DOI:
10.1039/D0RA09029H
(Paper)
RSC Adv., 2021,
11, 6259-6267
Bridging-arylene effects on spectroscopic and photophysical properties of arylborane–dipyrrinato zinc(II) complexes†
Received
23rd October 2020
, Accepted 23rd January 2021
First published on 3rd February 2021
Abstract
Novel bis(dipyrrinato)zinc(II) derivatives having 4-[bis(2,4,6-trimethylphenyl)boryl]phenyl (ZnBph) or 4-[bis(2,4,6-trimethylphenyl)boryl]-2,3,5,6-tetramethylphenyl groups (ZnBdu) at the 5-position of the dipyrrinato ligands were designed and synthesized. In ZnBph with the smaller dipyrrinate–arylene and arylene–dimesitylboryl dihedral angles, an intramolecular charge transfer arising from the presence of the vacant p orbital on the boron atom participates in the ππ* excited state in character in contrast to the pure ππ* excited state of ZnBdu. The synergistic ππ*/ILCT excited state was weakly fluorescent, and the fluorescence was enhanced upon binding of fluoride to the boron atom.
Introduction
Triarylborane derivatives exhibit a characteristic absorption/fluorescence owing to an intramolecular charge transfer (CT) transition from the π orbital of aryl group to the vacant p orbital on the boron atom (π(aryl)–p(B) CT) and, therefore, various triarylborane derivatives have been reported.1–11 Triarylboranes are also able to tune the spectroscopic and photophysical properties of a metal complex by being introduced to the periphery of its ligand(s). As a pioneering example reported by Kitamura and coworkers, a CHCl3 solution of [Pt(Bph-tpy)Cl]+ (Bph-tpy = 4′-{4-[bis(2,4,6-trimethylphenyl)boryl]phenyl}-2,2′:6′,2′′-terpyridine) showed intense phosphorescence even at room temperature (emission quantum yield = 0.011) whereas a metal-to-ligand CT (MLCT) excited state of a [Pt(tpy)Cl]+ (tpy = 2,2′:6′,2′′-terpyridine) derivative is typically nonemissive in a solution phase.12 The enhanced emission from [Pt(Bph-tpy)Cl]+ originates in synergistic CT interactions between MLCT in the Pt(tpy)Cl moiety and π(aryl)–p(B) CT in the triarylborane unit. Interestingly, an analogous complex, [Pt(Bdu-tpy)Cl]+ (Bdu-tpy = 4′-{4-[bis(2,4,6-trimethylphenyl)boryl]-2,3,5,6-tetramethylphenyl}-2,2′:6′,2′′-terpyridine), did not exhibit such room-temperature phosphorescence at all. The difference in the phosphorescence ability between the complexes was explained by a disconnection of the CT systems in [Pt(Bdu-tpy)Cl]+ due to the perpendicularly-oriented durylene (2,3,5,6-tetramethylphenylene) group. Thus, the triarylborane has become one of the choices to control photophysical/photochemical properties of a metal complex.13–37 However, the number of the reports focusing on effects of a linker between the triarylborane and metal-complex moieties is still limited.38–40
We recently reported synthesis, spectroscopic and photophysical properties of cyclometalated iridium(III)41 and platinum(II) complexes with an arylborane-appended dipyrrinato ligand(s).42 These complexes showed intense visible absorption (molar absorption coefficient ε ≥ 104 M−1 cm−1 (M = mol dm−3) at ∼490 nm) and visible–near-IR phosphorescence ascribed to the synergistic MLCT/ππ*/π(aryl)–p(B) CT transition. Dipyrrinato metal complexes exhibiting the CT-type excited states are characteristic at this stage since most of the excited states of dipyrrinato metal complexes are ascribed to ππ* transition in the dipyrrinato ligand.43–52 Especially, two stereoisomers of bis(5-{4-[bis(2,4,6-trimethylphenyl)boryl]phenyl}dipyrrinato)platinum(II), PtBph, possessing the square-planar and distorted tetrahedral geometries show different absorption/emission spectra not only in the MLCT/ππ*/π(aryl)–p(B) CT band but also in the ligand-centered (LC) band. Both crystallographic data and theoretical calculations suggest that a dihedral angle between the dipyrrinate and bridging phenylene moieties in the square-planar isomer is larger than that in the distorted tetrahedral one. The results remind us of the earlier Pt-tpy system and importance of further understanding of the ligand itself in an arylborane-appended metal complex.
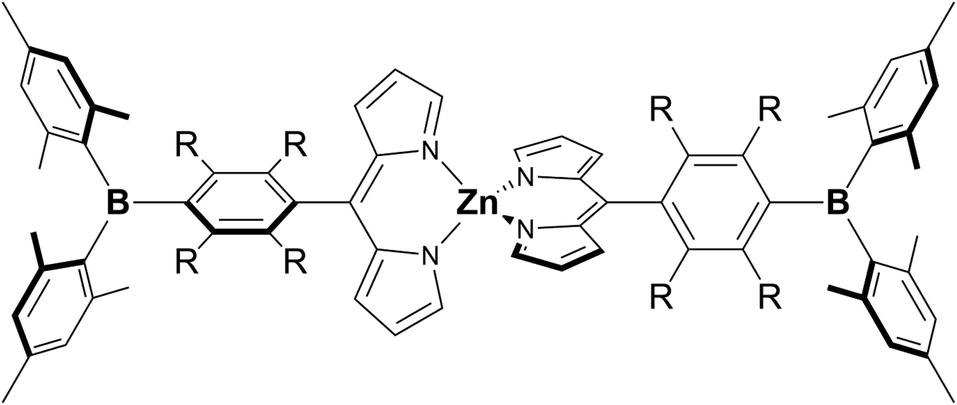
In order to extract the molecular/electronic effects of the ligand itself in an arylborane–dipyrronato metal complex, we chose dipyrrinato zinc(II) complexes. They exhibit intense visible absorption (absorption maximum wavelength λabs ≈ 470–500 nm; ε ≈ (5.0–10) × 104 M−1 cm−1) and fluorescence ascribed to pure LC ππ* excited states owing to the inert d10 nature of the zinc(II) center.53–61 Furthermore, the spectroscopic properties of a dipyrrinato zinc(II) complex are dependent on the substituent group at the 5-position of the dipyrrinato ligand. For example, Lindsey and coworkers revealed that bis[5-(2,4,6-trimethylphenyl)dipyrrinato]zinc(II) (Znmes) showed much higher fluorescence quantum yield (Φf = 0.36) than bis(5-phenyldipyrrinato)zinc(II) (Znph, Φf = 0.01) by effects of both fluorescent and nonfluorescent pathways.62 It has also been reported that the fluorescence from Znmes is quenched in a polar solvent by a thermal deactivation of the excited state via a CT-type state.58,63,64 These results indicate that structures of aryl groups at 5-position of the dipyrronato ligands and the introduction of intramolecular CT interactions are key points to control the spectroscopic/photophysical properties of a bis(dipyrrinato)zinc(II) complex. The π(aryl)–p(B) CT of an arylborane group is, therefore, one of possible candidates to tune the points. We synthesized novel bis(dipyrrinato)zinc(II) derivatives having 4-(dimesitylboryl)phenyl (ZnBph) or (dimesitylboryl)duryl group (mesityl = 2,4,6-trimethylphenyl and duryl = 2,3,5,6-tetramethylphenyl) at 5-position of the dipyrrinato ligands (ZnBdu), whose structures are shown in Chart 1. The bridging phenylene and durylene moieties in the complexes gave significant differences especially in the fluorescence and fluoride-binding properties. The characteristic photophysical properties of the former complex (ZnBph) can be explained by gained intraligand charge transfer (ILCT) character in the excited state and discussed in terms of the molecular geometries obtained by theoretical calculations.
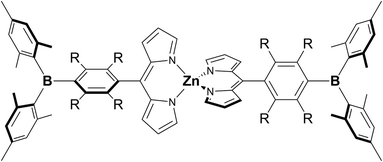 |
| | Chart 1 Chemical structures of ZnBph (R = H) and ZnBdu (R = CH3). | |
Experimental section
Chemicals
All the reagents including organic solvents were commercially available and used without further purification. 5-{4-[Bis(2,4,6-trimethylphenyl)boryl]phenyl}dipyrrin was prepared according to the literature methods.41 4-[Bis(2,4,6-trimethylphenyl)boryl]-2,3,5,6-tetramethylbenzaldehyde (2) was synthesized similarly to the phenyl analogue. All the synthetic reactions and subsequent workup manipulations were carried out under air unless otherwise noted.
Physical measurements and instrumentations
NMR spectra were recorded on a JEOL JNM-AL 400 Fourier-transform NMR spectrometers (400 MHz). The chemical shifts of the 1H, 13C{1H}, 11B{1H} and 19F{1H} NMR spectra determined in CDCl3 were given in ppm relative to tetramethylsilane (0.00 ppm for 1H and 13C{1H}) as an internal standard, or boron trifluoride diethyl-ether complex (0.00 ppm for 11B{1H}) and hexafluorobenzene (−164.9 ppm vs. CFCl3 for 19F{1H}) as external standards. High-resolution fast-atom bombardment mass spectroscopies (HR-FAB-MS) were carried out on a JEOL JMS-700N spectrometer. Elemental analyses (C, H, N) were performed by a Perkin Elmer 2400II elemental analyzer. UV-vis absorption spectra were recorded on a Jasco V-560 spectrophotometer. The corrected emission spectra were obtained by using a Jasco F-6500 spectrofluorometer (excitation wavelength = 365 nm). Fluorescence decay measurements were conducted by using a Hamamatsu Photonics picosecond fluorescence lifetime measurement system C11200 equipped with picosecond light pulser PLP-10 as a 405 nm excitation light source. Emission quantum yields were determined by using a Hamamatsu Photonic Absolute PL Quantum Yield Measurement System C9920-02 (excitation wavelength = 365 nm). For the photophysical measurements, spectrophotometric-grade toluene was used as supplied.
Synthesis of (4-iodo-2,3,5,6-tetramethylphenyl)bis(2,4,6-trimethylphenyl)borane (1)
Synthesis was performed with minor changes in the reported procedures.27 An oven-dried Schlenk tube was evacuated and filled subsequently with an argon gas. 1,4-Diiodo-2,3,5,6-tetramethylbenzene (2.8 g, 7.2 mmol) and dry diethyl ether (20 mL) was added, then, cooled to −78 °C in an acetone/dry-ice bath. n-Butyllithium in n-hexane (1.6 M, 5.2 mL, 8.3 mmol) was added to the reaction mixture at −78 °C. After stirring at −78 °C for 1 h, the reaction mixture became a pale-yellow suspension. Bis(2,4,6-trimethylphenyl)boron fluoride (2.1 g, 7.9 mmol) dissolved in dry diethyl ether (20 mL) was added to the reaction mixture, then, the mixture allowed to warm to room temperature and continuously stirred overnight. The reaction mixture became a yellow suspension. After an addition of HCl(aq) (1 M, 20 mL), the mixture was extracted with diethyl ether (30 mL × 2). The combined organic extract was washed with water (50 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was washed with n-hexane (50 mL) to give pure 1 (2.0 g, 59%) as a colorless solid. 1H NMR (400 MHz, CDCl3) δ: 6.73 (4H, s, m-Ar-H of mesityl), 2.43 (6H, s, 2,6-CH3 of duryl), 2.26 (6H, s, p-CH3 of mesityl), 2.09 (6H, s, 3,5-CH3 of duryl), 1.94 ppm (12H, s, o-CH3 of mesityl).
Synthesis of 4-[bis(2,4,6-trimethylphenyl)boryl]-2,3,5,6-tetramethylbenzaldehyde (2)
An oven-dried Schlenk tube was evacuated and filled subsequently with an argon gas. 1 (2.0 g, 4.9 mmol) and dry tetrahydrofuran (20 mL) was added, then, cooled to −78 °C in an acetone/dry-ice bath. n-Butyllithium in n-hexane (1.6 M, 4.4 mL, 7.0 mmol) was added to the reaction mixture at −78 °C. After stirring at −78 °C for 1 h, the reaction mixture became an orange suspension. Dry N,N-dimethylformamide (2.0 mL) was added to the reaction mixture at −78 °C. After stirring at −78 °C for 1 h, the reaction mixture became a yellow solution. Then, HCl(aq) (1 M, 50 mL) was added and stirred for another 4 h. The reaction mixture was extracted with ethyl acetate (30 mL × 2). The combined organic extract was washed with water (50 mL), dried over anhydrous MgSO4 and concentrated under reduced pressure. The crude product was purified by recrystallization from ethyl acetate to give pure 2 (1.3 g, 65%) as a pale-yellow solid. 1H NMR (400 MHz, CDCl3) δ: 10.7 (1H, s, CHO), 6.75 (4H, s, m-Ar-H of mesityl), 2.31 (6H, s, 2,6-CH3 of duryl), 2.27 (6H, s, p-CH3 of mesityl), 2.02 (6H, s, 3,5-CH3 of duryl), 1.95 ppm (12H, s, o-CH3 of mesityl).
Synthesis of 5-{4-[bis(2,4,6-trimethylphenyl)boryl]-2,3,5,6-tetramethylphenyl}dipyrrin (3)
2 (501.4 mg, 1.22 mmol) was dissolved in neat pyrrole (25 mL) and degassed by a nitrogen-gas bubbling for 30 min. Trifluoroacetic acid (20 μL, 0.23 mmol) was then added, and stirred at room temperature for 10 min. The reaction mixture was diluted with CH2Cl2 (50 mL), washed with NaOH(aq) (1 M, 50 mL) and dried over anhydrous MgSO4. After removing MgSO4 by filtration and evaporation of CH2Cl2 under reduced pressure, the remaining pyrrole was removed by vacuum distillation with heating (40 °C). Then, the product was added to a solution of p-chloranil (500 mg, 2.04 mmol) in CH2Cl2 (50 mL). The solution color changed from yellow to dark yellow-green. The solution was stirred overnight and, then, filtered and evaporated to remove resultant insolubles and CH2Cl2 respectively. The reaction mixture was processed by reprecipitation (CH2Cl2/n-hexane) and silica-gel column chromatography, eluting with CH2Cl2, to give the pure product (125 mg, 20%) as a pale yellow-green solid. 1H NMR (400 MHz, CDCl3) δ: 7.66 (2H, s, 1,9-Ar-H of dipyrrin), 6.78 (4H, s, m-Ar-H of mesityl), 6.39 (2H, dd, J = 1.1, 4.1 Hz, 2,8-Ar-H of dipyrrin), 6.36 (2H, dd, J = 1.6, 3.8 Hz, 3,7-Ar-H of dipyrrin), 2.29 (6H, s, p-CH3 of mesityl), 2.05 (6H, s, 3,5-CH3 of durylene), 2.01 (12H, d, J = 7.9 Hz, o-CH3 of mesityl), 1.94 ppm (6H, s, 2,6-CH3 of durylene). FAB-MS (CH2Cl2) m/z: 525 ([M + H]+).
Synthesis of bis(5-{4-[bis(2,4,6-trimethylphenyl)boryl]phenyl}dipyrrinato)zinc(II) (ZnBph)
To a CH2Cl2 solution (50 mL) of 5-{4-[bis(2,4,6-trimethylphenyl)boryl]phenyl}dipyrrin (100 mg, 0.21 mmol), a CH3OH solution (10 mL) of Zn(OAc)2·2H2O (138 mg, 0.63 mmol) was added and stirred at room temperature. After stirring overnight, the solvent was evaporated under reduced pressure. The crude product was purified by silica-gel column chromatography, eluting with CH2Cl2. Recrystallization from CH2Cl2/methanol afforded pure ZnBph (98.5 mg, 45%) as an orange solid. 1H NMR (400 MHz, CDCl3) δ: 7.60 (4H, d, J = 7.6 Hz, 2,6-Ar-H of phenylene), 7.55 (4H, d, J = 6.9 Hz, 3,5-Ar-H of phenylene), 7.54 (4H, s, 1,9-Ar-H of dipyrrinate), 6.86 (8H, s, m-Ar-H of mesityl), 6.68 (4H, d, J = 3.8 Hz, 2,8-Ar-H of dipyrrinate), 6.38 (4H, d, J = 3.8 Hz, 3,7-Ar-H of dipyrrinate), 2.33 (12H, s, p-CH3 of mesityl), 2.02 ppm (24H, s, o-CH3 of mesityl). 13C{1H} NMR (CDCl3) δ: 149.9, 148.6, 146.4, 142.3, 141.8, 140.9, 140.3, 139.0, 134.8, 132.8, 130.4, 128.3, 117.2, 23.5, 21.3 ppm. 11B{1H} NMR (CDCl3) δ: 75.7 ppm. HR-FAB-MS (CH2Cl2) m/z: calculated for C66H65B2N4Zn+ ([M + H]+), 999.4687; found, 999.4687. Anal. calcd (%) for C66H64B2N4Zn·CH3OH: C, 77.96; H, 6.64; N, 5.43. Found: C, 77.61; H, 6.63; N, 5.31.
Synthesis of bis(5-{4-[bis(2,4,6-trimethylphenyl)boryl]-2,3,5,6-tetramethylphenyl}dipyrrinato)zinc(II) (ZnBdu)
To a CH2Cl2 solution (30 mL) of 3 (125 mg, 0.24 mmol), a CH3OH solution (10 mL) of Zn(OAc)2·2H2O (158 mg, 0.72 mmol) was added and stirred at room temperature. After stirring overnight, the solvent was evaporated under reduced pressure. The crude product was purified by silica-gel column chromatography, eluting with CH2Cl2. Recrystallization from CH2Cl2/methanol afforded pure ZnBdu (80.8 mg, 61%) as a yellow-brown solid. 1H NMR (400 MHz, CDCl3) δ: 7.50 (4H, s, 1,9-Ar-H of dipyrrinate), 6.79 (8H, d, J = 6.8 Hz, m-Ar-H of mesityl), 6.57 (4H, d, J = 4.0 Hz, 2,8-Ar-H of dipyrrinate), 6.38 (4H, d, J = 4.2 Hz, 3,7-Ar-H of dipyrrinate), 2.30 (12H, s, p-CH3 of mesityl), 2.10 (12H, s, 2,6-CH3 of durylene), 2.05 (12H, s, 3,5-CH3 of durylene), 2.02 ppm (24H, s, o-CH3 of o-mesityl). 13C{1H} NMR (CDCl3) δ: 150.3, 149.0, 144.6, 141.0, 140.8, 140.3, 139.4, 138.9, 134.9, 132.1, 131.3, 129.0, 128.8, 117.0, 29.7, 23.3, 22.9, 21.2, 19.9, 17.4 ppm. 11B{1H} NMR (CDCl3) δ: 79.8 ppm. HR-FAB-MS (CH2Cl2) m/z: calculated for C74H81B2N4Zn+ ([M + H]+), 1111.5939; found, 1111.5938. Anal. calcd (%) for C74H80B2N4Zn·CH3OH: C, 78.71; H, 7.40; N, 4.90. Found: C, 78.89; H, 7.35; N, 4.77.
X-ray crystal structure determinations
Diffraction data were collected at −180 °C under a steam of cold N2 gas on a Rigaku RA-Micro7 HFM instrument equipped with a Rigaku Saturn724+ CCD detector by using graphite-monochromated Mo Kα radiation. The frame data were integrated using a Rigaku CrystalClear program package,65 and the data sets were corrected for absorption using a REQAB program. The calculation was performed with a CrystalStructure software package66 except for refinement, which was performed using SHELXL Version 2018/3.67 The structures were solved by direct methods and refined on F2 by the full-matrix least-squares methods. Anisotropic refinement was applied to all non-hydrogen atoms with the exception of the crystal solvents. All hydrogen atoms were put at calculated positions.
Computational methods
Theoretical calculations for the complexes were conducted with Gaussian 16W software (Revision A.03).68 Optimizations of the ground-state geometries of the complexes were performed by using the B3LYP density functional theory (DFT).69,70 The LanL2DZ71–73 and 6-31G(d,p)74 basis sets were used to treat the geometrical structures of the zinc and all other atoms, respectively. Time-dependent DFT (TD-DFT) calculations were then performed to estimate the energies and oscillator strengths f of electronic excitation transitions generating the 50 lowest-energy singlet excited states. All of the calculations were carried out as in toluene by using a polarizable continuum model (PCM).75 Optimized geometries, Kohn–Sham molecular orbitals and natural transition orbitals (isovalue = 0.03 e Å−3) were visualized by GaussView 5.76
Results and discussion
Synthesis and characterization
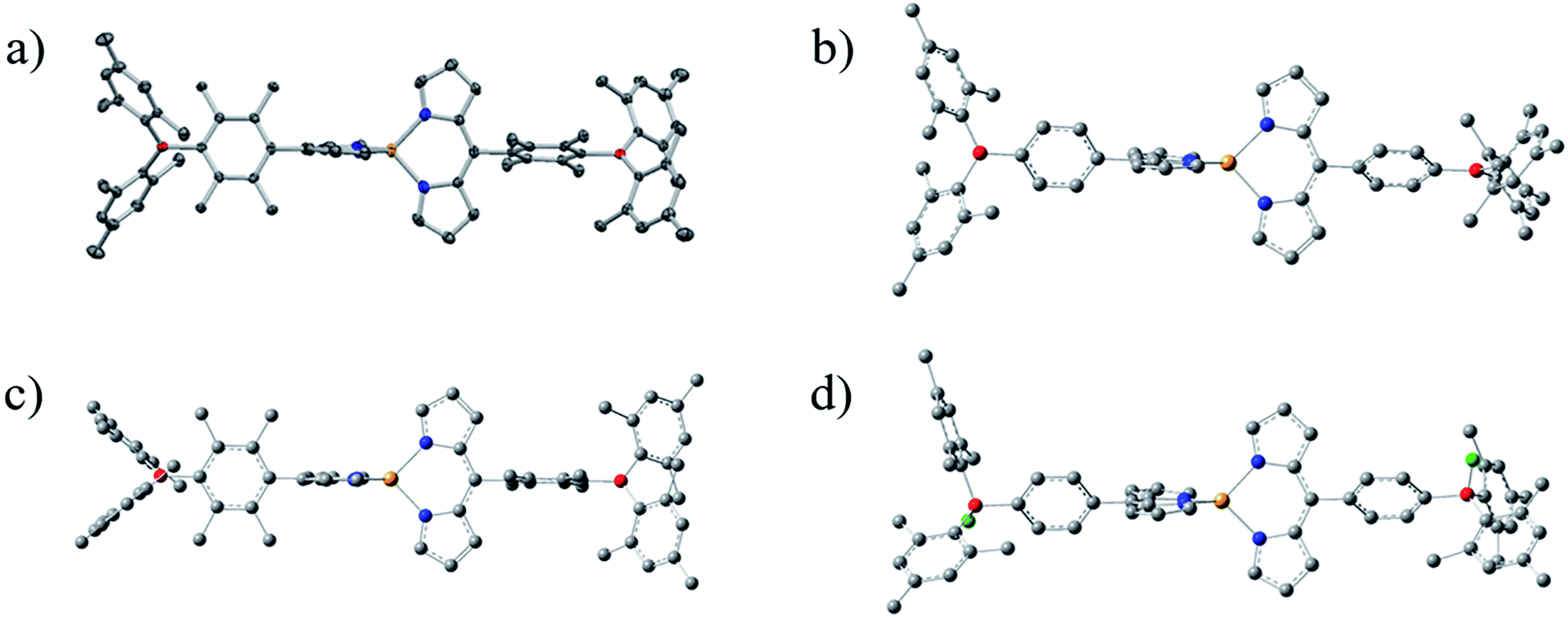
Novel dipyrrinato zinc(II) complexes ZnBph and ZnBdu were successfully synthesized in moderate yields (45% for ZnBph, 61% for ZnBdu) by mixing Zn(OAc)2·2H2O and relevant dipyrrin in a dichloromethane/methanol mixture at room temperature. The complexes were identified by the 1H and 13C{1H} NMR spectroscopies, HR-FAB-MS and elemental analysis. In the 1H NMR spectra of ZnBph and ZnBdu (see Fig. S3 and S4†), four protons at 1- and 9-positions of dipyrrinate moiety were observed as singlet at 7.50–7.54 ppm which was shifted to upfield upon the complexation. Two doublets ascribed to protons at 2,8- and 3,7-positions were observed at 6.38–6.68 ppm. The trends are similar to other bis(dipyrrinato)zinc(II) complexes.54 Single crystals of ZnBdu suitable for X-ray crystallographic analysis were obtained from CH2Cl2/n-pentane. The X-ray structure of ZnBdu is shown in Fig. 1(a), whose crystallographic data is summarized in Table S1.† The zinc center takes a tetrahedral geometry with a dihedral angle for two Zn–N2(dipyrrinato) planes being 90°. The distance between the zinc center and each pyrrolic nitrogen is 1.983 Å, which are similar to those in Znph (1.973–1.988 Å).54 The dihedral angle between a dipyrrinato ligating and bridging durylene moieties was 77.9°. The value is similar to those of the previous iridium(III)41 (71.1°) and platinum(II) complexes (80.7° and 68.7°).42 The boron atoms in the arylborane groups take a planar sp2-like configuration with B–C bond lengths being 1.57–1.58 Å and C–B–C angles being ∼120°. Dihedral angle between the durylene and BC3 plane was 54.1°. These structural characteristics of ZnBdu in the crystalline phase are indicative of the conjugation between the dipyrrinate moiety and boron atom in the ligand. The structures of the complexes were compared on the basis of the DFT calculations because single crystals of ZnBph suitable for X-ray structural analysis were not obtained in spite of many efforts for recrystallization from various conditions. Optimized geometries of ZnBph and ZnBdu are shown in Fig. 1(b) and (c), respectively. The zinc centers in both complexes take tetrahedral geometries, and zinc–nitrogen distances are in the range of 2.059–2.063 Å. The dihedral angle between dipyrrinato ligating moiety and bridging phenylene group (θ1) in ZnBph (64.1°–64.3°) is smaller than that in ZnBdu (89.0–89.2°). Similar tendency was also observed for the tilt angle between arylene group and the BC3 plane in the arylborane moiety (θ2 = 21.7°–25.6° and 57.1°–57.7° for ZnBph and ZnBdu, respectively). On the basis of these angles, (cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ1 × cosθ2) values as measures of the conjugation between the π orbital of the dipyrrinate moiety and the vacant p orbital on the boron atom via the bridging arylene moiety in ZnBph and ZnBdu were calculated to be 40 and 1%, respectively. The smaller dihedral angle in ZnBph than in ZnBdu strongly indicates enhanced electronic interactions between the dipyrrinato zinc(II) complex and the arylborane moieties, and therefore larger contribution of the arylborane moieties to the spectroscopic/photophysical properties of a complex are expected.
θ1 × cosθ2) values as measures of the conjugation between the π orbital of the dipyrrinate moiety and the vacant p orbital on the boron atom via the bridging arylene moiety in ZnBph and ZnBdu were calculated to be 40 and 1%, respectively. The smaller dihedral angle in ZnBph than in ZnBdu strongly indicates enhanced electronic interactions between the dipyrrinato zinc(II) complex and the arylborane moieties, and therefore larger contribution of the arylborane moieties to the spectroscopic/photophysical properties of a complex are expected.
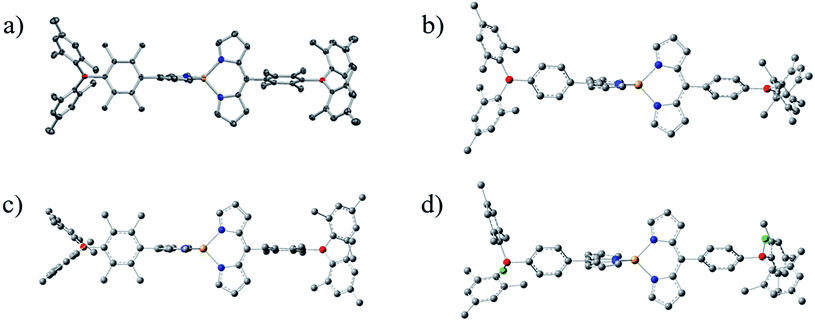 |
| | Fig. 1 Perspective view of crystal structure for ZnBdu ((a) 50% probability ellipsoids) and DFT optimized geometries of ZnBph (b), ZnBdu (c) and ZnBph·2F− (d): carbon (gray), nitrogen (blue), boron (red), zinc (orange) and fluorine (green). Hydrogen atoms are omitted for clarity. | |
Absorption spectra
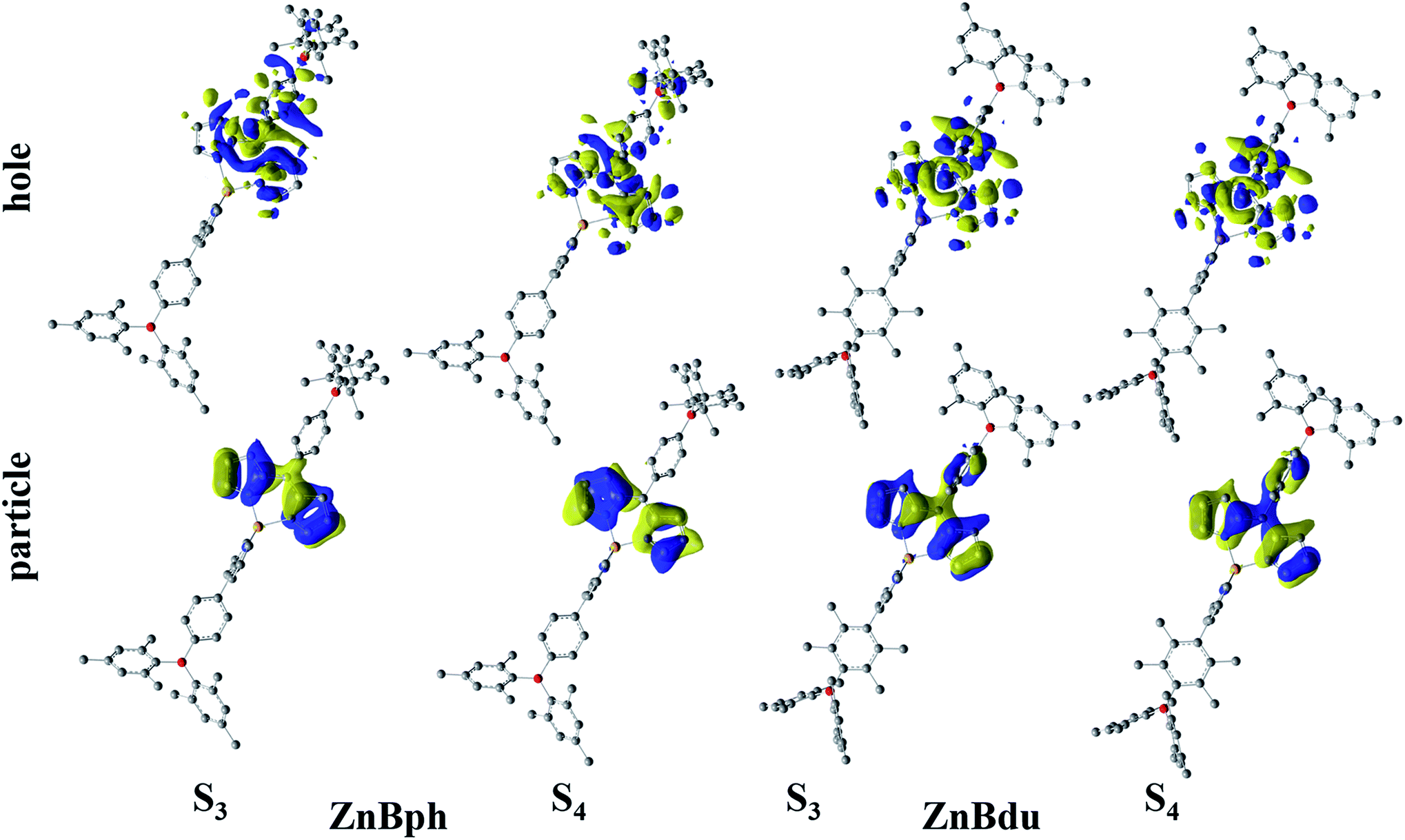
Fig. 2 shows absorption spectra of ZnBph and ZnBdu in toluene at 298 K, and the spectroscopic properties are summarized in Table 1. The complexes exhibited intense/narrow and weak/broad absorption bands at around 487 and 334 nm, respectively. According to the DFT calculations (vide infra), the former band is assigned to the typical ππ* transitions in a dipyrrinato ligand and the latter is ascribed to π(aryl)–p(B) CT transitions in an arylborane moiety, similarly to other arylborane–dipyrrinato metal complexes.41,42 The absorption maximum wavelengths of ZnBph and ZnBdu (λabs = 487 and 486 nm, respectively) are comparable with those of Znph and Znmes (λabs = 485 nm and 487 nm, respectively) without any arylborane substituents.62 Interestingly, the molar absorption coefficients (ε) at the maximum wavelength (λabs) of the low-energy band of the complexes (9.2 × 104 M−1 cm−1 for ZnBph and 1.32 × 105 M−1 cm−1 for ZnBdu) were larger than those of the arylborane–dipyrrinato iridium(III) and platinum(II) complexes (ε = 7.4 × 104 and 2.2 × 104 M−1 cm−1, respectively)41,42 whose ππ* transitions synergistically interact with MLCT and/or π(aryl)–p(B) CT ones. Such differences in the ε values indicates that an introduction of the charge-transfer character to the ππ* transition in a dipyrrinato–metal complex decreases the relevant oscillator strength. Thus, smaller ε value of ZnBph suggests the existence of the π interactions throughout the ligand presumably owing to the smaller dihedral angles between dipyrrinate–phenylene and phenylene–dimesitylboryl moieties. These discussions were theoretically supported by TD-DFT calculations as summarized in Tables S2–S5.† For both complexes, intense absorption bands at ∼487 nm appeared as electronic transitions generating third (S3) and forth excited states (S4). Fig. 3 shows natural transition orbitals (NTOs) for the S3 and S4 states of the complexes. Both S3 and S4 states of ZnBph originate in electronic transitions from the dipyrrinate moiety to the whole ligand, gaining the π(aryl)–p(B) CT character, whereas those of ZnBdu are assignable to the pure ππ* transitions in a 5-duryldipyrrinate moiety.
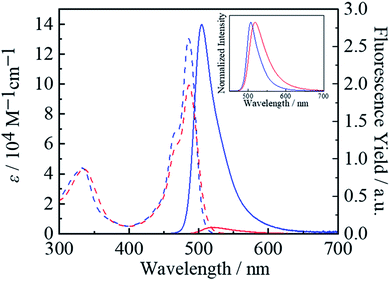 |
| | Fig. 2 UV-vis absorption (broken lines) and fluorescence spectra (solid lines) of ZnBph (red) and ZnBdu (blue) in toluene at 298 K. The integrations of the fluorescence spectra in a wavenumber scale correspond relatively to the fluorescence quantum yields of the complexes. Inset: normalized fluorescence spectra. | |
Table 1 Spectroscopic and photophysical properties of the dipyrrinato zinc(II) complexes in toluene at 298 K
| Complex |
λabs/nm (ε/104 M−1 cm−1) |
λf/nm |
Φf |
τf/ns |
kra/s−1 |
knra/s−1 |
| Calculated by the equation, Φf = kr/(kr + knr) = krτf. Data in toluene compiled from ref. 62. Data in CH2Cl2 compiled from ref. 54. |
| ZnBph |
334 (4.3), 487 (09.2) |
517 |
0.01 |
1.3 |
7.7 × 106 |
7.6 × 108 |
| ZnBdu |
333 (4.4), 486 (13.2) |
504 |
0.34 |
2.7 |
1.3 × 108 |
2.4 × 108 |
| ZnBph·2F− |
383 (1.6), 481 (09.4) |
503 |
0.02 |
0.2 |
1 × 108 |
5 × 109 |
| Znphb |
485 |
500 |
0.006 |
0.09 |
7 × 107 |
1 × 1010 |
| Znphc |
322 (1.4), 482 (11.5) |
500 |
|
|
|
|
| Znmesb |
487 |
501 |
0.36 |
2.7 |
1.3 × 108 |
2.4 × 108 |
| Znmesc |
345, 485 |
501 |
|
|
|
|
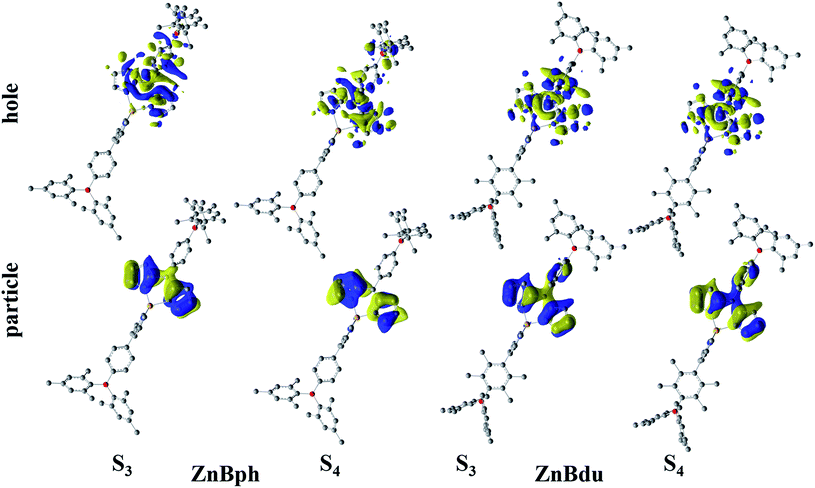 |
| | Fig. 3 NTOs for S3 and S4 states of the complexes in toluene. Hydrogen atoms are omitted for clarity. | |
Fluorescence spectra and photophysical properties
As shown in Fig. 2, the fluorescence from ZnBph (Φf = 0.01 in toluene at 298 K) was significantly weak compared with that from ZnBdu (Φf = 0.34). Furthermore, the fluorescence spectrum of ZnBph (maximum wavelength λf = 517 nm) was broadened and shifted to lower-energy compared with those of ZnBdu (504 nm), Znph (501 nm)62 and Znmes (500 nm)62 see Table 1. The spectroscopic data of ZnBph indicate a partial contribution of a CT character arising from the low-energy p(B) to the fluorescent excited state and energetic stabilization of the excited state by the solvation. In practice, the radiative rate constant (kr) of ZnBph (7.7 × 106 s−1, see Table 1) was 17-times smaller than that of ZnBdu (1.3 × 108 s−1) owing to a decreased wavefunction overlap between the excited and ground states. Furthermore, the CT character in the excited state accelerated thermal deactivation to the ground state as the nonradiative decay rate constant (knr) of ZnBph (7.6 × 108 s−1) was three-times larger than that of ZnBdu (knr = 2.4 × 108 s−1). It should be noted that the kr and knr values of ZnBdu are comparable to those of Znmes (kr = 1.3 × 108 s−1 and knr = 2.4 × 108 s−1).54 Thus, the strong fluorescence from ZnBdu would originate in the pure ππ* excited state and suppressed nonradiative decay processes owing to the presence of the bulky durylene bridging units. On the other hand, the kr and knr values of ZnBph are significantly smaller than those of Znph (kr = 7 × 107 s−1 and knr = 1 × 1010 s−1),54 suggesting the existence of CT interactions in the excited state. As results, the fluorescence from ZnBph was well characterized by the ππ*/ILCT excited state, and the participation of the ILCT character in the excited state of a complex was revealed by varying the extent of π-conjugation between the dipyrrinate and arylborane moieties.
Spectroscopic responses to fluoride
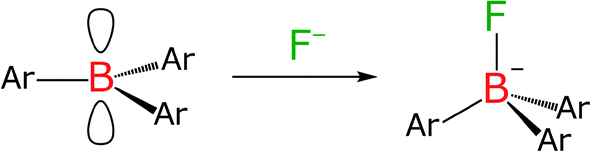
Since the electron-deficient boron atom in an arylborane derivative can bind with a small Lewis base such as fluoride (Chart 2) with a binding constant being ∼106 M−1,77,78 we carried out the fluoride-addition experiments for the complexes. Fig. 4 shows absorption and fluorescence spectra of the complexes in the absence and presence of tetra-n-butylammonium fluoride (TBAF) in toluene. The absorbances of each spectrum were divided by the total concentration of the complex (c0) and optical path length (l) so that the vertical axis corresponds to the molar absorption coefficient. Upon an addition of TBAF to ZnBph, the π(aryl)–p(B) CT absorption band at around 334 nm disappeared, and a broad absorption band at around 383 nm appeared with an isosbestic point at 360 nm. In addition, the fluoride binding shortened the maximum wavelength of the lowest-energy ππ*/ILCT absorption band of ZnBph from 487 nm to 481 nm, similarly to that observed for the relevant cyclometalated iridium(III) complex.41 Complete disappearance of the π(aryl)–p(B) CT band and the complicated spectral changes in the absorption band at ∼480 nm indicate successive bindings of two fluoride, affording a 1:1 adduct, followed by a 1:2 adduct, ZnBph·2F−. The spectroscopic changes arising from the binding of fluoride to the boron atom were strongly evidenced by 1H, 11B{1H} and 19F{1H} NMR measurements as shown in Fig. S8, S10 and S13,† respectively. The 11B NMR signal of ZnBph was drastically shifted from 75.7 ppm to 5.5 ppm upon the addition of fluoride as TBAF. The broad signal was also observed at −174.41 ppm for the 19F NMR spectrum of ZnBph in the presence of TBAF (4.0 eq.) owing to the coupling to 10B (I = 3) and 11B (I = 3/2), indicating the formation of B–F bond with ZnBph. In addition, the absence of the signal at −84.04 ppm (corresponding to the signal of CDClF2 generated by the reaction of fluoride with solvent molecule CDCl3)79 in the 19F NMR spectrum of ZnBph suggests that fluoride quickly binds to the boron atom before proceeding the exchange reaction of fluoride with chloride in the solvent molecule. Consequently, the binding of fluoride to the boron atom in ZnBph increased electron density around the boron atom.
 |
| | Chart 2 Structural change of an arylborane compound upon the binding of fluoride. | |
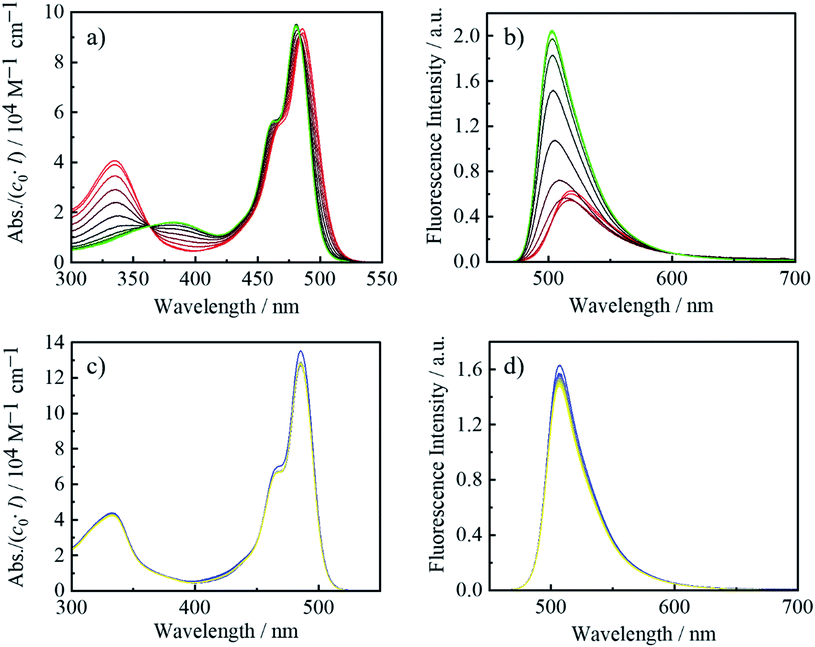 |
| | Fig. 4 UV-vis absorption and fluorescence spectral changes upon an addition of TBAF (0–4.0 equiv.) in toluene: ZnBph ((a and b) red to green) and ZnBdu ((c and d) blue to yellow). | |
The structure of ZnBph·2F− was theoretically investigated by the DFT calculation and the optimized geometry is shown in Fig. 1(d). Each boron atom possesses a tetrahedral geometry, and the dihedral angles between a dipyrrinato ligating moiety and bridging phenylene group (θ1) were reduced to be 56.8°–58.7°. The structural and electronic changes increased the transition energy of the lowest-energy absorption band through efficient electron donation from the fluorinated arylborane group to the dipyrrinato moiety, leading to the decrease of electron density on the dipyrrinato moiety and therefore the downfield-shift of proton signals in the pyrrole rings (see Fig. S15† for HOMO and HOMO−1). The fluorescence from ZnBph was shifted to higher-energy with a slight decrease in intensity and then enhanced largely upon a continuous addition of TBAF as shown in Fig. 4(b). The maximum wavelength (λf = 503 nm) and band shape in the presence of ≥3.0 equivalence of fluoride were almost identical to those of ZnBdu, and the kr value of ZnBph·2F− (1 × 108 s−1) was also similar to that of ZnBdu. Owing to these fluorescence characteristics, it can be expected that the fluorescent excited state of ZnBph·2F− possesses the pure ππ* character. Significantly smaller Φf (0.02) of ZnBph·2F− than that of ZnBdu can be explained by enhanced thermal deactivation (knr = 5 × 109 s−1) via a rotation of the phenylene moiety as reported for Znph and Znmes (knr = 1.1 × 1010 and 3.2 × 108 s−1 in toluene, respectively).54 On the other hand, there was no experimental evidence of the fluoride binding to ZnBdu in the absorption, fluorescence and 1H, 11B{1H} and 17F{1H} NMR spectra as shown in Fig. 4(c) and (d), S9, S11 and S14† presumably due to the steric hindrance of the durylene moieties. Thus, fluoride binding affinity of an arylborane–dipyrrinato zinc(II) complex was controllable by the bridging arylene moiety and, upon the fluoride binding, the excited-state electronic structure of ZnBph was switched from ππ*/ILCT to pure ππ*.
Conclusions
The bridging arylene moieties in novel bis(dipyrrinato)zinc(II) derivatives having the arylborane groups at 5-position of the dipyrrinato ligands had significant impacts on the absorption/fluorescence spectra and fluoride-binding affinity of the complex. The theoretical calculations suggest that ZnBph with the phenylene linkers possesses smaller dihedral angles between dipyrrinate–phenylene and phenylene–dimesitylboryl moieties than the relevant values of ZnBdu with the durylene linkers. The smaller dihedral angles in ZnBph afford the π-conjugation in the entire of the ligand and, therefore, the electron-withdrawing arylborane groups participate in the electronic structure of the complex. As a result, the excited state of ZnBph was best characterized by the synergistic ππ*/ILCT, whereas that of ZnBdu was the pure ππ*. ZnBph could bind with fluoride, and the excited state was switched from ππ*/ILCT to pure ππ* upon the fluoride binding owing to the disappearance of the electron-withdrawing ability of the dimesitylboryl moieties. Thus, we revealed the importance of the molecular design including the linker structure in metal complexes with an arylborane group(s). Tuning of the extent of the CT character in an excited-state metal complex will be an important factor to control spectroscopic and photophysical properties of metal complexes.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This research was partially funded by the Sasakawa Scientific Research Grant from the Japan Science Society. We are grateful to J. Nagaoka for the technical assistance and the X-ray measurements.
References
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS
 .
. - L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC .
- S. Y. Li, Z. B. Sun and C. H. Zhao, Inorg. Chem., 2017, 56, 8705–8717 CrossRef CAS .
- Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS .
- R. Misra, T. Jadhav, B. Dhokale and S. M. Mobin, Dalton Trans., 2015, 44, 16052–16060 RSC .
- S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2000, 122, 6335–6336 CrossRef CAS .
- S. Yamaguchi, T. Shirasaka and K. Tamao, Org. Lett., 2000, 2, 4129–4132 CrossRef CAS .
- A. Ito, T. Hiokawa, E. Sakuda and N. Kitamura, Chem. Lett., 2010, 40, 34–36 CrossRef .
- N. Kitamura and E. Sakuda, J. Phys. Chem. A, 2005, 109, 7429–7434 CrossRef CAS .
- N. Kitamura, E. Sakuda, T. Yoshizawa, T. Iimori and N. Ohta, J. Phys. Chem. A, 2005, 109, 7435–7441 CrossRef CAS .
- E. Sakuda, K. Tsuge, Y. Sasaki and N. Kitamura, J. Phys. Chem. B, 2005, 109, 22326–22331 CrossRef CAS .
- E. Sakuda, A. Funahashi and N. Kitamura, Inorg. Chem., 2006, 45, 10670–10677 CrossRef CAS .
- Y. Sun, N. Ross, S. Bin Zhao, K. Huszarik, W. L. Jia, R. Y. Wang, D. Macartney and S. Wang, J. Am. Chem. Soc., 2007, 129, 7510–7511 CrossRef CAS .
- S. Bin Zhao, T. McCormick and S. Wang, Inorg. Chem., 2007, 46, 10965–10967 CrossRef .
- G. Zhou, C. L. Ho, W. Y. Wong, Q. Wang, D. Ma, L. Wang, Z. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499–511 CrossRef CAS .
- N. You and S. Y. Park, Adv. Mater., 2008, 20, 3820–3826 CrossRef .
- Q. Zhao, F. Li, S. Liu, M. Yu, Z. Liu, T. Yi and C. Huang, Inorg. Chem., 2008, 47, 9256–9264 CrossRef CAS .
- Y. L. Rao and S. Wang, Inorg. Chem., 2009, 48, 7698–7713 CrossRef CAS .
- Z. M. Hudson, S. Bin Zhao, R. Y. Wang and S. Wang, Chem.–Eur. J., 2009, 15, 6131–6137 CrossRef CAS .
- S. T. Lam, N. Zhu and V. W. W. Yam, Inorg. Chem., 2009, 48, 9664–9670 CrossRef CAS .
- W. J. Xu, S. J. Liu, X. Y. Zhao, S. Sun, S. Cheng, T. C. Ma, H. Bin Sun, Q. Zhao and W. Huang, Chem.–Eur. J., 2010, 16, 7125–7133 CrossRef CAS .
- C. R. Wade and F. P. Gabbai, Inorg. Chem., 2010, 49, 714–720 CrossRef CAS .
- E. Sakuda, Y. Ando, A. Ito and N. Kitamura, Inorg. Chem., 2011, 50, 1603–1613 CrossRef CAS .
- A. Ito, T. Hiokawa, E. Sakuda and N. Kitamura, Chem. Lett., 2010, 40, 34–36 CrossRef .
- Z. M. Hudson and S. Wang, Organometallics, 2011, 30, 4695–4701 CrossRef CAS .
- Y. Sun, Z. M. Hudson, Y. Rao and S. Wang, Inorg. Chem., 2011, 50, 3373–3378 CrossRef CAS .
- A. Ito, Y. Kang, S. Saito, E. Sakuda and N. Kitamura, Inorg. Chem., 2012, 51, 7722–7732 CrossRef CAS .
- Z. M. Hudson, C. Sun, M. G. Helander, Y. L. Chang, Z. H. Lu and S. Wang, J. Am. Chem. Soc., 2012, 134, 13930–13933 CrossRef CAS .
- R. S. Vadavi, H. Kim, K. M. Lee, T. Kim, J. Lee, Y. S. Lee and M. H. Lee, Organometallics, 2012, 31, 31–34 CrossRef CAS .
- S. B. Ko, J. S. Lu, Y. Kang and S. Wang, Organometallics, 2013, 32, 599–608 CrossRef CAS .
- N. Wang, S. B. Ko, J. S. Lu, L. D. Chen and S. Wang, Chem.–Eur. J., 2013, 19, 5314–5323 CrossRef CAS .
- X. Wang, Y. L. Chang, J. S. Lu, T. Zhang, Z. H. Lu and S. Wang, Adv. Funct. Mater., 2014, 24, 1911–1927 CrossRef CAS .
- S. Sharma, H. Kim, Y. H. Lee, T. Kim, Y. S. Lee and M. H. Lee, Inorg. Chem., 2014, 53, 8672–8680 CrossRef CAS .
- Y. H. Lee, N. Van Nghia, M. J. Go, J. Lee, S. U. Lee and M. H. Lee, Organometallics, 2014, 33, 753–762 CrossRef CAS .
- A. Nakagawa, E. Sakuda, A. Ito and N. Kitamura, Inorg. Chem., 2015, 54, 10287–10295 CrossRef CAS .
- A. Nakagawa, A. Ito, E. Sakuda, S. Fujii and N. Kitamura, Eur. J. Inorg. Chem., 2017, 3794–3798 CrossRef CAS .
- N. Wang, M. Hu, S. K. Mellerup, X. Wang, F. Sauriol, T. Peng and S. Wang, Inorg. Chem., 2017, 56, 12783–12794 CrossRef CAS .
- L. F. Smith, B. A. Blight, H. J. Park and S. Wang, Inorg. Chem., 2014, 53, 8036–8044 CrossRef CAS .
- H. J. Park, S. B. Ko, I. W. Wyman and S. Wang, Inorg. Chem., 2014, 53, 9751–9760 CrossRef CAS .
- G. Rajendra Kumar and P. Thilagar, Inorg. Chem., 2016, 55, 12220–12229 CrossRef CAS .
- K. Takaki, E. Sakuda, A. Ito, S. Horiuchi, Y. Arikawa and K. Umakoshi, Inorg. Chem., 2019, 58, 14542–14550 CrossRef CAS .
- K. Takaki, E. Sakuda, A. Ito, S. Horiuchi, Y. Arikawa and K. Umakoshi, Chem. Lett., 2020, 49, 905–908 CrossRef CAS .
- T. E. Wood and A. Thompson, Chem. Rev., 2007, 107, 1831–1861 CrossRef CAS .
- Y. Wang, Z. Xue, Y. Dong and W. Zhu, Polyhedron, 2015, 102, 578–582 CrossRef CAS .
- K. Servaty, E. Cauët, F. Thomas, J. Lambermont, P. Gerbaux, J. De Winter, M. Ovaere, L. Volker, N. Vaeck, L. Van Meervelt, W. Dehaen, C. Moucheron and A. Kirsch-De Mesmaeker, Dalton Trans., 2013, 42, 14188–14199 RSC .
- T. M. McLean, D. M. Cleland, S. J. Lind, K. C. Gordon, S. G. Telfer and M. R. Waterland, Chem.–Asian J., 2010, 5, 2036–2046 CrossRef CAS .
- E. Zeini Jahromi and J. Gailer, Dalton Trans., 2010, 39, 329–336 RSC .
- S. Riese, M. Holzapfel, A. Schmiedel, I. Gert, D. Schmidt, F. Würthner and C. Lambert, Inorg. Chem., 2018, 57, 12480–12488 CrossRef CAS .
- T. M. McLean, J. L. Moody, M. R. Waterland and S. G. Telfer, Inorg. Chem., 2012, 51, 446–455 CrossRef CAS .
- C. Bronner, M. Veiga, A. Guenet, L. De Cola, M. W. Hosseini, C. A. Strassert and S. A. Baudron, Chem.–Eur. J., 2012, 18, 4041–4050 CrossRef CAS .
- C. Bronner, S. A. Baudron, M. W. Hosseini, C. A. Strassert, A. Guenet and L. De Cola, Dalton Trans., 2010, 39, 180–184 RSC .
- K. Hanson, A. Tamayo, V. V. Diev, M. T. Whited, P. I. Djurovich and M. E. Thompson, Inorg. Chem., 2010, 49, 6077–6084 CrossRef CAS .
- M. A. Filatov, A. Y. Lebedev, S. N. Mukhin, S. A. Vinogradov and A. V. Cheprakov, J. Am. Chem. Soc., 2010, 132, 9552–9554 CrossRef CAS .
- L. Yu, K. Muthukumaran, I. V. Sazanovich, C. Kirmaier, E. Hindin, J. R. Diers, P. D. Boyle, D. F. Bocian, D. Holten and J. S. Lindsey, Inorg. Chem., 2003, 42, 6629–6647 CrossRef CAS .
- M. Asaoka, Y. Kitagawa, R. Teramoto, K. Miyagi, Y. Natori, R. Sakamoto, H. Nishihara and M. Nakano, Polyhedron, 2017, 136, 113–116 CrossRef CAS .
- R. Matsuoka, R. Toyoda, R. Sakamoto, M. Tsuchiya, K. Hoshiko, T. Nagayama, Y. Nonoguchi, K. Sugimoto, E. Nishibori, T. Kawai and H. Nishihara, Chem. Sci., 2015, 6, 2853–2858 RSC .
- M. Tsuchiya, R. Sakamoto, M. Shimada, Y. Yamanoi, Y. Hattori, K. Sugimoto, E. Nishibori and H. Nishihara, Inorg. Chem., 2016, 55, 5732–5734 CrossRef CAS .
- S. Kusaka, R. Sakamoto, Y. Kitagawa, M. Okumura and H. Nishihara, Chem.–Asian J., 2012, 7, 907–910 CrossRef CAS .
- R. Sakamoto, T. Iwashima, J. F. Kögel, S. Kusaka, M. Tsuchiya, Y. Kitagawa and H. Nishihara, J. Am. Chem. Soc., 2016, 138, 5666–5677 CrossRef CAS .
- D. Tungulin, J. Leier, A. B. Carter, A. K. Powell, R. Q. Albuquerque, A. N. Unterreiner and C. Bizzarri, Chem.–Eur. J., 2019, 25, 3816–3827 CrossRef CAS .
- Q. Jiang, N. Desbois, S. Wang and C. P. Gros, J. Porphyrins Phthalocyanines, 2020, 24, 646–661 CrossRef CAS .
- I. V. Sazanovich, C. Kirmaier, E. Hindin, L. Yu, D. F. Bocian, J. S. Lindsey and D. Holten, J. Am. Chem. Soc., 2004, 126, 2664–2665 CrossRef CAS .
- C. Trinh, K. Kirlikovali, S. Das, M. E. Ener, H. B. Gray, P. Djurovich, S. E. Bradforth and M. E. Thompson, J. Phys. Chem. C, 2014, 118, 21834–21845 CrossRef CAS .
- N. Z. Alqahtani, T. G. Blevins and C. E. McCusker, J. Phys. Chem. A, 2019, 123, 10011–10018 CrossRef CAS .
- CrystalClear: Data Collection and Processing Software, Rigaku Corporation, Tokyo, Japan, 1998–2015 Search PubMed .
- CrystalStructure 4.3: Crystal Structure Analysis Package, Rigaku Corporation, Tokyo, Japan, 2000–2019 Search PubMed .
- SHELXL Version 2018/3: G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed .
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS .
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS .
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS .
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS .
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS .
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS .
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef .
- R. Dennington, T. Keith and J. Millam, Gauss View, Version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed .
- Y. Sun and S. Wang, Inorg. Chem., 2009, 48, 3755–3767 CrossRef CAS .
- Y. Sun and S. Wang, Inorg. Chem., 2010, 49, 4394–4404 CrossRef CAS .
- K. O. Christe and W. W. Wilson, J. Fluorine Chem., 1990, 47, 117–120 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C{1H} NMR spectra, crystallographic data of ZnBdu, detailed results of theoretical calculations, fluorescence decay profiles and 1H NMR spectra in the absence and presence of TBAF. CCDC 2035525. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09029h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Akitaka Ito
*a,
Akitaka Ito