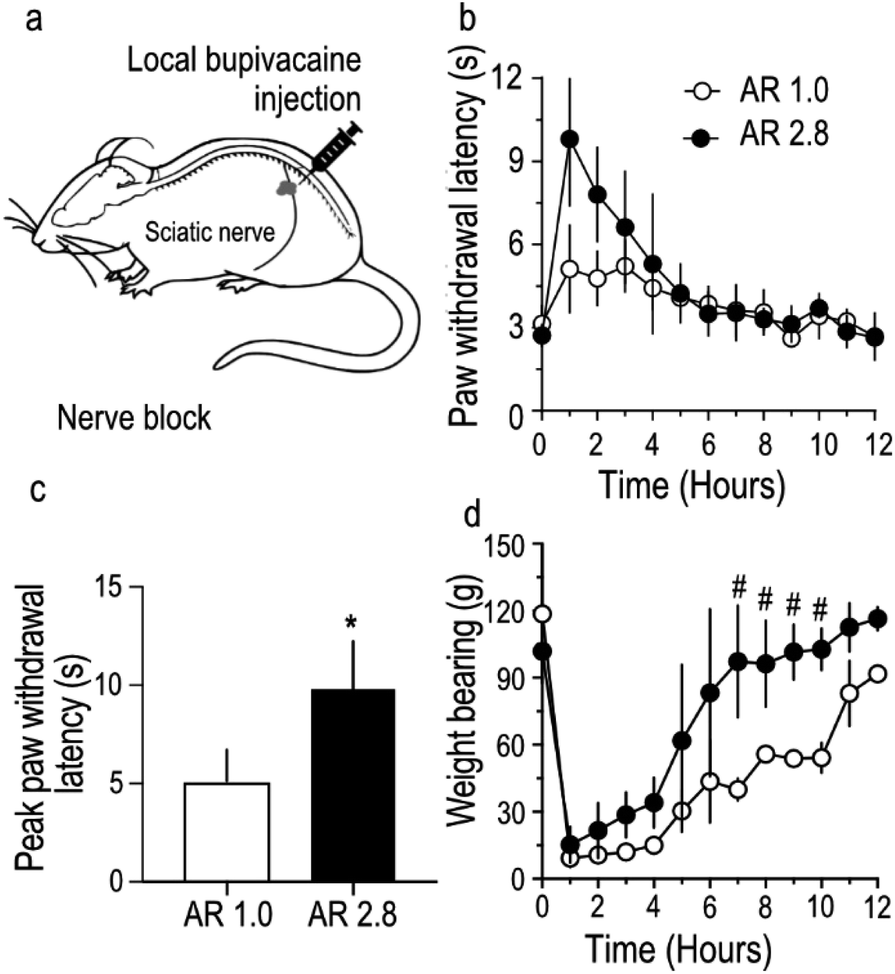 | ||
| Fig. 4 Stretched bupivacaine microparticles induced stronger sensory blocks. Stretched (AR 2.8), or un-stretched (AR 1.0) microparticle injectate, containing 27.5 mg of bupivacaine was administered around the sciatic nerve of SD rats as a local nerve block injection (a). Sensory nerve block duration (b) and peak effect (c), along with motor block duration (d) was recorded. Data shown are mean ± S.D., with n = 4 rats per group. * indicates P < 0.05, unpaired two-tailed Students t test. # indicates P < 0.05, unpaired Students t test, and corrected for multiple comparison using the Holm–Sidak method. | ||
Discussion
In this study, we wanted to examine if bupivacaine release from PLGA MPs can be altered by inducing particle anisotropy, and possibly utilized for modulating nerve blocks. The motivation for this approach was to demonstrate proof-of-concept principle that stretch-induced MP anisotropy could be utilized as a tunable parameter to modulate drug release pattern for a given condition.We chose to deform spherical drug-loaded PLGA MPs using the simple process of film stretching, mainly because this method is technically less challenging and allows for large scale production of deformed MPs that is relevant for in vivo applications.27,28 In principle, the process involves the formation of hydrogen bonds between the embedded PLGA MPs and the PVA film.28 By simultaneously maintaining the PLGA MPs above their glass-transition temperature, and applying a constant stretching force on the PVA film, the MPs undergo concomitant deformation along the axis of the stretching force. However, the drawback of the stretching technique is the variability in the aspect ratio of MPs that are obtained from each stretched-films. This variability was also noticed in our study where homogenously sized spherical microparticles, after stretching demonstrated approximately 20–30% variation in mean aspect ratio. The observed variability can be attributed to the uneven distribution in the stretching forces experienced by MPs distributed at different locations in the film. For instance, MPs located centrally in the film tend to have a higher AR compared to those in the periphery of the film. This observation may have to do with the formation of a ‘belly’ in the center of the film during the stretching process. During the course of this study, we found that by maintaining films at 80 °C, and stretching them at 50 mm min−1 minimized the belly formation in the film and reduced the variability in the AR of embedded MPs. Although the variability of particle AR within each stretched film was reduced, batch-to-batch variability (about 10–20%) still existed, and this is reflected in the slight differences in mean AR of MPs used in different experiments within the study. Furthermore, in alignment with previous reports,27–29 we observed a strong positive association between the aspect ratio of the embedded MPs and the final length of the stretched film.
Importantly, in comparison to the stretching technique used in this study, other particle-deformation strategies such as those using microfluidics,30 lithography, and micro-molding31 have been shown to produce homogeneously shaped-particles, but these processes are technically challenging with relatively low yield. In addition, we observed that both, bupivacaine- and ketoprofen-loaded stretched particles demonstrated higher release of loaded drug compared to unstretched particles. A plausible explanation for this observation could be due to an increased surface area to volume ratio, and the formation of stretch-induced pores and surface defects in the stretched PLGA MPs. As seen in SEM images, stretched bupivacaine-loaded MPs exhibited an increase in the number of micron-sized pores, while stretched ketoprofen-loaded MPs (Fig. S2†) developed a single large defect in the center. These observations have similarities with other reports, where ‘dimpling’ in MPs exposed to stretching conditions above 90 °C have been observed.29
It is quite plausible that water entry into MPs occur via such stretch-induced surface defects and enhance the erosion and hydrolytic degradation of the PLGA matrix resulting in increased rate of drug release, as observed in our study. This explanation is strongly supported by the drug-release pattern observed in stretched ketoprofen MPs (Fig. S2†), where the percentage of MPs with surface defects positively correlates with the rate of drug release.32 In contrast, moderately stretched bupivacaine particles (AR 1.9), with comparable pore density to un-stretched controls, exhibited a drug-release pattern that was remarkably quicker than controls. Interestingly, further stretching of bupivacaine particles (to AR 3.9), increased surface pore formation, but there was no change in the drug release pattern compared to moderately stretched (AR 1.9) particles. Though the reason for this observation is not completely clear, it is possible that the moderate stretching force was sufficient to distort the polymeric arrangement within the bupivacaine-loaded MPs, leading to enhanced bupivacaine release, but not sufficient enough to alter surface porosity. However, application of a stronger stretching force, in addition to probable matrix distortion produced visible surface pores in the MPs, but this did not seem to alter drug-release patterns any differently from MPs that were moderately stretched.
Since drug release from polymeric matrix is also determined by drug hydrophobicity, the difference in release rate between bupivacaine and ketoprofen could be attributed to their intrinsic chemical property. Even though both drugs are hydrophobic, ketoprofen is known to plasticize PLGA by forming hydrogen bonds with the polymer backbone, resulting in enhanced polymer hydration33 and quicker drug release compared to bupivacaine base, which has limited interaction with the polymer. In addition to the chemical nature of the drug, the amount of loaded drug is also known to modulate release. However, higher loading of hydrophobic drugs has been mostly shown to enhance release.34,35 This is quite opposite to what is observed in this study, which strongly suggests that the altered drug release from stretched-anisotropic particles is unlikely due to uneven drug content but more likely due to stretch-induced defects in the drug-loaded MPs. Furthermore, the surface defects seen in stretched bupivacaine MPs were sub-micron to micron sized pores, while stretched ketoprofen MPs exhibited a large central defect. It is likely that the larger defects in stretched ketoprofen MPs facilitated better water ingress and thereby promoted quicker drug release compared to bupivacaine MPs.
Another important feature of this study is the modulation of drug-induced functional effects by anisotropic drug-loaded particles. We chose to study the nerve-blocking effect of bupivacaine because behavioral tests to determine nerve function allows for a rapid, graded response to bupivacaine that can be quantified and compared.21,36,37 In good agreement with the bupivacaine-release pattern observed in our in vitro study, animals administered with bupivacaine-loaded anisotropic particles demonstrated a stronger sensory nerve block, most likely due to enhanced release of bupivacaine. However, the duration of sensory nerve-block in both spherical and anisotropic particles was similar, and quite brief, in comparison to the duration of drug release in vitro. This could be because tissue fluids typically induce rapid autocatalytic degradation of the polymer due to the presence of tissue enzymes,38 tissue-associated acidic components,39 and free radicals.40 In addition, even though the injectate comprising of bupivacaine MPs is deposited in the vicinity of the sciatic nerve, it is very likely that a significant proportion of the released drug is absorbed by the contiguous muscle layers, resulting in nerve block durations that are not-aligned with in vitro release patterns.
Further, we also noticed that the effect of motor nerve block was considerably shorter in animals administered with anisotropic particles compared to controls. This could be because, after the rapid phase of drug extravasation from anisotropic particles, the concentration of bupivacaine released from injected MPs may be insufficient to sustain motor block in large myelinated motor axons. In conclusion, bupivacaine-loaded anisotropic microparticles could be utilized to produce differential nerve blocks of varying intensity. The differential drug release from stretch-induced anisotropic particles was primarily attributed to the formation of surface defects within the polymeric matrix. Thus, by regulating anisotropy-associated polymeric defects, drug release kinetics could be potentially altered to suit specific therapeutic applications.
Conclusion
In this study, we show that bupivacaine-loaded anisotropic PLGA particles have a differential drug release pattern that produces a stronger nerve block compared to conventional spherical shaped-MPs. The importance of our results is the demonstration of MP anisotropy as a tunable parameter for drug release and its potential application in controlled therapeutic regimens. However, more systematic studies will be required to further examine how drug release patterns can be controlled by mixing different proportions of drug-loaded MPs with varying anisotropy.Materials and methods
Materials
Poly (lactide-co-glycolide) (PLGA) 50:50 (mol. wt = 150 KDa), and 75:25 (mol., wt = 97 KDa) were purchased from Polysciences, Inc. (Washington, PA). Poly (vinyl alcohol) (PVA) (87–89% hydrolyzed, mol. wt = 13–23 KDa) ketoprofen and bupivacaine hydrochloride were purchased from Sigma-Aldrich (USA). Bupivacaine hydrochloride was converted to bupivacaine free base by alkaline precipitation and filtration. Dichloromethane, methanol, tris, and hydrochloric acid were obtained from Merck (USA).
Animal care
Adult, female Sprague–Dawley rats weighing 225–275 g were housed in pairs, allowed standard rat diet and water ad libitum, and maintained on 10 h/14 h light/dark cycle. All protocols used in this study were approved by the Institutional Animal Ethical Committee (IAEC) (IAEC/2016/1/4), Amrita Institute of Medical Sciences, Kochi, India, in accordance with guidelines set forth by the Committee for Control and Supervision of Experiments on Animals (CPSCSEA), Government of India.PLGA microparticle synthesis and drug loading
Bupivacaine- and ketoprofen-loaded MPs were prepared by oil-in-water emulsion followed by solvent evaporation technique.26,41 In brief, bupivacaine free base (160 mg) or ketoprofen (50 mg) and PLGA (100 mg) were dissolved in methylene chloride (5 mL), and the mixture was added to 100 mL of 0.2% polyvinyl alcohol in 100 mM tris buffer (pH 8.5) under stirring condition at 1200 rpm and 600 rpm for 1 h and 3 h respectively. The synthesized microparticles were washed three times at 15161 g for 25 min at 4 °C. The final pellet was thoroughly resuspended in 3 mL of sterile water and lyophilized. Bare microparticles were prepared using the same method except for the drug. Drug loading of 45% and 17% were achieved for bupivacaine and ketoprofen encapsulated MPs respectively.
Controlled stretching of synthesized PLGA microparticles
Microparticles of varying aspect ratio were produced by physically stretching synthesized particles that were embedded in a PVA film,27 using a custom-made film-stretcher. Briefly, the thin-film was prepared by dissolving 10% (w/v) PVA at 60 °C, followed by the addition of 0.5% (v/v) glycerol at room temperature. ‘As-synthesized’ MPs (0.25 mg mL−1) were mixed in the PVA–glycerol solution, and poured as a thin layer into a tray measuring (9.2 × 11 cm), and allowed to dry at 37 °C. The resulting plasticized film was mounted on a custom-built film stretcher, immersed in paraffin oil (60–80 °C), and stretched one-dimensionally in the horizontal plane. The rate and extent of stretch were assessed along a graduated scale built within the stretcher, while the movement of the stretching platform was controlled manually by a calibrated screw-knob. Once the films were stretched to the desired length, the films were left to dry overnight. The embedded MPs were extracted by dissolving the films in water, washed at 15161 g for 25 min at 4 °C, and pellets resuspended in 3 mL of water, lyophilized and stored for later use.
Characterization studies
The surface morphology and particle size were determined using a scanning electron microscope (JEOL, JSM-6490LA, Japan) (SEM). Lyophilized MPs in powder form were placed on adhesive carbon tape and mounted on specimen stubs. Samples were sputter coated with gold before imaging at an accelerating voltage of 8 kV. SEM images were analyzed for size, aspect ratios, and surface pores and defects using an image analysis software (Image J, U.S. NIH, Bethesda, USA).Drug signatures in MPs was determined using Fourier-transform infrared spectroscopy (FTIR) and X-ray diffractometry (XRD). FTIR transmission spectra in the range from 400 to 4000 cm−1 were recorded using an FTIR spectrophotometer (IR Affinity-1s, Shimadzu, Japan) from lyophilized MP samples compacted using the KBr pellet method. Diffraction scans from lyophilized MP samples were recorded at 2θ range of 5–80° at a step size of 0.03°, using an X-ray diffractometer (XpertPro, PAN analytica, Germany).
Drug release studies
Ketoprofen, or bupivacaine loaded MPs of fixed weight (5 mg) were suspended in dialysis bags (10 KDa, cut off, Spectrum, USA), immersed in PBS, (pH 7.4) at 37 °C under constant stirring condition. Buffer conditions were optimized to mimic physiological temperature (37 °C) and pH (7.4). Dialysate samples (2 mL) were collected at fixed time intervals and stored at 4 °C until further use. Sample volume retrieved at each time interval was replaced with fresh PBS to maintain infinite sink conditions. Quantification of released ketoprofen in the dialysate fluid was performed by measuring the absorbance wavelength at 258 nm using a UV-spectrophotometer (Synergy H1, Biotek, USA). Drug concentrations from different experimental time-points were calculated using a ketoprofen standard curve. Similarly, the amount of released bupivacaine in the dialysate fluid was quantified using reverse-phase high performance liquid chromatography (RP-HPLC, 20AD, Shimadzu, Japan).26 Dialysate samples (20 μL) were injected into 4.6 × 250 mm Zorbax eclipse C18 5 μm column maintained at 30 °C. The HPLC column was eluted with an aqueous solution of 65:35 acetonitrile buffer (phosphate buffer, 10 mM, pH = 6.78) at 1 mL min−1, and bupivacaine peaks were detected by UV absorbance at 272 nm. Bupivacaine concentrations were calculated using standards composed of known concentrations.
Nerve block injections
Rats were anesthetized using 2% isoflurane mixed with 98% oxygen dispensed through a small-animal anesthesia manifold. The left hind-limb was shaved and nerve-block injection administered using a 23-gauge needle as described before.36 PLGA microparticles (AR 1.0, and AR 2.8), containing 27.5 mg of bupivacaine were suspended in 0.4 mL of 1% carboxymethyl cellulose and 0.1% Tween 80, and injected at the location of the sciatic nerve near the sciatic notch. Care was taken to administer the microparticle injectate around the nerve without injecting it into the muscle. Since stretched and unstretched MPs have uneven drug loading due to the stretching process, the amount of injected MPs were adjusted accordingly to contain 27.5 mg of bupivacaine (86.4 mg of stretched MPs and 55 mg of unstretched MP).Behavioral tests for thermal responsiveness and motor strength
To assess the effectiveness of the sensory nerve blockade, tactile responsiveness to thermal stimulus was elicited from the hind paw and measured at different time points. As mentioned before42,43 measurements were performed by gently placing the rat's hind paw on a pre-heated surface maintained at 55 °C. The time taken by the animal to withdraw its hind paw from the heater surface was recorded as the paw-withdrawal latency. The contralateral limb was used as internal control. Each recording was repeated thrice per animal and the mean value calculated.To assess motor blockade, each hind-limb of the animal was gently placed on a digital weighing scale (Health Sense, India) and the maximum weight borne by the tested limb was noted. Each measurement was repeated thrice per limb and the mean value calculated. The contralateral limb was used as internal control. The test was repeated at regular time intervals.
Statistics
All data are represented as mean ± standard deviation (SD). Difference in mean values among experimental and control groups was tested using one-way, or two-way ANOVA, using GraphPad Prism version 7.0 for Mac (GraphPad Software, La Jolla California USA).Data accessibility
Data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
SBS, VP, GR, SEM, and NK performed experiments, analyzed data, and wrote the first draft, DM analyzed data and edited the manuscript, SS conceived the idea, analyzed data, and wrote the manuscript.Funding
CSIR fellowship (DST, Govt. of India) to GR, Ramalingaswami fellowship grant (Department of Biotechnology, Government of India) & Grant SR/NM/NS-1153/2013 (G), Nano Mission, Department of Science and Technology, Government of India, to SS.Conflicts of interest
Authors declare no possible conflict of interest with any information in this study.Acknowledgements
CSIR fellowship (DST, Govt. of India) to GR, Ramalingaswami fellowship, Grant BT/PR24515/MED/30/1926/2017 (Department of Biotechnology, Government of India) & Grant SR/NM/NS-1153/2013 (G) (Nano Mission, Department of Science and Technology, Government of India) to SS.References
- D. Paul, S. Achouri, Y. Z. Yoon, J. Herre, C. E. Bryant and P. Cicuta, Biophys. J., 2013, 105, 1143–1150 CrossRef CAS.
- C. S. Schneider, J. G. Perez, E. Cheng, C. Zhang, P. Mastorakos, J. Hanes, J. A. Winkles, G. F. Woodworth and A. J. Kim, Biomaterials, 2015, 42, 42–51 CrossRef CAS.
- L. Florez, C. Herrmann, J. M. Cramer, C. P. Hauser, K. Koynov, K. Landfester, D. Crespy and V. Mailänder, Small, 2012, 8, 2222–2230 CrossRef CAS.
- R. A. Meyer and J. J. Green, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 191–207 CAS.
- A. Garapaty and J. A. Champion, Bioeng. Transl. Med., 2017, 2, 92–101 CrossRef CAS.
- S. Bale, A. Khurana, A. S. S. Reddy, M. Singh and C. Godugu, Crit. Rev. Ther. Drug Carrier Syst., 2016, 33, 309–361 CrossRef.
- X. Zhu, C. Vo, M. Taylor and B. R. Smith, Mater. Horiz., 2019, 6, 1094–1121 RSC.
- J. A. Champion, Y. K. Katare and S. Mitragotri, J. Controlled Release, 2007, 121, 3–9 CrossRef CAS.
- M. Cooley, A. Sarode, M. Hoore, D. A. Fedosov, S. Mitragotri and A. Sen Gupta, Nanoscale, 2018, 10, 15350–15364 RSC.
- K. Park, S. Skidmore, J. Hadar, J. Garner, H. Park, A. Otte, B. K. Soh, G. Yoon, D. Yu, Y. Yun, B. K. Lee, X. Jiang and Y. Wang, J. Controlled Release, 2019, 304, 125–134 CrossRef CAS.
- M. Hussain, J. Xie, Z. Hou, K. Shezad, J. Xu, K. Wang, Y. Gao, L. Shen and J. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 14391–14400 CrossRef CAS.
- S. Fredenberg, M. Wahlgren, M. Reslow and A. Axelsson, Int. J. Pharm., 2011, 415, 34–52 CrossRef CAS.
- F. Alexis, Polym. Int., 2005, 54, 36–46 CrossRef CAS.
- R. P. Batycky, J. Hanes, R. Langer and D. A. Edwards, J. Pharm. Sci., 1997, 86(12), 1464–1477 CrossRef CAS.
- H. Keles, A. Naylor, F. Clegg and C. Sammon, Polym. Degrad. Stab., 2015, 119, 228–241 CrossRef CAS.
- A. C. Doty, D. G. Weinstein, K. Hirota, K. F. Olsen, R. Ackermann, Y. Wang, S. Choi and S. P. Schwendeman, J. Controlled Release, 2017, 256, 19–25 CrossRef CAS.
- D. Ghosh Dastidar, S. Saha and M. Chowdhury, Int. J. Pharm., 2018, 548, 34–48 CrossRef CAS.
- H. Kim, H. Park, J. Lee, T. H. Kim, E. S. Lee, K. T. Oh, K. C. Lee and Y. S. Youn, Biomaterials, 2011, 32, 1685–1693 CrossRef CAS.
- S. A. Shankarappa and D. S. Kohane, Pain Management, 2013, 3, 91–93 CrossRef.
- S. Padmakumar, J. Joseph, M. H. Neppalli, S. Elizabeth, S. V Nair, S. A. Shankarappa and D. Menon, ACS Appl. Mater. Interfaces, 2016, 8, 6925–6934 CrossRef CAS.
- J. B. McAlvin, G. Reznor, S. A. Shankarappa, C. F. Stefanescu and D. S. Kohane, Anesth. Analg., 2013, 116, 794–803 CrossRef CAS.
- Y. S. Pek, P. Pitukmanorom and J. Y. Ying, J. Mater. Chem. B, 2014, 2, 8194–8200 RSC.
- A. A. da Silva-Junior, J. R. de Matos, T. P. Formariz, G. Rossanezi, M. V. Scarpa, E. S. T. do Egito and A. G. de Oliveira, Int. J. Pharm., 2009, 368, 45–55 CrossRef CAS.
- M. L. Martins, J. Eckert, H. Jacobsen, E. C. dos Santos, R. Ignazzi, D. R. de Araujo, M. C. Bellissent-Funel, F. Natali, M. Marek Koza, A. Matic, E. de Paula and H. N. Bordallo, Data in Brief, 2017, 15, 25–29 CrossRef.
- M. Jug, F. Maestrelli, M. Bragagni and P. Mura, J. Pharm. Biomed. Anal., 2010, 52, 9–18 CrossRef CAS.
- J. B. McAlvin, G. Reznor, S. A. Shankarappa, C. F. Stefanescu and D. S. Kohane, Anesth. Analg., 2013, 116, 794–803 CrossRef CAS.
- J. A. Champion, Y. K. Katare and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11901–11904 CrossRef CAS.
- C. C. Ho, A. Keller, J. A. Odell and R. H. Ottewill, Colloid Polym. Sci., 1993, 271, 469–479 CrossRef CAS.
- R. A. Meyer, R. S. Meyer and J. J. Green, J. Biomed. Mater. Res., Part A, 2015, 103, 2747–2757 CrossRef CAS.
- F. Ramazani, W. Chen, C. F. Van Nostrum, G. Storm, F. Kiessling, T. Lammers, W. E. Hennink and R. J. Kok, Int. J. Pharm., 2016, 499, 358–367 CrossRef CAS.
- J. P. Rolland, B. W. Maynor, L. E. Euliss, A. E. Exner, G. M. Denison and J. M. Desimone, J. Am. Chem. Soc., 2005, 127, 10096–10100 CrossRef CAS.
- D. Klose, F. Siepmann, K. Elkharraz, S. Krenzlin and J. Siepmann, Int. J. Pharm., 2006, 314, 198–206 CrossRef CAS.
- P. Blasi, A. Schoubben, S. Giovagnoli, L. Perioli, M. Ricci and C. Rossi, AAPS PharmSciTech, 2007, 8, 1–8 CrossRef.
- N. Leelarasamee, S. A. Howard, C. J. Malanga, L. A. Luzzi, T. F. Hogan, S. J. Kandzari and J. K. H. Ma, J. Microencapsulation, 1986, 3, 171–179 CrossRef CAS.
- H. S. Choi, S. A. Seo, G. Khang, J. M. Rhee and H. B. Lee, Int. J. Pharm., 2002, 234, 195–203 CrossRef CAS.
- S. A. Shankarappa, I. Sagie, J. H. Tsui, H. H. Chiang, C. Stefanescu, D. Zurakowski and D. S. Kohane, Reg. Anesth. Pain Med., 2012, 37, 483–489 CrossRef CAS.
- X. Garcia, E. Escribano, J. Domenech, J. Queralt and J. Freixes, J. Nanopart. Res., 2011, 13, 2213–2223 CrossRef CAS.
- D. da Silva, M. Kaduri, M. Poley, O. Adir, N. Krinsky, J. Shainsky-Roitman and A. Schroeder, Chem. Eng. J., 2018, 340, 9–14 CrossRef CAS.
- S. A. M. Ali, P. J. Doherty and D. F. Williams, Biomaterials, 1994, 15, 779–785 CrossRef CAS.
- M. A. Tracy, K. L. Ward, L. Firouzabadian, Y. Wang, N. Dong, R. Qian and Y. Zhang, Biomaterials, 1999, 20, 1057–1062 CrossRef CAS.
- R. Padera, E. Bellas, J. Y. Tse, D. Hao and D. S. Kohane, Anesthesiology, 2008, 108, 921–928 CrossRef CAS.
- S. A. Shankarappa, J. H. Tsui, K. N. Kim, G. Reznor, J. C. Dohlman, R. Langer and D. S. Kohane, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17555–17560 CrossRef CAS.
- S. E. Mathew, P. Madhusudanan and S. A. Shankarappa, J. Pain Res., 2020, 13, 1305–1313 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08386k |
| This journal is © The Royal Society of Chemistry 2021 |