DOI:
10.1039/D0RA08945A
(Paper)
RSC Adv., 2021,
11, 525-536
Multi-photon properties of branched chromophores derived from indenoquinoxaline and oxadiazole heterocyclic units†
Received
20th October 2020
, Accepted 24th November 2020
First published on 24th December 2020
Abstract
Two chromophoric congeners derived from indenoquinoxaline and oxadiazole are designed, synthesized and characterized for their multi-photon properties in the femtosecond time domain. These two model structures are experimentally found to exhibit strong and widely distributed two- and three-photon activities within the spectral range of 680–1500 nm and the larger congener manifests maximum two- and three-photon absorption cross-section values of 2120 GM (at 750 nm) and ∼85 × 10−80 cm6 s2 (at 1280 nm), respectively. Both two- and three-photon absorption-based optical power-limiting performance of a representative model compound are also evaluated and demonstrated.
1. Introduction
The last two decades have seen tremendous efforts in the development of materials that manifest strong two-photon absorption (2PA) properties due to many useful applications based on this nonlinear optical phenomenon including optical power-limiting, up-converted lasing, 3D data storage, noninvasive bio-imaging and photodynamic therapy.1–6 As scientists have realized the general guidelines for the molecular design toward efficient 2PA materials, the attention has gradually shifted to explore the structural parameters that link to the molecular three-photon absorption (3PA) efficiencies. Theoretically, it is comparatively much more difficult to trigger a 3PA process than a 2PA process for a given molecule due to the huge difference in cross-section values between these two nonlinear phenomena. As a fifth-order nonlinear optical phenomenon, the cubic power-dependence on the local intensity of a 3PA process can achieve a superior spatial confinement compared to that offered by a 2PA process. Therefore, a corollary that follows from this intrinsic characteristic is the potentiality to accomplish a higher spatial resolution and contrast in imaging-related applications. Moreover, a 3PA process also renders a greater optical-limiting and stabilization effect that covers a broader spectral range at equal nonlinear transmission level.7 For the past few years, we have been focused on searching new and useful heterocyclic π-units for the development of efficient 2PA-chromophores and have found that functionalized quinoxalinoids are beneficial moieties for the enhancement of molecular 2PA.8–12 Among these investigations, some of the quinoxalinoid systems were even found to manifest strong 3PA in both nanosecond and femtosecond time domain11,12 and such experience has lead us to consider that it may be worthy to explore the potentiality of using quinoxalinoids as the building units for 3PA materials. Following this line of concept, herein we would like to present our combined studies of degenerate 2PA and 3PA, up-converted emission and order of the absorption process (OAP) across a spectral range of 680–1500 nm based on a set of new multi-branched chromophores derived from 2,3,8-trifunctionalized indenoquinoxalines and oxadiazoles in the femtosecond time domain. Through the analysis of the relationship between molecular structures and 2PA/3PA properties, some molecular parameters that are useful for designing highly active 2PA/3PA materials may be retrieved.
2. Results and discussion
2.1 Design and synthesis of model molecules
Fig. 1 illustrates the molecular structures of the investigated model chromophores and the syntheses toward these target structures are presented in Schemes 1 and 2. Compound 1 manifests an asymmetric skeleton, which is constructed by attaching two identical electron-donating units (i.e. diphenylaminofluorene) and one electron-pulling group (i.e. oxadiazole) at C2, C3, and C8 positions of an indenoquinoxaline moiety. Therefore, compound 1 possesses a generic donor-π-acceptor (i.e. D-π-A) backbone. In contrast, compound 2 represents a conceptually “dimerized” version of compound 1 with a structural motif of donor-π-acceptor-π-donor (i.e. D-π-A-π-D).
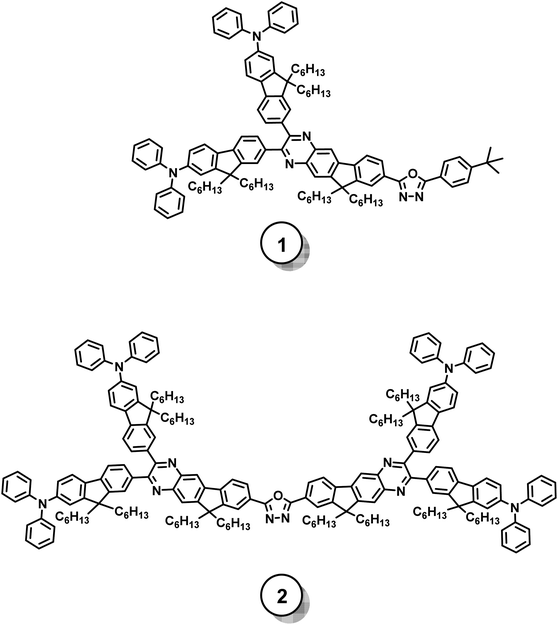 |
| | Fig. 1 Molecular structures of the studied model chromophores. | |
 |
| | Scheme 1 Synthetic routes for the preparation of the key intermediates. | |
 |
| | Scheme 2 Final coupling routes for the target model chromophores. | |
For the syntheses of designed model chromophores, the previously reported compounds 3 and 10 were utilized as the starting materials13 and consecutive transformation of functional groups were performed to prepare the essential diamine synthons (i.e. compounds 9 and 16) for the final coupling. In brief, the aforementioned functional group transformation starts from converting bromo groups to primary amino groups through an Ullmann-type amination followed by a reduction (i.e. 3 → 4 → 5 and 10 → 11 → 12). The obtained primary amino groups of compounds 5 and 12 were then acetylized followed by regioselective nitration processes (i.e. 5 → 6 → 7 and 12 → 13 → 14). Deacetylization were performed on compounds 7 and 14 to recover the primary amino groups (i.e. compounds 8 and 15) and the essential 1,2-diamines (i.e. 9 and 16) were obtained through simple reduction processes. Once these two diamines are in our hands, the same final coupling protocol can be conducted to prepare the targeted model chromophores 1 and 2 under an acid-catalyzed condition using a previously reported diketone (i.e. compound 17) as the corresponding reactant8 All the precursors and final compounds were carefully purified and their structures were confirmed by 1H NMR, 13C NMR and high resolution mass spectrometry.
2.2 Optical properties
2.2.1 Linear optical properties. In solution phase, these two model chromophores exhibit intensive linear absorption in the UV-visible region with their lowest-energy absorption maxima located at 428 nm (ε ∼ 6.08 × 104 cm−1 M−1) and 439 nm (ε ∼ 1.38 × 105 cm−1 M−1), respectively. In addition, very intensive bluish-green color of fluorescence from both compound solutions was readily observable when irradiated by a common UV lamp, agreeing to their fluorescence spectra as exhibited in the inset of Fig. 2. All the measured photophysical data are summarized in Table 1.
 |
| | Fig. 2 Linear absorption and fluorescence spectra (inset) of compounds 1 and 2 (experimental conditions: sample concentration = 1 × 10−6 M in toluene; both compound 1 and compound 2 are excited at 410 nm to record their fluorescence spectra). | |
Table 1 Photophysical properties of the studied model compounds in solution phasea
| Compound |
λ1PAmaxb (nm) |
ε (M−1 cm−1) |
λEmmaxc (nm) |
ΦFd |
δ2,maxe (GM) |
λ2PAmax (nm) |
δ3,maxf (10−80 cm6 s2) |
λ3PAmax (nm) |
| Sample solutions for linear and two-photon-related measurements were prepared in toluene whereas those for three-photon-related measurements were prepared in toluene-d8. Linear (one-photon) absorption maxima. One-photon-induced fluorescence emission maxima. Fluorescence quantum efficiency. Maxima of two-photon absorption at λ2PAmax; 1 GM = 1 × 10−50 cm4 s/photon-molecule. Maxima of three-photon absorption at λ3PAmax. |
| 1 |
312 |
7.9 × 104 |
498 |
0.51 |
970 |
750 |
50 |
1280 |
| 351 |
7.7 × 104 |
| 428 |
6.1 × 104 |
| 2 |
308 |
1.3 × 105 |
499 |
0.57 |
2120 |
750 |
85 |
1280 |
| 359 |
1.5 × 105 |
| 439 |
1.4 × 105 |
It is noted that both the studied model compounds manifest multi-maximum linear absorption bands that virtually cover identical spectral region in the UV-visible and the expansion of the π-framework from compound 1 to compound 2 has led to an overall promotion of the linear absorptivity. Furthermore, when analyzing the detailed spectral structures of their linear absorption bands, one can see that these two chromophores exhibit different tendency for the magnitude change of their local absorption maxima. That is, the molar absorption coefficients corresponding to the three local absorption maxima of compound 1 manifests a bathochromically descending manner whereas compound 2 shows a “fluctuating” trend of it. Since compounds 1 and 2 possess different symmetries in their molecular scaffolds, it should be reasonable that such intrinsic structural difference can lead to dissimilar linear absorption behaviors.
2.2.2 Two- and three-photon properties. For both fundamental research and application, it is important to grasp the combined information about the dispersion and band structures of two- and three-photon absorptivities across a wide spectral range for a novel material so that the molecular structure-2PA/3PA property relationship can be studied and the applicable wavelength range based on such a material can be determined. In general, mapping out the entire multi-photon absorption (MPA) spectrum for a given studied subject is not an easy task. Based on the accumulated knowledge and experience, one may select appropriate light sources with wavelengths located within the following spectral range for MPA studies:14| | |
λ1 ≪ λexc ≤ 2λ1 (for 2PA)
| (1) |
| | |
2λ1 ≪ λexc ≤ 3λ1 (for 3PA)
| (2) |
where λ1 represents the peak wavelength for one-photon absorption band of the studied nonlinear material.With this consideration of the wavelength range for multi-photon property studies in mind, we attempt to continuously probe OAP so that the entire dispersion pattern of 2PA and 3PA for the studied model chromophores can be realized. To do so, the detailed power-dependence of the up-converted emission intensity on the excitation intensity of the studied model chromophores in the range of 680–1500 nm should be investigated via a multi-photon-excited fluorescence (MPEF) technique.
It is worthy to note that for MPEF experiments, linear attenuation caused by the solvent used for the preparation of tested samples should be seriously taken into consideration particularly for higher-photon excitations as many organic solvents possess linear absorption in near-IR region due to the characteristic vibrational overtones and such phenomenon could suppress the incident photon flux and lead to a substantial decrease of the MPEF signal. It has been suggested that this detrimental effect can be alleviated if deuterated solvents are utilized.15 Among the commercially available deuterated solvents, we have selected toluene-d8 for this purpose since we have frequently used its non-deuterated analogue (i.e. the common toluene) as the solvent to prepare sample solutions for two-photon-related experiments in our other chromophore systems.
For the excitation light source, an integrated wavelength-tunable femtosecond laser system (Ti:sapphire oscillator + OPO, Coherent) was utilized to probe the detailed power-dependence of the multi-photon-induced emission intensity on the incident laser intensity of chromophores 1 and 2 in the range of 680–1500 nm and map out their OAP-spectra as illustrated in Fig. 3 (for compound 2) and Fig. S2† (for compound 1).
 |
| | Fig. 3 Measured OAP spectrum of compound 2. | |
Using compound 2 as a representative, three distinct regions representing different orders of the nonlinear absorption processes can be readily observed from Fig. 3 and can be interpreted as follows:
The observed second-order dependence in the region from 680 to 1000 nm indicates that all the transitions in this spectral range are caused by pure 2PA. When the excitation wavelengths moves bathochromically toward 1100 nm, intermediate values of OAP (i.e. ≈2.2–2.7) were obtained, implying the coexistence of 2PA and 3PA within this spectral section. Accordingly, 2PA processes would be more dominant when the excitation wavelengths are closer to 1000 nm because a comparatively lower threshold of excitation intensity is required to trigger a 2PA and the observed order of power-dependence is closer to 2. In contrast, when those excitation wavelengths near 1100 nm are utilized, the ascending 3PA may become competitive and observed OAP values are closer to 3. And lastly, when the excitation wavelengths were further tuned into the range of 1100–1500 nm, a cubic power-dependence was constantly held, verifying that pure 3PA is the major nonlinear process within this spectral region. Overall, this measured OAP-spectrum provides a very informative picture about the spectral coverage of pure 2PA and 3PA as well as the transition course from 2PA to 3PA.
With such investigation results as a guide, we have used the same laser system and MPEF technique to delineate the nonlinear absorption spectra of compounds 1 and 2 within their pure 2PA and 3PA region (i.e. 680–1000 nm for 2PA and 1100–1500 nm for 3PA). Both the 2PA-/3PA-induced fluorescence and corresponding logarithmic power-dependence for the specific nonlinear absorption process in this model chromophore system were also demonstrated at two representative excitation wavelengths. All these aforementioned experimental data are integrated and illustrated in Fig. 4 and 5. It is notable from Fig. 4 that these two chromophores possess nearly identical spectral dispersion of their two-photon activities with compound 2 manifests an overall ascending magnitude of 2PA. These features suggest that the expansion of π-domain size from a D-π-A framework (as for compound 1) to a D-π-A-π-D scaffold (as for compound 2) has led to the enhancement of molecular 2PA without changing the band position of major 2PA in this model system. Although the same structural expansion also leads to an intensified 3PA as one can see in Fig. 5, it should be noted that the extent of enhancement on maximal 2PA is larger than that on the maximal 3PA (i.e. δmax2PA(2)/δmax2PA(1) ≈ 2.2 whereas δmax3PA(2)/δmax3PA(1) ≈ 1.7). Although this discrepancy in the amplitude of enhancement is not too large in the current case, it may indicate the existence of some fundamental differences between the molecular designs for 2PA- and 3PA-active chromophores. In other words, those structural parameters that are beneficial for the promotion of molecular 2PA may or may not be necessary useful for the enhancement of 3PA for a given chromophore system. Nevertheless, the current observation is in agreement with previously reported studies based on different molecular systems, in which the structural expansion was also found to correlate with 2PA and 3PA cross-sections.16,17
 |
| | Fig. 4 (a) Normalized two-photon excited emission spectra of fluorophores 1 and 2 in toluene at 1 × 10−5 M. (b and c) Logarithmic plots of power-squared dependence of the 2PA-induced fluorescence intensities of these compounds on the input intensity (in toluene). (d) Measured degenerate 2PA-spectra of 1 and 2 in toluene solution at 1 × 10−4 M (experimental error ∼15%). | |
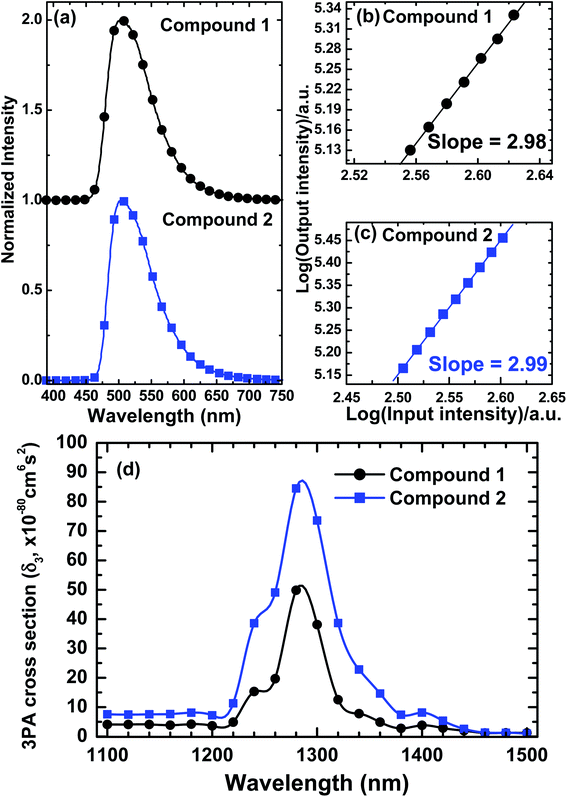 |
| | Fig. 5 (a) Normalized three-photon excited emission spectra of fluorophores 1 and 2 in toluene-d8 at 1 × 10−5 M. (b and c) Logarithmic plots of power-squared dependence of the 3PA-induced fluorescence intensities of these compounds on the input intensity (in toluene-d8). (d) Measured degenerate 3PA-spectra of 1 and 2 in toluene-d8 solution at 1 × 10−2 M (experimental error ∼15%). | |
It is also noted that the maximal 2PA and 3PA of these two dye molecules are located at wavelengths shorter than twice and three-times their lowest-energy linear absorption maxima (i.e. λ2PAmax < 2λ1PAmax and λ3PAmax < 3λ1PAmax for both studied compounds). This suggests that the most probable excited-states accessed by two- and three-photon processes are energetically higher than the lowest one-photon allowed excited-states of these compounds. In addition, the presently investigated compounds manifest significant multi-photon brightness (δ2 × ΦF or δ3 × ΦF), which is another essential concern for the molecular design specifically for those applications relied on the strength of multi-photon-induced fluorescence signals.
3. 2PA- and 3PA-based optical power-limiting
We have selected chromophore 2 for the demonstration of optical power-limiting properties based on 2PA and 3PA at 750 and 1280 nm in the femtosecond regime since this compound possesses larger overall 2PA and 3PA in this model system.
According to the nonlinear absorption theory, the attenuation of the incident laser intensity caused by multi-photon absorption processes can be described by the following phenomenological expression:14,18
| |
 | (3) |
Here
I(
z) is the local intensity of the incident light propagating along the
z-axis,
α,
β, and
γ are the one-, two-, and three-photon absorption coefficients of the given medium, respectively. When the appropriate wavelengths (
λ) are selected, there will be no linear absorption (
i.e. α = 0) and either a pure degenerate 2PA or a pure degenerate 3PA process that satisfies
eqn (3) can occur within the studied medium. The solutions to
eqn (3) for these two independent nonlinear processes should be:
| |
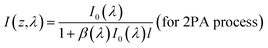 | (4) |
and
| |
 | (5) |
where
I0(
λ) is the incident light intensity with a top-hat pulse shape,
l is the propagation length in the medium, and
β(
λ) (in units of cm GW
−1) and
γ(
λ) (in units of cm
3 GW
−2) are the two-photon and three-photon absorption coefficients which are macroscopic parameters depending on the concentration of the studied sample. Practically, the
β and
γ values can be determined by nonlinear transmission experiments. Once
β and
γ values are found for a given wavelength the corresponding values of 2PA and 3PA cross-sections (
σ2 and
σ3) can be further retrieved from the following two equations:
| | |
β(λ) = σ2(λ)N0 = σ2(λ)NAd0 × 10−3
| (6) |
and
| | |
γ(λ) = σ3(λ)N0 = σ3(λ)NAd0 × 10−3
| (7) |
where
N0 is the molecular density,
NA is the Avogadro number,
d0 is the molar concentration of the absorbing molecules (in units of M).
Fig. 6 illustrates the measured data for the dependence of transmitted intensity on the input laser intensity at ∼750 nm (Fig. 6a) and ∼1280 nm (Fig. 6b), respectively. The blue and red solid lines in Fig. 6a and b represent the theoretical curves predicted by eqn (4) and (5) with best fitting parameters of β = 4.75 × 10−1 cm GW−1 and γ = 2.0 × 10−4 cm3 GW−2, respectively. These two parameters are the measured 2PA and 3PA coefficient values of the sample at corresponding probing wavelengths. It can be seen that both measured input/output curves exhibit none deviation from the linear transmission (gray solid lines in both figures) under very low input intensity and when the input intensity levels up, such deviation start to increase because the corresponding nonlinear absorption processes were triggered so that the output intensities were suppressed in each case accordingly. These observed nonlinear optical power-attenuation behaviors may suggest the possibility to develop a single-component material system which manifests strong 2PA and 3PA for efficient and broadband optical-limiting related applications based on appropriate molecular design. In addition, the 2PA and 3PA cross-sections values of this model compound can be calculated from the above experimental results to be ∼2090 GM and ∼80 × 10−80 cm6 s2 respectively. These values are very close to the results obtained from the MPEF method within the experimental error, indicating that 2PA and 3PA are the essential causes for the observed up-converted emission and optical power-limiting properties in this multi-branched chromophore system. Besides, it is worthy to mention here that for a designed material to be applicable in multi-photon-based technologies, other than the desired molecular parameters such as strong multi-photon-induced fluorescence and nonlinear optical attenuation, photostability is another important issue that needs to be considered for the molecular design since this character directly relates to the working lifetime of that material. For these two chromophores, neither salient decrease of the 2PA-/3PA-induced fluorescence strength nor observable decline of optical power-suppressing ability was found even under prolonged irradiation of femtosecond laser pulses in our experimental conditions, which suggests that the studied dye molecules in this work may manifest fairly acceptable robustness against ultrafast laser pulses. More sophisticated experiments should be designed to evaluate the photostability of these compounds and are undertaken.
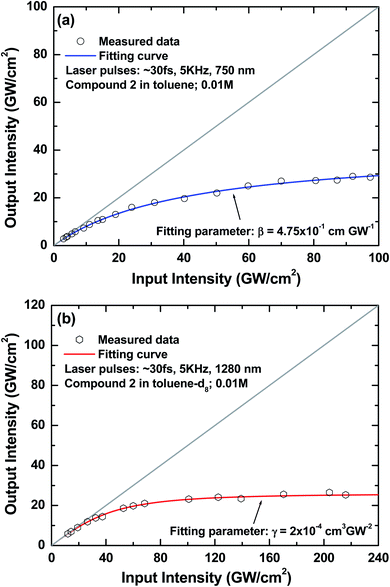 |
| | Fig. 6 (a) Measured characteristic 2PA-based optical power-limiting curve of compound 2 in toluene (0.01 M; blue solid line: theoretical curve using a best fitting 2PA coefficient of β = 4.75 × 10−1 cm GW−1); (b) measured characteristic 3PA-based optical power-limiting curve of compound 2 in toluene-d8 (0.01 M; red solid line: theoretical curve using a best fitting 3PA coefficient of γ = 2.0 × 10−4 cm3 GW−2). | |
4. Conclusions
In conclusion, a model compound set containing two multi-branched analogues based on indenoquinoxaline and oxadiazole moieties has been synthesized and investigated for their linear and nonlinear absorption as well as 2PA- and 3PA-based optical power-limiting properties in the femtosecond regime. Both model compounds were experimentally found to possess strong and wide range of 2PA and 3PA. It is also realized that although the structural expansion could lead to the promotion of molecular two- and three-photon absorptivities in this model chromophore system, the amplitude of promotion is not necessary to be identical due to the existence of some intrinsic differences in the requirements of molecular parameters for highly two- and three-photon active materials.
Moreover, by using multi-photon-excited fluorescence (MPEF) technique, we have been able to record and analyze the power-dependence relationship between the incident laser and up-converted fluorescence and map out the spectral dispersion of the order of absorption process so that the entire progression from pure 2PA to pure 3PA through a section of the coexistence of both processes can be clearly delineated (i.e. the OAP-spectrum) based on the studied model compounds. With the aid of the measured 2PA and 3PA-spectra, one may retrieve useful information for the future material design especially for those applications that requires highly active 2PA/3PA in a specific spectral region.
In addition, we have selected the larger congener (i.e. compound 2) to demonstrate 2PA- and 3PA-based optical power-limiting at the wavelength positions of its maximal 2PA and 3PA and the experimental results confirm that 2PA and 3PA are the major causes for the observed up-converted fluorescence and optical power-suppressing characteristics in this multi-branched chromophore system.
5. Experimental section
5.1 General
All the reagents utilized for the preparation of the intermediates and final structures were purchased from various commercial sources and were used as received. A 500 MHz NMR spectrometer was employed to measure the 1H NMR and 13C NMR spectra for all the intermediates and final structures using TMS or residual CHCl3 signals as the internal standards. The numbering of carbon and hydrogen atoms used for NMR signal assignment for the intermediates and the final chromophores are summarized in the ESI.† High-resolution mass spectra (HRMS) were measured by using an ESI-TOF mass spectrometer (Waters LCT) and MALDI-TOF MS spectra were measured by using a Voyager DE-PRO mass spectrometer (Applied Biosystem, USA).
5.2 Photophysical methods
The measurements of linear optical properties of the studied model chromophores were performed by utilizing the corresponding UV-VIS-NIR absorption and fluorescence spectrometers. For two- and three-photon-excited fluorescence properties, a Ti:sapphire oscillator was utilized as the major working tool for the measurements. On the other hand, a femtosecond regenerative amplifier system was employed to probe two- and three-photon-based nonlinear transmission properties of the studied compounds. The experimental conditions and the configuration of optical set-ups for the measurements of nonlinear optical properties are described in detail in the ESI.†
5.3 Synthesis
Compounds 3, 10 and 17 in Schemes 1 and 2 were employed as the starting compounds for the synthesis of intermediates and final chromophores. These compounds were prepared by following the reported procedures.8,13 The details for the synthesis of other key intermediates (i.e. compounds 4–9 and 11–16) and targeted model compounds are presented below:
5.3.1 2-(7-(5-(4-(tert-Butyl)phenyl)-1,3,4-oxadiazol-2-yl)-9,9-dihexyl-9H-fluoren-2-yl)isoindoline-1,3-dione (4). To a solution of compound 3 (1.3 g; 2.12 mmol) in DMAc (15 mL) was added potassium phthalimide (0.39 g; 2.12 mmol) and CuI (0.40 g; 2.12 mmol) and the whole system was heated to reflux for 24 h. After cooling to the room temperature, NH3(aq) (50 mL) was added to the reaction mixture and the resulting solution was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was further purified through column chromatography on silica gel using ethyl acetate/hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) as the eluent to give target product (0.79 g, 55%). 1H NMR (300 MHz, CDCl3) δ = 8.16 (d, J = 1.2 Hz, 1H, HK), 8.15 (dd, J1 = 1.2 Hz, J2 = 5.7 Hz, 1H, HI), 8.12 (dd, J1 = 3.0 Hz, J2 = 6.6 Hz, 2H, HP), 7.98 (dd, J1 = 3 Hz, J2 = 5.7 Hz, H1c), 7.90 (d, J = 5.4 Hz, 1H, HE), 7.88 (d, J = 5.4 Hz, 1H, HD), 7.81 (dd, J1 = 3 Hz, J2 = 5.4 Hz, H1d), 7.57–7.51 (m, 4H, HB, HH, HQ), 2.15–2.10 (m, 4H, Hf′), 1.37 (s, 9H, HT), 1.14–1.10 (m, 12H, Hc′, Hd′, He′), 0.78–0.74 (m, 10H, Ha′, Hb′); 13C NMR (75 MHz, CDCl3) δ = 166.93 (C1a), 164.67 (CM), 164.31 (CN), 155.00 (CR), 151.86 (CA), 143.55 (CL), 139.06 (CF), 134.20 (C1c), 133.55 (CG), 131.50 (CO), 131.41 (C1b), 126.54 (CP), 125.90 (CC), 125.79 (CQ), 125.01 (CJ), 123.40 (C1d), 122.82 (CE), 122.45 (CH), 121.10 (CI), 120.92 (CK), 120.55 (CD), 120.28 (CB), 55.53 (Cg′), 39.91 (Cf′), 34.79 (CS), 31.22 (Cc′), 30.85 (CT), 29.41 (Cd′), 23.63 (Ce′), 22.31 (Cb′), 13.74 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C45H49N3O3: 680.3852 [M]+; found: 680.3870.
:5) as the eluent to give target product (0.79 g, 55%). 1H NMR (300 MHz, CDCl3) δ = 8.16 (d, J = 1.2 Hz, 1H, HK), 8.15 (dd, J1 = 1.2 Hz, J2 = 5.7 Hz, 1H, HI), 8.12 (dd, J1 = 3.0 Hz, J2 = 6.6 Hz, 2H, HP), 7.98 (dd, J1 = 3 Hz, J2 = 5.7 Hz, H1c), 7.90 (d, J = 5.4 Hz, 1H, HE), 7.88 (d, J = 5.4 Hz, 1H, HD), 7.81 (dd, J1 = 3 Hz, J2 = 5.4 Hz, H1d), 7.57–7.51 (m, 4H, HB, HH, HQ), 2.15–2.10 (m, 4H, Hf′), 1.37 (s, 9H, HT), 1.14–1.10 (m, 12H, Hc′, Hd′, He′), 0.78–0.74 (m, 10H, Ha′, Hb′); 13C NMR (75 MHz, CDCl3) δ = 166.93 (C1a), 164.67 (CM), 164.31 (CN), 155.00 (CR), 151.86 (CA), 143.55 (CL), 139.06 (CF), 134.20 (C1c), 133.55 (CG), 131.50 (CO), 131.41 (C1b), 126.54 (CP), 125.90 (CC), 125.79 (CQ), 125.01 (CJ), 123.40 (C1d), 122.82 (CE), 122.45 (CH), 121.10 (CI), 120.92 (CK), 120.55 (CD), 120.28 (CB), 55.53 (Cg′), 39.91 (Cf′), 34.79 (CS), 31.22 (Cc′), 30.85 (CT), 29.41 (Cd′), 23.63 (Ce′), 22.31 (Cb′), 13.74 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C45H49N3O3: 680.3852 [M]+; found: 680.3870.
5.3.2 7-(5-(4-(tert-Butyl)phenyl)-1,3,4-oxadiazol-2-yl)-9,9-dihexyl-9H-fluoren-2-amine (5). To a mixture of compound 4 (0.8 g; 1.17 mmol) in methanol (25 mL) was added hydrazine monohydrate (0.5 g; 2.34 mmol) and the mixture was heated to reflux for 7 h. After cooling to the room temperature and removal of methanol by rotary evaporator, the residue was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was purified by column chromatography on silica gel using ethyl acetate/hexane (1:3) to afford the title compound (0.6 g, 93%). 1H NMR (300 MHz, CDCl3) δ = 8.10 (dd, J1 = 1.8 Hz, J2 = 6.9 Hz, 1H, HI), 8.07–8.03 (m, 3H, HD, HP), 7.65 (d, J = 7.8 Hz, 1H, HE), 7.56–7.51 (m, 3H, HH, HQ), 6.69 (s, 1H, HB), 6.67 (d, J = 1.8 Hz, 1H, HK), 3.97 (s, 2H, HNH2), 2.09–1.87 (m, 4H, Hf′), 1.36 (s, 9H, HT), 1.23–1.04 (m, 12H, Hc′, Hd′, He′), 0.77–0.63 (m, 10H, Ha′, Hb′); 13C NMR (75 MHz, CDCl3) δ = 165.23 (CM), 164.17 (CN), 154.98 (CR), 153.43 (CA), 150.48 (CL), 147.29 (CF), 145.36 (CG), 130.72 (CC, CO), 126.60 (CP), 125.88 (CQ), 121.31 (CJ), 121.17 (CE), 120.80 (CH), 120.25 (CI), 118.47 (CK), 114.04 (CD), 109.26 (CB), 54.99 (Cg′), 40.45 (Cf′), 34.92 (CS), 31.42 (Cc′), 30.99 (CT), 29.59 (Cd′), 23.60 (Ce′), 22.46 (Cb′), 13.87 (Ca′); HRMS (MALDI-FAB): m/z: calcd for C37H47N3O: 549.3719 [M]+; found: 549.3725.
5.3.3 N-(7-(5-(4-(tert-Butyl)phenyl)-1,3,4-oxadiazol-2-yl)-9,9-dihexyl-9H-fluoren-2-yl)acetamide (6). To a mixture of compound 5 (0.6 g; 1.09 mmol) in CH2Cl2 (20 mL) was added triethylamine (0.30 mL; 2.18 mmol) and acetic anhydride (0.20 mL; 2.18 mmol) and the whole system was stirred at room temperature for 20 min. After removal of the solvent by rotatory evaporator, the mixture was poured into water, and the precipitate was filtered and dried in vacuum oven to afford title compound (0.97 g, 90%). 1H NMR (300 MHz, CDCl3) δ = 8.22 (s, 1H, HNHAc), 8.11–8.08 (m, 4H, HB, HP, HI), 7.76 (d, J = 8.1 Hz, 1H, HE), 7.73 (s, 1H, HK), 7.69 (d, J = 8.1 Hz, 1H, HD), 7.58–7.55 (m, 3H, HH, HQ), 2.25 (s, 3H, HV), 2.08–1.98 (m, 4H, Hf′), 1.38 (s, 9H, HT), 1.12–0.97 (m, 12H, Hc′, Hd′, He′), 0.77–0.63 (m, 10H, Ha′, Hb′); 13C NMR (75 MHz, CDCl3) δ = 168.59 (CU), 165.11 (CM), 164.50 (CN), 155.30 (CR), 152.57 (CA), 151.47 (CL), 144.45 (CF), 138.66 (CG), 135.77 (CO), 126.72 (CP), 126.02 (CQ), 121.58 (CC), 121.05 (CJ), 120.81 (CE), 119.68 (CH, CI), 118.61 (CK), 114.23 (CB, CD), 55.55 (Cg′), 40.28 (Cf′), 35.03 (CS), 31.45 (Cc′), 31.06 (CT), 29.58 (Cd′), 24.66 (CV), 23.71 (Ce′), 22.49 (Cb′), 13.90 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C39H49N3O2: 592.3903 [M]+; found: 592.3924.
5.3.4 N-(7-(5-(4-(tert-Butyl)phenyl)-1,3,4-oxadiazol-2-yl)-9,9-dihexyl-3-nitro-9H-fluoren-2-yl)acetamide (7). To a mixture of compound 6 (1 g; 1.68 mmol) in AcOH (15 mL) and acetone (5 mL) was added nitric acid (1.4 mL; 33.79 mmol) and the whole system was stirred at room temperature. After the reaction was completed, the reaction mixture was poured into NaOH(aq) and the resulting solution was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was purified by column chromatography on silica gel using ethyl acetate/hexane (1:5) to give title product as a yellow powder (0.86 g, 80%). 1H NMR (300 MHz, CDCl3) δ = 10.66 (s, 1H, HNHAc), 8.91 (s, 1H, HE), 8.60 (s, 1H, HB), 8.19–8.10 (m, 4H, HI, HK, HP), 7.89 (d, J = 8.7 Hz, 1H, HH), 7.58 (d, J = 6.9 Hz, 2H, HQ), 2.36 (s, 3H, HV), 2.15–2.10 (m, 4H, Hf′), 1.38 (s, 9H, HT), 1.12–1.01 (m, 12H, Hc′, Hd′, He′), 0.77–0.67 (m, 10H, Ha′, Hb′); 13C NMR (75 MHz, CDCl3) δ = 169.03 (CU), 164.59 (CM), 164.46 (CN), 160.36 (CR), 155.32 (CA), 151.39 (CL), 141.82 (CF), 135.74 (CG), 135.25 (CO), 135.05 (CD), 126.69 (CP), 126.29 (CK), 125.93 (CQ), 123.32 (CC), 121.32 (CI), 120.81 (CJ), 120.51 (CH), 117.26 (CE), 116.14 (CB), 56.53 (Cg′), 40.05 (Cf′), 34.93 (CS), 31.27 (Cc′), 30.95 (CT), 29.33 (Cd′), 25.64 (CV), 23.72 (Ce′), 22.34 (Cb′), 13.78 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C39H48N4O4: 637.3753 [M]+; found: 637.3769.
5.3.5 7-(5-(4-(tert-Butyl)phenyl)-1,3,4-oxadiazol-2-yl)-9,9-dihexyl-3-nitro-9H-fluoren-2-amine (8). To a mixture of compound 7 (0.7 g; 0.74 mmol) in methanol (10 mL) was added sulfuric acid (2 mL) and the whole system was heated to reflux for 30 min. After cooled down to room temperature, the reaction mixture was poured into NaOH(aq) and the resulting solution was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was purified by column chromatography on silica gel using ethyl acetate/hexane (1:5) to afford a red powder as the title product (0.56 g, 60%). 1H NMR (300 MHz, CDCl3) δ = 8.50 (s, 1H, HE), 8.11–8.08 (m, 4H, HB, HI, HP), 7.78 (d, J = 8.7 Hz, 1H, HH), 7.58 (d, J = 8.7 Hz, 2H, HQ), 6.78 (s, 1H, HK), 6.33 (s, 2H, HNH2), 2.11–1.87 (m, 4H, Hf′), 1.38 (s, 9H, HT), 1.15–1.05 (m, 12H, Hc′, Hd′, He′), 0.78–0.65 (m, 10H, Ha′, Hb′); 13C NMR (75 MHz, CDCl3) δ = 164.82 (CM), 164.59 (CN), 160.27 (CR), 155.35 (CA), 150.18 (CL), 145.45 (CF), 142.90 (CG), 131.91 (CO), 130.57 (CC), 126.78 (CP), 125.39 (CK), 126.06 (CQ), 122.41 (CJ), 121.23 (CI), 121.10 (CD), 119.86 (CH), 117.75 (CE), 112.75 (CB), 55.73 (Cg′), 40.85 (Cf′), 35.09 (CS), 31.45 (Cc′), 31.11 (CT), 29.55 (Cd′), 23.82 (Ce′), 22.50 (Cb′), 13.92 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C37H46N4O3: 595.3648 [M]+; found: 595.3666.
5.3.6 7-(5-(4-(tert-Butyl)phenyl)-1,3,4-oxadiazol-2-yl)-9,9-dihexyl-9H-fluorene-2,3-diamine (9). To a mixture of compound 8 (0.56 g; 0.94 mmol) in ethyl acetate (20 mL) was added tin(II) chloride dihydrate (2.12 g; 9.4 mmol) and the whole system was heated to reflux for 3 h. After cooled down to room temperature, the reaction mixture was poured into NaHCO3(aq) and the resulting solution was filtered. The filtrate was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was directly used for the next step without further purification. 1H NMR (300 MHz, CDCl3) δ = 8.10 (d, J = 8.4 Hz, 2H, HP), 8.03–8.01 (m, 2H, HI, HK), 7.60 (d, J = 8.4 Hz, 1H, HH), 7.56 (d, J = 8.4 Hz, 2H, HQ), 7.10 (s, 1H, HE), 6.69 (s, 1H, HB), 3.66 (s, 4H, HNH2), 2.10–1.84 (m, 4H, Hf′), 1.37 (s, 9H, HT), 1.14–1.05 (m, 12H, Hc′, Hd′, He′), 0.78–0.72 (m, 10H, Ha′, Hb′); 13C NMR (75 MHz, CDCl3) δ = 165.34 (CM), 164.25 (CN), 155.07 (CR), 151.24 (CA), 145.58 (CL), 144.66 (CF), 136.43 (CG), 133.53 (CC), 132.01 (CD), 126.69 (CP), 125.97 (CQ), 125.87 (CO), 121.25 (CK), 120.76 (CJ), 120.31 (CI), 118.51 (CH), 110.65 (CE), 108.82 (CB), 54.73 (Cg′), 40.48 (Cf′), 35.02 (CS), 31.52 (Cc′), 31.08 (CT), 29.68 (Cd′), 23.69 (Ce′), 22.55 (Cb′), 13.95 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C37H48N4O: 564.3828 [M]+; found: 564.3836.
5.3.7 2,2′-((1,3,4-Oxadiazole-2,5-diyl)bis(9,9-dihexyl-9H-fluorene-7,2-diyl))bis(isoindoline-1,3-dione) (11). To a solution of compound 10 (1 g; 1.12 mmol) in DMAc (15 mL) was added potassium phthalimide (0.72 g; 3.92 mmol) and CuI (0.66 g; 3.46 mmol) and the whole system was heated to reflux for 24 h. After cooled down to room temperature, NH3(aq) (50 mL) was added to the reaction mixture and the resulting solution was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was purified through column chromatography on silica gel using ethyl acetate/hexane (1:5) as the eluent to give title product (0.59 g, 52%).1H NMR (300 MHz, CDCl3) δ = 8.20–8.17 (m, 4H, HI, HK), 7.99 (dd, J1 = 3 Hz, J2 = 5.4 Hz, 4H, H1c), 7.92 (d, J = 2.4 Hz, 2H, HH), 7.89 (d, J = 2.4 Hz, 2H, HE), 7.82 (dd, J1 = 3 Hz, J2 = 5.4 Hz, 4H, H1d), 7.53–7.50 (m, 4H, HB, HD), 2.12–2.07 (m, 8H, Hf′), 1.16–1.08 (m, 24H, Hc′, Hd′, He′), 0.78–0.74 (m, 20H, Ha′, Hb′); 13C NMR (75 MHz, CDCl3) δ = 166.95 (C1a), 164.83 (CM), 151.86 (CA), 143.61 (CL), 139.04 (CF), 134.22 (C1c), 133.78 (CG), 131.44 (C1b), 128.02 (CC), 126.76 (CI), 126.01 (CK), 125.02 (CJ), 123.40 (C1d), 122.36 (CH), 121.04 (CE), 120.55 (CD), 120.31 (CB), 55.53 (Cg′), 39.87 (Cf′), 31.19 (Cc′), 29.36 (Cd′), 23.61 (Ce′), 22.27 (Cb′), 13.72 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C68H72N4O5: 1025.5620 [M]+; found: 1025.5581.
5.3.8 7,7′-(1,3,4-Oxadiazole-2,5-diyl)bis(9,9-dihexyl-9H-fluoren-2-amine) (12). To a mixture of compound 11 (1.2 g; 1.17 mmol) in methanol (25 mL) was added hydrazine monohydrate (0.5 g; 2.34 mmol) and the mixture was heated to reflux for 7 h. After cooled down to the room temperature and removal of methanol by rotary evaporator, the mixed residue was extracted with ethyl acetate and the organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was purified by column chromatography on silica gel using ethyl acetate/hexane (1:3) to afford the title compound (0.8 g, 90%).1H NMR (300 MHz, CDCl3) δ = 8.11–8.09 (m, 4H, HI, HK), 7.67 (d, J = 8.4 Hz, 2H, HH), 7.54 (d, J = 8.4 Hz, 2H, HE), 6.69–6.67 (m, 4H, HB, HD), 3.97 (s, 4H, HNH2), 1.99–1.88 (m, 8H, Hf′), 1.14–1.05 (m, 24H, Hc′, Hd′, He′), 0.77–0.66 (m, 20H, Ha′, Hb′); 13C NMR (75 MHz, CDCl3) δ = 165.04 (CM), 153.38 (CA), 150.44 (CL), 147.25 (CF), 145.24 (CG) 130.69 (CC), 125.85 (CJ), 121.25 (CH), 120.74 (CE), 120.34 (CI), 118.43 (CK), 113.97 (CD), 109.19 (CB), 54.96 (Cg′), 40.42 (Cf′), 31.39 (Cc′), 29.54 (Cd′), 23.57 (Ce′), 22.42 (Cb′), 13.82 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C52H68N4O: 764.5393 [M]+; found: 764.5388.
5.3.9 N,N′-((1,3,4-Oxadiazole-2,5-diyl)bis(9,9-dihexyl-9H-fluorene-7,2-diyl))diacetamide (13). To a mixture of compound 12 (0.8 g; 0.94 mmol) in CH2Cl2 (20 mL) was added triethylamine (0.32 mL; 2.3 mmol) and acetic anhydride (0.22 mL; 2.3 mmol) and the whole system was stirred at the room temperature for 20 min. After removal of the solvent by rotatory evaporator, the mixed residue was poured into water, and the precipitate was filtered and dried in vacuum oven to afford title compound (0.84 g, 95%).1H NMR (300 MHz, CDCl3) δ = 8.14–8.12 (m, 4H, HI, HK), 8.00 (s, 2H, HNHAc), 7.78 (d, J = 8.4 Hz, 2H, HH), 7.72–7.69 (m, 4H, HB, HE), 7.56 (d, J = 8.4 Hz, 2H, HD), 2.18 (s, 6H, HO), 2.11–2.03 (m, 8H, Hf′), 1.10–1.04 (m, 24H, Hc′, Hd′, He′), 0.76–0.62 (m, 20H, Ha′, Hb′); 13C NMR (75 MHz, CDCl3) δ = 168.50 (CN), 165.11 (CM), 152.58 (CA), 151.47 (CL), 144.40 (CF), 138.52 (CG), 135.85 (CC), 126.07 (CJ), 121.65 (CH), 121.11 (CE), 120.82 (CI), 119.68 (CK), 118.57 (CD), 114.18 (CB), 55.57 (Cg′), 40.30 (Cf′), 31.46 (Cc′), 29.57 (Cd′), 24.68 (CO), 23.70 (Ce′), 22.48 (Cb′), 13.91 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C56H72N4O3: 849.5682 [M]+; found: 849.5710.
5.3.10 N,N′-((1,3,4-Oxadiazole-2,5-diyl)bis(9,9-dihexyl-3-nitro-9H-fluorene-7,2-diyl))diacetamide (14). To a mixture of compound 13 (0.8 g; 0.94 mmol) in AcOH (15 mL) and acetone (5 mL) was added nitric acid (0.78 mL; 18.84 mmol) and the whole system was stirred at room temperature. After the reaction was completed, the mixture was poured into NaOH(aq) and the resulting solution was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was purified by column chromatography on silica gel using ethyl acetate/hexane (1:5) to give the target product as a yellow powder (0.76 g, 87%). 1H NMR (300 MHz, CDCl3) δ = 10.67 (s, 2H, HNHAc), 8.89 (s, 2H, HE), 8.60 (s, 2H, HB), 8.22–8.18 (m, 4H, HI, HK), 7.90 (d, J = 8.1 Hz, 2H, HH), 2.36 (s, 6H, HO), 2.14–2.09 (m, 8H, Hf′), 1.14–1.05 (m, 24H, Hc′, Hd′, He′), 0.75 (t, 12H, Ha′), 0.62–0.60 (m, 8H, Hb′); 13C NMR (75 MHz, CDCl3) δ = 169.09 (CN), 164.90 (CM), 160.27 (CA), 151.58 (CL), 142.09 (CF), 135.80 (CG), 135.30 (CD), 129.14 (CC), 126.51 (CI), 123.37 (CJ), 121.53 (CH), 120.64 (CK), 117.44 (CE), 116.21 (CB), 56.72 (Cg′), 40.21 (Cf′), 31.42 (Cc′), 29.46 (Cd′), 25.85 (CO), 23.86 (Ce′), 22.47 (Cb′), 13.92 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C56H70N6O7: 939.5383 [M]+; found: 939.5404.
5.3.11 7,7′-(1,3,4-Oxadiazole-2,5-diyl)bis(9,9-dihexyl-3-nitro-9H-fluoren-2-amine) (15). To a mixture of compound 14 (0.7 g; 0.74 mmol) in methanol (10 mL) was added sulfuric acid (2 mL) and the whole system was heated to reflux for 30 min. After cooled down to room temperature, the reaction mixture was poured into NaOH(aq) and the resulting solution was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was purified by column chromatography on silica gel using ethyl acetate/hexane (1:5) to afford the target product as a red powder (0.51 g, 80%).1H NMR (300 MHz, CDCl3) δ = 8.48 (s, 2H, HE), 8.05 (d, J = 8.1 Hz, 2H, HI), 7.95 (s, 2H, HB), 7.68 (d, J = 8.1 Hz, 2H, HH), 6.78 (s, 2H, HK), 6.38 (s, 4H, HNH2), 2.06–1.85 (m, 8H, Hf′), 1.11–1.04 (m, 24H, Hc′, Hd′, He′), 0.78–0.73 (t, 12H, Ha′), 0.61 (m, 8H, Hb′); 13C NMR (75 MHz, CDCl3) δ = 167.32 (CM), 160.58 (CA), 149.30 (CL), 145.42 (CF), 144.00 (CC), 131.83 (CG), 130.57 (CD), 129.25 (CI), 128.58 (CE), 123.94 (CJ), 119.06 (CH), 117.93 (CK), 112.32 (CB), 55.49 (Cg′), 40.76 (Cf′), 31.41 (Cc′), 29.53 (Cd′), 23.75 (Ce′), 22.49 (Cb′), 13.92 (Ca′); HRMS (MALDI-TOF): m/z: calcd for C52H66N6O5: 854.5095 [M]+; found: 854.5098.
5.3.12 7,7′-(1,3,4-Oxadiazole-2,5-diyl)bis(9,9-dihexyl-9H-fluorene-2,3-diamine) (16). To a mixture of compound 15 (0.4 g; 0.46 mmol) in ethyl acetate (20 mL) was added tin(II) chloride dihydrate (2.11 g; 9.35 mmol) and the whole system was heated to reflux for 3 h. After cooled down to room temperature, the reaction mixture was poured into NaHCO3(aq) and the resulting solution was filtered. The filtrate was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was directly used for the next step without further purification. 1H NMR (300 MHz, CDCl3) δ = 7.97 (dd, J1 = 1.5 Hz, J2 = 8.1 Hz, 2H, HI), 7.91 (d, J = 1.5 Hz, 2H, HK), 7.50 (d, J = 8.1 Hz, 2H, HH), 7.07 (s, 2H, HE), 6.67 (s, 2H, HB), 3.51 (s, 8H, HNH2), 2.00–1.79 (m, 8H, Hf′), 1.13–1.01 (m, 24H, Hc′, Hd′, He′), 0.75 (t, 12H, Ha′), 0.66 (m, 8H, Hb′); 13C NMR (75 MHz, CDCl3) δ = 167.83 (CM), 150.39 (CA), 146.70 (CL), 145.07 (CF), 136.44 (CG), 133.46 (CC), 132.05 (CD), 128.76 (CJ), 126.56 (CH), 123.48 (CI), 117.74 (CI), 117.74 (CK), 110.63 (C1), 108.98 (CE), 54.51 (Cg′), 40.40 (Cf′), 31.52 (Cc′), 29.69 (Cd′), 23.65 (Ce′), 22.56 (Cb′), 13.96 (Ca′); HRMS (FAB): m/z: calcd for C52H70N6O: 794.5611 [M]+; found: 794.5611.
5.3.13 Compound 1. To a mixture of compound 9 (0.37 g; 0.65 mmol), AcOH (10 mL) in THF (20 mL) was added 1,2-bis(7-(diphenylamino)-9,9-dihexyl-9H-fluoren-2-yl)ethane-1,2-dione (17) (0.63 g, 0.59 mmol) and the whole system was heated to reflux for 12 h. After cooled down to room temperature, the reaction mixture was poured into NaOH(aq) and the resulting solution was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was purified by column chromatography on silica gel using THF/hexane (1:10) to afford title compound (0.7 g, 74%). 1H NMR (300 MHz, CDCl3) δ = 8.53 (s, 1H, HE), 8.22–8.20 (m, 2H, HK, HI), 8.14–8.12 (m, 3H, HB, HP), 8.07 (d, J = 8.1 Hz, 1H, HH), 7.63–7.51 (m, 10H, H9, H12, H13, H15, HQ), 7.27–7.22 (m, 8H, H2), 7.13–7.08 (m, 10H, H3, H6), 7.03–6.99 (m, 6H, H1, H8), 2.27–2.19 (m, 4H, Hf′), 1.75 (m, 8H, Hf), 1.39 (s, 9H, HT), 1.10–1.04 (m, 36H, Hc, Hc′, Hd, Hd′, He, He′), 0.82–0.66 (m, 30H, Ha, Ha′, Hb, Hb′); 13C NMR (75 MHz, CDCl3) δ = 164.76 (CM, CN), 155.40 (CR), 153.67 (Cm), 153.46 (Cn), 152.61 (C5), 152.27 (C16), 150.63 (CA), 150.56 (CL), 147.92 (C4), 147.46 (C11), 143.13 (CG), 143.02 (C10), 141.66 (CC), 141.59 (CD), 141.14 (C7), 137.44 (CF), 137.35 (C14), 135.52 (CO), 129.18 (CB), 129.14 (C3), 129.10 (CJ), 126.86 (CP), 126.36 (CE), 126.08 (CQ), 124.38 (CK), 124.04 (CH), 123.87 (C2), 123.39 (CI), 122.55 (C1), 121.60 (C15), 121.13 (C12), 120.85 (C13), 119.41 (C6), 119.11 (C8), 118.97 (C9), 55.60 (Cg′), 55.08 (Cg), 41.34 (Cf′), 40.12 (Cf), 35.11 (CQ), 31.55 (CR), 31.13 (Cc, Cc′), 29.66 (Cd′), 29.60 (Cd), 24.00 (Ce′), 23.85 (Ce), 22.62 (Cb′), 22.53 (Cb), 14.07 (Ca′), 13.93 (Ca); HRMS (MALDI-TOF): m/z: calcd for C113H128N6O: 1585.0149 [M]+; found: 1585.0203.
5.3.14 Compound 2. To a mixture of compound 16 (0.26 g; 0.33 mmol), AcOH (10 mL) in THF (20 mL) was added 1,2-bis(7-(diphenylamino)-9,9-dihexyl-9H-fluoren-2-yl)ethane-1,2-dione (17) (0.62 g, 0.59 mmol) and the whole system was heated to reflux for 12 h. After cooled down to room temperature, the reaction mixture was poured into NaOH(aq) and the resulting solution was extracted with ethyl acetate. The organic layer was collected and dried over MgSO4(s). After filtration, the filtrate was concentrated in vacuo to afford crude product, which was purified by column chromatography on silica gel using THF/hexane (1:20) to afford title compound (0.54 g, 58%). 1H NMR (300 MHz, CDCl3) δ = 8.56 (s, 2H, HE), 8.30–8.24 (m, 4H, HB, HI), 8.17 (s, 2H, HK), 8.12 (d, J = 8.1 Hz, 2H, HE), 7.65–7.52 (m, 16H, H9, H12, H13, H15), 7.27–7.22 (m, 16H, H2), 7.13–7.07 (m, 20H, H3, H6), 7.01–6.94 (m, 12H, H1, H8), 2.27–2.23 (m, 8H, Hf′), 1.76 (m, 16H, Hf), 1.14–1.04 (m, 72H, Hc, Hc′, Hd, Hd′, He, He′), 0.82–0.68 (m, 60H, Ha, Ha′, Hb, Hb′); 13C NMR (75 MHz, CDCl3) δ = 165.06 (CM), 153.69 (C5), 153.44 (C16), 152.60 (Cm), 152.35 (CA), 150.62 (CL), 150.55 (CL′), 147.90 (C4), 147.47 (C11), 143.30 (CG), 142.51 (CK), 141.68 (CD, CC), 141.13 (C7), 137.41 (CB), 137.33 (CE), 135.73 (C10), 135.48 (CJ), 129.13 (C3), 128.91 (C12), 126.49 (CH), 125.47 (C15), 124.37 (C14), 123.86 (C2), 123.38 (C13), 122.55 (C1), 121.67 (C6), 120.85 (C9), 119.46 (CF), 119.08 (CD), 118.98 (C8), 55.64 (Cg′), 55.07 (Cg), 41.34 (Cf′), 40.12 (Cf), 31.55 (Cc, Cc′), 29.59 (Cd′, Cd), 24.00 (Ce′), 23.85 (Ce), 22.61 (Cb′), 22.51 (Cb), 14.07 (Ca′), 13.93 (Ca); HRMS (MALDI-TOF): m/z: calcd for C204H230N10O: 2838.1519 [M]+; found: 2838.1650.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We acknowledge the financial support from the Ministry of Science and Technology (MOST), Taiwan under grant numbers: MOST 108-2113-M-008-001-, 109-2113-M-008-015-, 109-3111-8-008-001-, and 109-2124-M-033-001-.
References
- C. W. Spangler, J. Mater. Chem., 1999, 9, 2013–2020 RSC.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS.
- H. M. Kim and B. R. Cho, Acc. Chem. Res., 2009, 42, 863–872 CrossRef CAS.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS.
- S. Yao and K. D. Belfield, Eur. J. Org. Chem., 2012, 3199–3217 CrossRef CAS.
- Q. Zheng, H. Zhu, S.-C. Chen, C. Tang, E. Ma and X. Chen, Nat. Photonics, 2013, 7, 234–239 CrossRef CAS.
- G. S. He and P. N. Prasad, Proc. SPIE, 2003, 5211, 1–12 CrossRef CAS.
- T.-C. Lin, Y.-H. Lee, C.-Y. Liu, B.-R. Huang, M.-Y. Tsai and Y.-J. Huang, Chem.–Eur. J., 2013, 19, 749–760 CrossRef CAS.
- T.-C. Lin, C.-Y. Liu, M.-H. Li, Y.-Y. Liu, S.-Y. Tseng, Y.-T. Wang, Y.-H. Tseng, H.-H. Chu and C.-W. Luo, J. Mater. Chem. C, 2014, 2, 821–828 RSC.
- T.-C. Lin, J.-Y. Lin, B.-K. Tsai, N.-Y. Liang and W. Chien, Dyes Pigm., 2016, 132, 347–359 CrossRef CAS.
- T.-C. Lin, W. Chien, L. M. Mazur, Y.-Y. Liu, K. Jakubowski, K. Matczyszyn, M. Samoc and R. W. Amini, J. Mater. Chem. C, 2017, 5, 8219–8232 RSC.
- T.-C. Lin, W. Chien, S.-W. Dai, H.-W. Lin and Y.-C. Liu, Dyes Pigm., 2019, 168, 140–150 CrossRef CAS.
- T.-C. Lin, Y.-H. Lee, C.-L. Hu, Y.-K. Li and Y.-J. Huang, Eur. J. Org. Chem., 2012, 1737–1745 CrossRef CAS.
- G. S. He, Nonlinear optics and photonics, Oxford University Press, 2015 Search PubMed.
- A. Rebane and A. Mikhaylov, Proc. SPIE, 2018, 10498, 10498301–104983012 Search PubMed.
- Z. Suo, M. Drobizhev, C. W. Spangler, N. Christensson and A. Rebane, Org. Lett., 2005, 7, 4807–4810 CrossRef CAS.
- B. Gao, L. M. Mazur, M. Morshedi, A. Barlow, H. Wang, C. Quintana, C. Zhang, M. Samoc, M. P. Cifuentes and M. G. Humphrey, Chem. Commun., 2016, 52, 8301–8304 RSC.
- L. W. Tutt and T. F. Boggess, Prog. Quantum Electron., 1993, 17, 299–338 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08945a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc,
Shih-Huang Changa,
Hsiang-Yu Hsieha,
Zi-Ming Chena and
Chun-Yao Chua
*abc,
Shih-Huang Changa,
Hsiang-Yu Hsieha,
Zi-Ming Chena and
Chun-Yao Chua










